
Ultrafine Co2P nanorods wrapped by graphene enable a long cycle life performance for a hybrid potassium-ion capacitor†
Yixuan
Wang‡
a,
Zhongyu
Zhang‡
b,
Guangxia
Wang
a,
Xinyi
Yang
 *a,
Yongming
Sui
a,
Fei
Du
*b and
Bo
Zou
*a
*a,
Yongming
Sui
a,
Fei
Du
*b and
Bo
Zou
*a
aState Key Laboratory of Superhard Materials, College of Physics, Jilin University, Changchun, 130012, People's Republic of China. E-mail: yangxinyi@jlu.edu.cn; zoubo@jlu.edu.cn
bKey Laboratory of Physics and Technology for Advanced Batteries (Ministry of Education), College of Physics, Jilin University, Changchun, 130012, People's Republic of China. E-mail: dufei@jlu.edu.cn
First published on 11th June 2019
Abstract
Given their high theoretical capacities, metal phosphides are anticipated to be excellent charge-storage materials for high-efficiency potassium-ion batteries. However, one of the major problems is the shuttling of heavy and large K+ ions between electrodes, which triggers rapid capacity fading. Here, we demonstrate that sub-4 nm Co2P nanorods attached to reduced graphene oxide (Co2P@rGO) can operate at a lifespan exceeding thousands of cycles. By taking advantage of the high electronic conductivity and flexibility of reduced graphene oxide (rGO), the composite electrode delivers a high capacity (374 mA h g−1 at 20 mA g−1) and excellent C-rate capability (141 mA h g−1 at 2 A g−1), superior to its commercial counterpart. Impressively, the electrode maintains 54% of the capacity over 5000 cycles, and there is almost no capacity fading after the initial 200 cycles. In addition, a hybrid potassium-ion capacitor, assembled from a Co2P@rGO anode and activated carbon cathode, affords a high energy/power density (87 W h kg−1 and 4260 W kg−1) in a potential window of 1.0–4.0 V, as well as a long lifespan of over 1000 cycles. These extremes demonstrate the high-performance of the Co2P@rGO anode materials and an optimal synthesis strategy to boost K+ storage performance.
New conceptsPotassium-ion batteries (PIBs) are a promising alternative to lithium-ion batteries owing to the abundant natural resources of potassium. To date, the exploration of suitable materials for PIBs is in the early stages. Phosphide can deliver high reversible capacities but suffers from irreversible deterioration of its crystal structure resulting in unsatisfactory cycle stability. In this report, we synthesize sub-4 nm Co2P nanorods attached onto reduced graphene oxide using a colloidal mesostructural method. The ultrafine nanostructure not only effectively accommodates the strain to ensure the integrity of the electrode during cycling, but also reduces the solid-state diffusion length of potassium-ion and provides significantly more electrochemistry reaction sites. The flexible rGO can improve the electronic conductivity and provides a further buffering effect against volume changes during cycling, maintaining the integrity of the electrode. Benefiting from the design of the material, the electrode can deliver a high capacity and sustain a long lifespan of 5000 cycles. |
Introduction
Electrical energy storage has attracted world-wide interest owing to the electrification of transportation and renewable energy integration.1–10 Currently, lithium-ion batteries (LIBs) are considered to be one of the most promising technologies, largely owing to their high device efficiency enabled by their excellent electrical energy storage performance.11–19 Unfortunately, there is concern about the utilization of LIBs for large-scale energy storage owing to the low natural abundance of lithium (0.0017 wt%)20 and its uneven distribution on earth, which is likely to increase its manufacturing costs dramatically. Nowadays, exploitation of low-cost alternative electrochemical energy storage systems based on natural abundant elements, including Na+, K+, Al3+, Zn2+ and so forth, have gained significant attention.21–25 Among these, potassium-ion batteries (PIBs) are of particular interest because of the widespread terrestrial reserves on earth (2.09 wt%) and the low cost. Furthermore, potassium offers a lower standard hydrogen potential (−2.93 V vs. E°) compared to that of sodium (−2.71 V vs. E°) and closer to that of lithium (−3.04 V vs. E°),26 which is beneficial to an increase in the energy density of the battery. However, the larger ionic radius (1.38 Å) and heavy atomic weight of K+ ions are likely to result in a limited capacity and sluggish kinetic properties. So far, the main challenge that must be overcome for PIBs is rooted in the demand for suitable host materials for K+ storage.One promising class of anode materials for PIBs is phosphorus (P), which can provide a high reversible capacity27via an alloying–dealloying reaction,28–30 however, it still suffers from the irreversible deterioration of its crystal structure resulting in an unsatisfactory cycle stability and rate capability. To tackle this issue, one potential approach is hybridization of the carbonaceous materials with nanostructured active materials.31 The nanosized materials have large specific surface areas and a short ionic transfer pathway, facilitating fast ionic conductivity and kinetic properties. Moreover, the introduced carbonaceous materials are beneficial, improving the electronic conductivity and tolerance of the volume expansion during the reversible insertion and extraction of K+. So far, there have been few studies published which report the electrochemical properties of P-based anode materials for application in PIBs. Sultana et al. reported the K+ storage properties of hybrid black phosphorus/graphite, which delivered a high reversible capacity of almost 400 mA h g−1 at a current density of 25 mA g−1.32 Guo et al. prepared a binary compound, Sn4P3/C hybrid material, via a facile ball-milling method which had a stable capacity of ∼384 mA h g−1 at a current density of 50 mA g−1, which was superior to its unary compounds, Sn/C or P/C.27 Also, it was confirmed that both Sn and P alloy with the K+ ions to form K-Sn and K–P phases, with these phases acting as mutual buffers to alleviate the volume expansion. However, it is worth noting that, although the ball-milling method is convenient for achieving phosphorus/carbon composite materials, it is hard to control the particle sizes of P and the compositing manner between P and carbon. A serve agglomeration is usually found, preventing electrolyte infiltration and the transfer of K+ ions. In this respect, an optimal synthesis strategy to prepare the well-dispersed and homogeneously distributed P/carbonaceous nanocomposites is essential to achieving a long-term cycle stability, as well as the high-rate charge–discharge ability to satisfy the demands of large-scale energy storage.
Cobalt phosphides (e.g. Co2P) are described as being a promising anode material for rechargeable batteries because of their relatively low charge–discharge potential, metallic character and good thermal stability.33,34 Recent density functional theory calculations have suggested that Co2P with an orthorhombic phase better illustrates the electronic conductivity compared to other cobalt phosphides, which is helpful for the electron transportation and thus the improvement of alkali-metal storage properties.35 Here, we prepared a structure consisting of sub-4 nm diameter and ∼65 nm length ultrafine Co2P nanorods (NRs) attached onto reduced graphene oxide (abbreviated as U-Co2P@rGO-14) via a colloidal mesostructured method, and present the electrochemical properties as a viable anode material for PIBs. Benefiting from the large surface-to-volume ratio and ability to accommodate the strain of the as-prepared ultrafine NRs, the U-Co2P@rGO-14 hybrid electrode delivered a high reversible capacity of 374 mA h g−1 at a current density of 20 mA g−1, an excellent capacity retention of 54% after 5000 cycles, and a superior rate capability with a specific capacity of 141 mA h g−1 at 2 A g−1. Furthermore, an innovative hybrid potassium-ion capacitor (KIC) was constructed by coupling U-Co2P@rGO-14 and activated carbon (AC) as the anode and cathode, respectively, which could deliver a high energy density (maximum 87 W h kg−1), a high power density (maximum 4260 W kg−1), and a long lifespan (1000 cycles).
Results and discussion
The ultrafine rod-like nanostructure and its analogues offer several advantages, including a higher surface area and shorter ion transfer pathway.36 To ensure an ultrafine nanostructure, we fabricated the Co2P NRs using a colloidal mesostructural method. Briefly, ultrathin NRs were grown after injecting trioctylphosphine solution into a co-oleylamine (OLA) precursor at 300 °C, further details are provided in the experimental section. When the Co2P nuclei were formed, the OLA simultaneously assembled onto the Co2P nuclei and protected it from forming aggregates. Then, the mesostructure, with a long-range order, served as a highly confined and high-aspect ratio template for further growth of ultrafine Co2P NRs (Fig. 1a). To suppress the volume change during reversible K+ insertion/extraction and increase the electronic conductivity of the working electrode, we improved the engineering of the ultrafine Co2P NRs by combining with rGO. The carbon network has a high electronic conductivity and can improve the conductance of the active materials.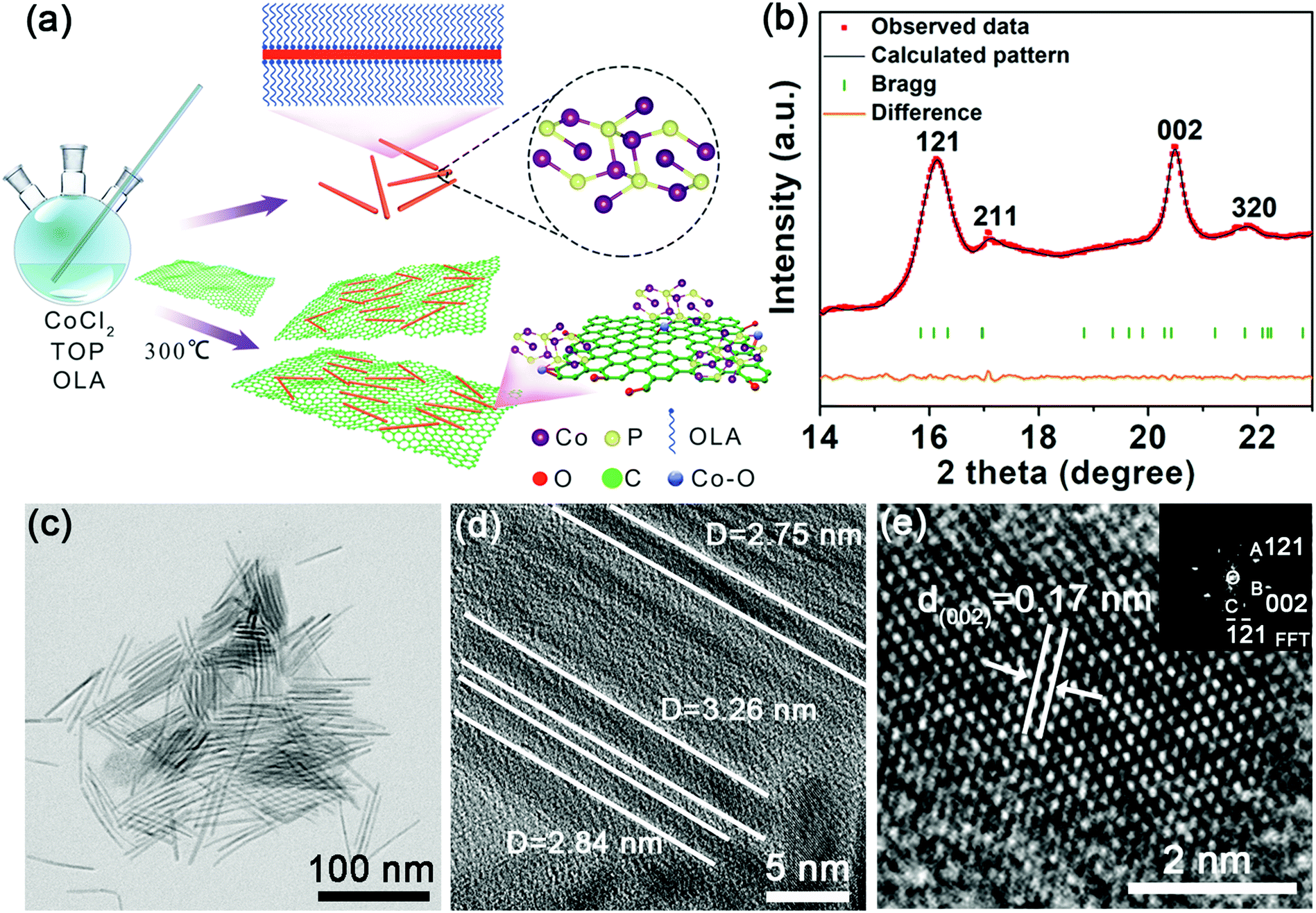 | ||
| Fig. 1 Morphology and structural features of the synthesized Co2P NRs. (a) Schematic representation of the fabrication of the ultrafine Co2P NRs and U-Co2P@rGO-14 nanocomposite. (b) ADXRD pattern of the as-prepared ultrafine Co2P NRs, in which red circles, black lines, orange lines and green bars show the observed diffraction plots, the refined patterns, the difference between the observed and calculated plots, and the positions of the Bragg reflections. (c) TEM image of the as-synthesized Co2P NRs. (d) Low-magnification HRTEM image of the Co2P NRs. (e) High-magnification HRTEM image of a typical Co2P nanorod and an FFT image taken from the corresponding dashed square in (e). | ||
The angle dispersive synchrotron X-ray diffraction (ADXRD) patterns of the ultrafine Co2P NRs were performed at Shanghai Synchrotron Radiation Facility (SSRF) with an incident monochromatic wavelength of 0.6199 Å37 (Fig. 1b). Rietveld refinement confirmed the orthorhombic polymorph with a space group of Pnma (a = 5.646 Å, b = 6.609 Å, and c = 3.513 Å). The narrow (002) peak is attributed to the preferred orientation growth along the length direction, while the broad (121) peak is due to the very small size in the width direction. These unusual characteristics of the ADXRD pattern are consistent with the unique geometry of the ultrathin NRs, which is expected to exhibit an overlap of extremely broad and sharp features owing to the extreme difference in the coherence lengths along different crystallographic axes. As shown in the transmission electron microscopy (TEM) image (Fig. 1c, d and Fig. S1, ESI†), these Co2P NRs have a highly uniform ultrafine morphology with an average diameter of less than 4 nm. The high-resolution TEM (HRTEM) image taken of one Co2P nanorod reveals distinct lattice fringes with a spacing of about 0.17 nm corresponding to the (002) plane of Co2P (Fig. 1e), which is consistent with the corresponding fast Fourier transform (FFT) pattern obtained (as shown in the inset of Fig. 1e).
After introducing the rGO sheets, the ultrafine Co2P NRs were tightly attached and confined in the flexible rGO framework (Fig. 2a). Fig. 2b shows the in-plane alignment of the ultrafine NRs in a stand-alone unit without aggregation. This low tortuosity arrangement is highly recommended for kinetically accessible storage electrodes.4,13 It should be noted that the Co2P NRs retained the ultrathin nature with an orthorhombic structure, as shown in Fig. 2c and d. As evidenced by the corresponding EDS elemental mappings, the Co, P and C elements are uniformly distributed throughout the U-Co2P@rGO-14 nanocomposite (Fig. 2e). The rGO content for the U-Co2P@rGO-14 nanocomposite is quantitatively estimated to be 14 wt%, according to the carbon, hydrogen and nitrogen analyzer (CHN) measurements. The chemical states of Co and P for Co2P were further characterized using X-ray photoelectron spectroscopy (XPS). We have measured the XPS of the pure Co2P nanorods, as shown in Fig. S2 (ESI†), the peaks at 797.3 and 781.7 eV are assigned to the Co 2p1/2 and Co 2p3/2 of the oxidized Co2+ species respectively.38–40 As shown in Fig. 2f, we also measured the XPS of U-Co2P@rGO-14, and the corresponding peak positions of 781.6 and 797.3 eV did not move significantly. It can be proven that the Co in Co2P is not chemically bonded to the oxygen in rGO. Therefore we propose that it is physically attached. The two apparent satellite peaks at 803.2 and 785.7 eV, are ascribed to the shakeup excitation of the high-spin Co2+ ions. Furthermore, the peaks at 793.6 and 778.7 eV are assigned to the Co 2p1/2 and Co 2p3/2 of Co species in U-Co2P@rGO-14, which have a partial positive charge from the Co metal.38 The high-resolution C 1s XPS peak of U-Co2P@rGO-14 can be compartmentalized into two peaks located around 284.6 and 286 eV. The former peak is assigned to the C–C bonding in rGO, while the latter peak is indexed to the C–O bonding species. Generally speaking, rGO exhibits the same oxygen-containing functionalities, but their intensities are much smaller than GO. Fig. 2f shows a lower peak intensity for the C–O bonding compared to the C–C bonding, indicating the presence of rGO.41 In the close-up P 2p spectrum of U-Co2P@rGO-14, the peak at 129.9 eV is close to the binding energy of P 2p3/2 of the P in U-Co2P@rGO-14, which shows a negative shift from that of the elemental P. In addition, the broad peak at 133.3 eV is assigned to the oxidized phosphorus species, resulting from the inevitable surface oxidation of U-Co2P@rGO-14.
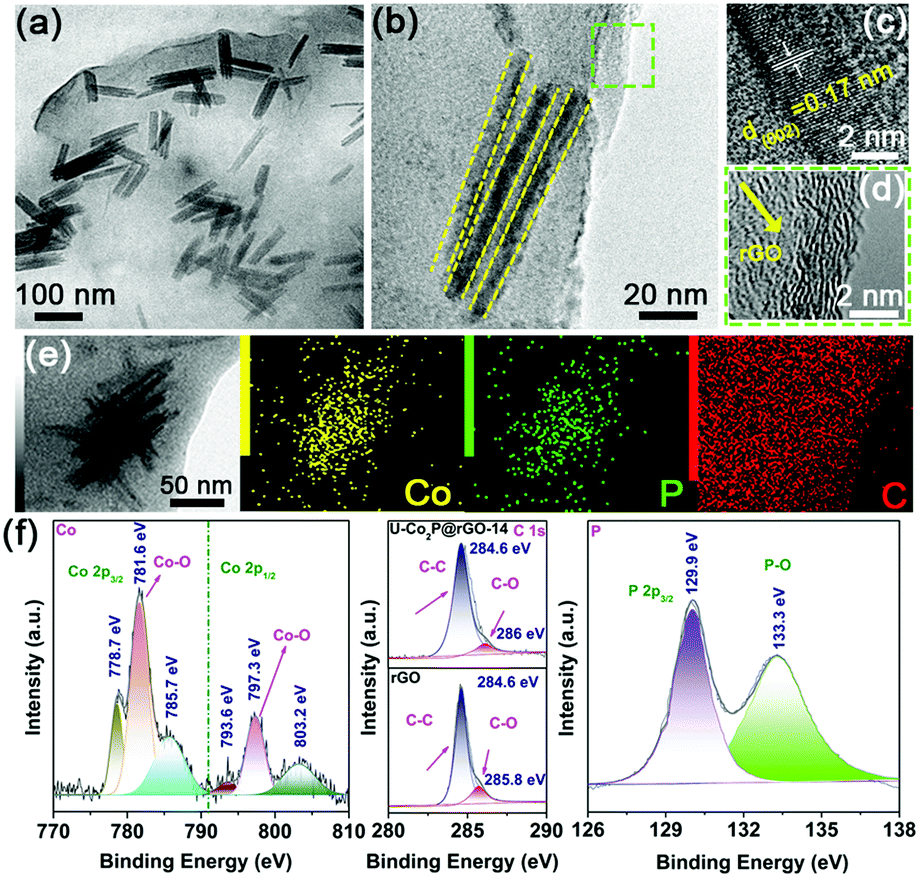 | ||
| Fig. 2 Synthesis and characterization of the U-Co2P@rGO-14 nanocomposite. (a) Low-magnification, and (b) high-magnification TEM images of the U-Co2P@rGO-14 nanocomposite. (c and d) High-magnification HRTEM images taken from the corresponding the dashed squares in (b). (e) STEM elemental mapping of the U-Co2P@rGO-14 nanocomposite. (f) XPS analysis of the U-Co2P@rGO-14 nanocomposite. | ||
Therefore, we propose that the ultrafine Co2P NRs are nucleated in situ and deposited on the amorphous carbon matrix (Fig. 1a) during the phosphorization process. The robust interfacial connection guarantees the structural stability of the composite. The N2 adsorption–desorption isotherms of the U-Co2P@rGO-14 are shown in Fig. S3 (ESI†), possessing a surface area of 47.57 m2 g−1 according to the Brunauer–Emmett–Teller (BET) method.42 The isotherms are type II, implying that U-Co2P@rGO-14 is nonporous or microporous.
To understand the advantages of the ultrafine nanostructure on the potassium storage performance, the U-Co2P@rGO-14 nanocomposite was investigated as an anode material for PIBs in comparison with its commercial counterpart (marked as C-Co2P@rGO) (Fig. S4, ESI†). As shown in Fig. S5 (ESI†), C-Co2P@rGO exhibits a sphere-like shape with an average particle size of around 50 nm, much larger than that of U-Co2P@rGO-14. Furthermore, we found a serve agglomeration of lots of Co2P particles and obvious separation between the active materials and rGO (Fig. S6, ESI†), preventing electrolyte infiltration and electron/ion transfer during the electrochemical reaction. The cyclic voltammetry of U-Co2P@rGO-14 was recorded as being between 3.0 and 0.01 V, as shown in Fig. 3a. The initial cathodic scan shows a reduced peak at ≈0.62 V, which is absent in subsequent cycles. This phenomenon is common in the studies of LIBs and sodium ion batteries (SIBs), possibly related to the decomposition of the electrolyte and the formation of a solid–electrolyte interface (SEI) layer on the surface of the anode materials.43,44 During subsequent cycles, stabilized curves exhibited a pair of weak but discernible peaks centred at about 0.71 V and 0.58 V, which can be attributed to the conversion reaction between K–P and Co2P.45 The lowest voltage pair at 0.01/0.3 V corresponds to the insertion/extraction processes of K+ into the rGO, which was further confirmed using the cyclic voltammetry (CV) profiles of rGO (Fig. S7, ESI†).46 Galvanostatic charge–discharge profiles of U-Co2P@rGO-14 were recorded at 20 mA g−1 (Fig. 3b). The U-Co2P@rGO-14 nanocomposite delivers an initial discharge and charge capacities of 686 and 374 mA h g−1 at a current density of 20 mA g−1, respectively. The low coulombic efficiency (CE) of 54.5% was attributed to the decomposition of the electrolyte to form SEI films. Encouragingly, the CE increases as the electrochemical reaction proceeds and stabilizes at almost 95% after 30 cycles, as shown in Fig. 3c. Moreover, it is worth noting that there is no obvious working plateau in the charge–discharge profiles, usually found in the conversion- or alloying-type anode materials for LIBs and SIBs.34,45 The possible reasons lie in the amorphisation of the working electrode upon insertion of a large amount of ions. To further demonstrate the advantage of the ultrafine nano-architecture, the rate capability of both samples was evaluated as the current densities increased from 0.02 to 2 A g−1 (Fig. 3d). At a low current density of 20 mA g−1, the U-Co2P@rGO-14 nanocomposite delivers a reversible capacity of 374 mA h g−1, considerably higher than that of the C-Co2P@rGO materials (143 mA h g−1). It is worth noting that the manifest capacity is still lower than its theoretical one based on the conversion reactions of three electrons (540 mA h g−1). This phenomenon can be understood in terms of the sluggish kinetic property of KIBs, which results in the incomplete electrochemical reaction. When the high current density is increased to 2 A g−1, the ultrafine nanocomposite electrode delivers a high capacity of 141 mA h g−1, a capacity up to 2.43 times higher than that of the C-Co2P@rGO electrode (58 mA h g−1). The excellent rate performance of the U-Co2P@rGO-14 electrode is due to the unique ultrafine nanostructure, which can improve the kinetics by offering a shorter solid-state diffusion length and further electrochemistry reaction sites. To separate the capacity contribution from rGO, its rate capability at the same current densities was also evaluated and is displayed in Fig. S8 (ESI†). rGO could only contribute 39.9 mA h g−1 at a current density of 20 mA g−1 and 11.2 mA h g−1 at current density of 2 A g−1 to the total reversible capacity as it accounts for 14% by weight. Furthermore, as listed in Table S1 (ESI†), the U-Co2P@rGO-14 nanocomposite demonstrates the excellent rate performance in the state-of-the-art conversion- and alloying-type anode materials for PIBs, which can be understood in terms of the unique ultrafine nanostructure of Co2P, not only providing significantly more electrochemistry reaction sites, but also reducing the solid-state diffusion length.
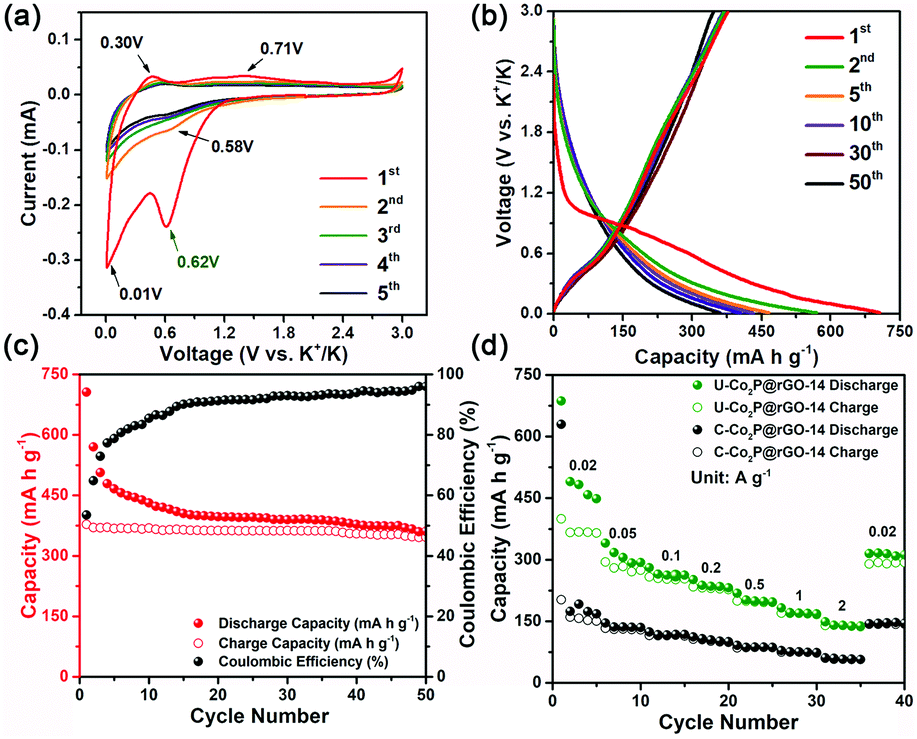 | ||
| Fig. 3 Electrochemical performance of U-Co2P@rGO-14 and C-Co2P@rGO electrodes. (a) Cyclic voltammograms for the first five cycles of U-Co2P@rGO-14. (b) Galvanostatic discharge–charge curves at a current density of 20 mA g−1 in the voltage range of 0.01–3.0 V. (c) Cycle performance of U-Co2P@rGO-14 at a current density of 20 mA g−1 for 50 cycles. (d) Rate performance of U-Co2P@rGO-14 and C-Co2P@rGO electrodes. | ||
As is well known, PIBs generally suffer from a problematic cycle life (typically not exceeding 1000 cycles) compared to their LIBs or SIBs counterparts, presumably owing to the larger size of the K+ ion.27,31,47,48 To assess the long-term response of the U-Co2P@rGO-14 electrode, the cycle performance was also evaluated at a current density of 200 mA g−1 (Fig. 4a). The electrode maintains 64.7% of its capacity during the first 200 cycles. This capacity degradation in the long-term cycle test at a high current density is usually found in studies of SIBs and PIBs,27,45 possibly related to the gradual decomposition of the electrolyte to form a stable SEI layer or activation of some cell components. Impressively, the electrode subsequently exhibits a high capacity retention rate of 83.6% from 200 to 5000 cycles, and there is no tendency of deterioration to date. As compared in Table S1 (ESI†), the U-Co2P@rGO-14 electrode demonstrated a remarkable cycle performance in terms of the high reversible capacity and the longest cycle life. Moreover, compared with U-Co2P@rGO-14 U-Co2P (Fig. S9, ESI†) delivers almost no capacity after 40 cycles, showing that the rGO provides a further buffering effect against volume changes during cycling, maintaining the integrity of the electrode. It also benefits from the moderate mass percent of rGO which could offer a high electronic conductivity, and the U-Co2P@rGO-14 could deliver the highest capacity among the other mass percentages as shown in Fig. S10 (ESI†). To elucidate the reason for the stable cycle properties found in the U-Co2P@rGO-14 electrode, we conducted SEM, TEM and HRTEM analyses to study the influence of potassiation and/or depotassiation on the morphology and microstructural stability of the U-Co2P@rGO-14 electrode. As shown in Fig. S11 (ESI†), the ultrafine nanostructure can effectively accommodate the strain to ensure integrity of the electrode during cycling. TEM analysis of the cycled U-Co2P@rGO-14 nanocomposite shows that its unique ultrafine structure is largely preserved after repetitive cycling, as shown in Fig. 4b and Fig. S12 (ESI†). The Co2P NRs, with a sub-4 nm diameter, remains attached onto the rGO, thereby attesting to the structural robustness of the ultrafine Co2P NRs (Fig. 4c). As shown in Fig. 4d, the HRTEM image of the depotassiated sample indicates that the single crystal structure of the ultrafine Co2P NRs could be maintained after charging to 3.0 V by contrast with its original crystal geometry. The corresponding EDS elemental mapping analysis confirms the coexistence and homogenous distribution of the Co, P, and C elements in the selected area (Fig. 4e). Moreover, the electrochemical impedance spectroscopy (EIS) results of U-Co2P@rGO-14 are shown in Fig. S13 (ESI†). The charge transfer resistance of U-Co2P@rGO-14 maintains almost the same value after the 10th cycle, which also suggests that the ultrafine nanostructure could be preserved, leading to a better cycling performance.49 In addition, the charge–discharge profiles for several selected cycles are displayed in Fig. S14 (ESI†), which shows no obvious difference from the 10th to the 5000th cycle, indicative of an excellent electrochemical reversibility.
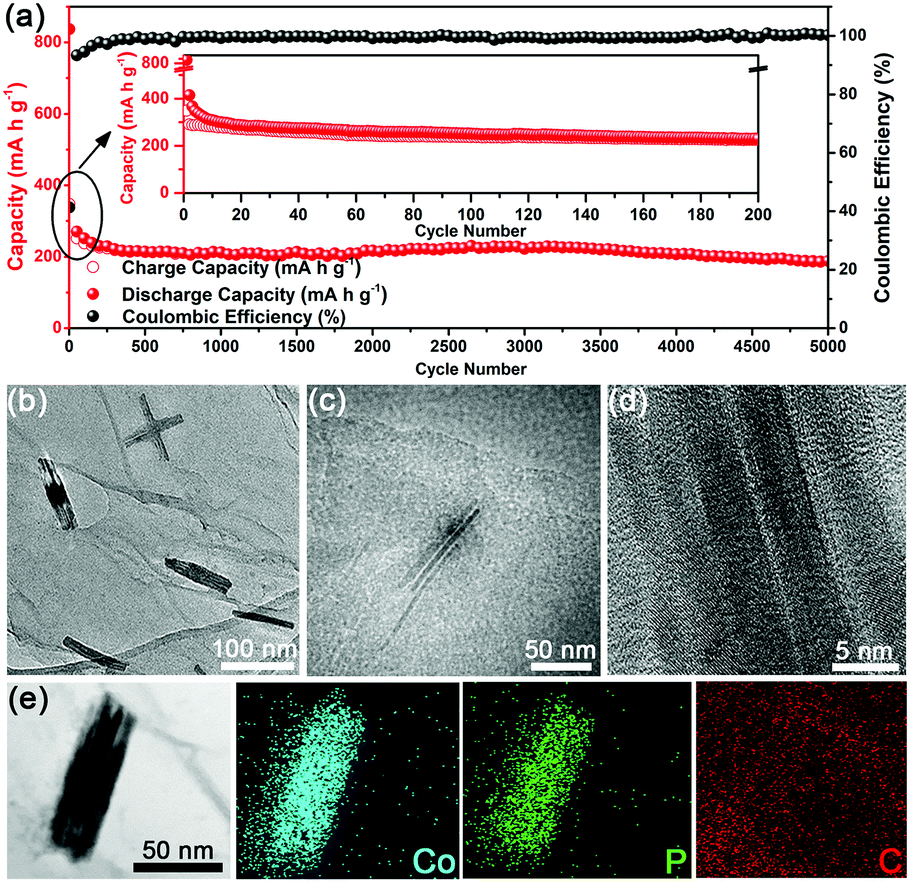 | ||
| Fig. 4 Long-term cycle performance of U-Co2P@rGO-14 and structural feature of the U-Co2P@rGO-14 electrodes after the 50th cycle. (a) Long-term cycling of U-Co2P@rGO-14 at a current density of 200 mA g−1. (b) Low-magnification TEM. (c) High-magnification TEM. (d) HRTEM images, and (e) STEM elemental mapping of U-Co2P@rGO-14 after the 50th cycle at a current density of 200 mA g−1. | ||
To further highlight the advantages of the U-Co2P@rGO-14 electrode, a hybrid KIC comprised of a U-Co2P@rGO-14 anode and a commercial activated carbon (AC) cathode was assembled. From a mechanistic point of view, charges are simultaneously and asymmetrically stored in the KIC by surface anion adsorption–desorption on the capacitive cathode and via potassiation/depotassiation in the anode, respectively. The charge–discharge processes of these two electrodes were performed in different potential ranges, which can enlarge the operating potential window in an efficient manner and result in an enhanced energy density. Before constructing the KIC, the activated carbon, with a surface area of 1368 m2 g−1 (Fig. S15, ESI†), was tested as the cathode with metallic potassium as the counter electrodes, the charge–discharge profiles, cycle, and rate performances are displayed in Fig. S16 in the ESI.† The working potential window of each electrode was first determined using a CV test in a half-cell configuration as shown in Fig. 5a, and the working voltage window of the full cell was set between 1.0 and 4.0 V. In Fig. 5b, the charging/discharging curves exhibit an almost triangular shape with relatively linear voltage-time plots, revealing a good capacitive behavior, mainly due to the combination of the charge storage mechanisms between the faradaic and non-faradaic reactions. On the basis of the galvanostatic charge–discharge measurements, the energy and power densities of U-Co2P@rGO-14//AC can be calculated based on the total sample mass of both the cathode and anode side. A high energy density of 87 W h kg−1 can be achieved at a power density of 12 W kg−1. This still delivers an energy density of 10 W h kg−1 at a high-power output of 4264.7 W kg−1. For the sake of evaluating the novel KIC configuration in the context, a Ragone plot was constructed (Fig. 5c), which demonstrates that the U-Co2P@rGO-14//AC device provides a high energy density and power density comparable to state-of-the-art supercapacitors, Na-based, and K-based hybrid energy storage devices, such as CNT-aerogel//CNT-aerogel,50 Ti3C2/Fe(OH)3//Ti3C2/Fe(OH)3,51 AC//Na-TNT,52 and AC//graphite.53 In addition, as shown in Fig. 5d, the U-Co2P@rGO-14//AC capacitor can be maintained up to 1000 cycles with a capacity retention of 68% at a current density of 1 A g−1, suggesting once again that it is a suitable energy storage device for large-scale applications.
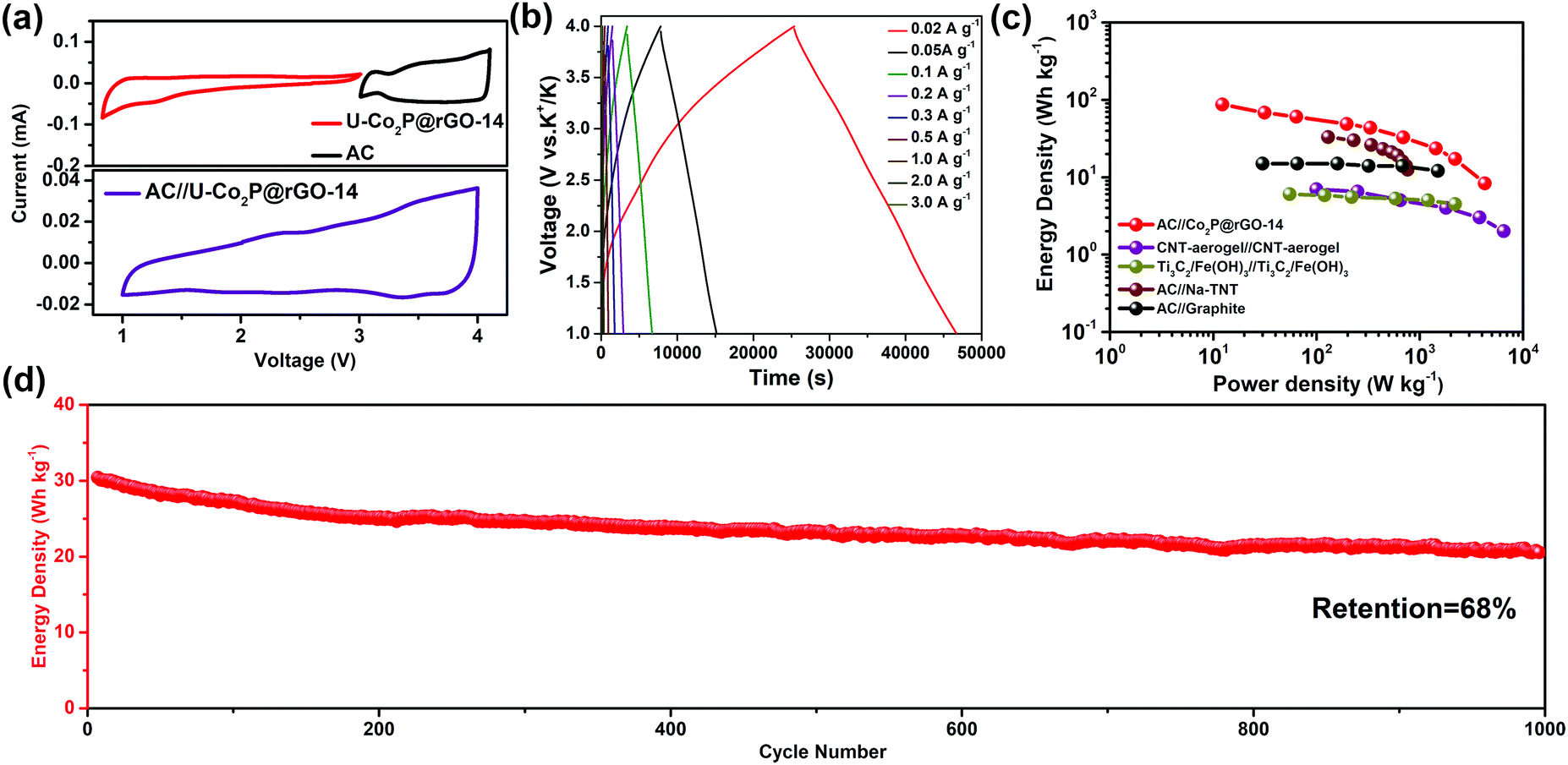 | ||
| Fig. 5 Electrochemical performance of the assembled hybrid potassium-ion capacitor. (a) CV curves of U-Co2P@rGO-14 and AC in half cells (top) and a full cell of KIC (bottom). (b) Profile of charge–discharge curves of full capacitors at different current densities with the mass ratio of 4. (c) Ragone plot of AC//U-Co2P@rGO-14 compared with other energy storage devices and another KIC device. (d) Long-cycle performance of the device. | ||
Conclusions
In conclusion, a U-Co2P@rGO-14 nanocomposite has been successfully prepared as a novel anode material for PIBs. The electrode exhibits a high K storage capacity of 374 mA h g−1, a remarkable rate capability of up to 141 mA h g−1 at 2 A g−1, a high cycle efficiency (≈100%), and an excellent cyclability with a 54% capacity retention over 5000 cycles. Impressively, there is almost no capacity fading after the initial 200 cycles. The key to the motif is the use of sub-4 nm Co2P NRs that not only possess an ultrafine orthorhombic nanostructure, but also embody a low tortuosity arrangement on rGO. Therefore, the kinetics are improved as the new architectures facilitate the transportation of K+ ions by offering a shorter solid-state diffusion length, along with better buffering of the active material volume expansion. We believe that improving the stability and electrochemical performance of the phosphorus-based alloy electrode materials through the strategy of an ultrafine nanostructure offers a new paradigm for developing various highly stable and effective alloy electrode materials for K-ions energy storage. When coupled with an AC cathode, an asymmetric K-ion capacitor is assembled, and it delivers a maximum energy density of ∼87 W h kg−1 and a power density of ∼4260 W kg−1.Experimental
Chemicals
All chemicals, including anhydrous CoCl2 (99.9%), trioctylphosphine (TOP) (97%) and oleylamine (OLA) (70%) were commercially purchased from Sigma-Aldrich without further purification. Commercial cobalt phosphide, ethanol, toluene, chloroform and acetone were of analytical grade (Shanghai Chemical Corp.). The rGO (TIME NANO Corp.) was used as received without any further purification.Synthesis of ultrafine Co2P NR
The ultrafine Co2P NRs were synthesized using a colloidal mesostructure method. In a typical synthesis, 52 mg CoCl2 was loaded into a 50 mL three-neck flash along with 6 mL OLA. The mixture was stirred and heated for 20 min at 120 °C under N2 until the solution became clear. Subsequently, 2 mL TOP was rapidly injected into the reaction mixture, and the mixed solution gradually become turbid. Then the solution was heated under N2 to 300 °C. After magnetic stirring, the resulting solution was kept at 300 °C for 60 min. After the reaction was completed, a black solution was obtained. This black product was precipitated by adding ethanol and acetone followed by centrifugation at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 5 min. The precipitate was washed several times and re-dispersed in toluene.
000 rpm for 5 min. The precipitate was washed several times and re-dispersed in toluene.
Fabrication of ultrafine Co2P@rGO (U-Co2P@rGO-5, U-Co2P@rGO-14, U-Co2P@rGO-25)
To synthesize the U-Co2P@rGO-5, U-Co2P@rGO-14, U-Co2P@rGO-25 the above described method was used, except 2.6 mg, 8.5 mg, 16.6 mg rGO were added into the mixture of CoCl2 and OLA.Preparation of commercial Co2P@rGO (C-Co2P@rGO)
To prepare C-Co2P@rGO, 8.5 mg rGO was added into a mixture of 500 mg commercial cobalt phosphide (Shanghai Chemical Corp, 98%) and 24 mL ethanol. After an hour of ultrasound, the product was precipitated by centrifugation at 12000 rpm for 5 min.
Characterization
The atomistic structural information and microtopography were characterized using TEM, HRTEM, selected area electron diffraction (SAED) and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM). Elemental mapping images were performed on a JEM-2200FS operating at an acceleration voltage of 200 kV, in which the corresponding FFT algorithms were analyzed. ADXRD patterns were measured with a monochromatic wavelength of 0.6199 Å at Beamline 15U1, Shanghai Synchrotron Radiation Facility (SSRF). CeO2 was utilized as the standard sample for the calibration. The pattern of intensity versus diffraction angle 2θ was plotted based on the FIT2D program, which integrated and analyzed the 2D images collected. The ADXRD experiments were conducted at room temperature. X-ray photoelectron spectroscopy (XPS) results were collected on a VG ESCALAB MKII spectrometer with Mg Kr excitation (1253.6 eV). The content of carbon was estimated using the vario EL cube. The nitrogen adsorption–desorption isotherm was measured at 77 K using a Kubo X1000 instrument (Beijing Builder Electronic Technology, China). The specific surface area calculation was performed using the BET method. The pore-size distribution (PSD) curve was calculated from the isotherm using the Barrett–Joyner–Halenda (BJH) algorithm.Electrochemical measurements
For the electrochemical measurements of the U-Co2P@rGO-14 and C-Co2P@rGO as anode materials, the electrodes were prepared by mixing the active material with Super P and sodium carboxymethyl cellulose (CMC-Na) binder in a weight ratio of 7:2:1. The obtained slurries were coated onto aluminum foil and dried in a vacuum oven at 120 °C overnight. The loading mass of the electrode was around 1 mg cm−2. For fabrication of the cathode electrodes, the active carbon and PTFE were mixed with a weight ratio of 9:1. CR2032 coin cells were fabricated inside an Ar filled glovebox, employing the glass fiber filter (Whatman GF/F) as the separator and K foil as both the counter and reference electrode. The electrolyte for PIBs was KPF6 (1.0 mol L−1) in an ethylene carbonate (EC)/diethyl carbonate (DEC)/ethyl methyl carbonate (EMC) solution (3:3:4, v/v/v). Hybrid K-ion capacitors (KICs) were likewise tested as coin cells, with pre-activated cathode and anode materials. For pre-activation, a half cell was first established using the active material as the working electrode, which was then charged–discharged for five cycles at 0.02 A g−1. Then the cells were disassembled inside the glove box, and a KIC was assembled by employing the pre-activated anode and cathode. The anode to cathode mass ratio was 4:1. All of the electrochemical tests were carried out at room temperature. Galvanostatic discharge–charge cycling was performed on a Land-2001A (Wuhan, China) automatic battery tester. CV was carried with a VSP multichannel potentiostatic–galvanostatic system (Bio-Logic SAS, France). The gravimetric energy and power densities of the KIC device were calculated by numerically integrating the galvanostatic discharge profiles using the equations below.5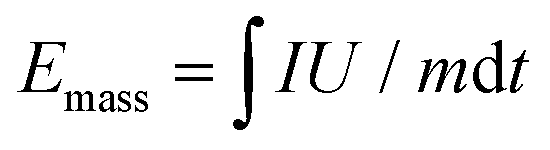 | (1) |
| Pmass = Emass/t | (2) |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 11874027, 21725304, and 11504126), the Chang Jiang Scholars Program of China (No. T2016051), Changbai Mountain Scholars Program (No. 2013007), and the Program for Innovative Research Team (in Science And Technology) in the University of Jilin Province, the China Postdoctoral Science Foundation (No. 2019T120233 and 2017M621198). F. D. thanks the Joint Project between Jilin Province and Jilin University (SXGJQY2017-10) and Science and Technology Development Project, Jilin Province (Grant No. 20180101211JC, 20170101168JC and 20180414004GH) for funding. Angle-dispersive XRD measurements were performed at the BL15U1 beamline, Shanghai Synchrotron Radiation Facility (SSRF). Y. W., X. Y., and B. Z. designed and performed experiments and analyzed data. Z. Z., G. W., and F. D. fabricated electrodes and conducted electrochemical testing. Y. S. provided advice about the electrochemical experiments. X. Y., F. D., and B. Z. supervised the work.Notes and references
- M. Lee, J. Hong, J. Lopez, Y. Sun, D. Feng, K. Lim, W. C. Chueh, M. F. Toney, Y. Cui and Z. Bao, Nat. Energy, 2017, 2, 861 CrossRef CAS.
- D. Wang, X. Bie, Q. Fu, D. Dixon, N. Bramnik, Y. Hu, F. Fauth, Y. Wei, H. Ehrenberg, G. Chen and F. Du, Nat. Commun., 2017, 8, 15888 CrossRef CAS PubMed.
- H. Wang, C. Zhu, D. Chao, Q. Yan and H. J. Fan, Adv. Mater., 2017, 29, 1702093 CrossRef PubMed.
- J. Billaud, F. Bouville, T. Magrini, C. Magrini and A. R. Studart, Nat. Energy, 2016, 1, 16097 CrossRef CAS.
- J. Yang, L. S. Tang, L. Bai, R. Y. Bao, Z. Y. Liu, B. H. Xie, M. B. Yang and W. Yang, Mater. Horiz., 2019, 6, 250 RSC.
- Z. Wang, C. Li and K. Domen, Chem. Soc. Rev., 2019, 48, 2109 RSC.
- J. Pang, R. G. Mendes, A. Bachmatiuk, L. Zhao, H. Q. Ta, T. Gemming, H. Liu, Z. Liu and M. H. Rummeli, Chem. Soc. Rev., 2019, 48, 72 RSC.
- C. Hu, M. Li, J. Qiu and Y. P. Sun, Chem. Soc. Rev., 2019, 48, 2315 RSC.
- Y. Zhao, G. I. Waterhouse, G. Chen, X. Xiong, L. Z. Wu, C. H. Tung and T. Zhang, Chem. Soc. Rev., 2019, 48, 1972 RSC.
- J. M. Giussi, M. L. Cortez, W. A. Marmisollé and O. Azzaroni, Chem. Soc. Rev., 2019, 48, 814 RSC.
- Z. Gao, H. Sun, L. Fu, F. Ye, Y. Zhang, W. Luo and Y. Huang, Adv. Mater., 2018, 30, 1705702 CrossRef PubMed.
- G. Wang, Y. Sui, M. Zhang, M. Xu, Q. Zeng, C. Liu, X. Liu, F. Du and B. Zou, J. Mater. Chem. A, 2017, 5, 18577 RSC.
- J. S. Sander, R. M. Erb, L. Li, A. Gurijala and Y. M. Chiang, Nat. Energy, 2016, 1, 16099 CrossRef CAS.
- K. Lu, H. Zhang, S. Gao, Y. Cheng and H. Ma, Nanoscale, 2018, 10, 20754 RSC.
- Y. Xu, F. Bahmani, M. Zhou, Y. Li, C. Zhang, F. Liang, S. H. Kazemi, U. Kaiser, G. Meng and Y. Lei, Nanoscale Horiz., 2019, 4, 202 RSC.
- C. Li, W. Yan, S. Liang, P. Wang, J. Wang, L. Fu, Y. Zhu, Y. Chen, Y. Wu and W. Huang, Nanoscale Horiz., 2019 10.1039/C8NH00484F.
- M. Samadi, N. Sarikhani, M. Zirak, H. Zhang, H. L. Zhang and A. Z. Moshfegh, Nanoscale Horiz., 2018, 3, 90 RSC.
- K. Moyer, J. Donohue, N. Ramanna, A. P. Cohn, N. Muralidharan, J. Eaves and C. L. Pint, Nanoscale, 2018, 10, 13335 RSC.
- X. Xu, K. S. Hui, D. A. Dinh, K. Hui and H. Wang, Mater. Horiz., 2019 10.1039/C8MH01375F.
- K. Kubota, M. Dahbi, T. Hosaka, S. Kumakura and S. Komaba, Chem. Rec., 2018, 18, 459 CrossRef CAS PubMed.
- J. Fu, Z. P. Cano, M. G. Park, A. Yu, M. Fowler and Z. Chen, Adv. Mater., 2017, 29, 1604685 CrossRef PubMed.
- A. Eftekhari and P. Corrochano, Sustainable Energy Fuels, 2017, 1, 1246 RSC.
- S. Wei, S. Xu, A. Agrawral, S. Choudhury, Y. Lu, Z. Tu, L. Ma and L. A. Archer, Nat. Commun., 2016, 7, 11722 CrossRef CAS PubMed.
- Q. Tan, P. Li, K. Han, Z. Liu, Y. Li, W. Zhao, D. He, F. An, M. Qin and X. Qu, J. Mater. Chem. A, 2019, 7, 744 RSC.
- L. Ma, P. Yan, S. Wu, G. Zhu and Y. Shen, J. Mater. Chem. A, 2019, 7, 843 RSC.
- A. Eftekhari, Z. Jian and X. Ji, ACS Appl. Mater. Interfaces, 2017, 9, 4404 CrossRef CAS PubMed.
- W. Zhang, J. Mao, S. Li, Z. Chen and Z. Guo, J. Am. Chem. Soc., 2017, 139, 3316 CrossRef CAS PubMed.
- N. Liu, X. Mamat, R. Jiang, W. Tong, Y. Huang, D. Jia, Y. Li, L. Wang, T. Wågberg and G. Hu, Chem. Eng. J., 2018, 343, 78 CrossRef CAS.
- D. Yu, Q. Pang, Y. Gao, Y. Wei, C. Wang, G. Chen and F. Du, Energy Storage Mater., 2018, 11, 1 CrossRef.
- X. Yang, R. Y. Zhang, J. Zhao, Z. X. Wei, D. X. Wang, X. F. Bie, Y. Gao, J. Wang, F. Du and G. Chen, Adv. Energy Mater., 2018, 8, 1701827 CrossRef.
- H. Gao, T. Zhou, Y. Zheng, Q. Zhang, Y. Liu, J. Chen, H. Liu and Z. Guo, Adv. Funct. Mater., 2017, 27, 1702634 CrossRef.
- I. Sultana, M. M. Rahman, T. Ramireddy, Y. Chen and A. M. Glushenkov, J. Mater. Chem. A, 2017, 5, 23506 RSC.
- Z. Zhang, J. Yang, Y. Nuli, B. Wang and J. Xu, Solid State Ionics, 2005, 176, 693 CrossRef CAS.
- D. Yang, J. Zhu, X. Rui, H. Tan, R. Cai, H. E. Hoster, D. Y. W. Yu, H. H. Hng and Q. Yan, ACS Appl. Mater. Interfaces, 2013, 5, 1093 CrossRef CAS PubMed.
- A. Lu, X. Zhang, Y. Chen, Q. Xie, Q. Qi, Y. Ma and D. L. Peng, J. Power Sources, 2015, 295, 329 CrossRef CAS.
- T. Shen, Z. Yao, X. Xia, X. Wang, C. Gu and J. Tu, Adv. Eng. Mater., 2018, 20, 1700591 CrossRef.
- L. Zhang, S. Yan, S. Jiang, K. Yang, H. Wang, S. He, D. Liang, L. Zhang, Y. He and X. Lan, Nucl. Sci. Technol., 2015, 26, 060101 Search PubMed.
- X. Chen, M. Cheng, D. Chen and R. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 3892 CrossRef CAS PubMed.
- Z. Huang, Z. Chen, Z. Chen, C. Lv, M. G. Humphrey and C. Zhang, Nano Energy, 2014, 9, 373 CrossRef CAS.
- V. V. T. Doan-Nguyen, S. Zhang, E. B. Trigg, R. Agarwal, J. Li, D. Su, K. I. Winey and C. B. Murray, ACS Nano, 2015, 9, 8108 CrossRef CAS PubMed.
- Q. Xie, D. Zeng, P. Gong, J. Huang, Y. Ma, L. Wang and D. L. Peng, Electrochim. Acta, 2017, 232, 465 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603 CAS.
- Y. Xu, C. Zhang, M. Zhou, Q. Fu, C. Zhao, M. Wu and Y. Lei, Nat. Commun., 2018, 9, 1720 CrossRef PubMed.
- X. Ren, Q. Zhao, W. D. McCulloch and Y. Wu, Nano Res., 2017, 10, 1313 CrossRef CAS.
- W. J. Li, Q. R. Yang, S. L. Chou, J. Z. Wang and H. K. Liu, J. Power Sources, 2015, 294, 627 CrossRef CAS.
- R. A. Adams, J. M. Syu, Y. Zhao, C. T. Lo, A. Varma and V. G. Pol, ACS Appl. Mater. Interfaces, 2017, 9, 17872 CrossRef CAS PubMed.
- J. Zhou, L. Wang, M. Yang, J. Wu, F. Chen, W. Huang, N. Han, H. Ye, F. Zhao and Y. Li, Adv. Mater., 2017, 29, 1702061 CrossRef PubMed.
- V. Lakshmi, Y. Chen, A. A. Mikhaylov, A. G. Medvedev, I. Sultana, M. M. Rahman, O. Lev, P. V. Prikhodchenko and A. M. Glushenkov, Chem. Commun., 2017, 53, 8272 RSC.
- Z. Liu, T. Lu, T. Song, X.-Y. Yu, X. W. Lou and U. Paik, Energy Environ. Sci., 2017, 10, 1576 RSC.
- Y. Li, Z. Kang, X. Yan, S. Cao, M. Li, Y. Guo, Y. Huan, X. Wen and Y. Zhang, Nanoscale, 2018, 10, 9360 RSC.
- Z. Fan, Y. Wang, Z. Xie, X. Xu, Y. Yuan, Z. Cheng and Y. Liu, Nanoscale, 2018, 10, 9642 RSC.
- J. Yin, L. Qi and H. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 2762 CrossRef CAS PubMed.
- A. L. Comte, Y. Reynier, C. Vincens, C. Leys and P. Azais, J. Power Sources, 2017, 363, 34 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nh00211a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |