
Poly(ethylene) glycols and mechanochemistry for the preparation of bioactive 3,5-disubstituted hydantoins†
L. Konnert,
M. Dimassi,
L. Gonnet,
F. Lamaty,
J. Martinez and
E. Colacino*
Institut des Biomolécules Max Mousseron (IBMM), UMR 5247 CNRS – UM – ENSCM, Green Chemistry and Enabling Technology Team, Université de Montpellier, Campus Triolet, Place E. Bataillon, 34095 Montpellier Cedex 5, France. E-mail: evelina.colacino@umontpellier.fr
First published on 8th April 2016
Abstract
Mechanochemistry was effective for the preparation of 3,5-disubstituted hydantoins from α-amino methyl esters, using either 1,1′-carbonyldiimidazole (CDI) or alkyl isocyanates. The preparation of the antimicrobial additives, 3-allyl-5,5′-dimethyl hydantoin (ADMH) and 1-chloro-3-ethyl-5,5′-dimethyl hydantoin (CEDMH) were performed by grinding. A chlorination reaction, never described before by mechanochemistry was achieved by Ca(ClO)2, while the preparation of the bioactive anticonvulsant marketed drug ethotoin was achieved by a novel approach based on poly(ethylene) glycol (PEGs) assisted grinding.
Introduction
Hydantoins or 2,4-imidazolidinediones are a well-known family of bioactive compounds with numerous therapeutic properties.1 Among the diversity of potential structures due to the high degree of possible substitutions of the hydantoin core, 3,5-disubstituted hydantoins show particularly interesting biological activities. Marketed drugs contain a hydantoin scaffold, notably the anticonvulsant drug ethotoin,2 the androgen receptor antagonist nilutamide,3 or the antimicrobial additives for polymer textiles and medical applications 1-chloro-3-ethyl-5,5-dimethyl hydantoin (CEDMH)4 and 3-allyl-5,5′-dimethylhydantoin (ADMH)5 (Fig. 1).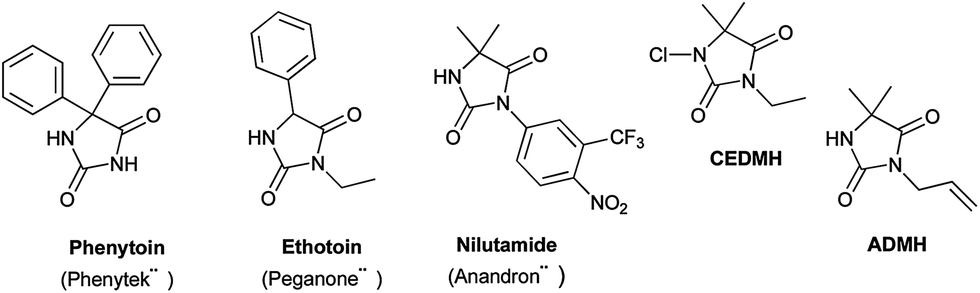 | ||
| Fig. 1 Drugs with N- and/or C-disubstituted hydantoin structure. | ||
Various methodologies have been applied to the synthesis of 3,5-disubstituted hydantoins. A straightforward pathway is the alkylation/arylation6–10 of the N-3 position of 5- or 5,5′-disubstituted hydantoins but most of the procedures describe the cyclisation of ureido derivatives of amino esters A, prepared by reaction with isocyanates (either in solution11–17 or on solid-support18–23) (Method A, Scheme 1), phosgene,24 or triphosgene.25–29
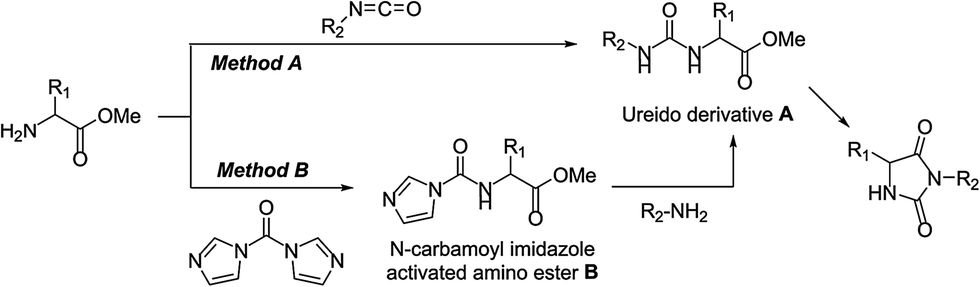 | ||
| Scheme 1 Possible routes to 3,5-disubstituted hydantoins via ureido derivatives of amino esters. | ||
We have recently demonstrated that mechanochemistry was successfully applied to the eco-friendly preparation of 5,5′-disubstituted hydantoins,30 including the antiepileptic drug phenytoin (Phenytek®, Fig. 1). We present herein a general and unprecedented mechanochemical approach to access 3,5-disubstituted hydantoins from α-amino esters, via an ureido derivative A (Method A, Scheme 1), obtained from the corresponding isocyanate. This ureido derivative A could also be obtained via an original route, from N-carbamoyl imidazole activated amino ester derivative B (Method B, Scheme 1), by reaction with various amines. Molecules such as 1-chloro-3-ethyl-5,5-dimethyl hydantoin (CEDMH),4 3-allyl-5,5′-dimethylhydantoin (AMDH)5 and the anticonvulsant drug ethotoin were synthesized. An unprecedented mechanochemical chlorination reaction was achieved with Ca(ClO)2 with better results when compared with solution syntheses. In the case of ethotoin, three synthetic strategies have been explored in a ball-mill (BM), including the poly(ethylene) glycols-assisted grinding approach,31,32 applied for the first time to access Active Pharmaceutical Ingredients (API).
Results and discussion
The mechanochemical preparation of 3-ethyl-5-benzyl hydantoin 2 was first investigated, according to a one-pot/two step procedure based on Method B (Scheme 1). Phenylalanine methyl ester hydrochloride was reacted with CDI in a planetary ball-mill (PBM), leading to the corresponding 1H-imidazole-carboxamido derivative 1 (Table 1). Milling the mixture in the presence of ethylamine hydrochloride – added in the second step – resulted in the unsymmetrical urea 3 that was converted into the hydantoin 2 by base-mediated intramolecular cyclization, using stoichiometric amounts of solid K2CO3. Selected data for the optimization of the reaction conditions are reported in Table 1.
|
|||||
|---|---|---|---|---|---|
| Entry | CDI (equiv.) | Balls (n) | Conditions (min) | Yieldb (%) | |
| Step 1 | Step 2 | ||||
| a The reactions were performed in a stainless steel jar (using stainless steel balls, 5 mm ∅) at 450 rpm under continuous milling except when otherwise stated.b Isolated yields.c (x′/y′): the reaction mixture was cycle-milled for 40 minutes during 8 cycles of 5 minutes followed by 2 minute standby, with reverse rotation between two cycles.d 400 μL (η = 0.37 μL mg−1) of EtOAc were added. | |||||
| 1 | 2 | 25 | 10 | 120 | 65 |
| 2 | 2 | 25 | 60 | 120 | 52 |
| 3 | 1.3 | 50 | 40 | 60 | 84 |
| 4 | 1.3 | 50 | 60, 8 × (5′/2′)c | 60 | 77 |
| 5 | 1.1 | 50 | 60, 8 × (5′/2′)c | 120 | 67 |
| 6 | 1.3 | 50 | 40, EtOAcd | 120 | 75 |
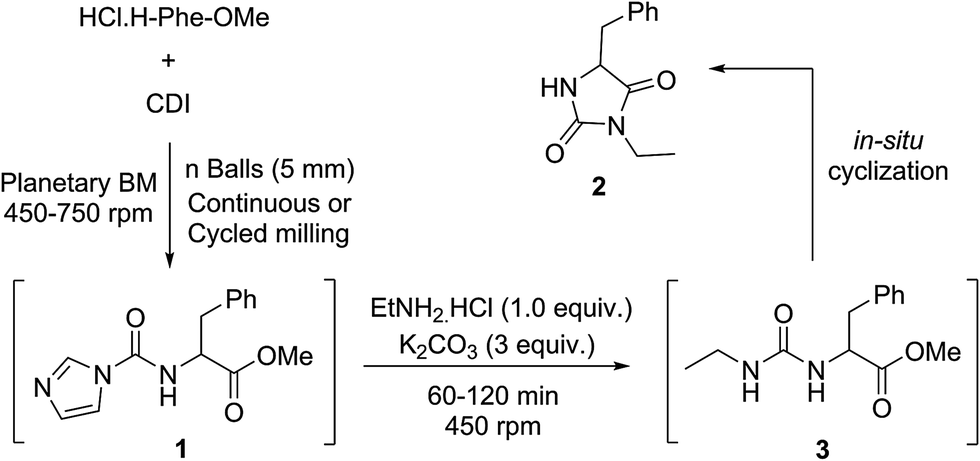
In the first trial (entry 1), the starting amino ester was not fully converted into the intermediate 1 and a symmetrical urea species of phenylalanine methyl ester was formed, resulting in a reduced yield (65%) of the desired product 3-ethyl-5-benzyl-hydantoin 2. Results were not improved when the milling speed was increased to 750 rpm (66%) or by adding ethylamine hydrochloride in excess (64%). Extending the reaction time during the first step (entry 2) resulted in a lower yield (52%) of 2, due to the formation of increased amounts of symmetrical urea. This trend was also observed when adding either CDI or amino ester portion-wise and seemed to be emphasized when CDI was present in excess (entries 1 and 2). When the amount of CDI was decreased (1.3 equiv.) and the number of balls in the jar was doubled (50), after precipitation in water, hydantoin 2 was obtained in a better yield (84%, entry 3). Neither cycled milling30,33,34 (entries 4 and 5) nor decreasing the amount of CDI (entry 5) or Liquid-Assisted Grinding (LAG) with ethyl acetate (entry 6), improved the yield of the reaction.
The reaction was investigated with other combinations of α-amino esters/amines (Table 2, Method B). Both allyl and benzylamine reacted smoothly with phenylalanine imidazole-carboxamido derivative 1 affording the corresponding hydantoins (13 and 18 respectively) in good yields. The reactivity of the system was not influenced by the physical state (solid or liquid) of the amine as also confirmed by leucine (compounds 4 and 19) and aminoisobutyric (Aib) methyl ester (compounds 11 and 16). However, it seemed that the steric and electronic nature of the entering amine drove the outcome of the reaction.
| R1 | R2 | Product | Yielda,b (%) | ||
|---|---|---|---|---|---|
| Bc | Ad | ||||
| NCe | RCe | NCe | |||
| a Isolated yields.b L-α-Amino esters were used except when otherwise stated.c Conditions: (step 1) α-amino ester (1 equiv.) and CDI (1.3 equiv.) at 450 rpm, 50 balls (5 mm, stainless steel, 5 mm ∅) for 40 min, (step 2) R2NH2 (1.6 equiv.) and K2CO3 (3.6 equiv.) at 450 rpm for 2 hours.d α-Amino ester (1 equiv.) R2NCO (3.0 equiv.) and K2CO3 (3.0 equiv.) at 30 Hz for 2 hours, 2 balls (5 mm, stainless steel, 5 mm ∅).e NC = not crystallized, RC = recrystallized.f Residual metals were analyzed by ICP-MS (see Experimental part).g Not performed.h The reaction was performed adding PEG-2000-(OMe)2 (455 mg mmol−1 substrate).i Quaternary α-amino ester HCl·H-Aib-OMe or HCl·H-Gly(Ph)2-OMe were used.j Yield reported within brackets is given for the ureido intermediate.k 1H NMR yield using CH2Br2 (20 μL) as internal reference. | |||||
| CH2Ph | CH2CH3 | 2f | 84 | n.p.g | 75 |
| CH2CH(CH3)2 | CH2CH3 | 4 | 61(70)h | 76(70)h | 83(90)h |
| CH2Ph(p-OtBu) | CH2CH3 | 5 | 56 | 82 | 82 |
| (CH2)4NHCbz | CH2CH3 | 6 | 31 | 61 | n.p.g |
| CH2CH2SCH3 | CH2CH3 | 7 | 40 | 57 | 78 |
| CH(CH3)CH2CH3 | CH2CH3 | 8 | 67 | 58 | 84 |
| CH2OtBu | CH2CH3 | 9 | 51 | 64 | 98 |
| CH2SCH2Ph | CH2CH3 | 10 | 38 | 50 | 61 |
| CH3/CH3i | CH2CH3 | 11 | 61 | 62 | 79 |
| Ph | CH2CH3 | 12 | 34 | 40 | 35 |
| CH2Ph | CH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 CH2 |
13 | 75 | n.p.g | n.p.g |
| CH2CH(CH3)2 | CH2CHCH2 |
14 | 57 | 58 | n.p.g |
| CH(CH3)OtBu | CH2CHCH2 |
15 | 51 | 66 | n.p.g |
| CH3/CH3i | CH2CHCH2 |
16 | 46 | 65 | n.p.g |
| Ph/Phi | CH2CHCH2 |
17 | 25 | n.p.g | n.p.g |
| CH2Ph | CH2Ph | 18 | 70 | n.p.g | 85 |
| CH2CH(CH3)2 | CH2Ph | 19 | 38 | 74 | 67 |
| CH2Ph | Ph | 20 | n.p.g | 0 | n.p.g |
| CH2OtBu | Ph | 21 | n.p.g | n.p.g | 30 |
| CH2Ph | CH(CH3)2 | 22 | n.p.g | n.p.g | 0(93)j |
| CH2Ph(p-OtBu) | CH(CH3)2 | 23 | n.p.g | 0 | n.p.g |
| CH2Ph(p-OtBu) | c-Hex | 24 | n.p.g | 49k | n.p.g |
Indeed, hydantoin 24 was obtained in moderate yield (49%) when cyclohexylamine (c-Hex-NH2) was tested, while no reaction was observed in the presence of the sterically hindered isopropylamine, or the less reactive and poorly nucleophilic aniline. These results suggested that the limiting step was the formation of the corresponding ureido intermediate (first step). Surprisingly, the yield of the reaction dropped with an α-amino ester component other than phenylalanine. Presuming that the difference in crystalline structure was the reason for this drop in the yield, the starting amino methyl esters were recrystallized to remove potential impurities.
After recrystallization, most of the desired hydantoins were obtained in much better yields (except for compound 8) especially in the case of compounds 5, 6, 10, 19. It can be hypothesized that the crystalline state and the steric and electronic nature of side chains of starting α-amino esters drive the mechanochemical reactivity. Indeed, similar side chains (compounds 2/5, 8/4 and 9/15) led to similar yields. Electronically rich α-amino esters, such as phenylglycine or diphenylglycine showed a moderate reactivity (12 and 17 respectively), while sterically hindered quaternary aminoisobutyric (Aib) methyl ester showed, as previously observed,30,33 good reactivity in mechanochemical processes (11 and 16), leading straightforwardly to 3-allyl-5,5′-dimethylhydantoin (ADMH, Fig. 1) 16 with a satisfying yield.
Overall, the mechanochemical preparation of CDI-mediated 3,5-disubstituted hydantoins is an environmentally friendly approach. The products were readily obtained from an easy-to-handle procedure, using cheap and commercially available substrates and reactants, allowing versatility of substituents in both C-5 and N-3 hydantoin ring positions. In most cases, the products were recovered by precipitation in water, the only side-products present in the reaction mixture being the non-toxic water-soluble imidazole, CO2 and inorganic salts. When precipitation of the expected compound was not possible, extraction with ethyl acetate and benign 10% aqueous citric acid35 was performed.
Mechanochemical preparation of 3,5-disubstituted hydantoins was also investigated using alkyl or aryl isocyanates (Method A, Scheme 1), for the sake of comparison and to provide an alternative route when Method B led to moderate yields. By grinding the α-amino ester, the isocyanate and the potassium carbonate, a cascade reaction sequence occurred, involving formation of the ureido derivative A, followed by in situ cyclization into hydantoins, with high atom economy (Scheme 1). In the case of benzyl hydantoin 2, no difference was observed when changing the jar material (stainless steel or tungsten carbide) or under different mechanical stress provided by a vibrational (VBM) or planetary (PBM) ball-mill (see Table S1 in ESI†). Vibrational ball-mill and stainless steel jars were used to prepare 3,5-disubstituted hydantoins, readily obtained in good to excellent yields, with generally improved results compared to the CDI-mediated Method B (Table 2).
It is worth noting here that isopropylisocyanate reacted smoothly with phenylalanine methyl ester, leading to the corresponding ureido derivative in high yield (93%). Cyclization was not observed, probably hampered by the steric hindrance around the nitrogen atom.
The study was completed by measuring the concentration of residual metals after ball-milling in the stainless-steel jar for compound 4 (see Experimental section). The organic product contamination was far below the threshold level of permitted daily exposures and uptake for elemental impurities in human. Overall, the two methods are a valuable alternative to solution syntheses, providing new or already commercialized compounds (see Table S2 in ESI†).
Method A was effective to access 3-ethyl-5,5′-dimethyl hydantoin (EDMH) 11 in good yield (79%), that was further transformed into the textile antimicrobial additive5 1-chloro-3-ethyl-5,5′-dimethylhydantoin (CEDMH) 25. A chlorination reaction using mechanochemistry – never described before – was performed in the planetary ball-mill using the swimming pool safe chlorinating agent trichloroisocyanuric acid (TCCA), which furnished 25 in 34% yield, similar to the synthesis in solution (Scheme 2). Better results were obtained when calcium hypochlorite was used: not only was compound 25 obtained in the very good yield of 87%, but the crude product was cleaner, generating only calcium salts as waste. This chlorination approach is a valuable and more powerful alternative to the use of TCCA either in solution or by grinding and it was successfully applied (99% yield) also to the preparation of compound 26, with high chemioselectivity, with the allyl moiety unreactive in these conditions (Scheme 2).
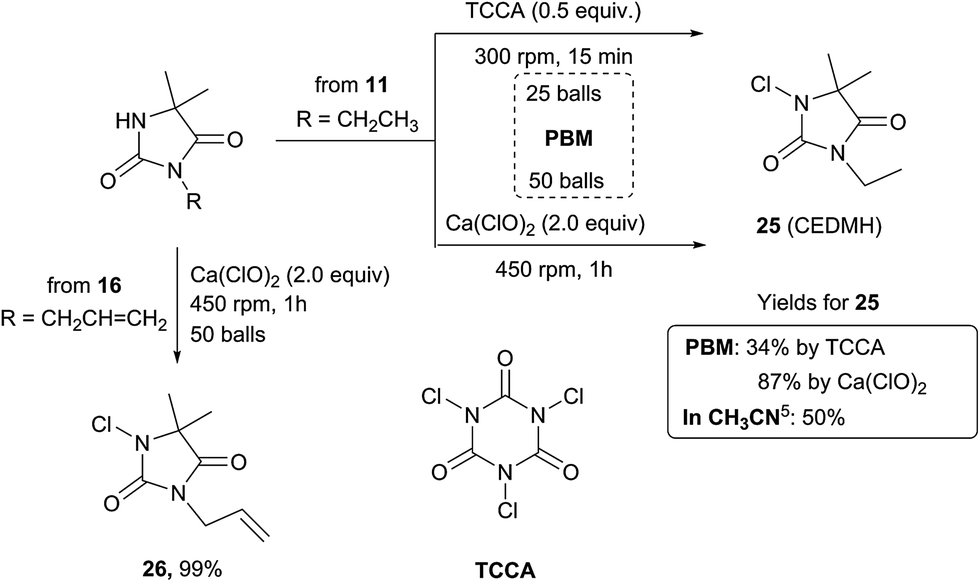 | ||
| Scheme 2 Mechanochemical routes to CEDMH 25 and 26. | ||
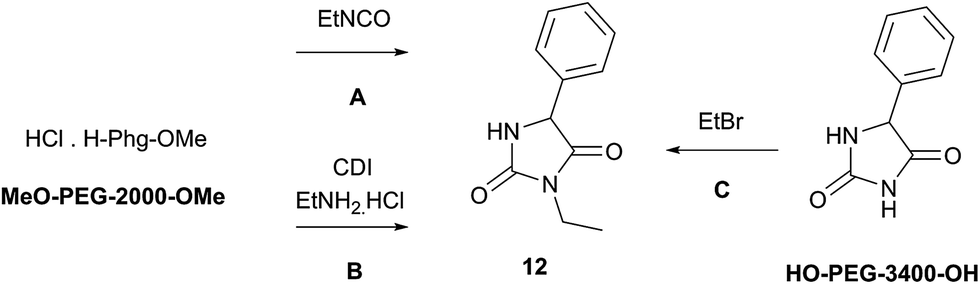
Synthesis of 3-ethyl-5-phenyl hydantoin (ethotoin, 12)
The preparation of ethotoin in solution is restricted to only two publications,36,37 involving either cyclization of 5-ethyl-2-phenylhydantoic acid36 or rearrangement of ethyl N-Boc-phenylglycinamide37 (70% and 60% yield respectively) under anhydrous conditions, both strongly acid-catalyzed conditions.N-3-alkylation of 5-phenylhydantoin, prepared by mechanochemistry following our previously described procedure,30 remained unsuccessful in ball-milling conditions, or neat under stirring, even with a large excess of alkyl halide. The N-alkylation (Method C) was improved when poly(ethylene) glycol (HO-PEG-400-OH) was used as the reaction solvent (entry 1, Table 3): ethotoin 12 was obtained in a 40% yield, a similar result to the previous methods A and B.
|
||||
|---|---|---|---|---|
| Entry | Methoda | RO-PEG-OR (av. MW) | Conditions | Yieldb (%) |
| a L-Phenylglycine methyl ester was used; PEG (455 mg mmol−1 substrate) was added in each experiment.b Isolated yields.c Both vibrational (VBM) and planetary (PBM) ball-mill were tested.d Determined by NMR.e The reaction medium was ground for 6 cycles of 15 min with 2 min pauses in between, with reverse rotation. | ||||
| 1 | Ac | — | 30 Hz 2 h or 450 rpm, 40/120 min | 35 |
| 2 | B | — | 450 rpm, 40/120 min | 40 |
| 3 | C | — | Stirring, rt, neat | Traces |
| 4 | C | HO-400-OH | Stirring, rt, overnight | 40d |
| 5 | C | HO-3400-OH | 300 rpm, 6 × (15/2)e | 45 |
| 6 | C | HO-2000-OH | 300 rpm, 6 × (15/2)e | 20 |
| 7 | C | MeO-2000-OMe | 300 rpm, 6 × (15/2)e | 20 |
| 8 | B | MeO-2000-OMe | 450 rpm, 40/120 min | 65 |
| 9 | A | MeO-2000-OMe | 450 rpm, 120 min | 63 |
Poly(ethylene) glycols are green solvents38 with low toxicity, finding applications in the biomedical field39 and catalysis.40 Very recently, their peculiar role was highlighted in metal-catalyzed processes in a ball-mill32 or by ultrasounds,41 having in common similar activation effects,42 or in Polymer-Assisted Grinding (POLAG) processes for co-crystal formation.31
Considering the preliminary result indicating that PEG polymer seemed to improve the efficiency of N-alkylation, the conditions were transposed to a ball-milled reaction (Table 3). Using solid HO-PEG-3400-OH (entry 5) in the mechanochemical conditions produced ethotoin 12 in 45% yield, compared with only traces when carried out under neat conditions (entry 3). The combination of mechanochemical forces and PEG matrix proved to be beneficial for N-alkylation when other approaches failed. Comparing several poly(ethylene) glycols (entries 5–7), it can be assumed that the length of the polymer might have an influence on the physical interactions of the system, more than its chemical composition.31 This suggests that the system would require a specific ‘polymer catalyst’ to be found, in agreement with the fact that mechanochemical forces can alter reaction pathways of synthetic polymers.43 In light of these results, MeO-PEG-2000-OMe was selected to grind the mixture (Table 3, entries 8 and 9) in the case of methods A and B, to avoid potential side reactions of HO-PEG-3400-OH end terminal hydroxyl groups with ethyl isocyanate or CDI.
As presumed, the mechanochemical productivity in the presence of a PEG matrix was increased as the reactions were by far more efficient. 3-Ethyl-5-phenylhydantoin 12 was obtained in much higher yield from both methods (Table 3, entries 8 and 9) compared with grinding in the absence of additives (Tables 2 and 3 entries 2 and 3).
It was reported that solid dispersion of ethotoin 12 in PEG solvent displayed improved solubility and diffusion properties.44 At a molecular level, this was explained by the existence of hydrogen bonding NH⋯O between the amide and the polyether chain.45 Similar interactions can be evoked with 5-phenyl hydantoin30 (Method C) or ureido derivative A (Methods A and B). In this case, liquid-assisted grinding would be responsible for the enhanced mechanochemical productivity observed for all the methods. However, POLAG cannot be excluded, not only because the existence of interactions with PEG polymers are known to induce changes in the physical state of the system,45 especially under mechanochemical stress,31 but also because PEG polymers act as a chelation matrix of potassium cations, as a result of its crown-ether-like effect.38 The direct consequence is that both reactivity and solubility of ‘naked’ CO32− counter ions under mechanochemical activation could be enhanced in the presence of PEG catalyst. Thus, two different effects acting in synergy and at different stages of the reaction mechanism could be responsible for the enhanced reactivity of the system in PEG: (i) improved solubility of reactants, reaction intermediates and final product; (ii) chelation effect of potassium cation (from K2CO3).
In view of these results, the PEG additive was also tested for preparation of compound 4 by using CDI- or isocyanate-based methods (Table 2). In the presence of MeO-PEG-2000-OMe, the yield was 70% when following Method B (27%) – independently of the crystalline state of the starting material – and slightly improved when following Method A (90%), however the recovery of the final pure product was more complicated. Instead of precipitation by water (when PEG was not used), liquid–liquid extraction and PEG-precipitation were needed, hampering the easiness of recovery and lessening the positive environmental impact of the methodology.
Compound 4 was analyzed to assess the chiral integrity for both A and B methods (Table 4). Chiral HPLC analyses (see ESI†) for compound 4 showed complete loss of enantiopurity. This might be due to the formation of ‘high concentration’ of potassium methanolate during cyclisation. However, the addition of MeO-PEG-2000-OMe proved to be beneficial, partially preserving the enantiopurity, probably because of the dilution effect of the medium. To the best of our knowledge, this is the first example showing that PEG-assisted grinding helped to retain optical purity to a certain extent. This unexpected behaviour deserves further investigations.
Conclusions
We described herein two mechanochemical routes to access diverse 3,5-disubstituted hydantoins from cheap and readily available α-amino esters, either by reaction with isocyanates or by activation of the N-terminal position with CDI. The versatility of these procedures allowed us to prepare bactericide monomers or additives for polymer textiles such as 3-allyl-5,5′-dimethylhydantoin (ADMH) 16 and 1-chloro-3-ethyl-5,5′-dimethylhydantoin (CEDMH) 25, obtained with highly increased yield compared with solution synthesis using Ca(ClO)2 as the chlorinating agent. Three novel and efficient procedures based on mechanochemistry and using polyoxygenated PEG solvents provided easy access to the anticonvulsant ethotoin 12 in good yields, with no use of strong base or acid and for which very few literature was available. Moreover, the use of a green solvent such as PEG has the advantage of being non-toxic, non-flammable and non-volatile, unlike solvents often used for LAG (ethyl acetate, nitromethane). The ball-mill technology afforded easily handling and mild reaction conditions and PEG solvents were applicable for all methods (A, B and C) where grinding without an additive failed. However, further detailed studies are needed to elucidate the mechanistic aspects of this process when PEG is used. This would also shed light on its role in optical purity preservation during milling, never investigated before. It is worth noticing that the drug examples prepared here are either achiral or racemic, so the loss of chirality does not affect their preparation, even in the absence of the PEG additive.The two mechanochemical methods herein described (Methods A and B) are a valuable alternative to solution synthesis to access to 3,5-dialkylhydantoins, but they were not suitable for the preparation of 3-aryl-5-alkyl hydantoin such as ethotoin 12, except when PEG was used. They have allowed the preparation of a library of hydantoins, some of which are already commercialized but whose synthesis has still not been divulged, but most have not been reported in the literature.
Experimental section
Method A
(Table 2, compounds 2, 4, 5, 7–12, 18–19 and 21) – Vibrational Ball Mill (VBM). The α-amino methyl ester (1.0 equiv.), alkyl isocyanate (3.0 equiv.) and K2CO3 (3.0 equiv.) were ground in a vibrational ball-mill at 30 Hz for 2 hours. Distilled water was added to the reaction mixture and the desired compound was precipitated and filtered over sintered glass, or extracted with ethyl acetate. The organic layer was washed with 10% aq. citric acid (×3) and brine (×1), dried on MgSO4 and concentrated. Flash chromatography, normal or reverse phase, was performed on the crude sample.Only for compound 12 (Table 3) – Planetary Ball Mill (PBM): in the case of PEG-assisted grinding the reaction was performed by adding MeO-PEG-2000-OMe (455 mg mmol−1 substrate), in a planetary ball-mill, at 450 rpm for 2 hours using 50 balls (5 mm diameter). The compound was recovered by extraction with ethyl acetate after addition of distilled water to the reaction mixture. The organic layer was washed with 10% aq. citric acid and brine, dried over MgSO4 and concentrated. The compound was purified by flash chromatography (linear gradient of EtOAc in cyclohexane: 0–80%).
Method B
(Table 2, compounds 2, 4, 5-19 and 24) – Planetary Ball Mill (PBM). The α-amino methyl ester (1.0 equiv.) and 1,1′-carbonylimidazole (CDI) (1.3 equiv.) were ground in a 12 mL stainless steel milling jar in a planetary ball-mill at 450 rpm for 40 minutes using 50 balls (5 mm diameter). The amine (1.6 equiv.) and K2CO3 (3.6 equiv.) were added and the mixture was ground at 450 rpm for two hours. Distilled water was added to the crude and the desired compound was precipitated and filtered over sintered glass, or extracted with ethyl acetate. The organic layer was washed with 10% aq. citric acid (×3) and brine (×1), dried on MgSO4 and concentrated. Flash chromatography, normal or reverse phase, was performed on the crude sample.Only for compound 12 (Table 3): in the case of PEG-assisted grinding the reaction was performed adding MeO-PEG-2000-OMe (455 mg mmol−1 substrate). The compound was recovered by extraction with ethyl acetate after addition of distilled water to the reaction mixture. The organic layer was washed with 10% aq. citric acid and brine, dried over MgSO4 and concentrated. The compound was purified by flash chromatography (linear gradient of EtOAc in cyclohexane: 0–80%).
Method C
(Table 3, compound 12). 5-Phenylhydantoin30 (50 mg, 1.0 equiv.), ethyl bromide (106.0 μL, 5.0 equiv.) and K2CO3 (40 mg 1.0 equiv.) were milled with HO-PEG-3400-OH (455 mg mmol−1 substrate) in a planetary ball-mill at 300 rpm for 6 cycles of 15 minutes with 2 minute pauses in between. Distilled water was added to the reaction mixture and the aqueous phase was extracted with ethyl acetate. The organic layer was washed 10% aq. citric acid (×3) and brine (×1), dried on MgSO4 and concentrated. Preparative TLC (cyclohexane: EtOAc 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 v/v) was performed on the crude sample.
O), 1693, 1605 (CO), 1496, 1462, 1427, 1380, 1227, 1194, 1981, 1095, 1069, 1009, 919, 934, 859, 786, 765, 755, 748, 696, 679; 1H NMR (300 MHz, CDCl3)46 δ (ppm): 7.32–7.18 (m, 5H, ArH), 5.87 (s, 1H, NH), 4.23–4.20 (m, 1H, C-5/CH), 3.51–3.43 (m, 2H, CH2), 3.23 (dd, J = 13.9 and J = 3.4, 1H, CH2/HA), 2.88 (dd, J = 13.9 and J = 8.1, 1H, CH2/HB), 1.06 (t, J = 7.1, 3H, CH3); 13C NMR (300 MHz, CDCl3) δ (ppm): 172.9, 157.0, 135.1, 129.3, 128.8, 127.4, 58.2, 37.9, 33.5, 13.2; MS ESI-(+): m/z 219 [M + H]+.
:40 v/v) was performed on the crude sample.
O), 1693, 1605 (CO), 1496, 1462, 1427, 1380, 1227, 1194, 1981, 1095, 1069, 1009, 919, 934, 859, 786, 765, 755, 748, 696, 679; 1H NMR (300 MHz, CDCl3)46 δ (ppm): 7.32–7.18 (m, 5H, ArH), 5.87 (s, 1H, NH), 4.23–4.20 (m, 1H, C-5/CH), 3.51–3.43 (m, 2H, CH2), 3.23 (dd, J = 13.9 and J = 3.4, 1H, CH2/HA), 2.88 (dd, J = 13.9 and J = 8.1, 1H, CH2/HB), 1.06 (t, J = 7.1, 3H, CH3); 13C NMR (300 MHz, CDCl3) δ (ppm): 172.9, 157.0, 135.1, 129.3, 128.8, 127.4, 58.2, 37.9, 33.5, 13.2; MS ESI-(+): m/z 219 [M + H]+.| Method | Cr [ppb] | Mn [ppb] | Fe [ppb] | Ni [ppb] | Cu [ppb] | Cd [ppb] | W [ppb] |
|---|---|---|---|---|---|---|---|
| A (VBM) | 28736.1 |
1871.0 | 224714.1 | 1736.5 | 1047.7 | 61.2 | 3.9 |
| B (PBM) | 38846.9 |
1561.1 | 219276.2 | 889.4 | 2200.6 | 12.6 | 26.7 |
O), 1697 (CO), 1450, 1423, 1350, 1314, 1256, 1216, 1147, 1092, 1007, 873, 841, 783, 764, 709; 1H NMR (300 MHz, CDCl3) δ (ppm): 5.74 (s, 1H, NH), 4.01 (m, 1H, C-5/CH), 3.55 (q, J = 7.1, 2H, CH2), 1.79 (m, 2H, CH2), 1.51 (m, 1H, CH), 1.21 (t, J = 7.2, 3H, CH3), 0.97 (m, 6H, CH3); 13C NMR (300 MHz, CDCl3) δ (ppm): 174.4, 157.6, 55.9, 41.0, 33.7, 25.4, 23.2, 21.8, 13.5; MS ESI-(+): m/z 185 [M + H]+; HRMS ESI-(+): calcd for C9H17N2O2 [M + H]+ 185.1290, found 185.1292.O), 1698 (CO), 1609, 1507, 1426, 1353, 1257 (C(CH3)3), 1222 (C(CH3)3), 1179, 1161, 1104, 897, 877, 840, 761, 707; 1H NMR (300 MHz, CDCl3) δ (ppm): 7.08 (d, J = 8.2, 2H, ArH3,5), 6.93 (d, J = 8.2, 2H, ArH2,6), 5.39 (s, 1H, NH), 4.19 (m, 1H, C-5/CH), 3.48 (m, 2H, CH2), 3.20 (dd, J = 13.9 and J = 3.3, 1H, CH2/HA), 2.83 (dd, J = 13.8 and J = 8.2, 1H, CH2/HB), 1.32 (s, 9H, OC(CH3)3), 1.08 (t, J = 7.1, 3H, CH3); 13C NMR (300 MHz, CDCl3) δ (ppm): 173.1, 157.0, 155.0, 130.0, 124.6, 78.8, 58.4, 37.4, 33.7, 29.0, 13.4; MS ESI-(+): m/z 291 [M + H]+, 235 [(M − tBu) + H]+; HRMS ESI-(+): calcd for C16H22N2O3 [M + Na]+ 313.1528, found 313.1526.O), 1686 (CO), 1540, 1463, 1436, 1272, 1141, 1108, 1026, 796, 765, 691; 1H NMR (300 MHz, DMSO-d6) δ (ppm): 8.19 (s, 1H, NH), 7.36–7.23 (m, 6H, ArH), 5.00 (s, 2H, OCH2), 4.02–3.99 (m, 1H, C-5/CH), 3.38–3.34 (m, 2H, CH2), 2.96 (q, J = 6.0, 2H, CH2), 1.67–1.23 (m, 6H, (CH2)3), 1.05 (t, J = 7.1, 3H, CH3); 13C NMR (300 MHz, DMSO-d6) δ (ppm): 174.1, 156.7, 156.1, 137.3, 128.4, 127.7, 65.1, 56.1, 32.5, 30.9, 21.3, 13.3; MS ESI-(+): m/z 334 [M + H]+, 290, 226, 356 [M + Na]+. HRMS ESI-(+): calcd for C17H23N3O4 [M + H]+ 334.1767, found 334.1765.O), 1684 (CO), 455, 1422, 1353, 1288, 1218, 1174, 1106, 1007, 999, 967; 833, 765, 708; 1H NMR (300 MHz, CDCl3) δ (ppm): 6.01 (s, 1H, NH), 4.14 (m, 1H, C-5/CH), 3.56 (q, J = 6.8, 2H, CH2), 2.64 (m, 2H, CH2), 2.29–2.20 (m, 1H, CH2/HA), 2.11 (s, 3H, SCH3), 2.00–1.88 (m, 1H, CH2/HB), 1.21 (t, J = 6.8, 3H, CH3); 13C NMR (300 MHz, CDCl3) δ (ppm): 173.7, 157.4, 56.6, 33.8, 30.7, 30.5, 15.4, 13.5; MS ESI-(+): m/z 203 [M + H]+, 174, 155. HRMS ESI-(+): calcd for C8H14N2O2S [M + H]+ 203.0854, found 203.0849.O), 1693 (CO), 1451, 1425, 1350, 1214, 1121, 1003, 956, 903, 764, 708; 1H NMR (300 MHz, DMSO-d6) δ (ppm): 8.15 (s, 1H, NH), 4.01 (m, 1H, C-5/CH), 3.39–3.35 (m, 2H, CH2), 1.80–1.76 (m, 1H, CH), 1.37–1.11 (m, 1H, CH2/HA), 1.04 (t, J = 7.1, 3H, CH3), 0.90–0.68 (m, 10H, CH3 and CH2/HB); 13C NMR (300 MHz, DMSO-d6) δ (ppm) (1:1 mixture of diastereoisomers): 173.9, 173.5, 157.2, 156.9, 60.8, 59.6, 38.7, 36.4, 36.1, 32.4, 25.5, 23.3, 14.9, 13.3, 12.9, 11.6; MS ESI-(+): m/z 185 [M + H]+; HRMS ESI-(+): calcd for C9H17N2O2 [M + H]+ 185.1290, found 185.1288.O), 1698 (CO), 1460, 1430, 1359, 1234 (C(CH3)3), 1213 (C(CH3)3), 1195, 1119, 1100, 1069, 887, 810, 755, 707, 676; 1H NMR (300 MHz, CDCl3) δ (ppm): 5.74 (s, 1H, NH), 4.10 (dd, J = 7.5 and J = 2.9, 1H, C-5/CH), 3.74 (dd, J = 9.2 and J = 3.0, 1H, CH2/HA), 3.59–3.44 (m, 3H, CH2/HB and CH2), 1.23–1.17 (m, 13H, CH3 and OC(CH3)3); 13C NMR (300 MHz, CDCl3) δ (ppm): 171.9, 157.5, 74.1, 62.0, 58.1, 33.8, 27.5, 13.5; MS ESI-(+): m/z 185 [M + H]+; HRMS ESI-(+): calcd for C8H18N3O3 [M + H]+ 215.0668, found 215.0670.O), 1700 (CO), 1451, 1405, 1346, 1237, 1216, 1115, 999, 878, 813, 776, 730, 693; 1H NMR (300 MHz, DMSO-d6) δ (ppm): 8.29 (s, 1H, NH), 7.31–7.24 (m, 5H, ArH), 4.35 (sbroad, 1H, C-5/CH), 3.77 (s, 2H, SCH2), 3.40–3.37 (m, 2H, CH2), 2.76 (m, 2H, CH2), 1.08 (t, J = 7.1, 3H, CH3); 13C NMR (300 MHz, DMSO-d6) δ (ppm): 172.9, 156.8, 138.2, 128.9, 128.4, 127.0, 56.3, 36.0, 32.7, 32.1, 13.3; MS ESI-(+): m/z 265 [M + H]+. HRMS ESI-(+): calcd for C13H16N2O2S [M + H]+ 265.1011, found 265.1007.O), 1700 (CO), 1449, 1420, 1349, 1285, 1204, 1180, 1094, 1067, 936, 879, 751, 705; 1H NMR (300 MHz, DMSO-d6) δ (ppm): 8.22 (s, 1H, NH), 3.39–3.32 (m, 2H, CH2), 1.26 (s, 6H, CH3), 1.06 (t, J = 7.1, 3H, CH3); 13C NMR (300 MHz, DMSO-d6) δ (ppm): 177.6, 155.7, 58.0, 33.0, 25.0, 13.7; MS ESI-(+): m/z 157.0 [M + H]+; HRMS ESI-(+): calcd for C7H12N2O2 [M + H]+ 157.0977, found 157.0978.O), 1691 (CO); 1454, 1427, 1340, 1288, 1211, 1179, 1080, 1002, 824, 760, 701; 1H NMR (300 MHz, DMSO-d6) δ (ppm): 8.68 (s, 1H, NH), 7.43–7.31 (m, 5H, ArH), 5.19 (s, 1H, C-5/CH), 3.40 (qd, J = 7.0 and J = 1.7, 2H, CH2), 1.08 (t, J = 7.1, 3H, CH3); 13C NMR (300 MHz, DMSO-d6) δ (ppm): 172.0, 157.7, 134.5, 129.3, 129.2, 126.6, 60.8, 34.0, 31.0, 13.5; MS ESI-(+): m/z 205 [M + H]+.O), 1698 (CO), 1496, 1450, 1418, 1359, 1312, 1254, 1207, 1119, 1000, 937, 915, 752, 738, 702, 669, 654; 1H NMR (300 MHz, DMSO-d6) δ (ppm): 8.28 (s, 1H, NH), 7.26–7.15 (m, 5H, ArH), 5.48 (td, J = 10.8 and J = 5.2, 1H, CHallyl), 4.86 (d, J = 10.3, 1H, CH2allyl/Hcis), 4.54 (d, J = 17.3, 1H, CH2allyl/Htrans), 4.44–4.42 (m, 1H, C-5/CH), 3.84–3.69 (m, 2H, CH2), 2.98 (d, J = 3.3, 2H, CH2); 13C NMR (300 MHz, DMSO-d6) δ (ppm): 173.0, 156.1, 135.2, 131.8, 129.8, 128.1, 126.7, 115.6, 57.1, 36.3; MS ESI-(+): m/z 231 [M + H]+; HRMS ESI-(+): calcd for C13H14N2O2 [M + H]+ 231.1134, found 231.1134.O), 1708 (CO) 1647, 1451, 1410, 1371, 1359, 1336, 1207, 1155, 996, 929, 875, 731, 693, 661; 1H NMR (300 MHz, CDCl3) δ (ppm): 5.85–5.76 (m, 2H, NH and CHallyl), 5.30–5.18 (m, 2H, CH2allyl), 4.11–4.05 (m, 3H, C-5/CH and CH2), 1.81–1.79 (m, 2H, CH2), 1.60–1.42 (m, 1H, CH), 0.98 (t, J = 4.4, 3H, CH3); 13C NMR (300 MHz, CDCl3) δ (ppm): 174.2, 157.3, 131.2, 118.1, 56.0, 41.1, 40.7, 25.4, 23.2, 21.8; MS ESI-(+): m/z 197 [M + H]+, 169.0; HRMS ESI-(+): calcd for C10H16N2O2 [M + H]+ 197.1290, found 197.1292.O), 1697 (CO), 1648, 1556, 1451 (C(CH3)3), 1415 (C(CH3)3), 1363, 1191, 1123, 1078, 969, 923, 771, 734; 1H NMR (300 MHz, DMSO-d6) δ (ppm) (3:1 mixture of diastereoisomers): 8.19 (2 × s, 1H, NH), 5.80–5.67 (m, 1H, CHallyl), 5.14–5.05 (m, 2H, CH2allyl), 4.03–3.90 (m, 4H, C-5/CH, OCH and CH2), 1.19–1.05 (m, 12H, CH3 and OC(CH3)); 13C NMR (300 MHz, DMSO-d6) δ (ppm) (3:1 mixture of diastereoisomers): 172.8, 172.2, 157.4, 157.1, 132.8, 132.7, 121.7, 116.6, 116.3, 74.2, 73.8, 67.6, 66.1, 63.2, 62.0, 28.8, 28.6, 20.9, 18.5; MS ESI-(+): m/z 241 [M + H]+, 185.0; HRMS ESI-(+): calcd for C12H20N2O3 [M + H]+ 241.1552, found 241.1554.O), 1690 (CO), 1645, 1527, 1444, 1393, 1329, 1265, 1178, 1015, 972, 916, 874, 777, 733; 1H NMR (300 MHz, CDCl3)47 δ (ppm): 5.93 (sbroad, 1H, NH), 5.90–5.77 (m, 1H, CHallyl), 5.20 (dq, J = 6.5 and J = 1.4, 1H, CH2allyl/Hcis), 5.17 (t, J = 1.4, 1H, CH2allyl/Htrans), 4.09 (dt, J = 5.5 and J = 1.5, 2H, CH2), 1.45 (s, 6H, CH3); 13C NMR (300 MHz, CDCl3) δ (ppm): 177.0, 156.2, 131.3, 117.7, 59.0, 40.6, 25.2; MS ESI-(+): m/z 241 [M + H]+.O), 1709 (CO), 1494, 1467, 1393, 1240, 1182, 1117, 1024, 801, 736, 702, 664; 1H NMR (300 MHz, CDCl3)47 δ (ppm): 7.36 (s, 10H, ArH), 6.29 (s, 1H, NH), 5.85 (qt, J = 10.2 and J = 5.6, 1H, CHallyl), 5.23 (dq, J = 11.2 and J = 1.5, 1H, CH2allyl/Hcis), 5.19–5.17 (m, 1H, CH2allyl/Htrans), 4.18 (dt, J = 5.7 and J = 1.5, 2H, CH2); 13C NMR (300 MHz, CDCl3) δ (ppm): 173.0, 156.1, 139.3, 131.0, 129.0, 128.8, 127.0, 70.3, 41.1; MS ESI-(+): m/z 293.0 [M + H]+, 265.0.49 and 145–14650); IR (cm−1): 3347, 3295 (NH), 3062, 3031, 2927, 1760 (CO), 1695 (CO), 1604, 1495, 1447, 1419, 1362, 1341, 116, 1251, 1183, 1140, 1026, 965, 930, 877, 753, 716, 669; 1H NMR (300 MHz, DMSO-d6)48 δ (ppm): 8.36 (s, 1H, NH), 7.25–7.17 (m, 8H, ArH), 6.78–6.76 (m, 2H, ArH), 4.50 (t, J = 4.5, 1H, C-5/CH), 4.36 (q, J = 18.5, 2H, CH2), 3.00 (d, J = 4.7, 2H, CH2); 13C NMR (300 MHz, DMSO-d6) δ (ppm): 173.2, 156.2, 136.3, 135.0, 129.9, 128.3, 128.1, 126.9, 126.8, 126.5, 57.2, 40.7, 36.1; MS ESI-(+): m/z 281.0 [M + H]+.O), 1709 (CO), 1448, 1415, 1348, 1334, 1209, 1153, 936, 891, 879, 763, 721, 658; 1H NMR (300 MHz, DMSO-d6)51 δ (ppm): 8.41 (s, 1H, NH), 7.33–7.22 (m, 5H, ArH), 4.51 (s, 2H, CH2), 4.17–4.13 (s, 1H, C-5/CH), 1.84–1.71 (m, 1H, CH), 1.58–1.35 (m, 2H, CH2), 0.89–0.86 (m, 6H, CH3); 13C NMR (300 MHz, DMSO-d6) δ (ppm): 174.6, 156.6, 136.8, 128.5, 127.3, 55.0, 41.0, 40.8, 24.1, 23.1, 21.4; MS ESI-(+): m/z 247 [M + H]+.Chlorination with TCCA. 3-Ethyl-5,5′-dimethyl-2,4-imidazolidinedione 11 (98 mg, 0.63 mmol) and trichloroisocyanuric acid (TCCA) (73 mg, 0.31 mmol) were placed into a 12 mL stainless steel jar with 25 stainless steel milling balls (5 mm diameter) and milled for 15 minutes at 300 rpm. Water was then added to the reaction medium and the aqueous layer was extracted with ethyl acetate. The organic layer was washed with 10% aq. citric acid and brine, dried over anhydrous MgSO4 and concentrated. The crude was purified by column chromatography (linear gradient of EtOAc in cyclohexane: 0–60%) affording 25 as white crystals (41 mg, 34% yield).
Chlorination with calcium hypochlorite. 3-Ethyl-5,5′-dimethyl-2,4-imidazolidinedione 11 (101 mg, 0.65 mmol) and calcium hypochlorite (185 mg, 1.29 mmol) were placed into a 12 mL stainless steel jar with 50 stainless steel milling balls (5 mm diameter) and milled for one hour at 450 rpm. Dichloromethane was added to the reaction medium. The precipitated inorganic salts were filtered over sintered glass and the filtrate was concentrated to yield 25 as a white solid (107 mg, 87% yield). Mp 72–74 °C (lit.5 96.6 °C); CAS [886757-26-2];5 IR (cm−1): 3468, 3231, 2983 (NH), 2938, 1770 (C
O); 1700 (CO); 1442, 1411, 1379, 1341, 1293, 1229, 1208, 1161, 1067, 1001, 943, 881, 833, 757, 729; 1H NMR (300 MHz, CDCl3)48 δ (ppm): 3.61 (q, J = 7.2, 2H, CH2), 1.45 (s, 6H, CH3), 1.22 (t, J = 7.2, 3H, CH3); 13C NMR (300 MHz, CDCl3) δ (ppm): 174.3, 154.7, 65.9, 34.9, 22.3, 13.5; MS ESI-(+): m/z for 35Cl/37Cl 191/193 [M + H]+; HRMS ESI-(+): calcd for C7H11N2O2Cl [M + H]+ 191.0587, found 191.0587.
O), 1714 (CO), 1648, 1413, 1398, 1327, 1226, 1165, 1135, 944, 912, 876, 824, 758, 728; 1H NMR (300 MHz, CDCl3) δ (ppm): 5.88–5.75 (m, 1H, CHallyl), 5.27–5.25, 5.25–5.23 (m × 2, 1H, CH2allyl/Hcis), 5.21–5.19 (m, 1H, CH2allyl/Htrans), 4.15 (dt, J = 6, 2H, CH2), 1.47 (s, 6H, 2 × CH3); 13C NMR (300 MHz, CDCl3) δ (ppm): 174.2, 130.8, 119.0, 66.2, 41.9, 22.5; MS ESI-(+): m/z for 35Cl/37Cl 203/205, 175, 169, 130; HRMS ESI-(+): calcd for C8H12N2O2Cl [M + H]+ 203.0587, found 203.0589.Notes and references
- M. Meusel and M. Gutschow, Org. Prep. Proced. Int., 2004, 36, 391–443 CrossRef CAS.
- W. J. Close, Anticonvulsant 3-ethyl-5-phenyl hydantoin unit dosages and method of using same, US 2793157 A, 1957.
- M. Moguilewsky, J. Fiet, C. Tournemine and J. P. Raynaud, J. Steroid. Biochem., 1986, 24, 139–146 CrossRef CAS PubMed.
- G. Xi, Y. Xiu, L. Wang and X. Liu, J. Appl. Polym. Sci., 2015, 132, 41821–41827 CrossRef.
- Z. Chen and Y. Sun, Ind. Eng. Chem. Res., 2006, 45, 2634–2640 CrossRef CAS PubMed.
- D. A. Babkov, A. O. Chizhov, A. L. Khandazhinskaya, A. Corona, F. Esposito, E. Tramontano, K. L. Seley-Radtke and M. S. Novikov, Synthesis, 2015, 47, 1413–1422 CrossRef CAS.
- A. Martinez, M. Alonso, A. Castro, I. Dorronsoro, J. L. Gelpi, F. J. Luque, C. Perez and F. J. Moreno, J. Med. Chem., 2005, 48, 7103–7112 CrossRef CAS PubMed.
- C. Pedregal, G. G. Trigo, M. Espada, J. Elguero, E. J. Vincent and R. Faure, J. Heterocycl. Chem., 1984, 21, 477–480 CrossRef CAS.
- J. H. Poupaert, C. Smeyers and P. Bottcher, Bull. Soc. Chim. Belg., 1985, 94, 431–434 CrossRef CAS.
- M. J. O. Anteunis, L. Spiessens, M. De Witte, R. Callens and F. Reyniers, Bull. Soc. Chim. Belg., 1987, 96, 459–465 CrossRef CAS.
- M. L. Sanders and I. O. Donkor, Synth. Commun., 2002, 32, 1015–1021 CrossRef CAS.
- N. Dieltiens, D. D. Claeys, V. V. Zhdankin, V. N. Nemykin, B. Allaert, F. Verpoort and C. V. Stevens, Eur. J. Org. Chem., 2006, 2649–2660 CrossRef CAS.
- D. Blanco-Ania, P. H. H. Hermkens, L. A. J. M. Sliedregt, H. W. Scheeren and F. P. J. T. Rutjes, J. Comb. Chem., 2009, 11, 527–538 CrossRef CAS PubMed.
- J. A. d. S. Luis, J. M. Barbosa Filho, B. F. Lira, I. A. Medeiros, L. C. S. Lima de Morais, A. F. Ernandes dos Santos, C. Soares de Oliveira and P. Filgueiras de Athayde-Filho, Molecules, 2010, 15, 128–137 CrossRef CAS PubMed.
- J. M. Fraile, G. Lafuente, J. A. Mayoral and A. Pallares, Tetrahedron, 2011, 67, 8639–8647 CrossRef CAS.
- H. S. G. Beckmann, F. Nie, C. E. Hagerman, H. Johansson, Y. S. Tan, D. Wilcke and D. R. Spring, Nat. Chem., 2013, 5, 861–867 CrossRef CAS PubMed.
- F. Fujisaki, K. Toyofuku, M. Egami, S. Ishida, N. Nakamoto, N. Kashige, F. Miake and K. Sumoto, Chem. Pharm. Bull., 2013, 61, 1090–1093 CrossRef CAS PubMed.
- W. Karnbrock, M. Deeg, J. Gerhardt and W. Rapp, Mol. Diversity, 2000, 4, 165–171 CrossRef.
- K. H. Park and M. J. Kurth, Tetrahedron Lett., 2000, 41, 7409–7413 CrossRef CAS.
- Z. Zhai, J. Chen, H. Wang and Q. Zhang, Synth. Commun., 2003, 33, 1873–1883 CrossRef CAS.
- G. J. T. Kuster, L. W. A. van Berkom, M. Kalmoua, A. Van Loevezijn, L. A. J. M. Sliedregt, B. J. Van Steen, C. G. Kruse, F. P. J. T. Rutjes and H. W. Scheeren, J. Comb. Chem., 2006, 8, 85–94 CrossRef CAS PubMed.
- E. Colacino, F. Lamaty, J. Martinez and I. Parrot, Tetrahedron Lett., 2007, 48, 5317–5320 CrossRef CAS.
- P. Y. Chong and P. A. Petillo, Tetrahedron Lett., 1999, 40, 2493–2496 CrossRef CAS.
- L. Hroch, M. Hruskova, J. Schmitz, G. Schnakenburg and M. Guetschow, Synthesis, 2012, 44, 1907–1914 CrossRef CAS.
- B. Tao and J. W. Timberlake, Synthesis, 2000, 1449–1453 CrossRef CAS.
- D. Zhang, X. Xing and G. D. Cuny, J. Org. Chem., 2006, 71, 1750–1753 CrossRef CAS PubMed.
- X. Teng, A. Degterev, P. Jagtap, X. Xing, S. Choi, R. Denu, J. Yuan and G. D. Cuny, Bioorg. Med. Chem. Lett., 2005, 15, 5039–5044 CrossRef CAS PubMed.
- P. M. Fresneda, P. Molina and M. A. Sanz, Tetrahedron Lett., 2001, 42, 851–854 CrossRef CAS.
- A. Bolognese, G. Correale, M. Manfra, A. Esposito, E. Novellino and A. Lavecchia, J. Med. Chem., 2008, 51, 8148–8157 CrossRef CAS PubMed.
- L. Konnert, B. Reneaud, R. M. de Figueiredo, J.-M. Campagne, F. Lamaty, J. Martinez and E. Colacino, J. Org. Chem., 2014, 79, 10132–10142 CrossRef CAS PubMed.
- D. Hasa, G. S. Rauber, D. Voinovich and W. Jones, Angew. Chem., Int. Ed., 2015, 54, 7371–7375 CrossRef CAS PubMed.
- V. Declerck, E. Colacino, X. Bantreil, J. Martinez and F. Lamaty, Chem. Commun., 2012, 48, 11778–11780 RSC.
- L. Konnert, A. Gauliard, F. Lamaty, J. Martinez and E. Colacino, ACS Sustainable Chem. Eng., 2013, 1, 1186–1191 CrossRef CAS.
- L. Konnert, F. Lamaty, J. Martinez and E. Colacino, J. Org. Chem., 2014, 79, 4008–4017 CrossRef CAS PubMed.
- M. Lanzillotto, L. Konnert, F. Lamaty, J. Martinez and E. Colacino, ACS Sustainable Chem. Eng., 2015, 3, 2882–2889 CrossRef CAS.
- K. H. Dudley and D. L. Bius, J. Heterocycl. Chem., 1973, 10, 173–180 CrossRef CAS.
- H. Liu, Z. Yang and Z. Pan, Org. Lett., 2014, 16, 5902–5905 CrossRef CAS PubMed.
- J. Chen, S. K. Spear, J. G. Huddleston and R. D. Rogers, Green Chem., 2005, 7, 64–82 RSC.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed.
- E. Colacino, J. Martinez, F. Lamaty, L. S. Patrikeeva, L. L. Khemchyan, V. P. Ananikov and I. P. Beletskaya, Coord. Chem. Rev., 2012, 256, 2893–2920 CrossRef CAS.
- G. Giachi, M. Frediani, W. Oberhauser, F. Lamaty, J. Martinez and E. Colacino, ChemSusChem, 2014, 7, 919–924 CrossRef CAS PubMed.
- G. Cravotto and P. Cintas, Chem. Sci., 2012, 3, 295–307 RSC.
- H. A. Klok and J. Genzer, ACS Macro Lett., 2015, 4, 636–639 CrossRef CAS.
- A. S. Geneidi, A. A. Ali and R. B. Salama, J. Pharm. Sci., 1978, 67, 114–116 CrossRef CAS PubMed.
- F. L. Guedes, B. G. de Oliveira, M. Z. Hernandes, C. A. De Simone, F. J. B. Veiga, M. d. C. A. de Lima, I. R. Pitta, S. L. Galdino and P. J. R. Neto, AAPS PharmSciTech, 2011, 12, 401–410 CrossRef CAS PubMed.
- S. M. Dumbris, D. J. Diaz and L. McElwee-White, J. Org. Chem., 2009, 74, 8862–8865 CrossRef CAS PubMed.
- G. Xi, Y. Xiu, L. Wang and X. Liu, J. Appl. Polym. Sci., 2015, 132, 41824 CrossRef.
- N. Trisovic, T. Timic, J. Divljakovic, J. Rogan, D. Poleti, M. M. Savic and G. Uscumlic, Monatsh. Chem., 2012, 143, 1451–1457 CrossRef CAS.
- H. Aspelund and P. Waselius, Resistance of hydantoins and 5-hydroxyhydantoins to alkali, Acta Acad. Abo., Ser. B, 1967,(6), 27 Search PubMed ; II.
- H. L. Finkbeiner, FR 1389841, 1965.
- D. Zhang, X. Xing and G. D. Cuny, J. Org. Chem., 2006, 71, 1750–1753 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General remarks, technical details and characterization of all compounds and spectral data (1H NMR, 13C NMR and IR). Chiral HPLC analyses of compounds 4. See DOI: 10.1039/c6ra03222b |
| This journal is © The Royal Society of Chemistry 2016 |