
Pt-based nanoparticles on non-covalent functionalized carbon nanotubes as effective electrocatalysts for proton exchange membrane fuel cells
Weiyong Yuan
a,
Shanfu Lu
b,
Yan Xiang
b and
San Ping Jiang
*a
aFuels and Energy Technology Institute & Department of Chemical Engineering, Curtin University, Perth, WA 6102, Australia. E-mail: s.jiang@curtin.edu.au
bKey Laboratory of Bio-Inspired Smart Interfacial Science and Technology of Ministry of Education, School of Chemistry and Environment, Beihang University, Beijing 100191, PR China
First published on 9th September 2014
Abstract
Due to their unique electronic and mechanical properties, carbon nanotubes (CNTs) have been attracting much attention as favourite catalyst supports for energy conversion and storage applications. However, CNTs require molecular engineering such as solubilization and surface modification or functionalization to tailor their surface properties for the catalyst support applications. Among various functionalization methods, non-covalent functionalization is preferred, because it enables the attachment of molecules, solvents or polyelectrolytes through π–π, CH–π or hydrophobic interactions, forming effective active sites for the uniform assembly and dispersion of Pt-based precursor and/or nanoparticles (NPs) and at the same time preserving the intrinsic electronic and structural integrity of CNTs. Non-covalent functionalization is also effective to incorporate a multi-component electrocatalyst system with significantly enhanced synergistic effects. Here, the progress in the synthesis and development of highly dispersed Pt, and Pt-based NPs such as PtRu, PtSn and PtPd on non-covalent functionalized CNTs will be presented. The significant effect of interactions between CNTs, Pt-based NPs and functionalization agents on the electrocatalytic activity of Pt-based NPs on non-covalent functionalized CNTs is discussed.
 Weiyong Yuan | Dr Weiyong Yuan received his PhD from Nanyang Technological University (NTU), Singapore in 2011. From 2011 to 2014, Dr Yuan did his postdoctoral research in NTU and Curtin University, Australia (working with Prof. Jiang), and was a visiting scholar in University of Kansas, US. His present position is an Associate Professor in Southwest University, China and his main research interest includes self-assembled nanomaterials and their applications in fuel cell electrocatalysts, water splitting, and biomedical devices. |
 Shanfu Lu | Dr Shanfu Lu received his PhD in Physical Chemistry (Electrochemistry) from Wuhan University, in 2008. He was a postdoctoral researcher at Nanyang Technological University, Singapore, working with Prof. S. P. Jiang (2008–2009). Then, he joined Beihang University. He is currently an Associate Professor in Electrochemistry and Energy Materials at School of Chemistry and Environment, Beihang University, China. His current research interests are focused on nanomaterials and polymer electrolytes for energy storage and conversion. |
 Yan Xiang | Dr Yan Xiang is a Professor and Dean of Basic Research Institute at Beihang University, Director of Beijing Key Laboratory of Bioinspired Energy Materials and Devices, and a Fellow of the Royal Society of Chemistry. Dr Xiang obtained her PhD from Sichuan University in 2001 and spent one year as post-doctor at Carnegie Mellon University. Her main current research interests involve key energy materials development and their applications. |
 San Ping Jiang | Dr San Ping Jiang obtained his BEng from South China University of Technology and PhD from The City University, London. He is a Professor at the Department of Chemical Engineering and Deputy Director of Fuels and Energy Technology Institute, Curtin University, Australia and Adjunct Professor of the University of Sunshine Coast, Australia. Before joining Curtin University in 2010, Dr Jiang worked at Essex University in UK, CSIRO Materials Science and Manufacturing Division, Ceramic Fuel Cells Ltd in Australia and Nanyang Technological University in Singapore. His research interests encompass fuel cells, water electrolysis, supercapacitors, electrocatalysis and nano-structured functional materials. |
1. Introduction
The proton exchange membrane fuel cell (PEMFC) is an energy conversion device to electrochemically convert the chemical energy of fuels such as hydrogen, methanol, ethanol and formic acid to electricity with high efficiency and low greenhouse gas emission.1–3 PEMFC has been identified as a clean energy source and has the potential to reduce energy dependence on fossil fuel.1,4,5 The development of a highly active, low cost and durable electrocatalyst is one of the critical challenges for the commercial viability of PEMFC technologies. Pt and Pt-based alloy is the most effective and commonly used electrocatalyst for reactions such as oxidation of hydrogen, methanol and formic acid and oxygen reduction reactions in PEMFCs.6–9 However, a major hindrance to the commercialization of the PEMFC technologies is the high cost associated with the Pt-based electrocatalysts due to scarce resources. Thus, much effort has been spent on enhancing the utilization efficiency of Pt to reduce its loading and to improve the fuel cell performance.6,7,10 Currently, a number of catalyst systems with reduced Pt-based catalyst loading and enhanced electrocatalytic activity have been proposed including Pt alloys,11–16 Pt nanoparticles (NPs),17,18 Pt nanotubes and nanowires19,20 core–shell Pt NPs21–23 and Pt-free electrocatalysts.24–27The size of nano-structured Pt-based electrocatalysts plays a key role in their electrocatalytic properties.9 In most cases, NPs of a small size are favourable to increasing the catalytic activity of Pt.28–30 However, since Pt NPs are easily aggregated in solution or during the fuel cell operations, unique surface modifications and/or a support material with good conductivity, high surface area, and strong interaction with the NPs are greatly desired.31–33 The most common approach is to use high surface area carbon-based supports to uniformly distribute and deposit nano-structured Pt catalysts to increase activity and stability. The ideal support materials for the fuel cell catalyst system should have good electrical conductivity, large surface area for electrochemical reaction, adequate hydrophilic properties for fast water and reactant transportation to and from the active sites, the strong interaction with metal catalyst precursor or NPs. The most common supports for Pt-based electrocatalysts are high surface area carbon, carbon nanotubes (CNTs), graphene and nano-structured carbon.34–36 The ordered mesoporous carbons (OMCs) also show a promising potential as catalyst supports in fuel cells due to their very high specific surface areas (up to 2000 m2 g−1), uniform pore size in the range of 2–10 nm and high thermal, chemical and mechanical stability.37,38 The structural, chemical and thermal stability of the carbon-based support is critical for the activity and stability of Pt-based electrocatalysts as carbon corrosion is considered to be one of the most important durability issues for fuel cells.39,40 Among them, high surface area carbon or carbon black (CB), especially Vulcan XC-72 is the most widely used support for Pt-based electrocatalysts of fuel cells. However, CB suffers from thermochemical instability and low electrochemical corrosion resistance under fuel cell operating conditions.41–44 As carbon corrodes, Pt NPs will detach from the CB supports and aggregate into large particles, resulting in the loss of electrocatalytic surface area and subsequently reducing the performance of fuel cells.
CNTs are promising supports of Pt-based NPs mainly because of their excellent conductivity, ultrahigh surface area and good chemical, thermal and structural stability.45,46 Graphitized CNTs are more intrinsically resistance to corrosion.42 However, due to their high aspect ratios and strong van der Waals interactions, pristine CNTs are difficult to disperse in water and the surface is very inert with few binding sites for homogeneously attaching metallic NPs and/or their precursors.47–49 Therefore, it is necessary to develop surface treatment methods to exfoliate bundled CNTs and modify the surface of CNTs to enhance the dispersion of Pt-based NPs.46,50–52 Covalent modifications based on surface oxidation treatment are perhaps the most widely used methods.18,53,54 Various oxidants can be used including HNO3, H2SO4, H2O2 and HF. The strong acid oxidation treatment introduces hydrophilic groups such as hydroxyl (–OH), carboxyl (–COOH) and carbonyl (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) groups to the CNT surface by the modification of the aromatic conjugate ring system of CNTs.55,56 The oxidized CNTs can be dispersed in water and the surface functionalized groups have strong attraction forces as the active sites for the synthesis or deposition of metallic NPs such as Pt18,57 and Pd58 in the presence of reducing agents such as NaBH4 and ethylene glycol (EG). Pt NP catalysts on covalently functionalized CNTs exhibit enhanced electrocatalytic activity and stability as compared to Pt NPs supported on high surface area carbon, Pt/C.18,55,59 Unfortunately, the strong acid treatment inevitably generates a number of defects, alters the electronic structure and therefore deteriorates the structural stability and corrosion resistance of CNTs.53,54 Moreover, these approaches introduce Pt-based NPs mostly at defects and ends of CNTs, resulting in non-uniform distribution and agglomeration.18
O) groups to the CNT surface by the modification of the aromatic conjugate ring system of CNTs.55,56 The oxidized CNTs can be dispersed in water and the surface functionalized groups have strong attraction forces as the active sites for the synthesis or deposition of metallic NPs such as Pt18,57 and Pd58 in the presence of reducing agents such as NaBH4 and ethylene glycol (EG). Pt NP catalysts on covalently functionalized CNTs exhibit enhanced electrocatalytic activity and stability as compared to Pt NPs supported on high surface area carbon, Pt/C.18,55,59 Unfortunately, the strong acid treatment inevitably generates a number of defects, alters the electronic structure and therefore deteriorates the structural stability and corrosion resistance of CNTs.53,54 Moreover, these approaches introduce Pt-based NPs mostly at defects and ends of CNTs, resulting in non-uniform distribution and agglomeration.18
In contrast, non-covalent or physical modification approach is a much more mild process, which preserves the intrinsic sp2 hybrid state and intrinsic properties of CNTs.60 There are large number of molecules including surfactants,61 aromatic compounds,16 polymers or polyelectrolytes,61–66 which can be used as non-covalent functionalization agents. The functional groups or charged sites of the functionalized molecules serve as the effective active sites for the assembly and anchoring metal precursors and NPs.54,67 For example, Li et al. assembled CdS and Pt NPs on 1-aminopyrene (1-AP)-functionalized CNTs.68 Ou et al. deposited gold NPs on CNTs by use of 1-pyrenemethylamine to functionalize the CNTs.69 We investigated extensively the use of a variety of polyelectrolyte and solvents such as poly(diallyldimethylammonium chloride) (PDDA),67,70–72 polyethylenimine (PEI),73 1-AP,16 tetrahydrofuran (THF),49 and chitosan (CS)74 to functionalize CNTs as Pt-based catalyst supports. In this paper, we will first review the current progress concerning the effect of Pt NP size on the electrocatalytic performance, followed by the main focus of this review on the progress in the non-covalent functionalization of CNTs, and their use as a support of Pt and Pt-based alloy NPs for fuel cell applications. For functionalization of CNTs and carbon-based materials by chemical, electrochemical, polyol-assisted methods, readers can refer to specific review articles.51,75–77
2. Electrocatalytic activity of Pt NPs
One of the main driving forces for the development of non-covalent functionalization methods is to create uniformly distributed binding/active sites for the deposition of finely dispersed Pt-based NPs on CNTs as the catalytic property of Pt NPs is closely related to the particle size. Thus before the discussion on the functionalization methods and the effect of interaction between the functionalization agents, Pt NPs and CNTs, it is helpful to briefly review the effect of Pt NP size and surface treatment on the activity and stability of Pt-based NPs.2.1. Effect of size of Pt NPs
For large Pt NPs, bulk of the material will not be available for the electrochemical reaction as the electrocatalytic reaction only occurs on the surface. With the reduction in size, more Pt atoms are exposed on the surface, leading to an increased active surface area and utilization efficiency of the Pt. However, studies have revealed that the intrinsic catalytic properties of the material change with particle size due to a combination of localized coordination effects, changes in surface site distribution and surface relaxation.78–80The effect of Pt NP size on the catalytic activity for the methanol and formic acid oxidation reaction was studied on carbon nanofiber (CNF) supported Pt NPs (see Fig. 1).28 CNF supported Pt NPs with different sizes were synthesized at different pH using ethylene glycol (EG) as reducing agent. The Pt NPs dispersed on platelet CNF are cuboctahedral particles, with the size spanning from 1.1 to 5.6 nm. The mass specific activities toward methanol and formic acid oxidation increase with the decrease of size from 5.6 to 1.8 nm, most likely due to an increase in electrochemically active surface area per unit mass. However, the activities of supported Pt NPs decrease when the particle size is below 1.8 nm. It has been found that after the electrochemical testing, the 1.5 nm and 1.8 nm Pt NPs seriously aggregated into large particles with very different morphology and shapes (Fig. 1(II)). The reduction of the electrocatalytic activity of Pt NPs with size below 1.8 nm is most likely due to the aggregation of Pt NPs, leading to the significantly reduced surface area.28
 | ||
| Fig. 1 (I) TEM images of Pt NPs dispersed on carbon nanofiber (CNF): (a) 5.6 nm, (b) 3.9 nm, (c) 1.8 nm, and (d) 1.1 nm. (II) TEM images of Pt NPs on CNF before and after electrochemical treatment for 30 min in H2SO4 solution: 1.5 nm Pt NPs (a) before and (b) after the electrochemical treatment and 1.8 nm Pt NPs (c) before and (d) after the electrochemical treatment (after ref. 28 reproduced with permission). | ||
The change in the particle size can also affect the crystallinity of the particles. Leontyev et al. studied the size effect on the oxygen reduction reaction on carbon-supported Pt NPs within the particle size range of 1.8–5.4 nm and Pt loading of 20% and observed a change of the particle shape and increasing fraction of the (111) crystal facets with the decrease of particle size.81 The mass specific activity of Pt catalysts for the oxygen reduction reaction first increased and then decreased again with the decrease in the particle size, with the maximum activity achieved on Pt NPs with a diameter 2.7 nm. In the case of extremely small particles, the shape or crystalline facets of Pt NPs might be dominant over other factors in terms of the catalytic activity. The effect of Pt particle size on the activities for the oxygen reduction reaction in HClO4 solutions was also studied by Shao et al.82 The surface area specific activity increased by 4-fold when the particles changed from 1.3 to 2.2 nm and increased slowly with the further increase in the particle size, while the mass specific activity increased by 2-fold from 1.3 to 2.2 nm and decreased again with the further increase in the particle size. DFT calculations showed that the oxygen binding energy is the weakest for the 2.2 nm Pt NPs. For particles smaller than 2.2 nm, all the surface sites show a very strong oxygen binding energy, resulting in a very low electrocatalytic activity for the O2 reduction reaction.82
There is a size limit of Pt NPs in terms of their electrocatalytic activities for fuel cells. Sun et al. studied the decline of the electrocatalytic activity of extremely small Pt NPs (∼1 nm) toward hydrogen electrooxidation using both experimental and simulation approaches.80 Pt NPs with diameter of ∼1 nm were synthesized using a high-surface-area mesoporous carbon black CMK-3 as a support, which is amorphous. MD simulation showed that this non-crystalline feature is a natural consequence of substantial reduction in the particle size (Fig. 2(I)). CO stripping results show that a positive shift in the peak potential is evident for Pt/CMK-3 (Pt NPs: 1 nm) compared to Pt/XC-72 (Pt NPs: 2.5 nm) and the peak of CO stripping on Pt/CMK-3 is wider than that on Pt/XC-72 (Fig. 2(II)), implying a stronger interaction of Pt/CMK-3 with CO, and slower kinetics and/or the surface nonuniformity. The 1 nm Pt NPs show a decrease in the reversibility of the electrode reaction and remarkable decline of the exchange current density toward hydrogen electrooxidation. DFT simulation indicates that the amorphous structure would lead to an enhancement in the interaction between the surface and adsorbates. Therefore, the Pt–Hads bond on Pt/CMK-3 is expected to be stronger than that on Pt/XC-72, adversely affecting the electrocatalytic activity.
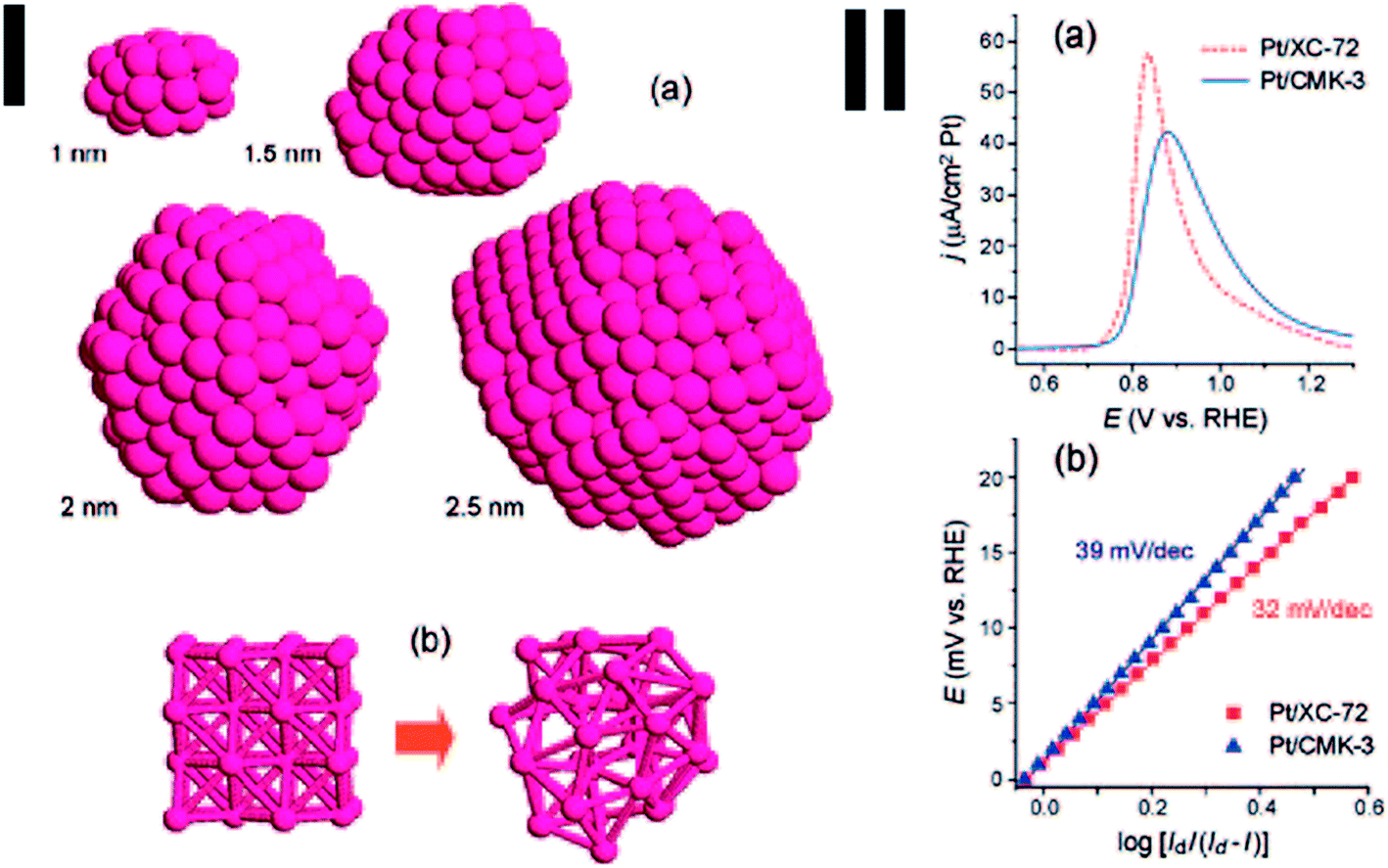 | ||
| Fig. 2 Molecular dynamic simulations using the effective medium theory: (I) (a) four Pt cubic particles with different diameters, defined in this work as the diameter of a sphere with a volume equal to the cubic, were heated in vacuum to 2000 K and then slowly cooled down to 300 K; (b) a virtual Pt cubic particle of 1 nm diameter was thermostatically kept at 300 K in vacuum for a period of time. (II) (a) CO stripping profiles for Pt/XC-72 and Pt/CMK-3, (b) E vs. log[Id/(Id − I)] plots for H2 oxidation reaction on Pt/XC-72 and Pt/CMK-3, where Id is the diffusion limiting current (after ref. 80 reproduced with permission). | ||
2.2. Effect of surface treatment of Pt NPs
One biggest challenge for the application of Pt-based NPs is the structural stability associated with aggregation, dissolution and growth, which will cause a significant loss of electrical, optical and catalytic properties.7,83 The agglomeration of Pt NPs during operation deteriorates the performance rapidly.32,33 For example, Pt NPs would migrate on carbon supports and subsequently aggregate. They can also grow through Ostwald ripening; i.e., surface Pt atoms in small Pt NPs are dissolved to form cationic Pt species and are then deposited on large Pt particles, which results in the grain growth of Pt NPs.11,12 Thus, surface treatment of Pt NPs can effectively enhance their electrocatalytic activity and stability.Takenaka et al. coated a layer of porous silica on CNT supported Pt NPs and found that the activity of SiO2/Pt/CNT catalyst for the oxygen reduction reaction is similar to Pt/CNT.84 However, SiO2/Pt/CNT has a much higher stability during potential cycling from 0.05 to 1.20 V vs. RHE, as compared to Pt/CNT electrocatalysts. The crystallite size of Pt NPs in SiO2/Pt/CNT did not change appreciably during the potential cycling, while Pt NPs in Pt/CNT seriously aggregated. Further study on the effect of SiO2 layer thickness on the catalytic activity showed that the thickness of the silica layer is important for the electrocatalytic activity of Pt NPs as a thick silica layer would significantly decrease the electrochemical surface area, ECSA.33 A dense silica layer with a thickness of ∼6 nm was found to be optimal for the ECSA with excellent durability.
Modification of Pt NPs with Au clusters also enhances the stability of Pt NPs for the O2 reduction reaction.85 More importantly, there is little change in the activity and surface area of Au-modified Pt under the oxidizing conditions of the O2 reduction reaction and potential cycling between 0.6 and 1.1 V (vs. RHE) over 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles, in contrast to the visible losses observed with the pure Pt NP catalyst tested under the same conditions. Therefore, Pt electrocatalysts can be stabilized by the Au clusters against dissolution under potential cycling conditions. Such stabilization could be due to the increase of Pt oxidation potential by the Au clusters as demonstrated by in situ X-ray absorption near-edge spectroscopy and voltammetry data.
000 cycles, in contrast to the visible losses observed with the pure Pt NP catalyst tested under the same conditions. Therefore, Pt electrocatalysts can be stabilized by the Au clusters against dissolution under potential cycling conditions. Such stabilization could be due to the increase of Pt oxidation potential by the Au clusters as demonstrated by in situ X-ray absorption near-edge spectroscopy and voltammetry data.
Wu et al. used the thermal initiation free radical polymerization of the ionic liquid (IL) monomer 1-vinyl-3-ethylimidazolium bromide to from an IL polymer (PIL) film on the surface of Pt NPs supported on 1-aminopyrene functionalized CNT (AP-CNTs) (Fig. 3(I)).86 The PIL film provides mechanical isolation between Pt NPs, electrostatic and coordinative action between Pt NPs and N-heterocyclic cations of ILs. Similar cyclic voltammograms of methanol oxidation have been observed for both PIL/Pt NPs/AP-CNTs and Pt NPs/AP-CNTs electrocatalysts. However, the stability of Pt NPs is greatly improved by the protection of PIL thin layer (Fig. 3(II)), due to the inhibiting effect of PIL thin films on the migration and agglomeration of Pt NPs.
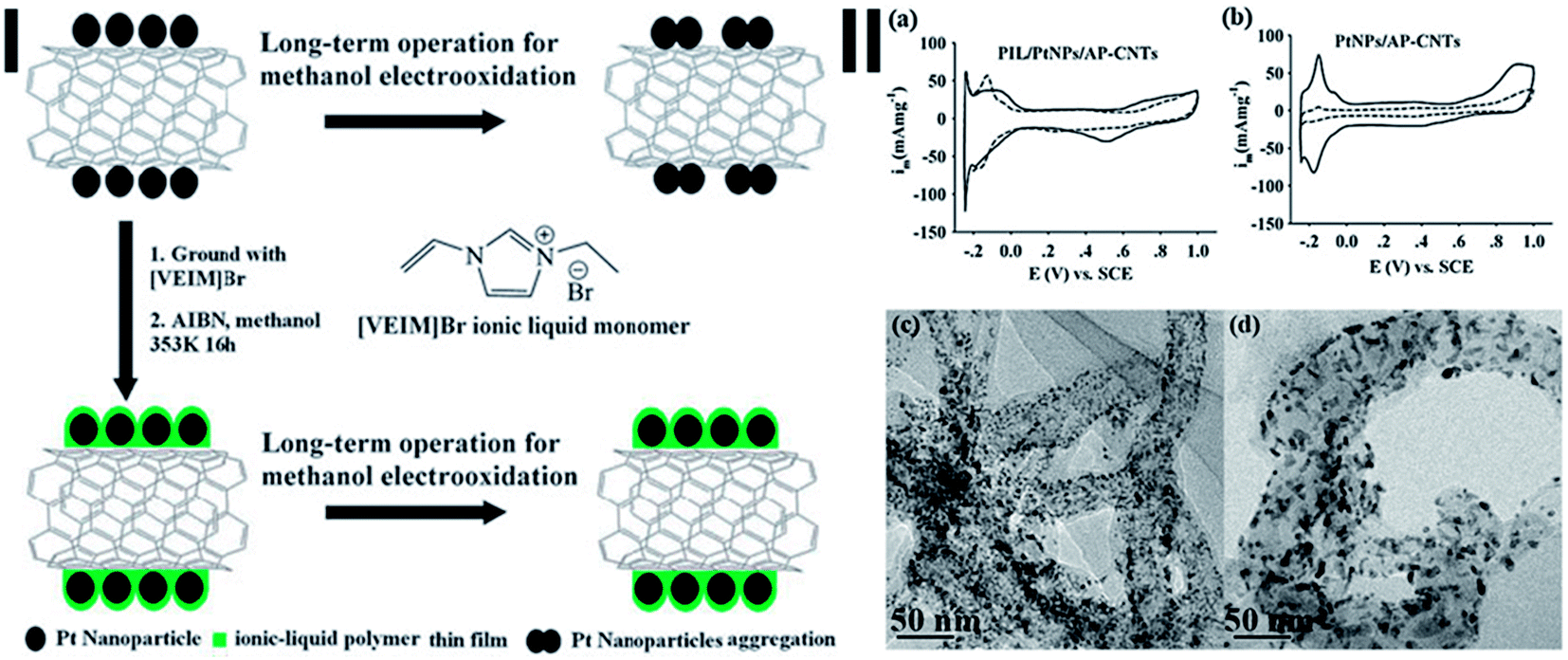 | ||
| Fig. 3 (I) Schematic diagram for stabilization of Pt NPs supported on CNTs by ionic liquid polymer (PIL). (II) Cyclic voltammograms of PIL/PtNPs/AP-CNTs (a) and PtNPs/AP-CNTs (b) electrocatalysts in nitrogen-saturated 0.5 M H2SO4 aqueous solution before (solid line) and after (dashed line) long-term chronoamperometry experiments (6 h); TEM images of PIL/PtNPs/AP-CNTs (c) and PtNPs/AP-CNTs (d) electrocatalysts after long-term chronoamperometry experiments (6 h) (after ref. 86 reproduced with permission). | ||
We studied in details the polyelectrolyte-stabilized Pt NPs as electrocatalysts for PEMFCs.87,88 Polyelectrolyte-stabilized Pt nanoparticles were synthesized by ethanol reduction of Pt precursor, H2PtCl6 in the presence of ionic polyelectrolyes such as PDDA, poly(allylamine hydrochloride) (PAH), poly(sodium 4-styrenesulfonate) (PSS), Nafion, poly(acrylic acid) (PAA), poly(2-acrylamido-2-methyl-1-propanesulfonic acid) (PAMP) and non-ionic polymer, poly(vinylpyrrolidone) (PVP). The best electrocatalytic activities were observed on PDDA-Pt and PSS-Pt NPs, indicating the significant enhancing effect of the NH4+ and SO3− functional groups of the polyelectrolytes on the electrocatalytic activities of Pt NPs for the methanol oxidation and O2 reduction reactions. Due to the charged nature of the polyelectrolyte-stabilized Pt NPs, the polyelectrolyte-stabilized Pt-based NPs can be self-assembled to the sulfonic acid group, SO3− at the Nafion membrane surface by the electrostatic interaction, forming a self-assembled monolayer (SAM). Further studies show that such SAM reduces the methanol crossover and enhances the power output of direct methanol fuel cells.89,90
The surface treatment can also lead to the increase of electrocatalytic activity of Pt-based NPs. Wlodarczyk et al. modified the Pt NPs with phosphotungstic acid (H3PW12O40, HPW) and produced highly reactive Pt-based electrocatalysts toward the oxygen reduction reaction.91 The overall reaction mechanism is unchanged but kinetic analysis have shown that HPW films on Pt NPs exhibit some enhancement effect on the oxygen reduction, which could be due to the ability of HPW to form strong electroactive adsorbates on solid surfaces, its high reductive reactivity and fast electron transfer (mediating) capabilities, as well as its acidic characteristics leading to the increased proton mobility. Barczuk et al. modified Pt, PtRu, and carbon (Vulcan)-supported PtSn NPs with ultrathin films of phosphomolybdic acid (H3PMo12O40, HPMo) and found that their electrocatalytic activities toward ethanol oxidation are enhanced.92 HPMo is electroactive itself and may act as mediators of interfacial electron transfer during ethanol oxidation. It may also contribute to the bifunctional mechanism to provide oxo groups that can remove poisoning species from Pt surface.
3. Electrocatalytic activity of Pt NPs supported on non-covalent functionalized CNT
As shown early, CNTs are attractive materials as electrocatalyst supports, due to their high electronic conductivity, large accessible surface area, and excellent chemical and electrochemical stability.15,16 However, one intrinsic disadvantage is that the surface of pristine CNTs is very inert with few binding sites for Pt precursors and/or Pt NPs. Therefore, surface modification or functionalization of CNTs is necessary to introduce functional groups onto the surface of CNTs to enhance the dispersion and to provide the active sites for the adsorption and deposition of Pt precursor and/or Pt NPs. As compared to covalent functionalization of CNTs, non-covalent functionalization by polyelectrolytes and solvent significantly improves the distribution and electrocatalytic activity of Pt and Pt based alloy NPs on CNTs and graphene.17,63,70,93–95 Here, non-covalent methods for modifying CNTs will be first summarized and the approaches for the synthesis of Pt and Pt-based alloy NPs on non-covalent functionalized CNTs will be described.3.1. Non-covalent functionalization of CNT
Non-covalent functionalization or physical modification of CNTs are achieved by the interaction between the functionalization molecules or agents and CNTs through various interaction modes such as π–π, hydrophobic, CH–π and charge transfer interaction.60 Since CNTs are a nanotubular structure composed of seamless single/multiple layered graphene sheets, many kinds of small molecules/polymers with a π–π conjugated structure can complex with CNT via π–π interaction. Conjugated polymers are a type of polymers with extensive π–π conjugated backbone. The hybrids of conjugated polymers and CNT can exhibit synergistic properties such as good stability, high mechanical strength, and high electrical conductivity of CNT and superior optoelectronic properties.96 Therefore several studies have been reported to use the non-covalent interaction of conjugated polymer to disperse and functionalize CNT. For example, polypyridinium salts have been synthesized and employed to form a strong π–π and cationic–π interaction with single-walled CNTs, SWCNT in dimethyl sulfoxide (DMSO).97 A carboxyl-containing conducting oligomer, poly(pyrenebutyric acid), has also been synthesized by direct oxidation of pyrenebutyric acid in boron trifluoride diethyl etherate containing SWCNTs for non-covalent functionalization of SWCNTs.98 The resulting hybrid can be well dispersed in THF, dimethylformamide (DMF), DMSO, and aqueous alkaline solution. Recently, a general strategy to disperse and functionalize pristine CNTs in a single-step process has been developed using conjugated block copolymers, which contain two blocks: a conjugated polymer block of poly(3-hexylthiophene), and a functional non-conjugated block with tunable composition.99 When the pristine CNTs are sonicated with the conjugated block copolymers, the poly(3-hexylthiophene) blocks bind to the surface of de-bundled CNTs through the π–π interactions, stabilizing the CNT dispersion, while the functional blocks locate at the outer surface of CNTs, rendering the CNTs with desired functionality. Two novel phenylenevinylenes having cyanobiphenyl terminates have been successfully synthesized and characterized.100 They have been found to form stable supramolecular complexes with SWCNTs through strong π–π interactions between the sidewall of SWCNTs and phenylenevinylene segment and wrapping around the SWCNTs, imparting excellent solubility in organic solvents.Pyrene is electronically similar to graphite and various pyrene containing polymers have been investigated for dispersion of CNTs via π–π interaction. Pyrene-functionalized monomers have been used to construct well-defined block copolymers and very stable polymer layers on multi-walled CNTs (MWCNTs) have been found. Pyrene-functionalized polymers have been prepared from maleic anhydride copolymers.101 The resulting polymers are water soluble and can form a strong non-covalent bonding with the nanotubes. A pyrene containing diblock copolymers based on poly(methyl methacrylate) have also been synthesized and their adsorption on CNTs has been investigated.102 The length of the anchor block and the types of anchor units were optimized for the maximum adsorption and best dispersion. Pyrene-linked glycosides have been employed to non-covalently functionalize SWCNTs and MWCNTs.103 These molecules can also be dissociated by tuning the solvent polarity. This reversibility could be useful as CNTs can be modified and regenerated in a controllable manner.
An amphiphilic molecule oligothiophene-terminated poly(ethylene glycol) (TN-PEG) was synthesized by Lee et al. (see Fig. 4(I) for synthesis procedures),104 and its ability to disperse and stabilize pristine CNTs in water has been examined. TN-PEGs can be adsorbed onto the CNT surface via strong π–π interaction, significantly enhancing the dispersion of CNT and increasing the long-term stability (Fig. 4(II)). In addition, the TN-PEG with a longer thiophene unit can effectively disperse the CNTs in water as compared to that with a shorter one (Fig. 4(II)). Yoo et al. described an evaluation method of the binding affinity of polyaromatic hydrocarbons with the surface of CNTs using an HPLC column packed with an SWCNT-coated silica gel.105 The results indicate that the shapes of the molecules also play a key role in the binding affinity. For example, linear-shaped polyacenes show a much higher affinity than the nonlinear counterparts due to the effective stacking along with the one-dimensional CNT structure.
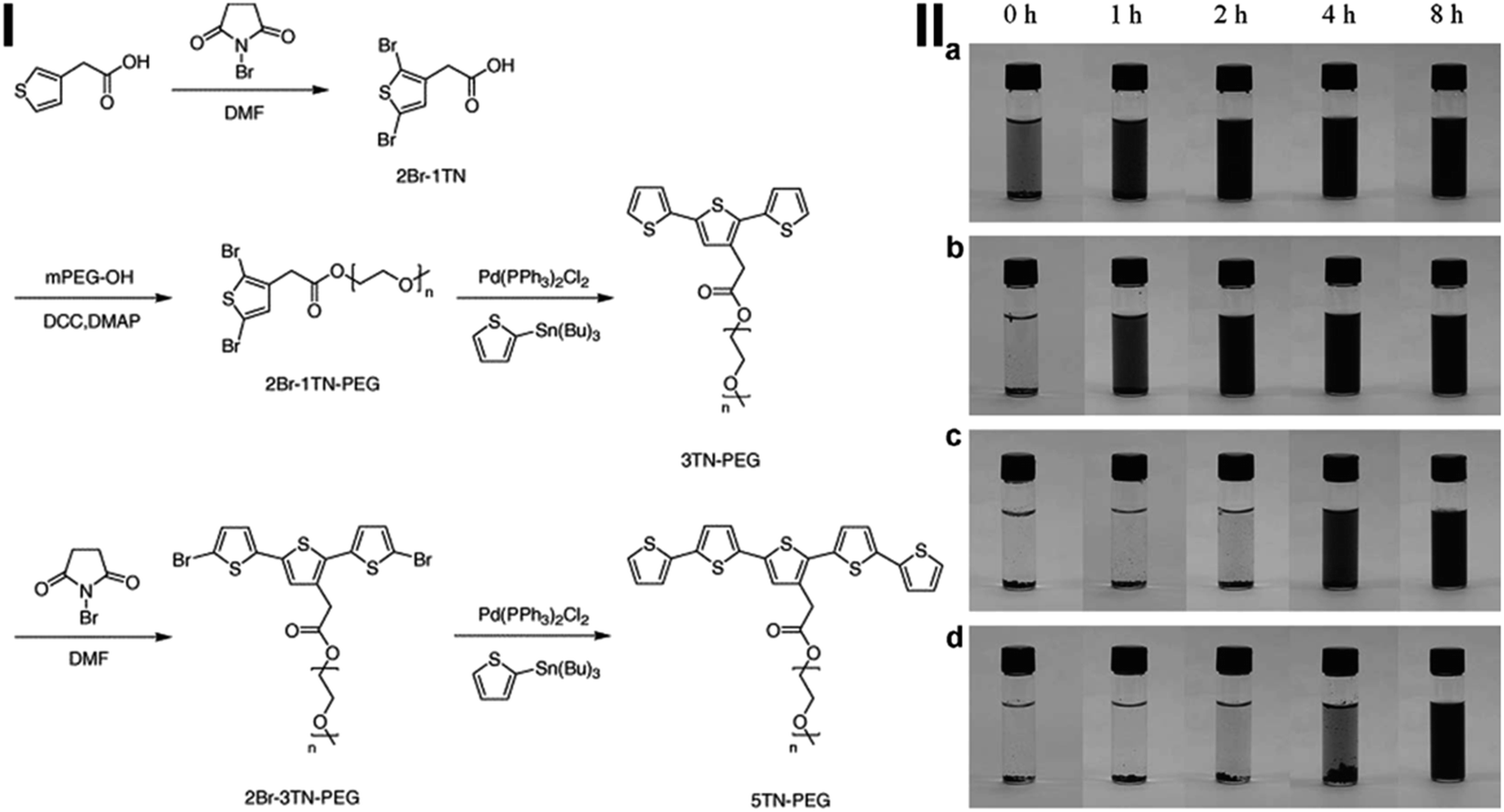 | ||
| Fig. 4 (I) Synthesis route of 3TN-PEG and 5TN-PEG; II. Dispersion state of SWCNTs in water as a function of sonication time with various surfactants: (a) 5TN-PEG, (b) 3TN-PEG, (c) Pluronic F127, and (d) SDS. All vials contain the same SWCNT concentration (0.05 mg mL−1) and the concentration of each dispersant in the dispersion samples is 0.5 mg mL−1 (after ref. 104 reproduced with permission). | ||
CNTs can also be non-covalently functionalized via the CH–π interaction. Xu et al. used hyperbranched polyethylene (HBPE) to functionalize and solubilize MWCNTs in organic solvents such as chloroform and THF (see the scheme in Fig. 5(I)).106 TEM results show that the HBPE functionalized MWCNTs in both chloroform and THF are well dispersed (Fig. 5(II)), in sharp contrast to the densely entangled nanotubes found with pristine MWCNTs in heptane and the poorly dispersed MWCNTs with HBPE in toluene. A high concentration up to 1235 mg L−1 and 920 mg L−1 has been achieved in chloroform and THF, respectively. The driving force could be the CH–π interactions between unique hyperbranched structure of HBPE and MWCNT sidewalls. The same approach has also been used by Petrie to significantly improve the dispersion of MWCNTs in an ethylene-octene copolymer matrix.107
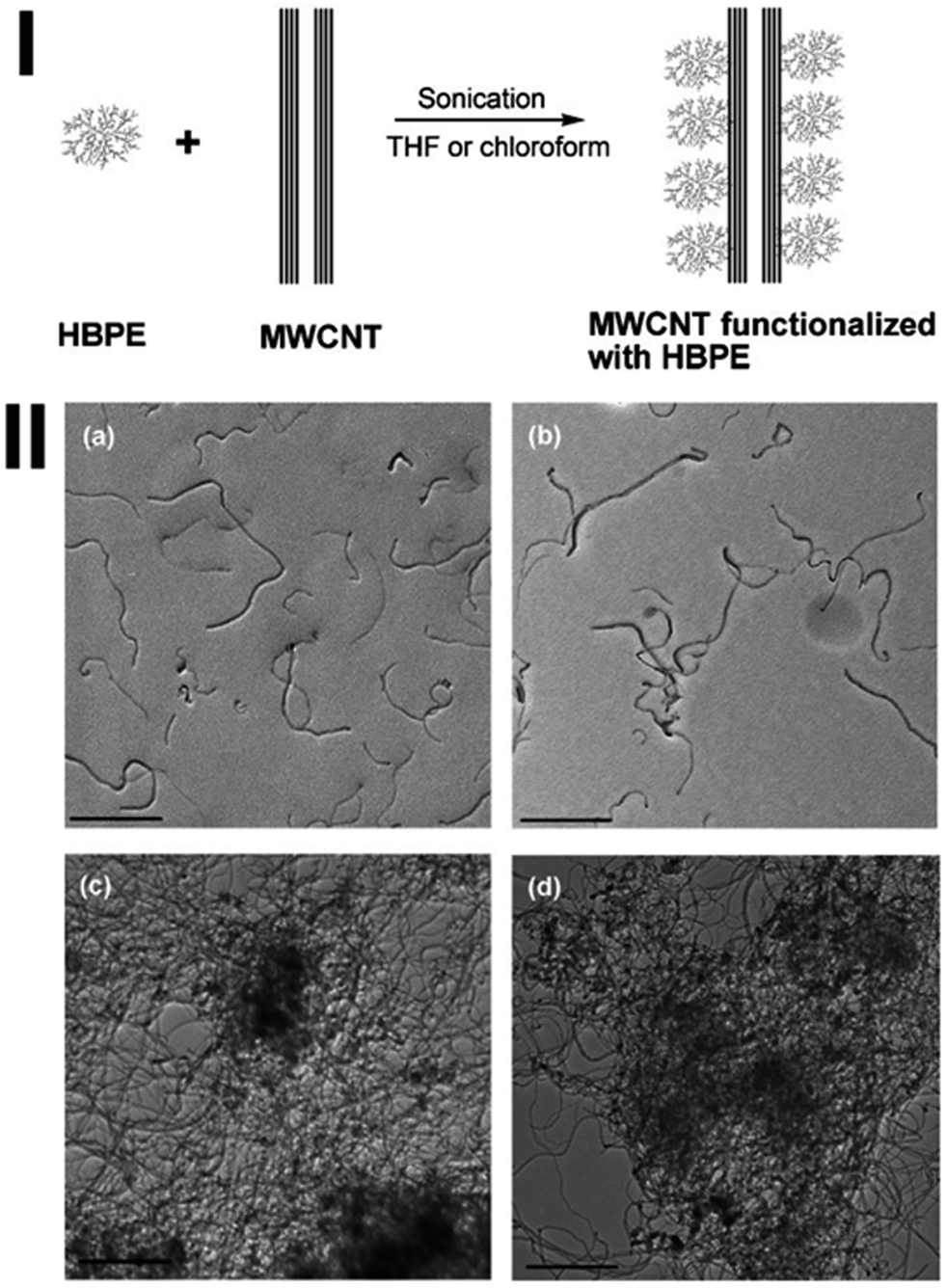 | ||
| Fig. 5 (I) Schematic noncovalent nonspecific functionalization and solubilization of MWCNTs with HBPE. (II) TEM images of (a) MWCNTs solubilized with HBPE in chloroform; (b) MWCNTs solubilized with HBPE in THF; (c) MWCNTs dispersed with HBPE in toluene; (d) pristine MWCNTs dispersed in heptane. In (a)–(c), the same HBPE/MWCNT mass ratio of 0.4 was used. Scale bar: 500 nm (after ref. 106 reproduced with permission). | ||
The hydrophobic force has also been used to disperse CNTs. Typical examples include the use of surfactants for the CNT dispersion. The hydrophilic parts of surfactants interact with water or other polar solvents and hydrophobic parts interact with CNTs.108,109 The nature of surfactant, its concentration, and type of interaction are known to play a crucial role in the phase behavior of CNTs. For the ionic surfactants, the dispersion can be stabilized by electrostatic repulsion, while for the non-ionic surfactants, the aggregation of CNTs can be prevented by the steric effect or solvation due to the hydrophilicity.108,109 Richard et al. studied the assembly of surfactants such as sodium dodecylsulfate (SDS) on CNTs by TEM.110 SDS formed rolled-up half-cylinders on nanotube surface. Rings, helices, or double helices were also observed depending on the symmetry and the diameter of the CNTs. Permanent assemblies have been produced from mixed micelles of SDS and different water-insoluble double-chain lipids after dialysis of the surfactant. The efficiency of various surfactants in dispersing SWCNT in water has been systematically studied.109 The length and the shape of the alkyl chain are very important for the dispersion, and longer and more branched alkyl chains are better than linear and straight ones.
Linton et al. investigated the formation of non-covalent electron donor–acceptor interactions between polymers and SWCNTs.111 Nanocomposites of SWCNTs and three sets of polymer matrices with varying compositions of electron donating 2-(dimethylamino)ethyl methacrylate (DMAEMA) or electron accepting acrylonitrile (AN) and cyanostyrene (CNSt) were prepared. The copolymers with 30 mol% DMAEMA, 45 mol% AN, 23 mol% CNSt and polyacrylonitrile homopolymer have the highest extent of intermolecular interaction, which is in agreement with quantum density functional theory calculations. Both experimental and computational results show that a sufficient distance between interacting functional groups is needed for the efficient intermolecular contact.
O'Connell et al. studied in detail the wrapping of SWCNT by hydrophilic linear polymers such as PVP, PSS, poly(1-vinyl pyrrolidone-co-vinyl acetate), poly(1-vinyl pyrrolidone-co-acrylic acid), poly(1-vinyl pyrrolidone-co-dimethylaminoethyl methacrylate), polyvinyl sulphate, poly(sodium styrenesulfonic acid-co-maleic acid), dextran, and dextran sulphate (see Fig. 6 for the case of PVP).112 The association between polymer and SWCNT is robust and independent of the excess polymers in solution. Such a wrapping by polymers has been explained thermodynamically and offers an alternative and new strategy for solution chemistry of pristine CNTs.
 | ||
| Fig. 6 (I) Tapping-mode AFM images of PVP-SWNTs on a functionalized substrate. 5 μm height (top left) and amplitude image (top right). 1 μm expanded height image (bottom left) and amplitude image (bottom right). (II) Some possible wrapping arrangements of PVP on an 8, 8 SWNT. A double helix (top) and a triple helix (middle). Backbone bond rotations can induce switch-backs, allowing multiple parallel wrapping strands to come from the same polymer chain (bottom) (after ref. 112 reproduced with permission). | ||
3.2. Pt-based NPs supported on non-covalent functionalized CNT
We reported a highly effective polyelectrolyte functionalization method for MWCNTs using PDDA.67 PDDA is a water-soluble quaternary ammonium and strong polyelectrolyte. During the functionalization process, NaCl salt is added to allow the PDDA chain to adopt a random configuration, thus leading to a high functionalization or wrapping of PDDA on MWCNTs, PDDA-MWCNTs (Fig. 7(I)). The strong adsorption of the positively charged PDDA on MWCNTs is believed due to the π–π interaction between PDDA and the basal plane of graphite of MWCNTs.113 Pt NPs are then in situ synthesized on the PDDA-MWCNTS via the self-assembly between negatively charged Pt precursors and positively charged functional groups of PDDA. This non-covalent polyelectrolyte functionalization not only leads to high density and homogeneous surface functional groups on MWCNTs, but also preserves the intrinsic properties of MWCNTs without damaging their surface structure for high loading of uniform and small Pt NPs (Fig. 7(II)). The Pt/PDDA-MWCNT electrocatalyst shows a much higher electrocatalytic activity for the methanol oxidation than Pt NPs on acid-treated MWCNT, Pt/AO-MWCNTs electrocatalyst, and the conventional Pt/C catalysts (Fig. 7(III)). Pt/PDDA-MWCNTs can also be used as template to assemble Pt NPs on CNTs with wide range Pt NPs loading of 10–93 wt% with controlled particle size.17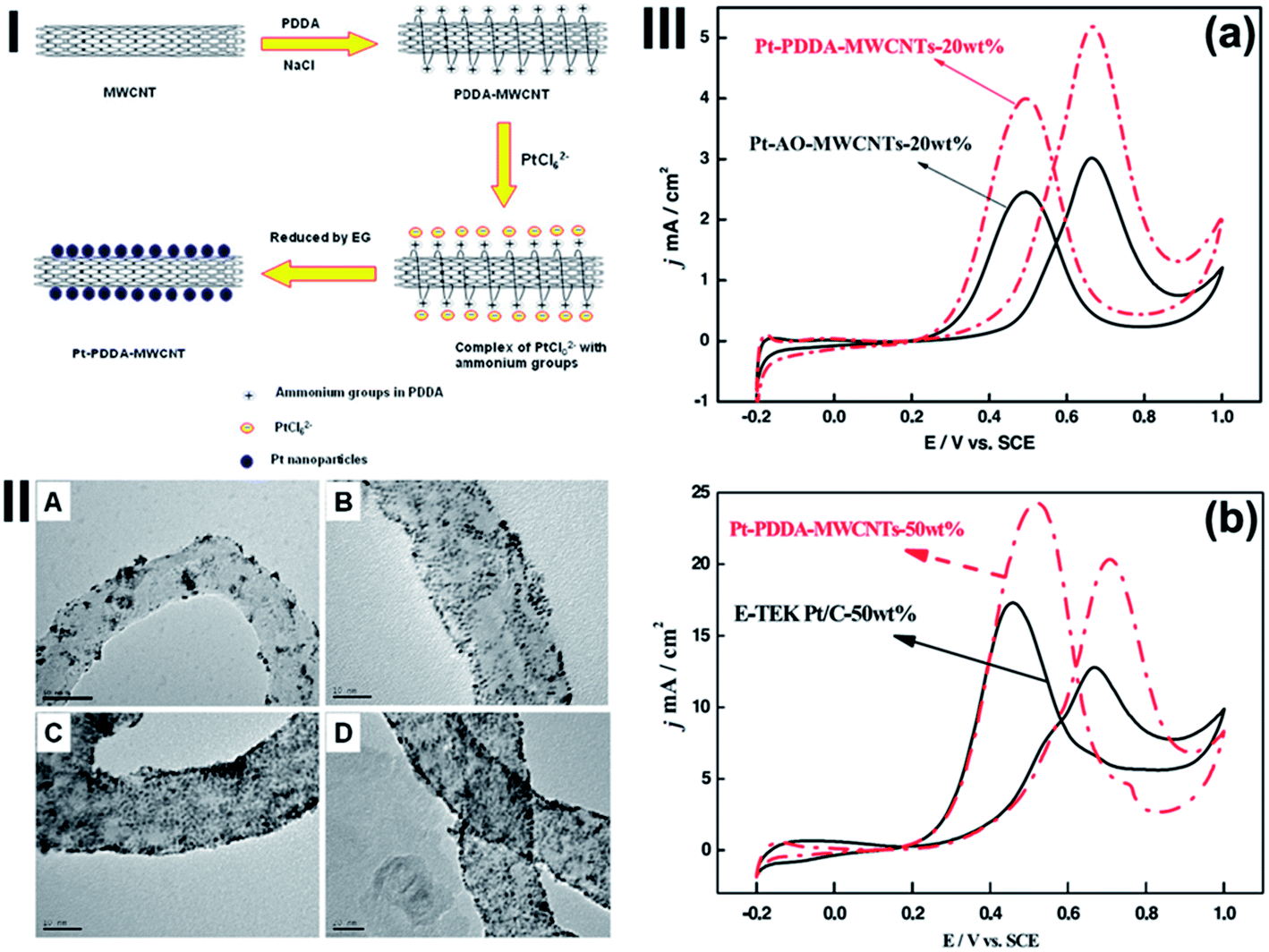 | ||
| Fig. 7 (I) Scheme of PDDA functionalization of MWCNTs and the in situ synthesis of Pt nanoparticles on PDDA-functionalized MWCNTs. (II) TEM images of (a) Pt-AO-MWCNTs-20 wt%, (b) Pt-PDDA-MWCNTs-20 wt%, (c) Pt-PDDA-MWCNTs-40 wt%, (d) Pt-PDDA-MWCNTs-60 wt%. (III) (a) CVs of Pt catalysts supported on PDDA-MWCNTs and AO-MWCNTs in nitrogen saturated 0.5 M H2SO4 + 1.0 M CH3OH. (b) CVs of Pt-PDDA-MWCNTs-50 wt% and E-TEK Pt/C (50 wt%) in nitrogen purged 0.5 M H2SO4 + 1.0 M CH3OH. The Pt loading was 0.05 mg cm−2 (after ref. 67 reproduced with permission). | ||
There are other electrolytes which have been used to effectively functionalize the CNTs for the dispersion of Pt-based electrocatalysts, including PAH, PSS, PAA.114,115 Zhang et al. studied the PAH functionalized CNTs as support for the self-assembly of Pt NPs.115 CNTs were first wrapped with PAH, followed by the self-assembly of Pt precursor and EG reduction. PAH modification introduces high density and homogeneous surface NH2 groups, which can combine with the Pt precursor by electrostatic interaction. This helps to form small, uniform and stable Pt NPs. The resulting catalysts show high electrochemically active surface area, higher onset potential of O2 reduction, and the higher half-wave potential. The oxygen reduction activity at 0.9 V on Pt/PAH-CNTs (32.4 A g−1 Pt) is much larger than that on Pt/COOH-CNTs (17.5 A g−1 Pt) and Pt/XC-72 (17.3 A g−1 Pt). Pd NPs can also be deposited uniformly on PDDA-CNTs in the presence of NaBH as reducing agent.116 In addition to the role of functionalization of CNTs, PDDA was also found to serve as reductant in the formation of Pt NPs on functionalized CNTs.117
Conducting polymer/monomer modified CNTs provide a great promise as an electrocatalyst support for Pt NPs. Shi et al. reported an in situ interfacial polymerization of aniline to form polyaniline (PANI) on acid-treated CNTs.118 Then the amine group of PANI was used to attach Pt precursors for the synthesis of Pt NPs in the presence of HCOOH. Consequently, fine and dispersed Pt NPs with controlled loading density were uniformly assembled on PANI-CNTs. The electrocatalysts showed an excellent catalytic activity and good stability toward both methanol oxidation and oxygen reduction in acid condition. He et al. adopted a one-pot aqueous solution-based approach to form a Pt NPs on PANI functionalized CNTs.119 Similarly, the PANI serves as a bridge between Pt NPs and CNT walls with the presence of platinum-nitride bonding and π–π bonding. Hsu et al. used aniline monomer to functionalize CNTs for the dispersion and deposition of Pt NPs.120 Aniline is adsorbed on CNTs via π–π interaction with amino group exposed to combine the Pt precursor. The interaction still remains after the reduction of Pt precursor because of the electron pair of N and the vacant orbital of Pt. The Pt NPs supported on aniline-functionalized CNTs have a higher oxygen reduction activity than the conventional Pt/C catalyst.
Fang et al. reported an efficient and reproducible approach to fabricate a MWCNT supported Pt NPs with high loading, using an anionic surfactant, SDS as functionalization agent.121 The SDS-modified MWCNTs were used to synthesize high loading of Pt NPs through a urea-assisted process, followed by reduction in the presence of EG (Fig. 8(I)). The urea provides an environment with an in situ pH adjustment for homogeneous deposition. The resulting Pt NPs are uniformly dispersed on SDS-MWCNT. A high loading of 60 wt% Pt was achieved due to the synergy of SDS, homogeneous deposition of Pt precursors and reduction of the precursor by EG (Fig. 8(IIa)). The electrocatalyst with a Pt loading of 60% has a significantly better performance than commercially available Pt/C catalyst with the same catalyst loading (Fig. 8(IIb and c)).
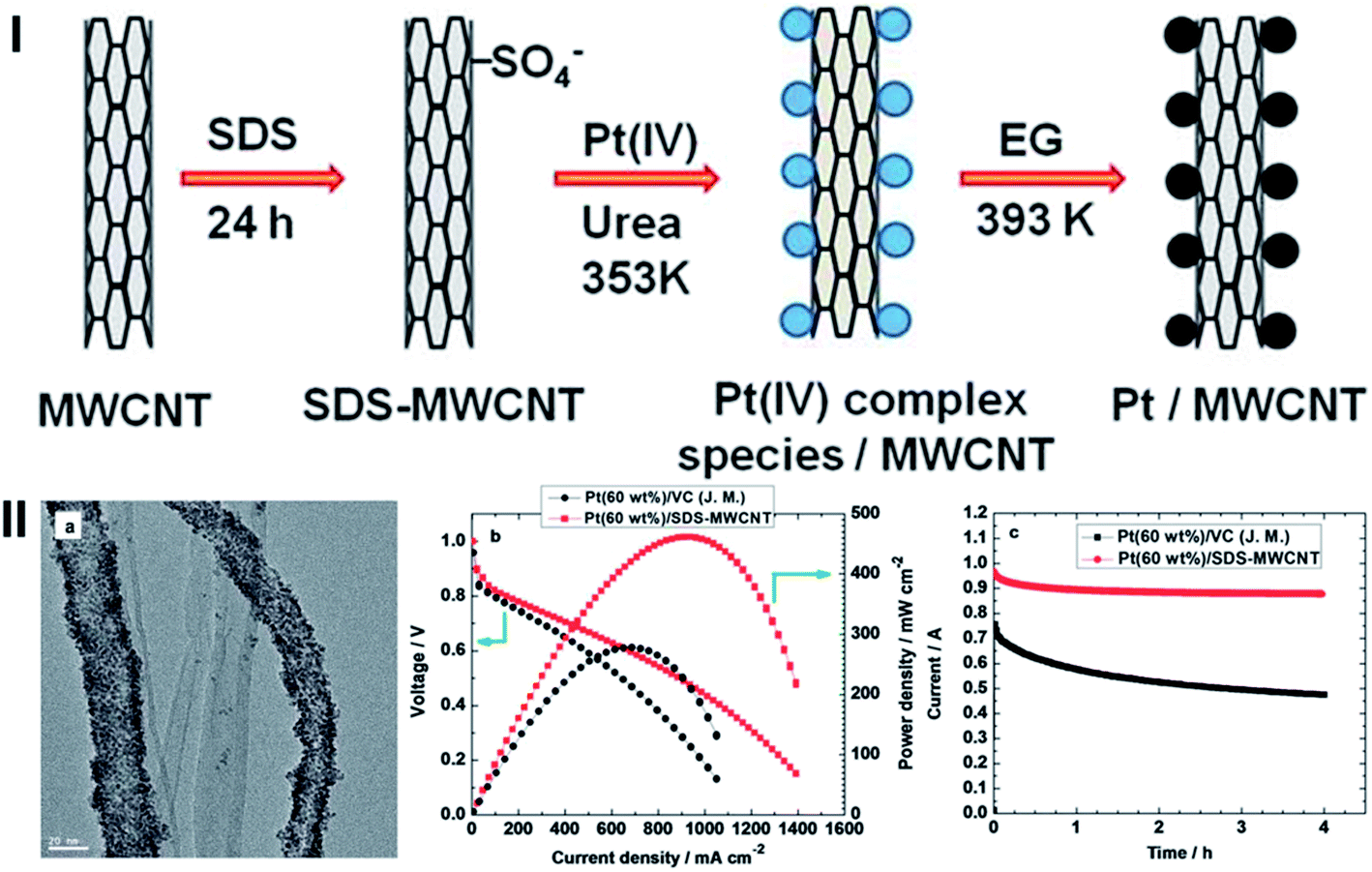 | ||
| Fig. 8 (I) A schematic diagram for the synthesis of a SDS-MWCNT supported Pt catalyst using a urea-assisted HD-EG strategy. II. (a) Representative TEM images for the SDS-MWCNT supported Pt (60 wt%) catalyst synthesized by the urea assisted HD-EG strategy. (b) Fuel cell polarization plots at 60 °C for the SDS-MWCNT-supported Pt (60 wt%) catalyst prepared by the HD-EG strategy and VC-supported Pt (60 wt%). (c) Chronoamperograms obtained at 0.75 V for the SDS-MWCNT-supported Pt (60 wt%) catalyst prepared by the HD-EG strategy and VC-supported Pt (60 wt%) (after ref. 121 reproduced with permission). | ||
Ionic-liquid polymer (PIL) based on thermal-initiation free radical polymerization of ionic liquid (IL) monomer (3-ethyl-1-vinylimidazolium tetrafluoroborate) can be used to effectively functionalize CNT.122 A large number of positively charged surface functional groups with uniform distribution are introduced, preventing the aggregation of CNTs and providing anchoring sites for Pt and PtRu NPs. The resulting PtRu/CNTs-PIL (or Pt/CNTs-PIL) electrocatalysts show better performance for the methanol oxidation than the PtRu/CNTs (or Pt/CNTs) electrocatalyst.
Nakashima's group studied in details the polybenzimidazole (PBI)-functionalized CNTs as catalyst supports for fuel cells.65,123 The attraction of PBI as the functionalization agent is that PBIs are known to have excellent proton conductivity after acid doping.124 The functionalization of PBIs on CNTs is primarily through π–π interaction.125 The loading of PBIs wrapped on CNTs was as high as 23 wt%, forming a 2.1 nm thick layer on the surface of MWCNTs. Pt NPs with average particle size of 4.0 nm were uniformly dispersed on the outer wall surface of CNTs and showed a peak current density of 480 mA mgPt−1 for the methanol oxidation reaction in acid solution. The proton conductivity of Pt supported on PBIs-CNTs electrocatalysts can be increased by acid doping, enhancing the power output of the fuel cells.126 An interesting extension of the PBI-functionalized CNTs is the application of KOH doped Pt/PBIs-CNTs as electrocatalysts for anion exchange membrane fuel cells.127 The KOH doping was carried out by immersing the Pt/PBIs-CNTs catalysts in a 6 M KOH solution. The KOH doped PBI-wrapping layers around the CNT surfaces function as a hydroxide-conducting path (Fig. 9a). The membrane-electrode-assembly (MEA) cells based on KOH doped Pt/PBIs-CNTs anode and cathode catalysts and an anion exchange membrane showed a maximum power density of 256 mW cm−2, significantly better than that of standard MEA (Fig. 9c). It is considered that the high hydroxide conduction of the KOH-doped PBIs functionalized on CNTs plays a key role in the high fuel cell performance.127
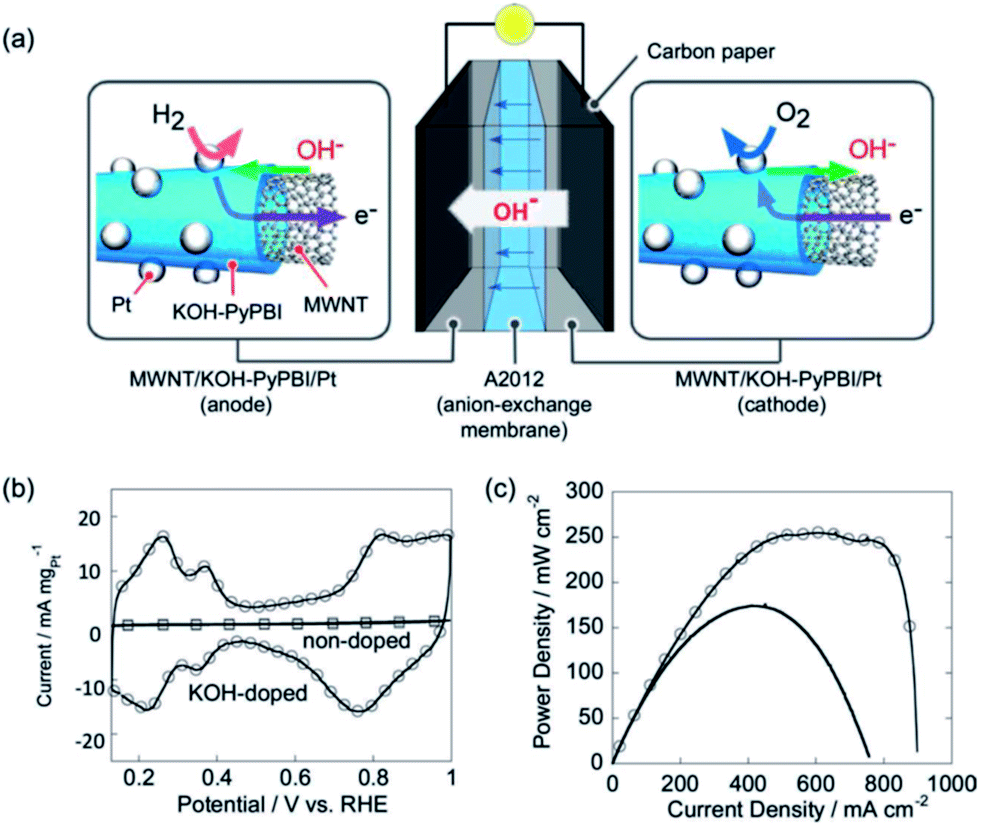 | ||
| Fig. 9 (a) Schematic illustration of the KOH doped PyPBI cell. Mass transfer in the cell is also shown. (b) In situ CVs of cathode in the KOH-doped MEA (circles) and non-doped MEA (squares) at a scan rate of 50 mV s−1. (c) Power density curves of the KOH-doped MEA (circles) and standard MEA (black line) (after ref. 127 reproduced with permission). | ||
Hsin et al. reported in situ polymerization of non-ionic PVP whose main chains are linked with MWCNTs through breaking the double bonds of MWCNTs, and then used this PVP-functionalized MWCNTs as supports to deposit Pt and PtRu NPs.66 CNTs were treated in concentrated HNO3–H2SO4 solution at 90 °C for 30 s. During the polymerization process, oligomeric vinylpyrrolidone radicals, because of their high reactivity, nonselectively attach to the unsaturated CC double bonds of carbon nanomaterials, leading to homogeneous surface functionalization. Strictly speaking, due to the PVP-grafting on the side walls of MWCNTs, this is not a non-covalent functionalization process. However, the Pt NPs supported on PVP modified carbon nanostructures showed a much better activity toward methanol oxidation as compared to Pt supported on acid oxidized MWCNTs. The conductivity results showed that PVP modified carbons have a much better conductivity than acid oxidized ones.
Non-covalent functionalization is also an effective way to synthesize multi-component electrocatalyst systems with significantly enhanced synergistic effect. A good example is the superior electrocatalytic activity of Pd NPs on HPW and PDDA co-functionalized CNTs though both Keggin-type HPW and Pd are not a prominent catalyst toward oxygen reduction reaction (ORR).72,128 The new multi-component catalyst was synthesized by self-assembly of HPW on CNTs via the electrostatic attraction between negatively charged HPW and positively charged PDDA-wrapped CNTs, followed by dispersion of Pd NPs onto the HPW-PDDA-CNT assembly (see Fig. 10(I)).128 The Pd catalysts are characterized by uniformly distributed Pd NPs on HPW and PDDA co-functionalized CNTs (Fig. 10(III)). HPW in the composite catalysts can act as an effective electron mediator and source of mobile protons at the electrocatalytic interface as has been reported elsewhere.129–131 A rich source of protons at the electrode surface makes the porous electrode extremely hydrophilic, facilitating the mass transfer process. The interaction of HPW molecules assembled on CNTs within a hybrid support may also change the interfacial electronic structure of Pd, as indicated by the negative shift in Pd3d binding energies for Pd/HPW-PDDA-CNTs relative to a Pd/acid treated-CNTs system. The existence of HPW in Pd/HPW-PDDA-CNTs results in a downward shift in the binding energy of the Pd3d level, possibly caused by a weakening of adsorption, substantially enhancing the electrocatalytic activity of Pd NPs for ORR (Fig. 10(II)).128
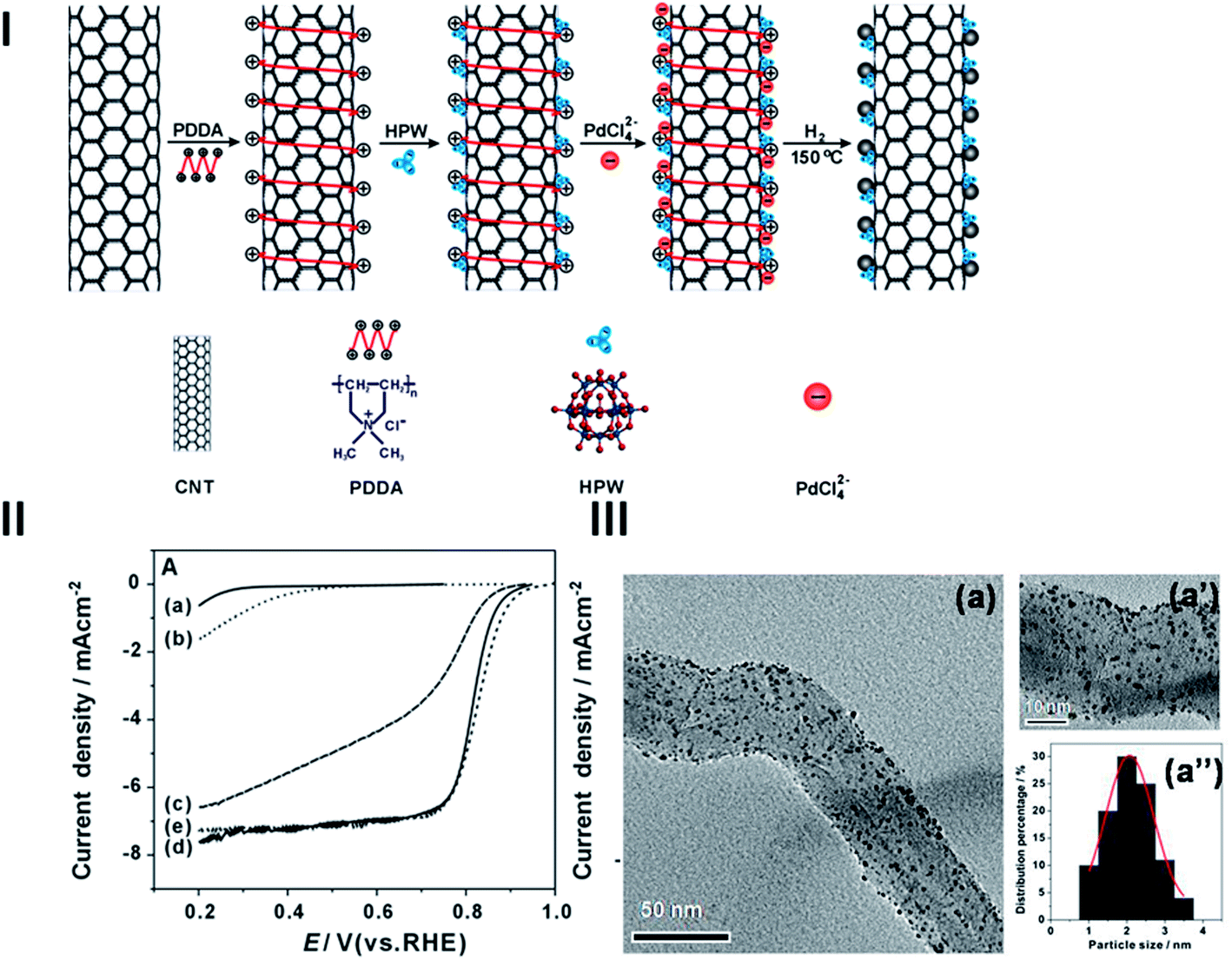 | ||
| Fig. 10 (I) A scheme for the self-assembly and synthesis of Pd NPs on HPW assembled PDDA-CNTs. (II) Polarization curves for the ORR on (a) pristine-CNT; (b) HPW-PDDA-CNTs; (c) Pd/acid treated-CNTs; (d) Pd/HPW-PDDA-CNTs and (e) E-TEK Pt/C electrodes in a 0.5 M O2-saturated H2SO4 solutions with a scan rate of 5 mV s−1 and a rotation speed of 2500 rpm. (III) TEM images and histograms of Pd/HPW-PDDA-MWCNTs at different magnifications with 20 wt% Pd loadings (after ref. 128 reproduced with permission). | ||
3.3. Other Pt alloy NPs supported on non-covalent functionalized CNT
Pt NPs based electrocatalysts are prone to the poisoning or deactivation of intermediate species such as CO formed during the oxidation of fuels such as methanol and ethanol, causing serious deterioration of their electrocatalytic activity.132 Pt-based alloyed NPs such as PtRu, PtSn, PtPd, Pt–W and Pt–Mo are the most effective for the enhancement of the electrocatalytic properties of Pt with increased tolerance towards the CO poisoning and thus the catalyst durability.7,133–135 Therefore, there are extensive studies on the use of non-covalent functionalized CNTs as a support to Pt-based alloy NPs for fuel cell applications. Here we will only focus on the well-studied binary alloy NPs supported on non-covalent functionalized CNTs. For other complex NPs such as ternary alloy NPs and core–shell nanoarchitectures, readers are recommended to refer to reviews on metal nanostructures/nanoarchitectures.7,135–137Among the Pt-based binary NPs, PtRu alloy NPs are the most widely studied electrocatalysts for the methanol oxidation reaction due to the combination of bifunctional and electronic mechanism of Ru for enhancing electrocatalytic performance.138–142 Kuang et al. modified CNTs with 1-pyrenecarboxaldehyde (PCA) via π–π interaction between CNTs and PCA as supports for the deposition of PtRu NPs.143 PCA not only serves as the reductant for the in situ formation of PtRu NPs, but also stabilizes the PtRu NPs on CNTs by the oxidation product of PCA. The PtRu/PCA-CNT catalysts exhibit higher electrocatalytic activities and better stability towards the methanol oxidation as compared to PtRu NPs supported on pristine CNTs. The same group has also functionalized CNTs with a perylene derivative, 3,4,9,10-perylene tetracarboxylic acid.144 The PtRu NPs supported on functionalized CNTs have a much better catalytic activity and stability toward the methanol oxidation than that supported on acid oxidized CNTs. Wu et al. functionalized CNTs with chitosan, which is a polycation with a high film-forming ability and good proton conductivity.145 Chitosan functionalization not only disperses CNT, but also introduces functional groups such as NH2 and OH groups for the attachment of metal NPs. Very fine PtRu NPs were grown uniformly on the modified CNTs and showed significantly enhanced activity toward the methanol oxidation reaction.
Water soluble heteropolyacids (HPAs) such as HPMo and HPW can be self-assembled on the chitosan (CS) functionalized CNTs to enhance the electrocatalytic activity and stability of supported PtRu NPs.146 The onset and peak potential for COad oxidation on PtRu/HPAs-CS-CNTs catalysts are more negative than that on PtRu/AO-CNTs, indicating that HPAs facilitate the electro-oxidation of CO. The PtRu/HPMo-CS-CNTs catalyst has a higher electrocatalytic activity for methanol oxidation and higher tolerance toward CO poisoning than that of PtRu/HPW-CS-CNTs. The better electrocatalytic enhancement of HPMo on PtRu/HPAs-CS-CNTs catalyst is most likely related to the fact that molybdenum-containing HPAs such as HPMo have more labile terminal oxygen to provide additional active oxygen sites while accelerating the CO and methanol oxidation in a similar way to that of Ru in the PtRu binary alloy system. HPMo self-assembled on CNTs followed by decomposition has been used to synthesize highly dispersed MoOx/CNTs as an effective support for Pt NP electrocatalysts for the methanol oxidation reaction with significantly enhanced activity and stability.147
PtSn alloys have been demonstrated to enhance the ethanol oxidation reaction.148 Our group developed a facile method to functionalize MWCNT with THF, a commonly used solvent.49 THF may interact with CNT through CH–π interaction. Furthermore, the oxygen atoms in THF are accessible to attract PtCl62− and Sn4+ (Fig. 11(I)). This concept has been demonstrated by uniform growth of Pt and PtSn on THF functionalized MWCNT (Fig. 11(II)). The formation of PtSn binary NPs has been demonstrated by the increase of lattice parameter of PtSn/MWCNTs with increasing in the Sn content. The catalyst shows a high and tunable electrocatalytic activity for the ethanol oxidation with excellent stability for fuel cells (Fig. 11(III)).
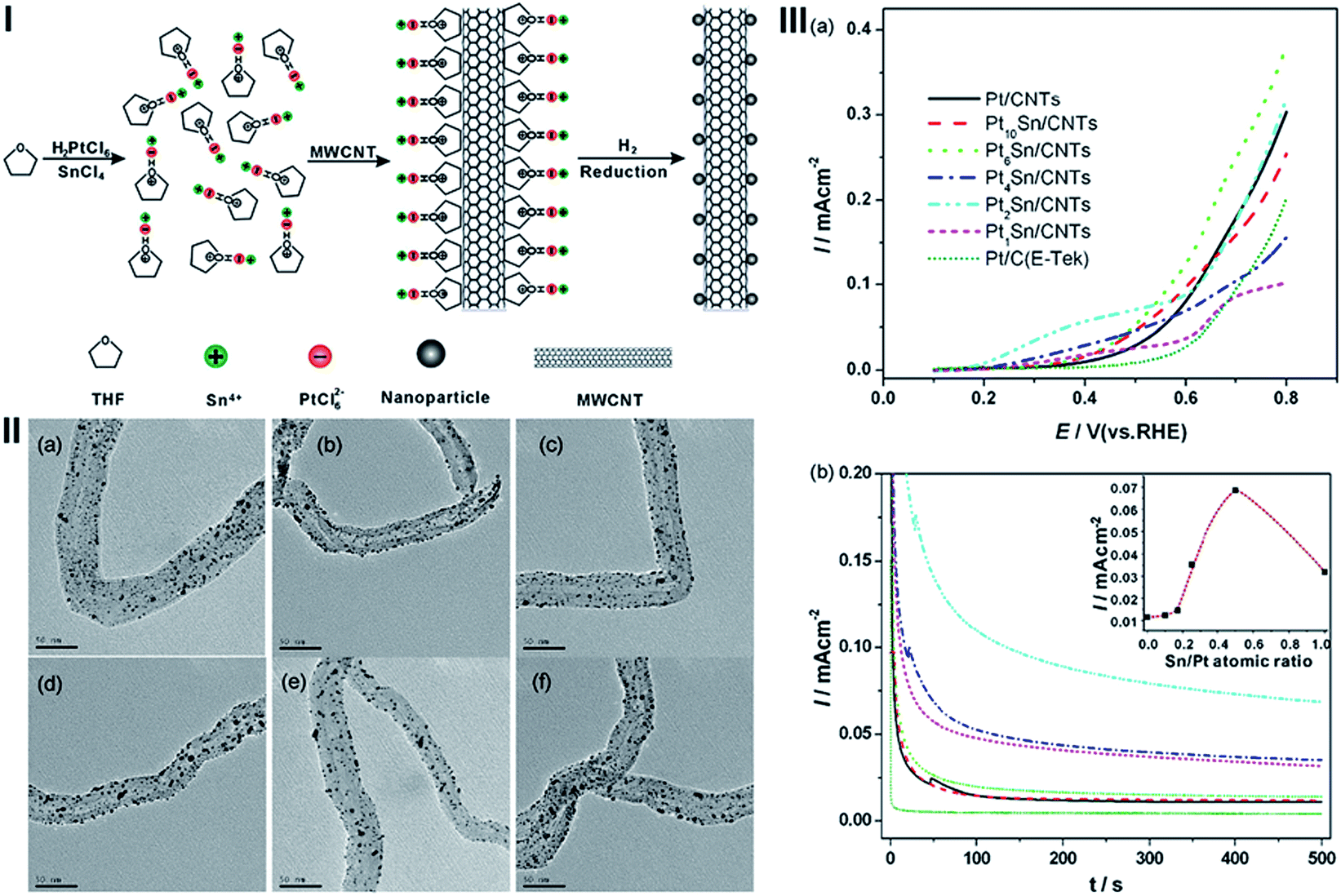 | ||
| Fig. 11 (I) Scheme of the synthesis of PtSn nanoparticle catalysts on THF-functionalized MWCNTs. (II) TEM micrographs of the PtSn/MWCNTs with different Pt/Sn atomic ratios; (a) Pt/MWCNTs, (b) Pt10Sn/MWCNTs, (c) Pt6Sn/MWCNTs, (d) Pt4Sn/MWCNTs, (e) Pt2Sn/MWCNTs and (f) Pt1Sn/MWCNTs. (III) (a) Linear sweep voltammetry and (b) chronoamperometry curves for the ethanol oxidation on Pt/MWCNTs and PtSn/MWCNTs with different Pt/Sn atomic ratios in a N2-saturated 0.5 M H2SO4 + 1.0 M ethanol solution at room temperature. LSV was measured at a scan rate of 1 mV s−1 and CA was measured at 0.5 V (RHE). Inset in (b) is the plots of current measured at 0.5 V (RHE) after 500 s test versus the Sn/Pt atomic ratios of PtSn/MWCNT catalysts (after ref. 49 reproduced with permission). | ||
We also explored the non-covalent functionalization method to immobilize water soluble HPW on Pt/C NPs via the electrostatic interaction between the negatively charged HPW and the positively charged functional groups of CS which has been attached to Pt/C NPs to provide positively charged sites for the self-assembly of HPW.74 The results indicate that HPW assembled on CS-functionalized Pt/C is very stable and Pt/C-CS-HPW catalyst has a higher utilization efficiency as compared to that of pristine Pt/C catalyst. Electrochemical activity of Pt/C-CS-HPW catalysts for methanol oxidation and oxygen reduction reaction (ORR) is significantly higher than that of Pt/C catalysts without assembled HPW. The enhanced electrocatalytic activities of HPW assembled Pt/C catalysts are most likely due to the synergistic effect between assembled HPW and Pt/C NPs and the presence of HPW leads to a downward shift in the d-band center of Pt catalyst and facilitates the oxidative removal of COads poisoning species for methanol oxidation and desorption of Oads species for ORR on Pt catalysts.
Both Pt and Pd have a face centered cubic structure with almost the same unit cell length, and therefore they can easily form an alloy.149,150 Ghosh et al. have explored the decoration of MWCNT with Pt–Pd alloy using the non-covalent polymer assisted impregnation.150 The composition of the catalyst can be controlled by tuning the molar ratio of the precursors during impregnation. The Pt46Pd54 catalyst has a superior electrocatalytic activity and durability toward the ORR with a very high tolerance toward methanol contamination.
Table 1 lists the most polyelectrolytes/solvents used in the functionalization of CNTs as supports for Pt-based NPs.
| Polyelectrolyte/agent | Structure | Metal NPs/size | Reducing method | Ref. |
|---|---|---|---|---|
| a In situ polymerization on acid-treated CNTs.b PVP is in situ grafted onto CNTs.c On CS and HPW or HPMo co-functionalized CNTs. | ||||
| Polybenzimidazole, PBI | 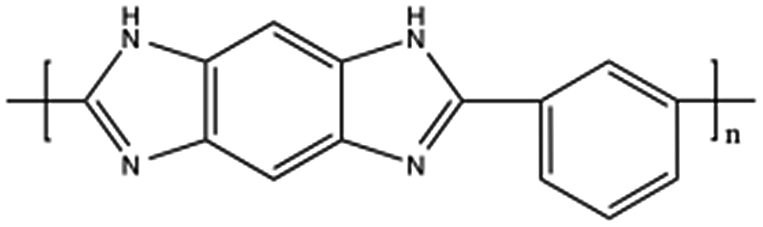 |
Pt/4.0 nm | EG, refluxing at 140 °C for 4 h | 65 |
| Poly(diallyldimethylammonium chloride), PDDA | 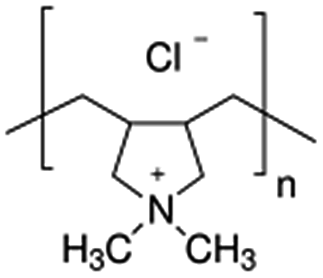 |
Pt/1.8 nm | EG, pH = 12.5, refluxing at 130 °C | 67 |
| As above | Pd/3.5 nm | NaBH4 | 116 | |
| As above | PtRu/3.1 nm | EG, pH = 6.5, microwave for 2 min | 152 | |
| Poly(sodium 4-styrenesulfonate), PSS | 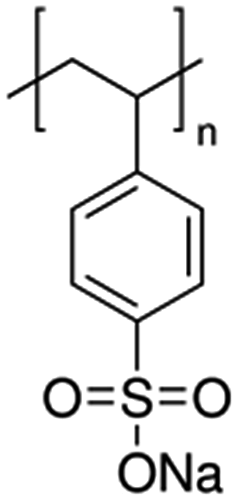 |
Pt/1.5 nm | EG, pH = 12, microwave for 60 s | 114 |
| Poly(allylamine hydrochloride), PAH | 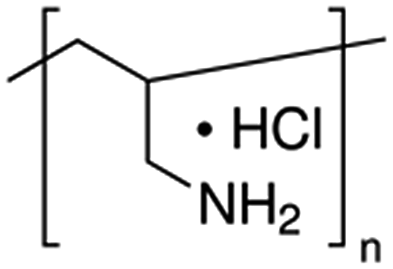 |
Pt/1.5 nm | EG, pH = 12, microwave for 60 s | 114 |
| Poly(acrylic acid), PAA | 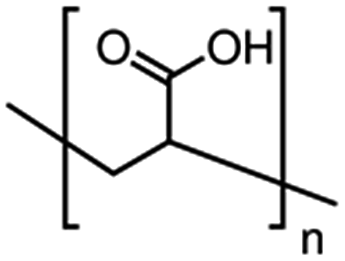 |
Pt/1.5 nm | EG, pH = 12, microwave for 60 s | 114 |
| Polyaniline, PANIa | 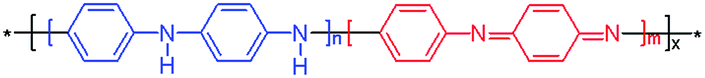 |
Pt/3.6 nm | HCOOH, stirred for 1 day | 118 |
| Sodium dodecylsulfate, SDS | 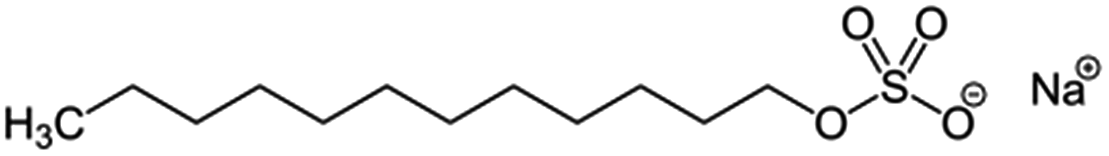 |
Pt/2.5–4.1 nm | Urea at 80 °C; EG at 120 °C | 121 |
| Aniline | 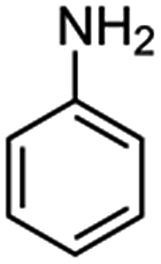 |
Pt/1.9–2.4 nm | Sodium citrate/H2 at 400 °C for 4 h | 120 |
| Polyvinyl pyrrolidone, PVPb | 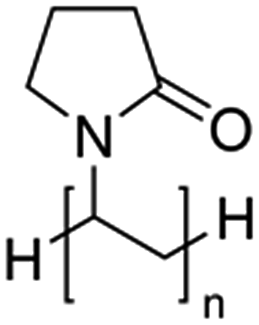 |
Pt/1.8 nm | EG, pH = 12.5, 140 °C for 3 h | 66 |
| 1-Aminopyrene, 1-AP | 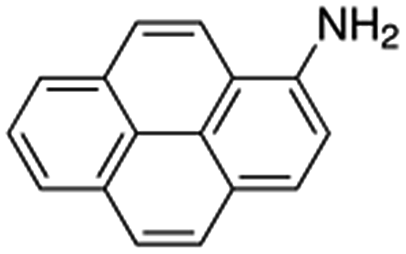 |
PtRu/2.0 nm | EG, pH = 6.5, microwave for 2 min | 16 |
| Poly(ethyleneimine), PEI | 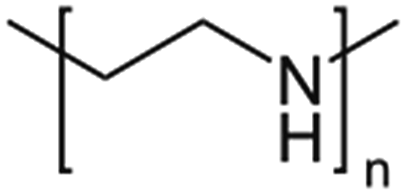 |
PtRu/2.5 nm | EG, pH = 6.5, microwave | 73 |
| Tetrahydrofuran, THF | 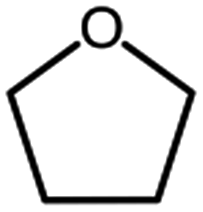 |
PtSn/4.2 nm | 300 °C under flowing H2 for 2 | 49 |
| As above | PtRu/3.3 nm | EG, pH = 6.5, microwave for 2 min | 152 | |
| Chitosan, CS | 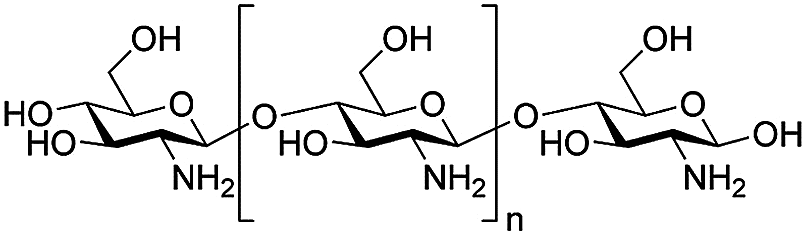 |
PtRu/3c nm | EG, 140 °C, 4 h | 146 |
4. Interaction between functionalization agents, Pt NPs and CNTs
4.1. Effect of functionalization agents
The electrocatalytic activity of Pt NPs is also affected by the nature of functionalization agents. We investigated the effect of polyelectrolytes on the electronic structure of Pt NPs on polyelectrolyte functionalized on CNTs.114 The spectroscopic and electrochemical characterization as well as DFT calculation revealed that polyanions such as PSS and PAA with electron-rich functional groups would donate electrons to Pt atoms which cause an increase in the electron density around Pt atoms and downshift d-band center of Pt (see Fig. 12).114 Based on the d-band theory,151 the downshift of the d-band center would result in a weaker chemisorptions of oxygen-containing species (e.g., CO), thus enhancing the electrocatalytic activity of Pt NPs. The nature of the functional groups in the interlinkers that are introduced via surface functionalization can alter the electronic and catalytic activities of Pt NPs.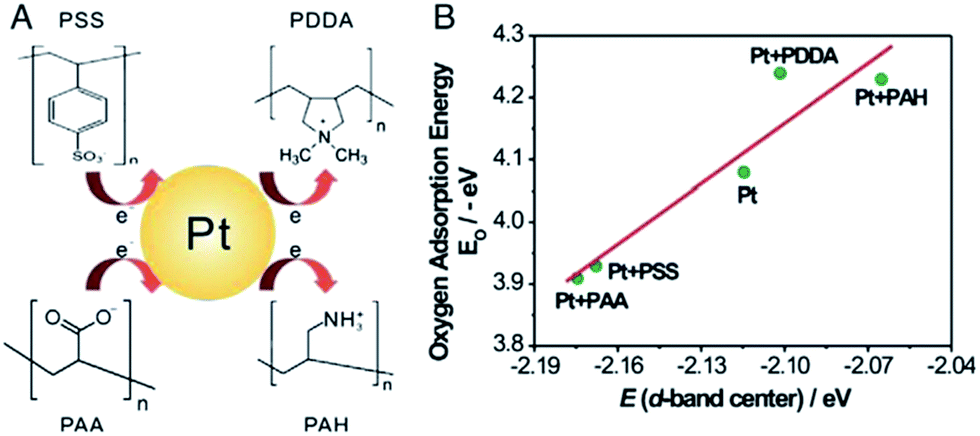 | ||
| Fig. 12 (A) The possible effect of the charged functional groups of polyanions (PSS and PAA) and polycations (PDDA and PAH) on the electron donor–acceptor behaviour of Pt NP; and (B) correlation between the adsorption energy of O and the d-band center of Pt slabs (after ref. 114 reproduced with permission). | ||
It is of significant importance to understand the influence of the polyelectrolyte and/or solvent on the morphology, distribution and electrocatalytic activity of the Pt-based NPs supported on CNTs in order to develop better and more efficient catalysts for fuel cells. Most recently, the effect of nitrogen-containing polyelectrolyte-functionalized CNTs was studied on the supported PtRu NPs using PDDA, PEI, 1-AP and THF as functioning agents.152 PtRu NPs uniformly dispersed on functionalized CNTs with average size of ∼3.0 nm. Most important, the results demonstrate that nitrogen-containing functional groups of the functionalization agents play a critical role in the electrocatalytic activity of PtRu NPs supported on CNTs. PtRu NPs supported on PEI, AP and in less extent PDDA functionalized CNTs exhibited significantly higher electrocatalytic activity and stability for the methanol oxidation reaction as compared with PtRu supported on THF-CNTs and conventional acid-treated CNTs. PEI is a cationic polymer with repeating unit composed of the amine group and two carbon aliphatic CH2CH2 spacers. 1-AP contains amino groups while PDDA is a water-soluble quaternary ammonium polyelectrolyte. THF is an oxygen-containing heterocycle with five-membered rings and no nitrogen-containing groups. The assembly of the PEI on CNTs would be strong due to the high molecular weight and the electrostatic attraction between the positively charged PEI and negatively charge CNTs, resulting in the attachment of large amount of N-containing amine group on the outer surface of CNTs. PDDA is also a cationic polymer with N-containing repeating unit, but the π–π interaction with the CNTs surface primarily occurs through the 2–3% of unsaturated contaminant in the PDDA chain.153,154 This may limit the attachment of N-containing functional groups to the surface of CNTs. In the case of 1-AP, the interaction with CNTs is via π–π stacking. However, due to the bulk size of benzene rings, the attached nitrogen functional groups on the surface of CNTs would be expected to be smaller than that of PEI but higher than that of PDDA. Fig. 13 shows schematically the assembly of PEI, 1-AP and PDDA on the outer wall surface of CNTs.152 The high density of N-containing functional groups assembled on CNTs in the case of PEI and 1-AP is consistent with the +0.23 eV positive shift of Pt4f binding energy of PtRu NPs supported on PEI and 1-AP functionalized CNTs as compared to that supported on PDDA-CNTs. The nitrogen in the amino functional groups of the attached functionalization agents can activate the π electrons of CNTs supports and promotes the methanol oxidation reaction, similar to that observed for the N-doped carbon materials like CNTs and grapheme.155–158 The superior activity of PtRu NPs supported on functionalized CNTs is most likely due to the strong interaction of the electron rich nitrogen-containing functional groups of the functionalization agents such as PEI, 1-AP and in less extend PDDA and the PtRu NPs assembled on CNTs.
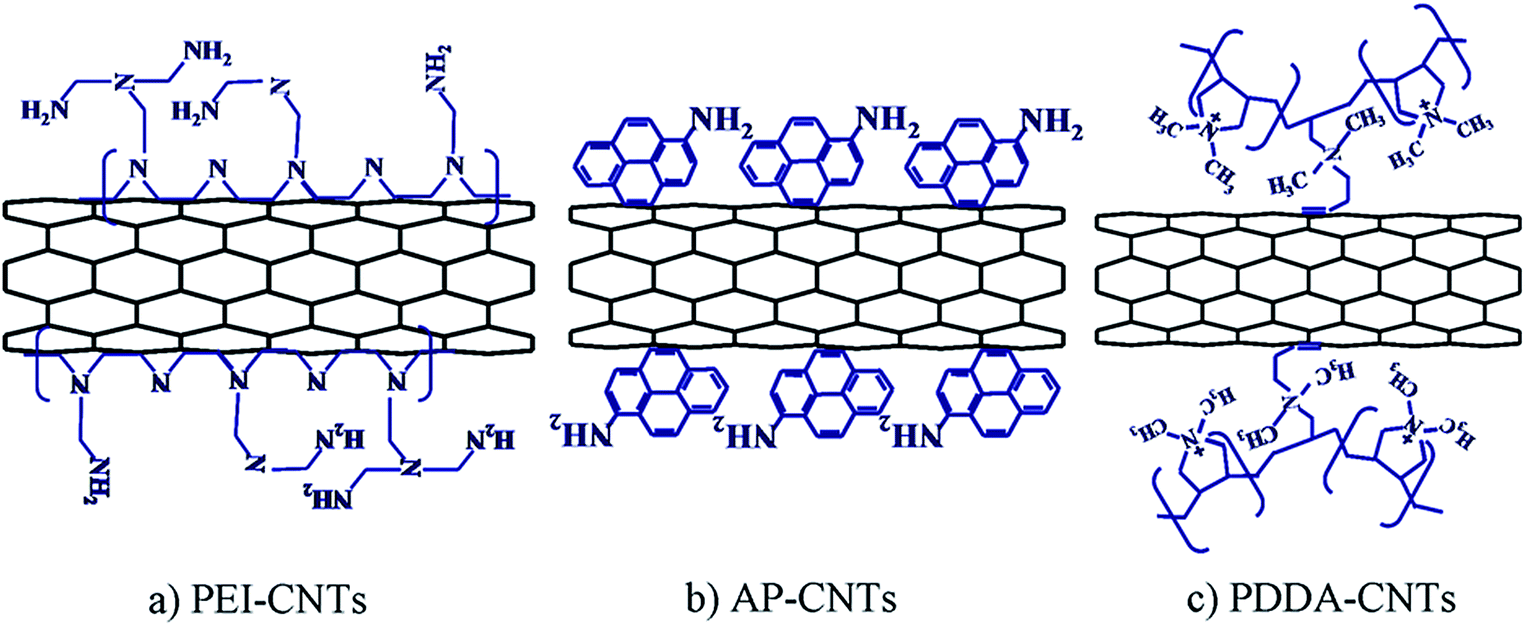 | ||
| Fig. 13 Schematic of the assembly and functionalization of PEI, 1-AP and PDDA on the surface of CNTs (after ref. 152 reproduced with permission). | ||
4.2. Interaction between Pt NPs and CNTs
The interaction or bonding between the Pt NPs and CNTs also play an important role in the electrocatalytic activity of supported Pt-based NPs. Zhou et al. investigated the interaction between Pt NPs and CNTs using X-ray absorption near edge structures (XAENES).159 They showed that the crystalline Pt NPs interact with CNTs through synergic binding involving charge redistribution between C 2p-derived states and Pt 5d bands due to the presence of unsaturation in the graphene sheets (delocalized π orbitals). Such bonding scheme would facilitate the uniform dispersion and immobilization of Pt NPs on the CNT surface, which would prevent lateral diffusion of Pt NPs under fuel cell operating conditions.The inner tubes of CNT support could also play an important role in the electrocatalytic activity of supported Pt-based NPs. We reported recently that pristine CNTs show a distinctive volcano-type dependence of electrocatalytic activity for the O2 evolution reaction (OER) in alkaline solutions as a function of number of concentric tubes or walls and the highest activities are observed on CNTs composed of between 2–7 concentric tubes or walls.160 Further studies showed that the Pd NPs supported on THF-functionalized CNTs for the ethanol oxidation reaction in alkaline solutions also show distinctive volcano-shaped dependence on the number of walls of CNTs (see Fig. 14).161 In this study, Pd NPs with diameter of 2.1 to 2.8 nm were deposited uniformly on THF-functionalized CNTs via self-assembly route with no agglomeration.49 The best activity was observed on Pd NPs supported on triple-walled CNTs (TWCNTs), similar to that observed for the OER on pristine CNTs in alkaline solution.160 Similar to OER on pristine CNTs, the adsorption and migration or diffusion of oxygen-containing species, OHads, on the outer wall surface of CNTs would lead to the fast stripping of ethoxy and CO species via the interaction with OHads, releasing acetate as final product.162 The charge transfer could occur on the inner tubes through the electron tunnelling between the outer wall and inner tubes under the electrochemical polarization potential driving force. Such effective charge transfer process can effectively reduce the PdO/Pd–OH and release the active sites on Pd NPs for the EOR. Such charge transfer through electron tunnelling would not be possible with SWCNTs and diminishes when the number of walls exceeds 12 for typical MWCNTs due to the reduced driving force (i.e., dc bias). This preliminary result indicates the possible activation effect of the inner tubes of CNT supports on the electrocatalytic activity of supported metal catalysts.
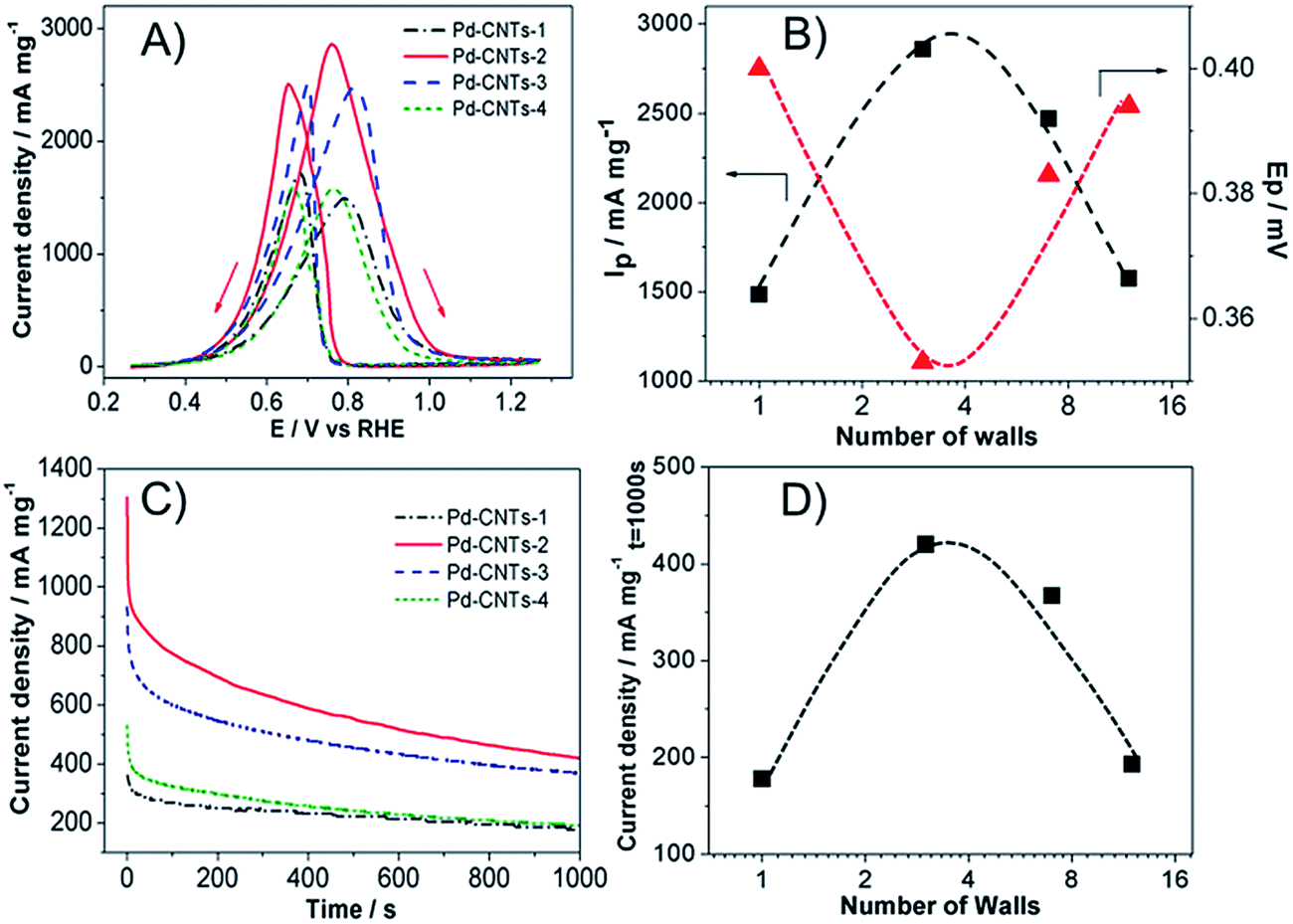 | ||
| Fig. 14 (A) Cyclic voltammetry curves of Pd-CNTs catalysts for ethanol oxidation reaction (EOR), measured in a 1.0 M ethanol + 1.0 M KOH solution at scan rate 50 mV s−1, (B) plots of peak current density and onset potential of EOR against wall numbers of CNTs, (C) chronoamperometry curves of Pd-CNTs catalysts for EOR, measured at 0.67 V in a 1.0 M ethanol + 1.0 M KOH solution, (D) plots of current density measured at t = 1000 s for EOR. Pd loading was 0.05 mg cm−2 (after ref. 161 reproduced with permission). | ||
In addition to the positive role of the interaction between Pt NPs and CNTs on the durability of the Pt-based NP catalysts, the interaction or interconnectivity of Pt NPs on CNTs is another fundamental reason for the observed much better electrochemical performance of Pt-based NPs supported on CNTs for the methanol oxidation and oxygen reduction reaction as compared to that on the Pt NPs supported on high surface area carbon.18,55,163–168 We studied in details the relationship between the interconnectivity of Pt NPs and the electrocatalytic activity for fuel cells.17 Pt NPs were assembled on PDDA-functionalized CNTs with loading of Pt NPs in the range from 10 to 93 wt% and controlled size (1.5 to 3 nm). The results show that the catalytic activity of Pt/CNTs for methanol oxidation, ORR and CO oxidation reactions increases with the Pt loading, following a characteristic S-shaped profile. Based on the concept of interconnectivity of Pt NPs defined as the ratio of the total number of interconnections between particles divided by the total number of particles involved (Fig. 15(I)), the electrocatalytic activities of Pt/CNTs catalysts show the linear dependence on the interconnectivity (Fig. 15(II)). The magnitude of the interconnectivity of Pt NPs is a critical factor influencing their electrocatalytic activity, and the interconnected Pt NPs are more active than the isolated Pt NPs. The high electrocatalytic activity of interconnected Pt NPs is related to the increased active intergrain boundaries. These grain boundaries provide large number of defect sites and generate discontinuities in the crystal planes of interconnected Pt NPs in Pt/CNTs, providing active sites for CO removal, methanol oxidation and O2 reduction reactions.
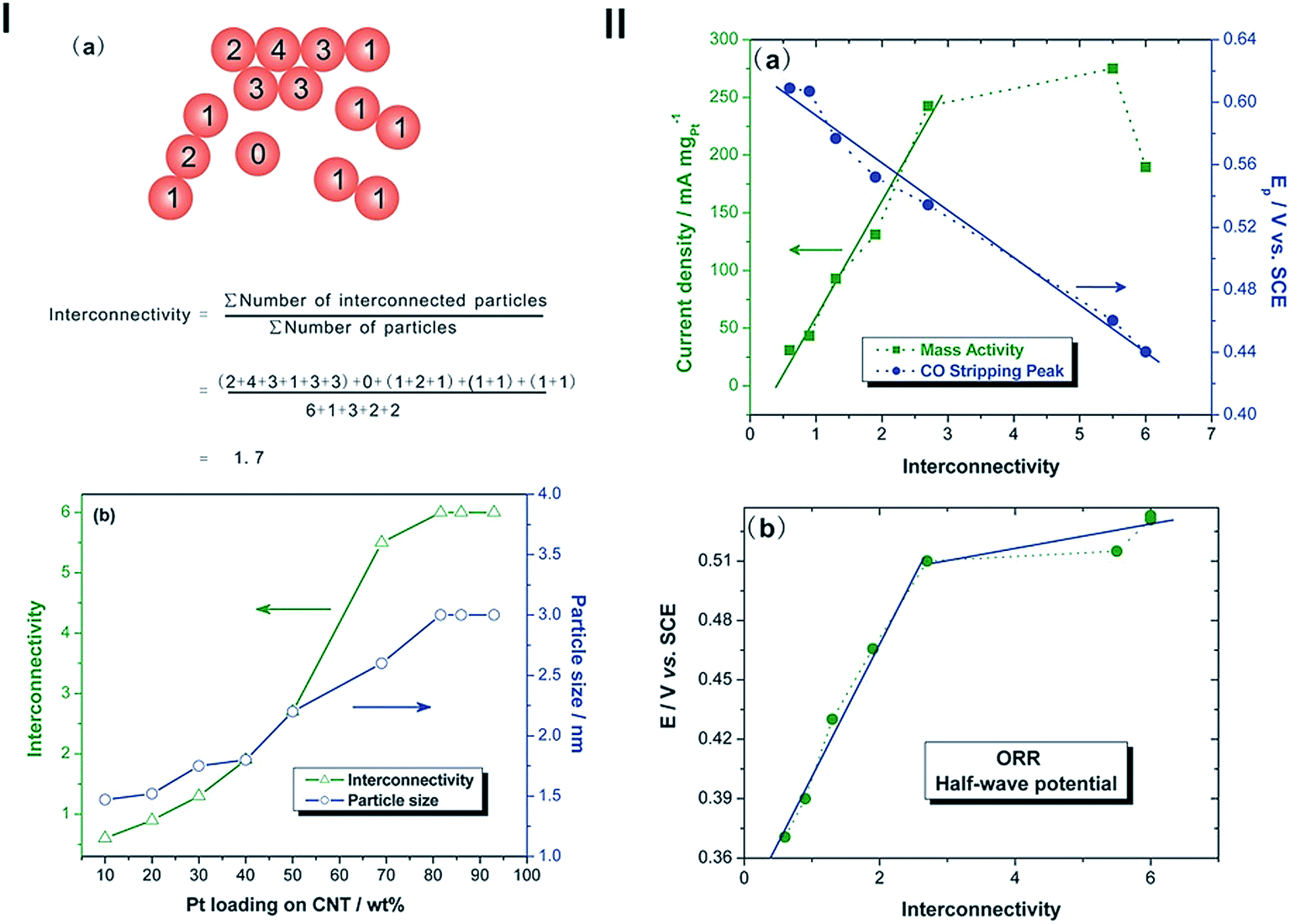 | ||
| Fig. 15 (I) (a) A calculation example of the interconnectivity for 14 spherical particles. The number on the spherical particle represents the number of interconnected particles with the particle concerned; and (b) dependence of interconnectivity and size of Pt NPs of Pt/MWCNTs catalysts on Pt loadings. (II) (a) Plots of the mass specific activity for the methanol oxidation reaction and the peak CO stripping potential as a function of the interconnectivity of Pt NPs on MWCNTs; (b) plots of the half-wave potential for ORR as a function of the interconnectivity of Pt NPs on CNTs (after ref. 17 reproduced with permission). | ||
5. Summary
Non-covalent functionalization is one of the most effective methods to surface modification of CNTs without detrimental effect on the characteristic π–electron system, the physical and structural integrity of CNTs. Non-covalent functionalization or wrapping by polyelectrolytes or solvents introduces large number of active sites uniformly on the surface of outer walls of CNTs without generating oxygenated functional groups or breaking the CC bonds of the graphene sheets of the CNTs. Pt-based NPs can then be uniformly dispersed on CNTs via self-assembly principle with significantly reduced agglomerates. Non-covalent functionalized CNTs are particularly attractive as effective supports for Pt-based NP catalysts for fuel cells due to the fact that (1) non-covalent functionalization method is versatile and there are large number of materials including solvents, surfactants, aromatic compounds, polymers or polyelectrolytes, which can be utilized as functionalization agents. (2) The electrocatalytic activity of Pt-based NPs can be manipulated or promoted by the nature of the functionalization agents as shown by Cheng et al.152 and Fujigaya and Nakashima.123 (3) The well-define geometry of outer wall surface of CNTs substantially promotes interconnectivity between Pt NPs, which leads to the significant increase in the grain boundaries between the NPs and thus active sites for fuel cell reactions,17 in addition to the synergistic interaction between Pt NPs and CNTs.159
Nevertheless, there are still challenges to further enhance their electrocatalytic performance. Since NP shapes also significantly affect the electrocatalytic activities of Pt-based catalysts, it would be desirable to control the shape or crystal facets of Pt NPs via non-covalent functionalization process. It is of technological and scientific importance to explore the effect of the structural parameters of CNTs such as diameter, length and in particularly number of walls on the electrocatalytic performance of CNT supported Pt-based NPs, as shown recently on the effect of inner tubes of CNT support on the electrocatalytic activity of Pd NPs for EOR.161 Fundamental mechanism for the performance enhancement of Pt NPs supported on non-covalent CNT has not been fully understood. Thus, substantial efforts are needed in the understanding of the underlying mechanism of the electrocatalytic activity of Pt-based NPs on non-covalent functionalized CNTs, particularly at the atomic scale, from both experimental and theoretical point of view.
Acknowledgements
This work was supported by the Australian Research Council under the Discovery Project funding scheme (project number: DP120102325, DP120104932). Beijing Higher Education Young Elite Teacher Project (29201493) and the Fundamental Research Funds for the Central Universities, China.References
- Y. Wang, K. S. Chen, J. Mishler, S. C. Cho and X. C. Adroher, Appl. Energy, 2011, 88, 981–1007 CrossRef CAS PubMed
.
- F. Lufrano, V. Baglio, P. Staiti, V. Antonucci and A. S. Arico, J. Power Sources, 2013, 243, 519–534 CrossRef CAS PubMed
- H. L. Tang, J. R. Li, Z. Wang, H. J. Zhang, M. Pan and S. P. Jiang, in Nanotechnology for Sustainable Energy, ed. Y. H. Hu, U. Burghaus and S. Qiao, 2013, vol. 1140, pp. 243–263 Search PubMed
- V. Mehta and J. S. Cooper, J. Power Sources, 2003, 114, 32–53 CrossRef CAS
- A. B. Stambouli, Renewable Sustainable Energy Rev., 2011, 15, 4507–4520 CrossRef CAS PubMed
- Z. Peng and H. Yang, Nano Today, 2009, 4, 143–164 CrossRef CAS PubMed
- V. Mazumder, Y. Lee and S. Sun, Adv. Funct. Mater., 2010, 20, 1224–1231 CrossRef CAS
- M. N. Cao, D. S. Wu and R. Cao, ChemCatChem, 2014, 6, 26–45 CrossRef CAS
- J. B. Wu and H. Yang, Acc. Chem. Res., 2013, 46, 1848–1857 CrossRef CAS PubMed
- N. Jha, P. Ramesh, E. Bekyarova, X. J. Tian, F. H. Wang, M. E. Itkis and R. C. Haddon, Sci. Rep., 2013, 3, 2257 Search PubMed
- K. G. Nishanth, P. Sridhar, S. Pitchumani and A. K. Shukla, Fuel Cells, 2012, 12, 146–152 CrossRef CAS
- C. Hiromi, M. Inoue, A. Taguchi and T. Abe, Electrochim. Acta, 2011, 56, 8438–8445 CrossRef CAS PubMed
- X. R. Jiang, T. M. Gur, F. B. Prinz and S. F. Bent, J. Electrochem. Soc., 2010, 157, B314–B319 CrossRef CAS PubMed
- R. S. Amin, K. M. El-Khatib, R. M. A. Hameed, E. R. Souaya and M. A. Etman, Appl. Catal., A, 2011, 407, 195–203 CrossRef CAS PubMed
- E. Antolini, J. R. C. Salgado and E. R. Gonzalez, Appl. Catal., B, 2006, 63, 137–149 CrossRef CAS PubMed
- S. Y. Wang, X. Wang and S. P. Jiang, Langmuir, 2008, 24, 10505–10512 CrossRef CAS PubMed
- S. Y. Wang, S. P. Jiang, T. J. White, J. Guo and X. Wang, J. Phys. Chem. C, 2009, 113, 18935–18945 CAS
- Z. Q. Tian, S. P. Jiang, Y. M. Liang and P. K. Shen, J. Phys. Chem. B, 2006, 110, 5343–5350 CrossRef CAS PubMed
- S. M. Alia, G. Zhang, D. Kisailus, D. S. Li, S. Gu, K. Jensen and Y. S. Yan, Adv. Funct. Mater., 2010, 20, 3742–3746 CrossRef CAS
- S. Y. Wang, S. P. Jiang, X. Wang and J. Guo, Electrochim. Acta, 2011, 56, 1563–1569 CrossRef CAS PubMed
- S. Y. Wang, N. Kristian, S. P. Jiang and X. Wang, Nanotechnology, 2009, 20, 25605 CrossRef PubMed
- V. Di Noto and E. Negro, Fuel Cells, 2010, 10, 234–244 CrossRef CAS
- N. V. Long, T. D. Hien, T. Asaka, M. Ohtaki and M. Nogami, Int. J. Hydrogen Energy, 2011, 36, 8478–8491 CrossRef PubMed
- K. P. Gong, F. Du, Z. H. Xia, M. Durstock and L. M. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed
- Y. G. Chen, L. Zhuang and J. T. Lu, Chin. J. Catal., 2007, 28, 870–874 CrossRef CAS
- L. B. Wu, Y. Nabae, S. Moriya, K. Matsubayashi, N. M. Islam, S. Kuroki, M. Kakimoto, J. Ozaki and S. Miyata, Chem. Commun., 2010, 46, 6377–6379 RSC
- R. Othman, A. L. Dicks and Z. H. Zhu, Int. J. Hydrogen Energy, 2012, 37, 357–372 CrossRef CAS PubMed
- C. K. Rhee, B.-J. Kim, C. Ham, Y.-J. Kim, K. Song and K. Kwon, Langmuir, 2009, 25, 7140–7147 CrossRef CAS PubMed
- Y. Xu, R. B. Getman, W. A. Shelton and W. F. Schneider, Phys. Chem. Chem. Phys., 2008, 10, 6009–6018 RSC
- Y. Zhou, K. Neyerlin, T. S. Olson, S. Pylypenko, J. Bult, H. N. Dinh, T. Gennett, Z. Shao and R. O'Hayre, Energy Environ. Sci., 2010, 3, 1437–1446 CAS
- X. Yu and S. Ye, J. Power Sources, 2007, 172, 133–144 CrossRef CAS PubMed
- X. Yu and S. Ye, J. Power Sources, 2007, 172, 145–154 CrossRef CAS PubMed
- H. Matsumori, S. Takenaka, H. Matsune and M. Kishida, Appl. Catal., A, 2010, 373, 176–185 CrossRef CAS PubMed
- S. L. Candelaria, Y. Y. Shao, W. Zhou, X. L. Li, J. Xiao, J. G. Zhang, Y. Wang, J. Liu, J. H. Li and G. Z. Cao, Nano Energy, 2012, 1, 195–220 CrossRef CAS PubMed
- W. Y. Wong, W. R. W. Daud, A. B. Mohamad, A. A. H. Kadhum, E. H. Majlan and K. S. Loh, Diamond Relat. Mater., 2012, 22, 12–22 CrossRef CAS PubMed
- Y. Xiang, S. Lu and S. P. Jiang, Chem. Soc. Rev., 2012, 41, 7291–7321 RSC
- S. H. Joo, S. J. Choi, I. Oh, J. Kwak, Z. Liu, O. Terasaki and R. Ryoo, Nature, 2001, 412, 169–172 CrossRef CAS PubMed
- S. Q. Song, Y. R. Liang, Z. H. Li, Y. Wang, R. W. Fu, D. C. Wu and P. Tsiakaras, Appl. Catal., B, 2010, 98, 132–137 CrossRef CAS PubMed
- P. J. Ferreira, G. J. la O, Y. Shao-Horn, D. Morgan, R. Makharia, S. Kocha and H. A. Gasteiger, J. Electrochem. Soc., 2005, 152, A2256–A2271 CrossRef PubMed
- D. A. Stevens and J. R. Dahn, Carbon, 2005, 43, 179–188 CrossRef CAS PubMed
- O. V. Cherstiouk, P. A. Simonov, V. B. Fenelonov and E. R. Savinova, J. Appl. Electrochem., 2010, 40, 1933–1939 CrossRef CAS PubMed
- C. C. Hung, P. Y. Lim, J. R. Chen and H. C. Shih, J. Power Sources, 2011, 196, 140–146 CrossRef CAS PubMed
- S. E. Jang and H. Kim, J. Am. Chem. Soc., 2010, 132, 14700–14701 CrossRef CAS PubMed
- M. X. Wang, Q. Liu, H. F. Sun, N. Ogbeifun, F. Xu, E. A. Stach and J. A. Xie, Mater. Chem. Phys., 2010, 123, 761–766 CrossRef CAS PubMed
- D. M. Guldi, G. M. A. Rahman, V. Sgobba and C. Ehli, Chem. Soc. Rev., 2006, 35, 471–487 RSC
- W. Zhang, P. Sherrell, A. I. Minett, J. M. Razal and J. Chen, Energy Environ. Sci., 2010, 3, 1286–1293 CAS
- P. Singh, S. Campidelli, S. Giordani, D. Bonifazi, A. Bianco and M. Prato, Chem. Soc. Rev., 2009, 38, 2214–2230 RSC
- V. Sanz, E. Borowiak, P. Lukanov, A. M. Galibert, E. Flahaut, H. M. Coley, S. R. P. Silva and J. McFadden, Carbon, 2011, 49, 1775–1781 CrossRef CAS PubMed
- D. L. Wang, S. F. Lu and S. P. Jiang, Electrochim. Acta, 2010, 55, 2964–2971 CrossRef CAS PubMed
- B. Wu, Y. Kuang, X. Zhang and J. Chen, Nano Today, 2011, 6, 75–90 CrossRef CAS PubMed
- M. F. Variava, T. L. Church, A. T. Harris and A. I. Minett, J. Mater. Chem. A, 2013, 1, 8509–8520 CAS
- J. H. Kim, D. S. Kim, H. H. Chun and Y. T. Kim, Electrochem. Commun., 2012, 19, 85–89 CrossRef CAS PubMed
- S. W. Kim, T. Kim, Y. S. Kim, H. S. Choi, H. J. Lim, S. J. Yang and C. R. Park, Carbon, 2012, 50, 3–33 CrossRef CAS PubMed
- D. A. Britz and A. N. Khlobystov, Chem. Soc. Rev., 2006, 35, 637–659 RSC
- Y. C. Xing, J. Phys. Chem. B, 2004, 108, 19255–19259 CrossRef CAS
- N. Rajalakshmi, H. Ryu, M. M. Shaijumon and S. Ramaprabhu, J. Power Sources, 2005, 140, 250–257 CrossRef CAS PubMed
- S. B. Yin, P. K. Shen, S. Q. Song and S. P. Jiang, Electrochim. Acta, 2009, 54, 6954–6958 CrossRef CAS PubMed
- D. J. Guo and H. L. Li, J. Colloid Interface Sci., 2005, 286, 274–279 CrossRef CAS PubMed
- X. Wang, W. Z. Li, Z. W. Chen, M. Waje and Y. S. Yan, J. Power Sources, 2006, 158, 154–159 CrossRef CAS PubMed
- T. Fujigaya and N. Nakashima, Polym. J., 2008, 40, 577–589 CrossRef CAS
- C. Y. Hu, Y. J. Xu, S. W. Duo, R. F. Zhang and M. S. Li, J. Chin. Chem. Soc., 2009, 56, 234–239 CAS
- K. R. Reddy, B. C. Sin, K. S. Ryu, J.-C. Kim, H. Chung and Y. Lee, Synth. Met., 2009, 159, 595–603 CrossRef CAS PubMed
- H.-Y. Lee, W. Vogel and P. P.-J. Chu, Langmuir, 2011, 27, 14654–14661 CrossRef CAS PubMed
- N. C. Cheng, S. C. Mu, X. J. Chen, H. F. Lv, M. Pan and P. P. Edwards, Electrochim. Acta, 2011, 56, 2154–2159 CrossRef CAS PubMed
- M. Okamoto, T. Fujigaya and N. Nakashima, Small, 2009, 5, 735–740 CrossRef CAS PubMed
- Y. L. Hsin, K. C. Hwang and C. T. Yeh, J. Am. Chem. Soc., 2007, 129, 9999–10010 CrossRef CAS PubMed
- S. Wang, S. P. Jiang and X. Wang, Nanotechnology, 2008, 19, 265601 CrossRef PubMed
- X. L. Li, Y. Q. Liu, L. Fu, L. C. Cao, D. C. Wei and Y. Wang, Adv. Funct. Mater., 2006, 16, 2431–2437 CrossRef CAS
- Y. Y. Ou and M. H. Huang, J. Phys. Chem. B, 2006, 110, 2031–2036 CrossRef CAS PubMed
- S. Wang, X. Wang and S. P. Jiang, Phys. Chem. Chem. Phys., 2011, 13, 7187–7195 Search PubMed
- S. Y. Wang, S. P. Jiang and X. Wang, J. Nanopart. Res., 2011, 13, 2973–2979 CrossRef CAS
- D. L. Wang, S. F. Lu and S. P. Jiang, Chem. Commun., 2010, 46, 2058–2060 RSC
- Y. Cheng and S. P. Jiang, Electrochim. Acta, 2013, 99, 124–132 CrossRef CAS PubMed
- D. L. Wang, S. F. Lu, Y. Xiang and S. P. Jiang, Appl. Catal., B, 2011, 103, 311–317 CrossRef CAS PubMed
- M. H. Chakrabarti, C. T. J. Low, N. P. Brandon, V. Yufit, M. A. Hashim, M. F. Irfan, J. Akhtar, E. Ruiz-Trejo and M. A. Hussain, Electrochim. Acta, 2013, 107, 425–440 CrossRef CAS PubMed
- D. Vairavapandian, P. Vichchulada and M. D. Lay, Anal. Chim. Acta, 2008, 626, 119–129 CrossRef CAS PubMed
- M. S. Saha and A. Kundu, J. Power Sources, 2010, 195, 6255–6261 CrossRef CAS PubMed
- L. Li, A. H. Larsen, N. A. Romero, V. A. Morozov, C. Glinsvad, F. Abild-Pedersen, J. Greeley, K. W. Jacobsen and J. K. Nørskov, J. Phys. Chem. Lett., 2012, 4, 222–226 CrossRef
- F. Yang, Q. Zhang, Y. Liu and S. Chen, J. Phys. Chem. C, 2011, 115, 19311–19319 CAS
- Y. Sun, L. Zhuang, J. Lu, X. Hong and P. Liu, J. Am. Chem. Soc., 2007, 129, 15465–15467 CrossRef CAS PubMed
- I. N. Leontyev, S. V. Belenov, V. E. Guterman, P. Haghi-Ashtiani, A. P. Shaganov and B. Dkhil, J. Phys. Chem. C, 2011, 115, 5429–5434 CAS
- M. Shao, A. Peles and K. Shoemaker, Nano Lett., 2011, 11, 3714–3719 CrossRef CAS PubMed
- M.-C. Daniel and D. Astruc, Chem. Rev., 2003, 104, 293–346 CrossRef PubMed
- S. Takenaka, H. Matsumori, H. Matsune, E. Tanabe and M. Kishida, J. Electrochem. Soc., 2008, 155, B929–B936 CrossRef CAS PubMed
- J. Zhang, K. Sasaki, E. Sutter and R. R. Adzic, Science, 2007, 315, 220–222 CrossRef CAS PubMed
- B. Wu, D. Hu, Y. Yu, Y. Kuang, X. Zhang and J. Chen, Chem. Commun., 2010, 46, 7954–7956 RSC
- Z. Q. Tian, S. P. Jiang, Z. C. Liu and L. Li, Electrochem. Commun., 2007, 9, 1613–1618 CrossRef CAS PubMed
- Z. Liu, Z. Q. Tian and S. P. Jiang, Electrochim. Acta, 2006, 52, 1213–1220 CrossRef CAS PubMed
- S. P. Jiang, Z. C. Liu, H. L. Tang and M. Pan, Electrochim. Acta, 2006, 51, 5721–5730 CrossRef CAS PubMed
- H. L. Tang, M. Pan, S. P. Jiang, Z. H. Wan and R. Z. Yuan, Colloids Surf., A, 2005, 262, 65–70 CrossRef CAS PubMed
- R. Wlodarczyk, M. Chojak, K. Miecnikowski, A. Kolary, P. Kulesza, K. Miecznikowski and R. Marassi, J. Power Sources, 2006, 159, 802–809 CrossRef CAS PubMed
- P. Barczuk, A. Lewera, K. Miecznikowski, A. Zurowski and P. Kulesza, J. Power Sources, 2010, 195, 2507–2513 CrossRef CAS PubMed
- Z. Bai, Y. Guo, L. Yang, L. Li, W. Li, P. Xu, C. Hu and K. Wang, J. Power Sources, 2011, 196, 6232–6237 CrossRef CAS PubMed
- C. Hu, Y. Cao, L. Yang, Z. Bai, Y. Guo, K. Wang, P. Xu and J. Zhou, Appl. Surf. Sci., 2011, 257, 7968–7974 CrossRef CAS PubMed
- D. Wang, S. Lu and S. P. Jiang, Chem. Commun., 2010, 46, 2058–2060 RSC
- D. Tuncel, Nanoscale, 2011, 3, 3545–3554 RSC
- T. S. Jo, H. Han, L. Ma and P. K. Bhowmik, Polym. Chem., 2011, 2, 1953–1955 RSC
- X. Wu and G. Shi, J. Mater. Chem., 2005, 15, 1833–1837 RSC
- J. Zou, S. I. Khondaker, Q. Huo and L. Zhai, Adv. Funct. Mater., 2009, 19, 479–483 CrossRef CAS
- M. Kimura, N. Miki, N. Adachi, Y. Tatewaki, K. Ohta and H. Shirai, J. Mater. Chem., 2009, 19, 1086–1092 RSC
- X. Chao-Hua, Z. Ren-Jia, S. Min-Min, G. Yan, W. Gang, Z. Xiao-Bin, C. Hong-Zheng and W. Mang, Nanotechnology, 2008, 19, 215604 CrossRef PubMed
- S. Meuer, L. Braun and R. Zentel, Macromol. Chem. Phys., 2009, 210, 1528–1535 CrossRef CAS
- C.-H. Andersson, M. Lahmann, S. Oscarson and H. Grennberg, Soft Matter, 2009, 5, 2713–2716 RSC
- J. U. Lee, J. Huh, K. H. Kim, C. Park and W. H. Jo, Carbon, 2007, 45, 1051–1057 CrossRef CAS PubMed
- J. Yoo, H. Ozawa, T. Fujigaya and N. Nakashima, Nanoscale, 2011, 3, 2517–2522 RSC
- L. Xu, Z. Ye, Q. Cui and Z. Gu, Macromol. Chem. Phys., 2009, 210, 2194–2202 CrossRef CAS
- K. Petrie, A. Docoslis, S. Vasic, M. Kontopoulou, S. Morgan and Z. Ye, Carbon, 2011, 49, 3378–3382 CrossRef CAS PubMed
- L. Vaisman, H. D. Wagner and G. Marom, Adv. Colloid Interface Sci., 2006, 128–130, 37–46 CrossRef CAS PubMed
- W. Wenseleers, I. I. Vlasov, E. Goovaerts, E. D. Obraztsova, A. S. Lobach and A. Bouwen, Adv. Funct. Mater., 2004, 14, 1105–1112 CrossRef CAS
- C. Richard, F. Balavoine, P. Schultz, T. W. Ebbesen and C. Mioskowski, Science, 2003, 300, 775–778 CrossRef CAS PubMed
- D. Linton, P. Driva, B. Sumpter, I. Ivanov, D. Geohegan, C. Feigerle and M. D. Dadmun, Soft Matter, 2010, 6, 2801–2814 RSC
- M. J. O'Connell, P. Boul, L. M. Ericson, C. Huffman, Y. Wang, E. Haroz, C. Kuper, J. Tour, K. D. Ausman and R. E. Smalley, Chem. Phys. Lett., 2001, 342, 265–271 CrossRef
- D. Q. Yang, J. F. Rochette and E. Sacher, J. Phys. Chem. B, 2005, 109, 4481–4484 CrossRef CAS PubMed
- S. Y. Wang, F. Yang, S. P. Jiang, S. L. Chen and X. Wang, Electrochem. Commun., 2010, 12, 1646–1649 CrossRef CAS PubMed
- S. Zhang, Y. Y. Shao, G. P. Yin and Y. H. Lin, J. Mater. Chem., 2010, 20, 2826–2830 RSC
- S. Y. Wang, S. P. Jiang, T. J. White and X. Wang, Electrochim. Acta, 2010, 55, 7652–7658 CrossRef CAS PubMed
- Y. Cui, Y. J. Kuang, X. H. Zhang, B. Liu and J. H. Chen, Acta Phys.–Chim. Sin., 2013, 29, 989–995 CAS
- L. Shi, R. P. Liang and J. D. Qiu, J. Mater. Chem., 2012, 22, 17196–17203 RSC
- D. He, C. Zeng, C. Xu, N. Cheng, H. Li, S. Mu and M. Pan, Langmuir, 2011, 27, 5582–5588 CrossRef CAS PubMed
- C. H. Hsu, H. Y. Liao and P. L. Kuo, J. Phys. Chem. C, 2010, 114, 7933–7939 CAS
- B. Z. Fang, M. S. Kim, J. H. Kim, M. Y. Song, Y. J. Wang, H. J. Wang, D. P. Wilkinson and J. S. Yu, J. Mater. Chem., 2011, 21, 8066–8073 RSC
- B. Wu, D. Hu, Y. Kuang, B. Liu, X. Zhang and J. Chen, Angew. Chem., Int. Ed., 2009, 48, 4751–4754 CrossRef CAS PubMed
- T. Fujigaya and N. Nakashima, Adv. Mater., 2013, 25, 1666–1681 CrossRef CAS PubMed
- Q. F. Li, R. H. He, J. O. Jensen and N. J. Bjerrum, Chem. Mater., 2003, 15, 4896–4915 CrossRef CAS
- M. Okamoto, T. Fujigaya and N. Nakashima, Adv. Funct. Mater., 2008, 18, 1776–1782 CrossRef CAS
- K. Matsumoto, T. Fujigaya, K. Sasaki and N. Nakashima, J. Mater. Chem., 2011, 21, 1187–1190 RSC
- K. Matsumoto, T. Fujigaya, H. Yanagi and N. Nakashima, Adv. Funct. Mater., 2011, 21, 1089–1094 CrossRef CAS
- D. L. Wang, S. F. Lu, P. J. Kulesza, C. M. Li, R. De Marco and S. P. Jiang, Phys. Chem. Chem. Phys., 2011, 13, 4400–4410 RSC
- R. Wlodarczyk, M. Chojak, K. Miecnikowski, A. Kolary, P. J. Kulesza and R. Marassi, J. Power Sources, 2006, 159, 802–809 CrossRef CAS PubMed
- S. A. Johnson, P. J. Ollivier and T. E. Mallouk, Science, 1999, 283, 963–965 CrossRef CAS
- J. Reguera, A. Fahmi, P. Moriarty, A. Girotti and J. C. Rodriguez-Cabello, J. Am. Chem. Soc., 2004, 126, 13212–13213 CrossRef CAS PubMed
- Z. Liu, J. E. Hu, Q. Wang, K. Gaskell, A. I. Frenkel, G. S. Jackson and B. Eichhorn, J. Am. Chem. Soc., 2009, 131, 6924–6925 CrossRef CAS PubMed
- K. Sundmacher, Ind. Eng. Chem. Res., 2010, 49, 10159–10182 CrossRef CAS
- O. Guillen-Villafuerte, R. Guil-Lopez, E. Nieto, G. Garcia, J. L. Rodriguez, E. Pastor and J. L. G. Fierro, Int. J. Hydrogen Energy, 2012, 37, 7171–7179 CrossRef CAS PubMed
- D. Wang and Y. Li, Adv. Mater., 2011, 23, 1044–1060 CrossRef CAS PubMed
- Q. Yuan and X. Wang, Nanoscale, 2010, 2, 2328–2335 RSC
- M. Sastry, A. Swami, S. Mandal and P. R. Selvakannan, J. Mater. Chem., 2005, 15, 3161–3174 RSC
- V. Neburchilov, J. Martin, H. Wang and J. Zhang, J. Power Sources, 2007, 169, 221–238 CrossRef CAS PubMed
- A. Chen and P. Holt-Hindle, Chem. Rev., 2010, 110, 3767–3804 CrossRef CAS PubMed
- Y. C. Zou, X. Li, Y. M. Huang and H. L. Liu, Chem. Res. Chin. Univ., 2011, 32, 150–154 CAS
- F. A. Viva, M. M. Bruno, M. Jobbagy and H. R. Corti, J. Phys. Chem. C, 2012, 116, 4097–4104 CAS
- S. Wang, X. Wang and S. P. Jiang, Langmuir, 2008, 24, 10505–10512 CrossRef CAS PubMed
- Y. Kuang, Y. Cui, Y. Zhang, Y. Yu, X. Zhang and J. Chen, Chem.–Eur. J., 2012, 18, 1522–1527 CrossRef CAS PubMed
- B. Wu, D. Hu, Y. Kuang, Y. Yu, X. Zhang and J. Chen, Chem. Commun., 2011, 47, 5253–5255 RSC
- B. Wu, Y. Zhang, Y. Kuang, Y. Yu, X. Zhang and J. Chen, Chem.–Asian J., 2012, 7, 190–195 CrossRef CAS PubMed
- Z. M. Cui, C. M. Li and S. P. Jiang, Phys. Chem. Chem. Phys., 2011, 13, 16349–16357 RSC
- Z. M. Cui, S. P. Jiang and C. M. Li, Chem. Commun., 2011, 47, 8418–8420 RSC
- F. Vigier, C. Coutanceau, F. Hahn, E. M. Belgsir and C. Lamy, J. Electroanal. Chem., 2004, 563, 81–89 CrossRef CAS PubMed
- H.-X. Zhang, C. Wang, J.-Y. Wang, J.-J. Zhai and W.-B. Cai, J. Phys. Chem. C, 2010, 114, 6446–6451 CAS
- S. Ghosh, R. K. Sahu and C. R. Raj, Nanotechnology, 2012, 23, 385602 CrossRef PubMed
- M. Mavrikakis, B. Hammer and J. K. Norskov, Phys. Rev. Lett., 1998, 81, 2819–2822 CrossRef
- Y. Cheng, C. W. Xu, P. K. Shen and S. P. Jiang, Appl. Catal., B, 2014, 158–159, 140–149 CrossRef CAS PubMed
- D.-Q. Yang, J.-F. Rochette and E. Sacher, J. Phys. Chem. B, 2005, 109, 4481–4484 CrossRef CAS PubMed
- C. Caddeo, C. Melis, L. Colombo and A. Mattoni, J. Phys. Chem. C, 2010, 114, 21109–21113 CAS
- B. Shan and K. Cho, Chem. Phys. Lett., 2010, 492, 131–136 CrossRef CAS PubMed
- R. Chetty, S. Kundu, W. Xia, M. Bron, W. Schuhmann, V. Chirila, W. Brandl, T. Reinecke and M. Muhler, Electrochim. Acta, 2009, 54, 4208–4215 CrossRef CAS PubMed
- G. Wu, R. Swaidan, D. Li and N. Li, Electrochim. Acta, 2008, 53, 7622–7629 CrossRef CAS PubMed
- B. Xiong, Y. K. Zhou, Y. Y. Zhao, J. Wang, X. Chen, R. O'Hayre and Z. P. Shao, Carbon, 2013, 52, 181–192 CrossRef CAS PubMed
- J. G. Zhou, X. T. Zhou, X. H. Sun, R. Y. Li, M. Murphy, Z. F. Ding, X. L. Sun and T. K. Sham, Chem. Phys. Lett., 2007, 437, 229–232 CrossRef CAS PubMed
- Y. Cheng, C. W. Xu, L. C. Jia, J. D. Gale, L. L. Zhang, C. Liu, P. K. Shen and S. P. Jiang, Appl. Catal., B, 2015, 163, 96–104 CrossRef CAS PubMed
- J. Zhang, Y. Cheng, S. F. Lu, L. C. Jia, P. K. Shen and S. P. Jiang, Chem. Commun. Search PubMed
- Z. X. Liang, T. S. Zhao, J. B. Xu and L. D. Zhu, Electrochim. Acta, 2009, 54, 2203–2208 CrossRef CAS PubMed
- D. Q. Yang, S. H. Sun, J. P. Dodelet and E. Sacher, J. Phys. Chem. C, 2008, 112, 11717–11721 CAS
- J. Prabhuram, T. S. Zhao, Z. K. Tang, R. Chen and Z. X. Liang, J. Phys. Chem. B, 2006, 110, 5245–5252 CrossRef CAS PubMed
- Y. Y. Mu, H. P. Liang, J. S. Hu, L. Jiang and L. J. Wan, J. Phys. Chem. B, 2005, 109, 22212–22216 CrossRef CAS PubMed
- Y. Chen, Y. W. Tang, L. Y. Kong, C. P. Liu, W. Xing and T. H. Lu, Acta Phys.–Chim. Sin., 2006, 22, 119–123 CAS
- D. Villers, S. H. Sun, A. M. Serventi, J. P. Dodelet and S. Desilets, J. Phys. Chem. B, 2006, 110, 25916–25925 CrossRef CAS PubMed
- D. Q. Yang and E. Sacher, J. Phys. Chem. C, 2008, 112, 4075–4082 CAS
| This journal is © The Royal Society of Chemistry 2014 |