
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePractical synthesis of 3-aryl anthranils via an electrophilic aromatic substitution strategy†
Yang
Gao
 *a,
Simin
Yang
a,
Minwei
She
a,
Jianhong
Nie
a,
Yanping
Huo
a,
Qian
Chen
a,
Xianwei
Li
a and
Xiao-Qiang
Hu
b
*a,
Simin
Yang
a,
Minwei
She
a,
Jianhong
Nie
a,
Yanping
Huo
a,
Qian
Chen
a,
Xianwei
Li
a and
Xiao-Qiang
Hu
b
aSchool of Chemical Engineering and Light Industry, Guangdong University of Technology, Guangzhou, 510006, China. E-mail: gaoyang@gdut.edu.cn
bKey Laboratory of Catalysis and Energy Materials Chemistry of Ministry of Education & Hubei Key Laboratory of Catalysis and Materials Science, School of Chemistry and Materials Science, South-Central University for Nationalities, Wuhan 430074, China
First published on 27th January 2022
Abstract
We report a practical route for the synthesis of valuable 3-aryl anthranils from readily available anthranils and simple arenes by using the classical electrophilic aromatic substitution (EAS) strategy. This transformation goes through an electrophilic substitution and rearomatisation sequence by employing Tf2O as an effective activator. A wide range of arenes were compatible in this transformation, delivering various structurally diversified 3-aryl anthranils in good yields and high regioselectivity. In addition, a variety of readily available feedstocks such as olefins, alkenyl triflates, silyl enolethers, carbonyl compounds, thiophenols and thiols could also participate in the reaction to achieve the C3 alkenylation, alkylation and thioetherification of anthranils. Of note, the synthesized 3-aryl anthranils proved to be a highly robust platform to access a series of biologically active compounds, drug derivatives and organic optoelectronic materials.
Introduction
Bi(hetero)aryls are important architectures that are widely found in a myriad of biologically active compounds, drugs, ligands, and organic functional materials.1 Among them, 3-aryl anthranils are privileged motifs with a broad spectrum of pharmacological activities (Fig. 1a).2 Remarkably, the facile reductive cleavage of the isoxazole rings to 2-aminodiaryl ketones renders them key synthetic precursors towards several marketed nonsteroidal anti-inflammatory drugs (NSAIDs) such as amfenac, bromfenac and nepafenac (Fig. 1b).3 Chlordiazepoxide, derived from 3-phenyl anthranil, is used for the treatment of anxiety, insomnia, and withdrawal symptoms (Fig. 1c).4 The clinically useful proquazone and fluproquazone as analgesics and NSAIDs can also be easily acquired from 3-aryl anthranils.5 In addition, anthranils are highly versatile synthons, which exhibit rich and tunable chemical reactivity in transition metal-catalysed process for the synthesis of valuable N-heterocycles (Fig. S1†).6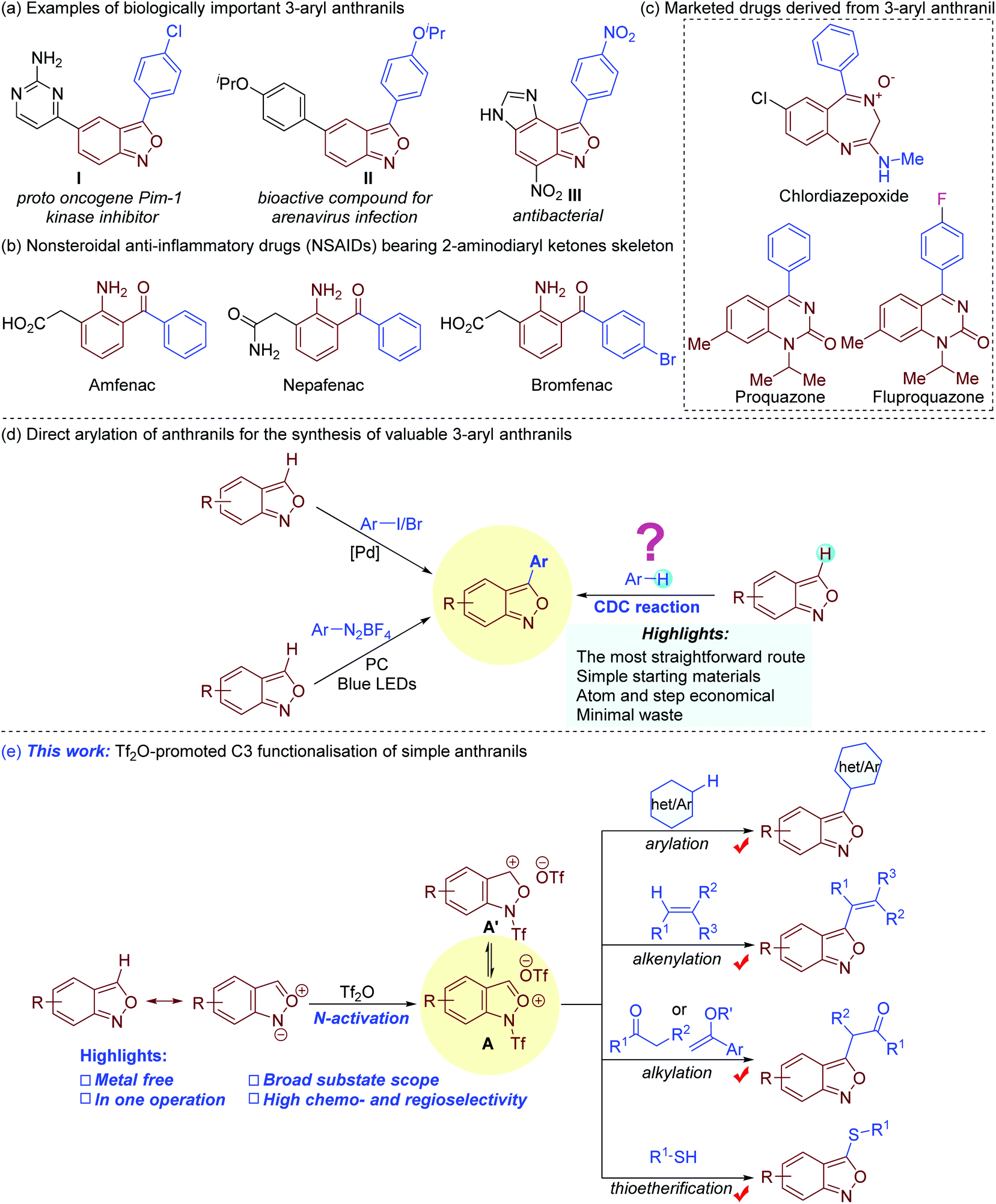 | ||
| Fig. 1 The importance of 3-aryl anthranils (a–c) and strategies to C3 functionalisation of anthranils (d and e). | ||
The 3-aryl anthranil frameworks were traditionally obtained through the de novo synthesis of the isoxazole ring,7 which suffered from limited substrate scope, toxic reagents, harsh conditions, and/or inconvenient substrates. Beyond these methods, the direct C–H arylation of simple anthranils has been established as a powerful strategy to access 3-aryl anthranils (Fig. 1d). In 2015, a palladium-catalysed C–H arylation of anthranils with aryl iodides was first achieved.8 In addition, the Hashmi group disclosed a photoredox C–H arylation of anthranils with aryl diazonium tetrafluoroborates as aryl sources.9 Despite these advances, the direct oxidative C–H/C–H cross-coupling (also known as cross-dehydrogenative-coupling)10 of anthranils and simple arenes should be the most straightforward and desirable route to assemble 3-aryl anthranil frameworks by avoiding the use of prefunctionalised aryl sources.
Electrophilic aromatic substitution (EAS) is a textbook organic reaction, which enables efficient acylation, alkylation, halogenation, nitration and sulfonation of simple arenes. With our continuous interest in anthranil chemistry,11 we recently envisioned the feasibility of synthesis of 3-aryl anthranils from anthranils and simple arenes by using the classical electrophilic aromatic substitution strategy. Given the resonance structure of anthranils (Fig. 1e),7a we presumed that the electrophilicity of anthranils at the C3 position might be further enhanced by an activator such as trifluoromethanesulfonic anhydride (Tf2O)12via the formation of a cation species A. This intermediate would undergo electrophilic substitution with electron-rich arenes, and the expected 3-aryl anthranils could be obtained through a subsequent rearomatisation. Though this proposal seems logical, several issues might be encountered: (1) competitive reactions caused by the reactive intermediate A; (2) regioselectivity of arenes with multiple nucleophilic centers; (3) the compatibility of arenes with mild nucleophilicity and other normal nucleophiles such as olefins, carbonyl compounds, alcohols, thiols, and sulphonamides etc. In addition, to avoid an additional rearomatisation step,12a,b one-pot operation also needs to be considered. Consequently, an appropriate activator and reaction system become essential to realize this novel transformation. Herein, we report our recent progress on a Tf2O-promoted formal oxidative cross-coupling of anthranils for the synthesis of C3 functionalised anthranils.
Results and discussion
The investigation commenced with anthranil 1a and anisole 2a as the model substrates. To our delight, the expected product 3a was obtained in 68% yield when Tf2O (1.0 equiv.) was used as a promoter in 1,2-dichloroethane (DCE) at room temperature (Table 1, entry 1). In sharp contrast, no desired product was detected when other anhydrides such as acetic anhydride (Ac2O), trifluoroacetic anhydride (TFAA) and methanesulfonic anhydride (Ms2O) were used (entries 2–4). In addition, trifluoromethanesulfonic acid (TfOH) and other Lewis acids such as In(OTf)3 and B(C6F5)3 proved to be ineffective for this transformation (entries 5–7). The yield of 3a increased to 81% when Tf2O was added at −20 °C and then slowly warmed the resulting mixture to room temperature (entry 8). The addition of 20 mol% pyridine or 20 mol% 2,6-di-tert-butylpyridine as an activator has an inferior effect on the yield (entries 9 and 10).12e 3 Å molecular sieves and MgSO4 were used to remove moisture, however, decreased yields were observed (entries 11 and 12). Finally, the desired product 3a was obtained in 86% yield when 1.1 equivalents of Tf2O and 1.2 equivalents of anisole were used (entry 13). Notably, all the fruitful conditions gave 3a in high para-selectivity probably because of the bulky secondary carbocation.|
|
|||||
|---|---|---|---|---|---|
| Entry | Promoter | Additive | T/°C |
p![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ob :ob |
Yield of 3a (%)c |
| a Reaction conditions: to a solution of 1a (0.2 mmol) in DCE (1.0 mL), the promoter (0.2 mmol) was added at a specified temperature. Subsequently, 2a (0.2 mmol) was added. The resulting mixture was slowly warmed to room temperature, and continued to stir under an air atmosphere for 5 h. b Ratio was determined by GC-MS analysis. c Yields were determined by 1H NMR analysis with 1,3,5-trimethoxybenzene as the internal standard. The yield in parentheses is the isolated yield. d Tf2O (0.22 mmol) and 2a (0.24 mmol) were used. | |||||
| 1 | Tf2O | — | rt | 13:1 |
68 |
| 2 | Ac2O | — | rt | — | 0 |
| 3 | TFAA | — | rt | — | 0 |
| 4 | Ms2O | — | rt | — | 0 |
| 5 | TfOH | — | rt | — | 0 |
| 6 | In(OTf)3 | — | rt | — | 0 |
| 7 | B(C6F5)3 | — | rt | — | 0 |
| 8 | Tf2O | — | −20 | 14:1 |
81 |
| 9 | Tf2O | Pyridine | −20 | 13:1 |
72 |
| 10 | Tf2O | 2,6-Di-tert-butylpyridine | −20 | 12:1 |
68 |
| 11 | Tf2O | 3 Å MS | −20 | 13:1 |
73 |
| 12 | Tf2O | Mg2SO4 | −20 | 13:1 |
72 |
| 13d | Tf2O | — | −20 | 14:1 |
86 (81) |
As shown in Fig. 2, a wide range of arenes and heteroarenes undergo oxidative cross-coupling with anthranil 1a to afford the desired 3-aryl anthranils in generally good yields with high regioselectivity. Remarkably, toluene (3b), naphthalene (3c) and 1,2,3,4-tetrahydronaphthalene (3d) that are mild nucleophiles reacted smoothly with anthranil 1a under the current conditions. Phenol (3e) and naphthalen-2-ol (3f) bearing a free –OH group were compatible in this reaction. Oxydibenzene (3g) and diphenylsulfane (3h) gave rise to the corresponding products in high para-selectivity. Triphenylamine (3i), a widely used electron donor in organic optoelectronic materials,13 was successfully converted to the desired product in 74% yield. Double functionalisation of oxydibenzene and triphenylamine (3g′ and 3i′) was also achieved by improving the amount of 1a to 2.5 equivalents. A variety of functional groups such as fluoro (3j), chloro (3k), bromo (3l), iodo (3m), phenyl (3n), ester (3q), benzoyl (3r) and allyloxy (3y) were well tolerated in this transformation. Electron withdrawing groups (EWGs) including cyano (3s), carbonyl (3t and 3u), acid (3v) and amide (3w) were also compatible in this transformation when they are not conjugated to the phenyl group. Notably, a 73% yield of 3x bearing 4,4,5,5-tetramethyl-1,3,2-dioxaborolane (Bipn) was obtained with a slight ipso deboronation by-product. Poly-substituted arenes also took part in the reaction (3z-3ad), and even the sterically hindered 1,2,3,4,5-pentamethylbenzene (3ae) delivered the expected product in 55% yield. Polycyclic aromatic hydrocarbons (PAHs) such as naphthalene (3af), 9H-fluorene (3ag and 3ah), pyrene (3ai) and anthracene (3aj) were successfully converted into the expected products in moderate to good yields (56–88%). Similar to the electrophilic bromination of PAHs, the reaction occurs selectively at the most nucleophilic carbon atom of the benzene ring with the lowest aromaticity (please see the ESI† for details).14 1,1,2,2-Tetraphenylethene (TPE) 3ak, an AIEgen molecule having wide application in materials science,15 was an active substrate in this reaction. Significantly, a large set of heterocycles such as indole (3al–3ao), furan (3ap), thiophene (3aq), benzothiophene (3ar and 3as), carbazole (3at), dibenzofuran (3au), 9H-xanthene (3av), dibenzothiophene (3aw), and 10-methylphenothiazine (3ax) proved to be suitable, furnishing the expected products in good yields with high regioselectivity. This protocol was applied in the late-stage modification of several readily available pharmaceutical derivatives. As outlined in Fig. 2 (bottom), the anthranil motif was successfully introduced into the derivatives of gemfibrozil (3ay), naproxen (3az), aspirin (3ba) and tocopherol (3bb) with high efficiency.
 | ||
| Fig. 2 Scope of arenes. Standard reaction conditions: to a solution of 1a (0.3 mmol) in DCE (1.0 mL), Tf2O (0.33 mmol) was added dropwise at −20 °C. Subsequently, 2 (0.36 mmol) was added. The resulting mixture was slowly warmed to room temperature, and continued to stir under an air atmosphere for 5 h. Isolated yields are given. a Ratio was determined by GC-MS analysis of the crude product. b 3.0 equivalents of toluene were used. c 2.5 equivalents of 1a and Tf2O were used. d The ratio indicates the yields of difuctionalisation and monofuctionalisation. e Deacetylated product was obtained. | ||
Next, the scope of anthranils was investigated in this novel oxidative cross-coupling reaction. As shown in Fig. 3, various substituents including F (3bc and 3bd), Cl (3be, 3bf and 3bg), Br (3bh and 3bi), OBz (3bj and 3bk), allyloxy (3bl and 3bm), TsO (3bn), Ph (3bo and 3bp), and CF3 (3bs) were well tolerated, giving rise to the desired poly-substituted 3-aryl anthranils in good yields (46–85%). When 5-(trifluoromethyl)benzo[c]isoxazole 1k was applied as a substrate, the rearomatisation process in the formation of 3bs is much slower than other tested anthranils probably because of the stabilisation of the strong electron-withdrawing group. Notably, C4 substituted anthranils such as 4-fluorobenzo[c]isoxazole (3bt) and 4-chlorobenzo[c]isoxazole (3bu) also showed high reaction efficiency in this transformation.
 | ||
| Fig. 3 Scope of anthranils. a Ratio was determined by GC-MS analysis of the crude product. b After the addition of 1-bromo-4-methoxybenzene for 2 h, the reaction temperature was increased to 60 °C for another 3 h. | ||
Apart from arenes, other readily available nucleophiles such as alkenes, ketones, alkenyl triflates, silyl enolethers, thiophenols and thiols are also compatible in this oxidative cross-coupling reaction. For instance, C3 alkenylation of anthranils was achieved with simple alkenes, furnishing the corresponding 3-alkenyl anthranils in satisfactory yields (Fig. 4a).16 When cyclohexene was used as a substrate, products with positional isomerism of the double bond were obtained (5f). The isomers might be formed via the migration of carbocation intermediates. In addition, 1-phenylvinyl trifluoromethanesulfonate and trimethyl((1-phenylvinyl)oxy)silane also served as effective nucleophiles in the reaction with anthranils to give the corresponding carbonyl compounds (Fig. 4b). Remarkably, similar products were obtained by employing simple carbonyl compounds 8 in the reaction system. Both 1,3-dicarbonyl compounds (7c, 7d and 7e) and simple ketones (7f–7i) could react with anthranil 1a to afford the corresponding 3-alkylated anthranils in 42–82% yields. Moreover, heteroatom nucleophiles such as alcohols, thiols, thiophenols and sulfonamides were tested in this transformation (Fig. 4c). Decane-1-thiol and 2-chlorobenzenethiol gave the desired C3 thioetherified anthranils 9a and 9b in 74% and 80% yields, respectively. However, MeOH, EtOH and TsNH2 could only afford the nucleophilic addition products 9c, 9d and 9e, and the aromatisation step is sluggish under the current reaction conditions.
 | ||
| Fig. 4 Nucleophile screening in C3 functionalisation of anthranils. a From cyclohexene. The marked a/b/c indicates the position of the double bond. | ||
2-Aminodiaryl ketones are core skeletons of nonsteroidal anti-inflammatory drugs (NSAIDs) and key intermediates for drug synthesis.3 They were facilely obtained through this oxidative cross-coupling and a subsequent reductive ring opening from anthranils and simple arenes in one pot (Fig. 5).
 | ||
| Fig. 5 Synthesis of 2-aminodiaryl ketones. | ||
A series of control experiments were conducted to gain more insight into the mechanism. First, anthranil 1c was rapidly converted to 12 and 13 once treated with Tf2O (Fig. 6a). The formation of 12 and 13 was presumably from the reaction of a trace amount of H2O with the proposed oxonium species A. As illustrated in Fig. 6a, these two species reacted smoothly with anisole to give the desired 3-aryl anthranil 3bg in the presence of TfOH, which revealed the intermediacy of compounds 12 and 13 in this transformation. The reaction of 5-(trifluoromethyl)benzo[c]isoxazole 1k and 1-bromo-4-methoxybenzene 2l was quenched in 30 min to obtain another fully characterized intermediate 14 in 87% yield (Fig. 6b). The aromatisation of 14 may go through two possible pathways, namely, direct elimination of CF3SO2H or amide hydrolysis/oxidative aromatisation cascade.12a Control experiments in Fig. 6b indicate that the aromatisation could occur under both acidic and basic conditions without any additional oxidants. Moreover, CF3SO2H was detected by 19F NMR when MsOH or TFA was used as an additive in the aromatisation of 14 (please see the ESI† for details). Therefore, the aromatisation of 14 is likely to be an elimination process of CF3SO2H. Subsequently, the observed KH/KD is 2.04 in parallel kinetic experiments of 14 and D-14 (Fig. 6c). In comparison, there is no significant kinetic isotope effect (KIE = 1.1) in competitive experiments of toluene and toluene-d8 (Fig. 6d). These results support that elimination of CF3SO2H is likely the rate-determining step in this oxidative cross-coupling reaction.
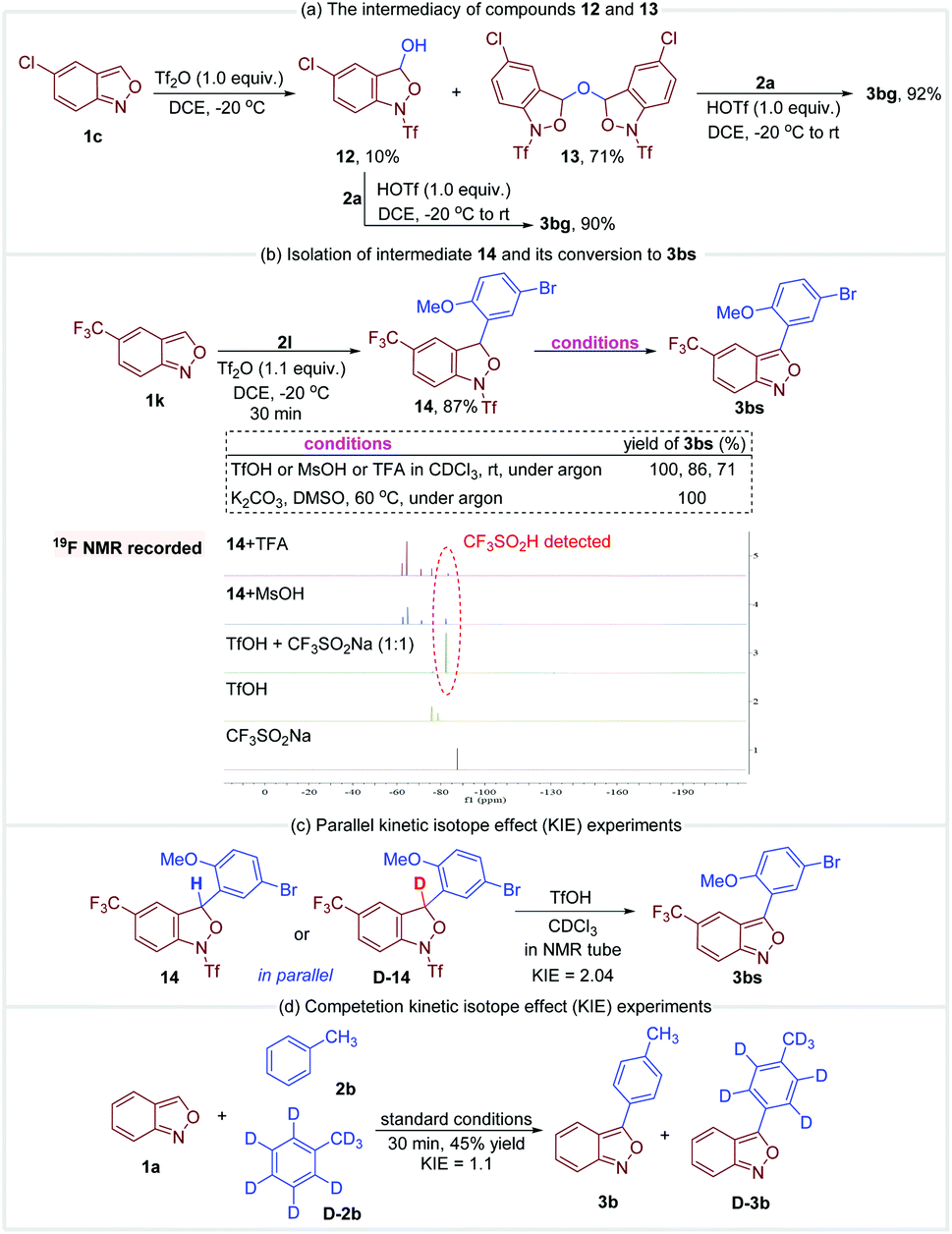 | ||
| Fig. 6 Mechanistic studies. | ||
According to the above results and the previous studies,12a a plausible mechanism is proposed in Fig. 7. The reaction begins with the activation of anthranil 1c by Tf2O, leading to reactive oxonium species 1A and its tautomers 1A′ and 1A″. Then, H2O attacks 1A to deliver intermediate 12, which further adds to another oxonium species giving rise to intermediate 13. In the presence of in situ-generated TfOH, electron-rich arene 2a undergoes electrophilic substitution with intermediate 12 and 13 to give 14a (path a and b). In addition, intermediate 14a may also generate from the direct reaction of arenes 2a and cation species 1A (path c). Finally, the elimination of CF3SO2H from 14a affords the expected 3-aryl anthranil product.12a,17
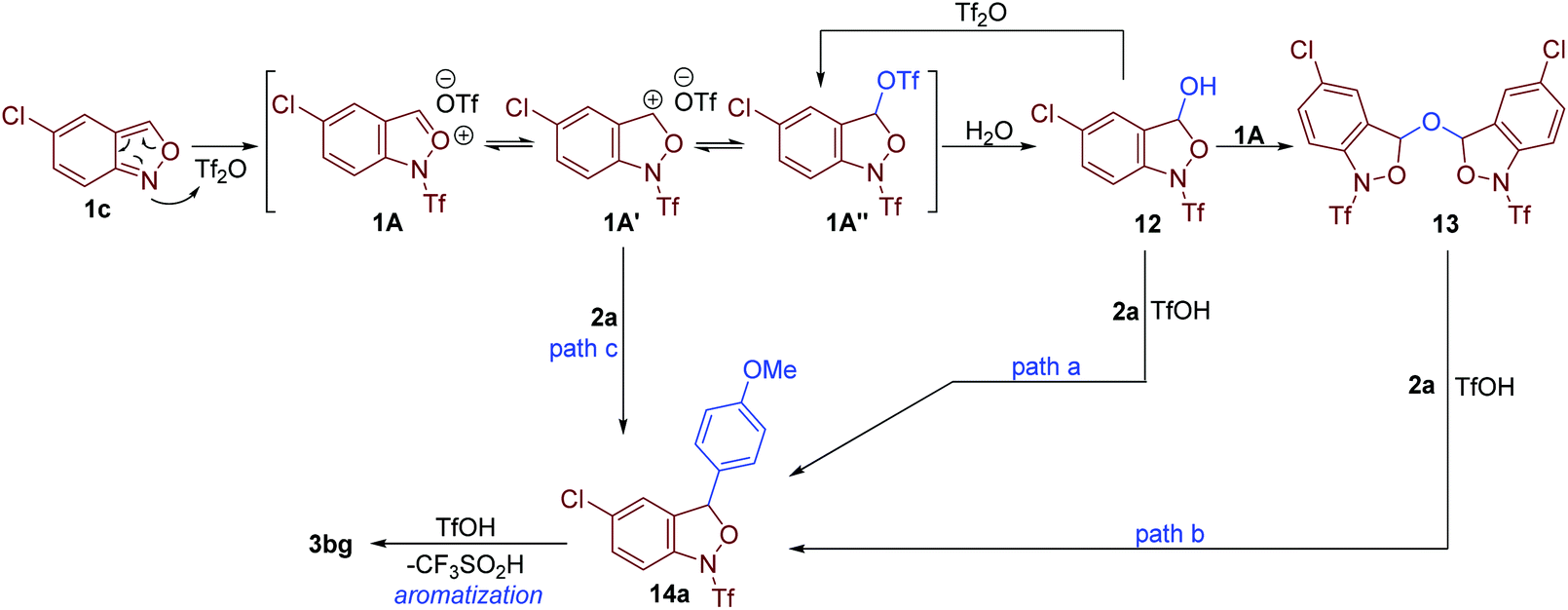 | ||
| Fig. 7 Plausible mechanism. | ||
A series of biologically active compounds and drug derivatives were successfully synthesized to demonstrate the practical utility of our method. 3-Aryl anthranil 3bw that is effective against arenavirus infection was obtained in good yield through the established oxidative cross-coupling and a subsequent Suzuki coupling (Fig. 8a).2d The key intermediate 15 for the synthesis of Amfenac derivative 16 was concisely prepared from 3b,3a in which Ir-catalyzed C–H alkylation with diazotized Meldrum's acid is a key step (Fig. 8b).18 In addition, chlordiazepoxide analogue 19 and proquazone derivative 20 were facilely obtained through the routes illustrated in Fig. 8c and d.4,5 These examples further demonstrate the importance of 3-aryl anthranils and the synthetic potential of this oxidative cross-coupling reaction.
 | ||
| Fig. 8 The construction of biologically active compounds and drug derivatives from 3-aryl anthranils. | ||
In addition, the anthranil backbone can be facilely converted to other useful motifs. As illustrated in Fig. 9, anthranil 3i was reduced by Fe powder to give 2-amino benzophenone 11h in 91% yield. 3i was applied as a robust aminating reagent in Rh-catalyzed sp2 C–H amination of 2-methylbenzoic acid and 2-methylbenzaldehyde, affording compounds 21 and 22.6e,11e Quinoline 23 was produced in 56% yield through a Ni-catalyzed hydroamination/cyclisation cascade of 3i and 1,2-diphenylethyne.11a Acridine derivative 24 was obtained through a Co-catalyzed electrophilic amination and tandem cyclisation,6b which was widely used as a fluorescent probe and a luminescence material in organic light emitting diode (OLED) emitters.13 In addition, 3i took part in a [4+3] cycloaddition with the azaoxyallyl cation, providing a biologically important benzodiazepine derivative 25 in 90% yield.6m
 | ||
| Fig. 9 Derivatisation of the anthranil framework. (a) 3i (0.1 mmol), Fe powder (0.2 mmol) in HOAc/H2O (2:1) at 90 °C. (b) 3i (0.1 mmol), 2-methylbenzoic acid (0.15 mmol), [RhCp*Cl2]2 (4 mol%), AgSbF6 (10 mol%), K2CO3 (0.15 mmol) in DCE (2.0 mL) at 100 °C. (c) 3i (0.1 mmol), 2-methylbenzaldehyde (0.15 mmol), [RhCp*Cl2]2 (4 mol%), AgSbF6 (10 mol%), HOAc (0.1 mmol) in DCE (2.0 mL) at 100 °C. (d) 3i (0.1 mmol), diphenylacetylene (0.1 mmol), Ni(BF4)2·6H2O (5 mol%), 6,6′-dimethyl-2,2′-bipyridin (5.5 mol%), Me(OEt)2SiH (0.2 mmol) in DMA (1.0 mL). (e) 3i (0.1 mmol), PhZnOPiv (0.12 mmol), CoCl2 (10 mol%) in THF (1.0 mL) at room temperature. After 5 h, the solvent was removed and TFA (1 mL) as added. The resulting mixture was stirred at 80 °C for 2 h. (f) 3i (0.1 mmol), 2-bromo-N-methoxy-2-methylpropanamide (0.2 mmol) and K2CO3 (0.2 mmol) in HFIP (1 mL) at room temperature. | ||
The photophysical properties of 3i, 22, 23 and 24 bearing a donor–acceptor (D–A) structure were further investigated and summarized in Table 2. The fluorescence quantum yield (ΦF,soln) values of 3i, 22, 23 and 24 in THF solutions (1 × 10−5 mol L−1) were measured to be 43.0%, 0.8%, 81.7%, 88.4%. All of these four compounds exhibited a marked solid luminescence phenomenon under the excitation of 365 nm ultraviolet light as shown in Fig. 10. Remarkably, compound 23 displays white-light emission in the solid state, and the photoluminescence of 23 is at 450 and 550 nm (Fig. 10d). This kind of white-light emitting material with a simple molecular structure is in great demand in organic optoelectronic materials.19
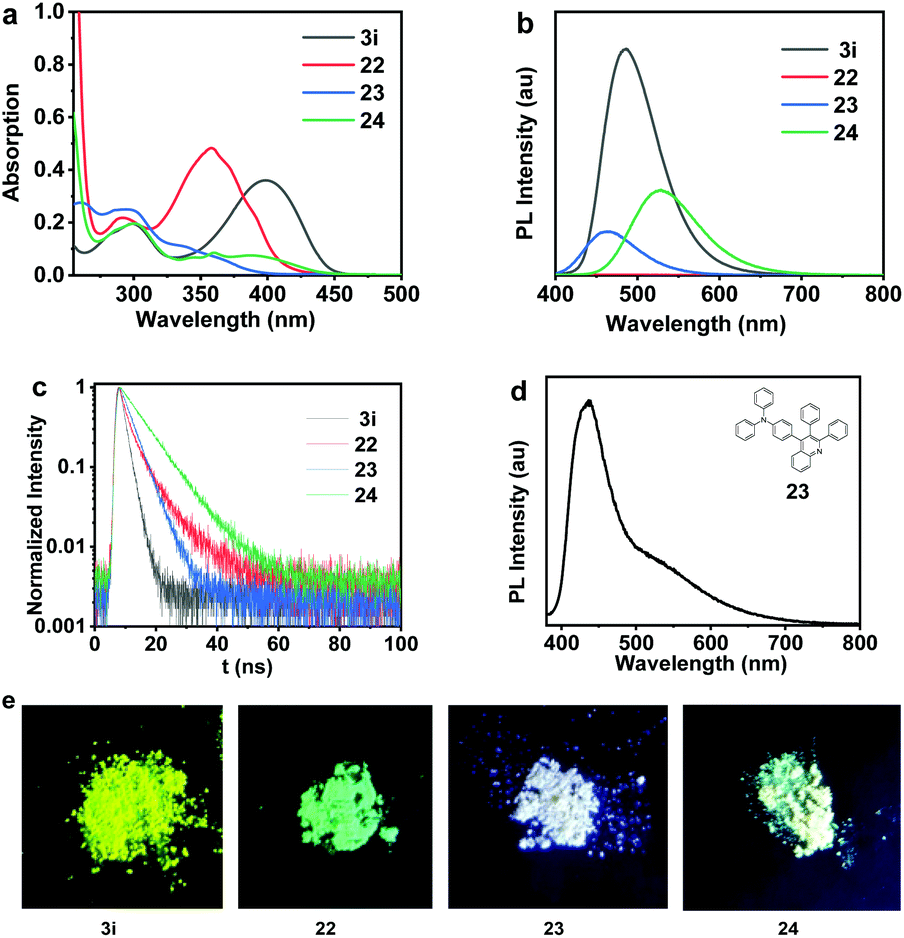 | ||
| Fig. 10 (a) The absorption spectra of 3i, 22, 23 and 24 in THF solutions (10−5 M). (b) The emission spectra of 3i, 22, 23 and 24 in THF solutions (10−5 M). (c) Time-correlated single photon counting in solution (10−5 M in THF). (d) The emission spectra of 23 (solid). (e) 3i, 22, 23 and 24 under the irradiation of 365 nm light. | ||
Conclusions
In summary, a Tf2O-mediated formal oxidative cross-coupling of readily available anthranils and simple arenes has been developed. This metal-free protocol is implemented in one operation under mild conditions, providing an efficient and environmentally benign route to acquire the privileged 3-functionalised anthranils. In addition, a wide array of olefins, alkenyl triflates, silyl enolethers, carbonyl compounds, thiophenols and thiols could serve as effective nucleophiles in this transformation. Mechanistic insights revealed that H2O induced intermediates are involved in this reaction and elimination of CF3SO2H is likely to be the rate-determining step. The synthetic utility of this protocol was demonstrated by the broad substrate scope (98 examples, up to 88% yield), the late-stage modification of pharmaceutical derivatives, and the diverse derivatization of the anthranil framework as well as the concise synthesis of biologically active compounds and drug derivatives. Moreover, through preliminary studies on photophysical properties, a novel white-light emitting material was synthesized from 3-aryl anthranils. Given the increasing importance of 3-aryl anthranils as synthetic precursors and building blocks, this protocol substantially streamlines their synthetic routes and would have broad applications in the construction of pharmaceuticals and optoelectronic materials.Data availability
Detailed experimental procedures and NMR spectra for all compounds are available in the ESI.†Author contributions
Y. G. supervised the project. S. Y., M. S. and J. N. performed all the experiments. Y. G. and X.-Q. H. prepared the draft and Y. H., X.L. and Q. C. revised the manuscript. All the authors analysed the data, discussed the results and contributed to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21901045, 21901258, U2001222), and the Technology Plan of Guangdong Province (2019A050510042).Notes and references
- (a) A. M. Boldi, Curr. Opin. Chem. Biol., 2004, 8, 281–286 CrossRef CAS PubMed; (b) G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563–639 CrossRef CAS PubMed; (c) D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338–6361 CrossRef CAS PubMed; (d) K.-H. Kim and J.-J. Kim, Adv. Mater., 2018, 30, 1705600 CrossRef PubMed.
- (a) A. Chaker, E. Najahi, O. Chatriant, A. Valentin, N. Téné, M. Treilhou, F. Chabchoub and F. Nepveu, Arabian J. Chem., 2017, 10, S2464–S2470 CrossRef CAS; (b) M. Rezazadeh, M. Pordel, A. Davoodnia and S. Saberi, Chem. Heterocycl. Compd., 2015, 51, 918–922 CrossRef CAS; (c) A. C. Pierce, M. Jacobs and C. Stuver-Moody, J. Med. Chem., 2008, 51, 1972–1975 CrossRef CAS PubMed; (d) M. Plewe, E. Brown, V. Gantla, G. Henkel, K. McCormack, N. V. Sokolova and Y.-J. Shin, PCT Int. Appl., 2018, 2018013430 Search PubMed.
- (a) D. A. Walsh, H. W. Moran, D. A. Shamblee, I. M. Uwaydah, W. J. Welstead, L. F. Sancilio and W. N. Dannenburg, J. Med. Chem., 1984, 27, 1379–1388 CrossRef CAS PubMed; (b) J. Koch-Weser, L. S. Simon and J. A. Mills, N. Engl. J. Med., 1980, 302, 1237–1243 CrossRef PubMed.
- (a) D. D. Morgan, J. D. Robinson and C. L. Mendenhall, Eur. J. Clin. Pharmacol., 1981, 19, 279–285 CrossRef CAS PubMed; (b) D. W. Choi, D. H. Farb and G. D. Fischbach, Nature, 1977, 269, 342–344 CrossRef CAS PubMed.
- S. P. Clissold and R. Beresford, Drugs, 1987, 33, 478–502 CrossRef CAS PubMed.
- For a review: (a) Y. Gao, J. Nie, Y. Huo and X.-Q. Hu, Org. Chem. Front., 2020, 7, 1177–1196 RSC ; for C-N coupling:; (b) J. Li, E. Tan, N. Keller, Y.-H. Chen, P. M. Zehetmaier, A. C. Jakowetz, T. Bein and P. Knochel, J. Am. Chem. Soc., 2019, 141, 98–103 CrossRef CAS PubMed ; for Rh/Co-catalyzed C-H amination:; (c) S. Yu, G. Tang, Y. Li, X. Zhou, Y. Lan and X. Li, Angew. Chem., Int. Ed., 2016, 55, 8696–8700 CrossRef CAS PubMed; (d) R.-H. Liu, Q.-C. Shan, X.-H. Hu and T.-P. Loh, Chem. Commun., 2019, 55, 5519–5522 RSC; (e) S. Kim, S. H. Han, N. K. Mishra, R. Chun, Y. H. Jung, H. S. Kim, J. S. Park and I. S. Kim, Org. Lett., 2018, 20, 4010–4014 CrossRef CAS PubMed ; for gold-catalyzed nitrene-transfer reactions:; (f) Z. Zeng, H. Jin, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2018, 57, 16549–16553 CrossRef CAS PubMed; (g) Z. Zeng, H. Jin, K. Sekine, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2018, 57, 6935–6939 CrossRef CAS PubMed; (h) R. L. Sahani and R. Liu, Angew. Chem., Int. Ed., 2017, 56, 12736–12740 CrossRef CAS PubMed; (i) H. Jin, B. Tian, X. Song, J. Xie, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 12688–12692 CrossRef CAS PubMed; (j) H. Jin, L. Huang, J. Xie, M. Rudolph, F. Rominger and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2016, 55, 794–797 CrossRef CAS PubMed for [4+n] cycloaddition reactions:; (k) C. Gao, X. Wang, J. Liu and X. Li, ACS Catal., 2021, 11, 2684–2690 CrossRef CAS; (l) Q. Cheng, J. Xie, Y. Weng and S. You, Angew. Chem., Int. Ed., 2019, 58, 5739–5743 CrossRef CAS PubMed; (m) J. Feng, M. Zhou, X. Lin, A. Lu, X. Zhang and M. Zhao, Org. Lett., 2019, 21, 6245–6248 CrossRef CAS PubMed.
- (a) R. K. Smalley, Sci. Synth., 2002, 11, 337–382 CAS; (b) J. Chauhan and S. Fletcher, Tetrahedron Lett., 2012, 53, 4951–4954 CrossRef CAS; (c) R. B. Davis and L. C. Pizzini, J. Org. Chem., 1960, 25, 1884–1888 CrossRef CAS; (d) B. J. Stokes, C. V. Vogel, L. K. Urnezis, M. Pan and T. G. Driver, Org. Lett., 2010, 12, 2884–2887 CrossRef CAS PubMed; (e) M. Zhang, Y. Meng, Y. Wu and C. Song, J. Org. Chem., 2021, 86, 7326–7332 CrossRef CAS PubMed.
- (a) M. Shigenobu, K. Takenaka and H. Sasai, Angew. Chem., Int. Ed., 2015, 54, 9572–9576 CrossRef CAS PubMed; (b) M. Aidene, F. Belkessam, J.-F. Soulé and H. Doucet, ChemCatChem, 2016, 8, 1583–1590 CrossRef CAS.
- T. Adak, C. Hu, M. Rudolph, J. Li and A. S. K. Hashmi, Org. Lett., 2020, 22, 5640–5644 CrossRef CAS PubMed.
- (a) Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787–8863 CrossRef CAS PubMed; (b) C. Liu, J. Yuan, M. Gao, S. Tang, W. Li, R. Shi and A. Lei, Chem. Rev., 2015, 115, 12138–12204 CrossRef CAS PubMed; (c) G. P. McGlacken and L. M. Bateman, Chem. Soc. Rev., 2009, 38, 2447 RSC; (d) C.-J. Li and Z. Li, Pure Appl. Chem., 2006, 78, 935–945 CAS; (e) C.-J. Li, Acc. Chem. Res., 2009, 42, 335–344 CrossRef CAS PubMed; (f) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed.
- (a) Y. Gao, S. Yang, Y. Huo, Q. Chen, X. Li and X.-Q. Hu, ACS Catal., 2021, 11, 7772–7779 CrossRef CAS; (b) Y. Gao, J. Nie, Y. Li, X. Li, Q. Chen, Y. Huo and X.-Q. Hu, Org. Lett., 2020, 22, 2600–2605 CrossRef CAS PubMed; (c) Y. Gao, Y. Cui, Y. Huo, J. Chen, M. She, X. Li, Q. Chen and X.-Q. Hu, J. Org. Chem., 2021, 86, 12107–12118 CrossRef CAS PubMed; (d) Y. Gao, S. Yang, Y. Li, Y. Huo, Z. Huang, Z. Chen and X.-Q. Hu, J. Org. Chem., 2020, 85, 10222–10231 CrossRef CAS PubMed; (e) Y. Gao, J. Nie, Y. Li, G. Liao, Y. Huo and X. Hu, ChemCatChem, 2020, 12, 2721–2725 CrossRef CAS.
- (a) P.-Y. Jiang, K.-F. Fan, S. Li, S.-H. Xiang and B. Tan, Nat. Commun., 2021, 12, 2384 CrossRef CAS PubMed; (b) E. J. Corey and Y. Tian, Org. Lett., 2005, 7, 5535–5537 CrossRef CAS PubMed; (c) T. Yanagi, S. Otsuka, Y. Kasuga, K. Fujimoto, K. Murakami, K. Nogi, H. Yorimitsu and A. Osuka, J. Am. Chem. Soc., 2016, 138, 14582–14585 CrossRef CAS PubMed; (d) S. Shaaban, V. Tona, B. Peng and N. Maulide, Angew. Chem., Int. Ed., 2017, 56, 10938–10941 CrossRef CAS PubMed; (e) K.-J. Xiao, J.-M. Luo, K.-Y. Ye, Y. Wang and P.-Q. Huang, Angew. Chem., Int. Ed., 2010, 49, 3037–3040 CrossRef CAS PubMed; (f) F. Berger, M. B. Plutschack, J. Riegger, W. Yu, S. Speicher, M. Ho, N. Frank and T. Ritter, Nature, 2019, 567, 223–228 CrossRef CAS PubMed; (g) I. L. Baraznenok, V. G. Nenajdenko and E. S. Balenkova, Tetrahedron, 2000, 56, 3077–3119 CrossRef CAS.
- (a) L. Xu, L. Ni, L. Sun, F. Zeng and S. Wu, Analyst, 2019, 144, 6570–6577 RSC; (b) C. Zhou, T. Zhang, S. Zhang, H. Liu, Y. Gao, Q. Su, Q. Wu, W. Li, J. Chen and B. Yang, Dyes Pigm., 2017, 146, 558–566 CrossRef CAS; (c) W. Li, Y. Pan, L. Yao, H. Liu, S. Zhang, C. Wang, F. Shen, P. Lu, B. Yang and Y. Ma, Adv. Opt. Mater., 2014, 2, 892–901 CrossRef CAS.
- (a) M. Murai, T. Ogita and K. Takai, Chem. Commun., 2019, 55, 2332–2335 RSC; (b) X. Xiong, F. Tan and Y.-Y. Yeung, Org. Lett., 2017, 19, 4243–4246 CrossRef CAS PubMed; (c) P. von R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed.
- (a) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC; (b) H. Tong, Y. Hong, Y. Dong, M. Häußler, J. W. Y. Lam, Z. Li, Z. Guo, Z. Guo and B. Z. Tang, Chem. Commun., 2006, 3705–3707 RSC.
- C. Gao, J. Xu, S. Zhu, K. Jian, Q. Xuan and Q. Song, Chem. Commun., 2021, 57, 2037–2040 RSC.
- For the role of Tf2O as an oxidizing agent, see: (a) V. G. Nenajdenko, P. V. Vertelezkij, A. B. Koldobskij, I. V. Alabugin and E. S. Balenkova, J. Org. Chem., 1997, 62, 2483–2486 CrossRef CAS PubMed; (b) X. Creary, J. Org. Chem., 1980, 45, 2727–2729 CrossRef CAS.
- P. Patel and G. Borah, Chem. Commun., 2017, 53, 443–446 RSC.
- B. Xu, H. Wu, J. Chen, Z. Yang, Z. Yang, Y.-C. Wu, Y. Zhang, C. Jin, P.-Y. Lu, Z. Chi, S. Liu, J. Xu and M. Aldred, Chem. Sci., 2017, 8, 1909–1914 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2094464. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc06565c |
| This journal is © The Royal Society of Chemistry 2022 |