
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLow-temperature activation of carbon black by selective photocatalytic oxidation†
Niels R.
Ostyn
 a,
Julian A.
Steele
a,
Michiel
De Prins
a,
Sreeprasanth Pulinthanathu
Sree
a,
C. Vinod
Chandran
a,
Wauter
Wangermez
a,
Gina
Vanbutsele
a,
Jin Won
Seo
b,
Maarten B. J.
Roeffaers
a,
Eric
Breynaert
a and
Johan A.
Martens
*a
a,
Julian A.
Steele
a,
Michiel
De Prins
a,
Sreeprasanth Pulinthanathu
Sree
a,
C. Vinod
Chandran
a,
Wauter
Wangermez
a,
Gina
Vanbutsele
a,
Jin Won
Seo
b,
Maarten B. J.
Roeffaers
a,
Eric
Breynaert
a and
Johan A.
Martens
*a
aCentre for Surface Chemistry and Catalysis, KU Leuven, Celestijnenlaan 200F, box 2461, 3001 Heverlee, Belgium. E-mail: johan.martens@kuleuven.be; Tel: +32 16 32 16 37
bDepartment of Materials Engineering, KU Leuven, Kasteelpark Arenberg 44, 3001 Heverlee, Belgium
First published on 22nd May 2019
Abstract
Carbon black is chemically modified by selective photocatalytic oxidation, removing amorphous carbon and functionalizing the graphitic fraction to produce porous, graphitized carbon black, commonly used as an adsorbent in chromatography. In contrast to pyrolytic treatments, this photocatalytic modification proceeds under mild reaction conditions using oxygen, nitric oxide, water vapor and a titanium dioxide photocatalyst at 150 °C. The photo-oxidation can be performed both with the photocatalyst in close proximity (contact mode) or physically separated from the carbon. Structural analysis of remotely photo-oxidized carbon black reveals increased hydrophilic properties as compared to pyrolysis at 700 °C in a N2 atmosphere. Carbon black photo-oxidation selectively mineralizes sp3-hybridized carbon, leading to enhanced graphitization. This results in an overall improved structural ordering by enriching carbon black with sp2-hybridized graphitic carbon showing decreased interplanar distance, accompanied by a twofold increase in the specific surface area. In addition, the photo-oxidized material is activated by the presence of oxygen functionalities on the graphitic carbon fraction, further enhancing the adsorptive properties.
1. Introduction
Carbon black (CB) represents a family of carbon powders produced on a large scale (over 12 million tons per year).1 Its structural properties, particle size and porosity, strongly depend on the primary feedstock and manufacturing process (thermal decomposition). Feedstocks primarily include oil, coal or natural gas. Next to its main application as a black pigment and electrical conductor, CB serves as an excellent reinforcing filler in tires and other rubber products because of its mechanical, chemical and electrical properties.1,2 CB is generally described as large agglomerates of graphitic and amorphous carbon aggregates consisting of spherical primary particles (Fig. 1). A distinguishing property is the carbon atom hybridization, where sp2-hybridization is characteristic of the graphite fraction, while sp3-hybridized carbon typically reflects the amorphous hydrocarbon fraction.3 CB particles, with sizes ranging from 10 to 100 nm,3–5 consist of a rather amorphous core and quasi-crystalline shell with graphitic carbon stacks.5–7 The carbon particles are often described as onion-like structures held together by van der Waals forces.8 These graphitic nanocrystallites typically exhibit turbostraticity, i.e. large structural disorder as a result of rotational misalignment. The coexistence of a disordered carbon fraction alongside ordered carbon led to the classification of CB as an intermediate between amorphous and crystalline carbon materials.3,5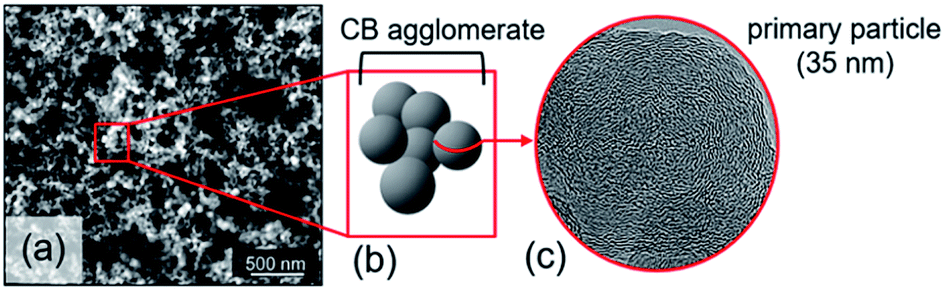 | ||
| Fig. 1 (a) High-resolution SEM image of CB nanoparticles. (b) Simplified representation of CB morphology. (c) High-resolution TEM micrograph of CB nanoparticle. | ||
Many applications require modification of pristine carbon black. Graphitized carbon black (GCB) is, for example, a widely used adsorbent offering large external surface area with excellent homogeneity and purity. This adsorbent is mainly suitable for selective purification processes in chromatography.9–11 Unfortunately, the production of GCB by thermal treatment of CB in anoxic conditions is energy intensive due to the required high temperatures.12–14 This has motivated research to increase the energy efficiency, for example, by catalytic or pressure induced graphitization. Though more sustainable, these processes still require fairly high temperatures (600–2500 °C) or pressures (0.5–5 GPa).15–18
In this work, low-temperature photocatalytic oxidation is presented for chemical modification of CB. This ultraviolet (UV) light driven process is compared to a classical thermal treatment of CB. Titanium dioxide (TiO2) materials are specifically known for their excellent photocatalytic activity in the decomposition of organic compounds.19,20 For automotive applications, TiO2-based photocatalysis has been studied for complete oxidation of CB into carbon dioxide at temperatures as low as 150 °C.21,22 While photo-oxidation is too slow for practical application in diesel particulate filter regeneration, TiO2 photocatalysis could be of interest for CB activation. Surface oxidation of CB, for example by acidic and electrochemical treatment, renders it as an active catalyst for the water and alcohol oxidation reactions.23 Many studies on photocatalytic mineralization of carbon materials by UV-illuminated TiO2 show the involvement of airborne oxidant species. These gas phase oxidants, photogenerated by TiO2, allow a remote oxidation at distances over 2 mm.24–30 Structural carbon modifications underpinning the photocatalytic oxidation were revealed using a combination of nuclear magnetic resonance (NMR) and Raman spectroscopies, X-ray diffraction (XRD) and transmission electron microscopy (TEM). The carbon black morphology and porosity have been investigated using scanning electron microscopy (SEM) and nitrogen physisorption. Energy dispersive X-ray spectroscopy (EDS) and electron energy loss spectroscopy (EELS) were particularly useful for revealing the structural and elemental composition.
2. Experimental section
2.1 Materials
Printex U carbon black (Degussa, now Orion Engineered Carbons) is a synthetic carbon produced from natural gas.31 It has a primary particle size of around 30 nm, specific surface area ranging from 100 to 141 m2 g−1 and a volatile organic compound fraction of 5 to 6 wt%. The carbon and oxygen contents are about 90 and 8 wt%, respectively.21,31–33 CristalACTIV™ PC-500 TiO2 (Cristal), an almost pure anatase phase (≥99.0 wt%), was used as the photocatalyst. This TiO2 material has a specific surface area of approximately 350 m2 g−1 and a primary particle size of about 10 nm. CB and TiO2 were slurried by magnetic stirring in isopropanol (Fisher Scientific) at 400 rpm for 15 minutes, at concentrations of 0.71 and 7.1 g L−1 respectively.2.2 Photocatalytic oxidation
Photocatalysis experiments were carried out using a custom-made, heated, flat rectangular photoreactor with internal volume of 82 cm3, sealed at atmospheric pressure by a borosilicate glass cover plate. Constant temperature during photoreaction is obtained by the stainless steel thermostatic support with temperature control (Fig. S1†). The flat photoreactor was illuminated from the top by UV light with a wavelength of 368 nm (Plus Lamp™, 15 W). Gases (alpha gas 1: O2 and NO, N2 and H2 (for leakage test)) were fed into the photoreactor through Teflon™ tubes using mass flow controllers (Bronkhorst) (see Table S1† for specifications). Water vapor was added to the gas flow by a thermostatic saturator. The reactor outlet gases were diluted with N2 carrier gas and transferred on-line to autocalibrated gas analyzers (ABB) for NH3, NO and NO2 (UV detector), and CO, CO2 and N2O (NDIR detector), ensuring quantitative analysis.The configuration of the TiO2 and CB layers in the photoreactor is presented in Fig. 2. Two different modes of operation were used, either with TiO2 in close proximity of carbon black (Fig. 2a), referred to as contact mode, or with TiO2 physically separated from the carbon layer (Fig. 2b), denoted as remote mode.
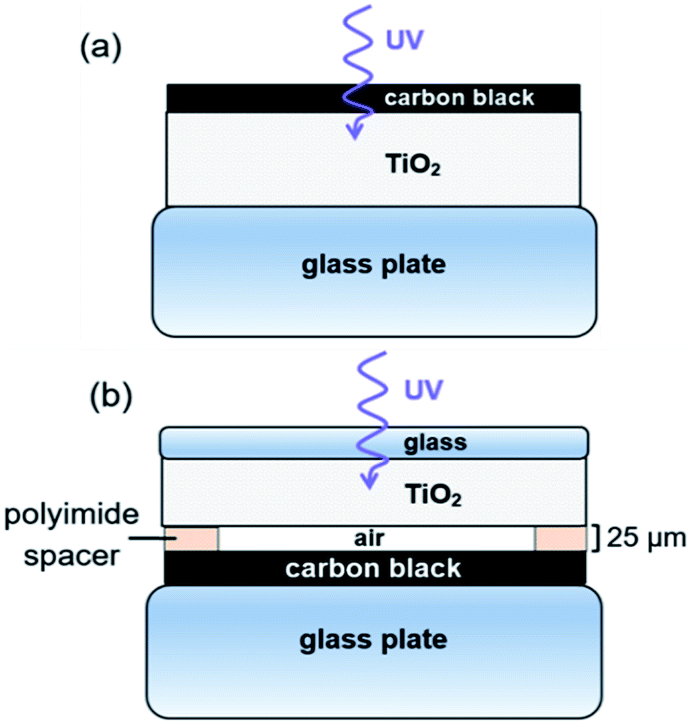 | ||
| Fig. 2 Configuration of CB and TiO2 layers. (a) Contact photocatalysis. (b) Remote photocatalysis experiments. | ||
For contact mode photo-oxidation, 430 μL of the TiO2 and CB suspensions were pipetted layer-by-layer on a glass support, with an intermediate drying step at room temperature. CB was pipetted on top of the dried TiO2 with a CB![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TiO2 weight ratio of 1:10. Supported films were further dried in an oven at 150 °C for at least 1 h. In contact mode, UV front irradiation was used, allowing suitable access for Raman scattering, SEM, TEM-EELS and EDS experiments of the treated CB. Contact photo-oxidation was not analyzed by NMR spectroscopy, XRD and nitrogen physisorption due to the carbon–TiO2 mix. For remote photo-oxidation, TiO2 and CB layers were deposited on different glass supports, separated by 25 μm-thick strips of polyimide to space the reaction (Kapton® HN). The CB:TiO2 weight ratio was 1:5 in remote experiments, that were performed with back UV irradiation to prevent absorption of light by CB.
:TiO2 weight ratio of 1:10. Supported films were further dried in an oven at 150 °C for at least 1 h. In contact mode, UV front irradiation was used, allowing suitable access for Raman scattering, SEM, TEM-EELS and EDS experiments of the treated CB. Contact photo-oxidation was not analyzed by NMR spectroscopy, XRD and nitrogen physisorption due to the carbon–TiO2 mix. For remote photo-oxidation, TiO2 and CB layers were deposited on different glass supports, separated by 25 μm-thick strips of polyimide to space the reaction (Kapton® HN). The CB:TiO2 weight ratio was 1:5 in remote experiments, that were performed with back UV irradiation to prevent absorption of light by CB.
Photo-oxidation experiments were initiated by purging the tubes and loaded photoreactor with dry nitrogen for about 15 minutes while heating to 150 °C. A continuous gas flow composed of 21 vol% O2, 5000 ppm NO and 1.8 vol% H2O, referred to as gas mixture 1, was subsequently sent through the photoreactor in the dark at a rate of 5.4 L h−1. Batch experiments were performed by closing the photoreactor inlet and outlet valves after stabilization of the gas concentrations in gas mixture 1, followed by switching on the UV lamp. Eventually, the product gas mixture was analyzed by sending it to the analyzers using nitrogen carrier gas. NOx conversion and N2O formation were calculated by the nitrogen atom balance.
2.3 Carbon black pyrolysis
Carbon black was thermally treated at 700 °C in an inert N2 atmosphere. The sample was heated at a rate of 1 °C min−1 until the set temperature was reached. This maximum pyrolysis temperature was maintained for 5 h, after which CB was cooled down to room temperature.2.4 Carbon black characterization
NMR experiments were carried out on a Bruker AVIII 500 spectrometer with 1H Larmor frequency at 500.13 MHz, operating at a static magnetic field of 11.74 T. Samples possessing the same sample weights were loaded in 1.9 mm zirconia rotors with vespel caps and with bottom and top columns of zirconia powder. Direct excitation 1H MAS NMR measurements were recorded at a MAS frequency of 40 kHz, using π/2 pulses of 200 kHz and recycle delays of 10 s (32 scans). Spectra were chemical shift calibrated versus TMS, using adamantane as a secondary reference.Raman scattering experiments were performed on a homemade system using a TriVista™ Spectrometer System with CCD detector from Princeton Instruments. Laser excitation was achieved using the 488 nm (2.54 eV) emission of an Ar+ laser, coupled into an optical microscope (IX 71) equipped with an ×20 magnification objective. A laser wavelength of 488 nm was selected to resonantly enhance Raman signals in the carbon materials, with laser powers kept relatively low (<6 mW) to exclude excessive sample heating. All Raman spectra were recorded within the range 600–3100 cm−1 and further analyzed using the Multipeak Fitting 2 package in the Igor Pro software program. An example of a deconvoluted spectrum is provided in the ESI (Fig. S2†).
XRD was performed using a STOE Powder Diffraction System (STOE) with Cu Kα radiation (1.54060 Å) in the 2θ range 0–90°. Samples were loaded in small capillary tubes. The diffraction patterns were analyzed with the STOE WinXPOW powder diffraction software.
High-Resolution (HR) SEM was carried out on a Nova NanoSEM 450 scanning electron microscope (FEI, Eindhoven). The carbon sample deposited on a small Al plate (before and after photo-oxidation) was attached to an Al stub with carbon tape, followed by imaging without any further sample modification. For the HR TEM measurement, the carbon samples were prepared by placing a droplet of the colloidal dispersion in isopropanol on a carbon-coated copper grid. TEM micrographs were taken using an ARM200F transmission electron microscope (JEOL) operating at 200 kV equipped with a cold field emission gun, energy dispersive X-ray detector and Gatan Image Filter (GIF, Tridiem 863 Gatan).
Nitrogen physisorption was performed using a TriStar Surface Area and Porosity Analyzer from Micromeritics. The CB samples were pretreated by heating the powders to 400 °C under N2 atmosphere using the SmartPrep™ Programmable Degas System from Micromeritics. BET surface areas and BJH cumulative pore volumes were obtained by TriStar 3000 software calculations. The micropore volumes were obtained by t-plot analysis.
3. Results and discussion
CB photo-oxidation at 150 °C using anatase TiO2 in presence of O2, NOx and H2O produces carbon oxides (COx), while reducing both NO and NO2 into chemically inert N2. Table 1 shows CO2 and CO production, NOx conversion and N2O byproduct formation, during two consecutive photocatalytic runs, starting from 5 mg of CB (CB:TiO2 weight ratio of 1:10). In between the two runs, the headspace of the photoreactor was renewed by gas mixture 1 (21% O2, 5000 ppm NO and 1.8% H2O). Overall, 30.2% of the initial CB converted into CO2 (97.7%) and CO (2.3%). Gas analysis after the second photocatalytic run shows a lower carbon oxidation rate compared to the initial run. This decrease indicates more reactive CB was present at the photo-oxidation onset, as also confirmed by higher NOx conversion in the first photocatalytic run. Next to a less easy oxidation of amorphous carbon to gaseous COx, the decreased CO2 formation in the second run could also be ascribed to an increased formation of stable oxygen functionalities on CB surface. Due to their CB oxidation performance, NOx species are largely reduced into N2, but a small amount of N2O forms as a photoreaction byproduct (see Table 1). In a 72 h control experiment at 150 °C in gas mixture 1, involving neither TiO2 nor UV light, 10.0% of CB converted into CO2 (80.1%) and CO (19.9%). In addition, the NOx conversion only reached 71.0%, emphasizing the essential contribution of anatase TiO2. A second 72 h control experiment at 150 °C without TiO2 was performed in inert N2 under UV light to show the effect of only heat and light. This experiment converted only 2.8% of CB into CO2 (89.1%) and CO (10.9%).
| UV illumination period | CO2 formed [%] | CO formed [%] | NOx conversion [%] | N2O formed [%] |
|---|---|---|---|---|
| 0–72 h (72 h) | 20.4 | 0.4 | 95.2 | 12.7 |
| 72–165 h (93 h) | 9.1 | 0.3 | 90.6 | 10.7 |
| Total | 29.5 | 0.7 | 92.9 | 23.4 |
Chemical modification of CB by photo-oxidation can be characterized using 1H NMR spectroscopy, especially by comparing remotely photo-oxidized CB (CBR72) with thermally treated (CB700) and pristine CB (CB). 1H MAS NMR spectra for these samples, recorded at 40 kHz using a 1.9 mm triple channel MAS probe, are shown in Fig. 3, before and after drying in vacuum at 150 °C. For these measurements, 1.9 mm zirconia rotors equipped with vespel top and bottom caps were used, packing small columns of zirconia powder at the bottom and top of the rotor to center the carbon samples and avoid contamination of the rotor caps.
 | ||
| Fig. 3 Magic angle spinning 1H NMR of (a) CBR72, (b) CB700 and (c) pristine CB, and their respective dried versions (d), (e) and (f) at a spinning frequency of 40 kHz and at a static magnetic field of 11.74 T. | ||
In the spectra in Fig. 3, the area highlighted in blue is the rotor background. The region highlighted with green represents water associated with zirconia powder, as has been confirmed with a rotor packed with zirconia powder only. All other resonances result from the presence of water in different environments. The region highlighted in red is assigned to the water clusters associated with the oxidized carbon. This signal is exclusively present in the photocatalytically oxidized sample CBR72 under both humid and dry conditions. The region with purple color is attributed to water trapped between with crystalline carbon fractions, similar to the case of diamagnetic shifts of water resonances in literature.34,35Fig. 4 illustrates different types of water contained in CBR72, as derived from the 1H NMR analysis. The higher water affinity of the photo-oxidized sample as compared to the pristine and thermally treated CB samples could be related to surface functionalization, but also to increased porosity in the sample. This high affinity is evident from the spectra of CB samples in humid and dry conditions. Upon drying however, complete removal of this water fraction was only observed for CB and CB700, while only partial removal was observed for CBR72. In the spectrum of humid CB, also a resonance around 4.7 ppm is observed, representing bulk water. Summarizing, 1H NMR points towards the photocatalytic formation of CB with oxidized graphitic fraction showing high water affinity.
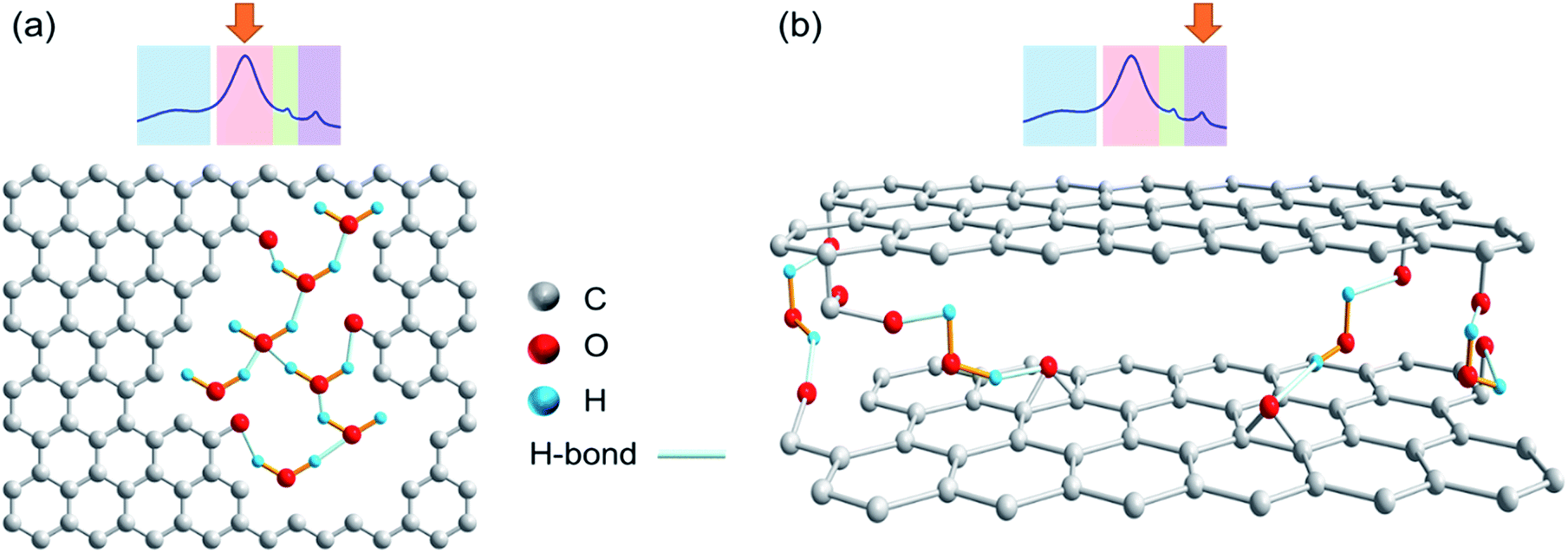 | ||
| Fig. 4 Chemical structures showing different water types present in the remotely photo-oxidized sample CBR72. (a) Intralayer or pore water. (b) Interlayer water. | ||
Raman spectra of pristine CB and CB photo-oxidized in contact and remote modes (CB, CBc165, CBC72, and CBR72 respectively) are shown in Fig. 5. Analyzing the Raman spectra in detail, multiple bands can be observed. The D band, at 1360 cm−1, is often denoted as the disorder band because it indicates the presence of defects in an sp2-hybridized carbon structure. The D band represents a breathing mode of six-membered carbon rings with A1g symmetry, corresponding to out-of-plane displacements, a process exclusively activated by defects. The G band, at 1600 cm−1, is referred to as the graphite band and represents planar sp2 carbon. It is ascribed to in-plane bond stretching vibrations of pairs of sp2-hybridized carbon atoms with E2g symmetry.36–38 The intensity ratio of the D and G bands (ID/IG) is therefore a suitable probe of the degree of disorder in the system.
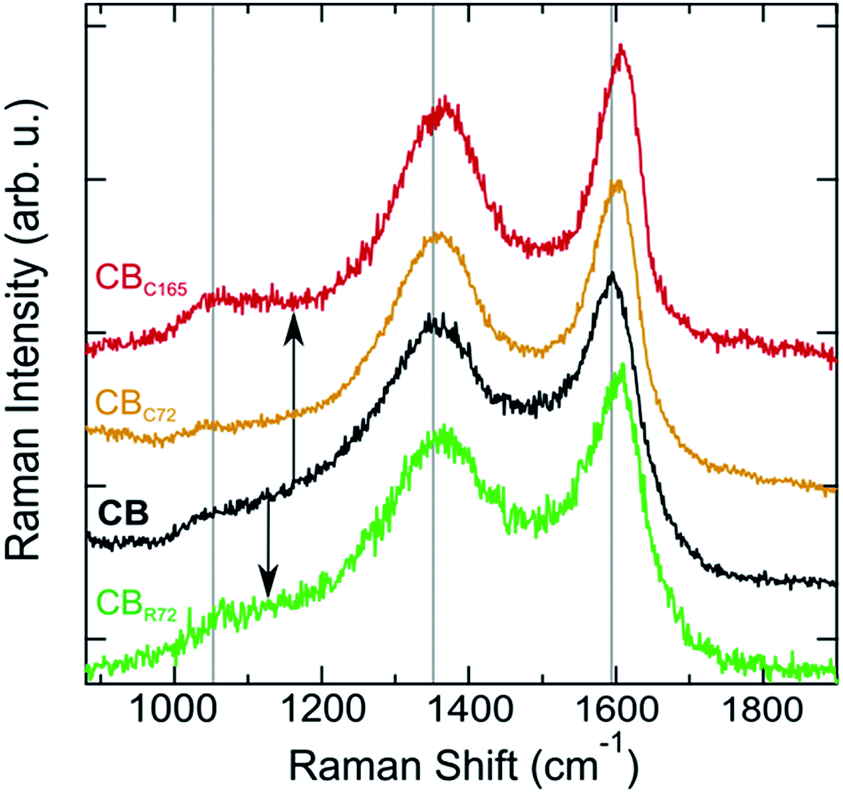 | ||
| Fig. 5 Comparison of Raman spectra of pristine CB, CBC72, CBC165 and CBR72. | ||
Upon contact mode and remote photo-oxidation, the Raman spectra show a similar spectral evolution for both samples. Comparing the spectra (Fig. 5), a more ordered quasi-crystalline CB structure can be observed, due to selective removal of amorphous carbon. This results in better resolved D and G bands by the gradual removal of the amorphous carbon band overlapping at 1530 cm−1.3–5,18,39,40 Overall, partial graphitization can be observed from the upward shift of both D and G bands, the decrease in D band width, the more symmetric G band and the overall ID/IG decrease. Higher D and G band frequencies and lower FWHMs are attributed to a higher degree of order in the CBs.41,42 Mean Raman parameters for the D and G bands allow quantifying the difference between the materials (Table 2). While the extent of spectral modification is lower for CBR72, as a result of the lower photo-oxidation rate at extended distances and the lower CB:TiO2 weight ratio (1:5) as compared to contact photocatalysis, both samples exhibit a tendency towards graphitization.
| Sample | D-band peak position [cm−1] | G-band peak position [cm−1] | D-band FWHM [cm−1] | G-band FWHM [cm−1] | I D/IG | ExpTau of G |
|---|---|---|---|---|---|---|
| CB | 1357 (3) | 1598 (2) | 254 (9) | 113 (5) | 2.9 (0.3) | −62 (6) |
| CBC72 | 1359 (1) | 1602 (1) | 187 (9) | 95 (2) | 2.4 (0.2) | −53 (1) |
| CBC165 | 1362 (2) | 1606 (1) | 178 (6) | 81 (3) | 2.7 (0.1) | −43 (1) |
| CBR72 | 1359 (2) | 1601 (2) | 237 (11) | 109 (3) | 2.8 (0.3) | −56 (3) |
Graphitization leads to a less asymmetric G band, as indicated by the reduced magnitude of the ExpTau values in Table 2. ExpTau describes the exponential decay of the exponentially modified Gaussian functions, so the larger the magnitude, the faster the decay and the more asymmetric the G band shape. After an extended photo-oxidation time, the graphitic CB fraction gradually becomes oxidized as can be seen from the continuous decrease in G band full width at half maximum (FWHM) and the ID/IG increase from 2.4 to 2.7 for CBC165. The gas analysis results (supra), on the one hand, indicate the partial graphitization occurs by conversion into CO2 and CO through the strong oxidative effect of ˙O2−, NOx radicals and ˙OH. An electron paramagnetic resonance (EPR) spectroscopy study showed the presence of the oxygen free radicals in similar reaction conditions.43 On the other hand, the strongly reduced CO2 formation at almost constant NOx conversion during the second run, confirms the gradual oxidation of graphitic CB (Table 1).
Further structural changes such as crystallinity and interlayer distance are resolved from X-ray diffraction, as shown in Fig. 6. In the diffraction patterns, two reflections can be observed at respectively, 24.1 and 43.1 degrees 2θ. The most intense reflection (at. 24.1° 2θ) arises from X-rays diffracted by the (002) graphitic planes in the CB.3,44–46 The second corresponds to the (100)/(101) reflections of the graphitic fraction of CB. Upon remote photo-oxidation the (002) diffraction peak becomes sharper, confirming the removal of amorphous carbon and gradually increasing contribution of graphitic carbon. Additionally, the (002) peak shifts towards higher angles, implying a reduction in interplanar distance (d002). Note that this inter-plane spacing in pure graphite is 3.35 Å as compared to 3.5 to 3.7 Å in the present samples.47 Selective mineralization of the amorphous carbon causes the transition of the turbostratic graphitic-like crystallite structure into more perfectly ordered ABA stacking of graphite layers. A similar decrease in d002 spacing was also observed by Asokan et al., upon treating CB with microwave irradiation.44
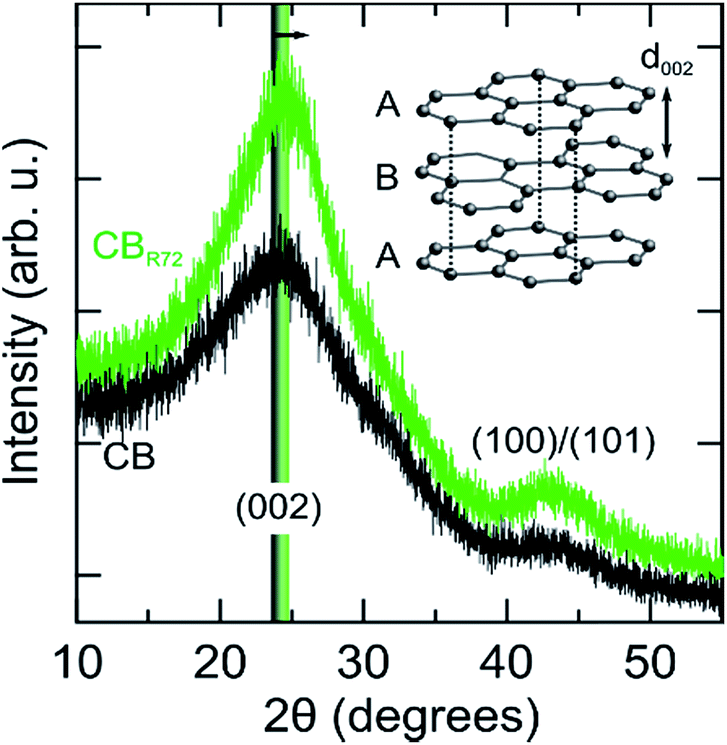 | ||
| Fig. 6 XRD patterns of pristine (CB) and remotely photo-oxidized (CBR72) carbon black. The inset shows the graphene sheet-like stacking giving rise to the (002) Bragg diffraction peak, which is shifted to higher scattering angles (smaller inter-plane spacing) following photo-oxidation (arrow). | ||
SEM analysis (Fig. 7) reveals the morphology of pristine and oxidized CB by both contact and remote photocatalysis. Photo-oxidized CB particles have become smaller and are less spherical in shape. CBC72 (Fig. 7e and f) consists of smaller, elongated, carbon structures relative to CBR72 shown in Fig. 7c and d. Contact photocatalysis has the most dramatic effect on the nanoparticles size and shape, due to the higher photo-oxidation rate. The larger void spaces in CBC72 and CBR72, highlighted at lower magnifications (Fig. 7e and c), also illustrate the conversion of solid carbon fractions into gaseous COx by the selective carbon mineralization. The observed effect on the outer appearance of CB is however rather subtle since many spherical particles are still intact. Apart from the uniform particle size reductions and slight particle deformations, CB photo-oxidation mainly affects the internal chemical structure as observed by NMR, Raman and XRD analyses.
 | ||
| Fig. 7 HR SEM images. (a and b) Pristine CB. (c and d) CBR72. (e and f) CBC72. Both contact and remote experiments were performed simultaneously in the same reactor at 150 °C for 72 h with gas mixture 1. | ||
As seen in Table 3, nitrogen physisorption indicates a CB porosity change resulting from the gas-assisted photo-oxidation, as suggested by 1H NMR and SEM. A large increase in BET specific surface area from 146 m2 g−1 to 277 m2 g−1 is measured after the remote photo-oxidation in gas mixture 1 at 150 °C. This porosity increase is partly caused by a larger number of micropores. Selectively removing the amorphous carbon from otherwise graphitic CB nanoparticles leads to a larger microporous volume, covering almost completely the increase in cumulative pore volume. These microporous volumes are determined using t-plot nitrogen adsorption isotherms (Harkins and Jura), shown in the ESI (Fig. S4†). Additionally, the increase in specific surface area is assumed to be due to the decreasing particle size and larger gaps between the particles upon photo-oxidation, indicated by SEM.1,48 The CB surface area is nearly doubled due to the remote photocatalytic treatment, which causes many small pores and interparticle gaps. Therefore, graphitic CB fractions become more accessible by the removal of conjunctive amorphous carbon and increase of total void fraction, shown in Fig. 7. Note that a porous CB with relatively higher graphitic carbon content is of great scientific interest for adsorbent applications. Strong evidence for the oxygen functionalization of the graphitic carbon is further provided.
| Sample | Surface area [m2 g−1] | Pore volume [cm3 g−1] | Micropore volume [mL g−1] |
|---|---|---|---|
| CB | 146 ± 2 | 0.157 | 0.043 |
| CBR72 | 277 ± 5 | 0.221 | 0.102 |
The structural and chemical analysis of the different CBs is summarized in Fig. 8 by means of TEM, EELS and EDS. In the HR TEM images, graphitic carbon structures with curved and facetted sections can be seen in all samples with interplanar spacing strongly varying between 0.3 nm and 0.43 nm. EELS spectra confirm the sp2-type graphitic structure with the presence of the characteristic peaks at 285 eV and 290 eV corresponding to transitions to the π* and σ* orbitals (Fig. 8a). The sharp π* peak at 285 eV, which is characteristic of sp2-hybridized carbon, is the highest for the CB-contact sample and the lowest for the CB-reference, confirming the increased graphitization degree after photocatalytic oxidation. However, it has to be noted that the EELS were taken not from one specific carbon particle but from a larger area in order to limit the irradiation damage. Therefore, the Ti- and O absorption edges are also visible in the CB-contact sample. Carbon materials are well known to be strongly sensitive to electron irradiation, in particular, at a high acceleration voltage due to the knock-on displacement of carbon atoms.49,50 Therefore, a quantitative comparison of the carbon structure is difficult.
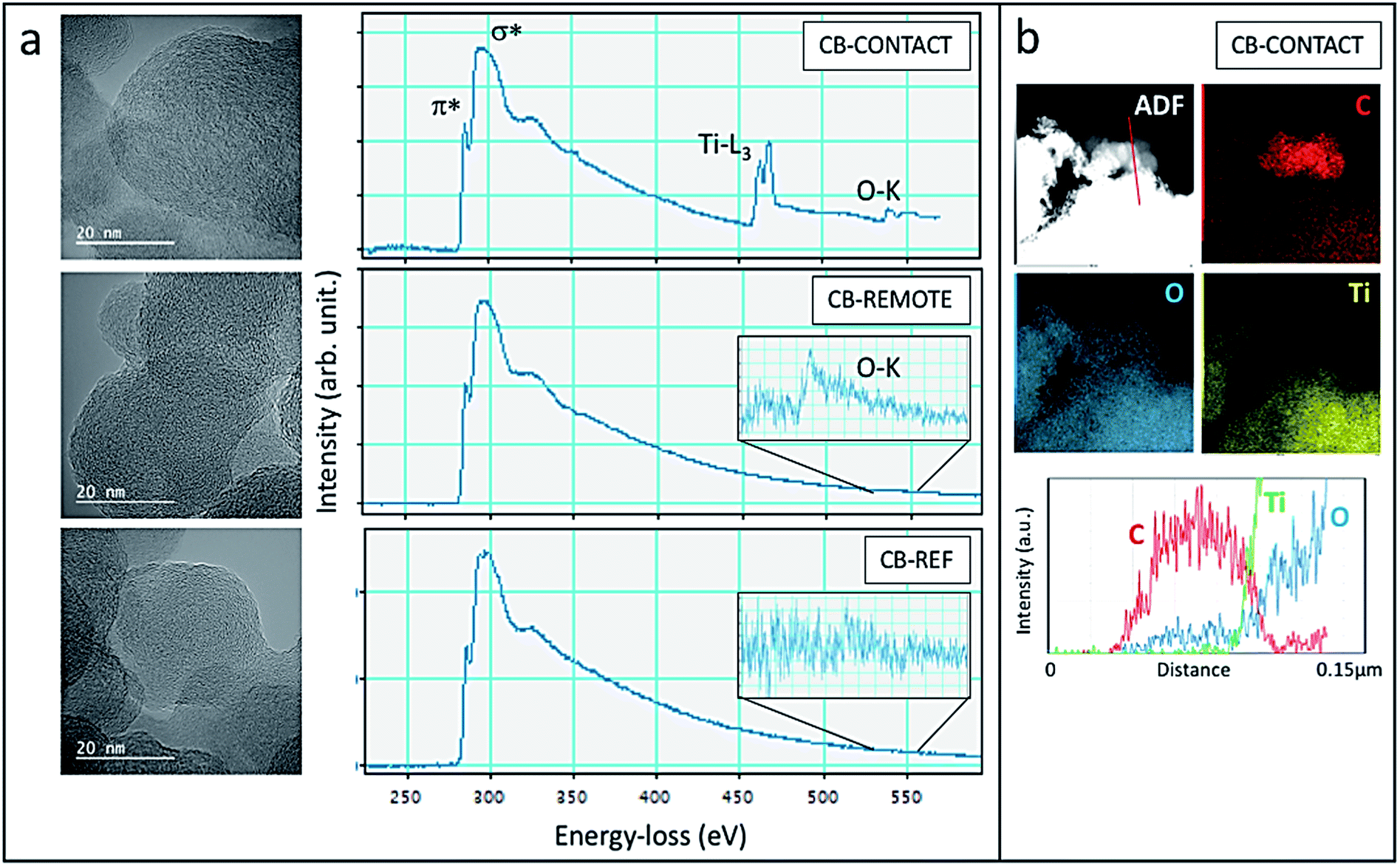 | ||
| Fig. 8 (a) HR TEM images and EELS spectra of CB-contact (top), CB-remote (middle) and CB-reference (bottom). The insets show magnified EELS spectra of the O–K peak. (b) The STEM-ADF image of the CB-contact sample with the respective EDS maps for C–K, O–K and Ti–K and the line-scan (bottom) across the red line indicates the presence of oxygen in CB. | ||
Upon contact and remote photocatalytic treatment, samples exhibit oxygen signals at 532 eV (see inset of Fig. 8a), which are absent in the CB-reference sample, demonstrating the photo-oxidation effectiveness. In the CB-contact sample, the oxidation also becomes visible in the EDS chemical map (Fig. 8b): at the location of carbon nanoparticles (red in the filtered C-map) a trace of oxygen can be seen whereas Ti-is absent. The line-scan across the carbon nanoparticles indeed confirms the presence of oxygen due to effective oxidation.
4. Conclusions
The amorphous carbon fraction present in carbon black is selectively mineralized using oxidative TiO2 photocatalysis in O2, NOx and H2O. Photocatalytic CB oxidation is performed by either physical contact or by remote photocatalyst control. In contrast to CB pyrolysis, the carbon is chemically modified at low temperature through this photocatalytic treatment. Easily oxidizable and refractory carbon fractions of CB react differently via the gas-assisted photocatalytic oxidation. A partially graphitized CB with high water affinity is obtained by preferentially removing the disordered sp3-hybridized carbon over the graphitic sp2-hybridized carbon fraction. The graphitic carbon product is activated by strongly increased porosity and oxygen functionalization due to the selective oxidation process. Photo-oxidation causes furthermore a size reduction of the elementary CB particles, and a shape change from spherical to slightly elongated. Photo-oxidation with anatase TiO2 offers an eco-friendly way of selectively removing the amorphous CB fraction, while functionalizing the graphitic carbon. The final porous CB enriched with oxidized graphitic carbon fractions could potentially serve as adsorbent material or be used in catalyst applications. From a materials perspective, remote photo-oxidation offers an easy and powerful avenue toward the production of activated CB.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by long-term structural funding by the Flemish government (Methusalem). The TEM equipment was funded by the Hercules project AKUL/13/19.References
- M. Gautier, V. Rohani and L. Fulcheri, Int. J. Hydrogen Energy, 2017, 42, 28140–28156 CrossRef CAS.
- J.-C. Huang, Adv. Polym. Technol., 2002, 21, 299–313 CrossRef CAS.
- T. Ungár, J. Gubicza, G. Ribárik, C. Pantea and T. W. Zerda, Carbon, 2002, 40, 929–937 CrossRef.
- P. Xue, J. Gao, Y. Bao, J. Wang, Q. Li and C. Wu, Carbon, 2011, 49, 3346–3355 CrossRef CAS.
- M. Pawlyta, J.-N. Rouzaud and S. Duber, Carbon, 2015, 84, 479–490 CrossRef CAS.
- J. B. Donnet, J. Schultz and A. Eckhardt, Carbon, 1968, 6, 781–788 CrossRef CAS.
- J. B. Donnet, Carbon, 1994, 32, 1305–1310 CrossRef CAS.
- P. A. Hartley, G. D. Parfitt and L. B. Pollack, Powder Technol., 1985, 42, 35–46 CrossRef CAS.
- A. V. Kiselev and Y. I. Yashin, Gas-Adsorption Chromatography, Plenum Press, 1969 Search PubMed.
- F. Bruner, G. Crescentini and F. Mangani, Chromatographia, 1990, 30, 565–572 CrossRef CAS.
- K. R. Kim, Y. J. Lee, H. S. Lee and A. Zlatkis, J. Chromatogr., 1987, 400, 285–291 CrossRef CAS PubMed.
- E. Charon, J.-N. Rouzaud and J. Aléon, Carbon, 2014, 66, 178–190 CrossRef CAS.
- T. Gruber, T. W. Zerda and M. Gerspacher, Carbon, 1994, 32, 1377–1382 CrossRef CAS.
- T. W. Zerda and T. Gruber, Rubber Chem. Technol., 2000, 73, 284–292 CrossRef CAS.
- H. Marsh and A. P. Warburton, J. Chem. Technol. Biotechnol., 1970, 20, 133–142 CAS.
- I. Mochida, R. Ohtsubo and K. Takeshita, Carbon, 1980, 18, 117–123 CrossRef CAS.
- I. Mochida, I. Ito, Y. Korai, H. Fujitsu and K. Takeshita, Carbon, 1981, 19, 457–465 CrossRef CAS.
- T. W. Zerda, W. Xu, A. Zerda, Y. Zhao and R. B. Von Dreele, Carbon, 2000, 38, 355–361 CrossRef CAS.
- L.-W. Zhang, H.-B. Fu and Y.-F. Zhu, Adv. Funct. Mater., 2008, 18, 2180–2189 CrossRef CAS.
- K. Lv, S. Fang, L. Si, Y. Xia, W. Ho and M. Li, Appl. Surf. Sci., 2017, 391, 218–227 CrossRef CAS.
- L. Liao, S. Heylen, B. Vallaey, M. Keulemans, S. Lenearts, M. B. J. Roeffaers and J. A. Martens, Appl. Catal., B, 2015, 166–167, 374–380 CrossRef CAS.
- L. Liao, S. Heylen, S. P. Sree, B. Vallaey, M. Keulemans, S. Lenaerts, M. B. J. Roeffaers and J. A. Martens, Appl. Catal., B, 2017, 202, 381–387 CrossRef CAS.
- B. H. R. Suryanto and C. Zhao, Chem. Commun., 2016, 52, 6439–6442 RSC.
- T. Tatsuma, S. Tachibana, T. Miwa, D. A. Tryk and A. Fujishima, J. Phys. Chem. B, 1999, 103, 8033–8035 CrossRef CAS.
- T. Tatsuma, S. Tachibana and A. Fujishima, J. Phys. Chem. B, 2001, 105, 6987–6992 CrossRef CAS.
- A. Mills, S. Hodgen and S. K. Lee, Res. Chem. Intermed., 2005, 31, 295–308 CrossRef CAS.
- U. I. Gaya and A. H. Abdullah, J. Photochem. Photobiol., C, 2008, 9, 1–12 CrossRef CAS.
- S. W. Verbruggen, K. Masschaele, E. Moortgat, T. E. Korany, B. Hauchecorne, J. A. Martens and S. Lenearts, Catal.: Sci. Technol., 2012, 2, 2311–2318 RSC.
- A. Fujishima, X. Zhang and D. A. Tryk, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef CAS.
- J.-M. Herrmann, Catal. Today, 1999, 53, 115–129 CrossRef CAS.
- C. J. Tighe, M. V. Twigg, A. N. Hayhurst and J. S. Dennis, Carbon, 2016, 107, 20–35 CrossRef CAS.
- N. Nejar, M. Makkee and M. J. Illán-Gómez, Appl. Catal., B, 2007, 75, 11–16 CrossRef CAS.
- Z. Meng, D. Yang and Y. Yan, J. Therm. Anal. Calorim., 2014, 118, 551–559 CrossRef CAS.
- A. M. Panich, V. Y. Osipov and K. Takai, New Carbon Mater., 2014, 29, 392–397 CrossRef.
- M. Concistrè, S. Mamone, M. Denning, G. Pileio, X. Lei, Y. Li, M. Carravetta, N. J. Turro and M. H. Levitt, Philos. Trans. R. Soc., A, 2013, 371, 1–11 CrossRef PubMed.
- F. Tuinstra and J. Koenig, J. Chem. Phys., 1970, 53, 1126–1130 CrossRef CAS.
- R. J. Nemanich, G. Lucovsky and S. A. Solin, Solid State Commun., 1977, 23, 117–120 CrossRef CAS.
- R. J. Nemanich and S. A. Solin, Phys. Rev. B: Solid State, 1979, 20, 392–401 CrossRef CAS.
- T. Jawhari, A. Roid and J. Casado, Carbon, 1995, 33, 1561–1565 CrossRef CAS.
- A. N. Mohan, B. Manoj and A. V. Ramya, Asian J. Chem., 2016, 28, 1501–1504 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- J. Robertson, Adv. Phys., 2011, 60, 87–144 CrossRef CAS.
- M. Smits, Y. Ling, S. Lenaerts and S. Van Doorslaer, ChemPhysChem, 2012, 13, 4251–4257 CrossRef CAS PubMed.
- V. Asokan, V. Venkatachalapathy, K. Rajavel and D. N. Madsen, J. Phys. Chem. Solids, 2016, 99, 173–181 CrossRef CAS.
- M. S. Seehra and A. S. Pavlovic, Carbon, 1993, 31, 557–564 CrossRef CAS.
- V. S. Babu, L. Farinash and M. S. Seehra, J. Mater. Res., 1995, 10, 1075–1078 CrossRef CAS.
- P. J. F. Harris, Interdiscip. Sci. Rev., 2001, 26, 204–210 CrossRef.
- L. Gao, Q. Li, Z. Song and J. Wang, Sens. Actuators, B, 2000, 71, 179–183 CrossRef CAS.
- B. W. Smith and D. E. Luzzi, J. Appl. Phys., 2001, 90, 3509–3515 CrossRef CAS.
- R. F. Egerton, Microsc. Res. Tech., 2012, 75, 1550–1556 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Temperature profile during photoreaction; gas specifications; example of deconvoluted Raman spectrum; wide Raman spectra of CB, CBC72 and CBC165 with bands in high-wavenumber range; Harkins–Jura t-plots of CB and CBR72. See DOI: 10.1039/c9na00188c |
| This journal is © The Royal Society of Chemistry 2019 |