
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel coating on plasmonic copper nanoparticles lowers cytotoxicity and enables colorimetric pH readout for antibacterial wound dressing application†
Bohan
Zhang
 a,
Sladjana
Slavkovic
abc,
Yumin
Qiu
d,
Chun
Peng
de and
Jennifer I-Ling
Chen
*a
a,
Sladjana
Slavkovic
abc,
Yumin
Qiu
d,
Chun
Peng
de and
Jennifer I-Ling
Chen
*a
aDepartment of Chemistry, York University, 4700 Keele Street, Toronto, M3J 1P3, Ontario, Canada. E-mail: jilchen@yorku.ca
bDepartment of Laboratory Medicine, Keenan Research Centre for Biomedical Science of St Michael's Hospital, Toronto, ON, Canada
cCanadian Blood Services Centre for Innovation, Toronto, ON, Canada
dDepartment of Biology, York University, 4700 Keele Street, Toronto, M3J 1P3, Ontario, Canada
eCentre for Research on Biomolecular Interactions, York University, 4700 Keele Street, Toronto, M3J 1P3, Ontario, Canada
First published on 2nd July 2024
Abstract
Wound infection poses a significant challenge to the natural healing process. It can impede various stages of wound healing, thereby hindering tissue regeneration and increasing the risk of systemic complications. Wound dressings emerged as a crucial option in the management of infections. Herein, we investigate fabrics coated with copper-based nanoparticles for potential wound dressing application. We synthesized copper and copper–nickel (Cu–Ni) core–shell nanoparticles via a polyol synthesis and investigated their particle growth dynamics and chemical stability. The nickel coating stabilized the nanoparticles against oxidation and dissolution, while dampening the localized surface plasmon resonance of copper. When coated on the fabrics, we found that Cu–Ni NPs were slightly less effective as an antibacterial agent than Cu NPs, however the cytotoxicity of Cu–Ni NPs was significantly reduced compared to pure Cu. Additionally, we show that the discoloration of nanoparticle-coated fabrics depended on pH, thus enabling the visualization of pH levels of simulated wound fluids which can provide information on the inflammatory state of the wound. Our work contributes to the understanding of copper-based nanoparticles and their potential applications in healthcare.
1. Introduction
Wound infections pose a significant healthcare challenge, particularly with surgical site infection that can develop in 5–20% of post-surgery cases, leading to extended hospital stays and increased morbidity, mortality rate, and costs.1,2 Wound infections can lead to life-threatening conditions like sepsis,3 while chronic wounds present further humanistic and economic burdens in healthcare. Therefore, rigorous infection prevention and timely treatment are crucial for patients' safety, and innovative solutions that merge materials science and biomedicine are sought-after.Traditional wound infection management methods, such as wound cleansing with antiseptic solutions and sterile dressings, have been crucial in reducing infections but come with drawbacks.4 The overuse or improper application of antiseptics can harm healthy tissues and impede wound healing. Of the dressings that incorporate antimicrobial agents, iodine and silver-based compounds are the common active ingredients. Iodine has broad-spectrum antimicrobial activity; however, it may cause staining and irritation at the wound site and should not be used on patients with thyroid disorders or iodine allergies. Silver exhibits high antibacterial efficacy because the released silver ions bind to proteins, cause oxidative stress via reactive oxygen species, induce structural changes in bacterial cell membranes, and inhibit DNA and RNA replication.5 An example of Ag-based dressing is acticoat; however, numerous studies have elucidated the cytotoxicity of silver that can prolong healing, such that their use should be limited in duration and the wound healing progress needs to be closely monitored.6,7
As a result, there is a growing interest in other nanomaterials for antimicrobial applications. Copper is an essential trace element for human health while it is toxic to many microorganisms, including bacteria, viruses, fungi and algae.8 When used as nanoparticles, the high surface area facilitates the release of copper ions which can disrupt the cellular integrity of microorganisms via the generation of reactive oxygen species, oxidation of proteins and lipids, and degradation of DNA and RNA.9 In wound healing, copper has been reported to accelerate tissue regeneration and wound closure by reducing the inflammatory response, stimulating the expression of vascular endothelial growth factor, which promotes angiogenesis within the wound,10 and enhancing collagen synthesis, which helps form and repair connective tissues, skin, and blood vessels.11,12 Hence, CuNPs may be a compelling choice for advanced wound care applications.13
Another fundamental interest of CuNPs is their plasmonic properties. CuNPs exhibit localized surface plasmon resonance (LSPR) arising from the collective oscillation of free electrons when excited by visible light.14 The unique optical properties are tunable by controlling the size and shape of nanoparticles and the local dielectric environment, thus allowing for colorimetric sensing and monitoring morphological changes to the nanoparticles.15,16 Although plasmon-mediated hot electron or photothermal effect can infer an antimicrobial effect, this light activation is not required in CuNPs because the main mode of action is through the released copper ions.9,17 Despite the promise of the dual functionality, the low stability of CuNPs poses challenges associated with their aggregation and oxidation, which can hinder consistencies in their performance, standardization and quality control for clinical use.18 It is therefore essential to address the stability issues of CuNPs for diverse healthcare applications.
Herein we investigate methods to improve the stability of colloidal CuNPs, which were synthesized via the ascorbic acid and polyol reduction route with ethylene glycol. We focus on plating CuNPs with nickel to form copper–nickel core–shell nanoparticles (Cu–Ni NPs). We evaluate the optical properties, chemical stability, antibacterial performance, and cytotoxicity of the nanoparticles in the form of coated dressing. We found that Cu–Ni NPs exhibit the desired low cytotoxicity while maintaining effective antibacterial performance. Our work may help address a critical safety aspect for potential biomedical applications of copper-based nanoparticles and make them an exciting avenue for further exploration and development.
2. Materials and methods
2.1. Materials
Copper sulfate pentahydrate (99%) was purchased from Factory Direct Chemicals. L-Ascorbic acid (reagent grade), polyvinylpyrrolidone (PVP, Mw = 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), anhydrous ethylene glycol (99.8%), TWEEN 80 and nickel(II) acetate tetrahydrate (Ni(OCOCH3)2·4H2O, 99.9% trace metals basis) and tris(hydroxymethyl)aminomethane (TRIS) were purchased from Sigma-Aldrich. Nonwoven polyester/cellulose fabric (DURX® 570) was purchased from Berkshire Corporation. Biological reagents including Dulbecco's modified eagle medium (DMEM) and penicillin–streptomycin were purchased from Gibco Inc, fetal bovine serum (FBS) was purchased from Corning Inc, Cell Counting Kit 8 (CCK-8) was purchased from Selleckchem Inc., and L-histidine (98%) was purchased from Bioshop. All reagents were used as-is.
000), anhydrous ethylene glycol (99.8%), TWEEN 80 and nickel(II) acetate tetrahydrate (Ni(OCOCH3)2·4H2O, 99.9% trace metals basis) and tris(hydroxymethyl)aminomethane (TRIS) were purchased from Sigma-Aldrich. Nonwoven polyester/cellulose fabric (DURX® 570) was purchased from Berkshire Corporation. Biological reagents including Dulbecco's modified eagle medium (DMEM) and penicillin–streptomycin were purchased from Gibco Inc, fetal bovine serum (FBS) was purchased from Corning Inc, Cell Counting Kit 8 (CCK-8) was purchased from Selleckchem Inc., and L-histidine (98%) was purchased from Bioshop. All reagents were used as-is.
2.2. Synthesis of CuNPs
A mass of 1.5 g of PVP was dissolved in 50 mL of ethylene glycol (EG) in a round-bottom flask with magnetic stirring and nitrogen gas protection. The temperature was raised to 130 °C, then 0.2346 g of L-ascorbic acid (AA) and 150 μL of 0.13 M (1.95 × 10−5 mol) copper sulfate pentahydrate stock solution in EG were separately added. The solution quickly became red and was reacted for 30 min, followed by cooling in an ice bath. The CuNPs in EG can be stored for 2 weeks without a noticeable change in the LSPR or can be purified by mixing 20 mL as-synthesized solution with 15 mL 95% ethanol and centrifuged at relative centrifugal force (RCF) = 16639 g for 30 minutes and then dried or redispersed in anhydrous ethanol depending on the subsequent experiment.
2.3. Synthesis of Cu–Ni NPs
A mass of 0.4 g PVP and 240 μL of 2 M NaOH(aq) was dissolved in 20 mL ethylene glycol and heated to 196 °C under nitrogen and reflux with stirring. Then, 3 mL of 0.19 M AA and 7.8 × 10−6 mol of purified CuNPs (0.4 fraction of the CuNP synthesis, resuspended in 1 mL of EG) were added to the reaction solution. Finally, a 1 mL EG solution containing 0.0018 M of nickel precursor was slowly injected at 2 mL h−1 using a syringe pump. The solution was allowed to react for an additional 30 min.2.4. Characterization of CuNPs and Cu–Ni NPs
The extinction spectra of CuNPs and Cu–Ni NPs were measured using a Lambda 950 UV/VIS/NIR spectrophotometer (PerkinElmer). Powder X-ray diffraction was performed with an AXRD Benchtop powder X-ray diffractometer with 0.3° min−1 scan rate, 10 s dwell time, and 0.1 mm slit width. Particle size and distribution of NPs were analyzed using dynamic light scattering on a Malvern Zetasizer Ultra instrument with a 0.63 mm beam diameter HeNe gas laser. The Fourier transform infrared spectra of PVP and CuNPs were acquired in the powder form using an ATR ALPHA II Compact FT-IR Spectrometer from Bruker Inc., covering a spectral range of 4000–400 cm−1. The morphology and composition of the CuNPs and Cu–Ni were examined using a scanning electron microscope with the model Thermofisher Quanta 3D, operating at an accelerating potential of 10 kV, and equipped with an energy dispersive X-ray analysis instrument. Scanning transmission electron microscopy analysis was performed using a Hitachi HF-3300 TEM/STEM equipped with a Bruker XFlash 6T160 for Energy-Dispersive X-ray Spectroscopy analyses. The microscope was operated at an accelerating voltage of 300 kV (and had an electron gun with an emission current of 17 μA). All measurements were performed at room temperature. The finite-difference time-domain simulation of the optical properties was carried out using Ansys Lumerical software (see the ESI†).2.5. Antibacterial activity of CuNP- and Cu–Ni NP-coated fabrics
NP-coated fabrics were tested against the Gram-negative bacterium (BL21) Escherichia coli (E. coli). The Japanese Industrial Standard (JIS) L 1902 antimicrobial test for textiles was implemented for quantitative analysis.Pieces of 2 cm × 2 cm nonwoven polyester fabric were cut. These fabrics were sterilized by soaking in ethanol and then dried. Select volumes of as-synthesized NPs (e.g., 2.5, 5, 10, 15, and 20 mL corresponding to theoretical amounts of 1, 2, 4, 6 and 8 μmol of Cu) were purified via centrifugation (RCF = 16639 g for 30 minutes) and resuspension in anhydrous ethanol. The final concentrated NP ethanolic solution (400 μL) was dropped onto the surface of the fabric and the fabric was dried for 30 minutes under nitrogen. The NPs were coated on the surface of the fabric upon evaporation.
E. coli was incubated in fresh Luria–Bertani broth for 24 hours, and a JENWAY 6300 model spectrophotometer was used to quantify the optical density of E. coli to determine its concentration. Then, 200 μL of cultured E. coli with a concentration between 1 × 105 and 3 × 105 cells per mL was added onto NP-coated fabric using a micropipette. The fabric was placed inside a 50 mL Falcon tube in an oven at 37 °C and incubated for 18 hours. After incubation, the E. coli was rinsed off the fabric using 40 μL of Tween 80 and 20 mL of 0.9% NaCl solution with vortexing for 30 seconds. The count of bacterial colonies was carried out via the serial dilution plate count method using a 0.9% NaCl solution after a one-day incubation period on the agar plate surface.
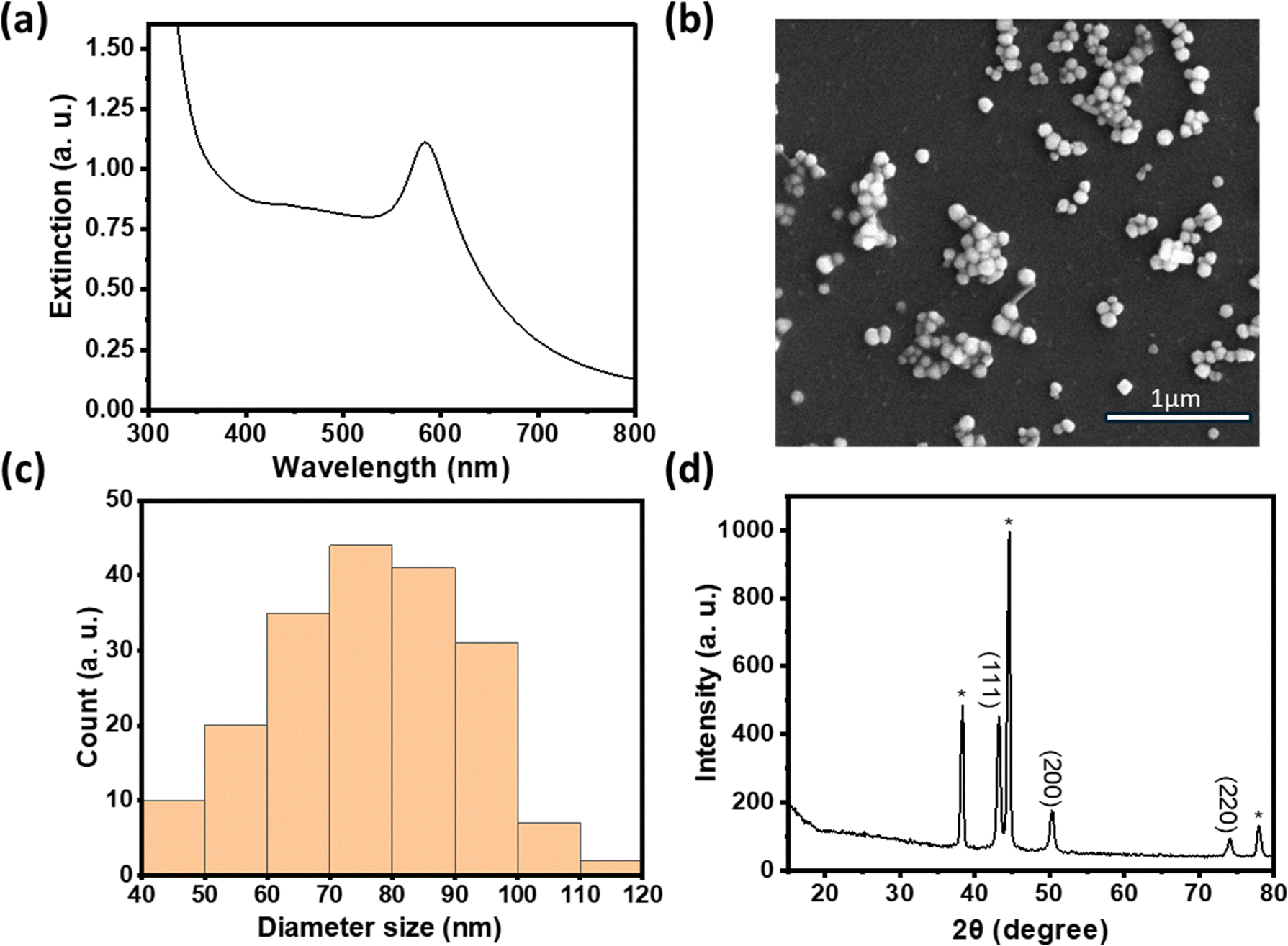
Antibacterial properties were quantified by calculating the bacterial colony reduction rate (antibacterial activity), which is defined as:
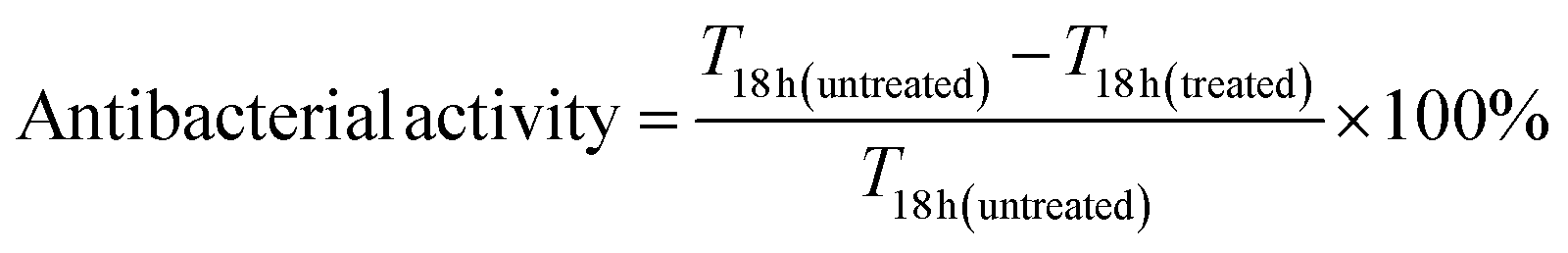
Additional quantitative analyses according to JIS L 1902/2008 (ref. 19) are in the ESI.†
2.6. Cytotoxicity assays of CuNP- and Cu–Ni NP-coated fabrics
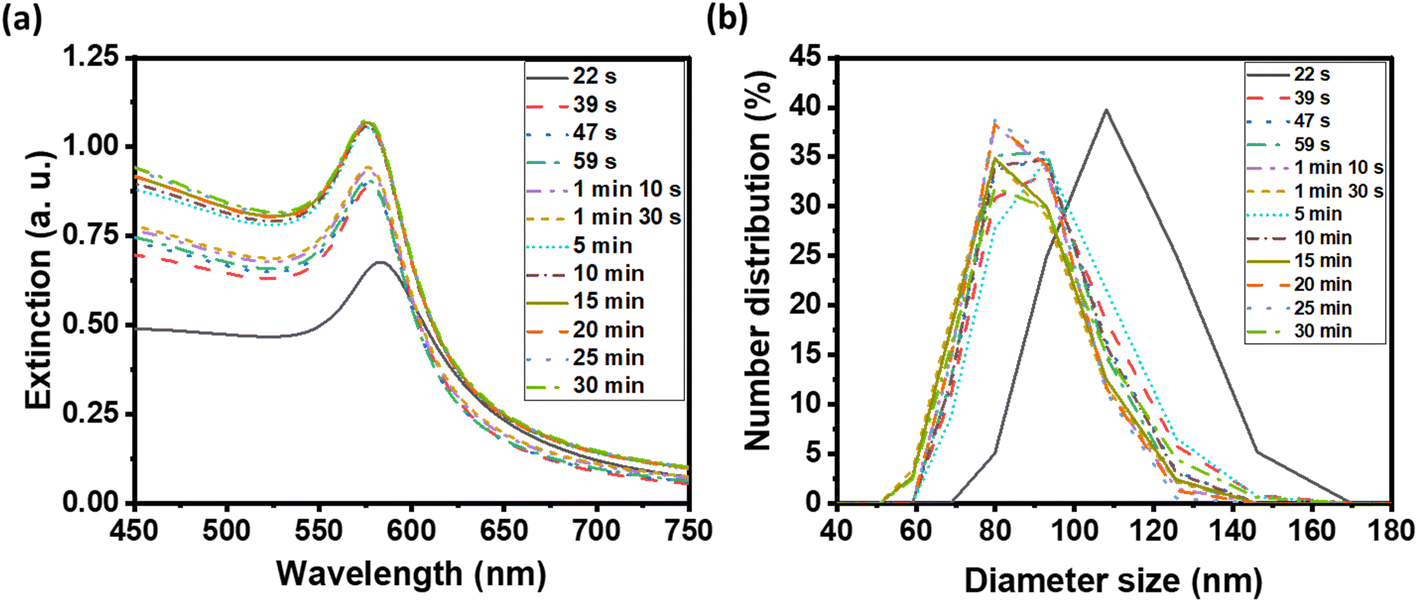
For the cytotoxicity assessment, BJ (ATCC CRL-2522) fibroblast cells were employed. Specifically, 6000 cells per well were seeded in 96-well plates and cultured in 100 μL of DMEM containing 20% FBS overnight. Concurrently, 6 μmol of CuNPs or Cu–Ni NPs were applied as coatings on 2 cm × 2 cm non-woven fabric substrates. Each piece of NP-coated fabric was immersed in 1 mL of phosphate-buffered saline (PBS) containing 1% penicillin–streptomycin for an 18 hours incubation period at room temperature. Then, the specified volumes of the resulting extracts were mixed with DMEM supplemented with 20% FBS to a final volume of 100 μL. This mixture was subsequently added to individual wells of the 96-well plate, each initially containing 6000 BJ cells. The cells were subsequently incubated at 37 °C with a 5% CO2 atmosphere for 2 days.After the 2 day incubation period, cell viability was determined using a Cell Counting Kit 8 (CCK-8) following manufacturer's suggested protocols. Briefly, 10 μL of the CCK-8 solution was added into each well and cells were incubated with the CCK-8 solution for 2 hours. During this incubation, viable cells with cellular dehydrogenases reduced the WST-8 dye within the CCK-8 solution, forming formazan and a color change from colorless or pale yellow to orange. The optical density (OD) of the CCK-8 solution was measured at 450 nm using an Agilent BioTek Synergy H4 hybrid microplate reader. Cell viability (CV) was calculated as follows:
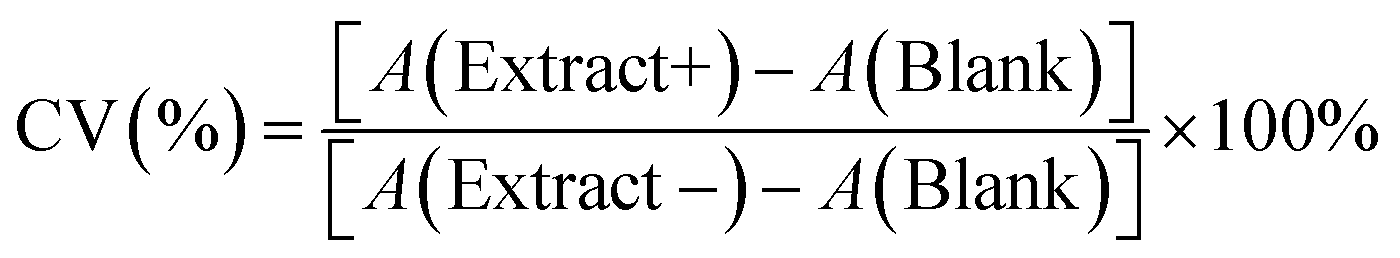
The OD of BJ cells incubated with the fabric extracts (with or without NPs) was compared to control cells (i.e., without extracts) to yield CV as a percentage. A high percentage of CV suggests greater cell viability, indicating that the NP-coated fabric that is used to incubate with BJ cells has low cytotoxicity.
2.7. Simulated wound exudate on the nanoparticle-coated fabric
A simulated wound exudate (SWE) was formulated by combining 0.142 M NaCl and 0.0025 M CaCl2·2H2O, with pH buffering agents including 0.025 M TRIS and 0.005 M L-histidine. The SWE was prepared at three different pHs: 3.5, 7, and 10. The fabrics coated with 6 μmol of CuNPs or Cu–Ni NPs were separately soaked in 3 mL of SWE for 105 minutes.2.8. Statistical analysis
Cytotoxic effects were analyzed using the Student's t-test to compare mean cell viability levels across groups. The differences were statistically significant at p < 0.05. Data were reported as mean ± standard error.3. Results and discussion
We synthesized CuNPs via polyol reduction.20 The EG solution with AA and PVP were heated to 130 °C under N2. The CuSO4 stock solution was rapidly added, turning the solution red in 22 s. Fig. 1a shows the UV-vis extinction spectrum of as-synthesized CuNPs with a sharp LSPR peak centered at 584 nm and a relatively low baseline at long wavelengths (>800 nm), suggesting that the CuNPs were dispersed with minimal aggregation. The LSPR peak wavelength agrees with theoretical simulation using the Finite-difference time-domain (FDTD) method for CuNPs (Fig. S1†). The scanning electron microscope (SEM) image in Fig. 1b shows that the CuNPs were predominantly spherical with an average diameter of 76.4 ± 15.3 nm; the distribution of the particle size is shown in Fig. 1c. Powder X-ray diffraction (PXRD) shown in Fig. 1d, collected using a cell holder sealed under nitrogen to limit oxidation, confirms the metallic phase of the CuNPs with diffraction peaks at 2θ values of 43.2°, 50.4°, and 74.1°, corresponding to the (111), (200), and (220) crystallographic planes of copper of space group fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m. The calculated crystallite size of the CuNPs was 14.7 nm using the Scherrer equation (see the ESI†), suggesting that CuNPs were polycrystalline.
m. The calculated crystallite size of the CuNPs was 14.7 nm using the Scherrer equation (see the ESI†), suggesting that CuNPs were polycrystalline.
 | ||
| Fig. 1 (a) UV-vis extinction spectrum of as-synthesized CuNPs. (b) SEM image of CuNPs and (c) their size distribution (N = 175). (d) XRD pattern of purified CuNPs (peaks labeled with * came from the sample holder). | ||
We examined the evolution of the nanoparticle growth during the reaction by monitoring the UV-vis extinction spectra (Fig. 2a) and dynamic light scattering (DLS) properties (Fig. 2b) over time. We observed an initial blueshift followed by increasing intensity in the LSPR peak. At the shortest time (22 s), the Cu nuclei or atoms were aggregated and clustered as detected in DLS. As the synthesis progressed, the CuNPs became dispersed and the hydrodynamic size decreased from 107.4 ± 4.8 to 87.8 ± 3.1 nm (Table S1†). The observed initial blue shift in the LSPR peak during the synthesis of CuNPs aligns with the concurrent decrease in size, as determined by DLS analysis. The increase in the intensity of the LSPR peak thereafter indicates a rise in the concentration of CuNPs while the size remained the same.
 | ||
| Fig. 2 (a) UV-visible extinction spectra of as-synthesized CuNP solution, and (b) distribution of CuNP hydrodynamic diameter by DLS at various reaction times. | ||
In the synthesis, we utilized PVP as a capping agent, which helped increase the stability of CuNPs by preventing aggregation, controlling the particle size and shape, and providing a protective coating on the nanoparticle surface. The reducing agent, AA, can also act as a capping agent to stabilize the CuNPs. However, the FT-IR spectrum in Fig. S2† shows that the CuNPs were mainly capped with PVP, where the stretching vibrational modes of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–N were observed at 1650 cm−1 and 1269 cm−1, respectively, and those of C–H and N–H were observed at 2800–3000 cm−1 and 3100–3500 cm−1, respectively.
O and C–N were observed at 1650 cm−1 and 1269 cm−1, respectively, and those of C–H and N–H were observed at 2800–3000 cm−1 and 3100–3500 cm−1, respectively.
It is critical to enhance the chemical stability of CuNPs due to their susceptibility to oxidation. While the as-synthesized CuNPs were stable in EG for an extended period of time, they oxidized upon purification and redispersion in aqueous or ethanolic solutions. Improving the chemical stability will prolong the duration of Cu ion release, thereby enhancing antimicrobial properties. The LSPR of CuNPs allows for facile monitoring of the changes and dissolution of the CuNPs spectroscopically. We first examined the incorporation of excess organic ligands and capping agents, such as AA and PVP, to improve the chemical stability of CuNPs. Fig. S3† shows the changes in the LSPR peak of CuNPs redispersed in water with the additives. Some improvements were seen with the addition of AA or PVP; however, they were ineffective as the LSPR peak of CuNPs rapidly decreased to 60% of the original intensity within 200 minutes for the most promising trial (Fig. S3a and b†). Motivated by cupronickel, a blend of copper and nickel in varying proportions that forms durable alloys with excellent corrosion resistance and is used in medical devices,21 we then explored Ni coating to increase the chemical stability of CuNPs. We hypothesized that Ni coating can reduce the oxidation and dissolution of copper and enable prolonged copper ion release, thereby addressing the limitations of copper-based nanoparticles.22
In comparison to copper, nickel has a much lower reduction potential.23,24 We first investigated the feasibility of reducing various Ni(II) compounds in the polyol synthesis by increasing the temperature to 196 °C to increase the reducing power. Interestingly, PXRD data in Fig. 3a show that metallic Ni was readily formed with nickel acetate as the precursor while a mixture of metallic Ni and various Ni hydroxides was formed when nickel sulfate was used as the precursor.25 Previous work examining the polyol reduction mechanism reported metal glycolates as the intermediate species from metal salts like sulfate and chloride.26,27 We hypothesize that nickel acetate remained as a complex and may be more easily reduced to Ni (0), hence minimizing the formation of by-products like nickel hydroxide.
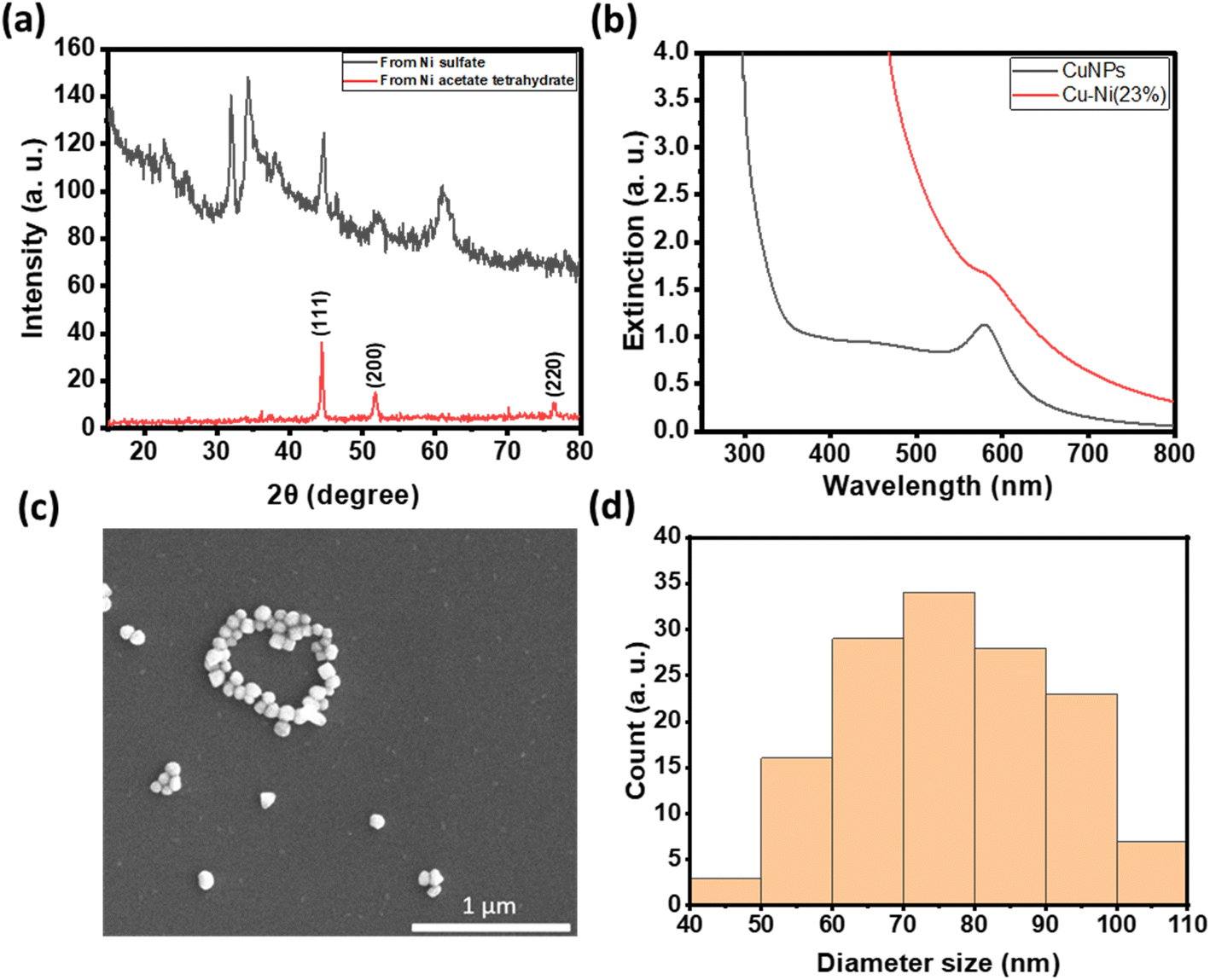 | ||
| Fig. 3 (a) X-ray diffraction patterns of metallic nickel synthesized from nickel acetate tetrahydrate (top red) and mixed nickel hydroxide and oxide from nickel sulfate (bottom black). (b) UV-vis extinction spectrum of as-synthesized CuNPs and Cu–Ni NPs. (c) SEM image of Cu–Ni NPs and (d) their size distribution (N = 126). | ||
We thus used nickel acetate as the precursor to incorporate a nickel shell on the CuNPs. The synthesis comprised two steps: first, CuNPs were synthesized, purified and redispersed in ethylene glycol growth solution, and then the nickel precursor was slowly added. We found the addition of a small amount of NaOH to be critical in keeping the colloidal stability of CuNPs at 196 °C; it may help with the electrostatic repulsion between negatively charged hydroxylated CuNPs. We examined Ni loadings of 23, 62, 94, and 154 mol% (Fig. S3c†), however, only that of 23 mol% yielded nanoparticles that could be centrifuged and purified. We denote them as Cu–Ni NPs. During the shell growth, the solution turned from red to dark brown, and the purified Cu–Ni NPs were black in colour. The Cu–Ni NPs exhibited a weaker LSPR as seen in the extinction spectrum in Fig. 3b, consistent with theoretical simulation (Fig. S1†), because Ni is not plasmonic and causes an overall dampening. The average size of Cu–Ni NPs was 77.3 ± 14.6 nm, similar to that of CuNPs. Fig. 4 shows scanning transmission electron microscopy (STEM) images of Cu–Ni NPs and their elemental mapping, which confirms the deposition of Ni on Cu (red and cyan, respectively in Fig. 4b and d) and the core–shell structure. On the other hand, the atomic % of Ni to Cu from energy-dispersive X-ray spectroscopy (EDS) analysis was much lower than the theoretical composition (1.6 mol% vs. 23 mol%). The discrepancy may be attributed to factors such as the incomplete reduction of the nickel precursor or the mismatch in the lattice and structure between the surface hydroxylated CuNPs and metallic Ni. Although Ni dampens the LSPR, the weak plasmonic peak was still seen and was used to monitor the stability of Cu–Ni NPs. Fig. S4a and b† show extinction spectra of CuNPs and Cu–Ni NPs redispersed in ethanol over time; after 250 minutes, the LSPR peak intensity remained at 80% for Cu–Ni NPs compared with CuNPs at 55%. The Ni coating also yielded better stability enhancement than with excess AA or PVP.
 | ||
| Fig. 4 (a and c) STEM images and (b and d) elemental mapping of Cu–Ni nanoparticles. Cu and Ni are shown, respectively, as cyan and red. The EDS atomic ratio of Ni:Cu was 1.6:100. | ||
For antibacterial and cytotoxicity analyses, we coated non-woven polyester textiles with varying loadings of CuNPs or Cu–Ni NPs. The colloidal particles were physisorbed onto the textiles after drying under nitrogen. Fig. 5c shows the photographs of CuNP- and Cu–Ni NP-coated fabrics. There was negligible loss of CuNPs or Cu–Ni NPs from the polyester fabric from rinsing, but some can be removed by sonication or when vigorously shaken in solution.
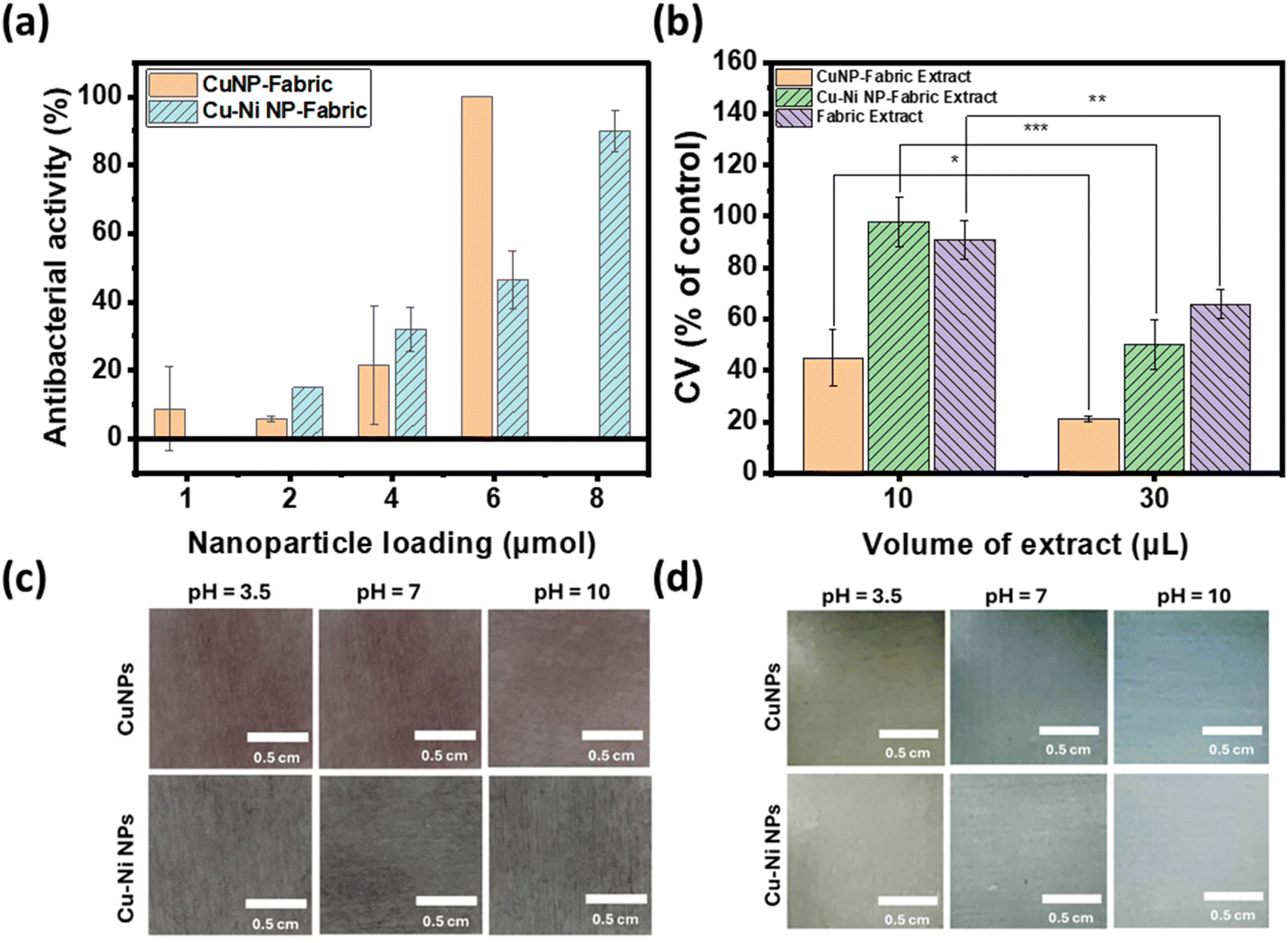 | ||
| Fig. 5 (a) Antibacterial activity of different amounts of CuNP- or Cu–Ni NP-coated fabrics. (b) Cell viability after incubation with extracts of CuNP- and Cu–Ni NP-coated fabrics. *: p < 0.05. **: p < 0.01. ***: p < 0.001. Photographs of CuNP- and Cu–Ni NP-coated fabrics in SWE with different pHs (c) at the beginning and (d) after 105 min of incubation. | ||
Fig. 5a summarizes the number of colonies of E. coli recovered from the NP-coated fabrics at different NP loadings. We observed a reduction in bacterial colonies with increasing amounts of CuNPs and Cu–Ni NPs on the fabrics. At 6 μmol, a remarkable decrease of up to 99.98 ± 0.04% in E. coli colony count was recorded for CuNP-coated fabric samples; in comparison, a decrease of up to 90.03 ± 6.04% was achieved for Cu–Ni NP loading of 8 μmol. The higher loading of Cu–Ni NP required may be related to the slower release of copper ions. We have summarized bacteriostatic (BS) and bactericidal (BC) values in Table S2.† The results show that CuNP loading of >6 μmol (i.e., 1.5 μmol·cm−2) on fabrics was effective at both prohibiting the growth and killing E. coli, based on the certification standards of antibacterial finished textile products.28 Although the minimum loading of Cu–Ni NPs required to meet certification standards is higher, Cu–Ni NP loading may retain the antibacterial properties for a longer period of time.
For wound dressing applications, it is desirable to have antimicrobial efficacy while exhibiting low cytotoxicity. Hence, we carried out cytotoxicity studies of extracts from CuNP- and Cu–Ni NP-coated fabrics on a model human fibroblast – BJ cells. Fig. 5b compares the viability of cells incubated with 10 or 30 μL extracts of CuNP- and Cu–Ni NP-coated fabrics and those of control, i.e., cells incubated with extracts of non-coated fabric and cultured in DMEM. We observed that an increase in the volume of the extracts (30 μL vs. 10 μL) resulted in a decrease in cell survival rates across all samples. The extracts of CuNP-coated fabric exhibited significant toxicity to BJ cells, and cell viabilities (CVs) of 44.8 ± 11.1 and 21.2 ± 1.0% were determined for 10 μL and 30 μL samples respectively. The extracts of Cu–Ni NP-coated fabric exhibited much lower cytotoxicity, as evidenced by a higher average CV at 97.9 ± 9.9 and 50.0 ± 9.7% for 10 μL and 30 μL of extracts, respectively. The extracts of the control – namely PBS incubated with non-coated fabric – showed some effect on cell viability as the growth media was diluted. Notably, the extracts from Cu–Ni NP-coated fabric yielded statistically the same results as the control at 10 μL volume (at p > 0.4). The low cytotoxicity of Cu–Ni NPs suggests a better biocompatibility than CuNPs.
Finally, we examined the colorimetric response of the NP-coated fabrics to SWE at different pHs. Wound exudate comprising plasma, proteins, and white blood cells is vital for healing as it provides nutrients and facilitates debris removal. Bacterial infections can raise the pH of the exudate, thereby negatively impacting the healing process.29 Therefore, it would be advantageous if a wound dressing could provide a visual cue to the state of the infection.
Fig. 5c and d show the photographs of CuNP (top) and Cu–Ni NP (bottom) coated fabrics in SWE with pH = 3.5, 7, and 10 (from left to right) at time 0 and after 105 minutes. After 105 minutes, the color of the CuNP- and Cu–Ni NP-coated fabrics turned orange/yellow in SWE of pH 3.5, which may be attributed to residual CuNPs and the formation of copper(I) oxide at acidic pH. In neutral or alkaline SWE, the NP-coated fabrics turned blue, and species such as Cu(II) hydroxide or other adsorbed Cu2+ complexes may contribute to the observed color change. The colors darkened and persisted after one month (Fig. S5†). These results show that CuNP- and Cu–Ni NP-coated fabrics not only combat bacterial infections but also provide a colorimetric response to pH. Since a normal exudate usually has an acidic pH ranging from 4 to 6,30 the color change to blue could point to an alkaline environment that may be associated with bacterial infection. Hence copper-based NPs may enable a diagnostic element to wound care without additional sensor technology integration.
4. Conclusions
We showed that copper nanoparticles with known antibacterial properties can be stabilized by plating a nickel shell. These copper-based nanoparticles exhibit localized surface plasmon resonance that provides a colorimetric handle to visually detect changes in their size and dissolution, such as in response to pH changes in wound exudates. Although installing a nickel shell decreased slightly the antibacterial efficacy, the resultant Cu–Ni NPs were significantly less cytotoxic than pure CuNPs – an important attribute of infection-control agents. Further studies on different compositions of Cu–Ni NPs, their large-scale production, and antimicrobial performance against a variety of species will help elucidate the feasibility of their use in healthcare.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
BZ carried out all experimental work and data analyses. SS designed antibacterial testing methodology and YQ and CP supported cytotoxicity measurement. JC conceptualized the project and provided supervision. BZ and JC wrote the manuscript with input and approval from all authors. Funding was secured by JC and CP.Conflicts of interest
The authors declare no financial or non-financial conflicts of interest that could influence the objectivity or integrity of the presented research.Acknowledgements
This research was supported by York University's COVID-19 rapid research fund, NSERC Research Tool and Instruments, and Discovery grants to JC, and a CIHR project grant (PJT-166079) to CP. We thank Dr Jaklewicz and Dr Nikbin at York University and University of Toronto, respectively, for technical support on EM imaging, Prof. Johnson for use of facility for bacterial work, Prof. Kitaev for helpful discussions, and Dr Malile for initial work.References
- B. M. Andersen, in Prevention and Control of Infections in Hospitals, Springer International Publishing, 2019, pp. 377–437 Search PubMed.
- J. M. Badia, A. L. Casey, N. Petrosillo, P. M. Hudson, S. A. Mitchell and C. Crosby, J. Hosp. Infect., 2017, 96, 1–15 CrossRef CAS PubMed.
- C. K. Sen, Adv. Wound Care, 2021, 10, 281–292 CrossRef PubMed.
- I. B. M. Ploegmakers, S. W. M. Olde Damink and S. O. Breukink, Br. J. Surg., 2017, 104, e24–e33 CrossRef CAS PubMed.
- A. Muhammad, in Nanomedicines, ed. N. Ivanova, V. Gugleva, M. Dobreva, I. Pehlivanov, S. Stefanov and V. Andonova, IntechOpen, London, 1st edn, 2018, ch. 4, vol. 5, pp. 71–92 Search PubMed.
- K. Nešporová, V. Pavlík, B. Šafránková, H. Vágnerová, P. Odráška, O. Žídek, N. Císařová, S. Skoroplyas, L. Kubala and V. Velebný, Sci. Rep., 2020, 10, 15216 CrossRef.
- A. Burd, C. H. Kwok, S. C. Hung, H. S. Chan, H. Gu, W. K. Lam and L. Huang, Wound Repair Regen., 2007, 15, 94–104 CrossRef PubMed.
- N. Cioffi and M. Rai, in Nano-Antimicrobials: Progress and Prospects, ed. D. Longano, N. Ditaranto, L. Sabbatini, L. Torsi and N. Cioffi, Springer-Verlag, Berlin Heidelberg, 1st edn, 2014, ch. 3, pp. 85–117 Search PubMed.
- A. K. Chatterjee, R. Chakraborty and T. Basu, Nanotechnology, 2014, 13, 135101 CrossRef.
- C. K. Sen, S. Khanna, M. Venojarvi, P. Trikha, E. Christopher Ellison, T. K. Hunt and S. Roy, Am. J. Physiol.: Heart Circ. Physiol., 2002, 5, H1821–H1827 CrossRef.
- S. K. Nethi, S. Das, C. R. Patra and S. Mukherjee, Biomater. Sci., 2019, 7, 2652–2674 RSC.
- A. P. Kornblatt, V. G. Nicoletti and A. Travaglia, J. Inorg. Biochem., 2016, 161, 1–8 CrossRef CAS PubMed.
- G. Borkow, J. Gabbay, R. Dardik, A. I. Eidelman, Y. Lavie, Y. Grunfeld, S. Ikher, M. Huszar, R. C. Zatcoff and M. Marikovsky, Wound Repair Regen., 2010, 18, 266–275 CrossRef PubMed.
- F. Parveen, B. Sannakki, M. V. Mandke and H. M. Pathan, Sol. Energy Mater. Sol. Cells, 2016, 144, 371–382 CrossRef CAS.
- J. Boken, P. Khurana, S. Thatai, D. Kumar and S. Prasad, Appl. Spectrosc. Rev., 2017, 52, 774–820 CrossRef CAS.
- K. A. Willets and R. P. Van Duyne, Annu. Rev. Phys. Chem., 2007, 58, 267–297 CrossRef CAS.
- A. D'Agostino, A. Taglietti, R. Desando, M. Bini, M. Patrini, G. Dacarro, L. Cucca, P. Pallavicini and P. Grisoli, Nanomaterials, 2017, 1, 7 CrossRef.
- R. L. Calabro, F. J. Burpo, S. F. Bartolucci and J. A. Maurer, J. Phys. Chem. C, 2023, 127, 15307–15315 CrossRef CAS.
- Japanese Standards Association, Testing for Antibacterial Activity and Efficacy on Textile Products. Jis L 1902, 2008, Japanese Standards Association, Tokyo, 2009 Search PubMed.
- F. Fievet, S. Ammar-Merah, R. Brayner, F. Chau, M. Giraud, F. Mammeri, J. Peron, J. Y. Piquemal, L. Sicard and G. Viau, Chem. Soc. Rev., 2018, 47, 5187–5233 RSC.
- M. S. Parvizi, A. Aladjem and J. E. Castle, Int. Mater. Rev., 1988, 33, 169–200 CrossRef.
- S. Zhao, F. Han, J. Li, X. Meng, W. Huang, D. Cao, G. Zhang, R. Sun and C. Wong, Small, 2018, 14, 1800047 CrossRef PubMed.
- G. Milazzo, S. Caroli and R. D. Braun, J. Electrochem. Soc., 1978, 125, 261C CrossRef.
- A. Bard, R. Parsons and J. Jordan, Standard Potentials in Aqueous Solution, Routledge, London, 2017 Search PubMed.
- D. S. Hall, D. J. Lockwood, C. Bock and B. R. MacDougall, Proc. R. Soc. A, 2015, 471, 20140792 CrossRef PubMed.
- K. J. Carroll, J. U. Reveles, M. D. Shultz, S. N. Khanna and E. E. Carpenter, J. Phys. Chem. C, 2011, 115, 2656–2664 CrossRef CAS.
- K. Takahashi, S. Yokoyama, T. Matsumoto, J. L. C. Huaman, H. Kaneko, J.-Y. Piquemal, H. Miyamura and J. Balachandran, New J. Chem., 2016, 40, 8632–8642 RSC.
- Japan Textile Evaluation Technology Council, JEC301, the Certification Standards of Antibacterial Finished Textile Products, Japan Textile Evaluation Technology Council, Osaka, 2012 Search PubMed.
- H. H. Leveen, G. Falk, B. Borek, C. Diaz, Y. Lynfield, B. J. Wynkoop, G. A. Mabunda, J. L. Rubricius and G. C. Christoudias, Ann. Surg., 1973, 6, 745 CrossRef.
- T. R. Dargaville, B. L. Farrugia, J. A. Broadbent, S. Pace, Z. Upton and N. H. Voelcker, Biosens. Bioelectron., 2013, 41, 30–42 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details and calculations, FDTD simulations, SEM, FT-IR, UV-vis spectra, DLS and quantitative antibacterial test and method. See DOI: https://doi.org/10.1039/d4na00244j |
| This journal is © The Royal Society of Chemistry 2024 |