
Cobalt(II)-catalysed transfer hydrogenation of olefins†
Guoqi
Zhang
*,
Zhiwei
Yin
and
Jiawen
Tan
Department of Sciences, John Jay College and Ph.D. Program in Chemistry, The Graduate Center of the City University of New York, New York, 10019 NY, USA. E-mail: guzhang@jjay.cuny.edu
First published on 22nd February 2016
Abstract
Catalytic transfer hydrogenation of olefins by isopropanol is achieved using an earth-abundant metal cobalt(II) complex based on a pincer-type PNP ligand. A range of olefins including aromatic and aliphatic alkenes as well as internal and cyclic alkenes have been transfer hydrogenated in good to excellent yields. The catalyst also showed good functional group and water tolerance to olefin transfer hydrogenation reactions.
Catalytic transfer hydrogenation (TH) that utilizes a mild and safe hydrogen source other than hydrogen gas provides a versatile and alternative way to reduce diverse multiple bonds, typically including polar C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CN bonds.1 Amongst the many methods for catalytic TH reaction developed thus far, transition metal catalytic TH is found to be the most efficient and practically applicable.2 Although precious metal Ru, Ir and Rh catalysts have been extensively explored and great advance has been achieved in terms of high turnover frequencies and efficacy,3–6 catalysts involving earth-abundant metals such as iron, cobalt and nickel are relatively less developed.7 It has been realized in recent years that it would be urgent to replace precious metal catalysts by abundant elements towards more practical and widespread industrial applications of TH in the future,1,8–12 as the high expense and scarcity of precious metals will largely limit their employment in sustainable chemical transformations.
O and CN bonds.1 Amongst the many methods for catalytic TH reaction developed thus far, transition metal catalytic TH is found to be the most efficient and practically applicable.2 Although precious metal Ru, Ir and Rh catalysts have been extensively explored and great advance has been achieved in terms of high turnover frequencies and efficacy,3–6 catalysts involving earth-abundant metals such as iron, cobalt and nickel are relatively less developed.7 It has been realized in recent years that it would be urgent to replace precious metal catalysts by abundant elements towards more practical and widespread industrial applications of TH in the future,1,8–12 as the high expense and scarcity of precious metals will largely limit their employment in sustainable chemical transformations.
Whereas most of reported TH catalysts were effective for polar double bonds, TH reactions of compounds that contain non-polar carbon–carbon multiple bonds were little explored.13 Compared to hydrogenation by H2, TH of alkenes and alkynes is more challenging, and known catalysts for such substrates involve mainly precious metal Ru, Rh and Pd complexes.13,14 To the best of our knowledge, only a few homogeneous nickel catalysts were reported for the asymmetric TH of amine-functionalized alkenes15 and a Fe(BF4)2/P4 system for the transfer semihydrogenation of terminal alkynes,16 except for heterogeneous nickel nanoparticle catalysts that have been extensively studied.17 Molecular cobalt catalysts capable of reducing such substrates under homogeneous TH conditions remain unprecedented, although cobalt-catalysed hydrogenation by H2 source has been previously reported by the Chirik and Peters groups.18 It is, therefore, quite desirable to develop new catalysts based on earth abundant metal cobalt for the TH of olefins.
Previously, we have reported on the discovery of an ionic cobalt(II) complex [(PNHPCy)Co(CH2SiMe3)][BArF4] (1, Scheme 1) built on a pincer-type PNP ligand that is efficient hydrogenation catalyst for a broad range of polar and non-polar double bonds under very mild conditions.19 The ability of this catalyst in carrying out effective “acceptorless” dehydrogenation was further established.19–23 Subsequently, homogeneous TH of a variety of polar double bonds including CO and CN bonds by the same cobalt catalyst was investigated using isopropanol as a hydrogen source.24 In this work, we noticed that except for simple ketones, two α,β-unsaturated ketones were fully hydrogenated to afford saturated alcohols, indicating the potential of this cobalt catalyst in reducing non-polar double bonds. Herein, we report for the first time a cobalt-catalysed TH of olefins by isopropanol under homogeneous conditions.
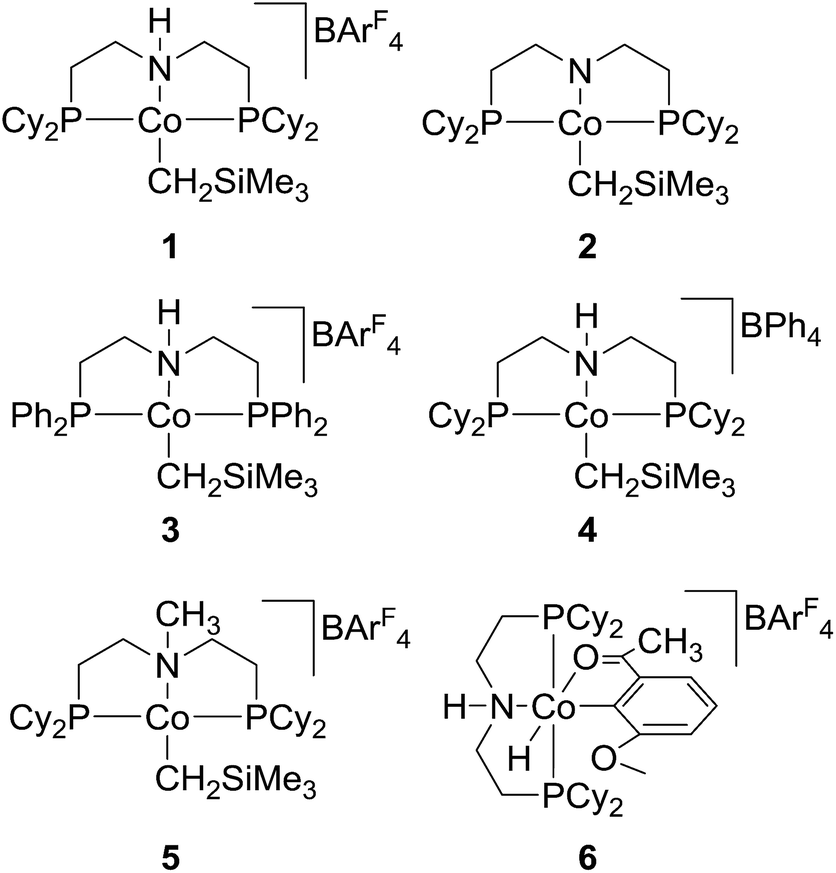 | ||
| Scheme 1 Cobalt complexes 1–6 studied for alkene transfer hydrogenation. | ||
Initial reactivity tests were conducted by using styrene as a model compound and the cobalt complexes 1 and 2 (Scheme 1) as catalysts, and the results are summarized in Table 1. Although 1 (2 mol%, generated in situ from equivalents of 2 and H[BArF4]·(Et2O)2) was known to readily hydrogenate a variety of alkenes at room temperature under 1 atm of dihydrogen, the TH test on styrene at 25 °C by using isopropanol (excess in THF) as a hydrogen source was unsuccessful (entry 1, Table 1). However, heating the reaction mixture to 80 °C resulted in the reduction of styrene to ethylbenzene in 75% yield (determined by GC analysis). To our delight, the reaction was observed to be almost completed at 100 °C, providing the desired product in 97% yield (entries 2 and 3, Table 1). In contrast, the neutral cobalt complex 2 was inactive to the TH of styrene under the same condition, and no reaction was detected without using a cobalt catalyst (entries 4 and 5). To evaluate the catalytic activity of related cobalt complexes in the TH of styrene, 3–6 were then examined for reactions under the same conditions (entries 6–9). It was observed that changing the substituents (from cyclohexyl to phenyl) on phosphorus atoms of the ligand has little influence on the catalytic activity, while a great reactivity difference was previously revealed for the H2 hydrogenation reactions.19 However, the counter anion in the cobalt catalyst was crucial for high reactivity, as replacing the BArF4− with BPh4− (in 4) drastically decreased the yield of ethylbenzene. In addition, ionic cobalt complex 5 built on the N-methylated ligand displayed the same activity as cobalt 1. However, when the Co(III) complex (6), an active reactive intermediate isolated during alcohol dehydrogenation reaction, was employed for the TH of styrene, only moderate yield was obtained, suggesting that Co(III) species was unlikely to be a catalyst resting state in the TH process, consistent with the results from TH of ketones catalysed by 6.24
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Solvent | Yieldb (%) |
| a Conditions: styrene (0.5 mmol), i-PrOH (0.5 mL), cobalt catalyst (2 mol%), solvent (1.5 mL) sealed in a 100 mL Schlenk tube under N2, 100 °C, 24 h. b Determined by GC with hexamethylbenzene as internal standard. c Reaction run at 25 °C. d Reaction run at 80 °C. e Reaction run under neat conditions (i-PrOH, 2 mL). f 1-Phenylethanol (0.5 mmol) was used as hydrogen source. g Ethanol (0.5 mL) was used as hydrogen source. h 1 mol% of 1 was used. | |||
| 1c | 1 | THF | 0 |
| 2d | 1 | THF | 75 |
| 3 | 1 | THF | 97 |
| 4 | 2 | THF | 0 |
| 5 | — | THF | 0 |
| 6 | 3 | THF | 95 |
| 7 | 4 | THF | 45 |
| 8 | 5 | THF | 96 |
| 9 | 6 | THF | 63 |
| 10 | 1 | Toluene | 98 |
| 11 | 1 | 1,4-Dioxane | 96 |
| 12 | 1 | CH3CN | 81 |
| 13 | 1 | Cyclohexane | 77 |
| 14e | 1 | — | 98 |
| 15f | 1 | THF | 99 |
| 16g | 1 | THF | 0 |
| 17h | 1 | THF | 81 |
Further reaction screening was carried out by using different solvents (entries 10–13, Table 1). Generally, solvents with high boiling point favored the reaction, furnishing the TH of styrene at 100 °C in high yields, while the use of volatile solvents such as acetonitrile and cyclohexane led to inferior results. The reaction also underwent smoothly under neat conditions without the addition of extra solvents (entry 14, Table 1). Finally, we tested the feasibility of utilizing other alcohols as hydrogen sources. It is worth noting that 1-phenylethanol (1.1 equivalent) could also act as a good hydrogen source for TH, stoichiometrically transferring dihydrogen to styrene. However, ethanol was not effective at all for the present TH reaction, despite it has found applications as a suitable hydrogen source in rhodium(I)-catalysed TH of both CC and CO bonds.14d
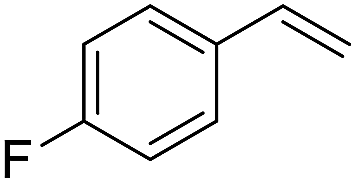
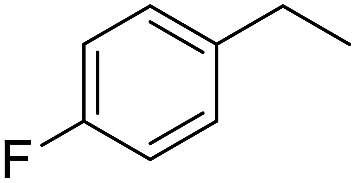
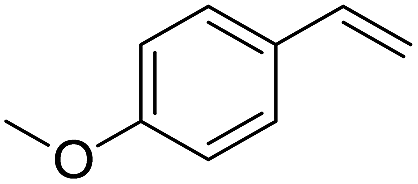
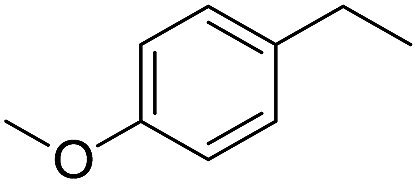
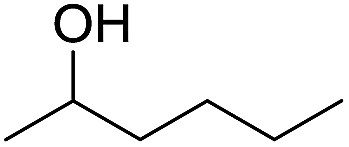
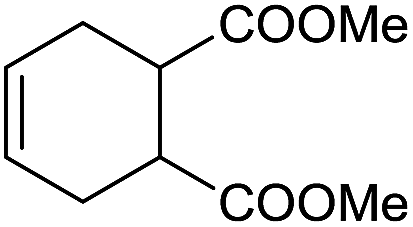
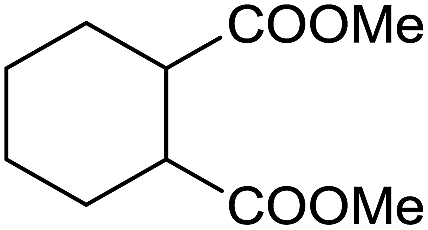
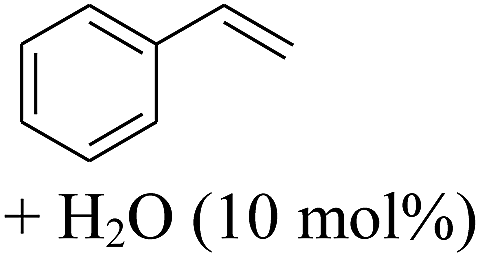
The finding of excellent catalytic activity of 1 in TH of styrene under the above mentioned conditions encouraged us to extend the substrate scope. Thus, a range of olefins were tested for the TH reactions under standard conditions (2 mol% 1, isopropanol/THF, 100 °C, 24 h), and the results are shown in Table 2. Substituted styrene with either electron-withdrawing or electron-donating group at the para-position was converted to the corresponding substituted ethylbenzene compound in quantitative yield (entries 2 and 3, Table 2). Likewise, the internal alkene, β-methylstyrene also proceeded well, affording propylbenzene in 98% yield (entry 4, Table 2). However, the relatively bulky internal alkenes, trans- and cis-stilbenes showed only moderate reactivity under the present TH conditions (entries 5 and 6, Table 2). In these cases, the hydrogenated product was observed after 48 h in 35% and 24% yields, respectively. In contrast, a precious metal ruthenium/N-heterocyclic carbene catalyst reported previously reduced both stilbenes in only 7% yield under TH conditions.14b Next, several aliphatic alkenes were also examined. Terminal alkenes such as 4-phenylbutene and 1-octene are suitable substrates for TH, affording the corresponding products in excellent yields (entries 7 and 8, Table 2). On the other hand, the TH with internal alkenes including cyclooctene and norbornylene exhibited equally high efficiency, producing both cyclic alkanes in quantitative yields (entries 9 and 10, Table 2). Furthermore, functional group-containing alkenes were also used to evaluate the functional group tolerance of the present cobalt catalyst under TH conditions (entries 11–13, Table 2). Interestingly, 3-butenoic acid bearing a carboxy group proceeded well for cobalt-catalysed TH, giving reduced product 1-butanoic acid in 98% yield. 5-Hexen-2-one bearing isolated CC and CO bonds was also fully reduced to 2-hexanol in high yield. However, the ester-containing internal alkene (dimethyl 4-cyclohexene-1,2-dicarboxylate) was found to be more challenging, providing the selectively hydrogenated product in moderate yield. Finally, it was found that cobalt catalyst 1 was water-tolerable under the TH reaction conditions. The addition of H2O (10 mol%) into the standard reaction system for styrene TH did not influence the conversion significantly (entry 14, Table 2), indicating the good stability of the cobalt complex against moisture. The functional group and water tolerance of cobalt 1 observed here is remarkable, as most of thus far reported base metal catalysts suited for hydrogenation or TH reactions showed either limited functional group tolerance or water stability.10–12
| Entry | Substrate | Product | Yieldb (%) |
|---|---|---|---|
a Conditions: substrate (0.5 mmol) and cobalt catalyst 1 (2 mol%, in situ formed by mixing 2 mol% of 2 and H[BArF4]·(Et2O)2) in i-PrOH/THF (2 mL, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (v/v), 100 °C, 24 h.
b Yields of products were determined by GC analysis using an internal standard method.
c Reaction run for 48 h. :3 (v/v), 100 °C, 24 h.
b Yields of products were determined by GC analysis using an internal standard method.
c Reaction run for 48 h.
|
|||
| 1 |
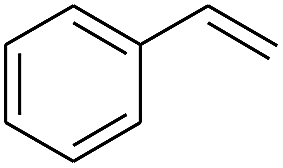
|
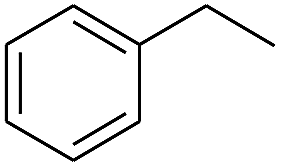
|
97 |
| 2 |
|
|
99 |
| 3 |
|
|
99 |
| 4 |
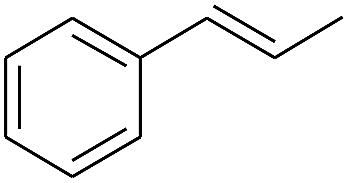
|
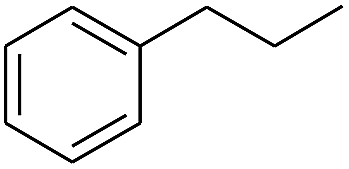
|
98 |
| 5c |
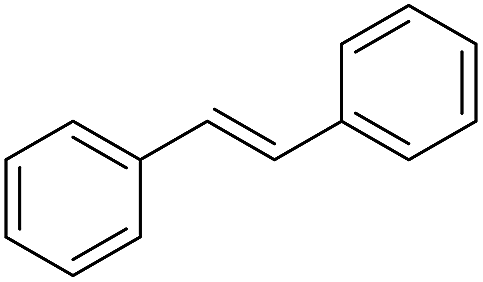
|
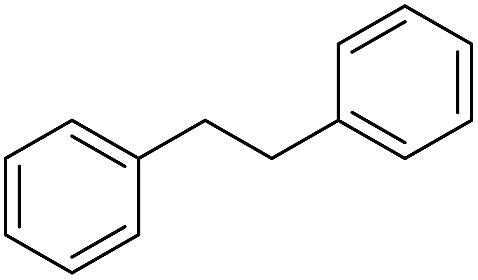
|
35 |
| 6c |
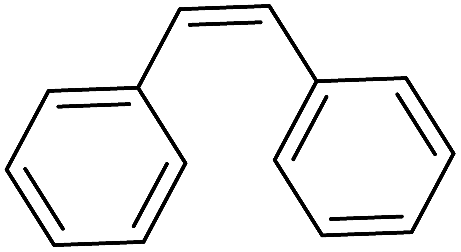
|
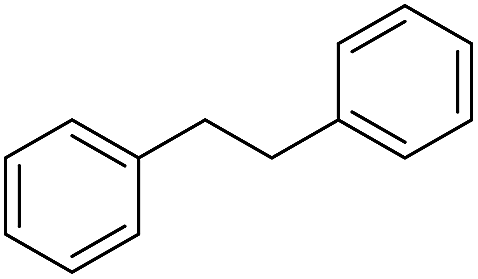
|
24 |
| 7 |
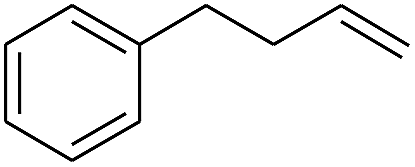
|
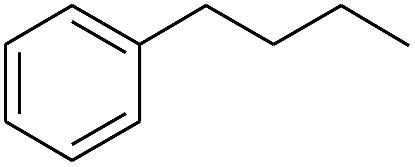
|
99 |
| 8 |
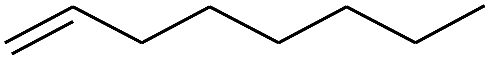
|

|
98 |
| 9 |
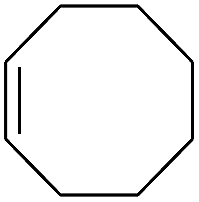
|
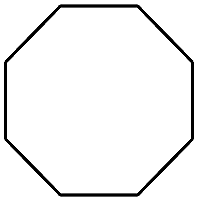
|
99 |
| 10 |
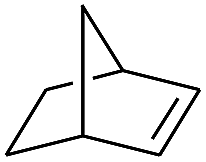
|
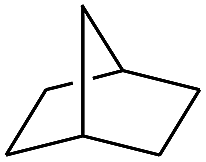
|
99 |
| 11c |
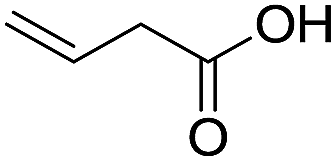
|
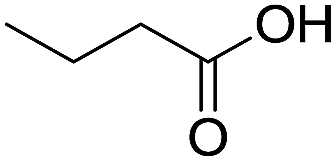
|
98 |
| 12 |
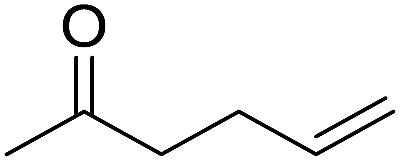
|
|
92 |
| 13 |
|
|
54 |
| 14 |
|

|
95 |
It was noticed that from the above results most of the alkene substrates studied showed excellent reactivity under the present TH conditions, similar to that observed under dihydrogen hydrogenation conditions, however, only low TH efficiency was revealed for the diaryl alkenes such as cis- and trans-stilbenes, which were not studied yet for the hydrogenation by H2 by using the same catalyst system.25 This urged us to further explore the reactivity difference between hydrogenation and TH reactions for relevant substrates. First, the hydrogenation reactions of both cis- and trans-stilbene were examined in the presence of 1 under 4 atm H2 gas (Scheme 2) at 25 or 100 °C, the optimal catalytic conditions for a plethora of CC, CO and CN bond hydrogenation.19 However, no reduction products were detected in both cases, indicating these olefins are reluctant to hydrogenation reaction, whereas they are moderately active for TH (entries 5 and 6, Table 2). Second, the same hydrogenation conditions were employed to the reduction of a related substrate, diphenylacetylene containing an internal carbon–carbon triple bond. Interestingly, the reaction was completed in the presence of 1 atm H2 and at room temperature. Effective semihydrogenation was observed (complete conversion to alkenes) and the resulting products cis- and trans-stilbenes were detected in a ratio of 34:66. Exactly same reactivity was also observed when using di-p-tolylacetylene as a substrate. It is worth mentioning that the semihydrogenation reaction here is significant although the stereoselectivity was modest. Such semihydrogenation of internal alkynes have been only carried out previously by using palladium complexes as catalysts, and (Z)-selective alkenes were exclusively obtained as the major products.13d In contrast, attempt to transfer hydrogenate diphenylacetylene under standard TH conditions gave no reduced product (Scheme 2). The different reactivity of cobalt catalyst in hydrogenating or transfer hydrogenating alkynes will deserve further investigations.25
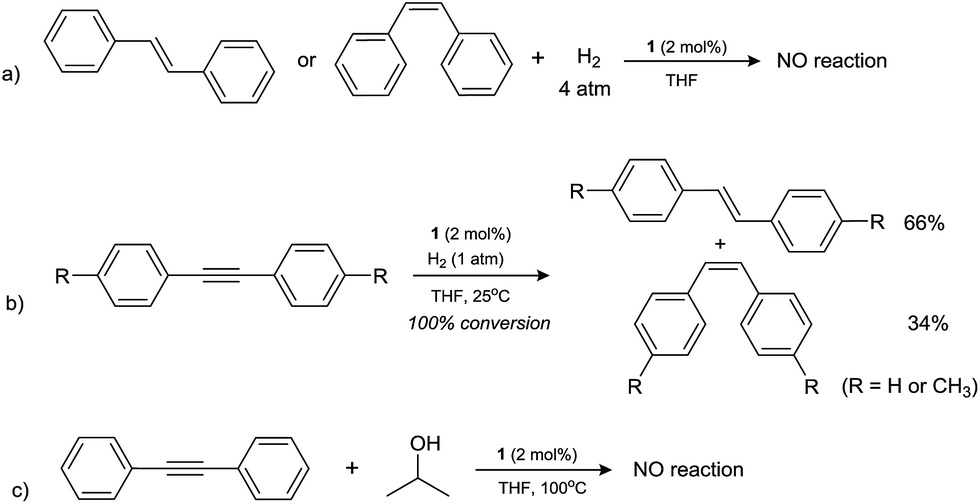 | ||
| Scheme 2 The comparison between hydrogenation and transfer hydrogenation of alkynes and stilbenes. | ||
In summary, we have reported an efficient cobalt-catalysed TH of olefins by using isopropanol as a hydrogen source. A variety of substituted terminal and internal alkenes as well as acid- and ester-functionalized alkenes have been successfully hydrogenated in moderate to high yields. Good functional group and water tolerance was disclosed. Different catalytic reactivity of the cobalt complex was also observed for the hydrogenation or TH of alkynes and stilbenes. This represents the first example of cobalt-catalysed transfer hydrogenation of olefins under homogeneous conditions. Further explorations leading to more effective and less expensive catalyst systems involving earth-abundant metals are currently underway.
Acknowledgements
Acknowledgment is made to the donors of the American Chemical Society Petroleum Research Fund for partial support of this research. We are grateful to the support from CUNY Collaborative Research Incentive Program, a Seed grant support from the Office for the Advancement of Research and the Program for Research Initiatives for Science Majors (PRISM) at CUNY John Jay College.Notes and references
- (a) D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621–6686 CrossRef CAS; (b) Y.-Y. Li, S.-L. Yu, W.-Y. Shen and J.-X. Gao, Acc. Chem. Res., 2015, 48, 2587–2598 CrossRef CAS.
- (a) M. J. Palmer and M. Wills, Tetrahedron: Asymmetry, 1999, 10, 2045–2061 CrossRef CAS; (b) K. Everaere, A. Mortreux and J.-F. Carpentier, Adv. Synth. Catal., 2003, 345, 67–77 CrossRef CAS; (c) T. Ikariya and A. J. Blacker, Acc. Chem. Res., 2007, 40, 1300–1308 CrossRef CAS PubMed; (d) S. Gladiali and E. Alberico, Chem. Soc. Rev., 2006, 35, 226–236 RSC.
- For ruthenium catalysts, see: (a) R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97–102 CrossRef CAS; (b) C. M. Moore and N. K. Szymczak, Chem. Comm., 2013, 49, 400–402 RSC; (c) R. Soni, J. M. Collinson, G. C. Clarkson and M. Wills, Org. Lett., 2011, 13, 4304–4307 CrossRef CAS PubMed; (d) R. Guo, C. Elpelt, X. Chen, D. Song and R. H. Morris, Chem. Commun., 2005, 3050–3052 RSC; (e) S. E. Clapham, A. Hadzovic and R. H. Morris, Coord. Chem. Rev., 2004, 248, 2201–2237 CrossRef CAS.
- For iridium catalysts, see: (a) M. Watanabe, Y. Kashiwame, S. Kuwata and T. Ikariya, Eur. J. Inorg. Chem., 2012, 504–511 CrossRef CAS; (b) A. Azua, S. Sanz and E. Peris, Chem.–Eur. J., 2011, 17, 3963–3967 CrossRef CAS; (c) H. Vázquez-Villa, S. Reber, M. A. Ariger and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 8979–8981 CrossRef PubMed; (d) F. E. Hahn, C. Holtgrewe, T. Pape, M. Martin, E. Sola and L. A. Oro, Organometallics, 2005, 24, 2203–2209 CrossRef CAS; (e) Z. R. Dong, Y. Y. Li, J. S. Chen, B. Z. Li, Y. Xing and J. X. Gao, Org. Lett., 2005, 7, 1043–1045 CrossRef CAS.
- (a) W. B. Cross, C. G. Daly, Y. Boutadla and K. Singh, Dalton Trans., 2011, 40, 9722–9730 RSC; (b) V. Gierz, A. Urbanite, A. Seyboldt and D. Kunz, Organometallics, 2012, 31, 7532–7538 CrossRef CAS.
- (a) E. Vega, E. Lastra and M. P. Gamasa, Inorg. Chem., 2013, 52, 6193–6198 CrossRef CAS PubMed; (b) D. Carmona, F. J. Lahoz, P. García-Orduña and L. A. Oro, Organometallics, 2012, 31, 3333–3345 CrossRef CAS.
- (a) Catalysis without precious metals, ed. R. M. Bullock, Wiley-VCH, Hoboken, NJ, 2010 Search PubMed; (b) T. Zell and D. Milstein, Acc. Chem. Res., 2015, 48, 1979–1994 CrossRef CAS; (c) S. Chakraborty, P. Bhattacharya, H. Dai and H. Guan, Acc. Chem. Res., 2015, 48, 1995–2003 CrossRef CAS.
- (a) S. Zhou, S. Fleischer, K. Junge, S. Das, D. Addis and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 8121–8125 CrossRef CAS PubMed; (b) S. Enthaler, B. Hagemann, G. Erre, K. Junge and M. Beller, Chem.–Asian J., 2006, 1, 598–604 CrossRef CAS; (c) S. Enthaler, G. Erre, M. K. Tse, K. Junge and M. Beller, Tetrahedron Lett., 2006, 47, 8095–8099 CrossRef CAS; (d) G. Wienhöfer, I. Sorribes, A. Boddien, F. Westerhaus, K. Junge, H. Junge, R. Llusar and M. Beller, J. Am. Chem. Soc., 2011, 133, 12875–12879 CrossRef PubMed.
- (a) C. Sui-Seng, F. N. Haque, A. Hadzovic, A. M. Pütz, V. Reuss, N. Meyer, A. J. Lough, M. Zimmer-De luliis and R. H. Morris, Inorg. Chem., 2009, 48, 735–743 CrossRef CAS; (b) N. Meyer, A. Lough and R. H. Morris, Chem.–Eur. J., 2009, 15, 5606–5610 CrossRef.
- (a) A. A. Mikhailine, M. I. Maishan, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2012, 134, 12266–12280 CrossRef CAS; (b) A. A. Mikhailine, M. I. Maishan and R. H. Morris, Org. Lett., 2012, 14, 4638–4641 CrossRef CAS; (c) P. Sues, A. Lough and R. H. Morris, Organometallics, 2011, 30, 4418–4431 CrossRef CAS; (d) P. O. Lagaditis, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2011, 133, 9662–9665 CrossRef CAS.
- W. Zuo and R. H. Morris, Nat. Protoc., 2015, 10, 241–257 CrossRef CAS.
- (a) D. Tavor, I. Gefen, C. Dlugy and A. Wolfson, Synth. Commun., 2011, 41, 3409–3416 CrossRef CAS; (b) J. Geboers, X. Wang, A. B. de Carvalho and R. Rinaldi, J. Mol. Catal. A: Chem., 2014, 388–389, 106–115 CrossRef CAS; (c) H. Xu, P. Yang, P. Chuanprasit, H. Hirao and J. Zhou, Angew. Chem., Int. Ed., 2015, 54, 5112–5116 CrossRef CAS PubMed.
- (a) A. M. Kluwer, T. S. Koblenz, T. Jonischkeit, K. Woelk and C. J. Elsevier, J. Am. Chem. Soc., 2005, 127, 15470–15480 CrossRef CAS; (b) J. W. Sprenger, J. Wassenaar, N. D. Clement, K. J. Cavell and C. J. Elsevier, Angew. Chem., Int. Ed., 2005, 44, 2026–2029 CrossRef; (c) P. Hauwert, G. Maestri, J. W. Sprengers, M. Catellani and C. J. Elsevier, Angew. Chem., Int. Ed., 2008, 47, 3223–3226 CrossRef CAS; (d) P. Hauwert, J. J. Dunsford, D. S. Tromp, J. J. Weigand, M. Lutz, M. Catellani and C. J. Elsevier, Organometallics, 2013, 32, 131–140 CrossRef CAS.
- (a) J. Broggi, V. Jurčík, O. Songis, A. Poater, L. Cavallo, A. M. Z. Slawin and C. S. J. Cazin, J. Am. Chem. Soc., 2013, 135, 4588–4591 CrossRef CAS PubMed; (b) S. Sabine and M. Albrecht, Chem. Commun., 2011, 47, 8802–8804 RSC; (c) J. M. Adriaan, B. L. Feringa, L. Lefort and J. G. de Vries, Acc. Chem. Res., 2007, 40, 1267–1277 CrossRef; (d) T. Zweifel, J. V. Naubron, T. Buttner, T. Ott and H. Grützmacher, Angew. Chem., Int. Ed., 2008, 47, 3245–3249 CrossRef CAS.
- P. Yang, H. Xu and J. Zhou, Angew. Chem., Int. Ed., 2014, 53, 12210–12213 CrossRef CAS.
- G. Wienhöfer, F. A. Westerhaus, R. V. Jagadeesh, K. Junge, H. Hunge and M. Beller, Chem. Commun., 2012, 48, 4827–4829 RSC.
- F. Alonso, P. Riente and M. Yus, Acc. Chem. Res., 2011, 44, 379–391 CrossRef CAS.
- (a) T.-P. Lin and J. C. Peters, J. Am. Chem. Soc., 2014, 136, 13672–13683 CrossRef CAS; (b) M. R. Friedfeld, M. Shevlin, J. M. Hoyt, S. W. Krska, M. T. Tudge and P. J. Chirik, Science, 2013, 342, 1076–1080 CrossRef CAS; (c) R. P. Yu, J. M. Darmon, C. Milsmann, G. W. Margulieux, S. C. E. Stieber, S. DeBeer and P. J. Chirik, J. Am. Chem. Soc., 2013, 135, 13168–13184 CrossRef CAS; (d) S. Monfette, Z. R. Turner, S. P. Semproni and P. J. Chirik, J. Am. Chem. Soc., 2012, 134, 4561–4564 CrossRef CAS.
- G. Zhang, B. L. Scott and S. K. Hanson, Angew. Chem., Int. Ed., 2012, 51, 12102–12106 CrossRef CAS.
- G. Zhang, K. V. Vasudevan, B. L. Scott and S. K. Hanson, J. Am. Chem. Soc., 2013, 135, 8668–8681 CrossRef CAS.
- G. Zhang and S. K. Hanson, Org. Lett., 2013, 15, 650–653 CrossRef CAS.
- G. Zhang, Z. Yin and S. Zheng, Org. Lett., 2016, 18, 300–303 CrossRef CAS.
- R. Xu, S. Chakraborty, H. Yuan and W. D. Jones, ACS Catal., 2015, 5, 6350–6354 CrossRef CAS.
- G. Zhang and S. K. Hanson, Chem. Commun., 2013, 49, 10151–10153 RSC.
- A step-wise, bifunctional mechanism for the hydrogenation of alkenes catalysed by FeII complexes based on the same pincer ligand has been recently suggested, see: R. Xu, S. Chakraborty, S. M. Bellows, H. Yuan, T. R. Cundari and W. D. Jones, ACS Catal., 2016 DOI:10.1021/acscatal.5b02674.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and spectroscopic data. See DOI: 10.1039/c6ra02021f |
| This journal is © The Royal Society of Chemistry 2016 |