
A high rate Li-rich layered MNC cathode material for lithium-ion batteries†
Mehmet Nurullah Ates,
Sanjeev Mukerjee and
K. M. Abraham*
Center for Renewable Energy Technology, Department of Chemistry and Chemical Biology, Northeastern University, Boston, MA 02115, USA. E-mail: kmabraham@comcast.net
First published on 12th March 2015
Abstract
We report a high rate Li-rich layered manganese nickel cobalt (MNC) oxide cathode material of the composition 0.5Li2MnO3·0.5LiMn0.5Ni0.35Co0.15O2, termed Li-rich MNC cathode material, with discharge capacities of 200, 250, and 290 mA h g−1 at C, C/4 and C/20 rates, respectively, for Li-ion batteries. This high rate discharge performance combined with little capacity fade during long term cycling is unprecedented for this class of lithium ion (Li-ion) cathode materials. The exceptional electrochemistry of the Li-rich MNC in Li-ion cells is attributed to its open porous morphology and high electronic conductivity. The structure of the material investigated by means of X-ray diffraction (XRD), High Resolution Transmission Electron Microscopy (HRTEM) and X-ray Absorption Spectroscopy (XAS) combined with electrochemical data revealed that the porous morphology was effective in allowing electrolyte penetration through the particle grains in tandem with its high electronic conductivity to provide high Li+ transport for high rate discharge. Extended cycling behavior and structural phase transition of the new material were further examined through Field Emission Scanning Electron Microscopy (FESEM), XRD, XAS and HRTEM. The new Li-rich MNC cathode material could provide the next generation Li-ion batteries with specific energy exceeding 400 W h kg−1 or energy density over 1000 W h l−1.
Introduction
A Li-ion battery cathode material that exhibits a capacity of >270 mA h g−1 (∼1 electron per metal utilization of the material) at 0.5 to 1C rates with little or no capacity fade for hundreds of discharge–charge cycles is of great practical interest. We wish to report such a material belonging to the family of Li-rich layered MNC composite oxides of the general formula xLi2MnO3·(1 − x)LiMO2 (M = transition metal)1–5 in which there is a layered lithium manganate (Li2MnO3) and a layered lithium metal dioxide (LiMO2) structural unit whose relative amounts can vary. Thus a large number of these materials with high capacities can be prepared. The transition metal (TM) in xLi2MnO3·(1 − x)LiMO2 is usually one or more metals selected from Mn, Co, and Ni. These lithium rich metal oxides are considered to be one of the most promising next generation cathode active materials for Li-ion batteries. These materials can deliver discharge capacities >250 mA h g−1 after they have been activated by charging first to a voltage of 4.6–4.9 V in a Li cell. In the initial charging of these materials Li is first extracted from the layered LiMO2 structure up to a voltage of about 4.4 V, and then the Li2MnO3 structural unit is activated with the extraction of Li2O as Li+, O2 and electrons from it at potentials between 4.6 and 4.9 V. The resulting layered cathode material cycles with >250 mA h g−1 capacities at low to medium rates of C/5 to C/20.1,6 The initial activation process involves a complex mechanism triggering transition metal migrations into Li and/or transition metal sites leading to voltage hysteresis in the following cycles. Several disadvantages of these materials still remain be resolved before they can be implemented in practical batteries and they include (i) the high irreversible capacity loss (ICL) along with oxygen generation in the initial activation charging;7 (ii) low discharge rate capability and high capacity fade during cycling3 (iii) low electronic conductivity leading to high resistance in Li-ion cells8,9 and (iv) voltage hysteresis10 and phase transformation after extended cycling. Here we report a cathode material of this family which has overcome some of these deficiencies.The material we report has the formula of 0.5Li2MnO3·0.5LiMn0.5Ni0.35Co0.15O2, which by tradition,11–13 can be written alternatively as Li1.2Mn0.6Ni0.14Co0.06O2. We synthesized this material using the “self-ignition combustion (SIC)” method, which to the best of our knowledge has not been reported for the preparation of Li-rich layered metal oxides. A similar method had been employed to synthesize Mn and Ti based spinel-structured cathode14 and anode15 materials. This new cathode material exhibits capacities of 290, 250, and 200 mA h g−1 at C/20, C/4 and C rates, respectively. Moreover, the new material exhibits high rate cycling ability with little or no capacity fade for over 100 cycles demonstrated at a series of rates from C/20 to 2C rates for electrodes loadings of 7–8 mg cm−2. Such exceptional electrochemical performance for a high capacity Li-rich layered metal composite oxide cathode material is reported for the first time here. The unprecedented cycling stability of as-synthesized 0.5Li2MnO3·0.5LiMn0.5Ni0.35Co0.15O2, achieved not only at high rates (1C, 2C) suitable for electric vehicle batteries but also at the moderate C/20 and C/4 rates desirable for powering portable electronic devices such as cell phones and laptops. The superior electrochemical properties of the new material are ascribed to its unique particle morphology and electronic conductivity achieved from the self-ignition combustion synthesis. A comprehensive account of electrochemical and structural results on the new cathode material is presented here.
Experimental
1. Preparation of cathode material
To prepare 0.5Li2MnO3·0.5LiMn0.5Ni0.35Co0.15O2 appropriate amounts of Mn(Ac)2·4H2O (Sigma Aldrich >99%), Ni(NO3)2·6H2O (Alfa Aesar-Puratronic), Co(NO3)2·6H2O (Alfa Aesar-Puratronic) were dissolved in distilled water at room temperature in a beaker. Nitric acid and glycine (Sigma Aldrich >99%) were added to the solution and heated it to 120 °C when the ignition combustion reaction took place. Different ratio of glycine, as a source of fuel, was used to optimize and control the combustion reaction. Glycine is known to be a complexing agent for transition metal ions due to the presence of both carboxylic acid and amino group in its structure. Acetate precursor was used in order to produce a large amount of gaseous by-product of the combustion reaction, whose evolution leads to a material with open porous microstructures. The material obtained from the combustion reaction was mixed in a mortar with stoichiometric amount of LiOH·H2O (Alfa Aesar >99.995%). This mixture was placed in a ceramic boat and fired at 480 °C for 3 h under air flow. Afterwards, the sample was slowly cooled, re-grounded, pelletized and fired at 900 °C for another 3 h in open air. We denote this material hereafter as SIC-MNC. For comparison, we also synthesized the same compound using the conventional co-precipitation method, designated hereafter as CP-MNC. Details of the co-precipitation method can be found elsewhere.1,9,16,17 To prepare a Ni4+ reference material for XAS experiments, LiNi0.85Co0.15O2 was synthesized via solid state reaction from appropriate amounts of LiOH·H2O, Ni(Ac)2·4H2O and Co(Ac)2·4H2O as reported previously.18 Charging of this material in a Li cell to 5.1 V produced the Ni4+ reference material.192. Characterization
The structure–property relationships of the high rate Li-rich MNC cathode material has been characterized by means of XRD, FESEM along with Energy Dispersive Spectroscopy (EDS), XAS, and HRTEM, combined with electrochemical discharge–charge cycling tests and Electrochemical Impedance Spectroscopy (EIS) of Li cells.Diffraction patterns of the materials were obtained using a Rigaku Ultima IV diffractometer with CuKα radiation. Unit cells of each sample were analyzed by PDXL software program provided by Rigaku Corporation. VESTA software20 were run to visualize unit cells in order to understand the reaction process. For ex situ XRD experiments, harvested electrodes from cycled Li cells were thoroughly rinsed with anhydrous dimethyl carbonate (DMC) to remove possible electrolyte residue before examination.
Morphological and structural investigations were observed by Hitachi S-4800 FESEM combined with its EDS attachment and a JEOL 2010F model HRTEM was utilized for structural investigations. The samples for HRTEM were prepared by dispersing powders into an ethanol solution followed by a few minutes of sonication, and then one or two drops of the dispersed solution was dropped onto Cu grid. Electron diffraction patterns were simulated using SingleCrystal software developed by CrystalMaker software Limited, Oxford, U.K.
Impedance measurements were performed in the range of 100 kHz to 10 mHz with a 5 mV amplitude AC sine wave on a Voltalab PGZ402 model potentiostat in order to evaluate and compare the impedance responses of the materials. Before running any EIS measurements, cells were rested for several hours to stabilize the voltage responses. The same instrument was used for cyclic voltammetry (CV) experiments with the electrode materials at a sweep rate of 100 μV s−1 at room temperature.
XAS measurements were performed at beam lines X-3A and X-18A of the National Synchrotron Light Source at Brookhaven National Laboratory located in New York. Electrodes from the Li coin cells, cycled at low rates (C/20), were rinsed with DMC and extracted, then sealed with Kapton tape and stored in glass vials, were taped with Teflon tape, and packed in moisture impermeable aluminized bags under argon to avoid any possible oxidation during transportation to the light source. The data were processed using the Athena software program.21 Scans were calibrated, aligned and normalized.
DC conductivity measurements were determined using pellets of the pristine materials. Special precaution was given to the pellet preparations of each material such that they had identical pellet densities. Details of the experimental setup and conductivity calculations can be found in our previous publication.9 The surface area and porosity properties of the material were determined using the Brunauer–Emmett–Teller (BET, Quantachrome Nova) method using nitrogen as adsorption gas.
Lithium anode-containing coin cells were fabricated for evaluating the MNC cathode electrochemistry. The cathode was prepared from a mixture of 80 wt% (weight-percent) of the MNC cathode material, 10 wt% Super P carbon black as electronic conductor and 10 wt% polyvinylidene fluoride (PVDF-Kynar® 2801) as binder. The cathode mixture was dissolved in N-methyl 2-pyrrolidone (NMP, Sigma Aldrich >99%) and the resulting slurry was coated onto an aluminum foil current collector using a doctor-blade technique. The cathode ribbon thus obtained was dried at 100 °C in vacuum. Coin cells were built with discs of this cathode and Li foil anode, separated by a porous propylene membrane separator, and filled with 1 M LiPF6/1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2 EC/DMC electrolyte. The cells were cycled between 2 and 4.9 V at room temperature with an Arbin Instrument BTZ2000 model cycler at a series of discharge currents and their corresponding C rates are mentioned in data figures presented throughout the paper. Theoretical capacity of the materials was calculated to be 280 mA h g−1 based on one Li utilization per MO2 formula.
:1.2 EC/DMC electrolyte. The cells were cycled between 2 and 4.9 V at room temperature with an Arbin Instrument BTZ2000 model cycler at a series of discharge currents and their corresponding C rates are mentioned in data figures presented throughout the paper. Theoretical capacity of the materials was calculated to be 280 mA h g−1 based on one Li utilization per MO2 formula.
Results and discussion
A comprehensive account of the electrochemistry of the new cathode material in Li cells is presented together with a detailed discussion of its morphology and structure.1. Electrochemical performance of 0.5Li2MnO3·0.5LiMn0.5Ni0.35Co0.15O2 in Li cells
The remarkable electrochemical behavior of the SIC-MNC cathode, and its outstanding cycling stability at 1C and other discharge rates are depicted in Fig. 1a. The initial discharge capacity at C-rate was around 220 mA h g−1 which after a few cycles stabilized at around 200 mA h g−1 and maintained this value even after 100 cycles with excellent columbic efficiency. The capacity fade rate between the 10th and the 100th cycle is less than 0.01% which for this type of materials is unprecedented. At this fade rate the cathode will lose less than 10% of its capacity after 1000 cycles. Even a 20% loss of capacity after 1000 cycles is exceptional for this family of next generation cathode materials. The material demonstrated impressive capacity retention of >99% at C/4 with a capacity of about 250 mA h g−1 after 90 cycles (see the inset of Fig. 1a which also shows capacities at different rates). Contrasting this performance is the recent report for an identical material, prepared by the conventional co-precipitation method,22 of a capacity of only 248 mA h g−1 after 75 cycles at 21 mA g−1 (∼C/10) rate, albeit a decent cycling stability. That material did not report any cycling data at the C rate. Our SIC-MNC exhibited 281 mA h g−1 after 75 cycles at the higher current density of 28 mA g−1. In the class of Li-rich “layered–layered’’ cathode materials, our SIC-NMC to our knowledge has demonstrated higher rate and better cycling abilities at both low and high discharge rates than any material reported.23–28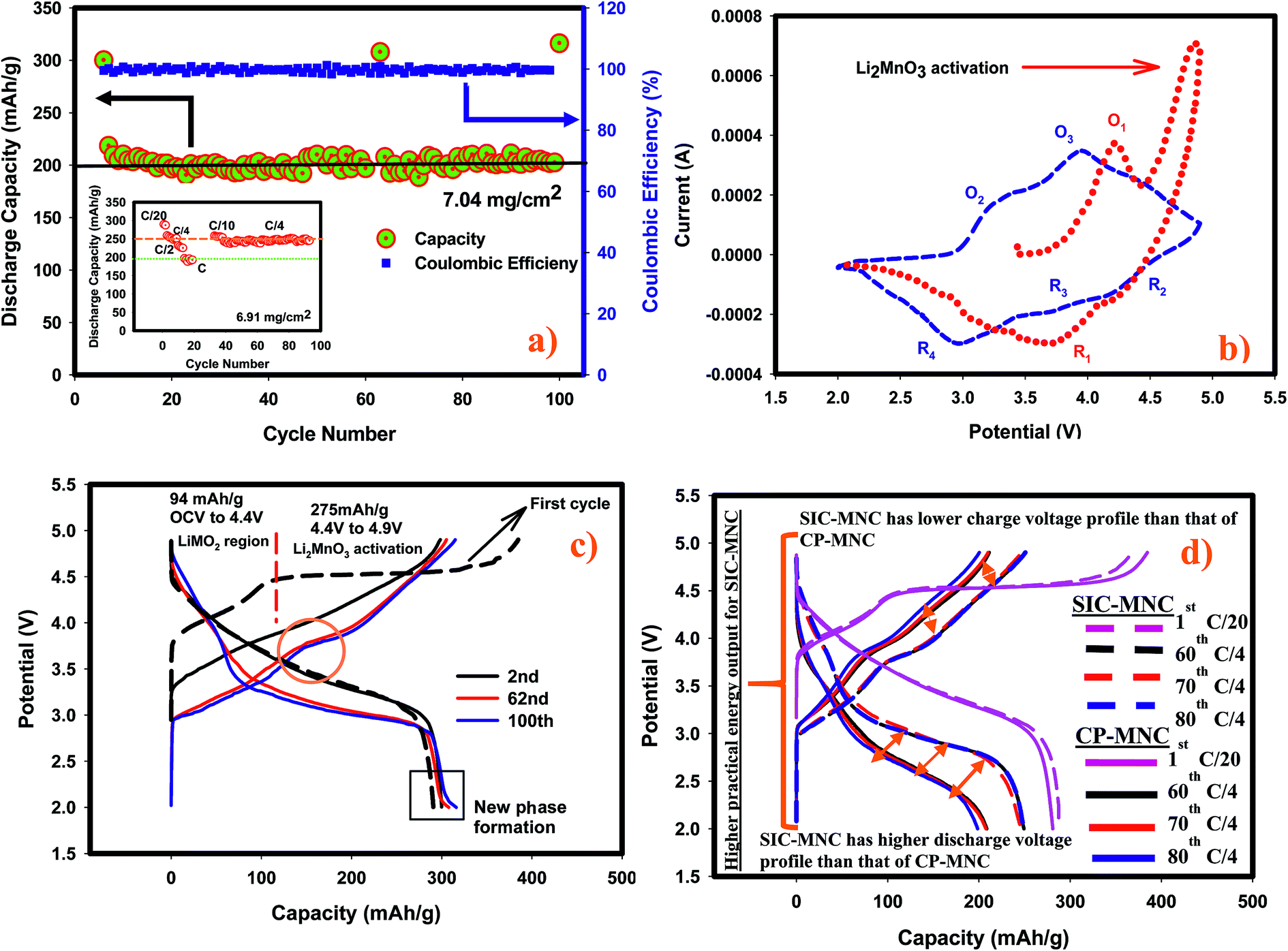 | ||
| Fig. 1 (a) Electrochemical cycling performance at C discharge rate (280 mA g−1) between 2–4.9 V. Inset shows different discharge rate and C/4 rate cycling performance. Each figure's data were obtained from different cells having similar loading. (b) Cyclic voltammograms of SIC-MNC recorded at a sweep rate of 100 μV s−1. (c) Charge–discharge curves at C/20 deduced from the data in a. (d) Charge–discharge profiles of both SIC-MNC and CP-MNC and their voltage fade performance. | ||
The high rate capability and exceptional capacity retention of our material with repeated discharge–charge cycling is attributed to the porous morphology and high electronic conductivity. We compared our new SIC-MNC material with the extensively studied CP-MNC in their rate and cycling performances. Fig. S1† shows the typical capacity fade during cycling and the poor rate capabilities of the CP-MNC material which clearly contrasts with the behavior of the new SIC-NMC presented in Fig. 1a.
The redox behavior of SIC-MNC was further investigated by cyclic voltammetry displayed in Fig. 1b. During the first charging process (1st chg), two dominant peaks appeared at around 4.2 V and above 4.5 V. The first peak, denoted as O1, can be ascribed to the oxidation of Ni2+ and Co3+ to Ni4+ and Co4+, respectively. The second peak above 4.5 V is due to Li2MnO3 activation where Li2O is released from the structure. During the subsequent first discharge (1st dchg), the reduction peak appearing around 3.6 V could be associated with Co4+ and Ni4+ reduction. After the activation of Li2MnO3, some new redox features were observed in the second cycle. For example, during the second charge, peak O2, appeared. This peak is probably due to the oxidation of reduced (lithiated) MnO2 (from Mn3+ to ∼Mn4+) created in the first discharge. Subsequently, peak O3 appeared due mainly to Ni2+/Ni4+ oxidation and a small peak just below 4.5 V was found which is ascribed to the oxidation of Co3+. These oxidation peaks are followed by reduction peaks R2 and R3 which could be Co4+ and Ni4+ reduction, respectively.28 Finally, peak R4 due to Mn reduction from partially oxidized Mn3+ appeared. In general, after the first cycle the peaks shifted to lower potentials, a strong indication of structural transformation. Fig. 1c shows the voltage versus capacity profile for the high rate cycling data presented in Fig. 1a. We observed several interesting features; the first feature is the plateau region during the first charging process after 4.3 V, attributed to Li2MnO3 activation where the ICL of 70 mA h g−1 originates. The charging in the second and subsequent cycles begin at lower voltages with upward sloping voltage profiles which are a clear indication of structural rearrangements as a result of the activation process in the first charge. Most importantly, the capacity attained in the second cycle was preserved after 100 cycles. A small plateau appeared at around 2 V region in later discharges, displayed with a box in Fig. 1c, suggesting a new phase formation with repeated cycling, most likely the spinel phase. The spinel peak associated with Mn reduction usually starting at 2.7 V but gradually moving to lower than 2.3 V24,29 augur well with this view. It is possible that such a phase transition at low rates can be inhibited by limiting the discharge voltage to about 2.5 V as no capacity contribution was observed under 2.5 V. Possible phase transformation is further elaborated later in this paper. Secondly, the voltage fade seen in Fig. 1c is a common problem encountered in Li rich layered cathode materials as a consequence of metal migration after several initial cycles following the Li2MnO3 activation. Fig. 1d shows the charge–discharge voltage profiles for each material with respect to their applied rate. It is readily seen from this figure that the cell utilizing SIC-MNC cathode mitigated the voltage fade which from the practical point of view will improve the overall specific energy of the Li-ion cell.
DC conductivity measurements of SIC-MNC and CP-MNC were determined using I–V curves plotted from galvanostatic excitation current data of pressed pellets as we reported previously for another material.9 It is known that conventional Li rich MNC materials suffer from low electronic conductivity.12 In contrast, we measured a higher electronic conductivity of 9 × 10−6 S cm−1 at 22 °C for the SIC-MNC than for the CP-MNC of 2 × 10−8 S cm−1, which partially accounts for its excellent rate capabilities. Complementing electronic conductivity EIS data showed that the SIC-MNC electrode has significantly lower impedance at open circuit voltage (OCP) than the CP-MNC electrode (see Fig. S2†). Normalized resistances were calculated to be 33 Ohm mg−1 for the SIC-MNC electrode while it was found to be 73 Ohm mg−1 for the CP-MNC electrode of identical loadings. As displayed in Fig. S2,† we also measured the EIS after 100 cycles for the cell utilizing SIC-MNC cathode, and the measured resistance of the cell (53 Ohm mg−1) is smaller than that of the fresh cell containing CP-MNC cathode. Plausible arguments to support this improvement are provided below from the FESEM data. These resistances are primarily a measure of charge transfer resistance (Rct), related to Li+ diffusion/migration through and/or at the surface of the electrode particles which is lower in the new material accounting for its higher rate capability.
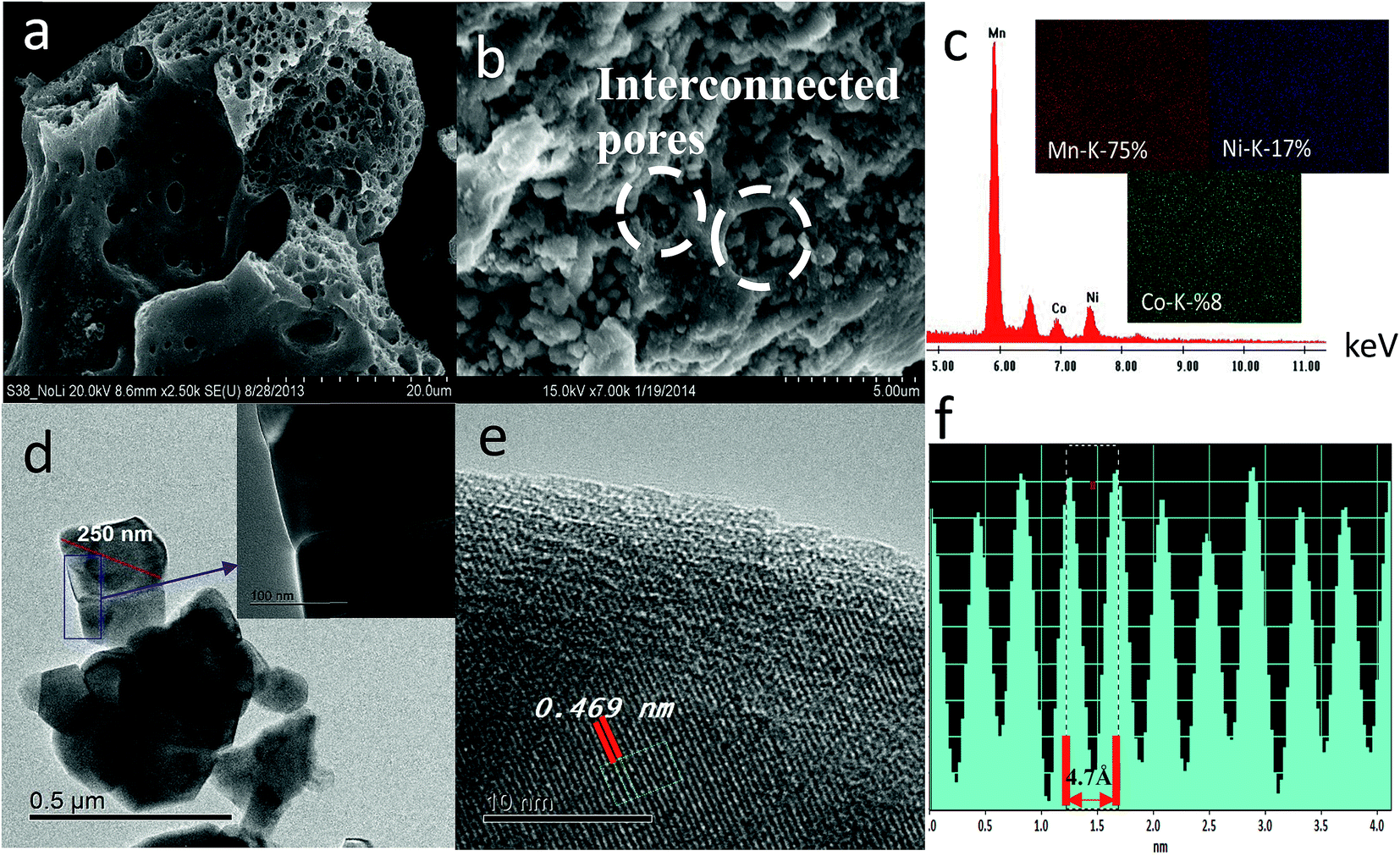 | ||
| Fig. 2 FESEM images of (a) MO sponge-like product and (b) SIC-MNC pristine material after final heat treatment. (c) EDS spectrum and mapping results of SIC-MNC. (d) Bright field images with magnified region where further investigation was performed. (e) HRTEM image revealing lattice fringes associated with (001) and/or (003) planes. (f) As generated cross-sectional profile of the HRTEM fringe presented in (e) to calculate the width between lattice fringes. | ||
Fig. S3† displays the FESEM images of cycled SIC-MNC cathodes at both high and low magnifications together with EDS analysis. Impedance of a cell and its capacity retention are greatly dependent upon the solid electrolyte interface (SEI) on the electrodes. The SEI thickness changes as cycling continue. Thick SEI layer, which is a consequence of continued reactions between electrode particles and electrolyte during cycling, is probably formed less in SIC-MNC due to interconnected particles which exposes less cathode surface to the electrolyte. This is seen from the FESEM data in Fig. S3† where after 100 cycles, interconnected particles are still seen. From EDS analysis, atomic ratios of each element was found to be little changed. In summary, lower cathode surface area of SIC-MNC is exposed to the electrolyte with an overall lower extent of reaction with the electrolyte resulting in excellent capacity retention; i.e. almost 100% capacity after 100 cycles.
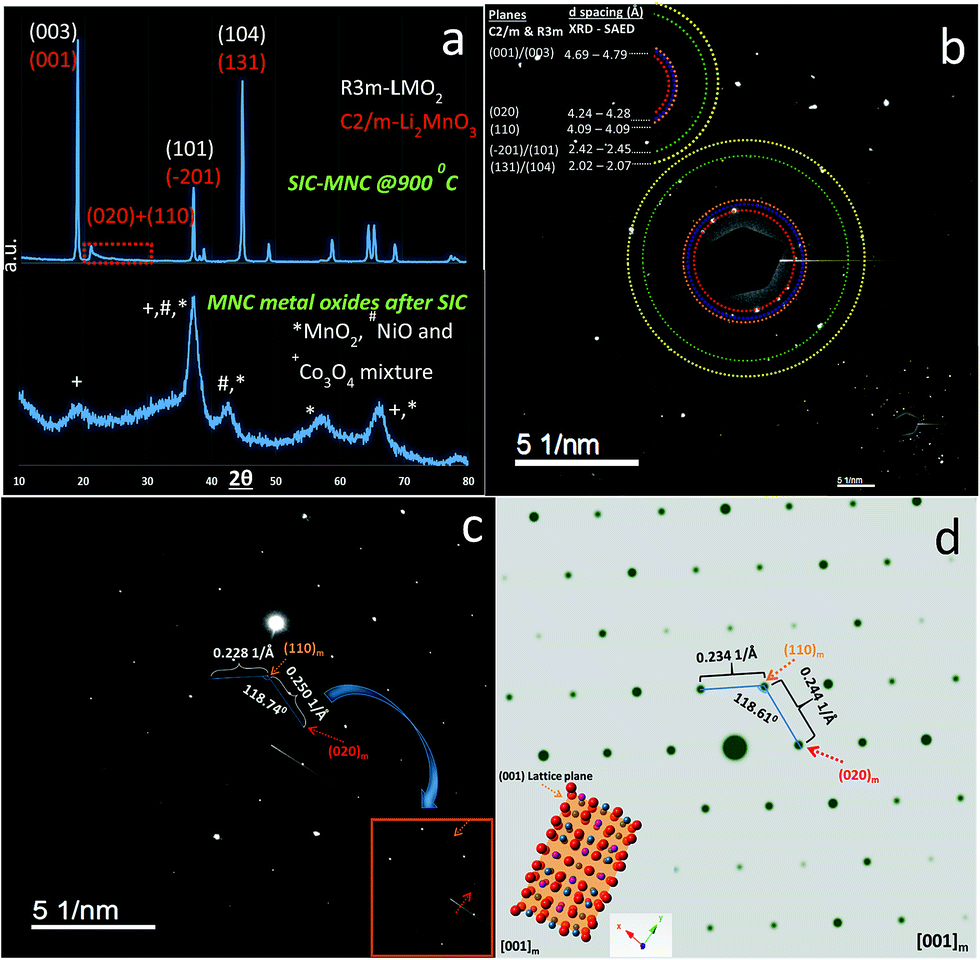 | ||
| Fig. 3 (a) XRD patterns associated with MO and final SIC-MNC at 900 °C are displayed including major planes based on two different space groups. (b) SAED ring-pattern unveiling polycrystalline nature of SIC-MNC along with calculated d-spacing values based on two different techniques. (c) SAED pattern of a single crystal of SIC-MNC yielding the existence of weak reflections corresponding to C2/m phase group. (d) Simulated SAED pattern of Li2MnO3 structure along with [001] zone directions. Structural demonstration in real-time was also generated from the pattern and shown at the bottom of the picture. | ||
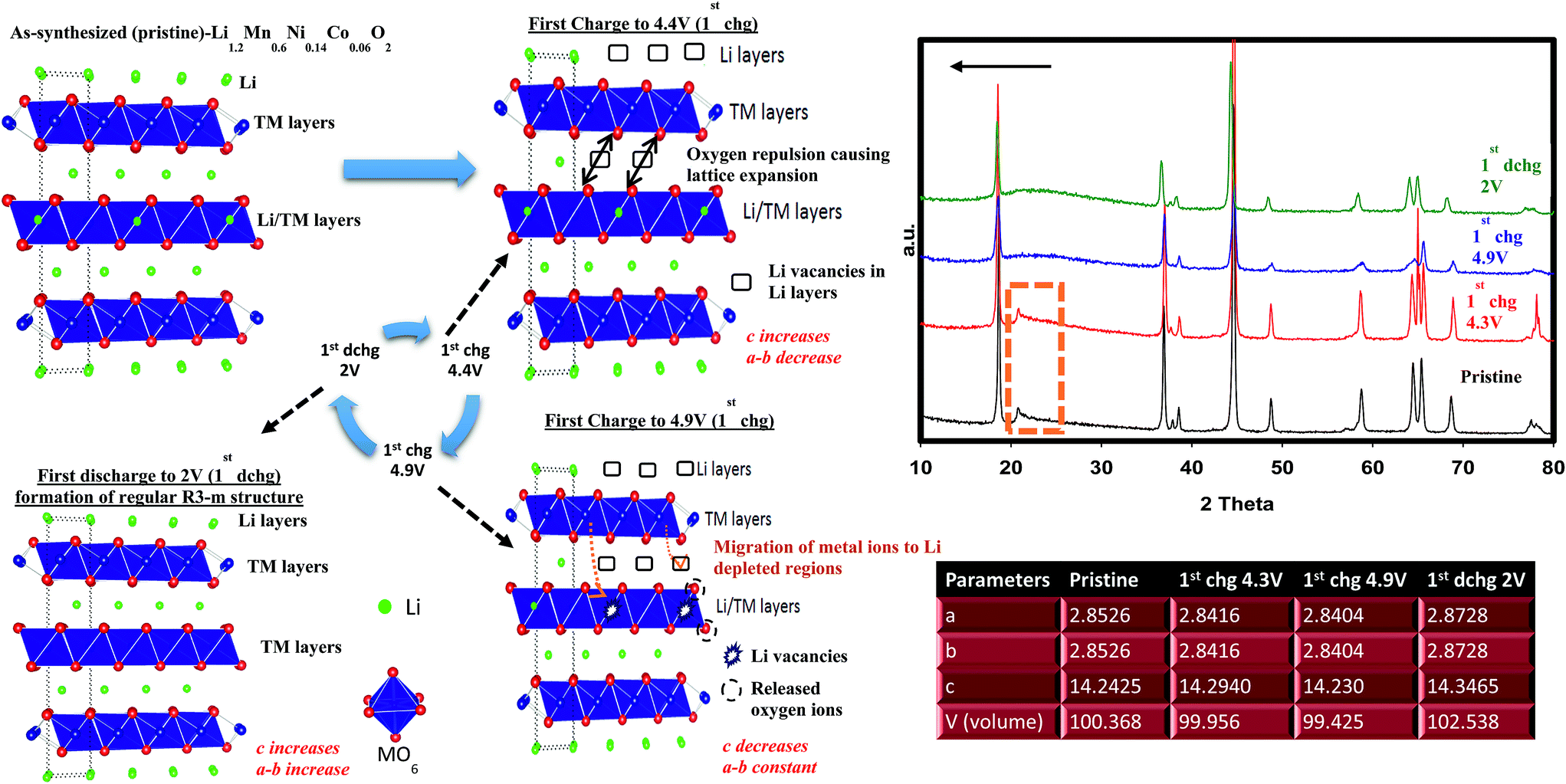 | ||
| Fig. 4 Ex situ XRD patterns of cycled and pristine cathode materials. Inset table shows unit cell parameters belonging to each region indicated in the text. Crystal lattice visualization was drawn to understand the reaction taking place in each voltage region. | ||
During charging process, we already determined that, at 4.9 V, the feature of superlattice ordering in Li2MnO3 vanished as revealed by XRD results. This can be further complemented by fast Fourier transformed (FT) patterns of an HRTEM image presented in Fig. S5a† where no trait of C2/m phases was observed in agreement with XRD results. Fig. S5a† also shows simulated SAED pattern of R3m space group along with crystal view in [010] zone axis which perfectly matches with observed SAED spots. Each HRTEM images shown in Fig. S5b–d† preserved lattice fringes with a significant expansion occurring during the first early charging process (4.3 V). This is due to the strong electrostatic repulsion among oxygen layers as discussed above. At 4.9 V, the lattice fringes showed contraction in the same way we observed in the XRD data. The sample discharged to 2 V exhibited lattice expansion again as supported by XRD. However, one should take into consideration that while XRD is a bulk technique, HRTEM images were taken from a single particle which is highly sensitive to atomic level investigations. That being said, HRTEM results for each particle may vary but here we studied at least 5–6 different particles and are reporting the average feature. Therefore, the slight discrepancy resulted from (001)/(003) plane or c parameter variations between two methods should be judged and interpreted accordingly.
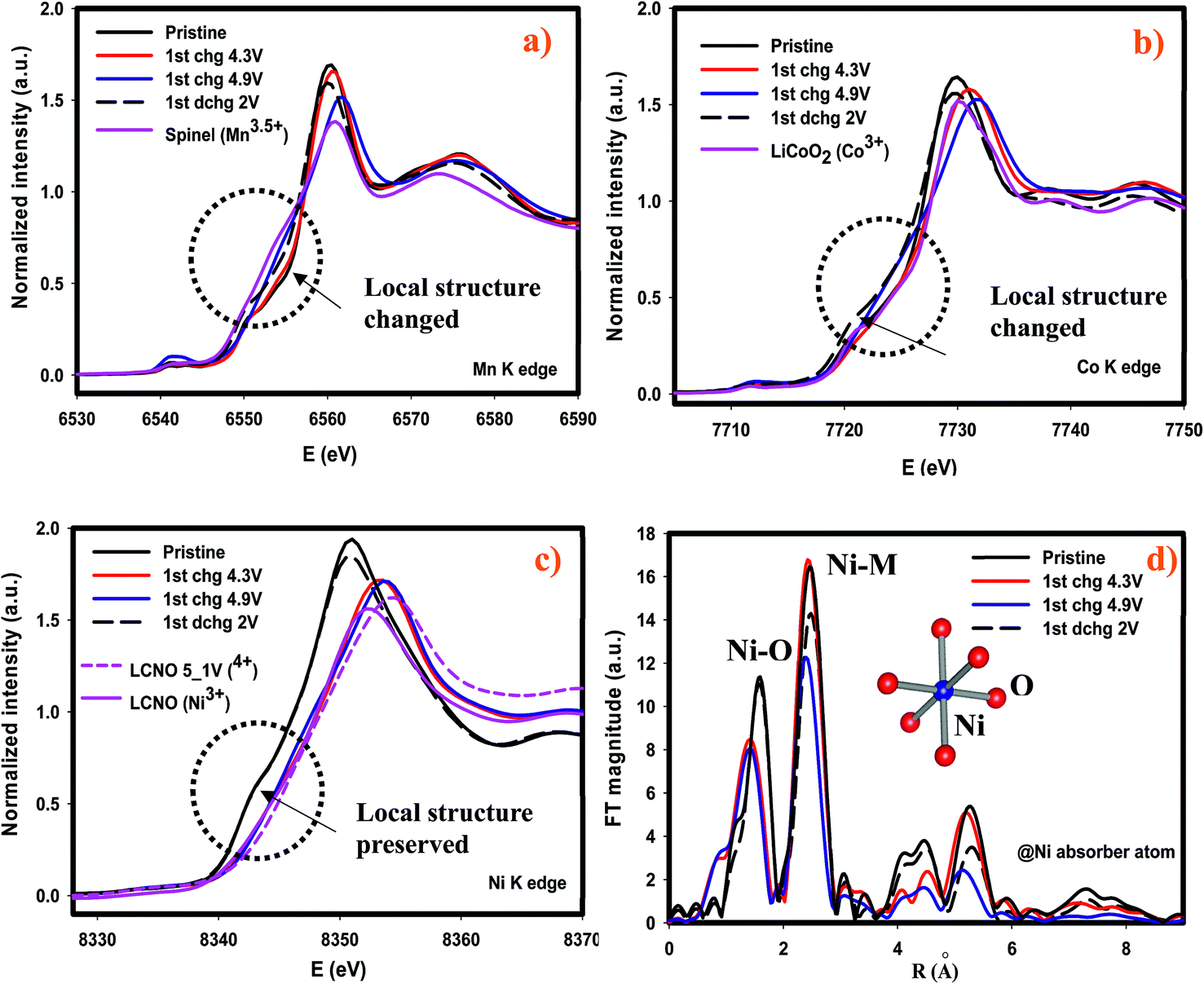 | ||
| Fig. 5 Selected ex situ XANES spectrums during the first cycles of (a) Mn-K edge (b) Co-K edge (c) Ni-K edge along with respected references and (d) magnitude of the Fourier Transformed (FT) Ni-K edge spectra along with metal oxygen framework. | ||
2. Extended cycling of SIC-MNC cathode
We have performed ex situ XRD, XAS and HRTEM studies for SIC-MNC cathodes in order to determine the origin of phase transformations after long-term cycling as mentioned earlier in this paper. Fig. 6 displays charge–discharge voltage versus capacity profiles and the corresponding dQ/dV plots of the cell utilizing SIC-MNC cathode at 1C discharge rate at room temperature. From Fig. 6a and b, one can easily observe that the major voltage decay takes place during early cycles going from the 1st to 50th cycle. After that the discharge profiles are stabilized as displayed in Fig. 6b. This is further seen from their differential capacity plots given in Fig. 6c and d. In Fig. 6c, the reduction peak corresponding to Ni and Co metals is shifting towards the Mn peak which we believe is the main cause of the voltage hysteresis seen in Fig. 6a. After these structural rearrangements the reduction peaks of all metals coalesce into a single dQ/dV peak as can be seen in Fig. 6d. This behavior yields the steady discharge voltage profiles versus constant capacity values displayed in Fig. 6b. To understand whether or not these metal migrations and voltage decay are reflected in the XRD patterns, we measured the XRD patterns of SIC-MNC cathode after 100 cycles at the 1C discharge rate. Fig. 7a shows the ex situ XRD patterns of three samples, the pristine SIC-MNC, the electrode after the first discharge, and after 100 cycles discharged to 2 V. Interestingly, we found no strong evidence for any phase change as evidenced by the absence of the spinel phase in these samples. Several points should be noted; firstly the doublet peak located at around 65° 2θ, a direct indication of perfect layered structure, was preserved after 100 cycles. Nevertheless, the ratio of the intensity of the two doublet peaks, which was approximately 1 before cycling and after the first discharge at 2 V, started to change which may be a sign of small phase transition. In addition, the volume of the crystal lattice remained almost the same as after the first discharge suggesting a robust structure was attained. This can be further explained by considering the void spaces in the crystal grains mentioned earlier in this paper which might play a key role against volume changes within the crystal framework. In an attempt to provide further support to the XRD results, the bulk technique, HRTEM was performed. Fig. 7b displays the HRTEM data after 100 cycles at the 1C rate and its corresponding FT patterns are embedded in the figure. Supported by the simulation of R3m phase, located on the right side of HRTEM figure, FT pattern of the cycled SIC-MNC revealed that the crystal maintained its layered feature. It should be noted that in the HRTEM experiments, we have run several other segments of the cathode but none of the acquired data revealed spinel-like feature.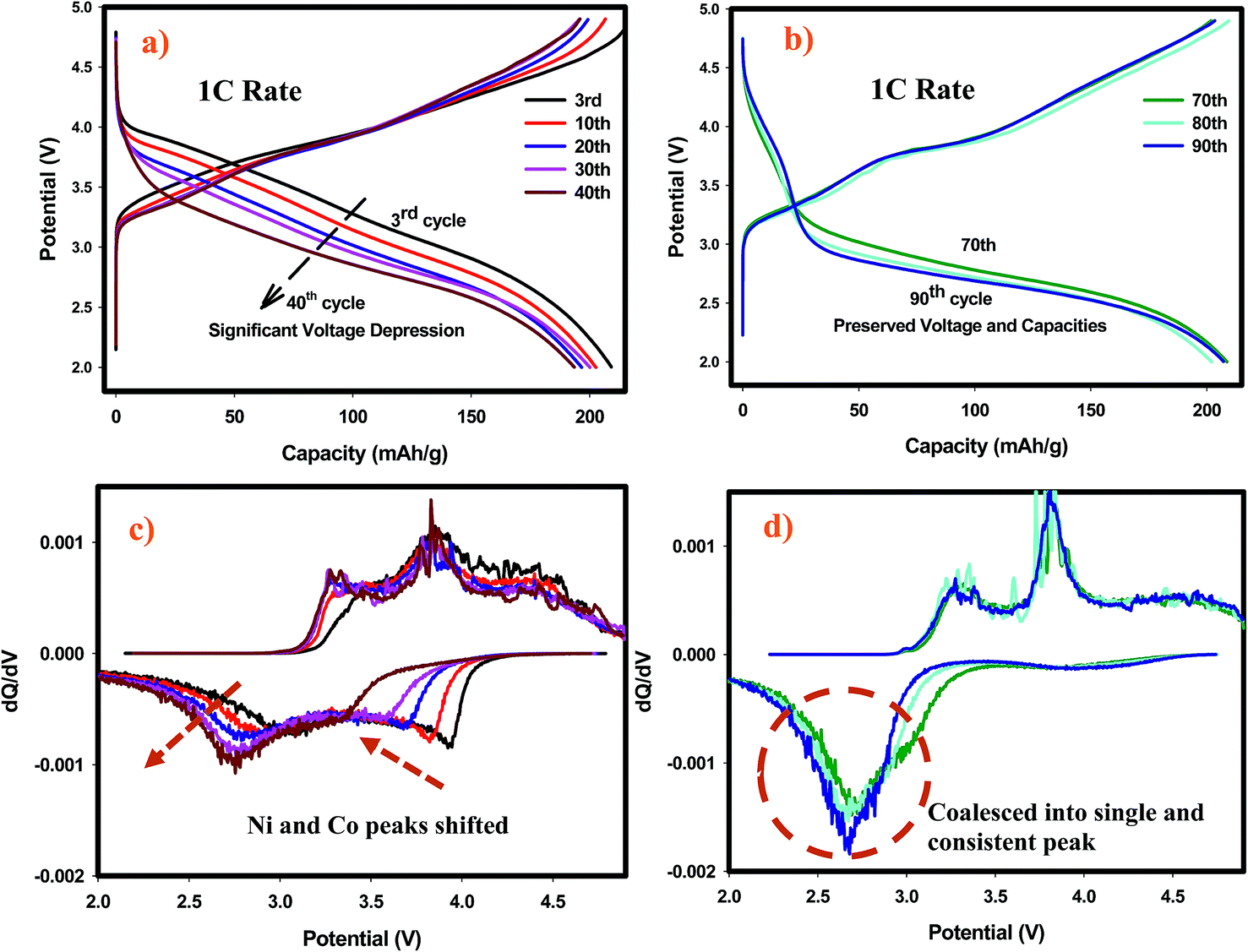 | ||
| Fig. 6 (a) Charge–discharge profiles of the first 40 cycles at 1C rate. (b) Charge–discharge profiles for rest of the cycles at 1C rate. (c) Differential capacity plots of the data presented in (a). (d) Differential capacity plots of the data presented in (b). All tests were performed under RT, between 2–4.9 V voltage range. | ||
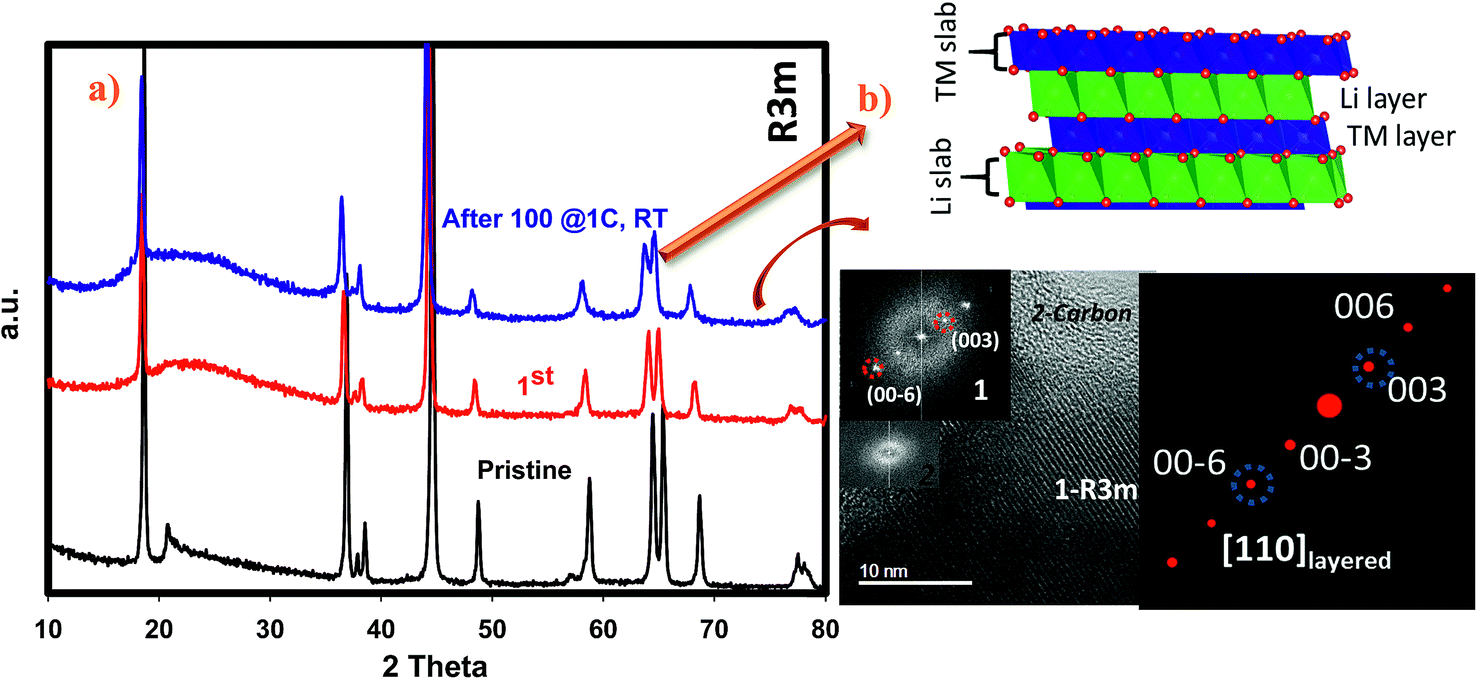 | ||
| Fig. 7 (a) Ex situ XRD patterns of pristine, after the first cycle and 100 cycles cutoff at 2 V. (b) HRTEM figure of SIC-MNC after 100 cycles and corresponding FT pattern along with the simulated R3m phase in approximately [110] zone axis. | ||
To understand rate and high temperature cycling effects on crystal structure and voltage fade phenomena, we have performed ex situ XRD and HRTEM on two samples, (i) after 130 cycles at 2C discharge rate at ambient temperature, and (ii) after 24 cycles at C/4 discharge rate at 50 °C. Contrasting to what was observed after 100 cycles at 1C discharge rate, we identified structural transformation as evidenced by XRD and HRTEM data. Fig. 8a and b display charge–discharge profiles of the SIC-MNC containing cell at the 2C discharge rate which show similar behavior as in Fig. 6a and b where voltage depression occurred in the early 50 cycles. This similarity suggests that voltage hysteresis commences irrespective of structural transformation. In fact, the high temperature cycling test results exhibited very low voltage fade after 20 cycles again advocating that voltage fade does not directly result from and/or contribute to the phase transition. The cycling behavior and charge–discharge profile of high temperature test results are plotted in Fig. S6.† From Fig. 8c one can easily surmise that both materials, one run at high temperature and the other at high rates, showed similar XRD patterns that can be indexed to a spinel phase. The doublet peak located at around 65° 2θ was converted into a singular peak ascribed to the spinel phase. This was further confirmed and supported by HRTEM figures, shown in Fig. 8d, where FT patterns of the SIC-MNC cathode after 130 cycles at the 2C rate revealed spinel character similar to simulated patterns based on LiMn2O4 material. Although this spinel feature was not observed throughout the sample, we can conclude that partial phase conversion occurs at harsh conditions i.e. high temperature and high rate. The crystal lattice changes are visualized in Fig. 8d.
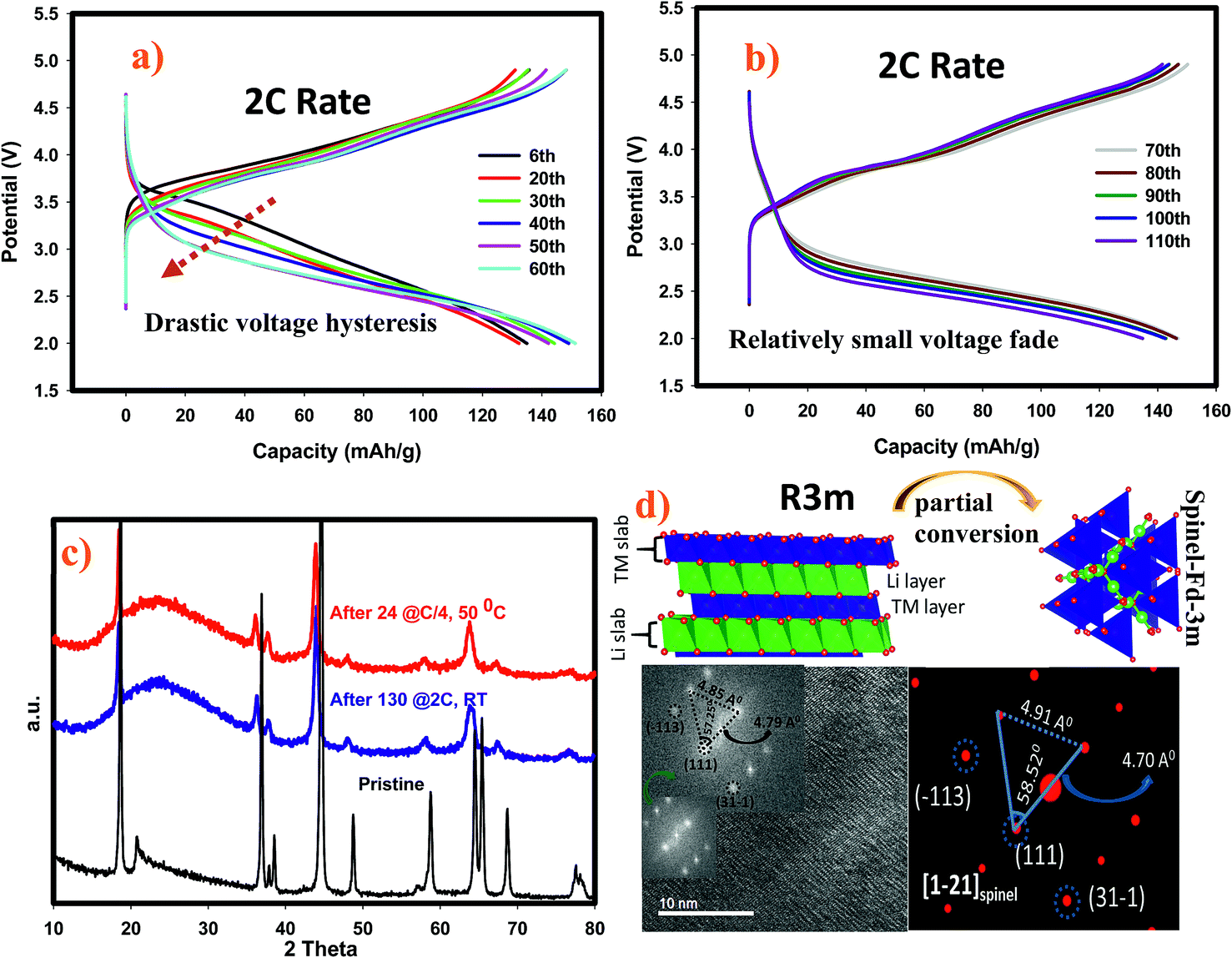 | ||
Fig. 8 (a) Charge–discharge profiles of the first 60 cycles at 2C rate. (b) Charge–discharge profiles for rest of the cycles at 2C rate. (c) Ex situ XRD patterns of pristine, after 100 cycles at 2C rate under ambient temperature and after 24 cycles at 50 °C with C/4 rate. (d) HRTEM figure of SIC-MNC after 130 cycles at 2C rate under ambient temperature and corresponding FT pattern along with the simulated Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m spinel phase in [31−1] zone axis. Crystal lattice visualization was drawn in order for readers to understand conversion phenomena. m spinel phase in [31−1] zone axis. Crystal lattice visualization was drawn in order for readers to understand conversion phenomena. | ||
We further investigated the cycled cathodes after 41 cycles through XAS, specifically the XANES region, in order to understand the redox behavior of each transition metal. Fig. 9 shows the XANES profiles of each transition metal after the first discharge and after the 41 cycles discharged to 2 V. In general, none of the XANES shapes were affected or altered implying that the coordination environment was preserved compared to those after the 1st discharge following the activation of Li2MnO3. Some changes occurred as cycling continued. In contrast to Ni and Co-K edges, Mn-K edge energy value in the white-line region was shifted to lower energy value indicating that Mn reduced more and more as cycling continues. This dynamic redox behavior of Mn might appear beneficial in terms of capacity, nonetheless this leads to voltage depression at the early cycles as demonstrated in Fig. 6a and 7a. Overall, it is found that the most robust transition metal during cycling appears to be Ni and to some extent Co in terms of redox behavior and chemical environment, while Mn atoms are more susceptible to changes during cycling as its chemical environment was partially destroyed after the first cycle and its redox behavior changes as cycling continues.
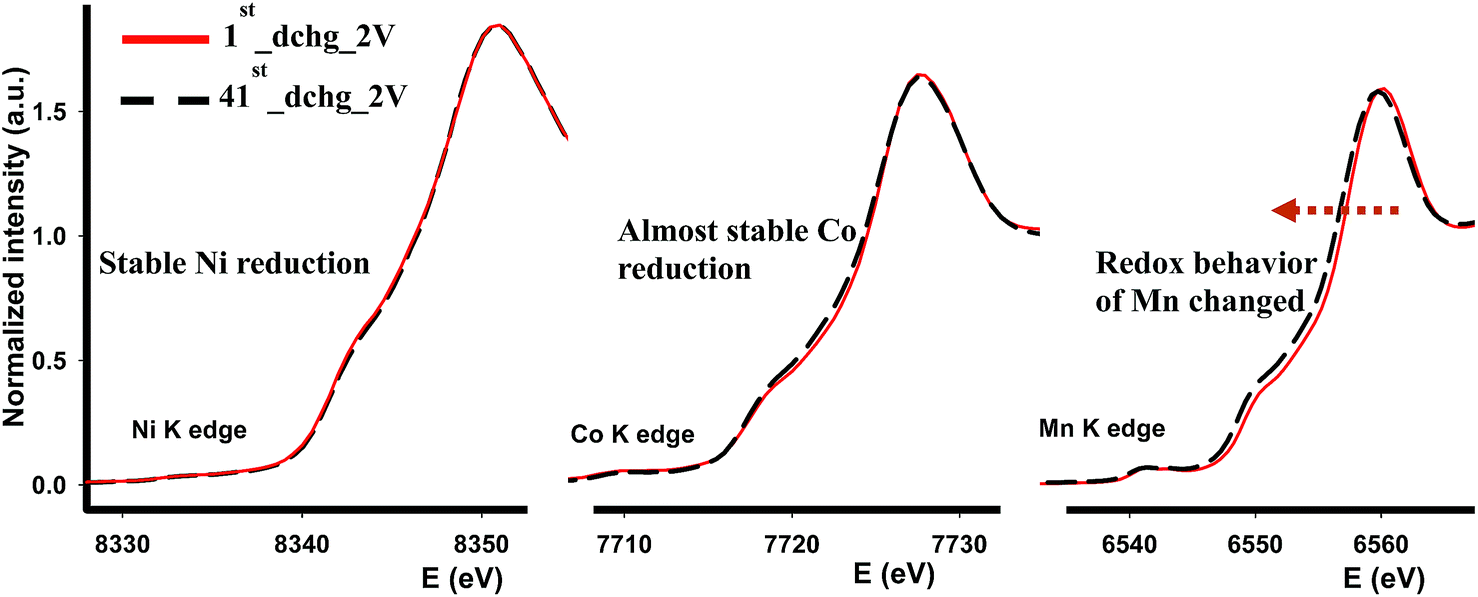 | ||
| Fig. 9 XANES profiles of each transition metal after the first cycle and 41 cycles at room temperature. | ||
Conclusion
The Li rich layered composite metal oxide of the formula 0.5Li2MnO3·0.5LiMn0.5Ni0.35Co0.15O2, or alternatively formulated as Li1.2Mn0.6Ni0.14Co0.06O2, was synthesized for the first time by the self-ignition combustion method as a high rate cathode material with a capacity as high as 290 mA h g−1. Its excellent rate capabilities with little or no capacity fade with cycling makes this material the best cathode candidate for 400 W h kg−1 Li-ion batteries suitable for powering electric vehicles and portable consumer products. Its open particle morphology combined with higher electronic conductivity provide favorable material attributes for use as a high rate cathode material with practically no capacity fade during long-term cycling. The highly desirable Li cell electrochemistry has been reinforced by structural information obtained from FESEM, XRD, HRTEM and XAS. The extensively cycled electrode material was investigated by means of ex situ FESEM, XRD, HRTEM, SAED and XAS. The voltage fade-phase transformation relationships elucidated indicated that they are not necessarily linked to each other. The XRD and HRTEM data suggested that partial structural transformations took place after large number (e.g. 100) of cycles at high rates whereas such structural changes occurred even at low rates after about 24 cycles at high temperature (50 °C).Acknowledgements
We are grateful to Erich Farqhuar and Syed Khalid for the assistance with XAS experiments at the hatches of X3A and X18A at Brookhaven National Lab. This publication was made possible by the Center for Synchrotron Biosciences grant, P30-EB-009998, from the National Institute of Biomedical Imaging and Bioengineering (NIBIB). We also thank Qingying Jia for the logistic and experimental support during XAS experiments at the light source. Partial financial support of this work was provided by US Army CERDEC through contract no. GTS-S-14-164.References
- Z. Lu, D. D. MacNeil and J. R. Dahn, Electrochem. Solid-State Lett., 2001, 4, A191–A194 CrossRef CAS PubMed.
- J.-S. Kim, C. S. Johnson, J. T. Vaughey, M. M. Thackeray, S. A. Hackney, W. Yoon and C. P. Grey, Chem. Mater., 2004, 16, 1996–2006 CrossRef CAS.
- F. Amalraj, D. Kovacheva, M. Talianker, L. Zeiri, J. Grinblat, N. Leifer, G. Goobes, B. Markovsky and D. Aurbach, J. Electrochem. Soc., 2010, 157, A1121–A1130 CrossRef CAS PubMed.
- K. A. Jarvis, Z. Deng, L. F. Allard, A. Manthiram and P. J. Ferreira, J. Mater. Chem., 2012, 22, 11550–11555 RSC.
- J. Liu and A. Manthiram, J. Mater. Chem., 2010, 20, 3961–3967 RSC.
- K. A. Jarvis, Z. Deng, L. F. Allard, A. Manthiram and P. J. Ferreira, Chem. Mater., 2011, 23, 3614–3621 CrossRef CAS.
- M. M. Thackeray, C. S. Johnson, J. T. Vaughey, N. Li and S. A. Hackney, J. Mater. Chem., 2005, 15, 2257–2267 RSC.
- D. Im, J. Kim, J. Yoon, K.-S. Park, Y.-G. Ryu, S. S. Lee, D. J. Lee and S.-G. Doo, Meet. Abstr., 2010, MA2010-01, 630 Search PubMed.
- M. N. Ates, Q. Jia, A. Shah, A. Busnaina, S. Mukerjee and K. M. Abraham, J. Electrochem. Soc., 2014, 161, A290–A301 CrossRef CAS PubMed.
- J. R. Croy, D. Kim, M. Balasubramanian, K. Gallagher, S.-H. Kang and M. M. Thackeray, J. Electrochem. Soc., 2012, 159, A781–A790 CrossRef CAS PubMed.
- D. Mohanty, A. S. Sefat, S. Kalnaus, J. Li, R. A. Meisner, E. A. Payzant, D. P. Abraham, D. L. Wood and C. Daniel, J. Mater. Chem. A, 2013, 1, 6249–6261 CAS.
- S. K. Martha, J. Nanda, Y. Kim, R. R. Unocic, S. Pannala and N. J. Dudney, J. Mater. Chem. A, 2013, 1, 5587–5595 CAS.
- Y. Li, M. Bettge, B. Polzin, Y. Zhu, M. Balasubramanian and D. P. Abraham, J. Electrochem. Soc., 2013, 160, A3006–A3019 CrossRef CAS PubMed.
- Y. Zhang, H.-C. Shin, J. Dong and M. Liu, Solid State Ionics, 2004, 171, 25–31 CrossRef CAS PubMed.
- D. I. Choi, H. Lee, D. J. Lee, K.-W. Nam, J.-S. Kim, R. A. Huggins, J.-K. Park and J. W. Choi, J. Mater. Chem. A, 2013, 1, 5320–5325 CAS.
- M. N. Ates, S. Mukerjee and K. M. Abraham, J. Electrochem. Soc., 2014, 161, A355–A363 CrossRef CAS PubMed.
- C. S. Johnson, N. Li, C. Lefief, J. T. Vaughey and M. M. Thackeray, Chem. Mater., 2008, 20, 6095–6106 CrossRef CAS.
- X. J. Zhu, H. H. Chen, H. Zhan, D. L. Yang and Y. H. Zhou, J. Mater. Sci., 2005, 40, 2995–2997 CrossRef CAS.
- M. Balasubramanian, X. Sun, X. Q. Yang and J. McBreen, J. Electrochem. Soc., 2000, 147, 2903–2909 CrossRef CAS PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- X. Yu, Y. Lyu, L. Gu, H. Wu, S.-M. Bak, Y. Zhou, K. Amine, S. N. Ehrlich, H. Li, K.-W. Nam and X.-Q. Yang, Adv. Energy Mater., 2014, 4, 1–11, DOI:10.1002/aenm.201300950.
- F. Cheng, Y. Xin, J. Chen, L. Lu, X. Zhang and H. Zhou, J. Mater. Chem. A, 2013, 1, 5301–5308 CAS.
- M. Gu, I. Belharouak, J. Zheng, H. Wu, J. Xiao, A. Genc, K. Amine, S. Thevuthasan, D. R. Baer, J.-G. Zhang, N. D. Browning, J. Liu and C. Wang, ACS Nano, 2012, 7, 760–767 CrossRef PubMed.
- W. He, D. Yuan, J. Qian, X. Ai, H. Yang and Y. Cao, J. Mater. Chem. A, 2013, 1, 11397–11403 CAS.
- J. Liu, L. Chen, M. Hou, F. Wang, R. Che and Y. Xia, J. Mater. Chem., 2012, 22, 25380–25387 RSC.
- S. Guo, H. Yu, P. Liu, X. Liu, D. Li, M. Chen, M. Ishida and H. Zhou, J. Mater. Chem. A, 2014, 2, 4422–4428 CAS.
- C.-H. Shen, Q. Wang, F. Fu, L. Huang, Z. Lin, S.-Y. Shen, H. Su, X.-M. Zheng, B.-B. Xu, J.-T. Li and S.-G. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 5516–5524 CAS.
- H. L. Bohang Song, Z. Liu, P. Xiao, M. O. Lai and L. Lu, Sci. Rep., 2013, 3, 1–12 Search PubMed.
- D. Mohanty, S. Kalnaus, R. A. Meisner, A. S. Safat, J. Li, E. A. Payzant, K. Rhodes, I. I. I. D. L. Wood and C. Daniel, RSC Adv., 2013, 3, 7479–7485 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Cycling data of CP-MNC, synthetic scheme of SIC-MNC, impedance data, FESEM images of cycled cathodes, XRD pattern-FESEM image evolution during synthesis, HRTEM images of the cycled cathodes, cycling data at high temperature for SIC-MNC. See DOI: 10.1039/c4ra17235c |
| This journal is © The Royal Society of Chemistry 2015 |