
Synergistic catalysis between atomically dispersed Fe and a pyrrolic-N-C framework for CO2 electroreduction†
Chaochen
Xu
 a,
Anthony
Vasileff
a,
Dan
Wang
b,
Bo
Jin
a,
Yao
Zheng
*a and
Shi-Zhang
Qiao
*a
a,
Anthony
Vasileff
a,
Dan
Wang
b,
Bo
Jin
a,
Yao
Zheng
*a and
Shi-Zhang
Qiao
*a
aCentre for Materials in Energy and Catalysis, School of Chemical Engineering and Advanced Materials, The University of Adelaide, Adelaide, SA 5005, Australia. E-mail: s.qiao@adelaide.edu.au; yao.zheng01@adelaide.edu.au
bInstitute of Process Engineering, Chinese Academy of Sciences, Beijing, 100190, China
First published on 16th July 2019
Abstract
Atomically dispersed Fe immobilized within N-doped carbon nanosheets (Fe-SA) is successfully synthesized. The optimal Fe-SA catalyst achieves a high faradaic efficiency of ca. 90% for CO2 electroreduction toward CO at a low overpotential of 0.47 V. A series of controlled tests show that there is a synergistic effect between the Fe centers and the pyrrolic-N-C framework which facilitates catalytic activity. Specifically, pyrrolic-N-C sites with high local electron density increase the initial CO2 adsorption while positively charged Fe enhances the water dissociation to provide a proton source for further CO2 reduction.
New conceptsSingle atom metal catalysts within a framework support present as an ideal platform for multi-step electron transfer processes like the electrocatalytic CO2 reduction reaction (CRR) because of the possibility for synergistic effects between two electroactive sites. Currently, the contribution of the metal atoms as active sites has been well-studied while the role of the supporting substrate in the catalytic process is scarcely reported. Herein, using atomically dispersed Fe immobilized within N-doped carbon nanosheets as a model, we carefully investigated the synergistic effect between the metal atom and its anchoring framework sites for efficient CRR. A series of controlled tests show that there is a synergistic effect between the Fe centers and the pyrrolic-N-C framework which facilitates catalytic activity. |
The electrochemical CO2 reduction reaction (CRR) is a promising technology for CO2 conversion toward carbonaceous products. Desirable catalyst properties for the CRR are high activity, selectivity, and robustness.1–5 However, a major hurdle for current CRR electrocatalysts is the inability to efficiently cleave the C
![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) O bond in order to form specific intermediates for further multi-step proton-assisted electron-transfer processes.6–10 This leads to a high overpotential and/or a low selectivity toward a specific product, consequently impeding the large scale applications of this CO2 conversion technology.
O bond in order to form specific intermediates for further multi-step proton-assisted electron-transfer processes.6–10 This leads to a high overpotential and/or a low selectivity toward a specific product, consequently impeding the large scale applications of this CO2 conversion technology.
Recently, single atom catalysts (SACs) have been explored by material science fields,2,11–14 offering new opportunities for electrocatalysis. Ideal SACs refer to metals that are uniformly distributed within a framework and chemically coordinated with specific species to form a new functional group. Atom-level utilization of metal catalysts provides a unique property that may trigger reactions through different pathways, resulting in enhanced performances. For instance, atomically dispersed 3d-transition metal catalysts trapped within N-doped carbon nanosheets have recently been highlighted as promising candidates for electrocatalytic applications, such as hydrogen- and oxygen-involving reactions and the CRR.11,15–18
A typical structure of SACs involves metal atoms that are anchored to the N sites of a N-containing carbon (N-C) framework.11,12,19 The interaction between the metal and N-C, denoted as a metal–N-C domain site, has been widely reported as the possible active site for facilitating the selective reduction of CO2 to CO.20–28 More interestingly, for a multi-step reaction, e.g., CRR, this metal–non-metal interaction provides two active sites (i.e., one metal site; one N-C site). In principle, they can work synergistically to break the inherent adsorption energy scaling relation of different reaction intermediates, leading to an enhanced selectivity towards a specific product.4,24,29,30 However, most research has focused on the role of the metal center itself or the metal–N-C domain; the function of the N-C framework and resultant synergistic effect is poorly known. Additionally, different from carbon-based metal-free materials, the effect of various N species (e.g. pyridinic-N, pyrrolic-N, and graphitic-N) in metal–N-C sites is scarcely reported in SAC studies. For example, most of the experimental and computational studies claim that the metal atoms are typically trapped at pyridinic-N-C sites.2,11,12,31–33 On the other hand, some metal complexes with similar π conjugated systems with metal–pyrrolic-N-C sites, such as heme, chlorophyll, and metal phthalocyanines, commonly exist and possess good stability. In fact, pyrrolic- and pyridinic-N-C have distinct electron structures. Compared to pyridinic-N, the unhybridized pz orbital in pyrrolic-N has a pair of electrons that contributes to increasing the electron density on the N-C sites.
Herein, to utilize this discussed synergistic effect and clarify the role of pyridinic/pyrrolic-N-C in catalysis, we successfully design and synthesize atomically dispersed iron catalysts immobilized within N-doped carbon nanosheets (Fe-SA). Iron nanoparticles on N-doped carbon nanosheets (Fe-NP) and N-doped carbon nanosheets (NS) are also prepared via a similar synthetic method and serve as controls. The optimal Fe-SA-900 sample (annealed at 900 °C) shows a high faradaic efficiency (FE) of ca. 90% under a low potential of −0.58 V vs. RHE (reversible hydrogen electrode) for CO2 reduction toward CO. This superior performance is attributed to the synergistic effect in the Fe–pyrrolic-N-C framework. Specifically, the adjacent pyrrolic-N-C sites serve as one active site which can activate CO2 molecules while the coordinated Fe centers serve as another active site which can generate protons (via water dissociation) for further CO2 reduction.
Fe-SA-900 was prepared by introducing Fe into a N-C framework via a facile thermal pyrolysis process (see the ESI† for Experimental details). Fe-NP-900 and NS-900 were prepared using the same method but with 10-fold Fe salt and no Fe salt added, respectively. Scanning electron microscopy (SEM) and high-resolution transmission electron microscopy (HRTEM) clearly confirm that ultrathin carbon nanosheets in Fe-SA-900 were successfully synthesized, and no visible particle phase was observed (Fig. S1a and b, ESI†). For further confirmation, Raman spectra display distinct D-band (1350 cm−1) and G-band (1580 cm−1) signals which correspond to the vibrational mode of sp2 and disordered sp2 carbon atoms within the nanosheet, respectively (Fig. S1c, ESI†).34 To identify atomic-level dispersed Fe, aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) imaging was applied. As seen in Fig. 1a, bright spots distributed in the carbon substrate are distinguishable from the carbon substrate. This indicates the presence of Fe atoms owing to the Z-contrast sensitivity for heavy elements. In contrast, Fe aggregated to form nanoparticles in Fe-NP-900 while the absence of Fe was observed for NS-900 (Fig. 1b and c). Energy-dispersive X-ray spectroscopy (EDS) results further confirmed the presence of Fe in Fe-SA-900 and Fe-NP-900 and its absence in NS-900 (Fig. S2, ESI†). X-ray diffraction (XRD) patterns exhibit a broad peak from 20° to 30° (intense crystalline peaks at 2θ = 24.5°) for each sample in Fig. 1d. This represents the (002) plane of hexagonal carbon as evidenced by the transmission electron microscopy (TEM) image and selected area electron diffraction (SAED) pattern (Fig. S1d, ESI†).35 The XRD pattern of Fe-NP-900 shows peaks related to Fe3C (PDF #35-0772) as Fe aggregation likely formed Fe carbide nanoparticles. No such peaks were observed for Fe-SA-900, indicating that Fe is atomically dispersed rather than aggregated.
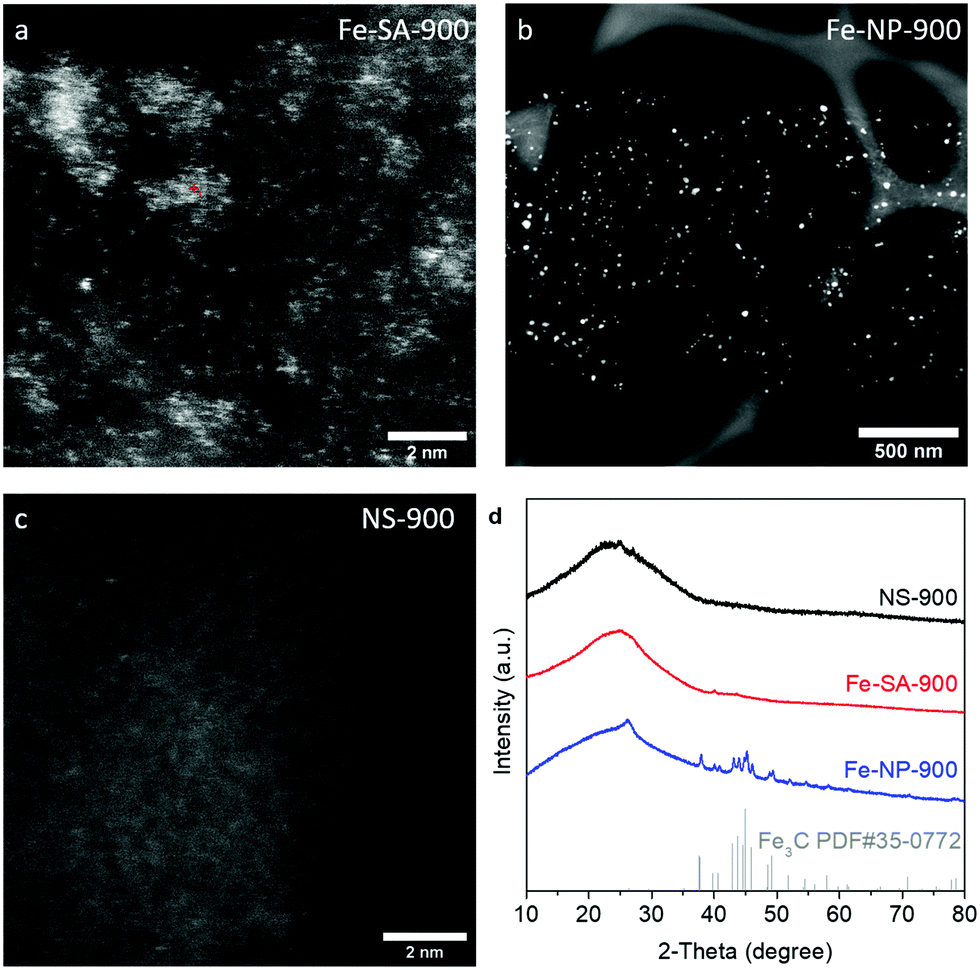 | ||
| Fig. 1 HAADF-STEM images of (a) Fe-SA-900, (b) Fe-NP-900, and (c) NS-900. (d) XRD patterns of NS-900, Fe-SA-900, and Fe-NP-900. | ||
X-ray photoelectron spectroscopy (XPS) survey spectra display distinct signals of C 1s, N 1s, and O 1s from all samples (Fig. 2a). The element quantification results show that Fe-SA-900 contains a trace amount of Fe at only 0.27 at% compared to 0.90 at% in Fe-NP-900 (Fig. 2b, see Table S1 (ESI†) for full elemental compositions). To investigate the chemical state of Fe, high-resolution XPS measurements were studied. In the Fe 2p region, the binding energy of the Fe 2p3/2 peak is located at 710.4 eV in Fe-SA-900, which is more positive than that of metallic Fe at 706.6 eV (Fig. 2c).36 This suggests that Fe exists in an oxidation state greater than zero in Fe-SA-900 where Fe atoms are likely coordinated via covalent bonds at N sites.2,11,12,31,32,37 A shoulder peak appears at a binding energy of 707.9 eV in Fe-NP-900 (Fig. S3, ESI†). This confirms that Fe aggregation leads to the formation of Fe compounds (e.g. Fe3C) in Fe-NP-900, which agrees with the XRD pattern of Fe-NP-900 in Fig. 1d. In the N 1s region, the signals are typically deconvoluted into four major N species: graphitic, pyridinic, pyrrolic, and oxidized N (Fig. 1d and Fig. S4, ESI†).38 In brief, graphitic N refers to a N atom that substitutes a carbon in the carbon framework, coordinating three carbon atoms. Pyrrolic and pyridinic N are assigned to N atoms located at the edge of five- and six-membered carbon rings, respectively.
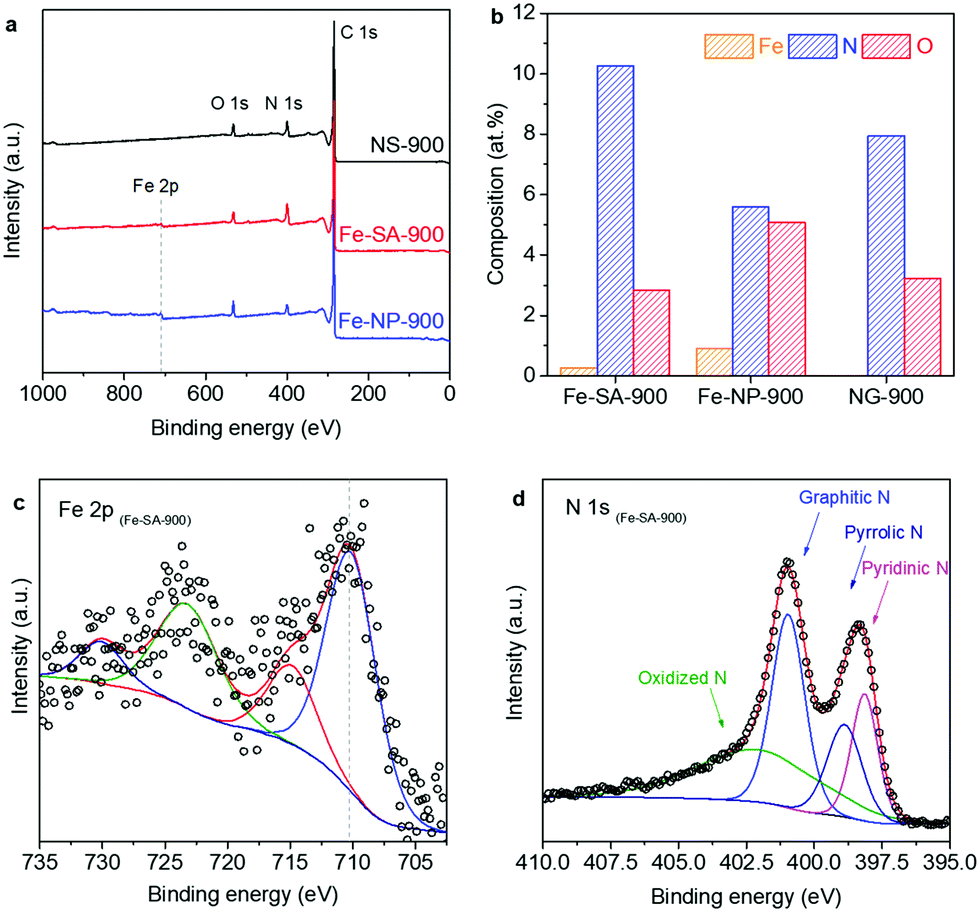 | ||
| Fig. 2 (a) XPS survey spectra of NS-900, Fe-SA-900, and Fe-NP-900. (b) XPS-based quantification of elemental compositions for all samples. (c) High-resolution Fe 2p spectra of Fe-SA-900. (d) Deconvoluted high-resolution N 1s spectra of Fe-SA-900. | ||
To explore the effects of atomic-level Fe dispersion on CRR activity, the electrochemical performances of the materials were evaluated in a CO2-saturated 0.1 M KHCO3 electrolyte (Fig. S5 and S6, ESI†). The generated products identified by gas chromatography (GC) were hydrogen, CO, and methane. No liquid products were detected by nuclear magnetic resonance spectroscopy (NMR) results. In Fig. 3a, a clear selectivity trend for CO can be seen with reduction potential increasing on Fe-SA-900. In the moderate reduction region, the FE of CO increases and approaches 89.8%. Meanwhile, the competitive hydrogen evolution reaction (HER) is effectively suppressed in this region. Similarly, Fe-NP-900 exhibits a similar trend but the HER rate is significantly greater (Fig. S7a, ESI†). NS-900 displays much lower CO2 reduction activity and instead significant HER activity is observed across all potentials (Fig. S7b, ESI†). Fig. 3b indicates that the FEs of CO formation for the Fe containing catalysts reach maxima as the reduction potential is decreased from −0.38 to −0.58 V vs. RHE. According to the measured Tafel slopes of ca. 136 mV dec−1 for CO formation on Fe-SA-900 and Fe-NP-900 in Fig. 3c, the rate-determining step (RDS) for CO2 reduction toward CO is identical on these two Fe containing catalysts, i.e., the CO2 molecule adsorbs at the active site with a rate limiting electron transfer to form the *CO2− intermediate.
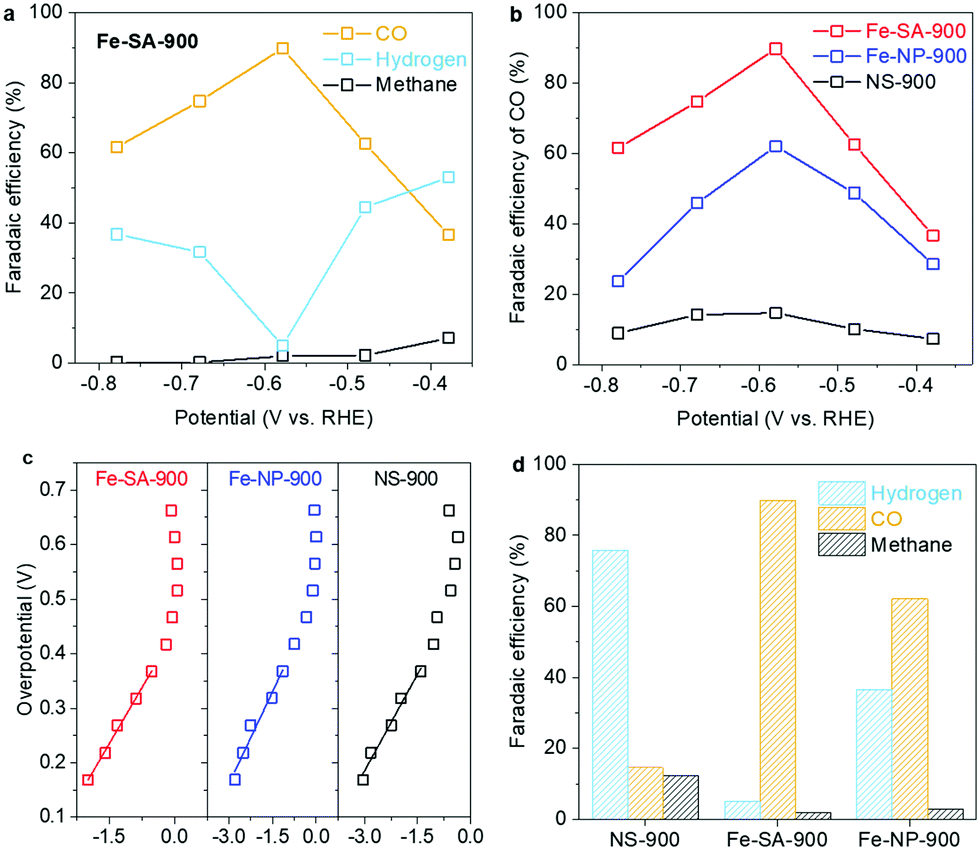 | ||
| Fig. 3 (a) The measured FEs for CO2 reduction on Fe-SA-900. (b) The measured FEs of CO on Fe-SA-900, Fe-NP-900, and NS-900. (c) Tafel plots for CO generation on Fe-SA-900, Fe-NP-900, and NS-900. (d) The distribution of FEs for CO2 reduction on NS-900, Fe-SA-900, and Fe-NP-900 at a constant applied potential of −0.58 V vs. RHE. | ||
Fig. 3d clearly demonstrates the selectivity trend across the samples. (i) The Fe containing catalysts have enhanced performances for CO formation. Therefore, Fe atoms are likely involved in the active sites for CO2 reduction. However, (ii) the catalytic activity for CO generation on the Fe-rich catalyst (Fe-NP) is inferior to that of the Fe-lean catalyst (Fe-SA). This finding suggests that Fe is not the only active component which determines catalytic activity and selectivity for CO on Fe-SA-900. Instead, it may be attributed to dual active sites rather than single active sites. (iii) The Fe-rich catalyst shows much higher HER activity than that of the Fe-lean catalyst. From this evidence, we hypothesize that it is likely the N-C sites that are the secondary active sites.
To identify the second active sites in the Fe-based samples, Fe-SA and Fe-NP were also synthesized at 700 °C and 800 °C. Changing the annealing temperature results in various levels of both overall N content and specific N species (Table S1 and Fig. S8, ESI†). The three catalysts contained the same Fe content but the ratio of pyrrolic-to-pyridinic-N (Npyr/Npyd) increased with annealing temperature. Raman spectroscopy showed that the degree of graphitization hardly changes with annealing temperature, thus it is not likely to be an influencing factor here (Fig. S9, ESI†). Importantly, as shown in Fig. 4a, there is a clear trend showing that the FE toward CO is correlated with the value of Npyr/Npyd. Specifically, Fe-SA-900 has the largest Npyr/Npyd of 0.96 and the highest FE toward CO among the Fe-SA samples. However, no obvious relationship between Npyr/Npyd and CO selectivity was observed for the Fe-NP samples (Fig. 4b). This further confirms that the atomically dispersed Fe coordinated to pyrrolic-N-C sites can synergistically promote CO2 reduction.
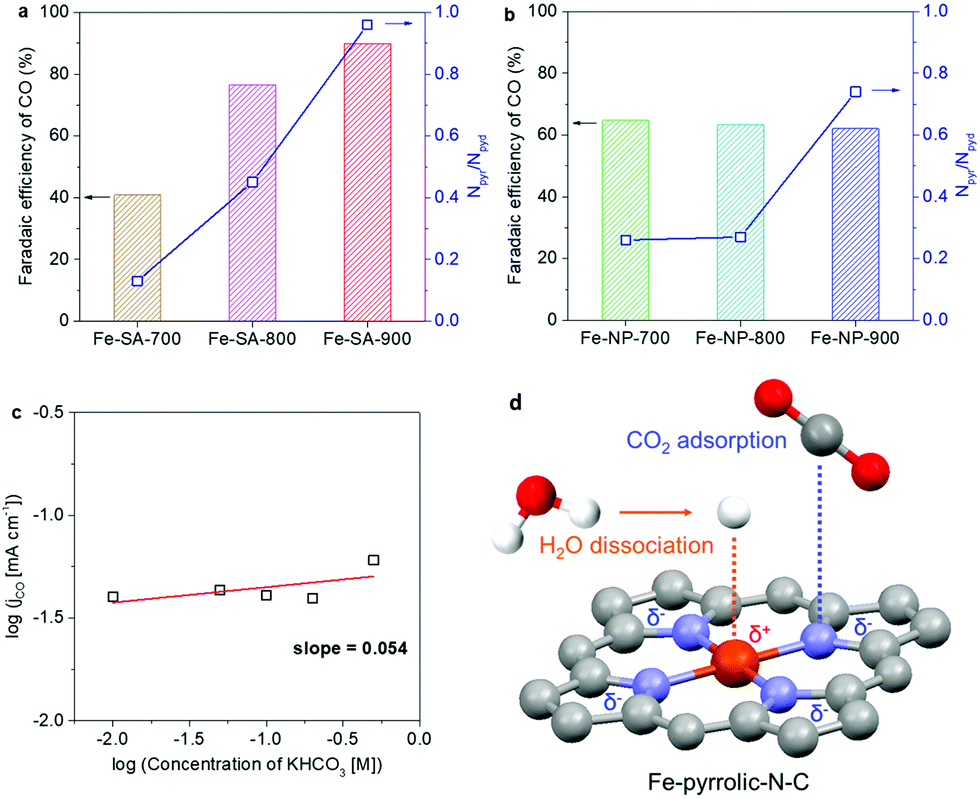 | ||
| Fig. 4 The measured FEs of CO on (a) Fe-SA samples and (b) Fe-NP samples with the corresponding ratios of pyrrolic-to-pyridinic-N species. (c) Rate dependency on [HCO3−] for CO on Fe-SA-900 at −0.38 V vs. RHE. (d) Scheme of synergetic catalysis between the Fe atom and pyrrolic-N-C sites. At the metal site, the positively charged (δ+) Fe atom further enhances water dissociation to form protons for CO2 reduction. While, at the N-C sites, the pyrrolic N contributes a pair of pz electrons toward π bonds, resulting in high electron density (δ−) at N-C sites that improves CO2 adsorption. | ||
Towards the reaction mechanism, from the previous analysis of Tafel slopes (Fig. 3c), the first electron transfer step (CO2 + e− + * = *CO2−) is likely the RDS of the entire catalytic process. Therefore, improving the adsorption of CO2 on the active sites can facilitate the reaction kinetics and enhance activity.23,24,26 Also, it should be noted that the protons participating in CO2 reduction can also affect the reaction rate, e.g. insufficient protons lead to hindering the following proton-assisted reduction steps (e.g., *CO2− + H+ = *COOH, *COOH + H+ + e− = *CO + H2O, etc.).39,40 From the zero-order dependency of CO generation on [HCO3−] in the electrolyte (Fig. 4c), it is shown that the protons for consequent hydrogenation steps are provided from water dissociation instead of HCO3− ionization.41,42 Thus, optimization of water dissociation can also facilitate CO2 reduction. At this stage, we identify that CO2 adsorption and water dissociation are key processes for optimization of catalysts. From Fig. 3d, Fe-NP-900 contains a high Fe content that leads to obvious competition from the HER against CO2 reduction. This implies that Fe as the sole active site plays a role in facilitating water dissociation. The other active site is indicated in Fig. 4a. Given their identical Fe content, the significant increase in performance across the Fe-SA samples derives from the increase in Npyr/Npyd. It supports that the N-C sites adjacent to the Fe atom contribute to CO2 adsorption.
At the atomic level, the pyrrolic N has a pair of electrons in the unhybridized pz orbital that leads to increased electron density on the N-C sites. In contrast, the lone pair of electrons from pyridinic N form bonds and do not participate in the π system on N-C sites. Therefore, pyrrolic N induces greater electron density at the N-C sites, which favors CO2 adsorption and electron transfer. This can be also reflected by the more negatively charged N-C sites resulting in positively charged Fe. From the Fe 2p spectra, the binding energy of the Fe 2p3/2 peak shifts to higher energy as the annealing temperature is increased (Fig. S10 and S11, ESI†), indicating that the oxidation state of Fe increases. This confirms that more positively charged Fe is achieved in a pyrrolic-N-rich structure, which has been widely reported in facilitating water dissociation for proton generation.43,44
To summarize, we successfully obtained Fe-SA-900 using a simple pyrolysis method. The atomically dispersed Fe coordinated to the pyrrolic-N-C framework sites synergistically enhanced CO2 reduction to CO. As one of the active sites, the Fe promotes water dissociation to form sufficient protons for CO2 reduction. Furthermore, the Fe atom is also affected by the local electron structure of adjacent N-C sites. Consequently, the Fe exists in a positively charged state which favors water dissociation. The pyrrolic-N-C site acts as the other active site which synergistically promotes CO2 reduction. Pyrrolic-N-C sites provide regions of higher electron density that facilitate CO2 adsorption and electron transfer, which activates the first reaction step. Through a combination of atomically dispersed Fe and pyrrolic-N-C framework sites (Fig. 4d), a near 90% FE toward CO is achieved on Fe-SA-900 at a low overpotential of 0.47 V (i.e. −0.58 V vs. RHE). The effect of pyrrolic-N-C sites in metal–N-C catalysts could be extended to further study CO2 reduction toward products like methane and C2 products.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support from the Australian Research Council (ARC) through Discovery Project programs (DP160104866, DP170104464, DE160101163, and FL170100154). We thank Dr Ashley Slattery at Adelaide Microscopy for TEM and STEM imaging and Dr Haolan Xu at the University of South Australia for XPS measurements. Chaochen Xu acknowledges the support from the Beacon of Enlightenment PhD Scholarship.Notes and references
- Z. Y. Sun, T. Ma, H. C. Tao, Q. Fan and B. X. Han, Chem, 2017, 3, 560–587 CAS.
- C. Zhu, S. Fu, Q. Shi, D. Du and Y. Lin, Angew. Chem., Int. Ed., 2017, 56, 13944–13960 CrossRef CAS PubMed.
- R. Francke, B. Schille and M. Roemelt, Chem. Rev., 2018, 118, 4631–4701 CrossRef CAS PubMed.
- A. Vasileff, C. Xu, Y. Jiao, Y. Zheng and S.-Z. Qiao, Chem, 2018, 4, 1809–1831 CAS.
- T. Zheng, K. Jiang and H. Wang, Adv. Mater., 2018, 30, 1802066 CrossRef PubMed.
- L. Zhang, Z. J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2017, 56, 11326–11353 CrossRef CAS PubMed.
- Y. Wang, J. Liu, Y. Wang, A. M. Al-Enizi and G. Zheng, Small, 2017, 13, 1701809 CrossRef PubMed.
- J. L. Liu, C. X. Guo, A. Vasileff and S. Z. Qiao, Small Methods, 2017, 1, 1600006 CrossRef.
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423–3452 CrossRef CAS PubMed.
- Y. Zheng, A. Vasileff, X. Zhou, Y. Jiao, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2019, 141, 7646–7659 CrossRef CAS PubMed.
- Y. J. Chen, S. F. Ji, C. Chen, Q. Peng, D. S. Wang and Y. D. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- Y. Wang, J. Mao, X. Meng, L. Yu, D. Deng and X. Bao, Chem. Rev., 2019, 119, 1806–1854 CrossRef CAS PubMed.
- A. Han, B. Wang, A. Kumar, Y. Qin, J. Jin, X. Wang, C. Yang, B. Dong, Y. Jia, J. Liu and X. Sun, Small Methods, 2019, 1800471 CrossRef.
- B.-W. Zhang, Y.-X. Wang, S.-L. Chou, H.-K. Liu and S.-X. Dou, Small Methods, 2019, 1800497 CrossRef.
- T. T. Zheng, K. Jiang, N. Ta, Y. F. Hu, J. Zeng, J. Y. Liu and H. T. Wang, Joule, 2019, 3, 265–278 CrossRef CAS.
- Y. Chen, S. Ji, Y. Wang, J. Dong, W. Chen, Z. Li, R. Shen, L. Zheng, Z. Zhuang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 6937–6941 CrossRef CAS PubMed.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- H. J. Qiu, Y. Ito, W. Cong, Y. Tan, P. Liu, A. Hirata, T. Fujita, Z. Tang and M. Chen, Angew. Chem., Int. Ed., 2015, 54, 14031–14035 CrossRef CAS PubMed.
- L. Zhao, Y. Zhang, L. B. Huang, X. Z. Liu, Q. H. Zhang, C. He, Z. Y. Wu, L. J. Zhang, J. Wu, W. Yang, L. Gu, J. S. Hu and L. J. Wan, Nat. Commun., 2019, 10, 1278 CrossRef PubMed.
- Z. Zhang, J. Xiao, X. J. Chen, S. Yu, L. Yu, R. Si, Y. Wang, S. Wang, X. Meng, Y. Wang, Z. Q. Tian and D. Deng, Angew. Chem., Int. Ed., 2018, 57, 16339–16342 CrossRef CAS PubMed.
- L. Zhang, F. Mao, L. R. Zheng, H. F. Wang, X. H. Yang and H. G. Yang, ACS Catal., 2018, 8, 11035–11041 CrossRef CAS.
- A. S. Varela, N. Ranjbar Sahraie, J. Steinberg, W. Ju, H. S. Oh and P. Strasser, Angew. Chem., Int. Ed., 2015, 54, 10758–10762 CrossRef CAS PubMed.
- A. Vasileff, Y. Zheng and S. Z. Qiao, Adv. Energy Mater., 2017, 7, 1700759 CrossRef.
- Y. Jiao, Y. Zheng, P. Chen, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2017, 139, 18093–18100 CrossRef CAS PubMed.
- X. Li, W. Bi, M. Chen, Y. Sun, H. Ju, W. Yan, J. Zhu, X. Wu, W. Chu, C. Wu and Y. Xie, J. Am. Chem. Soc., 2017, 139, 14889–14892 CrossRef CAS PubMed.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang, W. Huang, J. Yang, X. Hong, S. Wei, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 CrossRef CAS PubMed.
- C. H. Zhang, S. Z. Yang, J. J. Wu, M. J. Liu, S. Yazdi, M. Q. Ren, J. W. Sha, J. Zhong, K. Q. Nie, A. S. Jalilov, Z. Y. Li, H. M. Li, B. I. Yakobson, Q. Wu, E. L. Ringe, H. Xu, P. M. Ajayan and J. M. Tour, Adv. Energy Mater., 2018, 8, 1703487 CrossRef.
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS PubMed.
- M. Zhu, J. Chen, L. Huang, R. Ye, J. Xu and Y. F. Han, Angew. Chem., Int. Ed., 2019, 58, 6595–6599 CrossRef CAS PubMed.
- W. Zhong, R. Sa, L. Li, Y. He, L. Li, J. Bi, Z. Zhuang, Y. Yu and Z. Zou, J. Am. Chem. Soc., 2019, 141, 7615–7621 CrossRef CAS PubMed.
- W. Bi, X. Li, R. You, M. Chen, R. Yuan, W. Huang, X. Wu, W. Chu, C. Wu and Y. Xie, Adv. Mater., 2018, 30, 1706617 CrossRef PubMed.
- Y. Cheng, S. Zhao, B. Johannessen, J. P. Veder, M. Saunders, M. R. Rowles, M. Cheng, C. Liu, M. F. Chisholm, R. De Marco, H. M. Cheng, S. Z. Yang and S. P. Jiang, Adv. Mater., 2018, 30, 1706287 CrossRef PubMed.
- W. Ju, A. Bagger, G. P. Hao, A. S. Varela, I. Sinev, V. Bon, B. Roldan Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Nat. Commun., 2017, 8, 944 CrossRef PubMed.
- M. Liu, R. Zhang and W. Chen, Chem. Rev., 2014, 114, 5117–5160 CrossRef CAS PubMed.
- Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 6717–6731 RSC.
- P. L. Tan, J. Catal., 2016, 338, 21–29 CrossRef CAS.
- H. Fei, J. Dong, C. Wan, Z. Zhao, X. Xu, Z. Lin, Y. Wang, H. Liu, K. Zang, J. Luo, S. Zhao, W. Hu, W. Yan, I. Shakir, Y. Huang and X. Duan, Adv. Mater., 2018, 30, 1802146 CrossRef PubMed.
- Y. Zheng and S. Z. Qiao, Nat. Chem., 2018, 10, 900–902 CrossRef CAS PubMed.
- M. Dunwell, W. Luc, Y. S. Yan, F. Jiao and B. J. Xu, ACS Catal., 2018, 8, 8121–8129 CrossRef CAS.
- A. Wuttig, Y. Yoon, J. Ryu and Y. Surendranath, J. Am. Chem. Soc., 2017, 139, 17109–17113 CrossRef CAS PubMed.
- W. Luo, J. Zhang, M. Li and A. Züttel, ACS Catal., 2019, 9, 3783–3791 CrossRef CAS.
- M. Ma, B. J. Trzesniewski, J. Xie and W. A. Smith, Angew. Chem., Int. Ed., 2016, 55, 9748–9752 CrossRef CAS PubMed.
- H. Y. Jin, X. Liu, Y. Jiao, A. Vasileff, Y. Zheng and S. Z. Qiao, Nano Energy, 2018, 53, 690–697 CrossRef CAS.
- J. Staszak-Jirkovsky, C. D. Malliakas, P. P. Lopes, N. Danilovic, S. S. Kota, K. C. Chang, B. Genorio, D. Strmcnik, V. R. Stamenkovic, M. G. Kanatzidis and N. M. Markovic, Nat. Mater., 2016, 15, 197–203 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nh00361d |
| This journal is © The Royal Society of Chemistry 2019 |