
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDFT and AFIR study on the copper(I)-catalyzed mechanism of 5-enamine-trisubstituted-1,2,3-triazole synthesis via C–N cross-coupling and the origin of ring-opening of 2H-azirines†
Fan Yua,
Zhaoman Zhoua,
Jiajia Songa and
Yanying Zhao *ab
*ab
aDepartment of Chemistry, Key Laboratory of Surface & Interface Science of Polymer Materials of Zhejiang Province, Zhejiang Sci-Tech University, Hangzhou 310018, China
bState Key Laboratory of Advanced Textiles Materials and Manufacture Technology, Ministry of Education, Zhejiang Sci-Tech University, Hangzhou 310018, China
First published on 13th January 2021
Abstract
Understanding the synthesis mechanism of substituted 1,2,3-triazoles is an important and state-of-the-art research area of contemporary copper(I)-catalyzed terminal alkyne and organic azide click reaction (CuAAC), which has invoked increasing close collaborations between experiment and theory including copper catalyzed interrupted click reaction. In this study, the mechanism of Cu(I)-catalyzed 5-enamine-functionalized fully substituted 1,2,3-triazole synthesis was rationalized via density functional theory (DFT) and multicomponent artificial force-induced reaction (MC-AFIR) methods. The reasonable reaction route consists of (a) di-copper catalyzed ring-opening of 2H-azirines, (b) alkyne hydrogen atom transfer, (c) [3 + 2] ring cycloaddition, and (d) C–N bond formation through reductive elimination. The MC-AFIR method was used for the systematic determination of transition states for the C/N–Cu bond formation, C–N bond coupling and crossing points between singlet and triplet states. Our survey on the prereactant complexes suggested that the dicopper-catalyzed 2H-azirine ring-opening and alkyne hydrogen activation are both thermodynamically feasible via a singlet/triplet crossing point. This explains why Et3N is critical for alkyne hydrogen transfer (HT) before the [3 + 2] cycloaddition reaction, and the C–N cross-coupling product instead of the click product (byproduct). Our DFT results indicate that the transmetalation process is the rate determination step along the triplet state potential energy surface. This study provides important mechanistic insights for the interrupted CuAAC reaction to form 5-enamine-fully-substituted-1,2,3-triazoles. Further insight prediction interprets that solvent and extra strong ligand coordination play a certain role in competitive reactions.
Introduction
Fully substituted 1,2,3-triazoles play a significant role in chemical, biological, and pharmaceutical fields, and are widely applied in chemical medicine and materials science.1–3 The importance of 1,2,3-triazoles has inspired chemists to develop more and more economically and effectively synthetic methods.4–7 The synthesis among the substituted 1,2,3-triazoles can be traced back to 1960s, when Huisgen reported a simple and widely useful approach to synthesize a mixture of 1,4- and 1,5-disubstituted 1,2,3-triazole mixture via the 1,3-dipolar cycloaddition reaction of azides and alkynes.8,9 However, until 2002, Sharpless et al. reported a copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction leading to the efficient and regioselective synthesis of 1,4-disubstituted 1,2,3-triazoles separated successfully from mixtures.10 To date, copper is the superior catalyst in the exclusive 1,4-disubstituted 1,2,3-triazole formation although other transition metals were developed for the click reaction.11–17 However, it is still a challenge to unravel the reaction mechanism of this catalytic cycle to direct the synthesis. Understanding the path(s) of the reaction along with numerous possible intermediates and transition states is crucial for both the deep development and further generalization of the-state-of-art application.To obtain the above goal, either experimental or theoretical investigations have been explored for trapping the active intermediates and optimizing the transition states connecting between the reactant and product. Initially, Rodionov et al.18 ever proposed experimentally a mononuclear mechanistic model including a six-membered mononuclear copper(III) intermediate. However, quantum mechanical investigations predicated another idea, demonstrating that the reaction path with a binuclear structure has an energy barrier lower than that of a mononuclear structure. Subsequent experiments showed that the nuclearity of this structure was highly debatable and more than one copper species may be actively involved in the reaction.19–25 In 2013, Fokin et al.26 directly verified the realization of the CuAAC reaction by the X-ray structures of two key binuclear Cu(I) intermediates, as shown in Scheme 1(a) and (b). It is also revealed theoretically that a cycloaddition step is dicopper process for CuAAC reaction via a six-membered binuclear copper structure intermediate, as shown in Scheme 1(c). Thus, a series of trapping dinuclear copper intermediates have been reported to form 1,4-disubstituted 1,2,3-triazoles via traditional CuAAC reaction.27–32 Its click mechanism is also constantly updated.33–37
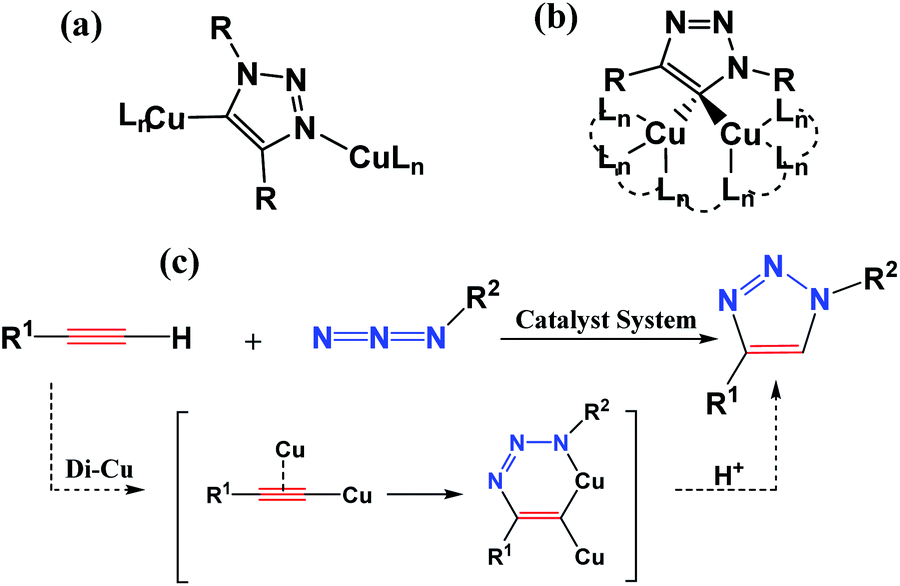 | ||
| Scheme 1 Captured key intermediates (a) and (b), and proposed the dicopper-catalyzed CuAAC reaction mechanism (c) (from ref. 22). | ||
However, the efficient synthesis of 1,4,5-trisubstituted 1,2,3-triazoles via the CuAAC reaction is a state-of-the-art goal for the strategy named “Cu-catalyzed interrupted click reaction”.38 Electronegative groups, such as N-tosylhydrazones, and S-, Se-, N-, CF3-, SCF3-, have been applied to efficiently synthesize trisubstituted-1,2,3-triazoles.39–43 It is worth mentioning that 1,4-disubstituted by-product generated is still highly required in atom-economic point of view. There is the most direct approach in intercepting 5-cuprous-1,2,3-triazole intermediates in situ by an electron-withdrawing trapping reagent rather than proton reductive elimination to give 1,4-disubstituted-5-functionalized 1,2,3-triazole.44–46
In 2016, Chen et al.47 reported a one-pot three-component synthesis of fully substituted enamine-functionalized 1,2,3-triazoles with about 97% yield via copper(I)-catalyzed 2H-azirine ring opening. In addition, there is no yield of the 1,4-disubstituted 1,2,3-triazole byproduct, which is significantly different from achieving the goal product via the interrupted click reaction. According to their experimental results and control experiments in Scheme 2b and c, the mechanism should involve 2H-azirine ring opening to copper nitrene, [3 + 2] cycloaddition reaction, and the C–N bond formation. It is necessary to obtain an explicit understanding of the reaction path(s) with high yield and selectivity to search more efficient synthetic strategies. Herein, we present our quantum chemical calculations on the reaction intermediates and transition states aiming to generalize and interpret it to a greater extent, as shown in Scheme 3.
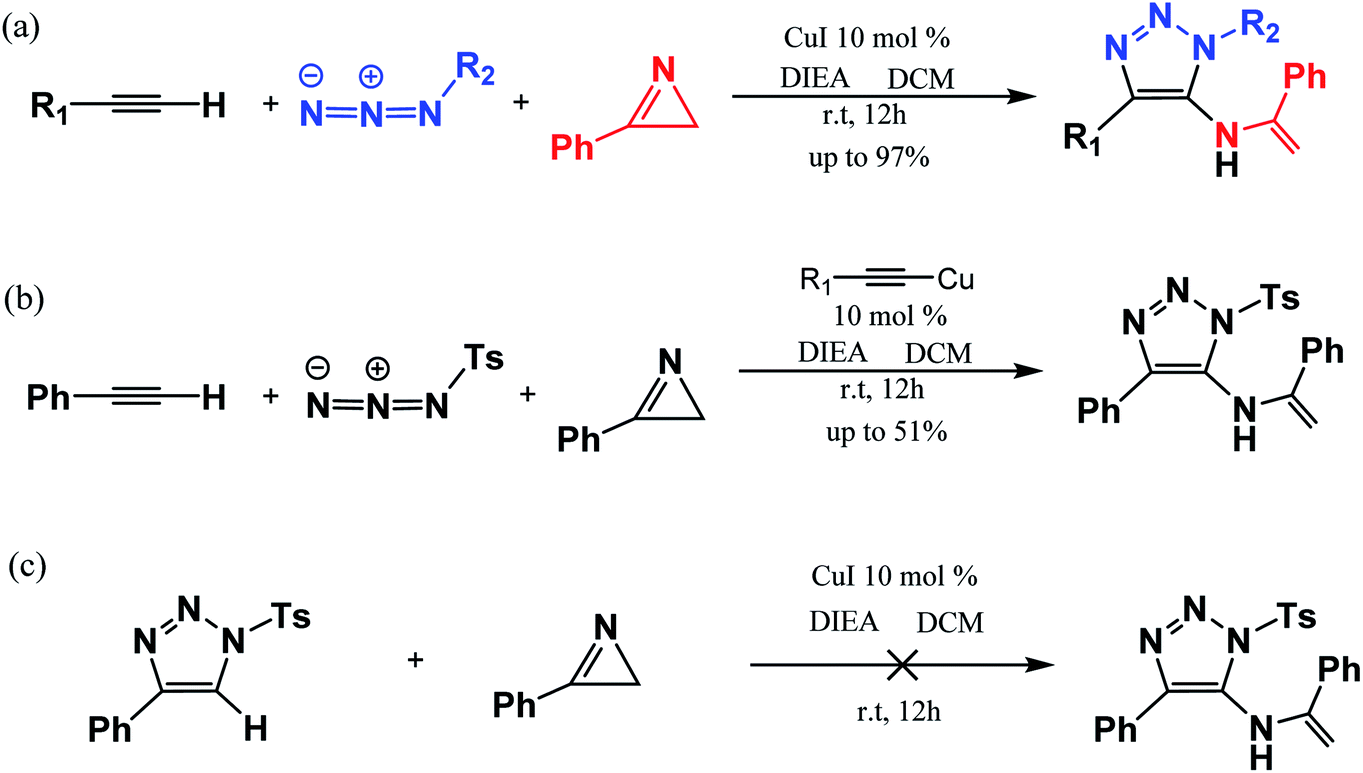 | ||
| Scheme 2 Three-component reaction reported experimentally (a) and controlled experiments (b and c) by Chen et al. (ref. Org. Lett. 2017, 19, 10).47 | ||
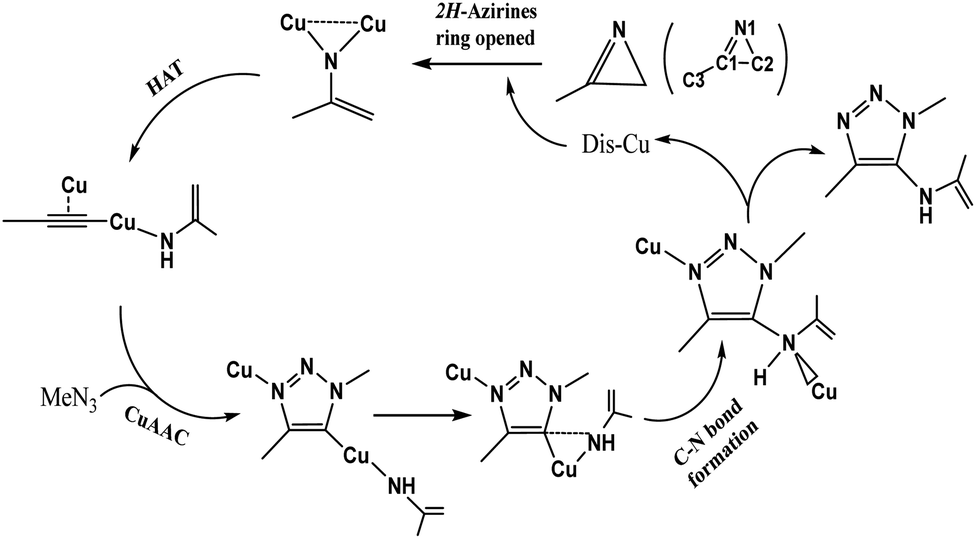 | ||
| Scheme 3 Proposed catalytic cycle by the dicopper(I) catalyst. | ||
Computational details
Density functional theory, as implemented in Gaussian 09 program,48 was used to optimize all structures. The M06-L functional, including the empirical dispersion corrections, with the Becke–Johnson damping (D3BJ), was employed.49 The optimization of closed shell singlet spin states was performed with restricted-M06-L (RM06-L), and the triplet states were calculated by unrestricted-M06L (UM06L). The LANL2TZ+ effective core potential (ECP) was for copper atoms and iodine atoms. The 6-31+G(d,p) basis sets were used for all other atoms. The integral equation formalism-polarizable continuum model (IEF-PCM) was used as the implicit solvation model for geometry optimizations, where dichloromethane (DCM) was used as the solvent (ε = 10.125).50 The nature of the stationary points, minima or transition states (TS) was confirmed by performing vibrational frequency calculations at 298.15 K and 1 atm.All minima stationary points have no imaginary frequencies and each transition state (TS) correctly linking with two adjacent intermediates by intrinsic reaction coordinate (IRC) calculations have confirmed with only one imaginary frequency. The minimum energy crossing points (MECPs) were investigated. The automated reaction path searing methods, namely the global reaction route mapping (GRRM)51 strategy and the artificial force induced reaction (AFIR),52 were applied to the lowest PESs of singlet and triplet crossing points. By these methods, critical points, that is minima, and transition states for the ground (S0), the lowest triplet (T1) PESs and S0/T1 crossing minima, were systematically explored. The GRRM strategy and AFIR method were employed for the singlet states and triplets, respectively. Furthermore, the wave function analysis was performed by a Multiwfn 3.5 program,53 and the relaxed force constant (RFC) was obtained by the Compliance 3.0.2 procedure.54,55
Results and discussion
Initially, dicopper nitrene was obtained from the ring-opening of 2H-azirine ring catalyzed by cuprous iodide, which played a key role in the hydrogen atom transfer (HAT) of the terminal alkyne to form di-copper acetylene. After HAT, the CuAAC reaction occurred by dicopper acetylene and azide. Finally, the C–N bond was formed on the C5 position of 1,2,3-triazole.2H-Azirine ring-opening
The ring-opening of 2H-azirine is generally catalyzed by copper(I) species to form copper nitrenes, which are of interest as intermediates in the catalytic amination of C–H bonds. However, to the best of our knowledge, despite the advances in the isolation of multiply bonded late-transition-metal complexes, a structurally characterized terminal copper nitrene has not been reported. DFT calculations are further applied to explore the activation of 2H-azirines by mono- and di-cuprous copper cores, as shown in Fig. 1. Initially, one Cu(I) coordinating to 2H-azirine decreases ∼27 kcal mol−1 in energy to form the singlet P2-1s intermediate. Otherwise, the triplet P2-1t intermediate increased by ∼32 kcal mol−1 in energy. The following C–N single bond cleavage passed a singlet P2-TSmonos transition state with an energy barrier of 37.9 kcal mol−1 to produce a triplet P2-monot intermediate via singlet and triplet crossing structure Mono-CP1. The triplet P2-monot intermediate is consistent with the reported results from Cundari et al.56 However, the barrier energy is above 48 kcal mol−1 from the triplet P2-monot via the P2-TSmonot transition state. Thus, it is too difficult to cross the energy barrier for either a singlet or triplet intermediate in the mono-copper catalyzed process at room temperature. So, we performed the di-copper catalytic mechanism.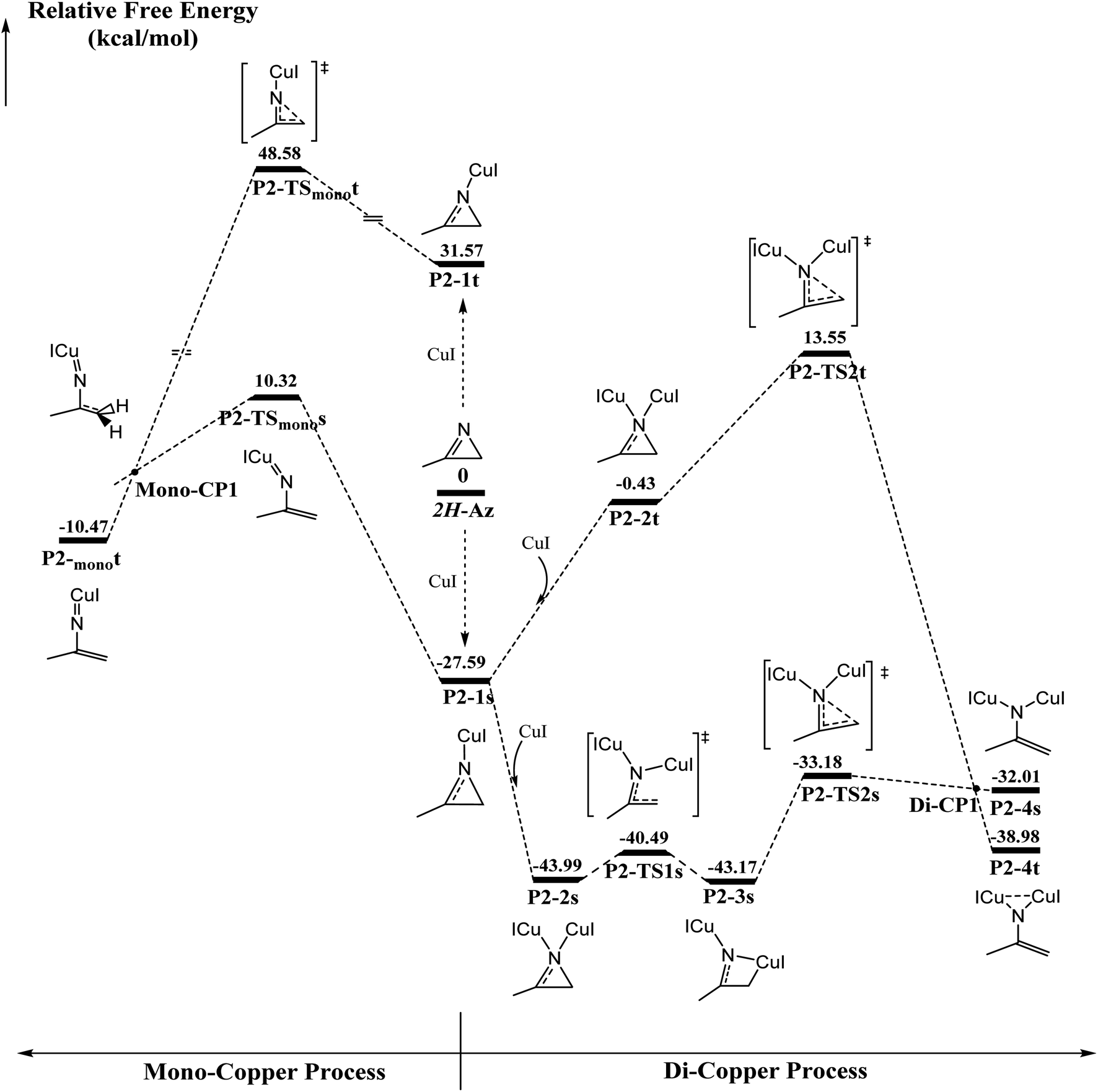 | ||
| Fig. 1 Calculated energy profiles of mononuclear and binuclear pathways for 2H-azirine ring opening. The relative Gibbs energies (kcal mol−1) are given in kcal mol−1 calculated at the (U)M06-L-D3BJ-IEFPCM/6-31+G(d,p)/Lanl2TZ+ level in dichloromethane. | ||
Initially, the singlet P2-2s with the C2v symmetry is formed due to the second copper coordination to P2-1s, whose energy further decreases 16.4 kcal mol−1. Next, the C–N single bond cleavage of P2-2s gives rise to a four-member-ring P2-3s intermediate with an energy barrier 3.5 kcal mol−1. Finally, the ring is opened across over the P2-TS2s transition state with an energy barrier of 10 kcal mol−1 via a singlet/triplet crossing point Di-CP1 direct to triplet P2-4t. As shown in Fig. S1,† 2H-azirine ring is opened for the transition state P2-TS1s due to the light blue in the C–N single bond region of valence layer density and without zero value point in the region of reduced density gradient (RDG) diagram. The AIM analysis of P2-3s revealed a ring critical point (Rcp) and a bond critical point (Bcp) between C2 atom and Cu1 atom, as shown in Fig. S2(a).† On the other hand, the orange region (Red arrow) indicated the lone pair electrons on the C2 atom.
The electron local function (ELF), as shown in Fig. S2(b),† suggests that the P2-3s intermediate is the nitrogen heterocyclic copper(I) complex. Single di-copper nitrene P2-4s is formed via P2-TS2s with an energy barrier of 9.99 kcal mol−1. There is a significant difference in localizing molecular orbital (LMO) among N1 and C2 atoms, as shown in Fig. S3.† The LMO of P2-3s is a heart-shaped σ orbital, formed by the “head-to-head” mode of 2p orbital of N1 and C2 atom and 3d orbital of the Cu1 atom, and the LMO of P2-TS2s is a typical π orbital, formed by the “side by side” mode due to the 2p orbital of N1, C1 and C2 atoms. That indicates that the hybridization type of C2 is transformed from sp3 to sp2 via P2-TS2s. Furthermore, that the triplet di-copper nitrene P2-4t was found to be 6.97 kcal mol−1 lower in energy than P2-4s. P2-4t was formed via P2-TS2t with an energy barrier of about 41 kcal mol−1 starting from P2-1s. Also, there must be a MECP Di-CP1 about di-copper nitrene. Natural Population Analysis (NPA) for P2-4t on N1 and C2 is 0.80 and 0.70, and it indicated that two single electrons are respectively populated on N1 and C2. The P2-4t geometry is very similar to the synthesis study of the di-copper nitrene intermediate via {[Me3NN]Cu}2(toluene)57 and azide N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NN–Ar reported by Warren et al.58 Fig. S4† indicates that the localized orbital locator (LOL) of P2-4s and P2-4t is obviously different in the region of Cu1–N1–Cu2. For P2-4s, the lone pair of N1 atom is mainly located at the Cu1–N1 and Cu2–N1 regions (red arrows as shown in Fig. S4 (a)†). As shown in Fig. S5(a) and (b),† there are two opposite spin p–d π bonds. The larger LOL value (black arrows in Fig. S4†) of P2-4t than that of P2-4s indicates that the lone pair electrons of N1 are distributed on the Cu1–N1–Cu2 region. In Fig. S5(c),† the localized molecular orbital (LMO) map shows that the electron density center is attributed to the n orbital from Cu1 and Cu2. The n orbital is mainly contributed to the weak interaction in the region, as shown in Fig. S6.†
NN–Ar reported by Warren et al.58 Fig. S4† indicates that the localized orbital locator (LOL) of P2-4s and P2-4t is obviously different in the region of Cu1–N1–Cu2. For P2-4s, the lone pair of N1 atom is mainly located at the Cu1–N1 and Cu2–N1 regions (red arrows as shown in Fig. S4 (a)†). As shown in Fig. S5(a) and (b),† there are two opposite spin p–d π bonds. The larger LOL value (black arrows in Fig. S4†) of P2-4t than that of P2-4s indicates that the lone pair electrons of N1 are distributed on the Cu1–N1–Cu2 region. In Fig. S5(c),† the localized molecular orbital (LMO) map shows that the electron density center is attributed to the n orbital from Cu1 and Cu2. The n orbital is mainly contributed to the weak interaction in the region, as shown in Fig. S6.†
The ellipticity of RPC for P2-1s and P2-2s is −5.184, −4.355 and −5.217, respectively, which indicates that the ring stability is P2-1s > 2H-Az > P2-2s. ELF diagrams of 2H-Az and P2-1s show that the value of C2–N1 for P2-1s is significantly higher than that of 2H-Az, as shown in Fig. S7.† The AdNDP analysis shows that only the C1–C2–N1 three-center π orbits in Fig. S8(a)† are found for P2-1s, but the C1–C2–N1 three-center π orbits of 2H-Az are not to be found. It is concluded that the mono-copper coordination to 2H-AZ makes the πC1N1 bond delocalization and strengths the σC2-N1 bond conjugation effect to enhance the stability of the triatomic heterocycle.
The AdNDP analysis of P2-2s displays a C1–C2–N1 three-center π orbital, as shown in Fig. S8(b).† Table S1† lists the atom contributions by the Beche method for P2-1s and P2-2s. It was found that the atom contributions from P2-1s to P2-2s, N1, C1 and C2 totally decreased by ∼9%, corresponding to the increase in nearly 9% for both coppers. These results indicate that di-copper coordination can induce π electrons flow to the copper center, which can reduce greatly the conjugation effect and deteriorate the stability of ring. Next, the relaxed force constant (RFC) analysis indicates the values of the N1–C2 and C1–C2 bonds, as shown in Table S2.† For the N1–C2 bond, there is a decrease according to the order P2-1s > 2H-Az > P2-2s, which indicates that the mono-copper coordination increases and di-copper coordination can weaken the covalent effect of the N1–C2 bond. These results exactly interpret the lower barrier for di-copper ring-opening than mono-copper one.
Hydrogen transfer (HT)
Catalytic C–H amination could be achieved by converting the di-copper nitrene intermediate to mono-copper nitrene.59,60 We calculated the reaction path of the hydrogen activation of terminal alkyne via di-copper nitrene P2-4t, as shown in Fig. 2.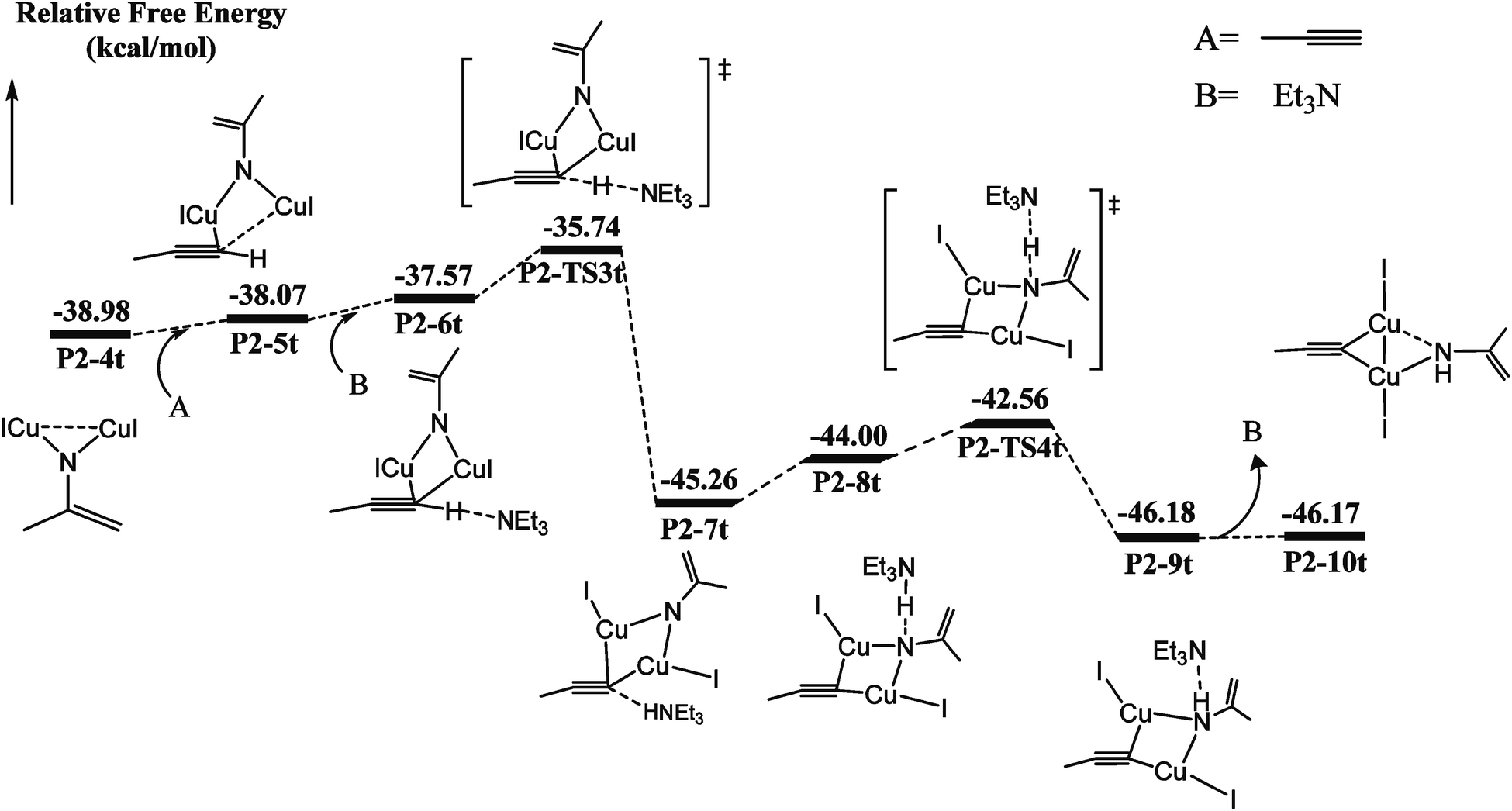 | ||
| Fig. 2 Calculated energy profiles of the hydrogen atom transfer (HAT) of the terminal alkyne process starting from the ring opening 2H-azirine intermediate P2-4t. The relative Gibbs energies (kcal mol−1) are given in kcal mol−1 calculated at the (U)M06-L-D3BJ-IEFPCM/6-31+G(d,p)/Lanl2TZ+ level in dichloromethane. | ||
A copper atom of P2-4t coordinates with C5 to form a P2-5t intermediate with a potential energy increase of 0.91 kcal mol−1. Then, Et3N and terminal acetylene H form a hydrogen bond of C–H⋯N with a bond distance of 1.800 Å. Both Cu coordinate with C5 to form a P2-6t. Compared with P2-4t, the potential energy increases by 1.41 kcal mol−1, and HOMO indicates the bonding character between H1 and N2, as shown in Fig. S9,† so the first barrier is only 1.83 kcal mol−1. Transition state P2-TS3t activates the C–H bond of terminal acetylene and obtains a bis-copper acetylene P2-7t intermediate with H fixed by Et3N. The potential energy decreases to −45.26 kcal mol−1. P2-8t is the intermediate of Et3N-carrying terminal acetylene H in P2-7t, which forms the N–H⋯N hydrogen bond between terminal acetylene H and nitrogen binder N atom after isomerization. The potential energy increases to −44.00 kcal mol−1. The transition state P2-TS4t with a potential barrier of 2.70 kcal mol−1 transfers H atom from Et3N to nitrogen binder N atom, and the intermediate P2-9t is obtained. The potential energy decreases to −46.18 kcal mol−1. The intermediate P2-10t is obtained with the loss of Et3N.
As shown in Fig. 3, from P2-5t to P2-7t, Cu1–N1 and Cu2–N1 bond lengths also gradually increase from 1.886 and 1.844 to 1.943 and 1.962 Å, and the natural charges of N1 becomes negative from −0.120 to −0.39. Moreover, the Cu1–C5 and Cu2–C5 bond lengths with 2.222 Å and 2.304 Å in P2-6t become short to 2.077 and 1.939 Å in P2-8t, which demonstrates that the strong coordination of Cu1 and Cu2 with C5 induces the weak coordination of Cu1 and Cu2 with N1, and further enhances the nucleophilicity of the N atom of nitrene. Therefore, the small barrier 1.44 kcal mol−1 is easily crossed for the H1 transfer from N2 (Et3N) to N1. The BCP parameters of Cu1–N1 in P2-10t indicate a closed-shell interaction, not an obvious covalent interaction due to the ρ(r) = 0.073, ∇2(r) = 0.419, E(r) = 0.001, respectively. It is concluded that transformation from di-copper nitrene to mono-copper nitrene completed at P2-10t.
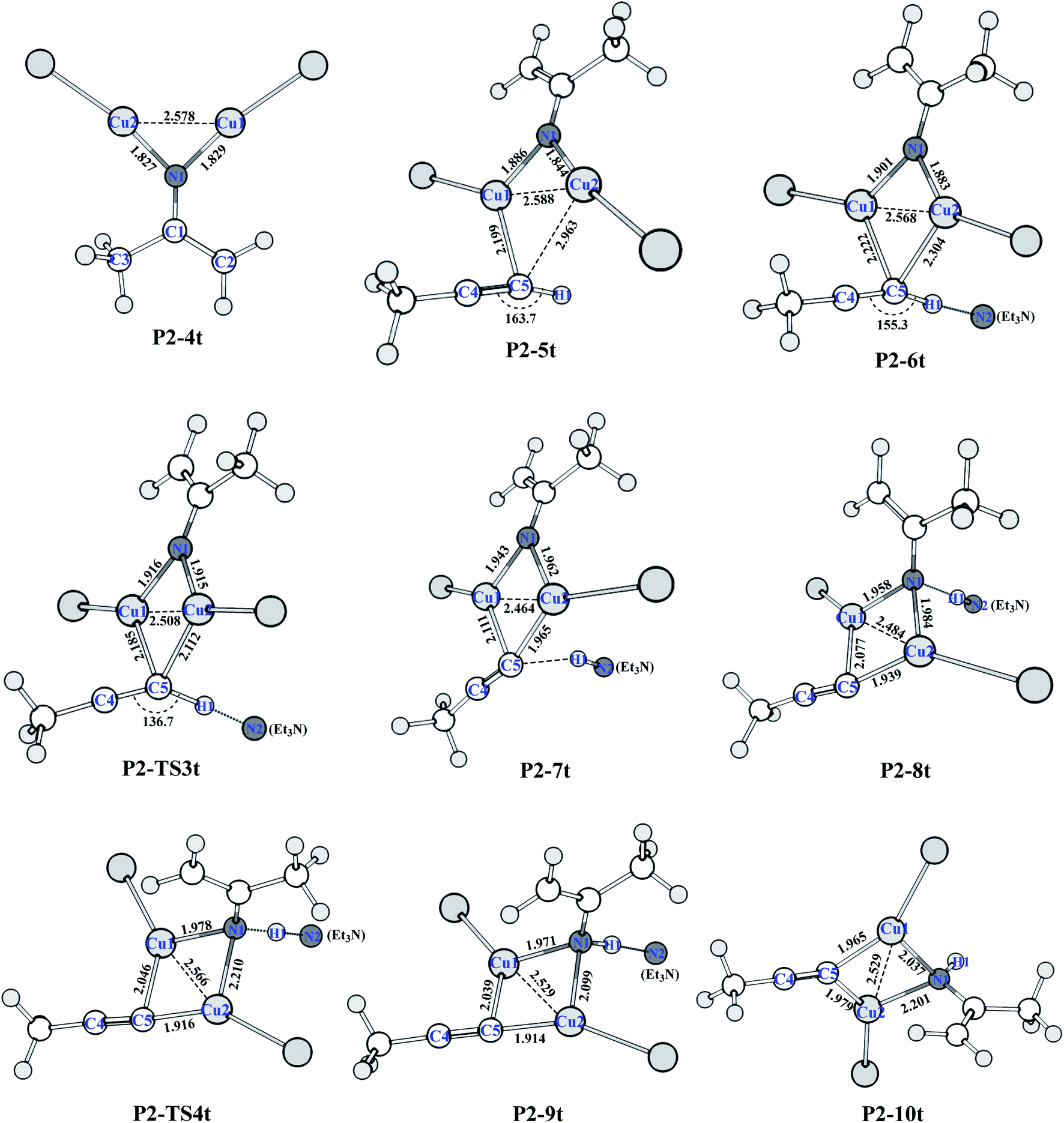 | ||
| Fig. 3 Optimized structures of HAT of the terminal alkyne process (bond distances in Å) at the (U)M06-L-D3BJ-IEFPCM/6-31+G(d,p)/Lanl2TZ+ level in dichloromethane. | ||
[3 + 2] cycloaddition
As shown in Fig. 4, N3Me reacts with P2-10t to form P2-11t with 3.40 kcal mol−1 increasing. The activation barrier for C–N coupling from the P2-11t complex through P2-TS5t is 10.67 kcal mol−1 to the six-membered P2-12t intermediate. However, it is more important to note that the activation barrier is 3.44 kcal mol−1 for the second C–N coupling to produce five-membered P2-13t, which is ∼7 kcal mol−1 lower than the activation barrier for the first C–N cross-coupling. The 5-dicuprous 1,2,3-triazole formation lowers to −84.74 kcal mol−1 in energy, which is in good agreement with previous reports.26,61,62 Therefore, the 3 + 2 cycloaddition reaction can easily occur along one certain reaction potential energy surface profile.33–37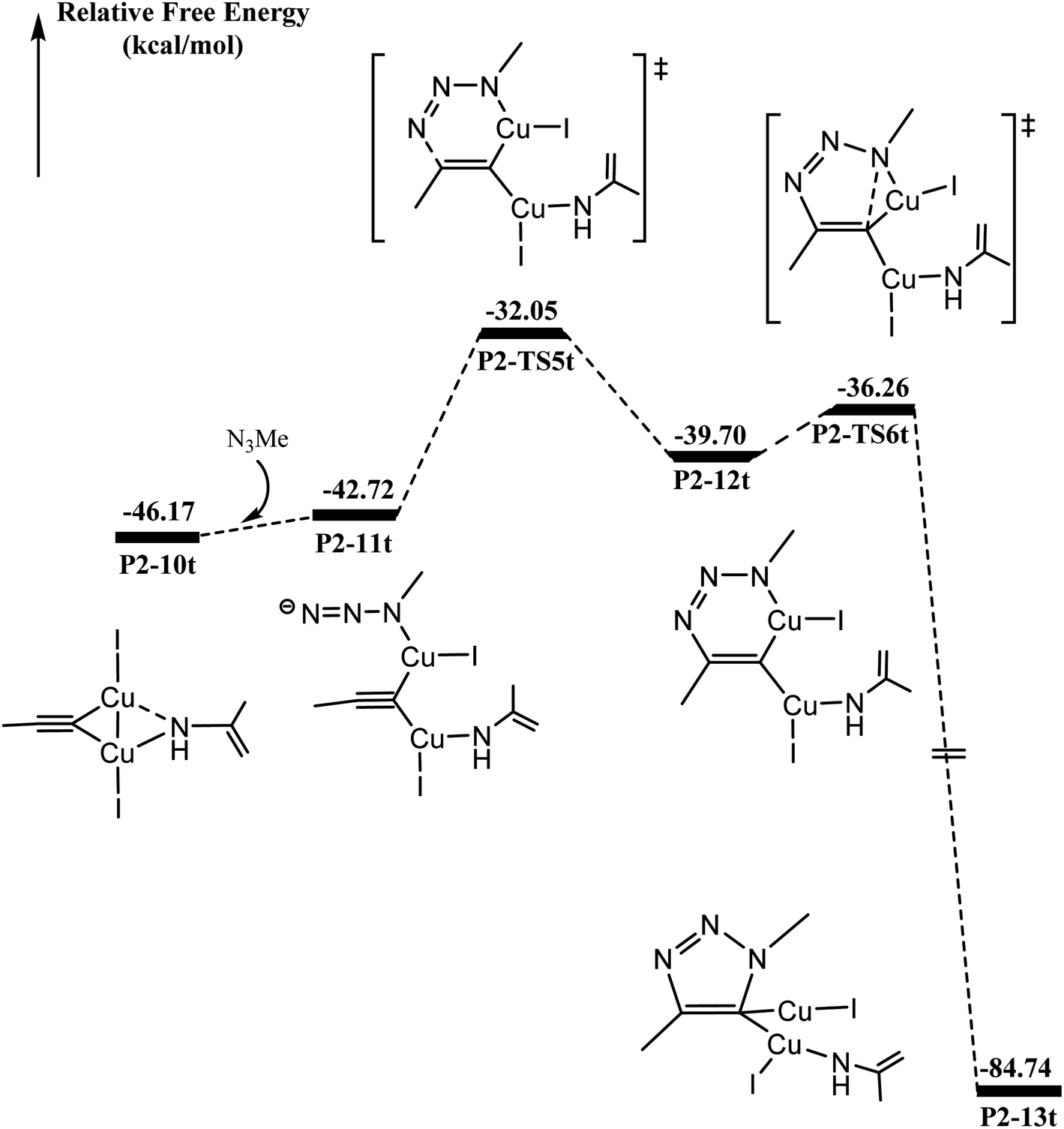 | ||
| Fig. 4 Calculated energy profiles of binuclear [3 + 2] cycloaddition in the CuAAC reaction. The relative Gibbs energies calculated at the (U)M06-L-D3BJ-IEFPCM/6-31+G(d,p)/Lanl2TZ+ level in dichloromethane are given in kcal mol−1. | ||
The origin of C–N bond coupling
It has been proved that the Cu(I) complexes mainly contributed to the catalytic effect for the aromatization of amides.63–67 Here, we calculated the C–N coupling reaction on the C5 position of 1,2,3-triazole by Cu(I), as shown in Fig. 5. According to our results, the direct dissociation of CuI from P2-13t to P2-mono14t leads to an increase in energy from −84.44 to −53.02 kcal mol−1. However, the interchangeable ligand reaction needs an energy barrier of only 14.37 kcal mol−1 to P2-14t intermediate with −84.85 kcal mol−1 in energy. P2-14t further isomerized to P2-15t intermediate. The C5–N1 distance was shortened from 3.795 Å to 3.027 Å, while their energies were close. Triplet P2-16t is 30.32 kcal mol−1 higher than P2-15t. It is speculated that the singlet P2-15s is easily formed through the singlet P2-TS8s transition state with an energy barrier of 9.81 kcal mol−1, and the potential energy is reduced to −110.95 kcal mol−1. The weak interaction in the P2-TS7s transition state is further confirmed by the analysis of noncovalent interactions (using Multiwfn program). As shown in Fig. 6, blue indicates a strong electrostatic interaction between Cu1 and N1. Furthermore, we calculated the reaction mechanism proposed by Chen et al.,47 as shown in Fig. S10.† The rate-determining step (RDS) barrier is up to 18 kcal mol−1, which is higher than any energy barrier in Fig. S11† (summed potential energy profiles). Furthermore, the potential energy increases along the ring-opening of 2H-azirine after the [3 + 2] cycloaddition reaction. In addition, no hydrogen reduction product was observed in Chen's experiments. The above-mentioned factors conclude that 2H-azirine ring-opening is prior to the [3 + 2] CuAAC reaction. The reasonable reaction mechanism should be carried out along path 2 in Fig. S11.† However, in general, 2H-azirine is a three-membered species with strong ring tension, and its ring-opening easily occurs to produce copper nitrene due to the cleavage of the C–N single bond, which may be the fundamental origin leading to the interrupted CuAAC reaction without hydrogen reduction products.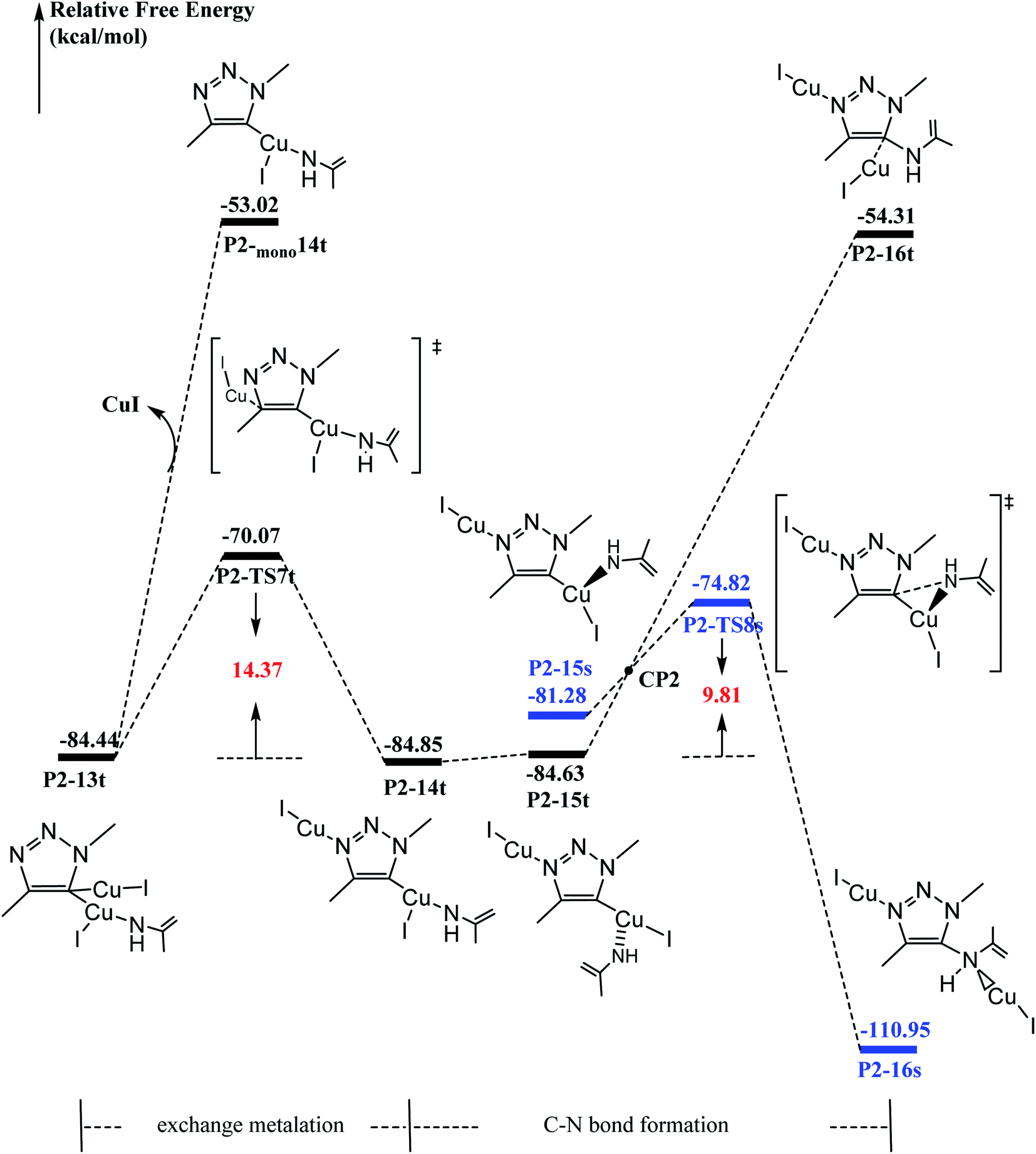 | ||
| Fig. 5 Calculated energy profiles of binuclear [3 + 2] cycloaddition in the CuAAC reaction. The relative Gibbs energies calculated at the (U)M06-L-D3BJ-IEFPCM/6-31+G(d,p)/Lanl2TZ+ level in dichloromethane are given in kcal mol−1. | ||
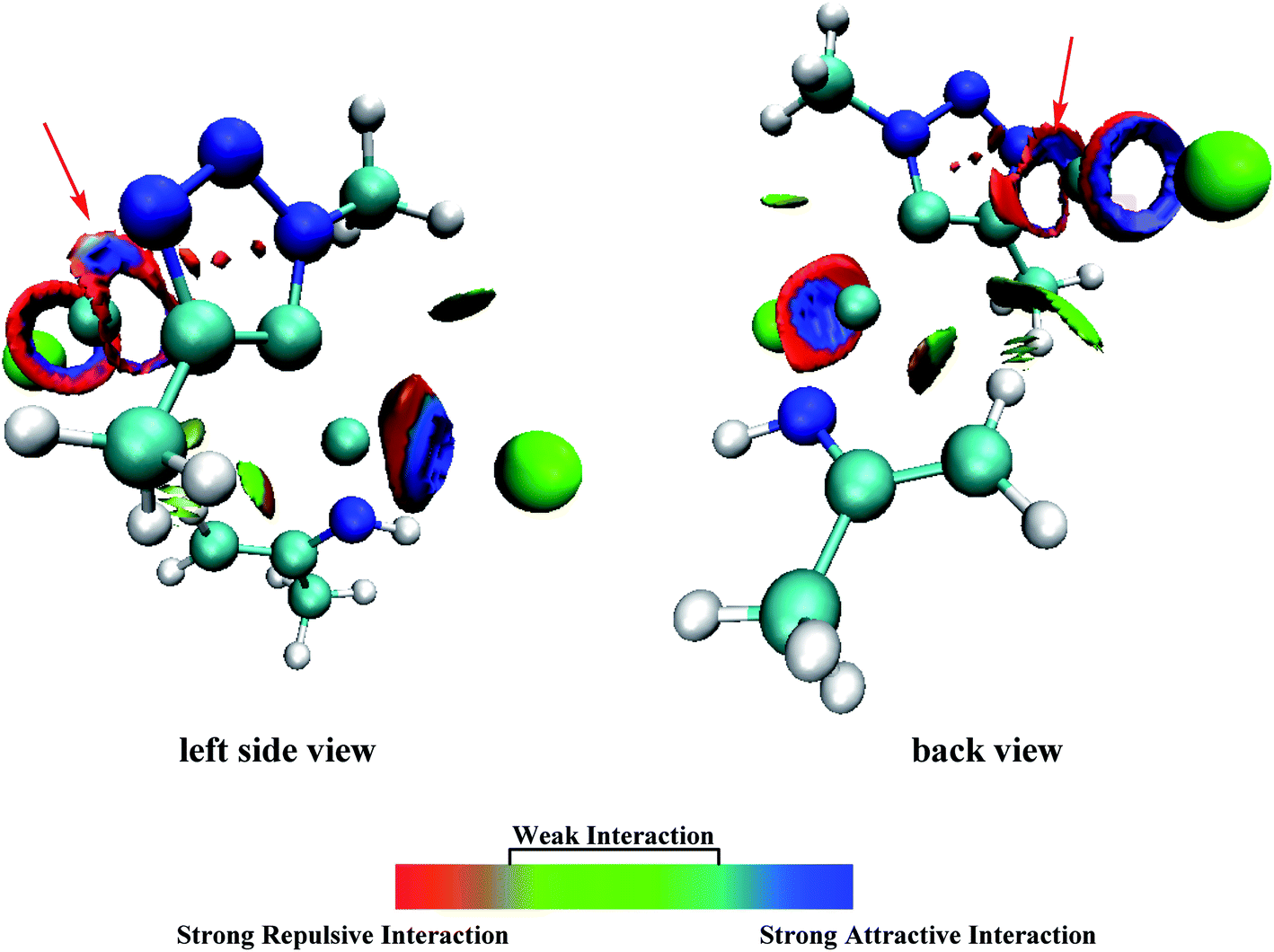 | ||
| Fig. 6 Gradient Isosurfaces (s = 0.5 au) for P2-TS7t, the surfaces are colored on a blue-green-red scale according to values of sign(λ2)ρ, ranging from −0.04 to +0.02 au. Blue indicates strong attractive interactions, and red indicates a strong nonbonded overlap. | ||
Furthermore, acetonitrile (MeCN), tetrahydrofuran (THF) and N-heterocyclic pentadiene carbene (NHC) decrease the potential barrier of 5-dicopper(I)-1,2,3-triazole to 5-mono-copper(I)-1,2,3-triazole. This indicates that the strong coordination ligands could lead to the lower reaction barrier on the Cu–C bond cleavage step for P1-7s (Fig. S10†) and P2-13t (Fig. S11†) intermediates. Thus, their coordination is in favor of hydrogen reduction by the CuAAC reaction, and prohibits the interrupted click reaction, as shown Fig. S12 and S13.† This may be a good explanation for why Chen's experiments were carried out in dichloromethane (DCM) rather than in MeCN, THF, etc. Thus, the coordination of MeCN, THF etc., prohibits the interrupted click reaction in favor of hydrogen reduction by the CuAAC reaction.
In general, DFT calculations rationalize the mechanism of the full catalytic cycle for the Cu(I)-catalyzed interrupted click reaction involving the ring-opening cross-coupling of 2H-azirines with terminal alkynes and organic azides. The calculation results clearly interpreted the mechanism of the dicopper-catalyzed 2H-azirine ring-opening, alkyne hydrogen activation, [3 + 2] cycloaddition and C–N cross-coupling reaction by optimizing the reaction intermediates, transition states etc., as shown in Fig. S14.† Moreover, when phenylacetylene, TsN3, and 3-phenyl-2H-azirine were used to be calculated, the similar potential surface profiles are also concluded in Fig. S15.†
Based on the above-mentioned calculations, as shown in Fig. S16,† the third route seems also reasonable starting with the formation of dinuclear Cu(I)-acetylide from alkyne and copper iodide, followed by the ring-opening of 2H-azirines to form P3-5t and finally [3 + 2] cycloaddition, considering that alkyne is more nucleophilic than azirine and azide, while copper iodide has an electrophilic character.33
Conclusions and outlook
In summary, we present the full Cu(I)-catalyzed mechanism to rationalize the three synthetic paths of 5-enamined-functionalized fully substituted 1,2,3-triazole via azide, terminal alkyne, and 2H-azirine by density functional theory and multicomponent artificial force-induced reaction (MC-AFIR) methods. We concluded that the binuclear mechanism was preferred because once the 2H-azirine was coordinated by one copper(I), it operates as another efficient copper(I) scavenger. Formation of the binuclear Cu complex further stabilizes the system, making the ring opening step accessible with a much lower activation barrier, which is further confirmed by AIM analysis. The AIM analysis showed that the ring opening of 2H-azirine became more difficult because the stability of the 2H-azirine three-membered-ring was enhanced when the coordinated by mono-copper(I). However, due to bimetallic copper(I) coordination, the three-membered-ring of 2H-azirine became not stable. Secondly, during the activation of the C–H bond in terminal alkynes, Et3N plays a fundamental important role in the hydrogen atom transfer process. Ring-opened dicopper nitrene is initially coordinated to alkyne, and subsequently, terminal hydrogen is pulled away from alkyne and stabilized by Et3N with a low energy barrier. Et3N is considered as a strong Lewis base that can fix active hydrogen. We believed that this was one of the main reasons for the absence of CuAAC products in the whole reaction. Furthermore, the hydrogen was given to the nitrogen atom of dicopper nitrene. The AIM analysis indicated that the coordination of bis-copper with alkyne was stronger than that of single-copper, which weakened the covalent interactions of terminal C–H. Thirdly, the 3 + 2 cycloaddition as well as C–N cross-coupling was the determination step including ring reducing, transmetalation and reductive elimination reactions. It is a barrierless process for the six-membered heterocyclic cuprated intermediate to turn into triazole. The rate determination step of the whole reaction was from 5-bis-copper(I) −1,2,3-triazole to 5-mono-copper(I) −1,2,3-triazole with a potential barrier of up to 14 kcal mol−1, which can further be decreased if acetonitrile (MeCN), tetrahydrofuran(THF) and N-heterocyclic pentadiene carbene (NHC) are coordinated to P2-13t. This may be a good explanation for why Chen's synthesis experiments were carried out in dichloromethane (DCM) rather than in MeCN, THF, etc.Overall, our results provide a better understanding of the interrupted click reaction from the dicopper-catalyzed terminal alkyne and organic azide reaction when adding the third reaction component. Organometallic P2-13t, P2-14t and P2-15t intermediates are powerfully potential to be captured experimentally in the calculated reasonable reaction potential energy surface profiles. Furthermore, solvent and extra strong ligand coordination plays an important role in C–N coupling and Cu–C cleavage competitive reactions. Thus, the synthesis of these reactions should prefer the solvents with weak and without coordination, such as dichloromethane and chloroform.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (grant no. 21473162). Y. Zhao is grateful to the Project Grants 521 Talents Cultivation of Zhejiang Sci-Tech University. This work was also supported by Zhejiang Provincial Top Key Academic Discipline of Chemical Engineering and Technology. Thanks to Prof. Wanzhi Chen from Zhejiang University for the beneficial discussion.References
- F. Amblard, J. H. Cho and R. F. Schinazi, Cu(I)-catalyzed Huisgen azide-alkyne 1,3-dipolar cycloaddition reaction in nucleoside, nucleotide and oligonucleotide chemistry, J. Chem. Rev., 2009, 109(9), 4207–4220 CrossRef CAS.
- H. Cai, S. Shukla, C. Wang, H. Masarapu and N. F. Steinmetz, Heterologous prime-boost enhances the antitumor immune response elicited by plant-virus-based cancer, J. Am. Chem. Soc., 2019, 141, 6509–6518 CrossRef CAS.
- P. Z. Li, X. J. Wang and Y. Zhao, Click chemistry as a versatile reaction for construction and modification of metal–organic frameworks, Coord. Chem. Rev., 2019, 380, 484–518 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Click chemistry: diverse chemical function from a few good reactions, Angew. Chem., Int. Ed., 2001, 40(11), 2004–2021 CrossRef CAS.
- P. Xiao, C. X. Li and W. H. Fang, Mechanism of the visible-light-mediated copper-catalyzed coupling reaction of phenols and alkynes, J. Am. Chem. Soc., 2018, 140(44), 15099–15113 CrossRef CAS.
- M. A. Morozova, M. S. Yusubov, B. Kratochvil, V. Eigner, A. A. Bondarev, A. Yoshimura, A. Saito, V. V. Zhdankin, M. E. Trusova and P. S. Postnikov, Regioselective Zn(OAc)2-catalyzed azide–alkyne cycloaddition in water: the green click-chemistry, Org. Chem. Front., 2017, 4(6), 978–985 RSC.
- J. E. Moses and A. D. Moorhouse, The growing applications of click chemistry, Chem. Soc. Rev., 2007, 36(8), 1249–1262 RSC.
- R. Huisgen, 1,3-Dipolar cycloadditions. Past and future, Angew. Chem., Int. Ed., 1963, 2(10), 565–598 CrossRef.
- R. Huisgen, Kinetics and mechanism of 1,3-dipolar cycloadditions, Angew. Chem., Int. Ed., 1963, 2(10), 633–696 CrossRef.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, A stepwise Huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes, Angew. Chem., Int. Ed., 2002, 41(14), 2596–2599 CrossRef CAS.
- W. Song and N. Zheng, Iridium-catalyzed highly regioselective azide–ynamide cycloaddition to access 5-amido Fully substituted 1,2,3-triazoles under mild, air, aqueous, and bioorthogonal conditions, Org. Lett., 2017, 19(22), 6200–6203 CrossRef CAS.
- Y. Liao, Q. Lu, G. Chen, Y. Yu, C. Li and X. Huang, Rhodium-catalyzed azide-alkyne cycloaddition of internal ynamides: regioselective assembly of 5-amino-triazoles under mild conditions, ACS Catal., 2017, 7(11), 7529–7534 CrossRef CAS.
- L. Zhang, X. Chen, P. Xue, H. H. Y. Sun, I. D. Williams, K. B. Sharpless, V. V. Fokin and G. Jia, Ruthenium-catalyzed cycloaddition of alkynes and organic azides, J. Am. Chem. Soc., 2005, 127(46), 15998–15999 CrossRef CAS.
- W. G. Kim, M. E. Kang, J. B. Lee, M. H. Jeon, S. Lee, J. Lee, B. Choi, P. M. S. D. Cal, S. Kang, J. Kee, G. J. L. Bernardes, J. U. Rohde, W. Choe and S. Y. Hong, Nickel-catalyzed azide–alkyne cycloaddition to access 1,5-disubstituted 1,2,3-triazoles in air and water, J. Am. Chem. Soc., 2017, 139(35), 12121–12124 CrossRef CAS.
- G. Fang and X. Bi, Silver-catalysed reactions of alkynes: recent advances, Chem. Soc. Rev., 2015, 44(22), 8124–8173 RSC.
- J. R. Johansson, T. Beke-Somfai, A. S. Said Stålsmeden and N. Kann, Ruthenium-catalyzed azide alkyne cycloaddition reaction: scope, mechanism, and applications, Chem. Rev., 2016, 116(23), 14726–14768 CrossRef CAS.
- P. Destito, J. R. Couceiro and H. Faustino, et al., Ruthenium-catalyzed azide–thioalkyne cycloadditions in aqueous media: A mild, orthogonal, and biocompatible chemical ligation, Angew. Chem., Int. Ed., 2017, 56(36), 10766–10770 CrossRef CAS.
- V. O. Rodionov, V. V. Fokin and M. G. Finn, Mechanism of the ligand-free CuI-catalyzed azide–alkyne cycloaddition reaction, Angew. Chem., Int. Ed., 2005, 44(15), 2210–2215 CrossRef CAS.
- B. R. Buckley, S. E. Dann and H. Heaney, Experimental evidence for the involvement of dinuclear alkynyl copper(I) complexes in alkyne–azide chemistry, Chem. - Eur. J., 2010, 16(21), 6278–6284 CrossRef CAS.
- B. R. Buckley, S. E. Dann, D. P. Harris, H. Heaney and E. C. Stubbs, Alkynylcopper(I) polymers and their use in a mechanistic study of alkyne–azide click reactions, Chem. Commun., 2010, 46(13), 2274–2276 RSC.
- S. I. Presolski, V. Hong, S. H. Cho and M. G. Finn, Tailored ligand acceleration of the Cu-catalyzed azide–alkyne cycloaddition reaction: practical and mechanistic implications, J. Am. Chem. Soc., 2010, 132(41), 14570–14576 CrossRef CAS.
- C. Nolte, P. Mayer and B. F. Straub, Isolation of a copper(I) triazolide: a “click” intermediate, Angew. Chem., Int. Ed., 2007, 46(12), 2101–2103 CrossRef CAS.
- V. O. Rodionov, S. I. Presolski, S. Gardinier, Y. H. Lim and M. G. Finn, Benzimidazole and related ligands for Cu-catalyzed azide–alkyne cycloaddition, J. Am. Chem. Soc., 2007, 129(42), 12696–12704 CrossRef CAS.
- V. O. Rodionov, S. I. Presolski, D. D. Díaz, V. V. Fokin and M. G. Finn, Ligand-accelerated cu-catalyzed azide-alkyne cycloaddition: a mechanistic report, J. Am. Chem. Soc., 2007, 129(42), 12705–12712 CrossRef CAS.
- Q. Wang, T. R. Chan, R. Hilgraf, V. V. Fokin, K. B. Sharpless and M. G. Finn, Bioconjugation by copper(I)-catalyzed azide-alkyne [3 + 2] cycloaddition, J. Am. Chem. Soc., 2003, 125(11), 3192–3193 CrossRef CAS.
- B. T. Worrell, J. A. Malik and V. V. Fokin, Direct evidence of a dinuclear copper intermediate in Cu(I)-catalyzed azide–alkyne cycloadditions, Science, 2013, 340(6131), 457–460 CrossRef CAS.
- L. Jin, D. R. Tolentino, M. Melaimi and G. Bertrand, Isolation of bis(copper) key intermediates in Cu-catalyzed azide-alkyne “click reaction”, Sci. Adv., 2015, 1(5), e1500304 CrossRef.
- C. Iacobucci, S. Reale and J. F. Gal, et al., Dinuclear copper intermediates in copper(I)-catalyzed azide–alkyne cycloaddition directly observed by electrospray ionization mass spectrometry, Angew. Chem., Int. Ed., 2015, 54(10), 3065–3068 CrossRef CAS.
- V. K. Tiwari, B. B. Mishra and K. B. Mishra, et al., Cu-catalyzed click reaction in carbohydrate chemistry, Chem. Rev., 2016, 116(5), 3086–3240 CrossRef CAS.
- M. Meldal and C. W. Tornøe, Cu-catalyzed azide–alkyne cycloaddition, Chem. Rev., 2008, 108(8), 2952–3015 CrossRef CAS.
- J. E. Hein and V. V. Fokin, Copper-catalyzed azide–alkyne cycloaddition (CuAAC) and beyond: new reactivity of copper(I) acetylides, Chem. Soc. Rev., 2010, 39(4), 1302–1315 RSC.
- C. Wang, D. Ikhlef, S. Kahlal, J. Y. Saillard and D. Astruc, Metal-catalyzed azide-alkyne “click” reactions: mechanistic overview and recent trends, Coord. Chem. Rev., 2016, 316, 1–20 CrossRef CAS.
- S. Calvo-Losada, M. S. Pino-González and J. J. Quirante, Rationalizing the catalytic activity of copper in the cycloaddition of azide and alkynes (CuAAC) with thetTopology of ∇2ρ(r) and ∇∇2ρ(r), J. Phys. Chem. B, 2015, 119(4), 1243–1258 CrossRef CAS.
- Y. Özkılıç and N. Ş. Tüzün, A DFT study on the binuclear CuAAC reaction: mechanism in light of new experiments, Organometallics, 2016, 35(16), 2589–2599 CrossRef.
- B. E. A. Hicham, L. Bahsis, H. Anane, L. R. Domingo and S. E. Stiriba, Understanding the mechanism and regioselectivity of the copper(I) catalyzed [3 + 2] cycloaddition reaction between azide and alkyne: a systematic DFT study, RSC Adv., 2018, 8(14), 7670–7678 RSC.
- R. Roohzadeh, B. Nasiri, A. Chipman, B. F. Yates and A. Ariafard, Disclosure of some obscure mechanistic aspects of the copper-catalyzed click reactions involving N2 elimination promoted by the use of electron-deficient azides from a DFT perspective, Organometallics, 2019, 38(2), 256–267 CrossRef CAS.
- Y. C. Lin, Y. J. Chen, T. Y. Shih, Y. H. Chen, Y. C. Lai, M. Y. Chiang, G. C. Senadi, H. Y. Chen and H. Y. Chen, Mechanistic study in click reactions by using (N-heterocyclic carbene)copper(I) complexes: anionic effects, Organometallics, 2019, 38(2), 223–230 CrossRef CAS.
- C. Spiteri and J. E. Moses, Copper-catalyzed azide–alkyne cycloaddition: Regioselective synthesis of 1,4,5-trisubstituted 1,2,3-triazoles, Angew. Chem., Int. Ed., 2010, 49(1), 31–33 CrossRef CAS.
- Z. E. Blastik, S. Voltrová, V. Matoušek, B. Jurásek, D. W. Manley, B. Klepetářová and P. Beier, Azidoperfluoroalkanes: synthesis and application in copper(I)-catalyzed azide–alkyne cycloaddition, Angew. Chem., Int. Ed., 2017, 56(1), 346–349 CrossRef CAS.
- W. Wu, J. Wang, Y. Wang, Y. Haung, Y. Tan and Z. Weng, Trifluoroacetic anhydride promoted copper(I)-catalyzed interrupted click reaction: from 1,2,3-triazoles to 3-trifluoromethyl-substituted 1,2,4-triazinones, Angew. Chem., Int. Ed., 2017, 56(35), 10476–10480 CrossRef CAS.
- K. P. S. Cheung and G. C. Tsui, Copper(I)-catalyzed interrupted click reaction with TMSCF3: Synthesis of 5-trifluoromethyl 1,2,3-triazoles, Org. Lett., 2017, 19(11), 2881–2884 CrossRef CAS.
- F. Wei, T. Zhou, Y. Ma, C. Tung and Z. Xu, Bench-stable 5-stannyl triazoles by a copper(I)-catalyzed interrupted click reaction: bridge to trifluoromethyltriazoles and trifluoromethylthiotriazoles, Org. Lett., 2017, 19(8), 2098–2101 CrossRef CAS.
- W. Wang, F. Wei, Y. Ma, C. H. Tung and Z. Xu, Copper(I)-catalyzed three-component click/alkynylation: one-pot synthesis of 5-alkynyl-1,2,3-triazoles, Organic Letters, 2016, 18(17), 4158–4161 CrossRef CAS.
- L. Deng, X. Ca, Y. Liu and J. Wan, In-water synthesis of 5-thiolated 1,2,3-triazoles from β-thioenaminones by diazo transfer reaction, The Journal of Organic Chemistry, 2019, 84(21), 14179–14186 CrossRef CAS.
- S. Lal, H. S. Rzepa and S. Díez-González, Catalytic and computational studies of N-heterocyclic carbene or phosphine-containing copper(I) complexes for the synthesis of 5-iodo-1,2,3-triazoles, ACS Catalysis, 2014, 4(7), 2274–2287 CrossRef CAS.
- W. Wang, X. Peng, F. Wei, C. H. Tung and Z. Xu, Copper(I)-catalyzed interrupted click reaction: Synthesis of diverse 5-hetero-functionalized triazoles, Angew. Chem., Int. Ed., 2016, 55(2), 649–653 CrossRef CAS.
- W. Zhou, M. Zhang, H. Li and W. Chen, One-pot Three-component synthesis of enamine-functionalized 1,2,3-triazoles via Cu-catalytic azide–alkyne click (CuAAC) and Cu-catalyzed vinyl nitrene transfer sequence, Org. Lett., 2017, 19(1), 10–13 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. B. Robb, V. Cheeseman, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, S. S. Kobayashi, J. Rormand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision D.01, Inc., Wallingford CT, 2013 Search PubMed.
- Y. Zhao and D. G. Truhlar, A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions, J. Chem. Phys., 2006, 125, 194101 CrossRef.
- G. Scalmani and M. J. Frisch, Continuous surface charge polarizable continuum models of solvation. I. General formalism, J. Chem. Phys., 2010, 132(11), 114110 CrossRef.
- S. Maeda, K. Ohno and K. Morokuma, An automated and systematic transition structure explorer in large flexible molecular systems based on combined global reaction route mapping and microiteration methods, J. Chem. Theory Comput., 2009, 5(10), 2734–2743 CrossRef CAS.
- S. Maeda, K. Ohno and K. Morokuma, Systematic exploration of the mechanism of chemical reactions: the global reaction route mapping (GRRM) strategy using the ADDF and AFIR methods, Phys. Chem. Chem. Phys., 2013, 15(11), 3683–3701 RSC.
- T. Lu and F. Chen, Multiwfn: a multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33(5), 580–592 CrossRef CAS.
- K. Brandhorst and J. Grunenberg, How strong is it? The interpretation of force and compliance constants as bond strength descriptors, Chem. Soc. Rev., 2008, 37(8), 1558–1567 RSC.
- K. Brandhorst and J. Grunenberg, Efficient computation of compliance matrices in redundant internal coordinates from Cartesian Hessians for nonstationary points, J. Chem. Phys., 2010, 132(18), 184101 CrossRef.
- T. R. Cundari, A. Dinescu and A. B. Kazi, Bonding and structure of copper nitrenes, Inorg. Chem., 2008, 47(21), 10067–10072 CrossRef CAS.
- (a) Y. M. Badiei and T. H. Warren, Electronic structure and electrophilic reactivity of discrete copper diphenylcarbenes, J. Organomet. Chem., 2005, 690(24–25), 5989–6000 CrossRef CAS; (b) Y. M. Badiei, A. Krishnaswamy and M. M. Melzer, et al., Transient terminal Cu–nitrene intermediates from discrete dicopper nitrenes, J. Am. Chem. Soc., 2006, 128(47), 15056–15057 CrossRef CAS.
- Y. M. Badiei, A. Dinescu, X. Dai, R. M. Palomino, F. W. Heinemann, T. R. Cundari and T. H. Warren, Copper–nitrene complexes in catalytic C–H amination, Angew. Chem., Int. Ed., 2008, 47(51), 9961–9964 CrossRef CAS.
- M. J. B. Aguila, Y. M. Badiei and T. H. Warren, Mechanistic insights into C–H amination via dicopper nitrenes, J. Am. Chem. Soc., 2013, 135(25), 9399–9406 CrossRef CAS.
- M. S. Ziegler, K. V. Lakshmi and T. D. Tilley, Dicopper Cu(I) Cu(I) and Cu(I) Cu(II) complexes in copper-catalyzed azide–alkyne cycloaddition, J. Am. Chem. Soc., 2017, 139(15), 5378–5386 CrossRef CAS.
- C. Iacobucci, A. Lebon, F. De Angelis and A. Memvoeuf, CuAAC click reactions in the gas phase: unveiling the reactivity of bis-copper intermediates, Chem. - Eur. J., 2016, 22(52), 18690–18694 CrossRef CAS.
- A. J. Paine, Mechanisms and models for copper mediated nucleophilic aromatic substitution. 2. Single catalytic species from three different oxidation states of copper in an Ullmann synthesis of triarylamines, J. Am. Chem. Soc., 1987, 109(5), 1496–1502 CrossRef CAS.
- S. L. Zhang, L. Liu, Y. Fu and Q. X. Guo, Theoretical study on copper(I)-catalyzed cross-coupling between aryl halides and amides, Organometallics, 2007, 26(18), 4546–4554 CrossRef CAS.
- C. Ryan, V. Anh, V. V. Fokin and J. E. Hein, Catalyst activation, chemoselectivity, and reaction rate controlled by the counterion in the Cu(I)-catalyzed cycloaddition between azide and terminal or 1-iodoalkynes, ACS Catal., 2018, 8(9), 7889–7897 CrossRef.
- Z. Liu, H. Ji, W. Gao, G. Zhu and B. Tang, Copper(I)-mediated carboamination of vinyl azides by aryldiazonium salts: synthesis of N2-substituted 1,2,3-triazoles, Chem. Commun., 2017, 53(46), 6259–6262 RSC.
- K. M. Carsch, I. M. Dimucci, D. A. Iovan, A. Li and T. A. Betley, Synthesis of a copper-supported triplet nitrene complex pertinent to copper-catalyzed amination, Science, 2019, 365, 1138–1143 CrossRef CAS.
- F. Sebest, K. Lachhani, C. Pimpasri, L. Casarrubios and S. Díez-González, Cycloaddition reactions of azides and electron-deficient alkenes in deep eutectic solvents: Pyrazolines, aziridines and other surprises, Adv. Synth. Catal., 2020, 362(9), 1877–1886 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures, tables, and schemes, Cartesian coordinates of optimized geometries, additional discussion, details of X-ray crystal structure, and the synthesis procedure. See DOI: 10.1039/d0ra07498e |
| This journal is © The Royal Society of Chemistry 2021 |