
Green synthesis of noble metal nanoparticles using cysteine-modified silk fibroin: catalysis and antibacterial activity†
Soumen Das and
Basab Bijayi Dhar*
Chemical Engineering and Process Development Division, CSIR-National Chemical Laboratory, Dr Homi Bhabha Road, Pune – 411 008, India. E-mail: bb.dhar@ncl.res.in; basabbijayi@gmail.com
First published on 17th September 2014
Abstract
Noble metal nanoparticles (NPs) have shown remarkable potential for numerous applications. In this work, a simple, one-pot, green method for the synthesis of gold, silver, palladium, and platinum NPs by using thiol-modified silk fibroin (SF-SH) has been described. The incorporation of thiol groups into silk fibroin (SF) yields small, mono-dispersed metal nanoparticles with good colloidal stability. UV-Vis, transmission electron microscopy (TEM), and X-ray powder diffraction (XRD) analyses show the formation of NPs, and thermo-gravimetric analysis (TGA) data reveal interaction of the NPs with thiol-modified SF. We also show that all the NP–SF conjugates catalyse the reduction of p-nitrophenol to p-aminophenol in the presence of NaBH4 at room temperature. The NP–SF conjugate materials were processed into different material formats like porous scaffolds and films without compromising their individual properties. The Au–SF–SH composite scaffold was used successfully in the heterogeneous catalysis of p-nitrophenol reduction using NaBH4 while the Ag–SF–SH conjugated film showed good antibacterial activity.
Introduction
Metal (Au, Ag, Pd, and Pt) nanoparticles (NPs) have unique physicochemical properties that are different from those of bulk materials.1,2 In the past two decades, metal NPs have been extensively explored in the context of physics, chemistry, and biology.3–6 Particularly, metal NPs have attracted considerable attention because of their potential application in catalysis,7,8 fuel cells,9 labelling,10 sensing,11 photonics,12,13 biomedicine,14 and nanoelectronics.15 As the properties of NPs and hence their application are crucially dependent on their size, size-controlled synthesis of NPs has been envisaged as very important.16,17 In general, metal NPs are synthesized by the chemical reduction of high valent metal ions using a reducing agent in the presence of one or more surfactants or capping agents to avoid aggregation and to control size.18–20 Nowadays, considerable research effort is being directed towards the synthesis of NPs using biogenic materials because of the possibility of reducing and/or eliminate toxic substances during the synthesis of NPs. This possibility supports the fundamental principle of green chemistry.21,22 But most biogenic materials (plant extracts, for example) are not chemically well characterized. Therefore, it is difficult to know the purity of the biomaterial or which functional groups present in the biomaterial are responsible for reduction and stabilization, although the processes are eco-friendly and sometimes considered economic too.23 Moreover, the major drawback of these procedures is the inadequacy of control over the size and stability of NPs probably due to the lack of functional group (like –SH, –NH2 etc.).24,25 In addition, these procedures require high temperature and their reaction time is long; these make NPs inappropriate for practical applications. Hence, naturally occurring, well-characterized materials that have both reducing and stabilizing power and functionalized easily would be preferable; we thought SF from Bombyx mori silkworms would help make noble metal NPs in an environmentally friendly way. The high molecular weight SF biopolymers are already characterized by repetitive hydrophobic and hydrophilic peptide sequences. In SF, the hydrophobic empires, consisting mainly of Gly–Ala–Gly–Ala–Gly–Ser repeats, self-assemble into an anti-parallel β-sheet structure responsible for the insolubility, high mechanical strength, and thermal stability of silk fibers.26 Other than these three major constituent amino acids (glycine, alanine, and serine), many tyrosine residues (5.3 mol%) are equally distributed throughout the protein sequence and are available for chemical modification on SF.27 It is already reported that tyrosine moiety could be used as a reducing agent in the synthesis of metal NPs.28,29 In addition, the methanolic –OH group of serine moiety can act as a potential reducing agent, especially for the preparation of silver NPs.30 Nowadays, SF is regarded as an excellent biomaterial due to its versatile properties. Coupled with its excellent mechanical strength, biocompatibility, biodegradability, and its ability to process into multiple material formats such as gels, sponge, and films, is attracting greater attention from researchers.31 In 2001, Y. Chujo and co-workers reported that tyrosine residue present in the SF can act as a reducing agent in the formation of Au NPs.32 But the catalytic activity and detail study of the composite material has not been explored at all to the best of our knowledge. Furthermore, they only report the formation of colloidal dispersions in their study.It is well known that proper functionalization of metal NPs is a prerequisite for every possible application that determines their interaction with the environment. These interactions ultimately affect the particles' colloidal stability, and may yield to controlled assembly. It has been reported that thiol moiety (–SH) can efficiently improve the stability and dispersity of colloidal noble metal NPs, especially to gold NPs in solution, in comparison to other functional groups (–NH2, –COOH, etc.).33 This is because thiol (–SH) groups bind covalently to the surface of Au NPs.34 Numerous reports support the interaction of thiol moiety with Ag, Pd, and Pt NPs too. In 2001, Sastry et al. reported the presence of strong electrostatic interaction in between Ag NPs and cysteine molecule.35 Tsukada et al. already clarified that cysteine and cysteine thiolate were adsorbed on the surface of Pd NPs and increased the material's biocompatibility.36 Fang et al. showed that a Pt nano-crystal can easily be loaded on the graphene surface by introducing thiol functionality.37 Based on this, we envisioned that the introduction of sufficient amounts of thiols (–SH) into SF can improve its stabilizing power against NPs aggregation.
Accordingly, in this study, Au, Ag, Pd, and Pt NPs have been synthesized by in situ redox technique using cysteine-modified SF for the first time. The first step in this endeavour was the modification of aspartic and glutamic acid residues in SF with cysteine Me-ester. Subsequently, this modified SF was used for NP synthesis (NP–SF). These NP–SF conjugates are well characterized by UV-Vis, TEM, XRD, X-ray photoelectron spectroscopy (XPS), microwave plasma-atomic emission spectrometer (MP-AES), and TGA. We show that the Au, Ag and Pd NPs conjugate materials efficiently catalyse the p-nitrophenol reduction using NaBH4 at room temperature following first-order kinetics. We also demonstrate that the NP–SF conjugate materials could be easily processed into self-standing three-dimensional (3D) porous scaffolds. Our studies also disclose that the physical property of SF and catalytic activity of NPs remain intact in the NP–SF composite. We conclude that the interaction between thiols and NPs help make the 3D scaffold robust against leaching. Ag–SF–SH conjugate film also showed antibacterial activity.
Experiment section
Materials and methods
Cocoons from Bombyx mori were obtained from the Central Sericulture Research and Training Institute, Mysore. Cysteine methyl ester was supplied by Aldrich. HAuCl4, AgNO3, H2PtCl6 Na2PdCl4 and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimidehydrochloride (EDC-Cl), and N-hydroxysuccinimide (NHS) were obtained from Aldrich. All other chemicals used were obtained from Merck, India.Preparation of aqueous silk solution
The Bombyx mori silk cocoons were boiled in 0.5% (w/v) solution of NaHCO3 twice for about 30 min to remove the sericin coating. The resulting cottony mass of fibroin fibers was then dissolved in 9.3 M LiBr solution using a concentration of 1 g/10 mL at 60 °C for 45 minutes. This solution was then dialyzed against distilled water for 48 h at 4 °C using a dialysis bag of cellulose acetate membrane (molecular weight cut-off 12 kDa). The water for the dialysis was changed at least six times; first after three hours, then six hours, and later twelve hours each. The solution was centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 20 min at 4 °C and the supernatant was collected. This regenerated SF solution had a final concentration of 3.5–4 wt% (wt/vol) and could be stored in the refrigerator for about 3–4 months. Then cysteine was incorporated into SF by using EDC-NHS coupling (ESI Fig. 1†).
000 rpm for 20 min at 4 °C and the supernatant was collected. This regenerated SF solution had a final concentration of 3.5–4 wt% (wt/vol) and could be stored in the refrigerator for about 3–4 months. Then cysteine was incorporated into SF by using EDC-NHS coupling (ESI Fig. 1†).
Synthesis of nanoparticles by SF–SH
The noble metal (Au, Ag, Pd, and Pt) NPs fabricated SF–SH was synthesized using the in situ redox technique. For example, 2 mL of freshly prepared SF–SH aqueous solution (0.5 wt%) at pH ∼ 10 was added drop-wise into 2 mL of an aqueous solution of metal precursor (HAuCl4, AgNO3, and Na2PdCl4) of 2 mM at room temperature under slow stirring. The pH of the reaction mixture was adjusted to 10 with addition of aqueous NaOH (0.5 N) to prevent cross-linked aggregation of the SF (the isoelectric point of the SF is 5.4). Then the reaction mixture was stirred for three hours at room temperature. The same procedure was followed at 85 °C in the case of Pt. After the reaction was complete, the solution of NPs was transferred into a dialysis bag of a molecular weight cut-off 12 kDa. The solution was dialyzed against deionized water for three days, with water changes once every four hours for the first day and then thrice per day.Preparation of NP–SF scaffolds
Porous 3D self-standing NP–SF protein scaffolds were prepared by the salt leaching method.31 The solid NP–SF obtained after lyophilizing the NP–SF aqueous solution was dissolved in hexafluroisopropanol (HFIP) (8 wt%). 4.0 g of granular NaCl was added into 2 mL of HFIP solution of NP–SF and mixed thoroughly. The containers were covered and the solvent was evaporated at room temperature for 3 days. This provided sufficient time for homogeneous distribution of the porogen. The solid matrices were then treated in methanol for 1 h, followed by immersion in water for 2 days to remove the NaCl. The porous silk scaffolds were then freeze-dried.Characterization of NPs
UV-Vis spectra and reaction kinetics of NPs were monitored on Carry-300 UV-Vis spectrometer using 1 cm quartz cuvette at 25 °C. Samples for HR-TEM and TEM were prepared by evaporating a droplet of solution onto a carbon-coated copper mesh 200 grid. FEI Tecnai TF-30 and T-20 electron microscopes, operating at 300 kV and 200 kV respectively, were used for normal and high resolution TEM and HR-TEM, respectively, to determine size, shape, and fringes. The formation of metallic NPs was also confirmed by the XRD pattern. TGA analysis was done in Perkin Elmer STA 6000 simultaneous thermal analyser in the temperature range of 70–840 °C under nitrogen atmosphere. The amounts of Au, Ag, Pd, and Pt present in the NP–SF composite materials were estimated by Agilent 4100 MP-AES. XPS measurements were taken with a custom-built ambient-pressure XPS system from Prevac equipped with a VG Scienta SAX 100 emission controller monochromator using an Al Kα anode (1486.6 eV) in transmission lens mode. The photoelectrons were energy-analysed using VG Scienta's R3000 differentially pumped analyser.Catalytic reduction of p-nitrophenol by NP–SF in presence of NaBH4
In the presence of metal NPs, reduction of 4-nitrophenol to 4-aminophenol was done by using excess of NaBH4. Reaction kinetics was monitored by UV-Vis spectroscopy. The decrease in peak intensity at 400 nm with time was used to calculate the pseudo first order rate constant of this reaction. Typically, reaction mixtures were made by taking 960 μL of 90 μM stock solution of p-nitrophenol into that 30 μL of 1.3 M stock solution NaBH4 and 10 μL of NP–SF solution (dissolving lyophilized NP–SF powder into water to make a stock solution of 0.5 wt%) to make total volume of 1.0 mL.Preparations of Ag–SF–SH composite film and its antibacterial study
Solutions containing Ag–SF–SH (2 wt% in water) were cast onto a cover slip and dried in a laminar flow hood for 24 h. The films were then placed in vacuum for another 24 h. To make them water-insoluble, these films were treated with 70% methanol in water for 1 h and dried in vacuum overnight.Antibacterial activities of this Ag–SF–SH coated cover slip against Escherichia coli (E. coli) were determined using the agar diffusion assay method. Stock culture of E. coli was grown separately in liquid nutrient broth medium. The bacterial suspension was spread on nutrient agar in Petri plate to create a confluent lawn of bacterial growth. The cover slip coated with only SF–SH was treated as control. These plates were incubated for 12 h at 35 °C and examined for zones of inhibition, which appeared as a clear area around the wells.
Results and discussions
NPs synthesis by SF–SH
In SF, there are only ∼5 residues of cysteine present.26 However, SF contains 55 acid residues (aspartic and glutamic acid),26 which lets us easily incorporate cysteine moiety into the acid residues of SF via EDC–NHS coupling chemistry (ESI Fig. 1†). The concentration of free –SH in SF–SH was found to be six times higher than the normal SF by the spectrophotometric technique using Ellman's reagent (ESI Fig. 2†).During the synthesis of Au, Ag, and Pd NPs at RT, pH of the reaction mixture was maintained at ∼10. The high basic reaction condition helps the reaction by preventing cross-linked aggregation of SF; it also helps to achieve the highest reduction power of tyrosine residue present in SF towards high valent metal salts. It is well known that the pKa value of phenol in water is 9.95. As a result, at higher pH (10.0), tyrosine phenolic moiety represents as a phenoxide anion and provides higher electron density in the π → π* transition of the tyrosine residue, which results in energetically favourable electron transfer from tyrosine to high valent metal ions. During the synthesis of Au NPs, the colour of the reaction mixture gradually changed from yellow to light purple to purple to wine red, indicating the formation of gold nanoparticles and the reduction of AuIII salt by SF–SH. After three hours, no colour change was observed from wine red. This suggests that the reaction was complete. However, in the case of Ag and Pd, the reaction mixtures instantaneously turned colourless to blackish brown and yellow to colourless respectively. No absorption peak of PdII precursor was observed after the reaction was complete (ESI Fig. 3†). From the standard electrode potential of Ag+/Ag0 (0.80 V vs. SCE) and [PdCl4]2−/Pd0 (0.60 V vs. SCE), the reduction of Ag+ and [PdCl4]2− is expected to be more difficult and slower than Au3+ (E0 of Au3+/Au0 = 1.50 V vs. SCE) and Pt4+ (E0 of Pt4+/Pt0 = 1.43 V vs. SCE). However, our results showed the opposite trend, indicating that the reduction of high valent Ag and Pd metal precursor by SF–SH is not a straightforward process. It might be possible that the peptide sequences in SF favour the reduction process of high valent Ag and Pd precursor in an enzymatic way and, during the process, tyrosine, serine (methanolic –OH group), and cysteine (–SH group) present in SF–SH could play the crucial role. In 2008, Mukherjee et al. explained cysteine was responsible in the controlled biosynthesis of Ag NP by Trichoderma asperellum.38 In 2009, Donati et al. reported how the polyol reduces the Ag+ ion to Ag NP.30 The contradictory reduction rate for different metal precursor could be explained in terms of hard–soft interaction between high valent metal precursor (which acts as a Lewis acid) and –SH moiety of SF–SH (which acts as a Lewis base). According to R. G. Pearson, the hardness parameter (η) for both Ag+ and Pd2+ is same (η ∼ 6.8), and both are considered soft Lewis acids.39 In comparison, due to high positive charge, Au3+ and Pt4+ are considered relatively hard Lewis acids (η > 8.0).39 Therefore, possibly, soft–soft interaction between Ag+ and Pd2+ ions with –SH moiety of SF–SH favours bringing the metal centre closer enough with the reducing centre (tyrosine, serine, etc.) and helps achieving faster electron transfer by decreasing the activation energy barrier. In nature, electron transfer generally occurs in outer sphere fashion, and it is well known that changing the distance between the redox partners makes a huge difference in the activation energy of the overall electron transfer process. In the case of Au, simultaneous reduction of Au(III) to Au(0) was followed by thiol adsorption on Au NP surface.
Our above contention was further supported by the observation that Pt NPs were not formed at RT by using the same reaction procedure. It is possible that the proximity of redox centres was not achieved at RT due to less interaction between very hard Pt4+ metal ion and soft base thiol (–SH) to form a stable adduct or intermediate. However, at a higher temperature, SF–SH is able to cross the activation energy barrier for electron transfers from tyrosine of SF and facilitates the reductions of Pt4+ with SF–SH.
MP-AES analysis of NP–SF conjugates was performed to analyse the amount of metal NP present in the composite materials. It showed 5.5 wt% metallic Au, 2.9 wt% metallic Ag, 1.02 wt% metallic Pd and 1.10 wt% metallic Pt with respect to SF–SH were present in NP–SF conjugate materials. Whereas 15.7 wt% Au salt, 6 wt% Ag salt, 10 wt% Pd salt and 16.3 wt% Pt salt with respect to SF–SH used for synthesis of metal NP.
UV-Vis spectral studies of NPs
The UV-Vis absorption spectrum of an aqueous solution of pure SF–SH displayed a strong absorption band at 276 nm, assigned mainly to the π → π* transition of the tyrosine residue in the SF chain. However, after the reaction was complete, it was found that the characteristic peak of tyrosine at 276 nm decreased and strongly red-shifted to 288 nm. The shifting of the tyrosine peak may correspond to the electron transfer from the tyrosine to the metal ions to have altered electron density in the tyrosine moiety during the reduction process.28,29 The solution of the Au–SF–SH conjugate shows a strong absorption band in the UV-Vis spectra at 524 nm (Fig. 1A) due to the localized surface-plasmon-resonance (SPR) in gold nanoparticles. Similarly, Ag–SF–SH conjugate showed an absorption band at 410 nm (Fig. 1B). The UV-Vis spectra of thus synthesized Au and Ag–SF–SH conjugates were measured to be intact after a month, which indicates that the biosynthetic nanomaterial were very stable for a long time in solution. For Pd and Pt NPs, the absorption band of the salt disappears, and there is no new peak generation that may confirm the formation of zero valent metal NPs (ESI Fig. 3†).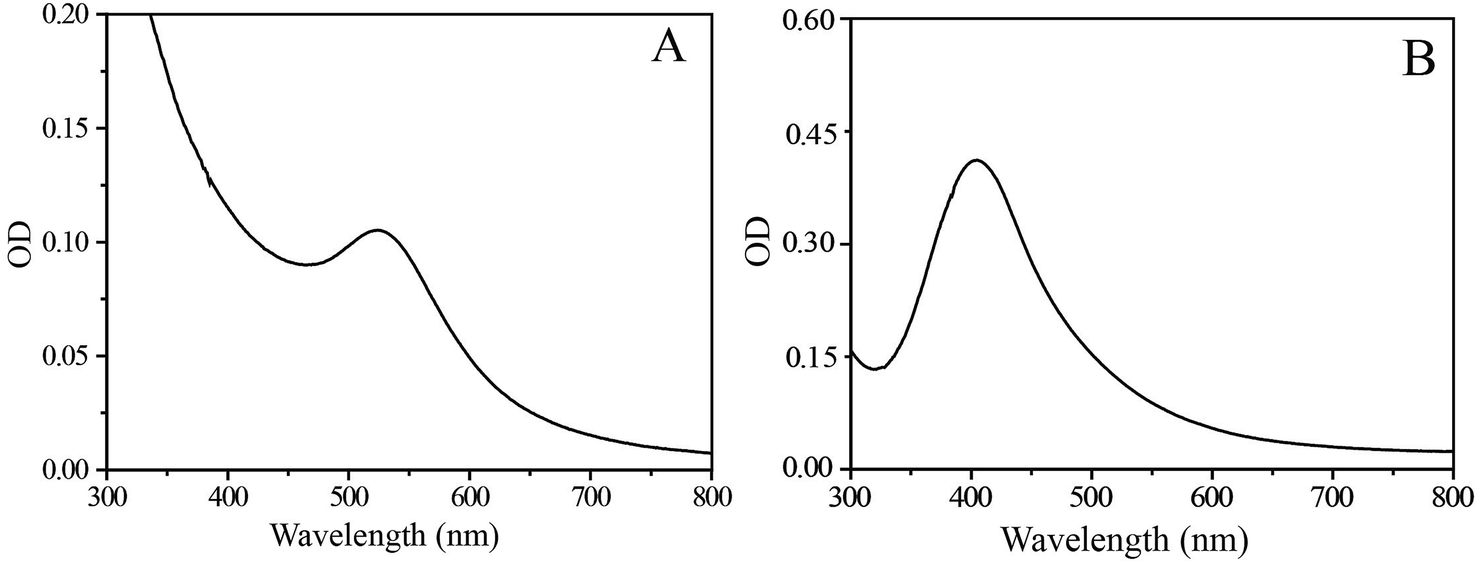 | ||
| Fig. 1 UV-Vis spectra of Au (A) and Ag (B) NPs as synthesized. | ||
Thermo-Gravimetric Analysis (TGA) of NPs
TGA is a very useful technique to estimate the thermal stability as well as organic content of organic–inorganic hybrid materials. To understand the extra stabilization offered by thiol moiety present in SF–SH towards metal NPs, TGA analysis of lyophilized powder of Au NPs synthesized by SF–SH (Fig. 2, brown) was compared with normal SF synthesized Au NPs (Fig. 2, black). For control experiment, TGA of SF–SH was also carried out (Fig. 2, red). In case of SF–SH, ∼5% weight loss was observed at 150 °C and second weight loss was found at 450 °C (∼70%). However, in both Au–SF hybrid material, the first weight loss was observed to take place at ∼270–280 °C (∼10% weight loss) while the second weight loss was observed to take place at ∼450 °C and ∼650 °C (∼65% weight loss) respectively for Au–SF and Au–SF–SH, corresponding to a total weight loss of ∼75%. Probably, in both curves, the first region was not only responsible for water loss. Actually, in SF and SF–SH (except thiol), other functional groups like –COOH, –NH2, and phenolic–OH were present. They are also acting as a potential stabilizing agent for NPs. Thermal decomposition of those functionalities probably merged with water loss in case of NPs containing SF. However, sluggish second weight loss was observed in the case of Au–SF–SH compared to Au–SF. This suggests that extra stability might be gained from Au–S bond in the Au–SF–SH, although other NPs (ESI Fig. 4†) show less thermal stability during the second phase of weight loss than Au–SF–SH.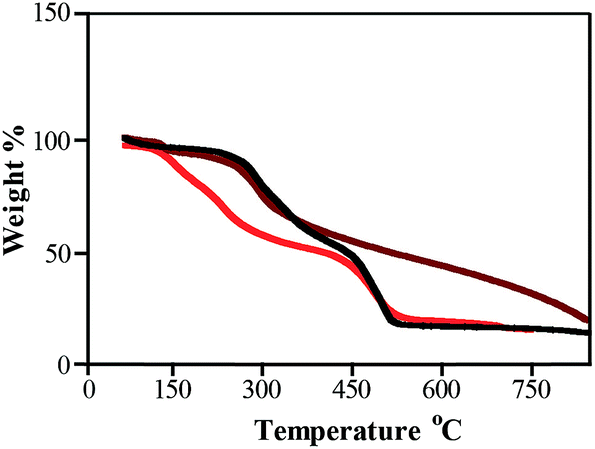 | ||
| Fig. 2 TGA analysis of SF–SH (red), Au–SF (black) and Au–SF–SH (brown). | ||
XRD and XPS studies of NPs
X-ray diffraction study was used to confirm the crystalline nature of the particle. The XRD patterns clearly show that the Au and Ag NPs formed by the reduction of SF–SH (ESI Fig. 5†). The XRD analysis showed intense peaks corresponding to (111), (200), and (220) Bragg's reflection based on the face-centred cubic (fcc) structure of Au and Ag NPs. In case of Pd (ESI Fig. 5†), only 200 peak was prominent. However, Pt (ESI Fig. 6†) NPs prepared at 85 °C showed 111 and 200 peaks prominently enough. Compared to Au and Ag NPs, not all the characteristic XRD peaks were present in Pd and Pt NPs probably because the size of their particles was very small. XPS studies were conducted to analyse the electronic states of metals in NP–SF conjugates. The XPS spectra of Au–SF–SH and Ag–SF–SH composite materials display a doublet peak with binding energies of 84.5 (Au0 4f7/2), 87.8 eV (Au0 4f5/2) and 367 (Ag0 4d5/2), 373 eV (Ag0 4d3/2) respectively (ESI Fig. 7†).40 However, XPS spectra of Pd–SF–SH composite showed double peaks with binding energies 337 (PdII 4d5/2) and 342 eV (PdII 4d3/2) along with 334 (Pd0 4d5/2) and 341 eV (Pd0 4d3/2) suggesting presence of very small Pd NP and some Pd NP converted to Pd(II)oxide (ESI Fig. 7†).41Characterization of NPs by TEM and HR-TEM
Shape, lattice fringes, crystal plane, and particle size distribution of all the nanoparticles synthesized, as part of the study was determined from their TEM images (Fig. 3 and 4). The Au and Ag NP synthesized by using SF–SH show a narrow distribution particle size (13.0 ± 2.5) and (5.0 ± 1.0) respectively. We also made Au and Ag NP by using normal SF to realize the effect of thiol moiety in the synthesis of NP. The TEM image of these synthesized NPs show a large size with broad distribution (ESI Fig. 8†). Therefore, it correlates to our hypothesis that introduction of thiol moiety in SF imparts greater stability and thus narrows size distribution. The Pd and Pt NP synthesized by using SF–SH also show a narrow distribution particle size (3.0 ± 1.0), and (2.0 ± 0.2), respectively. The Au and Ag NPs show inter-fringe spacing of 0.234 nm corresponding to the (111) plane and assigned to the fcc lattice. Inter-fringe spacing of 0.195 nm corresponds to 200 plane for Pd NP and 111 plane for Pt NP were observed respectively. | ||
| Fig. 3 TEM micrograph of Au NPs (A), HR-TEM micrograph of Au NPs (B) synthesized by reduction of HAuCl4 with SF–SH; Histogram for Au NPs size distribution obtained from TEM image, according to image J software (C). | ||
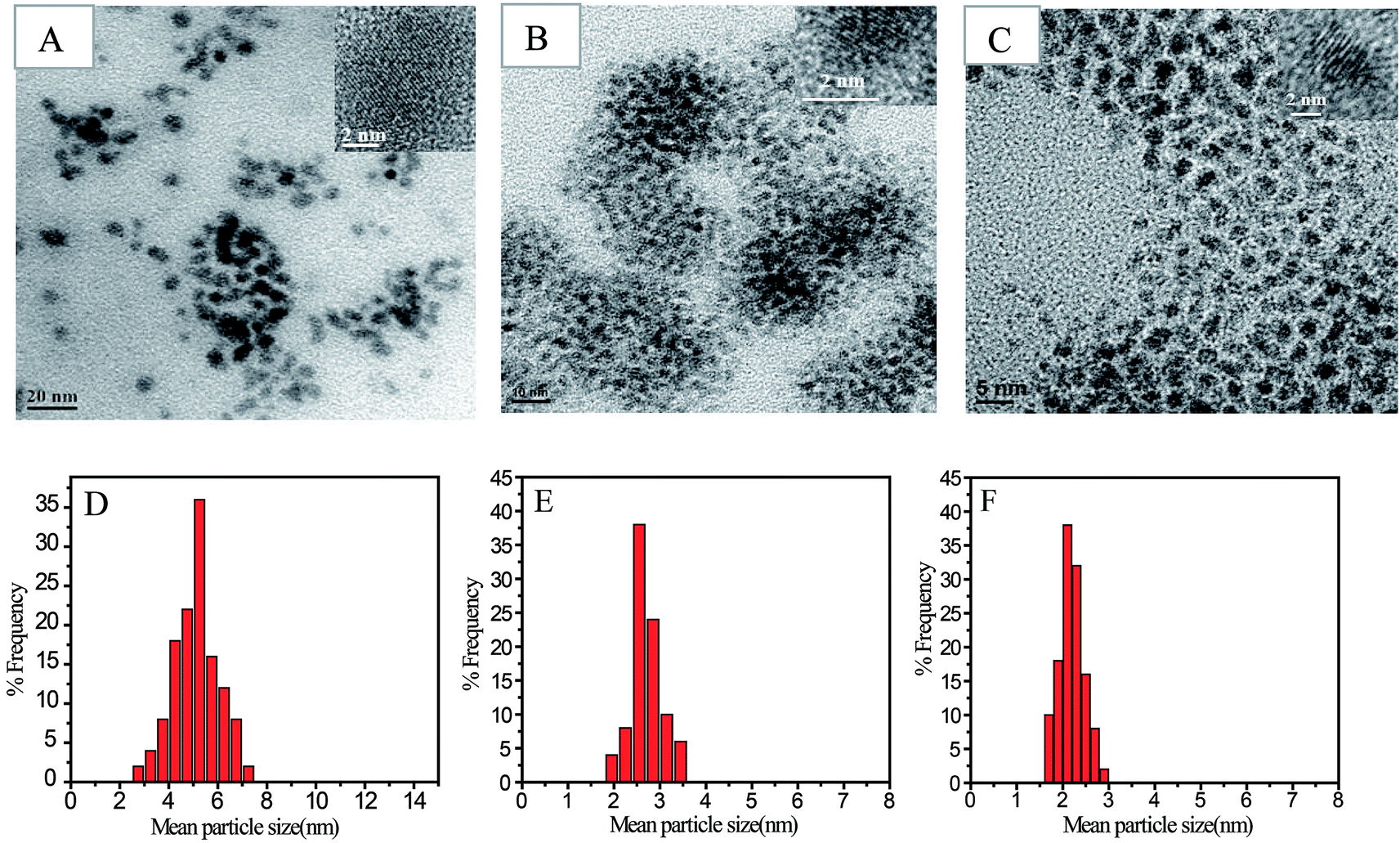 | ||
| Fig. 4 TEM micrograph of Ag NPs (A), Pd NPs (B) and Pt NPs (C) synthesized by reduction of metal salts with SF–SH. Picture in inset showing HR-TEM image of the corresponding NPs. Histogram for Ag, Pd and Pt NPs size distribution obtained from TEM image according to image J software (D–F). | ||
Catalytic property studies of NPs and scaffold
The reduction of 4-nitrophenol to 4-aminophenol using aqueous NaBH4 is thermodynamically favourable (E0 for 4-nitrophenol/4-aminophenol = −0.76 V and H3BO3/BH4− = −1.33 V vs. NHE), but the control reaction carried out in the absence of NPs showed that the colour of the reaction mixture remain unaltered even after two days. It might be due to the kinetic barrier produced by the large potential difference between the donor and acceptor molecules. However, in the presence of metal NPs, the kinetic barrier is overcome by the easy relay of electrons from the donor BH4− to the acceptor 4-nitrophenol. During the reaction, the liberated hydrogen purged air from the reaction mixture, thereby preventing the reduced product of 4-nitrophenol from aerial oxidation. In the presence of NaBH4, the absorbance peak of 317 nm for 4-nitrophenol red shifted to 400 nm due to the formation of 4-nitrophenolate ion and, therefore, we monitored the catalytic activity of SF–SH fabricated NPs by tracking the change in absorption spectra of 4-nitrophenolate ion. Excess NaBH4 helps to maintain pseudo first order condition and, side by side, an increase in the pH of the reacting mixture retards the degradation of the BH4−. ESI Fig. 9† shows the successive absorption spectra of 4-nitrophenolate ion for different metal NPs with time. The appearance of a new peak at 298 nm confirms the formation of 4-aminophenol (ESI Fig. 9†) for all NPs. Rate constants (kobs) were calculated from slope of the ln[(At − Ainfi)/(Ao − Ainfi)] vs. time plot where At, Ainfi, and Ao stand for absorbance at time t, infinity, and zero respectively for different NPs (Table 1, Fig. 5). Rate of 4-nitrophenol reduction by excess NaBH4 followed the reactivity trends of Au < Ag < Pd. Pt NP and showed catalytic reduction of 4-nitrophenol. However, we did not compare its rate with other NPs because its synthetic procedure was different from that of other nanoparticles.| Metal NPs | Size of the NPs (nm) | Rate constant (kobs)/s−1 |
|---|---|---|
| Au | 13.0 ± 2.5 | (4.76 ± 0.01) × 10−3 |
| Ag | 5.0 ± 1.0 | (7.70 ± 0.04) × 10−3 |
| Pd | 3.0 ± 1.0 | (13.3 ± 0.05) × 10−3 |
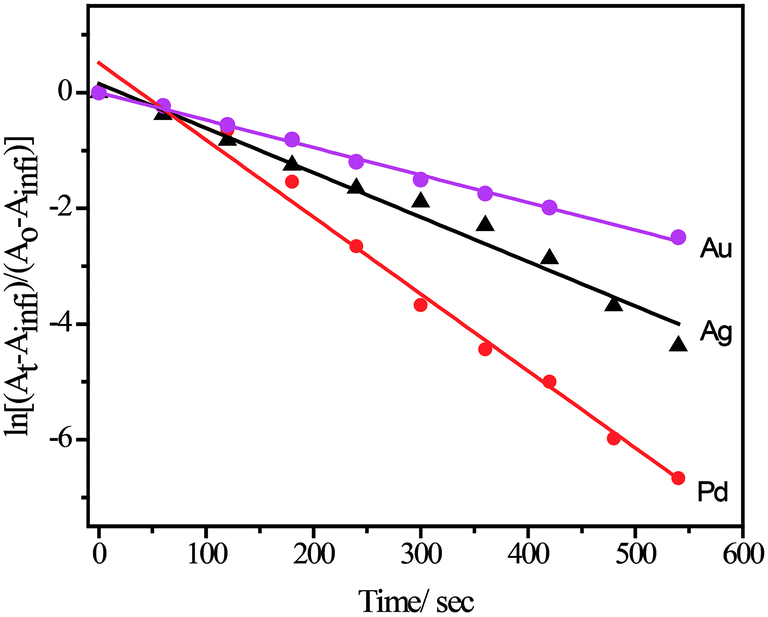 | ||
| Fig. 5 Pseudo first order rate was calculated from ln[(At − Ainfi)/(Ao − Ainfi)] vs. time plot for use various NPs acting as catalyst for 4-nitrophenol reduction by NaBH4. | ||
For a particular metal, small NPs that have greater surface area can assist fast interfacial electron transfer during catalysis. However, it is difficult to generalize the catalytic rate comparison of a particular reaction using various NPs of different metals of different sizes. The catalytic process involves three major steps: (1) diffusion of 4-nitrophenol to the NP surfaces; (2) interfacial electron transfer from NPs to 4-nitrophenol; and (3) diffusion of 4-aminophenol away from the surface. Any of the above three steps can influences the catalytic rate of the electron transfers at the metal surface. The observed rate constant may be explained by the equation
| 1/kobs = [1/(4πR2)][(1/ket) + (R/D)], |
In order to expand the applicability of NP–SF conjugate materials, we prepared self-standing 3D porous scaffold (Fig. 6) for Au–SF–SH. The porous Au–SF–SH scaffold was found to possess very high mechanical strength. Dynamic rheological tests indicate the gel-like nature of the Au–SF–SH conjugate materials. The frequency sweep measurements was done to evaluate the mechanical response of the gel, we observe that the solid modulus (G′) exceeded the loss modulus (G′′) by about an order of magnitude over the entire experimental frequency range 1–100 rad s−1 (ESI Fig. 10†). This indicates a largely elastic response to small deformations, which is a unique property of the network structure of a gel. The G′ shows weak frequency dependence, and is approximately frequency-independent, which is characteristic of a gel. The solid modulus is around 31000 Pa, indicating that the SF network forms a stiff hydrogel. We tested the catalytic activity of this scaffold against the same reaction, reduction of 4-nitrophenol to 4-aminophenol using aqueous NaBH4. 4-Nitrophenol (90 μM) and NaBH4 (1.3 M) were added to the Au–SF–SH scaffold (2.0 mg) in water. The colour of the reaction mixture changed from straw yellow to almost colourless indicating the formation of 4-aminophenol. It was confirmed spectrophotometrically by the disappearance of peaks at 400 nm and generation of new peaks at 298 nm. The recovered scaffold from the first cycle was then used for another two more cycles; there was no change in catalytic activity. This suggests that the degree of leaching of NP from the scaffold is minimal and the catalytic activity is not affected after each consecutive cycle. The porous scaffolds made from Pd–SF–SH conjugate also showed catalytic activity similar that of Au–SF–SH scaffold. Taken together, these data suggest that the SF–NP scaffolds can be used in reusable heterogeneous catalysis.
 | ||
| Fig. 6 SEM image of Au–SF–SH scaffold and the photograph of the scaffold. | ||
Preparations of Ag–SF–SH conjugate film and its antibacterial activity study
Silk films were casted from 2 wt% aqueous solution of Ag–SF–SH by slow evaporation of water. Exposure to methanol induces the formation of a high percentage of β-sheets, which leads to the formation of insoluble films. An antimicrobial test against E. coli was performed using the Ag–SF–SH composite film. The film showed excellent antimicrobial activity whereas SF–SH film showed no zone of inhibition (ESI Fig. 11†), which indicates that bactericidal activity is exclusively due to Ag NP (Fig. 7).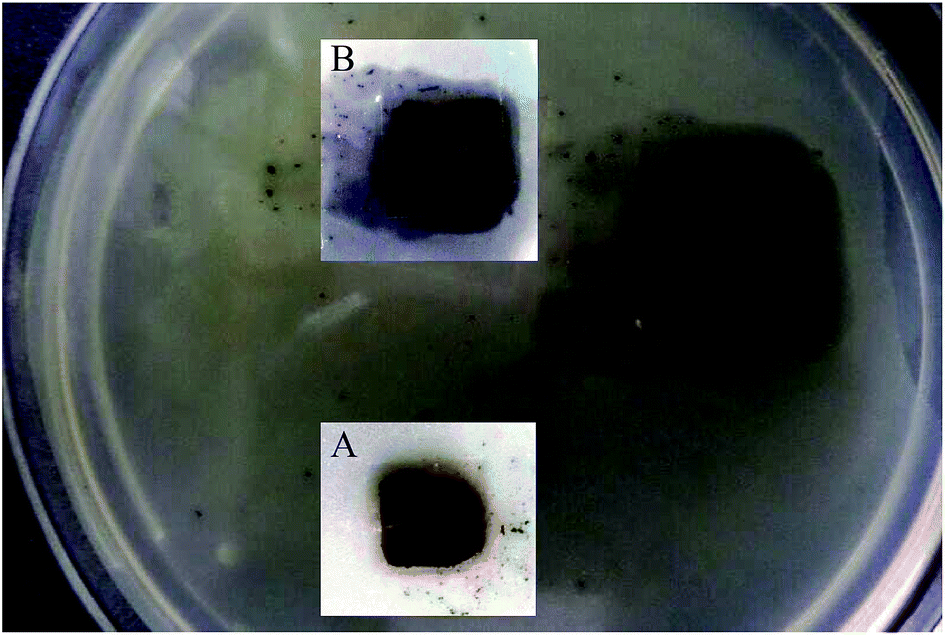 | ||
| Fig. 7 Zone of inhibition (antimicrobial effect) by Ag–SF–SH film. Insets are zoomed image at the time of addition (A) and after 10 h (B). | ||
Conclusions
In summary, we have successfully shown that cysteine-modified SF can act as a reducing agent, stabilizing agent, and material matrix in synthesis of noble metal NPs. In our strategy, the synthesis of all metal NPs (Au, Ag, Pd, and Pt) was performed by a simple one-pot procedure by using SF–SH, without the addition of any external reagents, which supports the fundamental principle of green chemistry. Incorporation of thiols (–SH) into SF helps to get almost mono-dispersed metal NPs. From TEM and HR-TEM analysis, the size of metal NPs was found to follow the order Au > Ag > Pd ≈ Pt. This biosynthetic method of synthesizing NPs is a promising result and can be useful for various catalytic applications. The availability of catalytic site of NPs in these bio-hybrid materials was demonstrated by 4-nitrophenol reduction by using NaBH4. The pseudo first order rate constants followed the order Au < Ag < Pd; this supports the hypothesis that a comparatively larger surface area of small size Pd nanoparticles enhances the relay electron transfer from BH4− ion to 4-nitrophenol. We are also able to obtain 3D porous scaffold of high mechanical strength from the Au–SF–SH and Pd–SF–SH conjugate materials. Using this scaffold allows us to facilitate heterogeneous catalysis. This demonstration highlights several practical advantages of solution-phase NP-based catalysis, like (1) the exclusion of laborious chromatographic purification and (2) the high stability of embedded metal NPs against leaching under the reaction conditions. This would enable the use of this scaffold in a continuous flow system. Moreover, the Ag NP composite film showed very good antibacterial activity. Therefore, the use of these NP–SF conjugate materials in catalysis and biomedicine is promising for future investigations.Acknowledgements
The authors acknowledge Dr B. L. V. Prasad for valuable discussion on this manuscript. We thank Soumyajyoti Chatterjee for TGA measurements at NCL, Pune. S. D. thanks CSIR, New Delhi for fellowship and B. B. D. thanks CSIR SRA-ship. B. B. D. acknowledges DST for funding (Grant no. SERB/F/3945/2013-14). Authors thank Mr Surit Das for editing the manuscript.References
- P. K. Jain, X. Huang, I. H. El-sayed and M. A. El-sayed, Acc. Chem. Res., 2008, 41, 1578–1586 CrossRef CAS PubMed.
- M. A. El-sayed, Acc. Chem. Res., 2001, 34, 257–264 CrossRef CAS PubMed.
- R. A. Sperling, P. R. Gil, F. Zhang, M. Zanella and W. J. Parak, Chem. Soc. Rev., 2008, 37, 1896–1908 RSC.
- Y. Ofir, B. Samanta and V. M. Rotello, Chem. Soc. Rev., 2008, 37, 1814–1825 RSC.
- R. Wilson, Chem. Soc. Rev., 2008, 37, 2028–2045 RSC.
- P. V. Kamat, J. Phys. Chem. B, 2002, 106, 7729–7744 CrossRef CAS.
- A. Roucoux, J. Schulz and H. Patin, Chem. Rev., 2002, 102, 3757–3778 CrossRef CAS PubMed.
- A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096–2126 RSC.
- V. Mazumder, Y. Lee and S. Sun, Adv. Funct. Mater., 2010, 20, 1224–1231 CrossRef CAS.
- S. Guoa and E. Wanga, Nano Today, 2011, 6, 240–264 CrossRef.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS PubMed.
- D. A. Genov, A. K. Sarychev, V. M. Shalaev and A. Wei, Nano Lett., 2004, 4, 153–158 CrossRef CAS.
- W. L. Barnes, A. Dereux and T. W. Ebbesen, Nature, 2003, 424, 824–830 CrossRef CAS PubMed.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- C. N. R. Rao, G. U. Kulkarni, P. J. Thomas and P. P. Edwards, Chem. Soc. Rev., 2000, 29, 27–35 RSC.
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef CAS.
- E. Roduner, Chem. Soc. Rev., 2006, 35, 583–592 RSC.
- L. D. Pachon and G. Rothenberg, Appl. Organomet. Chem., 2008, 22, 288–299 CrossRef.
- M. Brust, J. Fink, D. Bethell, D. J. Schiffrin and C. Kiely, J. Chem. Soc., Chem. Commun., 1995, 1655–1656 RSC.
- I. P. Santos and L. M. Liz-Marzán, Langmuir, 2002, 18, 2888–2894 CrossRef.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, Inc., New York, 1998 Search PubMed.
- R. S. Varma, Green Chem., 2014, 16, 2027–2041 RSC.
- R. Y. Parikh, S. Singh, B. L. V. Prasad, M. S. Patole, M. Sastry and Y. S. Shouche, ChemBioChem, 2008, 9, 1415–1422 CrossRef CAS PubMed.
- A. Gole, C. Dash, V. Ramakrishnan, S. R. Sainkar, A. B. Mandale, M. Rao and M. Sastry, Langmuir, 2001, 17, 1674–1679 CrossRef CAS.
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607–609 CrossRef CAS PubMed.
- A. R. Murphy and D. L. Kaplan, J. Mater. Chem., 2009, 19, 6443–6450 RSC.
- S. Das, D. Pati, N. Tiwari, A. Nisal and S. S. Gupta, Biomacromolecules, 2012, 13, 3695–3702 CrossRef CAS PubMed.
- S. Si, R. R. Bhattacharjee, A. Banerjee and T. K. Mandal, Chem.–Eur. J., 2006, 12, 1256–1265 CrossRef CAS PubMed.
- S. Si, E. Dinda and T. K. Mandal, Chem.–Eur. J., 2007, 13, 9850–9861 CrossRef CAS PubMed.
- I. Donati, A. Travan, C. Pelillo, T. Scarpa, A. Coslovi, A. Bonifácio, V. Sergo and S. Paoletti, Biomacromolecules, 2009, 10, 210–213 CrossRef CAS PubMed.
- D. N. Rockwood, R. C. Preda, T. Yücel, X. Wang, M. L. Lovett and D. L. Kaplan, Nat. Protoc., 2011, 6, 1612–1631 CrossRef CAS PubMed.
- Y. Zhou, W. Chen, H. Itoh, K. Naka, Q. Ni, H. Yamane and Y. Chujo, Chem. Commun., 2001, 2518–2519 RSC.
- A. C. Templeton, S. Chen, S. M. Gross and R. W. Murray, Langmuir, 1999, 15, 66–76 CrossRef CAS.
- H. Häkkinen, Nat. Chem., 2012, 4, 443–455 CrossRef PubMed.
- S. Mandal, A. Gole, N. Lala, R. Gonnade, V. Ganvir and M. Sastry, Langmuir, 2001, 17, 6262–6268 CrossRef CAS.
- C. Tsukadaa, S. Ogawaa, T. Mizutania, G. Kutlukb, H. Namatameb, M. Taniguchib and S. Yagia, Appl. Surf. Sci., 2012, 262, 184–187 CrossRef.
- Z. Fang, Y. Wang, J. Song, Y. Sun, J. Zhou, R. Xu and H. Duan, Nanoscale, 2013, 5, 9830–9838 RSC.
- P. Mukherjee, M. Roy, B. P. Mandal, G. K. Dey, P. K. Mukherjee, J. Ghatak, A. K. Tyagi and S. P. Kale, Nanotechnology, 2008, 19, 1–7 Search PubMed.
- R. G. Pearsons, Inorg. Chem., 1998, 27, 734–740 CrossRef.
- A. Samanta, B. B. Dhar and R. N. Devi, J. Phys. Chem. C, 2012, 116, 1748–1754 CAS.
- A. Samanta and R. N. Devi, ChemCatChem, 2013, 5, 1911–1916 CrossRef CAS.
- R. Isono, T. Yoshimura and K. Esumi, Langmuir, 2004, 20, 237–243 CrossRef.
- T. Taguchi, K. Isozaki and K. Miki, Adv. Mater., 2012, 24, 6462–6467 CrossRef CAS PubMed.
- M. Moreno, F. J. Ibanez, J. B. Jasinski and F. P. Zamborini, J. Am. Chem. Soc., 2011, 133, 4389–4397 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra06179a |
| This journal is © The Royal Society of Chemistry 2014 |