
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombining metal–metal cooperativity, metal–ligand cooperativity and chemical non-innocence in diiron carbonyl complexes†
Cody B.
van Beek
a,
Nicolaas P.
van Leest
 b,
Martin
Lutz
c,
Sander D.
de Vos
a,
Robertus J. M.
Klein Gebbink
a,
Bas
de Bruin
b and
Daniël L. J.
Broere
*a
b,
Martin
Lutz
c,
Sander D.
de Vos
a,
Robertus J. M.
Klein Gebbink
a,
Bas
de Bruin
b and
Daniël L. J.
Broere
*a
aOrganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Faculty of Science, Utrecht University Universiteitsweg 99, 3584 CG, Utrecht, The Netherlands. E-mail: d.l.j.broere@uu.nl
bHomogeneous, Supramolecular and Bio-Inspired Catalysis Group, Van 't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands
cStructural Biochemistry, Bijvoet Centre for Biomolecular Research, Faculty of Science, Utrecht University Universiteitsweg 99, 3584 CG, Utrecht, The Netherlands
First published on 18th January 2022
Abstract
Several metalloenzymes, including [FeFe]-hydrogenase, employ cofactors wherein multiple metal atoms work together with surrounding ligands that mediate heterolytic and concerted proton–electron transfer (CPET) bond activation steps. Herein, we report a new dinucleating PNNP expanded pincer ligand, which can bind two low-valent iron atoms in close proximity to enable metal–metal cooperativity (MMC). In addition, reversible partial dearomatization of the ligand's naphthyridine core enables both heterolytic metal–ligand cooperativity (MLC) and chemical non-innocence through CPET steps. Thermochemical and computational studies show how a change in ligand binding mode can lower the bond dissociation free energy of ligand C(sp3)–H bonds by ∼25 kcal mol−1. H-atom abstraction enabled trapping of an unstable intermediate, which undergoes facile loss of two carbonyl ligands to form an unusual paramagnetic (S = 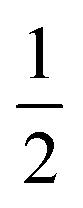 ) complex containing a mixed-valent iron(0)–iron(I) core bound within a partially dearomatized PNNP ligand. Finally, cyclic voltammetry experiments showed that these diiron complexes show catalytic activity for the electrochemical hydrogen evolution reaction. This work presents the first example of a ligand system that enables MMC, heterolytic MLC and chemical non-innocence, thereby providing important insights and opportunities for the development of bimetallic systems that exploit these features to enable new (catalytic) reactivity.
) complex containing a mixed-valent iron(0)–iron(I) core bound within a partially dearomatized PNNP ligand. Finally, cyclic voltammetry experiments showed that these diiron complexes show catalytic activity for the electrochemical hydrogen evolution reaction. This work presents the first example of a ligand system that enables MMC, heterolytic MLC and chemical non-innocence, thereby providing important insights and opportunities for the development of bimetallic systems that exploit these features to enable new (catalytic) reactivity.
Introduction
Various active sites in metalloenzymes feature cofactors with a multinuclear assembly that catalyze challenging chemical transformations at ambient conditions.1 A notable example is [FeFe]-hydrogenase which contains an active site comprising two iron centers that are connected by a bridging azadithiolate ligand.1b,2 This enzyme rivals the efficiency of the platinum group metals for both H2 oxidation and H2 production from protons and electrons.3 The extraordinary catalytic activity of [FeFe]-hydrogenase is the result of a combination of structural features in the inner and outer coordination sphere of the active site.4 These include the azadithiolate mediated heterolytic cleavage of H2 through metal–ligand cooperativity (MLC),5 the close proximity between the Fe centers that allows for metal–metal cooperativity (MMC),2 and long-range concerted proton–electron transfer (CPET) steps.6Inspired by nature's multinuclear cofactors, a renewed interest in the synthesis and study of bimetallic systems is observed in recent years.7 The utilization of synthetic complexes wherein multiple metals work together to activate chemical bonds is a promising avenue to stabilize reactive species or develop new chemical transformations.8 The increased popularity for studying multimetallic complexes is evident by a wide range of ligand scaffolds designed to accommodate multiple metal centers that have recently been reported.7–9 Ligands based on 1,8-naphthyridine have been shown to be highly suitable for the synthesis of bimetallic complexes capable of MMC.10 However, even with these ‘privileged’ platforms binding of two metals in close proximity can present challenges on its own.
A more established avenue wherein chemists have drawn inspiration from nature involves the application of ligand systems containing design features for MLC. Such cooperative substrate activation through MLC is a well-developed concept in homogeneous catalysis.11 The two main strategies involve proton-responsive ligands that facilitate bond heterolysis over the ligand and metal12 or redox non-innocent ligands that can participate in electron transfer processes.13 In recent years researchers have developed ligand systems that can enable both MMC and MLC for the activation of chemical bonds. Most prominently, the group of Uyeda has demonstrated how dinickel complexes of a redox non-innocent 1,8-naphthyridinediimine-based ligand can catalyze chemical transformations with superior activity and selectivity or distinct reactivity from what is possible with mononuclear analogues.14 This demonstrates the potential of ligand systems that can both accommodate two metal atoms in close proximity and display redox non-innocence (Fig. 1, top). Along these lines our group has recently reported a proton-responsive 1,8-naphthyridine-derived ‘expanded pincer’ ligand, tBuPNNP, which can bind two metal atoms and enables heterolytic cooperative H2 cleavage on a dicopper(I) complex (Fig. 1, middle).15
 | ||
| Fig. 1 Different methods of metal–ligand cooperativity in bimetallic complexes. | ||
A less-explored MLC strategy involves the use of chemically non-innocent ligands that can act as both a proton and electron donor or acceptor through concerted proton–electron transfer (CPET) processes.16 The utilization of CPET steps in catalytic cycles is a strategy exploited by various natural systems and circumvents the formation of high-energy charged intermediates that result from sequential electron and proton transfer steps.17 Systems that can combine MMC for the binding and activation of substrates together with CPET steps through ligand chemical non-innocence could enable new or more efficient catalytic chemical transformations. However, to the best or our knowledge, no ligand systems that can combine these elements have been reported to date.
Herein, we describe the synthesis and coordination chemistry of dinuclear iron(0) carbonyl complexes of a new iPrPNNP ligand. Like the related dicopper analogues, the ligand methylene linkers in these complexes can be deprotonated concomitant with partial dearomatization of the naphthyridine core. In addition, we show that the expanded pincer ligand displays chemical non-innocence in CPET reactivity, and that the binding mode of the ligand drastically affects the C(sp3)–H bond strength. This feature can be leveraged to enable binding of two iron-carbonyl centers in close proximity, which proved challenging via other methods. Finally, we show that the diiron complexes enable the same three features that are key to the function of [FeFe]-hydrogenase – i.e. proton-responsive MLC, MMC and CPET (albeit long range) steps – and are also active catalysts for the electrochemical hydrogen evolution reaction (HER).
Results & discussion
Synthesis and reactivity
The 2,7-bis((diisopropylphosphaneyl)methyl)-1,8-naphthyri-dine (iPrPNNP) ligand was prepared as an air-sensitive off-white solid in 41% yield through a procedure analogous to that previously reported for the tBuPNNP ligand (see ESI† for more detail).15 Reacting iPrPNNP with two equivalents of Fe2(CO)9 in THF at ambient temperature (Scheme 1) results in the formation of a dark green solution. Analysis of the reaction mixture after 3 h by 31P{1H} and 1H NMR spectroscopy shows the formation of an approximately equimolar mixture of nonsymmetric [Fe2(iPrPNNP)(CO)7] (1) and (on average) C2v-symmetric [Fe2(iPrPNNP)(CO)8] (2). Prolonged reaction times or heating the reaction mixture to reflux in THF did not change the ratio between 1 and 2. Nonsymmetric complex 1 was isolated as an air-sensitive dark green solid in 26% yield by crystallization from toluene at −40 °C. The 31P{1H} NMR spectrum of 1 in C6D6 at 298 K shows two singlets at δ = 93.6 and 86.5 ppm, which are shifted downfield compared to free iPrPNNP. This implies coordination of both phosphorous atoms to iron centers.18 The 13C{1H} NMR spectrum features the expected number of resonances for a nonsymmetric species, but only two resonances corresponding to CO ligands at δ = 225.8 and 214.2 ppm are found. This implies that the axial and basal carbonyl ligands on each metal center are in rapid exchange with each other, causing averaging to a single resonance.19 The IR spectrum of complex 1 shows six bands in the carbonyl region, consistent with the incorporation of more than one iron carbonyl fragment. Three CO bands are assigned to the iron tricarbonyl fragment20 and three are expected for a R1Fe(CO)4 fragment,21 which is consistent with the NMR spectra and solid-state structure of 1 (see Fig. 3 and ESI† for detail). Although C2v-symmetric complex 2 was not isolated, it could be obtained as the major species in a 0.85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.15 mixture with complex 1 following a series of extractions. Spectroscopic analyses of 2 (see ESI†) and its reactivity (see below) agree with its proposed structure (Scheme 1).
:0.15 mixture with complex 1 following a series of extractions. Spectroscopic analyses of 2 (see ESI†) and its reactivity (see below) agree with its proposed structure (Scheme 1).
 | ||
| Scheme 1 Synthesis of complexes 1 and 2 by reaction of iPrPNNP with Fe2(CO)9. | ||
To investigate the proton-responsive behavior of the iPrPNNP ligand in 1, its reactivity towards bases was investigated. The addition of 1.1 equiv. KOtBu to a dark green solution of 1 in THF at ambient temperature results in immediate formation of a dark red solution due to the formation of complex K(THF)x[Fe2(iPrPNNP*)(CO)7] (3). Complex 3 was isolated as a red-brown air- and water sensitive solid in 67% yield (Scheme 2). The 31P{1H} NMR spectrum of 3 in THF-d8 at 298 K displays two closely spaced singlets at δ = 82.6 and 82.4 ppm. The deprotonation of one of the ligand methylene linkers in 3 is evident from the two characteristic doublets at δ = 4.01 ppm (2JH,P = 3.2 Hz) and δ = 3.40 ppm (2JH,P = 8.9 Hz) with an integral ratio of 1:2, respectively. Additionally, an upfield shift of the naphthyridine 1H NMR resonances is observed, consistent with partial dearomatization of the ligand backbone.15 Similar to 1, the 13C{1H} NMR spectrum of 3 in THF-d8 at 298 K features only two resonances for the carbonyl ligands at δ = 231.6 and 216.1 ppm. Compared to 1 and 2 the CO stretching vibrations in 3 are less well separated in the IR spectrum, but show a largely unaffected Fe(CO)4 fragment and slightly red-shifted vibrational energies for the Fe(CO)3 fragment. The latter is consistent with the expected increase in π-backdonation in the Fe(CO)3 fragment. Together these observations show that there is no change in binding to both iron centers beyond the change from a neutral to a mono-anionic ligand bound to the tricarbonyl iron fragment, and this is in agreement with the solid-state structure (see Fig. 3). In the solid state, 3 forms a one-dimensional coordination polymer with the anionic complex linked by potassium in a chain (Fig. S88†). The potassium ion is stabilized by coordination of two bound THF molecules, three carbonyl O atoms, and the π-system of the naphthyridine. Interestingly, the potassium ion is almost equidistant to both the oxygen atom (K1–O1 = 3.050(4) Å) and carbon atom (K1–C23 = 3.172(5) Å) of one of the carbonyl ligands. This is a rare case of a side-on coordinated carbonyl ligand of which only a handful of examples have been reported.22 Partial dearomatization of the iPrPNNP ligand is evident by shortening of the C1–C2 bond (1.364(7) Å), which is 0.131(7) Å shorter than in 1 and the C9–C10 bond (1.514(6) Å) within the same molecule. In addition, localized double bonds in the dearomatized ring (Table S4†) and the shortened C1–P1 bond length of 1.781(5) Å are observed, which are distinct features of a partially dearomatized iPrPNNP* ligand.15
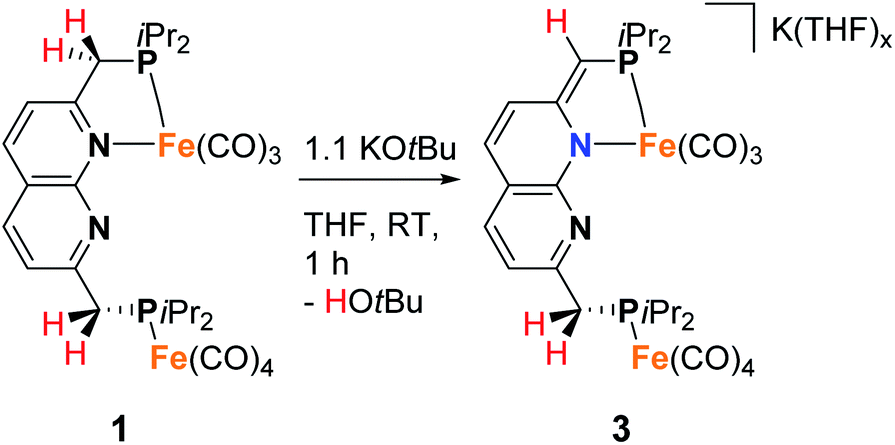 | ||
| Scheme 2 Synthesis of 3 by deprotonation of 1. | ||
As we were interested in studying the chemistry of diiron complexes wherein both metal centers are bound in the naphthyridine binding pocket, we pursued various strategies to remove CO ligands from isolated 1. Routes involving the use of N-oxides (TEMPO or Me3NO),23 refluxing in toluene or anisole or different synthesis conditions all gave inseparable mixtures of unidentified products containing free iPrPNNP or insoluble precipitates (see ESI†). In contrast, we found that exposure of a solution of 2 (containing 15% 1) in benzene to a high-pressure Hg-arc lamp (125 W) for 15 min leads to a color change from yellow–green to dark green. 31P{1H} NMR analysis of the mixture (Fig. S36†) showed selective conversion of 2 to 1 (Scheme 3). Surprisingly, irradiation of the reaction mixture for a total of 120 min leads to a dark amber-colored solution. 1H and 31P NMR analysis of the reaction mixture showed the loss of diamagnetic signals displaying only a few broad resonances in the aliphatic region (Fig. S36 and S37†) due to formation of paramagnetic complex [Fe2(iPrPNNP*)(CO)5] (4), which was isolated as a dark-brown solid in 86% yield.
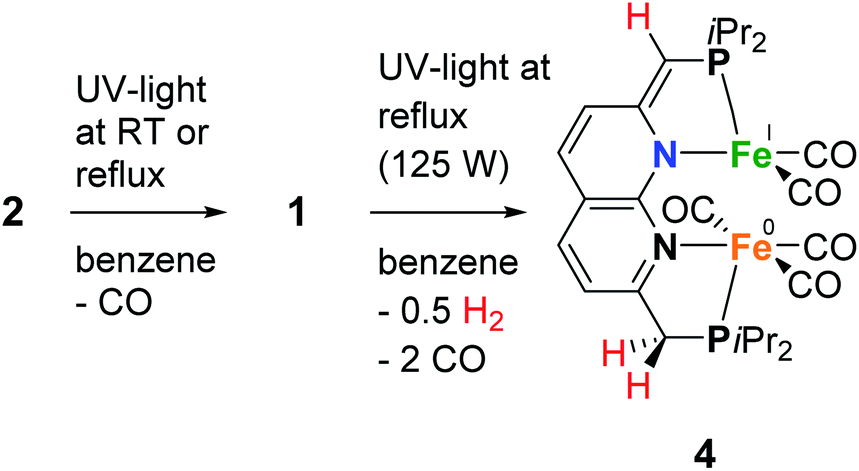 | ||
| Scheme 3 The photochemical synthesis of complexes 1 and 4. | ||
Despite numerous attempts, no single-crystals of 4 suitable for X-ray diffraction or satisfactory combustion analysis were obtained. However, based on spectroscopic analysis and follow-up reactivity (see below), we found that 4 comprises a diiron pentacarbonyl core contained within the dinucleating binding pocket of the singly dearomatized iPrPNNP* ligand as depicted in Scheme 3. A balanced equation for the formation of 4 from 1 would require the loss of 2 CO ligands and one H atom (i.e. 0.5 equiv. of H2), which could originate from a process involving homolytic cleavage of a C–H bond on the ligand side-arms. Indeed, headspace GC analysis of the reaction vessel after photolysis of 1 qualitatively confirmed the formation of both CO and H2 (see Fig. S85–S87†).
The IR spectrum of 4 shows four bands in the CO region at νCO of 1990, 1929, 1893 and 1871 cm−1. Although a weak band at 1633 cm−1 is observed, which is in the range of where bridging CO ligands can be found,24 we assign this band to a vibration of localized double bonds in the dearomatized ring (see ESI† for detail). The X-band EPR spectrum of a toluene solution of 4 at room temperature displays an isotropic singlet (giso = 2.0475) without resolved hyperfine interactions (HFIs) (Fig. 2A), indicative of a compound with a doublet electronic ground state (S = 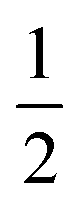 ). Performing low temperature EPR measurements at 30 K in a toluene glass revealed a rhombic signal with g11 = 2.0081, g22 = 2.0282, g33 = 2.1028 without resolved HFIs present according to simulation (Fig. 2B). These EPR results indicate that the paramagnetic center in 4 is a low spin Fe(I) complex.25 The calculated spin density plot of a DFT optimized (B3LYP/def2-TZVP) geometry of 4 (S =
). Performing low temperature EPR measurements at 30 K in a toluene glass revealed a rhombic signal with g11 = 2.0081, g22 = 2.0282, g33 = 2.1028 without resolved HFIs present according to simulation (Fig. 2B). These EPR results indicate that the paramagnetic center in 4 is a low spin Fe(I) complex.25 The calculated spin density plot of a DFT optimized (B3LYP/def2-TZVP) geometry of 4 (S = 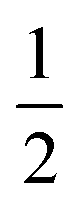 ) shows 94% of the positive Mulliken spin density on the iron center (SOMO on dz2 orbital) (Fig. 2C). The DFT-calculated EPR parameters (B3LYP/def2-TZVP: giso = 2.0245, g11 = 2.0032, g22 = 2.0221, g33 = 2.0483, A31P1 = −19.0 MHz, A31P2 = −12.4 MHz) are in good agreement with the simulated EPR spectrum of 4 at both room temperature and 30 K, although the calculated HFIs with phosphorus are overestimated while the rhombicity is somewhat underestimated.
) shows 94% of the positive Mulliken spin density on the iron center (SOMO on dz2 orbital) (Fig. 2C). The DFT-calculated EPR parameters (B3LYP/def2-TZVP: giso = 2.0245, g11 = 2.0032, g22 = 2.0221, g33 = 2.0483, A31P1 = −19.0 MHz, A31P2 = −12.4 MHz) are in good agreement with the simulated EPR spectrum of 4 at both room temperature and 30 K, although the calculated HFIs with phosphorus are overestimated while the rhombicity is somewhat underestimated.
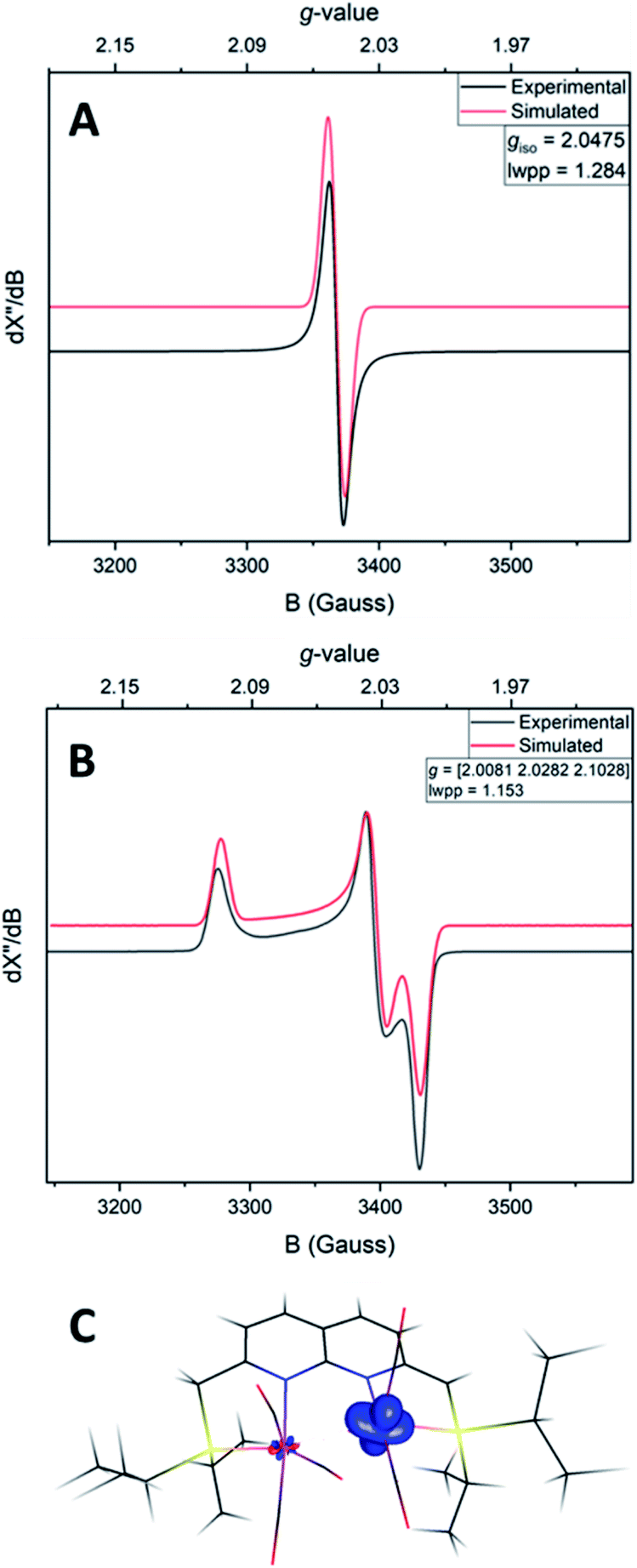 | ||
| Fig. 2 Experimental (black) and simulated (red) X-band EPR spectrum of 4 in toluene at room temperature (A) or in a toluene glass at 30 K (B). (A): microwave freq. 9.650747 GHz, mod. amp. 4.000 G, power 0.6325 mW. Simulation parameters: giso = 2.0475, Gaussian line broadening 1.284 mT. (B): microwave freq. 9.641384 GHz, mod. amp. 4.000 G, power 0.6325 mW. Simulation parameters: g [2.0081, 2.0282, 2.1028], Gaussian line broadening 1.153 mT. (C): the DFT optimized structure of 4 (B3LYP/def2-TZVP) and its spin density plot (isosurface value of 0.02 e Bohr−3). | ||
Cyclic voltammetry (CV) of 4 in THF solution using [Bu4N]PF6 as supporting electrolyte, displayed a reversible reduction at E1/2 = −1.74 V vs. Fc+/Fc (Fig. S67†). Accordingly, the addition of one equiv. of potassium naphthalenide (K(C10H8)) or potassium graphite (KC8) to a brown THF solution of 4 at −40 °C results in instantaneous formation of a dark yellow–brown solution. NMR analysis of the resulting mixture revealed a single major diamagnetic species, which was characterized as K(THF)x[Fe2(iPrPNNP*)(CO)5] (5) and was isolated as a brown solid material in 64% yield (Scheme 4). In contrast to 4, complex 5 is poorly soluble in benzene, and we found that using 0.95 equiv. K(C10H8) enabled facile separation from the starting material without the formation of side products. It should be noted that the synthesis of 4via the photochemical route depicted in Scheme 3 is poorly scalable above 10 mg, which is evident by the observation of an inseparable unknown diamagnetic byproduct upon one-electron reduction (see ESI† for more details). Therefore, a different more scalable method for the synthesis of 5 was developed (see below). The IR spectrum of 5 contains four bands assigned to terminal CO ligands at νCO of 1993, 1930, 1899, 1850 cm−1 and a band assigned to a bridging CO ligand at 1635 cm−1. The terminal CO bands are at similar energies as those observed in 4 (1990, 1929, 1893 and 1871 cm−1). This contradicts the larger degree of π-backdonation that would be expected upon one-electron reduction of 4. We reason that this is due to the coordination of the potassium cation to carbonyl ligands, which is observed in the solid-state structure of 5 (Fig. 3 and S89†) and has been reported to result in shorter C–O bonds22a and lower the C–O stretching frequency.26 The 31P{1H} NMR spectrum of 5 in THF-d8 at 298 K displays two doublets at δ = 96.6 ppm (J = 5.2 Hz) and 86.6 ppm (J = 5.2 Hz), indicating that the phosphorous nuclei are magnetically coupled. As in 3, downfield shifted naphthyridine resonances and two methine/methylene doublets are observed at δ = 4.26 ppm (2JH,P = 2.8 Hz) and 3.18 ppm (2JH,P = 8.6 Hz), which are characteristic for a partially dearomatized iPrPNNP* ligand. The observation of a single methylene resonance that integrates for two protons is indicative of a on average Cs-symmetric species in solution.27 Although the solid-state structure of 5 displays a bridging carbonyl ligand orthogonal to the naphthyridine plane (Fig. 3), we reason that the μ-carbonyl undergoes rapid exchange on the NMR timescale giving on average a Cs-symmetric species in solution.
 | ||
| Scheme 4 The reduction of complex 4 to complex 5 and its protonation to 6. | ||
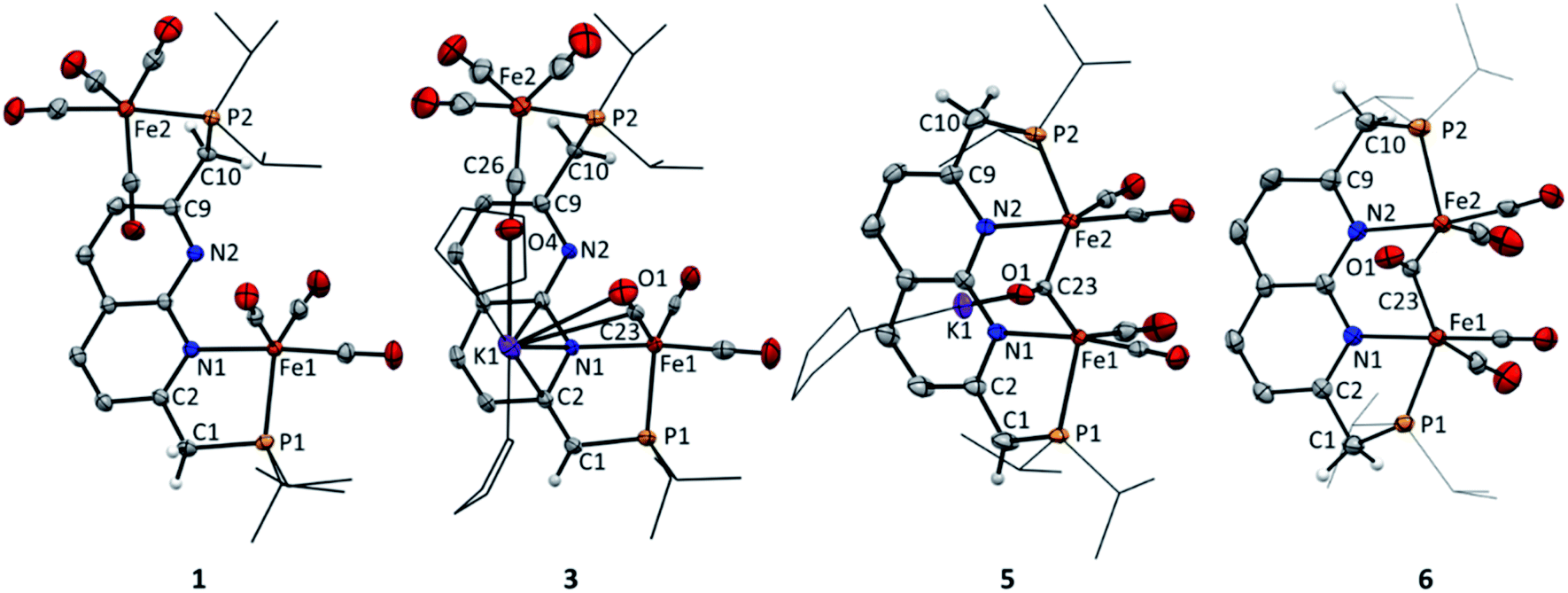 | ||
| Fig. 3 Displacement ellipsoid plots (50% probability) of the asymmetric units of complex 1, 3, 5 and 6. Most hydrogen atoms and the benzene molecules in 6 are omitted and iPr groups on P and the THF molecules in 3 and 5 are depicted as wireframe for clarity. For 5 and 6 only the major disorder component is shown. | ||
This is supported by the absence of carbonyl resonances in the 13C{1H} NMR spectrum, which could be due to line broadening because of rapid exchange of the terminal and bridging carbonyl ligands.28 The solid-state structure features an anionic complex wherein a diironpentacarbonyl core is bound within the expanded pincer binding pocket, and a potassium cation (Fig. 3). Both iron centers display heavily distorted trigonal bipyramidal geometries, likely caused by the rigid chelating ligand environment. All carbonyl ligands are coordinated to potassium ions through their oxygen atoms, with the terminal carbonyl ligands coordinated to potassium ions from other unit cells (see Fig. S89†). The Fe1–Fe2 distance (2.5585(4) Å) is typical for carbonyl bridged diiron complexes2b29 and smaller than the sum of their covalent radii30 (low spin iron(0): 2.64 Å). However, quantum chemical analysis has shown that in such systems Fe–Fe bonding is very weak and that a covalent bond is not present.31 NBO calculations of a DFT (BP86/def2-TZVP) gas-phase optimized structure of 5 also indicate that there is no significant bonding interaction between the iron centers (Wiberg bond index 0.1781, atom–atom net linear NLMO/NPA bond orders 0.285).
Although 5 is stable at room temperature under inert atmosphere, it is highly susceptible to protonation. Filtrating small amounts of yellow–brown THF solutions of 5 over a Celite pad (note that Celite can serve as a mild proton source), causes a color change to reddish brown. 31P NMR analysis of the resulting mixture showed quantitative conversion to a new complex. In line with this observation, reacting 5 with one equiv of Et3NHCl in THF affords complex [Fe2(iPrPNNP)(CO)5] (6) as a light brown solid in quantitative yields (see Scheme 4). The 1H, 13C and 31P NMR spectra of 6 in THF-d8 at 298 K show the expected number of resonances for a C2v-symmetric species. As for 5, this could indicate that rapid exchange of both terminal and bridging carbonyl ligands takes place or that the bridging carbonyl ligand is in the naphthyridine plane. The 31P{1H} NMR spectrum contains a single resonance at δ = 100.9 ppm and the resonances observed in the 1H NMR spectrum are consistent with the presence of an aromatic naphthyridine backbone and two symmetric methylene linkers. Similar to 5, the 13C{1H} NMR spectrum of 6 does not contain resonances corresponding to the carbonyl ligands, which is attributed to rapid exchange between the carbonyl ligands. This would also explain the apparent observed C2v symmetry of 6 in solution that is not observed in the solid-state (Fig. 3). The solid-state structure of 6 reveals a neutral complex that displays the same diiron pentacarbonyl core as complex 5, but features a symmetric aromatic naphthyridine ligand backbone. Although the iPrPNNP ligand in 6 is less rigid than the iPrPNNP* ligand in 5, both iron centers still have heavily distorted trigonal bipyramidal geometries. The Fe1–Fe2 distance (2.5538(7) Å) in 6 is almost identical to that observed in the solid-state structure of 5. Although 6 contains five carbonyl ligands, its IR spectrum only displays four CO bands located at νCO = 1992, 1945, 1886, and 1695 cm−1. This feature is also observed in the Hessian vibrational spectrum of the DFT-optimized geometry of 6 (see Fig. S93–S96†).
Since it is proposed that the synthesis of complex 4 from complex 1 involves the homolytic cleavage of a C–H bond, its reactivity towards hydrogen atom transfer (HAT) using 2,4,6-tri-tert-butylphenoxyl radical (TBP) (BDFEO–H = 80.6 kcal mol−1 in DMSO)36 as the HAT reagent was studied. Directly upon addition of one equiv of TBP to a solution of 1 in THF at ambient temperature a color change from dark green to brown was observed. 1H and 31P{1H} NMR analysis of the reaction mixture showed the disappearance of all resonances of 1 and the formation of 2,4,6-tri-tert-butylphenol (TBP–H), which was also confirmed by IR spectroscopy. This is due to the formation of the paramagnetic [Fe2(iPrPNNP*)(CO)7] (7, Scheme 5). Although complex 7 is readily prepared by an H-atom abstraction from 1, we were surprised to discover it is unstable in solution and in the solid state, even when stored inside a N2-filled glovebox. EPR and IR spectroscopy and headspace GC analysis show that this is due to facile loss of CO ligands upon exposure to visible light concomitant with the formation of complex 4 (see ESI† for details). Nonetheless, this process does not proceed quantitatively even when applying a dynamic vacuum or in refluxing THF. Fortunately, overnight exposure of a THF solution of 7 to a weak (TLC) UV source results in quantitative conversion to 4 (see ESI† and Scheme 5). Moreover, this transformation was found to be insensitive to the scale of the reaction – unlike the harsh photochemical route starting from 1 (Scheme 3) – thereby providing a reproducible route for the synthesis of mixed-valent 4.
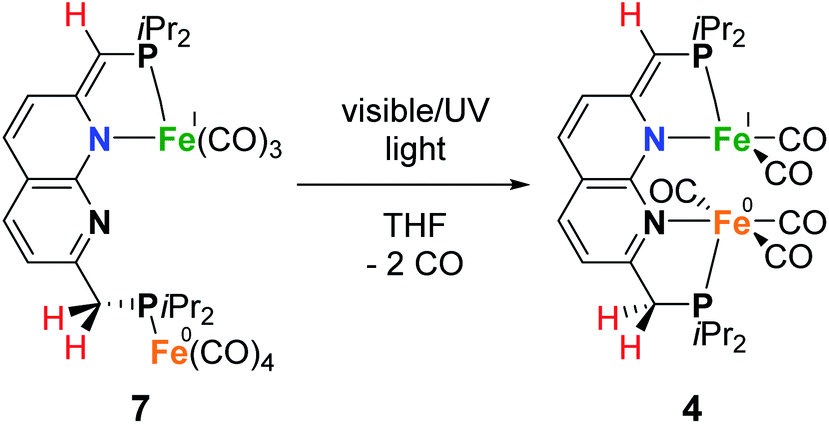 | ||
| Scheme 5 Light induced conversion of 7 to 4. | ||
Despite the sensitive nature of 7, with strict exclusion of light and appropriate precautions spectroscopically clean samples of 7 can be obtained. The ATR-IR spectrum of a freshly prepared reaction mixture containing 7 and TBP–H in THF displays CO bands νCO at 2043, 1965 and 1923 (with a shoulder at approx. 1930) cm−1 together with a weak band at 1883 cm−1. The CO band at νCO = 2043 cm−1 is solely observed in complexes 1–3 and is characteristic for the P-bound Fe(CO)4 outside the naphthyridine pocket, showing that this fragment is present in 7. The addition of TBP to a toluene solution of 1 at room temperature followed by rapid freezing in liquid nitrogen after 5 minutes enabled obtaining a clean EPR spectrum of 7 at both room temperature and 30 K (Fig. 4). Note that freezing the samples after 10 minutes already resulted in observable formation of complex 4. The isotropic EPR spectrum of in situ prepared 7 at room temperature (Fig. 4A) reveals a doublet (giso = 2.0418) due to HFI with phosphorus (Aiso31P = 58.3 MHz). The EPR spectrum of 7 collected in a toluene glass at 30 K displayed a rhombic signal with g11 = 2.0027, g22 = 2.0478, g33 = 2.0684, and HFIs with a single P-nucleus of A1131P = 40.2 MHz, A2231P = 71.2 MHz and A3331P = 62.9 MHz. In contrast to 4, HFIs with phosphorus are clearly present and resolved for 7. A minor contribution of 0.1% weight is attributed to unreacted TBP (giso = 2.005). Similarly to complex 4, these EPR results indicate that the paramagnetic center in complex 7 is a low spin Fe(I) complex. The DFT calculated (B3LYP/def2-TZVP) g values and HFIs of 7 correlate well with the simulated EPR spectrum at 30 K, with exception of the A1131P HFI that is overestimated by 20 MHz. Interestingly, the gas-phase optimized geometry of 7 with an S =  ground state adopts a distorted square pyramidal geometry around the iron tricarbonyl fragment (Fig. S91†), which has been observed before for other low-spin Fe(I) complexes.25a,32 The positive Mulliken spin density is primarily centered on iron (89%) with the SOMO in the dz2 orbital oriented towards phosphorus and the empty sixth coordination site (Fig. 4C). The spin density on 7 is orthogonally oriented compared to 4, which might explain the large differences in phosphorus HFIs between the two compounds.
ground state adopts a distorted square pyramidal geometry around the iron tricarbonyl fragment (Fig. S91†), which has been observed before for other low-spin Fe(I) complexes.25a,32 The positive Mulliken spin density is primarily centered on iron (89%) with the SOMO in the dz2 orbital oriented towards phosphorus and the empty sixth coordination site (Fig. 4C). The spin density on 7 is orthogonally oriented compared to 4, which might explain the large differences in phosphorus HFIs between the two compounds.
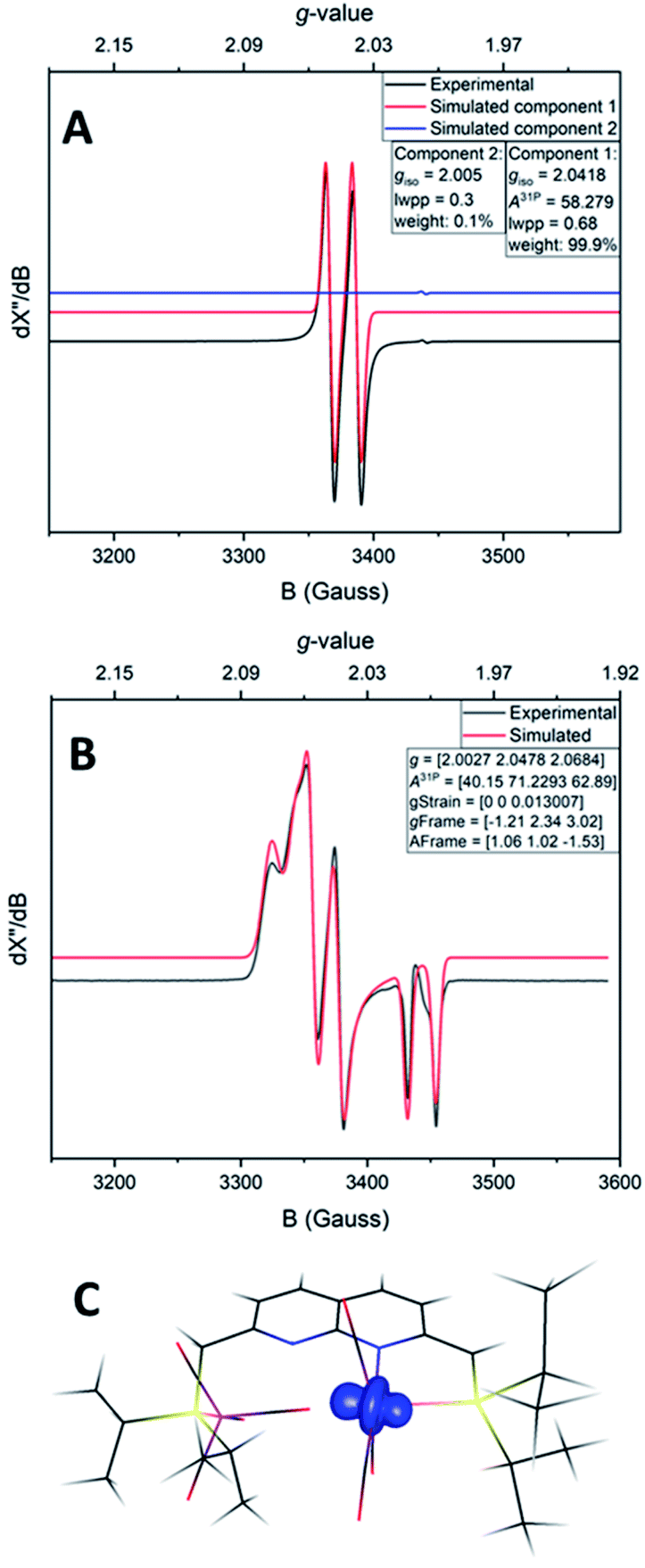 | ||
| Fig. 4 Experimental (black) and simulated (red) X-band EPR spectrum of 7 in toluene at room temperature (A) or in a toluene glass at 30 K (B). (A) Microwave freq. 9.649849 GHz, mod. amp. 4.000 G, power 0.6325 mW. Simulation parameters component 1 (7): giso = 2.0418, A31P = 58.279 MHz, Gaussian line broadening 0.68 mT, weight = 99.9%. Simulation parameters component 2 (TBP): giso = 2.005, Gaussian line broadening 0.3 mT, weight = 0.1%. (B) Microwave freq. 9.650852 GHz, mod. amp. 2.000 G, power 0.2000 mW. Simulation parameters: g [2.0027, 2.0478, 2.0684], A31P [40.15, 71.2293, 62.89], g-strain [0, 0, 0.013007], g-frame [−1.21, 2.34, 3.02], A-frame [1.06, 1.02, −1.53]. (C) The structure of 7 and its spin density plot (isosurface value of 0.02 e Bohr−3). | ||
Despite numerous attempts, we were unsuccessful in growing crystals of 7 that were suitable for X-ray diffraction. Nonetheless, the spectroscopic data, which are supported by computations, are in line with 7 being a low spin paramagnetic Fe(I) complex, as depicted in Scheme 5. This is further supported by the observation that the addition of 1.9 equiv. of KC8 to a freshly prepared solution of complex 7 (from a reaction of 1 with TBP) (excess KC8 for formed TBP–H), yields complex 3 as the main product based on NMR spectroscopy (see ESI† for detail). This shows that hydrogen atom abstraction from 1 is not associated with simultaneous CO ligand loss.
BDFE determination and intermediate trapping
Although mononucleating lutidine-derived PNL (L = P, N, S or other) pincer ligands are mainly known for their MLC involving dearomatization–aromatization through Brønsted acid–base chemistry, their CPET chemical non-innocence reactivity is not unprecedented.33 Pioneering work by Milstein and co-workers showed spontaneous H-atom loss from the methylene linker in [(RPNP)Co(I)-X] (X = CH3 or H) complexes in solution at room temperature, resulting in paramagnetic species (S = ) featuring a proposed ligand-centered radical.34 Chirik and co-workers further investigated this and showed that the resulting paramagnetic complex is best described as a low spin Co(II) complex bearing a monoanionic ligand with a dearomatized pyridine core.35 The facile C–H cleavage was proposed to be inherent to the energetically accessible one-electron Co(I)/Co(II) redox couple paired with the ability for metal-to-ligand single-electron transfer.
) featuring a proposed ligand-centered radical.34 Chirik and co-workers further investigated this and showed that the resulting paramagnetic complex is best described as a low spin Co(II) complex bearing a monoanionic ligand with a dearomatized pyridine core.35 The facile C–H cleavage was proposed to be inherent to the energetically accessible one-electron Co(I)/Co(II) redox couple paired with the ability for metal-to-ligand single-electron transfer.
To probe the feasibility of the proposed homolytic C(sp3)–H bond cleavage in the synthesis of 4 – and the potential of the PNNP platform to engage in cooperative bond activation through pathways involving chemical non-innocence – we were interested to study the bond dissociation free energy (BDFE) of the pseudobenzylic C(sp3)–H bonds in 1. In a thermochemical square scheme, the acidity of a reduced/oxidized species, the reduction potential of protonated/deprotonated species and the BDFE are thermodynamically related in free energies.36 Using the partial square scheme depicted in Scheme 6, the pseudobenzylic C(sp3)–H BDFE can be determined using the Bordwell equation (eqn (1)) with the (C–H) pKa, the E0, as estimated from a reversible electrochemical wave E1/2, and the H+/H standard reduction potential in a certain solvent (CG,sol).
| BDFEsol(C–H) = 1.37 pKa + 23.06E0 + CG,sol | (1) |
 | ||
| Scheme 6 Partial thermochemical square scheme of complex 1 to determine the BDFEC–H. | ||
In non-aqueous media, the pKa of a compound can be determined by a bracketing approach.37 To this end, 3 was dissolved in THF together with equimolar amounts with a series of acids with known pKa values, and the equilibrium mixtures were analyzed by 1H, 19F and 31P NMR spectroscopy.38 Serendipitously, we found that the addition of one equiv. of the C–H acid 9-(perfluorophenyl)-9H-fluorene (pKaDMSO = 14.7) to 3 in THF-d8 at 298 K led to a near 1:1 mixture of 1 and 3. Given that some acid/base reactions can take days or weeks to equilibrate, even with strong bases,39 we also investigated the reaction of 1 with the potassium salt of the 9-(perfluorophenyl)-9H-fluorenide anion. This reaction also provided a similar near 1:1 mixture of 1 and 3. This shows that thermodynamic equilibrium was established and that 1 and 9-(perfluorophenyl)-9H-fluorenene have a near equal pKa in THF. Since the pKaTHF of this 9-(perfluorophenyl)-9H-fluorene has not been reported, we used the relative acidity scale, pKαTHF,40 established by Morris to estimate a pKaTHF of 28 for complex 1 (see ESI† for more detail).39 Cyclic voltammetry of 3 in THF/[Bu4N]PF6 displays a reversible redox couple at E1/2 = −1.28 V (vs. Fc+/Fc) when scanned in the positive direction (Fig. S68†). Together with the estimated pKa, the BDFEC–H in 1 was calculated as 70 kcal mol−1 using eqn (1) with CG,THF = 61 kcal mol−1.41 Using gas phase DFT calculations (B3LYP/def2-TZVP), BDFEs of 90 kcal mol−1 and 66 kcal mol−1 were calculated for the pseudobenzylic C(sp3)–H bonds in iPrPNNP and 1, respectively (see ESI† for details). The calculated value for 1 is in good agreement with the experimentally determined value. The difference in BDFEC–H between the free ligand and complex is comparable to what Chirik and coworkers computationally determined for tBuPNP (80 kcal mol−1) and [(tBuPNP)Co(I)-X] complexes (43–50 kcal mol−1).35 Interestingly, an even lower BDFE of 56 kcal mol−1 was calculated for the pseudobenzylic C(sp3)–H bonds in 6, showing that binding both Fe centers in the PNNP ligand even further weakens these bonds.
In contrast to the reactivity of 1, one equiv of TBP at ambient temperature in THF does not abstract a H-atom from 2 or iPrPNNP, suggesting that the coordination mode influences the pseudobenzylic C(sp3)–H bond strength. This lack of reactivity agrees with the computationally determined BDFEs (B3LYP/def2-TZVP) of 91 kcal mol−1 and 90 kcal mol−1 for the pseudobenzylic C–H bonds in 2 and iPrPNNP, respectively. Compared to the low (calculated) BDFEC–H of 66 kcal mol−1 for 1, this suggests that the coordination of the iron carbonyl fragment in the ligand PN binding pocket leads to significant decrease of the C(sp3)–H bond strength. We reason that this can be attributed to a stabilization of the partially dearomatized iPrPNNP* (i.e., the conjugate base) and Fe(I) center in this coordination mode, thereby enabling facile metal-to-ligand single-electron transfer associated with H-atom abstraction.
Electrochemistry
Considering our observations that the PNNP platforms enables MMC, CPET and heterolytic MLC like processes – three features that are key to [FeFe]-hydrogenase's activity – we decided to investigate complex 4 for the electrochemical hydrogen evolution reaction (HER). Accordingly, CV experiments of complex 4 with phenol as proton source (pKaDMSO = 18.0)42 were performed in THF/[Bu4N]PF6. Although no visible reaction is observed upon the addition of one equiv. phenol to a solution of 4, the one-electron reduction event at −1.74 V vs. Fc+/Fc associated with the formation of 5 loses its reversibility (Fig. S69†). This suggests that upon reduction of 4 to 5 the latter is protonated by phenol to give 6, which is in line with the facile protonation observed during the synthesis of 5. This is further supported by the observation of a second partially reversible reductive event with an onset potential at −1.95 V vs. Fc+/Fc. This potential is identical to the E1/2 potential of the first reductive event observed in the cyclic voltammogram of 6, and this event also loses its reversibility in the presence of one equiv phenol (Fig. 5 and 6).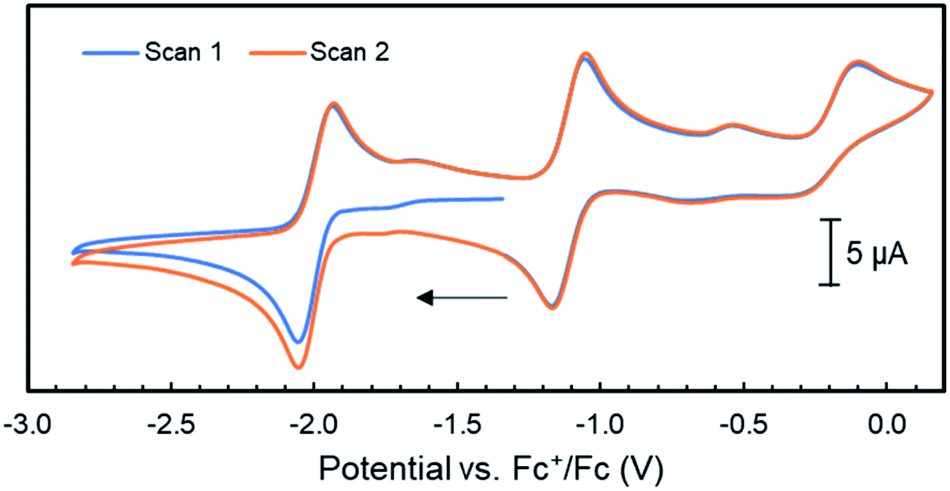 | ||
| Fig. 5 Cyclic voltammograms of complex 6 (1 mM) in THF/[Bu4N]PF6 (scan rate = 0.1 V s−1); the arrow indicates the scan direction. | ||
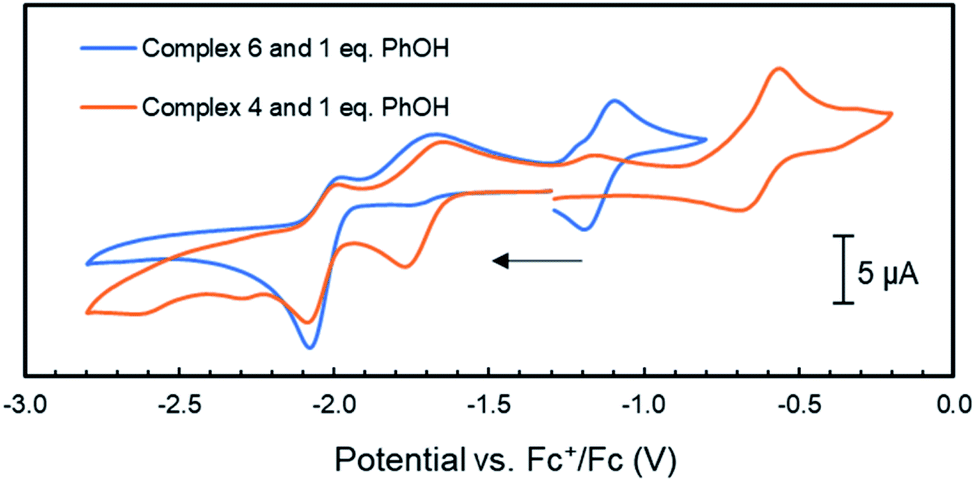 | ||
| Fig. 6 Cyclic voltammograms of complex 4 and 6 (1 mM) with the addition 1 mM of phenol in THF/[Bu4N]PF6 (scan rate = 0.1 V s−1); the arrow indicates the scan direction. | ||
Unfortunately, no indications in the voltammograms of electrocatalytic reduction are observed in the presence of additional equivalents of phenol (Fig. S70†). In contrast, CV experiments of 4 (1 mM) with triethyl ammonium hexafluorophosphate (TEA) as proton source (pKaDMSO = 9.0)43 in THF/[Bu4N]PF6 gave different results. Already upon addition of one equiv. TEA (1 mM) to 4, a color change from amber to orange–green was observed. We propose that this stronger acid protonates the iPrPNNP* ligand of 4 prior to reduction. Nonetheless, the cyclic voltammogram (Fig. S71†) shows an irreversible reductive event with an onset potential at −1.95 V vs. Fc+/Fc, identical to the potential observed in the same experiments of 4 or 6 with phenol (Fig. 6). In contrast to the experiments with phenol, an additional small reductive event with an onset potential at −2.30 V vs. Fc+/Fc feature is observed in the CVs of either 4 or 6 with 1 equiv. TEA (Fig. S71† and 7, respectively). Interestingly, for both 4 (Fig. S72†) and 6 (Fig. 7), higher [TEA] shifts of the onset potential of this event towards less negative potentials and results in increased cathodic current. Both observations are clear signs of electrocatalytic proton reduction.44 This shows that these diiron complexes are catalysts for electrochemical proton reduction, like [FeFe]-hydrogenase, albeit with inferior catalytic performance45 and faradaic efficiency (54%) as observed in an initial bulk electrolysis experiment (see ESI† for details).
 | ||
| Fig. 7 Cyclic voltammograms of complex 6 (1 mM) with the addition of 1–10 mM of TEA in THF/[Bu4N]PF6 (scan rate = 0.1 V s−1); the arrow indicates the scan direction. | ||
The catalytic wave in Fig. 7 is observed at slightly more negative potentials than the reversible reduction of 6. Together with the loss of reversibility of the latter event in the presence of acid (Fig. 6), we hypothesize that the catalytic wave corresponds to the catalytic reaction of a reduced and protonated species. Experiments involving chemical reduction of 6 followed by protonation and vice versa show that H2 is formed in either pathway. Notably, we found that stoichiometric protonation of complex 6 results in formation of diiron hydride complexes (see ESI† for detail), but we have been unable to isolate or characterize these species thus far. Although it is likely that the ligand methylene C–H bonds will be even weaker in more reduced diiron complexes that are accessed at these negative potentials, it is unclear whether the chemical non-innocence of the PNNP ligand described above plays a role in the electrocatalytic pathway. Future research will focus on obtaining mechanistic insights into the operating reaction mechanism and optimizing the catalytic activity of these diiron systems in the HER.
Summary & conclusion
This work describes the preparation of the iPrPNNP ‘expanded pincer’ ligand and its rich coordination chemistry with iron carbonyls. Similar to our previous reports on related dicopper systems, the ligand's methylene linkers can be deprotonated concomitant with partial dearomatization of the naphthyridine core. Interestingly, we show that the methylene linkers in the PNNP ligand are also susceptible to CPET reactivity concomitant with the formation of dihydrogen. The CPET chemistry was evaluated both experimentally and computationally, leading to the conclusion that coordination of the iron carbonyl center within the ligand PN binding pocket weakens the pseudobenzylic C(sp3)–H bond dissociation free energy (BDFE) by ∼25 kcal mol−1 with respect to a P-bound analogue or the free ligand. H-atom abstraction using 2,4,6-tri-tert-butylphenoxyl radical enabled trapping of an unstable potential intermediate, which undergoes facile loss of two carbonyl ligands. EPR analysis and computational methods demonstrate that hydrogen atom abstraction proceeds via a net CPET process and yields low spin Fe(I) complexes with a closed-shell anionic iPrPNNP* ligand. This reactivity was leveraged to develop a reliable synthetic route toward complexes wherein both iron centers are bound in close proximity within the expanded pincer ligand. Finally, we demonstrated that the diiron carbonyl complexes are catalysts for the electrochemical hydrogen evolution reaction.In conclusion, the PNNP expanded pincer ligand platform allows the synthesis of bimetallic first row transition metals and is capable of metal–ligand cooperativity (MLC) through both proton-responsiveness and chemical non-innocence. Both pathways involve partial dearomatization of the naphthyridine core, but the latter is enabled by metal-to-ligand single-electron transfer. To the best of our knowledge, this work demonstrates the first example of ligand chemical non-innocence in naphthyridine-based bimetallic complexes. The low bond-dissociation free energy of the pseudobenzylic C(sp3)–H bonds in the PNNP ligand should be carefully considered when these dinucleating PNNP ligands are combined with metals that undergo facile single-electron redox changes. Future research in our group will focus on obtaining mechanistic understanding of the reported electrochemical HER catalysis and on exploiting the described two-sided MLC strategies for bimetallic bond activation processes of relevance to small molecule activation and homogeneous catalysis.
Data availability
Experimental procedures and details, NMR spectra, and computational and crystallographic computational details can be found in the ESI.† CCDC 2110888–2110891 contain the supplementary crystallographic data for this paper.Author contributions
Synthesis, characterization and DFT calculations were done by C. B. B. All crystallographic measurements and the corresponding data refinement was performed by M. L. EPR measurements and the corresponding simulations and analysis thereof were performed by C. B. B., N. P. L. and B. B. Electrochemical experiments were performed by C. B. B. and S. D. V. Project design, funding acquisition, administration and oversight were done by D. L. J. B. supervision was done by D. L. J. B and R. J. M. K. G. The original draft was written by C. B. B., and reviewing and editing was done by D. L. J. B. with contributions by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by The Netherlands Organization for Scientific Research (START-UP grant 740.018.019 to D. L. J. B.). Access to supercomputer facilities was sponsored by NWO Exacte en Natuurwetenschappen (Physical Sciences). Jim de Ruiter is acknowledged for help with headspace GC analyses. Marc-Etienne Moret is acknowledged for valuable discussions and suggestions. The X-ray diffractometer was financed by the NWO. Emily Monkcom is greatly acknowledged for her artwork for the TOC graphic.References
- (a) M. Can, F. A. Armstrong and S. W. Ragsdale, Chem. Rev., 2014, 114, 4149–4174 CrossRef CAS PubMed; (b) W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed; (c) M. D. Wodrich and X. Hu, Nat. Rev. Chem., 2017, 2, 0099 CrossRef; (d) D. Sippel, M. Rohde, J. Netzer, C. Trncik, J. Gies, K. Grunau, I. Djurdjevic, L. Decamps, S. L. A. Andrade and O. Einsle, Science, 2018, 359, 1484–1489 CrossRef CAS PubMed.
- (a) Y. Li and T. B. Rauchfuss, Chem. Rev., 2016, 116, 7043–7077 CrossRef CAS PubMed; (b) J. A. Birrell, V. Pelmenschikov, N. Mishra, H. Wang, Y. Yoda, K. Tamasaku, T. B. Rauchfuss, S. P. Cramer, W. Lubitz and S. DeBeer, J. Am. Chem. Soc., 2020, 142, 222–232 CrossRef CAS PubMed.
- (a) H. Land, M. Senger, G. Berggren and S. T. Stripp, ACS Catal., 2020, 10, 7069–7086 CrossRef CAS; (b) J. T. Kleinhaus, F. Wittkamp, S. Yadav, D. Siegmund and U.-P. Apfel, Chem. Soc. Rev., 2021, 50, 1668–1784 RSC.
- H. Tai, S. Hirota and S. T. Stripp, Acc. Chem. Res., 2021, 54, 232–241 CrossRef CAS PubMed.
- J. Duan, M. Senger, J. Esselborn, V. Engelbrecht, F. Wittkamp, U.-P. Apfel, E. Hofmann, S. T. Stripp, T. Happe and M. Winkler, Nat. Commun., 2018, 9, 4726 CrossRef PubMed.
- O. Lampret, J. Duan, E. Hofmann, M. Winkler, F. A. Armstrong and T. Happe, Proc. Natl. Acad. Sci., 2020, 117, 20520–20529 CrossRef CAS PubMed.
- For recent reviews see: (a) M. Iglesias, E. Sola and L. A. Oro, in Homo- and Heterobimetallic Complexes in Catalysis: Cooperative Catalysis, ed. P. Kalck, Springer International Publishing, Cham, 2016, pp. 31–58 Search PubMed; (b) N. P. Mankad, Chem.–Eur. J., 2016, 22, 5822–5829 CrossRef CAS PubMed; (c) D. R. Pye and N. P. Mankad, Chem. Sci., 2017, 8, 1705–1718 RSC; (d) C. M. Farley and C. Uyeda, Trends Chem., 2019, 1, 497–509 CrossRef CAS; (e) N. Xiong, G. Zhang, X. Sun and R. Zeng, Chin. J. Chem., 2020, 38, 185–201 CrossRef CAS; (f) S. Rey, H. Tsurugi and K. Mashima, Coord. Chem. Rev., 2018, 355, 223–239 CrossRef; (g) R. Maity, B. S. Birenheide, F. Breher and B. Sarkar, ChemCatChem, 2021, 13, 2337–2370 CrossRef CAS; (h) Q. Wang, S. H. Brooks, T. Liu and N. C. Tomson, Chem. Commun., 2021, 57, 2839–2853 RSC.
- Selected recent examples (a) A. Z. Spentzos, M. R. Gau, P. J. Carroll and N. C. Tomson, Chem. Commun., 2020, 56, 9675–9678 RSC; (b) S. Zhang, P. Cui, T. Liu, Q. Wang, T. J. Longo, L. M. Thierer, B. C. Manor, M. R. Gau, P. J. Carroll, G. C. Papaefthymiou and N. C. Tomson, Angew. Chem., 2020, 132, 15327–15331 CrossRef; (c) M. C. Eaton, V. J. Catalano, J. Shearer and L. J. Murray, J. Am. Chem. Soc., 2021, 143, 5649–5653 CrossRef CAS PubMed; (d) A. Nicolay, M. S. Ziegler, D. W. Small, R. Grünbauer, M. Scheer and T. D. Tilley, Chem. Sci., 2020, 11, 1607–1616 RSC; (e) O. Rivada-Wheelaghan, A. Comas-Vives, R. R. Fayzullin, A. Lledós and J. R. Khusnutdinova, Chem.–Eur. J., 2020, 26, 12168–12179 CrossRef CAS PubMed.
- Selected additional examples (a) M. Li, S. K. Gupta, S. Dechert, S. Demeshko and F. Meyer, Angew. Chem. Int. Ed., 2021, 60, 14480–14487 ( Angew. Chem. , 2021 , 133 , 14601–14608 ) CrossRef CAS PubMed; (b) A. Brinkmeier, R. A. Schulz, M. Buchhorn, C.-J. Spyra, S. Dechert, S. Demeshko, V. Krewald and F. Meyer, J. Am. Chem. Soc., 2021, 143, 10361–10366 CrossRef CAS PubMed; (c) T. Ouyang, H.-J. Wang, H.-H. Huang, J.-W. Wang, S. Guo, W.-J. Liu, D.-C. Zhong and T.-B. Lu, Angew. Chem. Int. Ed., 2018, 57, 16480–16485 ( Angew. Chem. , 2018 , 130 , 16718–16723 ) CrossRef CAS PubMed; (d) A. R. Scheerder, M. Lutz and D. L. J. Broere, Chem. Commun., 2020, 56, 8198–8201 RSC; (e) R. C. Cammarota, M. V. Vollmer, J. Xie, J. Ye, J. C. Linehan, S. A. Burgess, A. M. Appel, L. Gagliardi and C. C. Lu, J. Am. Chem. Soc., 2017, 139, 14244–14250 CrossRef CAS PubMed; (f) W. Tao, J. K. Bower, C. E. Moore and S. Zhang, J. Am. Chem. Soc., 2019, 141, 10159–10164 CrossRef CAS PubMed.
- (a) A. N. Desnoyer, A. Nicolay, P. Rios, M. S. Ziegler and T. D. Tilley, Acc. Chem. Res., 2020, 53, 1944–1956 CrossRef CAS PubMed; (b) C. He, A. M. Barrios, D. Lee, J. Kuzelka, R. M. Daavydov and S. J. Lippard, J. Am. Chem. Soc., 2000, 122, 12683–12690 CrossRef CAS; (c) C. He, J. L. Dubois, B. Hedman, K. O. Hodgson and S. J. Lippard, Angew. Chem. Int. Ed., 2001, 40, 1484–1487 ( Angew. Chem. , 2001 , 113 , 1532–1535 ) CrossRef CAS; (d) J. K. Bera, N. Sadhukhanb and M. Majumdar, Eur. J. Inorg. Chem., 2009, 4023–4038 CrossRef CAS; (e) M. Sarkar, H. Doucet and J. K. Bera, Chem. Commun., 2013, 49, 9764–9766 RSC.
- (a) G. Van Koten, K. Kirchner and M.-E. Moret, Metal–Ligand Co-operativity, Topics in Organometallic Chemistry, 2021, vol. 68 CrossRef; (b) M. R. Elsby and R. T. Baker, Chem. Soc. Rev., 2020, 49, 8933–8987 RSC; (c) T. Higashi, S. Kusumoto and K. Nozaki, Chem. Rev., 2019, 119, 10393–10402 CrossRef CAS PubMed; (d) L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2019, 119, 2681–2751 CrossRef CAS PubMed; (e) J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 ( Angew. Chem. , 2015 , 127 , 12406–12445 ) CrossRef CAS PubMed.
- (a) D. L. DuBois, Inorg. Chem., 2014, 53, 3935–3960 CrossRef CAS PubMed; (b) D. Milstein, Philos. Trans. R. Soc., A, 2015, 373, 20140189 CrossRef PubMed; (c) J. I. van der Vlugt and J. N. H. Reek, Angew. Chem., Int. Ed., 2009, 48, 8832–8846 ( Angew. Chem. , 2009 , 121 , 8990–9004 ) CrossRef CAS PubMed; (d) C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588–602 CrossRef CAS PubMed.
- (a) V. Lyaskovskyy and B. de Bruin, ACS Catal., 2012, 2, 270–279 CrossRef CAS; (b) R. Arevalo and P. J. Chirik, J. Am. Chem. Soc., 2019, 141, 9106–9123 CrossRef PubMed; (c) O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440–1459 RSC; (d) J. I. van der Vlugt, Chem.–Eur. J., 2019, 25, 2651–2662 CrossRef CAS PubMed; (e) N. P. van Leest, F. J. de Zwart, M. Zhou and B. de Bruin, JACS Au, 2021, 1, 1101–1115 CrossRef CAS PubMed.
- (a) Y.-Y. Zhou, D. R. Hartline, T. J. Steiman, P. E. Fanwick and C. Uyeda, Inorg. Chem., 2014, 53, 11770–11777 CrossRef CAS PubMed; (b) Y.-Y. Zhou and C. Uyeda, Science, 2019, 363, 857–862 CrossRef CAS PubMed; (c) J. M. Andjaba, C. J. Rybak, Z. Wang, J. Ling, J. Mei and C. Uyeda, J. Am. Chem. Soc., 2021, 143, 3975–3982 CrossRef CAS PubMed; (d) A. K. Maity, A. E. Kalb, M. Zeller and C. Uyeda, Angew. Chem., Int. Ed., 2021, 60, 1897–1902 ( Angew. Chem. , 2021 , 133 , 1925–1930 ) CrossRef CAS PubMed.
- (a) E. Kounalis, M. Lutz and D. L. J. Broere, Organometallics, 2020, 39, 585–592 CrossRef CAS; (b) E. Kounalis, M. Lutz and D. L. J. Broere, Chem.–Eur. J., 2019, 25, 13280–13284 CrossRef CAS PubMed.
- (a) F. Schneck, M. Finger, M. Tromp and S. Schneider, Chem.–Eur. J., 2017, 23, 33–37 CrossRef CAS PubMed; (b) K. E. Rosenkoetter, M. K. Wojnar, B. J. Charette, J. W. Ziller and A. F. Heyduk, Inorg. Chem., 2018, 57, 9728–9737 CrossRef CAS PubMed; (c) M.-C. Chang, K. A. Jesse, A. S. Filatov and J. S. Anderson, Chem. Sci., 2019, 10, 1360–1367 RSC; (d) A. J. McNeece, K. A. Jesse, A. S. Filatov, J. E. Schneider and J. S. Anderson, Chem. Commun., 2021, 57, 3869–3872 RSC.
- (a) B. Meunier, S. P. de Visser and S. Shaik, Chem. Rev., 2004, 104, 3947–3980 CrossRef CAS PubMed; (b) J. W. Whittaker, Chem. Rev., 2003, 103, 2347–2364 CrossRef CAS PubMed.
- H. Inoue, T. Nakagome, T. Kuroiwa, T. Shirai and E. Fluck, Z. Naturforsch., B: J. Chem. Sci., 1987, 42, 573–578 CrossRef CAS.
- (a) R. Bramley, B. N. Figgis and R. S. Nyholm, Trans. Faraday Soc., 1962, 58, 1893–1896 RSC; (b) F. A. Cotton, A. Danti, J. S. Waugh and R. W. Fessenden, J. Chem. Phys., 1958, 29, 1427–1428 CrossRef CAS; (c) J. P. Jesson and P. Meakin, J. Am. Chem. Soc., 1973, 95, 1344–1346 CrossRef CAS; (d) P. Portius, M. Bühl, M. W. George, F.-W. Grevels and J. J. Turner, Organometallics, 2019, 38, 4288–4297 CrossRef CAS.
- (a) H. T. Dieck and G. Hahn, Z. Anorg. Allg. Chem., 1989, 577, 74–82 CrossRef; (b) Y.-M. Wuu, J. G. Bentsen, C. G. Brinkley and M. S. Wrighton, Inorg. Chem., 1987, 26, 530–540 CrossRef CAS.
- D. J. Darensbourg, H. H. Nelson and C. L. Hyde, Inorg. Chem., 1974, 13, 2135–2145 CrossRef CAS.
- For iron: (a) A. K. Hickey, C. H. Chen, M. Pink and J. M. Smith, Organometallics, 2015, 34, 4560–4566 CrossRef CAS; (b) A. K. Hickey, W.-T. Lee, C.-H. Chen, M. Pink and J. M. Smith, Organometallics, 2016, 35, 3069–3073 CrossRef CAS; (c) A. V. Polezhaev, C. J. Liss, J. Telser, C.-H. Chen and K. G. Caulton, Chem.–Eur. J., 2018, 24, 1330–1341 CrossRef CAS PubMed; (d) For cobalt: S. Aldridge, R. J. Baker, N. D. Coombs, C. Jones, R. P. Rose, A. Rossin and D. J. Willock, Dalton Trans., 2006, 3313–3320 RSC; (e) For manganese: T. K. Mukhopadhyay, N. L. Maclean, M. Flores, T. L. Groy and R. J. Trovitch, Inorg. Chem., 2018, 57, 6065–6075 CrossRef CAS PubMed.
- (a) T.-Y. Luh, Coord. Chem. Rev., 1984, 60, 255–276 CrossRef CAS; (b) J. A. S. Howell, G. Walton, M.-C. Tirvengadum, A. D. Squibb and M. G. Palin, J. Organomet. Chem., 1991, 401, 91–123 CrossRef CAS; (c) Y. Shvo and E. Hazum, J. Chem. Soc., Chem. Commun., 1975, 829–830 RSC; (d) A. J. Birch and L. F. Kelly, J. Organomet. Chem., 1985, 286, c5–c7 CrossRef CAS.
- (a) I. S. Butler, S. Kishner and K. R. Plowman, J. Mol. Struct., 1978, 43, 9–15 CrossRef CAS; (b) I. A. Oxton, J. Mol. Struct., 1982, 78, 77–84 CrossRef CAS; (c) D. T. Moore, J. Oomens, J. R. Eyler, G. Meijer, G. von Helden and D. P. Ridge, J. Am. Chem. Soc., 2004, 126, 14726–14727 CrossRef CAS PubMed.
- For example: (a) Y. Ohki, R. Hoshino and K. Tatsumi, Organometallics, 2016, 35, 1368–1375 CrossRef CAS; (b) R. E. Cowley, M. R. Golder, N. A. Eckert, M. H. Al-Afyouni and P. L. Holland, Organometallics, 2013, 32, 5289–5298 CrossRef CAS; (c) C. J. Adams, R. B. Bedford, E. Carter, N. J. Gower, M. F. Haddow, J. N. Harvey, M. Huwe, M. Á. Cartes, S. M. Mansell, C. Mendoza, D. M. Murphy, E. C. Neeve and J. Nunn, J. Am. Chem. Soc., 2012, 134, 10333–10336 CrossRef CAS PubMed; (d) M. Glatz, B. Stöger, B. Bichler, G. Bauer, L. F. Veiros, M. Pignitter and K. Kirchner, Eur. J. Inorg. Chem., 2020, 1101–1105 CrossRef CAS.
- J. P. Shupp, A. R. Rose and M. J. Rose, Dalton Trans., 2017, 46, 9163–9171 RSC.
- In several unpublished compounds wherein both faces of the naphthyridine plane are inequivalent we always see two independent resonances with a large 2JH,H coupling constant due to the diastereotopic nature of these protons .
- (a) L. Kruczynski and J. Takats, Inorg. Chem., 1976, 15, 3140–3147 CrossRef CAS; (b) J. A. S. Howell, G. Walton, M.-C. Tirvengadum, A. D. Squibb and M. G. Palin, J. Organomet. Chem., 1991, 401, 91–123 CrossRef CAS; (c) D. C. Harris, E. Rosenberg and J. D. Roberts, J. Chem. Soc., Dalton Trans., 1974, 2398–2403 RSC; (d) G. W. Grynkewich and T. J. Marks, Inorg. Chem., 1976, 15, 1307–1314 CrossRef CAS.
- R. H. Duncan Lyngdoh, H. F. Schaefer and R. B. King, Chem. Rev., 2018, 118, 11626–11706 CrossRef CAS PubMed.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC.
- H. Chevreau, C. Martinsky, A. Sevin, C. Minot and B. Silvi, New J. Chem., 2003, 27, 1049–1053 RSC.
- (a) M. J. Therien and W. C. Trogler, J. Am. Chem. Soc., 1986, 108, 3697–3702 CrossRef CAS; (b) J. H. MacNeil, A. C. Chiverton, S. Fortier, M. C. Baird, R. C. Hynes, A. J. Williams, K. F. Preston and T. Ziegler, J. Am. Chem. Soc., 1991, 113, 9834–9842 CrossRef CAS; (c) A. M. Tondreau, C. Milsmann, E. Lobkovsky and P. J. Chirik, Inorg. Chem., 2011, 50, 9888–9895 CrossRef CAS PubMed.
- (a) Y. Gloaguen, C. Rebreyend, M. Lutz, P. Kumar, M. Huber, J. I. van der Vlugt, S. Schneider and B. de Bruin, Angew. Chem., Int. Ed., 2014, 53, 6814–6818 ( Angew. Chem. , 2014 , 126 , 6932–6936 ) CrossRef CAS PubMed; (b) C. Rebreyend, V. Mouarrawis, M. A. Siegler, J. I. van der Vlugt and B. de Bruin, Eur. J. Inorg. Chem., 2019, 4249–4255 CrossRef CAS.
- E. Khaskin, Y. Diskin-Posner, L. Weiner, G. Leitus and D. Milstein, Chem. Commun., 2013, 49, 2771–2773 RSC.
- S. P. Semproni, C. Milsmann and P. J. Chirik, J. Am. Chem. Soc., 2014, 136, 9211–9224 CrossRef CAS PubMed.
- J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS PubMed.
- (a) A. E. Carpenter, C. Chan, A. L. Rheingold and J. S. Figueroa, Organometallics, 2016, 35, 2319–2326 CrossRef CAS; (b) S. Tretiakov, L. Witteman, M. Lutz and M. Moret, Angew. Chem., Int. Ed., 2021, 60, 9618–9626 ( Angew. Chem. , 2021 , 133 , 9704–9712 ) CrossRef CAS PubMed.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS.
- K. Abdur-Rashid, T. P. Fong, B. Greaves, D. G. Gusev, J. G. Hinman, S. E. Landau, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2000, 122, 9155–9171 CrossRef CAS.
- The known pKaDMSO value was converted to pKαTHF using Morris' empirical relationship: pKaDMSO(HA) = 0.85 pKαTHF (HA) − 9.6 .
- F. Schneck, M. Finger, M. Tromp and S. Schneider, Chem.–Eur. J., 2017, 23, 33–37 CrossRef CAS PubMed.
- F. G. Bordwell, R. J. McCallum and W. N. Olmstead, J. Org. Chem., 1984, 49, 1424–1427 CrossRef CAS.
- (a) I. M. Kolthoff, M. K. Chantooni and S. Bhowmik, J. Am. Chem. Soc., 1968, 90, 23–28 CrossRef CAS; (b) M. R. Crampton and I. A. Robotham, J. Chem. Res., 1997, 22–23 RSC.
- C. P. Andrieux, C. Blocman, J. M. Dumas-Bouchiat, F. M'Halla and J. M. Savéant, J. Electroanal. Chem. Interfacial Electrochem., 1980, 113, 19–40 CrossRef CAS.
- R. Zaffaroni, R. J. Detz, J. I. van der Vlugt and J. N. H. Reek, ChemSusChem, 2018, 11, 209–218 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2110888–2110891. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc05473b |
| This journal is © The Royal Society of Chemistry 2022 |