
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical shaping of CPO-27-M (M = Co, Ni) through an in situ crystallization within chitosan hydrogels†
Yassine
Khadiri
 ab,
Christophe
Volkringer
a,
Sébastien
Royer
a,
Abdelkrim
El Kadib
b,
Thierry
Loiseau
a and
Jérémy
Dhainaut
*a
ab,
Christophe
Volkringer
a,
Sébastien
Royer
a,
Abdelkrim
El Kadib
b,
Thierry
Loiseau
a and
Jérémy
Dhainaut
*a
aUniv. Lille, CNRS, Centrale Lille, Univ. Artois, UMR 8181 - UCCS - Unité de Catalyse et Chimie du Solide, F-59000 Lille, France. E-mail: jeremy.dhainaut@univ-lille.fr
bEuromed University of Fes, UEMF, Morocco
First published on 1st July 2024
Abstract
The preparation of MOF composites is considered as an effective method to address the challenges of shaping MOFs and to create porous solids with enhanced properties and broader applications. In this study, CPO-27-Co was crystallized via a simple strategy within porous chitosan beads. The resulting CS@CPO-27-Co composites were tested for CO2 sorption and they demonstrated promising performances by exceeding 3 mmol(CO2) g−1. The versatility of this strategy was further demonstrated by replacing cobalt(II) ions with nickel(II), also leading to the isostructural CPO-27 framework.
Engineering and manufacturing solid materials occupy a forefront position in materials science, owing to their utility in various domains and their strong impact on human life. Hitherto, various porous materials have been developed including zeolites, clays, activated carbons, mesoporous metal oxides and more recently COFs (covalent-organic frameworks) and MOFs (metal–organic frameworks).1 The latter are described as a recent class of hybrid crystalline materials formed by coordination bonds between metal-based nodes and organic linkers.2,3 In addition to their diversity, structural flexibility, versatility, and various active sites,3 their ultra-high porosity extending up to 6000 m2 g−1 is the most characteristic property of MOFs that ranks them as strong candidates for a wide range of applications.4 However, the as-prepared powdered form of MOFs limits their commercialization for practical use at industrial scale due to challenges in handling, recovery, and regeneration. Additionally, using an unshaped MOF can lead to clogging and further damage, making the shaping step a critical aspect in the transitioning process from laboratory use to commercial scale. Several solutions were investigated, including classical mechanical techniques such as pelletization and extrusion, and hybridization with polymer matrices including those of natural origin (e.g. chitosan, alginate…).5 The latter is favored over mechanical techniques, which often result in significant damage to the properties of MOFs.6
There are usually two methods for shaping MOFs with a polymer: the in situ crystallization method begins by dispersing metal ions within the polymer hydrogel, followed by adding organic ligands to interact with the metal ions and ultimately creating the desired MOF structure within the polymer matrix.7 The second approach is ex situ incorporation, which includes blending a pre-synthesized MOF with a polymer solution (direct mixing) or immersing a shaped polymeric matrix into a solution of MOF particles (immersion coating).8,9 Nonetheless, in situ crystallization is preferred for its simplicity, the uniform distribution of MOF crystals within the polymer matrix, and their enhanced interaction with the polymer.8
One of the widely researched MOF materials is CPO-27-M (M = divalent metal), also recognized as M-MOF-74, first discovered in 2005 by Rosi et al.10 It is formed by combining 2,5-dihydroxyterephthalic acid (H4dhtp) as the organic ligand and one of various transition metal ions (M = Zn2+, Cu2+, Co2+, Ni2+, Fe2+) as nodes.10 Due to its easy synthesis and tunable crystal size and morphology, large surface areas, and abundant unsaturated metal sites, CPO-27 presents a great potential in various applications, such as heterogeneous catalysis, drug delivery, and gas adsorption and separation.11 In particular, the relative chemical stability of CPO-27 compared to other types of MOFs (e.g. HKUST-1), along with their exceptional structural characteristics, such as their high density of open metal sites (OMS), reaching up to 4.51 sites nm−3 for CPO-27-Co, makes them one of the most promising MOFs for CO2 capture.12,13 Apart from being shaped using classical techniques, only a few studies have reported processing through combination with polymer matrices.14,15 Moreover, most of them are based on ex situ incorporation methods, with the exception of the study by J. Lim et al.,15 where Zn2+ was used to cross-link alginate followed by introducing the resulting hydrogels in a linker (H4dhtp) solution. However, in the latter case, the ligand diffusional process limited the crystallization of the MOF only at the surface of the hydrogels.15 To solve this issue, alginate was first mixed with H4dhtp, and next introduced in a solution containing zinc salt precursor.
Another way to circumvent this issue is to replace alginate with another biopolymer presenting lower bonding energy with metal ions such as chitosan (CS), a biopolymer derived from crustacean waste chitin. It is known for its biocompatibility, biodegradability, and the presence of randomly distributed glucosamine and acetylglucosamine units in its backbone that ensures its solubilization in aqueous acidic conditions and allows its chemical modification and shaping.16
To prepare CS@CPO-27-Co composite beads, we developed a simple in situ crystallization method involving the coordination of cobalt(II) ions with CS functional groups, followed by shaping the viscous mixture into beads after its dropwise introduction in a bath of caustic soda as illustrated in Fig. S1 (ESI†). Subsequently, the beads are neutralized and soaked in the organic linker aqueous solution (H4dhtp). The composite beads (Fig. S2, ESI†) with different R (where R represents the molar ratio n(Co)/n(NH2)CS applied during synthesis) are obtained after atmospheric drying (specific synthesis conditions are outlined in ESI†). Clearly, this parameter has a dramatic influence over the homogeneity of the beads as well as over their sizes.
The powder X-ray diffraction (XRD) pattern (Fig. 1) of CS@CPO-27-Co (R = 2) shows a unique crystalline phase, displaying characteristic Bragg peaks identical to those observed for the pristine CPO-27-Co powder.12,17 This suggests the successful crystallization of CPO-27-Co within the CS matrix. The purity of this crystalline phase and the morphology of the hydrogels are significantly influenced by the molar ratio R = (Co)/n(NH2). Indeed, when the molar ratio falls below 2, new peaks characteristic of cobalt oxides (notably Co3O4) appear,18 while higher ratios (R ≥ 3) result in non-viscous solutions preventing the formation of spherical beads, revealing the drastic role of controlling the molar ratio R during the synthesis process to achieve composite beads with a pure CPO-27 crystalline phase. Fourier-transform infrared (FTIR) spectroscopy shows the presence of CPO-27-Co characteristic bond vibrations in the composites (Fig. S3, ESI†). This includes vibrational Co–O interactions (477 cm−1 and 586 cm−1), out-of-plane C–H bending (814 and 882 cm−1),17 C–O stretching vibration band (1198 cm−1), and symmetric and asymmetric stretching vibrations of the carboxyl group (1417 cm−1 and 1554 cm−1, respectively) of the organic linker.19 One thus observes a strong correlation between band intensities and the R ratio: the intensity of the characteristic bands of CPO-27 decreases as the ratio R decreases, in contrast to the characteristic bands of CS (Table S1, ESI†), which become increasingly intense.
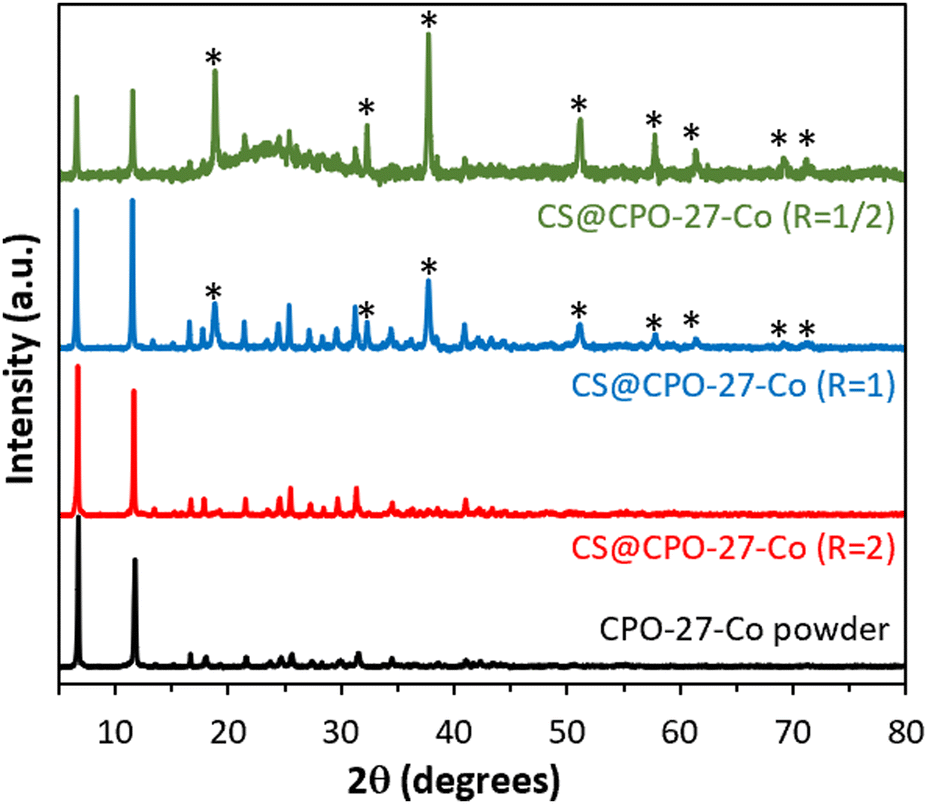 | ||
| Fig. 1 Powder XRD patterns of the as-synthesized CS@CPO-27-Co (R) beads and CPO-27-Co powder. * = Co3O4. | ||
Scanning electron microscopy (SEM) images of CS@CPO-27-Co (R = 2), shown in Fig. 2, exhibit a consistent external surface free from crystalline particles. On the contrary, the cross-section of the beads displays crystals with a morphology similar to the rod-like form observed in the pristine powder. Interestingly, CPO-27 crystals within the beads are prominently visible and concentrated near the surface, contrasting with the denser core. This gradient could be attributed to the rapid formation of CPO-27 particles near the surface, where the organic linker is more concentrated, as reported by Lim et al.15 Composite beads with lower R display similar MOF particle morphology within the polymer matrix (Fig. S4, ESI†). However, it can clearly be seen that composites with higher CS contents present dense cores with encapsulated MOF particles. The chemical composition determined by EDS analysis (Fig. S5, ESI†), shows the good dispersion of the MOF building elements (Co, C, O) within the beads.
 | ||
| Fig. 2 SEM images of CPO-27-Co powder (a), CS@CPO-27-Co (R = 2) external surface (b), and CS@CPO-27-Co (R = 2) cross-section (c) and (d). | ||
Thermogravimetric analysis (TGA) of the prepared materials under an air atmosphere shows a weight loss curve (Fig. S6, ESI†) similar to that of CPO-27-Co, exhibiting three distinct steps. The initial weight loss up to 120 °C is likely due to the removal of residual solvents such as methanol, water, and DMF. The second weight loss step, starting at 290 °C, is attributed to the beginning of chitosan degradation,20 and the collapse of the MOF structure as a result of the combustion of the organic linker and the formation of cobalt oxide(II, III).21 The final weight loss corresponds to the reduction of Co3O4 to CoO at approximately 900 °C.22 In contrast, pure CS beads undergo complete combustion at 600 °C. Owing to the final CoO residue content, and by knowing the CPO-27-Co molecular formula [C8H6Co2O8], a MOF loading of 55 wt% can be estimated within the composite beads presenting a pure crystalline phase (CS@CPO-27-Co (R = 2)).
The BET surface area measured from the N2 physisorption isotherm of CS@CPO-27-Co (R = 2), shown in Fig. S7 (ESI†), is 254 m2 g−1. Comparatively, the BET surface area of the CPO-27-Co powder is 1052 m2 g−1. Such difference is attributed to the low porosity of the biopolymer, the bead densification following atmospheric drying (Fig. S8 and Table S2, ESI†), and partial pore blockage by the polymer matrix owing to its high content.7,20,23 Pore blockage is even more marked at lower MOF loadings (lower molar ratios R), as the crystals become fully embedded with the biopolymer matrix, thus hindering N2 accessibility to the pores and preventing the application of the BET equation (Fig. S7, ESI†). This observation aligns with the dense morphology observed through SEM analysis. To minimize densification and increase the specific surface area, the hydrogel beads were frozen with liquid nitrogen and freeze-dried. Their corresponding PXRD patterns and N2 sorption isotherms are presented in Fig. S9 (ESI†). Notably, the measured specific surface areas were 210, 300, and 567 m2 g−1, for CS@CPO-27-Co (R = 1/2)-C, CS@CPO-27-Co (R = 1)-C, and CS@CPO-27-Co (R = 2)-C (C standing for cryogels).
The obtained cryogels were evaluated for CO2 adsorption under static conditions at 298 K, across a pressure range of 0–760 mmHg. As depicted in Fig. 3, the maximum CO2 uptakes reached 0.84, 1.85, and 3.10 mmol g−1 for composites with molar ratios R of 1/2, 1, and 2, respectively. As CPO-27-Co accounts for about 55% of the CS@CPO-27-Co (R = 2)-C composite, and CS cryogels have a neglectable CO2 sorption capacity (0.1 mmol g−1), the CPO-27-Co within the beads can adsorb up to 5.85 mmol g−1 (MOF). This value falls behind the reported CO2 adsorption capacity for the pristine CPO-27-Co (∼6.90 mmol g−1 at 298 K) found in the literature,24 but it is surprisingly higher than pristine CPO-27-Co as-synthesized herein (3.35 mmol g−1), which may underline a favorable effect of chitosan over the crystallization of the CPO-27-Co MOF under the applied conditions. Also, it is significantly higher than what would have been expected from the BET surface area measurement. Additionally, the consistent upward slope of the adsorption curve for the three samples suggests that there may be a potential for increased adsorption capacity at higher pressures.25 Xerogels were similarly tested for CO2 sorption at 298 K. The maximum uptake measured for these composites (0.44, 1.50, and 3.16 mmol g−1 for R = 1/2, 1, and 2, respectively) is in line with those of the cryogels for the same MOF loadings (Fig. S10, ESI†). Thus, the accessibility of CO2 to the pores at 298 K post-atmospheric drying is mostly unaltered compared to the accessibility of N2 to the pores at liquid nitrogen temperature, where pore blockage is more marked.
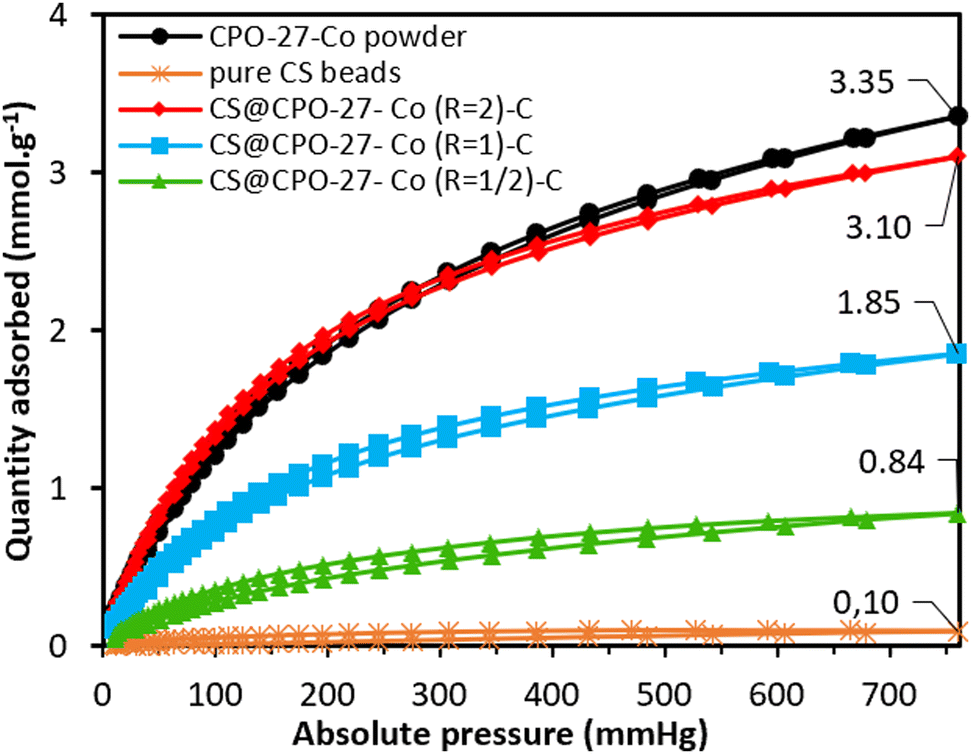 | ||
| Fig. 3 CO2 adsorption isotherms of the as-synthesized CPO-27-Co powder, pure CS beads, and CS@CPO-27-Co (R) beads measured at 298 K. | ||
It is worth noting that the CO2 sorption capacity of CS@CPO-27-Co (R = 2) xerogels and cryogels exceeds by more than 50% all reported CS@MOF composites prepared via an in situ crystallization route. For instance, CS@ZIF-67 aerogels (supercritical CO2 dried) demonstrated an adsorption capacity of only 0.76 mmol g−1,26 while CS@ZIF-8 and CS@HKUST-1 cryogels (freeze-dried) exhibited capacities of 0.68 and 1.98 mmol g−1 at 298 K, respectively.20,27 To investigate the order of magnitude of CO2 adsorption energy on our composites, CO2 isotherms of CS@CPO-27-Co (R = 2)-C were measured at 273, 283, and 298 K (Fig. S11, ESI†). The averaged adsorption isosteric enthalpy |ΔadsH|, measured according to the Clausius–Clapeyron equation,28 is 33.3 kJ mol−1. This closely aligns with the value of 33.9 kJ mol−1 reported by Yoo et al. at 298 K29 and suggests that the adsorption of CO2 with CS@CPO-27-Co (R = 2)-C is driven by physical interactions.30 Importantly, chitosan does not impede the use of CS@MOF composites for CO2 sorption over multiple cycles.26 Indeed, the uptake capacity of CS@CPO-27-Co (R = 2)-C was constant during five cycles of adsorption–desorption (Fig. S12, ESI†).
To prove the versatility of this approach to produce CS@CPO-27-M composite beads, the cobalt(II) precursor was replaced by nickel(II) precursors. Fig. 4 illustrates that the obtained CS@CPO-27-Ni (R = 2) exhibits the same characteristic Bragg peaks as its respective pristine CPO-27-Ni powder. Moreover, N2 physisorption highlights its microporous nature (Fig. S13 and Table S3, ESI†), with an SBET of 315 m2 g−1. Thus, the use of chitosan as a matrix to crystallize the CPO-27-M MOF is well-adapted for the production of CS@CPO-27-M composites.
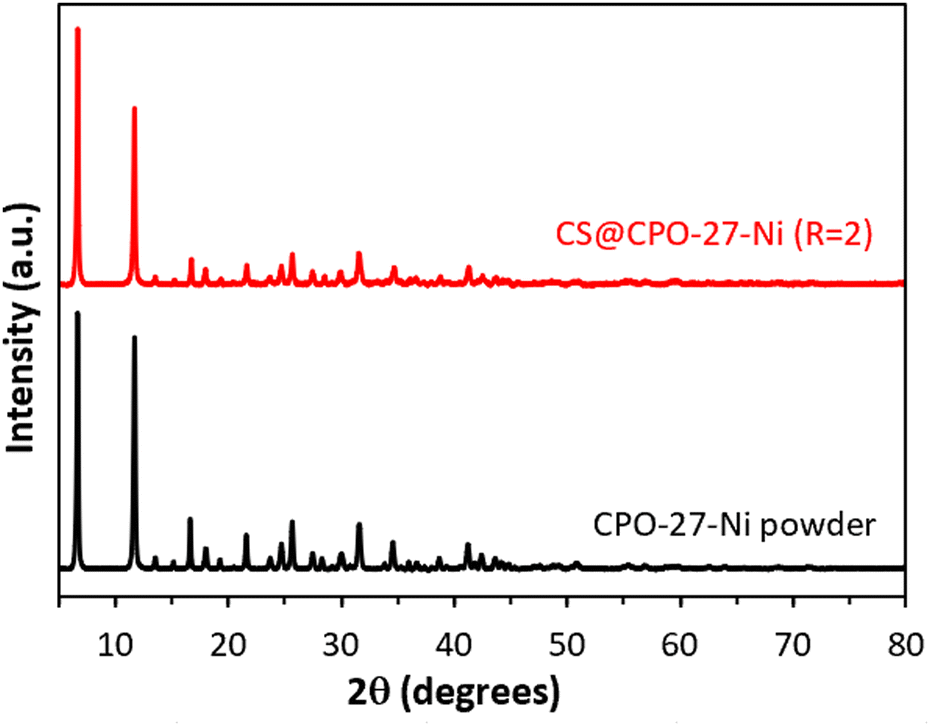 | ||
| Fig. 4 PXRD patterns of CS@CPO-27-Ni (R = 2) xerogels (red) and its pristine CPO-27-Ni powder (black). | ||
In summary, an in situ crystallization route was successfully adapted to produce porous chitosan@CPO-27-Co shaped in the form of beads, loading up to 55 wt.% of MOF particles. Such composite beads present a high potential for CO2 uptake, with an adsorption capacity of 3.10 mmol g−1 at 298 K, surpassing all the reported MOF composites prepared via an in-situ crystallization approach so far. The calculation of the adsorption isosteric enthalpy further confirmed the physical nature of CO2 sorption within the composite beads, highlighting their potential for regeneration. Finally, cobalt ions (Co2+) were replaced by nickel ions (Ni2+) to produce a series of well-crystallized CS@CPO-27-M composites.
Y. K., J. D. and T. L. conceived and designed the project. Y. K. prepared the materials and conducted most characterization. Y. K. and C. V. conducted CO2 sorption measurements. Y. K. wrote the original manuscript. All authors analysed the data, discussed the results and commented on the manuscript.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- A. G. Slater and A. I. Cooper, Science, 2015, 348, aaa8075 CrossRef PubMed.
- A. J. Howarth, A. W. Peters, N. A. Vermeulen, T. C. Wang, J. T. Hupp and O. K. Farha, Chem. Mater., 2017, 29, 26–39 CrossRef CAS.
- S. Gulati, S. Vijayan, M. S. Kumar, B. Harikumar, M. Trivedi and R. S. Varma, Coord. Chem. Rev., 2023, 474, 214853 CrossRef CAS.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. Ö. Yazaydin, R. Q. Snurr, M. O’Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424–428 CrossRef CAS PubMed; H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef PubMed.
- X.-M. Liu, L.-H. Xie and Y. Wu, Inorg. Chem. Front., 2020, 7, 2840–2866 RSC; T. Kitao, Y. Zhang, S. Kitagawa, B. Wang and T. Uemura, Chem. Soc. Rev., 2017, 46, 3108–3133 RSC.
- S. Gaikwad and S. Han, J. Environ. Chem. Eng., 2023, 11, 109593 CrossRef CAS.
- R. Zhao, T. Ma, S. Zhao, H. Rong, Y. Tian and G. Zhu, J. Chem. Eng., 2020, 382, 122893 CrossRef CAS.
- A. Balakrishnan, M. M. Jacob, N. Dayanandan, M. Chinthala, M. Ponnuchamy, D.-V. N. Vo, S. Appunni and A. S. Gajendhran, Mater. Adv., 2023, 4, 5920–5947 RSC.
- Q. Miao, L. Jiang, J. Yang, T. Hu, S. Shan, H. Su and F. Wu, J. Water Process Eng., 2022, 50, 103348 CrossRef.
- N. L. Rosi, J. Kim, M. Eddaoudi, B. Chen, M. O’Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 1504–1518 CrossRef CAS PubMed.
- T. Xiao and D. Liu, Micro. Meso. Mater., 2019, 283, 88–103 CrossRef CAS; H. He, R. Li, Z. Yang, L. Chai, L. Jin, S. I. Alhassan, L. Ren, H. Wang and L. Huang, Catal. Today, 2021, 375, 10–29 CrossRef.
- H.-Y. Cho, D.-A. Yang, J. Kim, S.-Y. Jeong and W.-S. Ahn, Catal. Today, 2012, 185, 35–40 CrossRef CAS.
- T. Kim, D. H. Kim, S. Kim, Y. D. Kim, Y.-S. Bae and C. Y. Lee, Polyhedron, 2015, 90, 18–22 CrossRef CAS.
- A. I. Spjelkavik, A. S. Divekar, T. Didriksen and R. Blom, Chem. – Eur. J., 2014, 20, 8973–8978 CrossRef CAS PubMed; S. Dasgupta, S. Divekar, A. I. Spjelkavik, T. Didriksen, A. Nanoti and R. Blom, Chem. Eng. Sci., 2015, 137, 525–531 CrossRef; S. Krishnamurthy, R. Blom, M. C. Ferrari and S. Brandani, Adsorption, 2020, 26, 711–721 CrossRef; X. Peng, J. Zhang, J. Sun, X. Liu, X. Zhao, S. Yu, Z. Yuan, S. Liu and X. Yi, ACS Appl. Nano Mater., 2023, 6, 16694–16701 CrossRef; G. Mondino, A. I. Spjelkavik, T. Didriksen, S. Krishnamurthy, R. E. Stensrød, C. A. Grande, L. O. Nord and R. Blom, Ind. Eng. Chem. Res., 2020, 59, 7198–7211 CrossRef.
- J. Lim, E. J. Lee, J. S. Choi and N. C. Jeong, ACS Appl. Mater. Interfaces, 2018, 10, 3793–3800 CrossRef CAS PubMed.
- S. El Hankari, M. Bousmina and A. El Kadib, Prog. Mater. Sci., 2019, 106, 100579 CrossRef CAS.
- X. Liang, P. Wang, C. Li, M. Yuan, Q. Shi and J. Dong, Micro. Meso. Mater., 2021, 320, 111109 CrossRef CAS.
- J. Mujtaba, H. Sun, G. Huang, K. Mølhave, Y. Liu, Y. Zhao, X. Wang, S. Xu and J. Zhu, Sci. Rep., 2016, 6, 20592 CrossRef CAS PubMed.
- S. Lu, Y. Xiao, Q. Zhao, W. Zhao and G. He, Sep. Purif. Technol., 2023, 306, 122665 CrossRef CAS; C. Huang, Z. Zhao, E. Ping, L. Zhang, Y. Zhou and L. Qin, Micro. Meso. Mater., 2021, 323, 111241 CrossRef.
- N. Hammi, M. Bonneau, A. El Kadib, S. Kitagawa, T. Loiseau, C. Volkringer, S. Royer and J. Dhainaut, ACS Appl. Mater. Interfaces, 2023, 15, 53395–53404 CrossRef CAS PubMed.
- M. Díaz-García, Á. Mayoral, I. Díaz and M. Sánchez-Sánchez, Cryst. Growth Des., 2014, 14, 2479–2487 CrossRef.
- J. M. Rami, C. D. Patel, C. M. Patel and M. V. Patel, Mater. Today: Proc., 2021, 43, 655–659 CAS.
- A. El Kadib, Chem. Rec., 2020, 20, 753–772 CrossRef CAS PubMed.
- S. Mahajan and M. Lahtinen, J. Environ. Chem. Eng., 2022, 10, 108930 CrossRef CAS.
- A. K. Adhikari and K.-S. Lin, Chem. Eng. J., 2016, 284, 1348–1360 CrossRef CAS.
- N. Hammi, N. Couzon, T. Loiseau, C. Volkringer, A. El Kadib, S. Royer and J. Dhainaut, Mater. Today Sustainable, 2023, 22, 100394 CrossRef.
- J. Yao, R. Chen, K. Wang and H. Wang, Micro. Meso. Mater., 2013, 165, 200–204 CrossRef CAS.
- F. Rouquerol, J. Rouquerol and K. Sing, Adsorption by Powders and Porous Solids: Principles, Methodol. Appl. 2nd Ed., 2014, 25–56 Search PubMed.
- G. Y. Yoo, W. R. Lee, H. Jo, J. Park, J. H. Song, K. S. Lim, D. Moon, H. Jung, J. Lim, S. S. Han, Y. Jung and C. S. Hong, Chem. – Eur. J., 2016, 22, 7444–7451 CrossRef CAS PubMed.
- A. Samanta, A. Zhao, G. K. H. Shimizu, P. Sarkar and R. Gupta, Ind. Eng. Chem. Res., 2012, 51, 1438–1463 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental protocols, photographs, and additional characterization (XRD, IR, TGA, N2 and CO2 isotherms, SEM, EDS). See DOI: https://doi.org/10.1039/d4cc02082k |
| This journal is © The Royal Society of Chemistry 2024 |