
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances in the synthesis of anticancer pyrazole derivatives using microwave, ultrasound, and mechanochemical techniques
Diana Becerra* and
Juan-Carlos Castillo
 *
*
Escuela de Ciencias Químicas, Universidad Pedagógica y Tecnológica de Colombia, Avenida Central del Norte 39-115, Tunja, Colombia. E-mail: diana.becerra08@uptc.edu.co; juan.castillo06@uptc.edu.co
First published on 4th March 2025
Abstract
Pyrazole and its derivatives have attracted considerable attention in pharmaceutical and medicinal chemistry, as reflected in their presence in numerous FDA-approved drugs and clinical candidates. This review presents a comprehensive analysis of articles published between 2014 and 2024, focusing on the microwave-, ultrasound-, and mechanochemical-assisted synthesis of pyrazole derivatives with anticancer activity. It explores synthetic methodologies, anticancer efficacy, and molecular docking studies, underscoring the significance of pyrazole derivatives in drug discovery and medicinal chemistry. Notably, microwave irradiation stands out as the most widely employed technique, providing high efficiency by significantly reducing reaction times while maintaining moderate temperatures. Ultrasound irradiation serves as a valuable alternative, particularly for processes that require milder conditions, whereas mechanochemical activation, though less frequently employed, offers distinct advantages in terms of sustainability.
 [Left to right]: Juan-Carlos Castillo and Diana Becerra | Juan-Carlos Castillo, born in Cali, Colombia, completed his undergraduate studies in Chemistry at the Universidad del Valle, where he also earned his PhD in Chemical Sciences in 2013 under the supervision of Prof. Rodrigo Abonía. From 2013 to 2015, he conducted postdoctoral research at the Institute des Sciences Moléculaires de Marseille in France, collaborating with Prof. Jean Rodríguez and Prof. Yoann Coquerel. Between 2015 to 2017, he served as a postdoctoral researcher at the Universidad de los Andes in Bogotá, Colombia, under the supervision of Prof. Jaime Portilla. In 2018, he began his academic career at the Universidad Pedagógica y Tecnológica de Colombia. His current research focuses on medicinal chemistry, supramolecular chemistry, and the application of catalysis in organic synthesis. Diana Becerra, born in Florida, Colombia, completed her undergraduate studies in Chemistry at the Universidad del Valle in Cali, Colombia. She earned her PhD in Chemical Sciences in 2014 under the supervision of Prof. Braulio Insuasty. From 2014 to 2015, she conducted postdoctoral research at the Institut des Sciences Moléculaires de Marseille in France, working in collaboration with Prof. Jean Rodríguez and Prof. Damien Bonne. In 2017, she began her academic career at the Universidad Pedagógica y Tecnológica de Colombia. Her current research focuses on organic chemistry and medicinal chemistry. |
1. Introduction
Pyrazole, a five-membered heterocycle with two adjacent nitrogen atoms, displays unique reactivity in organic chemistry: nucleophilic attacks are favored at positions 3 and 5, while electrophilic substitution reactions predominantly occur at position 4.1 Within the azole family, pyrazole and its derivatives have attracted considerable attention due to their diverse applications across multiple fields, including medicine,2,3 agriculture,4 catalysis,5 ion detection sensors,6 supramolecular,7 coordination,8 and polymer chemistry,9 as well as in the food,10 cosmetic,11 and petrochemical industries.12 Notably, pyrazole derivatives are considered privileged scaffolds in drug discovery programs and medicinal chemistry due to their extensive range of pharmacological properties, such as antibacterial, antifungal, antioxidant, neuroprotective, anti-inflammatory, antimycobacterial, antimalarial, anticonvulsant, and antiviral activities, among others.13–18 Several FDA-approved tyrosine kinase inhibitors (TKIs) incorporate a pyrazole scaffold, emphasizing its pivotal role in the development of effective cancer therapies. Examples include Crizotinib and Pralsetinib, both used for the treatment of non-small cell lung cancer (NSCLC),19,20 Avapritinib, indicated for the management of multidrug-resistant gastrointestinal tumors,21 and Asciminib and Rebastinib, which are employed in the treatment of chronic myeloid leukemia (Fig. 1).22,23 Moreover, pyrazole derivatives have demonstrated multiple mechanisms of anticancer action by interacting with diverse targets such as tubulin,2,24 epidermal growth factor receptor (EGFR),25 cyclin-dependent kinase (CDK),26 DNA,27 topoisomerase,28 and human carbonic anhydrase (hCA) IX.29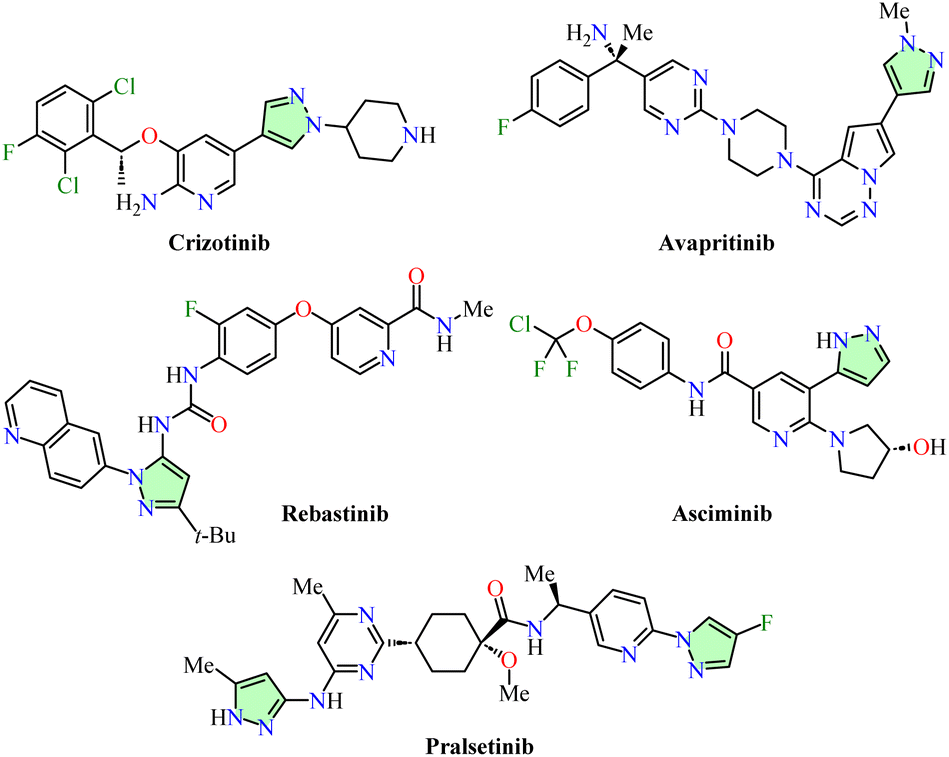 | ||
| Fig. 1 FDA-approved pyrazole-based drugs for cancer treatment. | ||
The extensive applications of pyrazole derivatives have driven the synthetic community to develop time-efficient and eco-friendly methodologies.30–35 Among these, microwave-, ultrasound-, and mechanochemical-assisted synthesis have emerged as highly effective approaches for facilitating multiple bond formations under solvent-free conditions. These innovative methodologies not only reduce reaction times and temperatures but also achieve higher yields compared to conventional heating methods.36–38 Such advancements constitute a significant contribution to the progress of sustainable chemistry, aligning closely with the fundamental principles of green chemistry.
This review provides a comprehensive analysis of articles published from 2014 to 2024, focusing on the synthesis of pyrazole derivatives with anticancer activity using microwave, ultrasound, and mechanochemical techniques (Fig. 2). Notably, microwave irradiation was employed in 68% of the reviewed articles, employing both ovens and reactors, while 22% of studies relied on ultrasound irradiation, including baths and reactors, and 10% adopted mechanochemical approaches, primarily involving grinding with a mortar and pestle. Accordingly, the review is structured into three sections: the first explores the microwave-assisted synthesis of pyrazole derivatives, followed by sections examining ultrasound- and mechanochemical-assisted methods, each evaluating their respective anticancer efficacy.
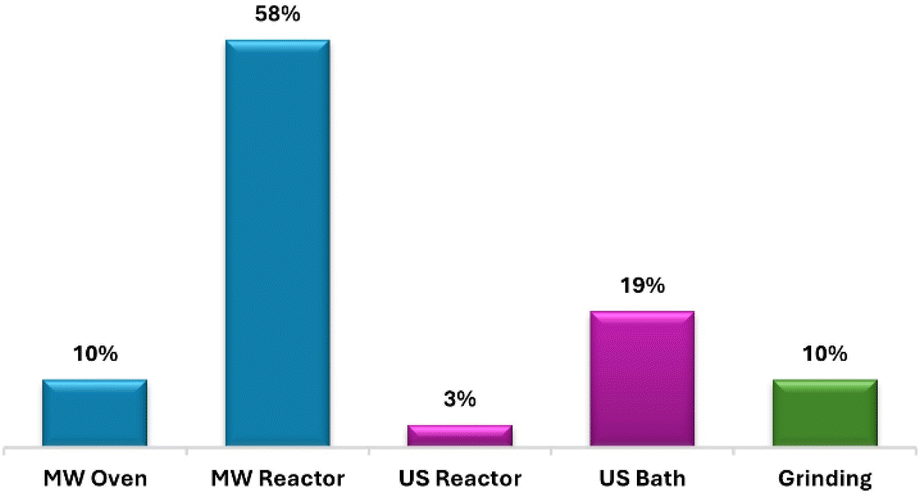 | ||
| Fig. 2 Bibliometric graph illustrating the percentage of articles related to microwave-, ultrasound-, and mechanochemical-assisted synthesis of pyrazole derivatives with anticancer activity from 2014 to 2024 [data were collected from a Scopus search using the keywords: “pyrazole derivatives”, “anticancer activity”, “microwave”, “ultrasound”, and “mechanochemical”]. | ||
2. Microwave-assisted synthesis of pyrazole derivatives
Microwave chemistry has transformed organic synthesis by introducing more sustainable protocols that overcome the limitations of conventional heating methods. Microwave-assisted organic synthesis (MAOS) enables the use of environmentally friendly solvents or even solvent-free conditions.39 Selective dielectric heating drastically reduces reaction times and energy consumption, resulting in enhanced electivity and higher yields compared to traditional reflux-based processes.40–42 Despite its advantages, MAOS has certain limitations, including (i) uneven heating, which may lead to localized overheating, side reactions, or thermal degradation, (ii) the requirement for specialized and costly reactors, which limits accessibility and presents challenges for industrial scalability, and (iii) poor microwave absorption in certain solvents, reducing heating efficiency and restricting its broader applicability.In this context, Sankaran et al. reported the microwave-assisted synthesis of quinolin-2(1H)-one-based pyrazole derivatives 3 in 68–86% yields via a reaction between quinolin-2(1H)-one-based α,β-unsaturated ketones 1 and arylhydrazines 2 in acetic acid using a microwave reactor set at 360 W and 120 °C for 7–10 min (Scheme 1).43 Additionally, the protocol was extended to hydrazine hydrate 4 using ethanol under identical experimental conditions, affording quinolin-2(1H)-one-based pyrazoles 5 with yields ranging from 71% to 75%. This procedure is notable for its short reaction times, high yields, minimal solvent use, and broad substrate scope. Additionally, select compounds 3 and 5 were evaluated for anticancer activity against cervical (HeLa) and colon (HCT-116 and HCT-8) cancer cell lines using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, with Adriamycin as the reference drug. Notably, compound 3i (R = C6H5, R1 = H, R2 = 4-MeO, R3 = H) exhibited the highest potency against HeLa cells, with an IC50 value of 2.4 ± 0.14 μM, making it 2.6-fold more potent than Adriamycin (IC50 = 6.3 ± 0.22 μM). Similarly, compound 3i demonstrated 4.0-fold greater potency against HCT-116 cells (IC50 = 2.2 ± 0.12 μM) and 1.3-fold greater potency against HCT-8 cells (IC50 = 5.6 ± 0.16 μM) compared to Adriamycin (IC50 = 8.7 ± 0.20 μM and 7.2 ± 0.32 μM, respectively).
 | ||
| Scheme 1 Synthesis and anticancer evaluation of quinolin-2(1H)-one-based pyrazole derivatives 3 and 5. | ||
Ashok et al. reported the efficient synthesis of pyrazolyl-substituted benzochroman-4-ones 8 and 10 in good yields through the reaction of pyrazole 4-carbaldehydes 6 with acetylnaphthols 7 and 9, respectively, in the presence of pyrrolidine and ethanol (Scheme 2).44 The process was optimized using a microwave reactor set at 180 W for 5–7 min. Under reflux conditions, the same reaction resulted in lower yields (59–71%) and significantly longer reaction times (10–12 h). Moreover, compounds 8 and 10 were evaluated for their anticancer activity against breast (MCF-7), colon (Colo-205), and lung (A549) cancer cell lines using the MTT assay, with Adriamycin as the reference drug. Among them, compound 10b (R = Me, GI50 < 0.1 μM) exhibited potency comparable to Adriamycin (GI50 = 0.13 μM) against MCF-7 cells. However, against Colo-205 and A549 cells, compound 10b displayed GI50 values of 2.7 μM and 2.9 μM, respectively, indicating lower potency than Adriamycin (GI50 < 0.1 μM in both cell lines).
 | ||
| Scheme 2 Synthesis and anticancer evaluation of pyrazolyl-substituted benzochroman-4-ones 8 and 10. | ||
Desai et al. reported the Claisen–Schmidt condensation of 1-(1,3,4-oxadiazol-3(2H)-yl)ethan-1-one 11 with various aromatic aldehydes 12 in the presence of ethanolic potassium hydroxide using a microwave reactor at 400 W for 5–8 min, resulting in the formation of pyrazole-containing 1,3,4-oxadiazoles 13 in good yields (Scheme 3).45 Conducting the same reaction under reflux conditions led to lower yields (59–66%) and longer reaction times (6–9 h). The mouse embryonic fibroblast cell line (NIH 3T3) and the cervical cancer cell line (HeLa) were used to assess the anticancer activity of compounds 13a–n. However, data for the reference drug were not available. Among them, compounds 13a (R = H) and 13d (R = 3-Me) exhibited the highest potency against HeLa cells, with IC50 values of 58.47 μM and 56.60 μM, respectively, while displaying significantly lower potency against NIH 3T3 cells (IC50 > 100 μM).
 | ||
| Scheme 3 Synthesis and anticancer evaluation of pyrazole-containing 1,3,4-oxadiazoles 13. | ||
The multicomponent reaction (MCR) strategy has proven highly effective in the pharmaceutical industry due to its high bond-forming efficiency and streamlined processes, enabling the rapid assembly of complex molecular architectures.46,47 For example, Gomha et al. described a three-component reaction of 4-acetylpyrazole 14, dimethylformamide dimethylacetal 15, hydroximoyl chlorides 16, and triethylamine in toluene using a microwave oven set at 150 °C and 500 W for 6 min, resulting in the formation of isoxazoles 17 in good yields (Scheme 4).48 Moreover, a multicomponent reaction involving 4-acetylpyrazole 14, thiosemicarbazide 18, arylcarbohydrazonoyl chlorides 19, and triethylamine in dioxane under similar microwave conditions led to the formation of 1,3,4-thiadiazoles 20 in good yields. Additionally, pyrazole-based azoles 17 and 20 were evaluated for their anticancer activity against lung (A549) and hepatocellular carcinoma (HepG2) cell lines using the MTT assay, with Cisplatin as the reference drug. Among them, compounds 17a (R = 4-NO2C6H4, R1 = 2-naphthyl) and 17b (R = 4-NO2C6H4, R1 = 2-furanyl) exhibited the highest potency against the A549 cell line with IC50 values of 4.47 ± 0.3 and 3.46 ± 0.6 μg mL−1, making them 4.7- and 3.6-fold less potent than Cisplatin (IC50 = 0.95 ± 0.23 μg mL−1). Similarly, compounds 17b and 20b (R = 4-NO2C6H4, R2 = R3 = C6H5) demonstrated superior potency against the HepG2 cell line with IC50 values of 4.67 ± 0.9 and 5.67 ± 1.7 μg mL−1, making them 3.3- and 4.1-fold less potent than Cisplatin (IC50 = 1.4 ± 0.37 μg mL−1).
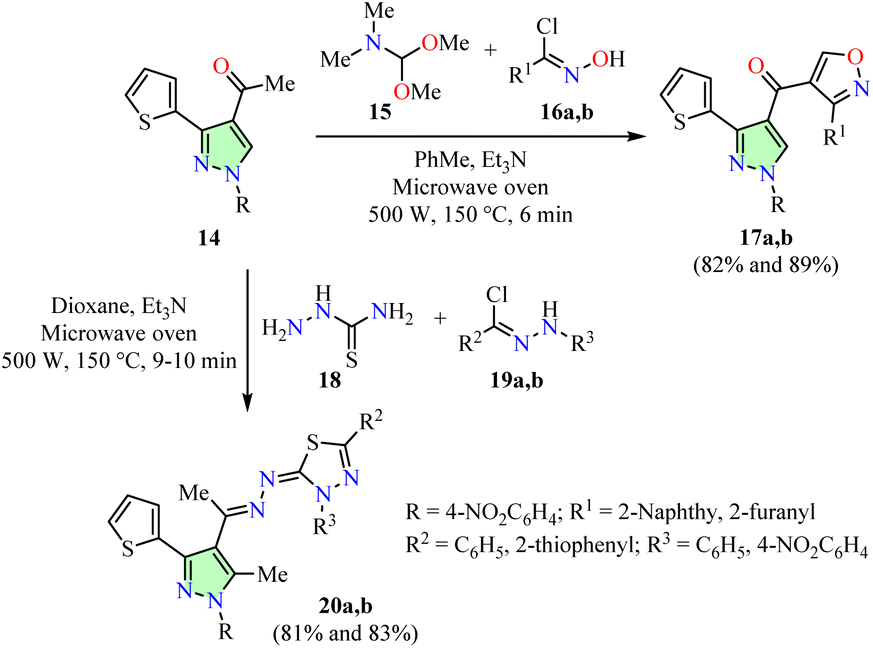 | ||
| Scheme 4 Three-component synthesis and anticancer evaluation of pyrazole-based azoles 17 and 20. | ||
Arasakumar et al. reported the synthesis of 8-nitroquinoline derivatives 22 with good yields via a microwave-assisted reaction of arylhydrazines 2 with 3-arylidene-2,3-dihydro-8-nitro-4-quinolones 21 in ethanol using a microwave reactor set at 80 °C and 110 W for 7–10 min (Table 1).49 The anticancer activity of compounds 22a–i was evaluated against breast (MCF-7) and lung (A549) cell lines using the MTT assay, with Doxorubicin as the reference drug. Among them, compound 22g (R = Cl, R1 = 4-ClC6H4, R2 = MeO) exhibited the highest potency against MCF-7 cells with an IC50 value of 25.76 μM, making it 1.7-fold less potent than Doxorubicin (IC50 = 15.12 μM). Similarly, compound 22b (R = H, R1 = 4-FC6H4, R2 = H) showed the strongest activity against A549 cells with an IC50 value of 23.21 μM, making it 1.2-fold less potent than Doxorubicin (IC50 = 18.56 μM).
|
||||||
|---|---|---|---|---|---|---|
| Compound | R | R1 | R2 | Yield 22 | IC50b (μM) | |
| MCF-7 | A549 | |||||
| a Reaction conditions: arylhydrazines 2 and 3-arylidene-2,3-dihydro-8-nitro-4-quinolones 21 in EtOH using a microwave reactor set at 80 °C and 110 W for 7–10 min.b The half-maximal inhibitory concentration (IC50) of each compound was determined by treating cells for 72 h, with untreated cells serving as controls. Cell viability was assessed using the MTT assay. | ||||||
| 22a | H | C6H5 | H | 87 | 53.38 | 59.78 |
| 22b | H | 4-FC6H4 | H | 90 | 45.98 | 23.21 |
| 22c | H | 4-BrC6H4 | H | 89 | 34.72 | 29.57 |
| 22d | H | 4-ClC6H4 | H | 89 | 41.35 | 28.66 |
| 22e | H | 4-MeOC6H4 | H | 91 | 31.31 | 40.17 |
| 22f | H | 4-MeC6H4 | H | 87 | 72.80 | 45.79 |
| 22g | Cl | 4-ClC6H4 | MeO | 85 | 25.76 | 31.98 |
| 22h | H | 3,4-(MeO)2C6H3 | H | 83 | 58.54 | 46.36 |
| 22i | H | 3,4-(EtO)2C6H3 | H | 81 | 65.73 | 50.91 |
| Doxorubicin | — | — | — | — | 15.12 | 18.56 |

Subsequently, a molecular docking simulation was performed to analyze the interactions between compound 22g and the tyrosine kinase domain of the epidermal growth factor receptor (EGFR) (PDB ID: 1M17) (Fig. 3).49 The docking analysis revealed that the NH group in the tetrahydroquinoline ring forms a hydrogen bond with the ASN818 residue.
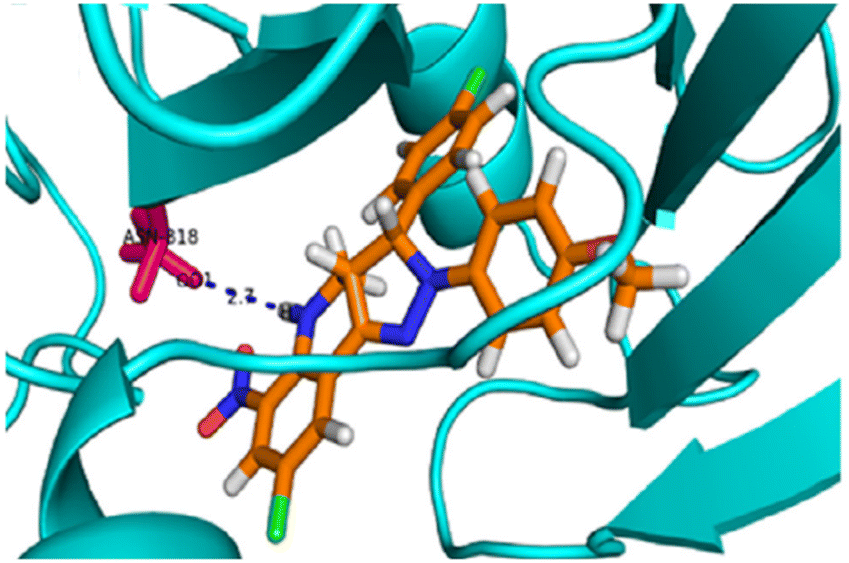 | ||
| Fig. 3 3D representation of the interactions between compound 22g and the epidermal growth factor receptor (EGFR) tyrosine kinase domain (PDB ID: 1M17). Reproduced with permission from ref. 49. Copyright Elsevier Inc., 2024. | ||
Mótyán et al. reported an iodine-mediated oxidative cyclization of dihydrotestosterone (DHT) 23 with various arylhydrazines 2 in ethanol using a microwave reactor set at 100 °C for 2 min, resulting in the formation of DHT-derived pyrazoles 24 in 80–89% yields (Scheme 5).50 This protocol is notable for its reduced reaction times and the elimination of the need to isolate pyrazoline intermediates. The anticancer activity of the synthesized compounds 24a–j was evaluated against prostate (PC-3 and DU 145), breast (MCF-7 and MDA-MB-231), and cervical (HeLa) cancer cell lines, as well as non-cancerous MRC-5 fibroblasts, using the MTT assay. However, data for the reference drug were not available. The IC50 values of compounds 24a–j were consistently lower in all cancer cell lines compared to non-cancerous MRC-5 cells, suggesting a selective cytotoxic effect against malignant cells. Among them, compound 24e (R = H, R1 = MeO) exhibited the highest potency against PC-3 and DU 145 cells, with IC50 values of 4.2 ± 1.1 μM and 3.6 ± 1.2 μM, respectively. Similarly, compound 24e showed the greatest potency against MCF-7 and MDA-MB-231 cells, with IC50 values of 5.5 ± 0.6 μM and 6.6 ± 0.9 μM, respectively, as well as against HeLa cells with an IC50 value of 8.5 ± 0.6 μM.
 | ||
| Scheme 5 Synthesis and anticancer evaluation of DHT-derived pyrazoles 24. | ||
The effects of DHT-derived pyrazole 24e on apoptosis and necrosis were evaluated in p53-deficient PC-3 cells, using cisplatin as a ref. 50. Treatment with 24e significantly induced apoptosis, as evidenced by a high percentage of annexin V-positive cells (Q2 + Q3) at 33.96%, compared to less than 1% in the untreated control (Fig. 4a). Under the same experimental conditions, cisplatin also induced significant apoptosis. To further confirm apoptosis in PC-3 cells, quantitative real-time PCR revealed a significant increase in caspase 3 and Bax mRNA levels (Fig. 4b and c, respectively), indicating activation of the apoptotic pathway in both 24e and cisplatin-treated PC-3 cells compared to the untreated control.
 | ||
| Fig. 4 (a) Quantification of apoptotic cells and relative mRNA expression levels of (b) caspase 3 and (c) Bax in PC-3 cells. Data are expressed as mean ± SD from three independent experiments. ****p < 0.0001 (Fisher's LSD test). This is an open-access article distributed under the terms of the Creative Commons CC BY license from ref. 50. | ||
The remarkable biological and photophysical properties of functionalized pyrazolo[1,5-a]pyrimidines have generated increasing interest in their synthesis.6,51 For instance, Fouda et al. reported a pyridine-catalyzed synthesis of pyrazolo[1,5-a]pyrimidines 29–31 in good yields via a cyclocondensation reaction of 3,5-diaminopyrazoles 25 with hydrazonoyl dicyanides 26–28 in ethanol using a microwave oven set at 50% power and 140 °C for 3–8 min (Scheme 6).52 The anticancer activity of compounds 29–31 was evaluated against breast (MCF-7), hepatocellular carcinoma (HepG2), and colon (HCT-116) cancer cell lines using the MTT assay, with Doxorubicin as the reference drug. Among them, compound 31 (R = CF3, R1 = thiazol-2-yl) exhibited the greatest potency against MCF-7 cells with an IC50 value of 4.2 ± 0.2 μg mL−1, which was 3.5-fold lower in potency compared to Doxorubicin (IC50 = 1.2 ± 0.2 μg mL−1). Additionally, compound 30b (R = CF3, R1 = 4,6-dimethylpyrimidin-2-yl) demonstrated the highest potency against HepG2 and HCT-116 cells, with IC50 values of 3.9 ± 0.4 μg mL−1 and 2.7 ± 0.6 μg mL−1, respectively. However, it was 4.3- and 1.7-fold less potent than Doxorubicin (IC50 = 0.9 ± 0.3 μg mL−1 and 1.6 ± 0.2 μg mL−1, respectively).
 | ||
| Scheme 6 Synthesis and anticancer evaluation of pyrazolo[1,5-a]pyrimidines 29–31. | ||
Shekarrao et al. reported an efficient palladium-catalyzed solvent-free synthesis of pyrazolo[1,5-a]pyrimidines 34 in good yields via the reaction of β-bromovinyl/aryl aldehydes 32 with 5-aminopyrazoles 33 using a microwave reactor set at 700 W and 120 °C for 15 min (Scheme 7).53 The compounds 34a–k were evaluated for their anticancer activity against cervical (HeLa) and prostate (DU-145) cell lines using the MTT assay, with Doxorubicin as the reference drug. Among them, compound 34d exhibited the highest potency against HeLa and DU-145 cell lines, with IC50 values of 10.41 ± 0.217 μM and 10.77 ± 0.124 μM, respectively. However, it was 1.1- and 1.2-fold less potent than Doxorubicin, which exhibited IC50 values of 9.76 ± 0.114 μM and 9.00 ± 0.721 μM, respectively.
 | ||
| Scheme 7 Synthesis and anticancer evaluation of pyrazolo[1,5-a]pyrimidines 34. | ||
Aydın et al. reported the microwave-assisted synthesis of 1-aroyl-3,5-dimethyl-1H-pyrazoles 37 in 82–98% yields through a cyclocondensation reaction of carbohydrazide derivatives 35 with 2,4-pentanedione 36 in ethanol using a microwave oven set at 270 W for 3–5 min (Table 2).54 The National Cancer Institute (NCI) evaluated the anticancer activity of compounds 37c, 37d, and 37f across a panel of 60 human cancer cell lines using the Sulforhodamine B (SRB) assay at a concentration of 10 μM. However, data for the reference drug were not available. Interestingly, compound 37c exhibited the highest growth inhibition percentage, achieving 69.95% against the K-562 leukemia cell line. Additionally, it demonstrated an IC50 value of 4.0 μM against the K-562 cell line, as determined by the MTT assay. Its apoptotic effects were further analyzed at concentrations of 1, 10, and 1000 μM over 24 h and 48 h, using TUNEL and Annexin V analyses. Notably, the addition of 37c to K-562 cells induced apoptosis most effectively at a concentration of 10 μM, resulting in apoptosis rates of 12.0% and 22.5% after 24 h and 48 h, respectively.
|
|||
|---|---|---|---|
| Compound | R | R1 | Yield 37 |
| a Reaction conditions: carbohydrazide derivatives 35 and 2,4-pentanedione 36 in ethanol using a microwave oven set at 270 W for 3–5 min. | |||
| 37a |  |
Me | 98 |
| 37b |  |
Me | 97 |
| 37c |  |
Me | 86 |
| 37d |  |
Me | 82 |
| 37e |  |
Me | 82 |
| 37f |  |
Me | 98 |

Thalassitis et al. reported the one-pot synthesis of (2-pyrazolin-5-yl)methyl-9H-purines 40 in acceptable yields via a 1,3-dipolar cycloaddition reaction between 9-allyl-6-chloro-9H-purine 38 and nitrile imines, generated in situ from the corresponding hydrazones 39 in the presence of N-bromosuccinimide and triethylamine, using a microwave reactor set at 80 °C for 80 min (Scheme 8).55 Interestingly, (pyrazol-5-yl)methyl-9H-purine 41 was obtained in 3% yield through the in situ oxidation of 40b under microwave conditions. The National Cancer Institute (NCI) evaluated the anticancer activity of compounds 40 and 41 across a panel of 60 human cancer cell lines using the SRB assay. However, data for the reference drug were not available. Notably, (pyrazol-5-yl)methyl-9H-purine 41 demonstrated the highest potency with GI50 values of 25.8, 36.1, and 36.8 μM against prostate (PC-3), renal (SN12C), and non-small cell lung (A549) cancer cell lines, respectively.
 | ||
| Scheme 8 Synthesis and anticancer evaluation of 9-substituted purines 40 and 41. | ||
Reddy et al. reported an oxidative coupling reaction between various (hetero)aromatic aldehydes 12 and 4-amino-1-methyl-3-propyl-1H-pyrazole-5-carboxamide 42 using K2S2O8 as the oxidizing agent in a DMSO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (1:1) mixture. The reaction was conducted in a microwave reactor set at 100 °C and 350 W for 3 min, resulting in the synthesis of 5-substituted-1H-pyrazolo[4,3-d]pyrimidin-7(6H)-ones 43 with yields ranging from 80% to 98% (Scheme 9).56 The anticancer activity of compounds 43a–t was evaluated against cervical (HeLa), renal (CAKI-I), prostate (PC-3), pancreatic (MiaPaCa-2), and lung (A549) cancer cell lines using the MTT assay. However, data for the reference drug were not available. Among them, compound 43m (R = 3,5-(MeO)2C6H3) exhibited the highest potency with IC50 values of 19 μM in HeLa cells, 17 μM in CAKI-I cells, 37 μM in PC-3 cells, 24 μM in MiaPaCa-2 cells, and 14 μM in A549 cells. Moreover, compound 43m exhibited anticancer activity through an apoptotic mechanism and demonstrated mTOR inhibition with an IC50 value of 203 nM.
:H2O (1:1) mixture. The reaction was conducted in a microwave reactor set at 100 °C and 350 W for 3 min, resulting in the synthesis of 5-substituted-1H-pyrazolo[4,3-d]pyrimidin-7(6H)-ones 43 with yields ranging from 80% to 98% (Scheme 9).56 The anticancer activity of compounds 43a–t was evaluated against cervical (HeLa), renal (CAKI-I), prostate (PC-3), pancreatic (MiaPaCa-2), and lung (A549) cancer cell lines using the MTT assay. However, data for the reference drug were not available. Among them, compound 43m (R = 3,5-(MeO)2C6H3) exhibited the highest potency with IC50 values of 19 μM in HeLa cells, 17 μM in CAKI-I cells, 37 μM in PC-3 cells, 24 μM in MiaPaCa-2 cells, and 14 μM in A549 cells. Moreover, compound 43m exhibited anticancer activity through an apoptotic mechanism and demonstrated mTOR inhibition with an IC50 value of 203 nM.
 | ||
| Scheme 9 Synthesis and anticancer evaluation of 5-substituted-1H-pyrazolo[4,3-d]pyrimidin-7(6H)-ones 43. | ||
A molecular docking simulation was performed to analyze the interactions between compound 43m and the active site of mTOR (PDB ID: 4JT5) (Fig. 5).56 The results revealed that the oxygen atom of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group in compound 43m forms a hydrogen bond with the Val2240 residue, while the hydrophobic environment provided by Leu2185, Trp2239, Met2345, and Ile2356 residues further enhances the stability of 43m within the binding pocket.
O group in compound 43m forms a hydrogen bond with the Val2240 residue, while the hydrophobic environment provided by Leu2185, Trp2239, Met2345, and Ile2356 residues further enhances the stability of 43m within the binding pocket.
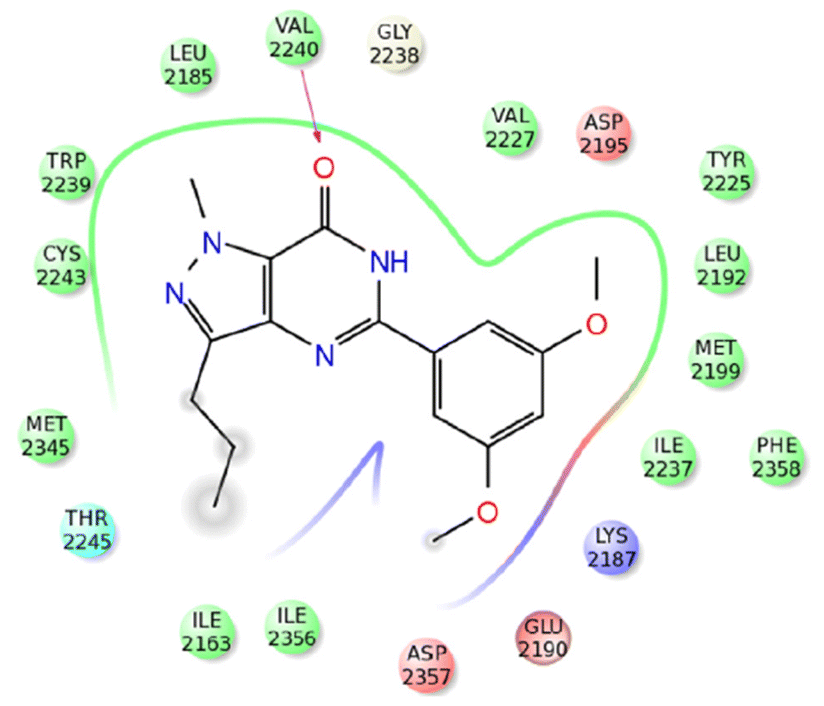 | ||
| Fig. 5 2D representation of the interactions of compound 43m with active site of mTOR (PDB ID: 4JT5). Reproduced with permission from ref. 56. Copyright Elsevier Inc., 2024. | ||
Alnaja et al. reported the efficient synthesis of pyrazolo[1,5-a]pyrimidines 45 in 75–94% yields via a cyclocondensation reaction between 3,5-diaminopyrazoles 25 and β-enaminone 44 in acetic acid using a microwave reactor set at 110 °C and 800 W for 20–30 min (Scheme 10).57 The anticancer activity of compounds 45a–g was evaluated against colon (HCT-116), breast (MCF-7), and hepatocellular carcinoma (HepG2) cell lines using the MTT assay, with Doxorubicin as the reference drug. Among them, compound 45b (R = 3-ClC6H4) exhibited the highest potency with IC50 values of 0.053 μM in HCT-116 cells, 0.126 μM in MCF-7 cells, and 0.039 μM in HepG2 cells, corresponding to 2.2-, 126.0-, and 19.5-fold lower potency, respectively, compared to Doxorubicin (IC50 = 0.024 μM, 0.001 μM, and 0.002 μM, respectively). The inhibitory activity of compound 45b against CDK2/cyclin A3 was evaluated and compared to Lapatinib as the reference drug. Interestingly, Lapatinib (IC50 = 0.122 μM) exhibited 1.4-fold greater inhibitory activity than compound 45b (IC50 = 0.178 μM).
 | ||
| Scheme 10 Synthesis and anticancer evaluation of pyrazolo[1,5-a]pyrimidines 45. | ||
A molecular docking simulation was performed to analyze the interactions between compound 45b and the crystal structure of inhibitor B within the CDK2/A3 complex (PDB ID: 1H0V). The results revealed that the amino group of the pyrazolo[1,5-a]pyrimidine ring acted as a hydrogen bond donor to the carboxylic group of Asp86, while the nitrogen atom in the diazenyl moiety formed a strong hydrogen bond with the amino group of Lys89. Furthermore, the stability of compound 45b within the binding pocket was enhanced by hydrophobic interactions, including two arene-H interactions between the pyrazolo[1,5-a]pyrimidine ring and the Ile10 and Val18 residues.57
Filho et al. reported the synthesis of ferrocene-pyrazole hybrids 47 in 58–75% yields via a cyclocondensation reaction between phenylhydrazine 2 and chalcones 46 in a mixture of acetic acid and water, using a microwave reactor set at 100 °C and 150 W for 5–30 min (Scheme 11).58 The synthesized compounds 47a–c were evaluated for anticancer activity against colon (HCT-116), prostate (PC-3), promyelocytic leukemia (HL60), and astrocytoma (SNB19) cancer cell lines using the MTT assay. However, data for the reference drug were not available. Notably, the ferrocene-pyrazole hybrid 47c (R = NH2) exhibited the highest anticancer activity with IC50 values of 3.12 μM in HCT-116 cells, 124.40 μM in PC-3 cells, 6.81 μM in HL60 cells, and 60.44 μM in SNB19 cells.
 | ||
| Scheme 11 Synthesis and anticancer evaluation of ferrocene-pyrazole hybrids 47. | ||
Molecular docking studies were performed to evaluate the interactions of compound 47c with the tyrosine kinase domain of the epidermal growth factor receptor (EGFR) (PDB ID: 1M17) and the active site of the human IDH1 mutant (PDB ID: 5LGE) (Fig. 6a and b, respectively). The binding energies of compound 47c with EGFR and IDH1 were determined to be −8.3 and −7.4 kcal mol−1, respectively. Notably, the ferrocene moiety exhibited effective interactions within both the hydrophobic and hydrophilic cavities of these molecular targets.
 | ||
| Fig. 6 3D representation of the interactions of compound 47c with (a) the tyrosine kinase domain of the epidermal growth factor receptor (EGFR) (PDB ID: 1M17) and (b) the active site of the human IDH1 mutant (PDB ID: 5LGE). Reproduced with permission from ref. 58. Copyright Elsevier Inc., 2024. | ||
Parikh et al. reported the one-pot multicomponent synthesis of pyrano[2,3-c]pyrazoles 50 under solvent-free conditions (Scheme 12).59 Initially, a mixture of aryl hydrazine 2, β-ketoesters 49, and zinc triflate (10 mol%) was heated in a microwave reactor at 80 °C for 10 min. After cooling the reaction mixture to room temperature, aromatic aldehyde 12 and malononitrile 48 were added, and the mixture was further heated under microwave irradiation at 120 °C for 15 min. Recrystallization of the crude product in ethanol afforded pyrano[2,3-c]pyrazoles 50 in 92–99% yields. The synthesized compounds 50a–j were evaluated for their anticancer activity against renal (786-0), epidermal (A431), breast (MCF-7), and glioblastoma (U-251) cancer cell lines using the MTT assay, with Doxorubicin as the reference drug. Among them, compound 50h (R = 3-NO2C6H4, R1 = 4-MeOC6H4, R2 = Me) exhibited the highest potency against the 786-0 and MCF-7 cell lines, with IC50 values of 9.9 ± 1.33 μg mL−1 and 31.87 ± 8.22 μg mL−1, respectively. However, it was 10.0- and 15.2-fold less potent compared to Doxorubicin, which displayed IC50 values of 0.99 ± 0.17 μg mL−1 and 2.1 ± 0.79 μg mL−1, respectively. Moreover, compound 50j (R = 3-NO2C6H4, R1 = 5-Br-2-OHC6H3, R2 = Me) exhibited the highest potency against the A-431 and U-251 cell lines, with IC50 values of 19.98 ± 6.01 μg mL−1 and 25.78 ± 8.47 μg mL−1, respectively. However, it was 12.5- and 13.6-fold less potent compared to Doxorubicin, which displayed IC50 values of 1.6 ± 0.82 μg mL−1 and 1.9 ± 0.68 μg mL−1, respectively.
 | ||
| Scheme 12 Synthesis and anticancer evaluation of pyrano[2,3-c]pyrazoles 50. | ||
Dahal et al. reported the synthesis of 1,3-diarylpyrazolones 51 in 66–93% yields through a cyclocondensation reaction between arylhydrazine hydrochlorides 2 and β-ketoesters 49 in a 1:1 mixture of water and glycerol, using a microwave reactor set at 100 °C and 300 W for 20 min (Table 3).60 The anticancer activity of the synthesized compounds was evaluated against two human lung adenocarcinoma cell lines (A549 and NCI-H522) using the MTT assay, with Afatinib and Gefitinib as reference drugs. Interestingly, compound 51d (R = C6H5, R1 = H, R2 = 4-ClC6H4) exhibited an IC50 value of 1.98 ± 1.10 μM against A549 cells, showing 4.3- and 7.2-fold greater potency compared to the clinically approved drugs Afatinib (IC50 = 8.46 ± 2.03 μM) and Gefitinib (IC50 = 14.27 ± 4.20 μM), respectively. Conversely, compound 51m (R = R2 = 4-BrC6H4, R1 = H) displayed an IC50 value of 2.41 ± 0.57 μM against NCI-H522 cells, demonstrating 2.2- and 5.8-fold higher potency compared to Afatinib (IC50 = 5.34 ± 0.94 μM) and Gefitinib (IC50 = 13.86 ± 2.99 μM), respectively. In addition, the effect of compound 51j (R = 4-CF3C6H4, R1 = H, R2 = C6H5) on cell cycle progression in NCI-H522 cells was analyzed using flow cytometry. Treatment of NCI-H522 cells with compound 51j at a concentration of 5 μM for 48 h resulted in a significant increase in the G0/G1 phase (72.73 ± 3.01%), while the S phase (11.23 ± 2.52%) and G2/M phase (11.4 ± 1.10%) decreased accordingly.
|
|||||
|---|---|---|---|---|---|
| Compound | R | R1 | R2 | IC50b (μM) | |
| A549 | NCI-H522 | ||||
| a Reaction conditions: arylhydrazine hydrochlorides 2 and β-ketoesters 49 in a 1:1 mixture of water and glycerol using a microwave reactor set at 100 °C and 300 W for 20 min.b The half-maximal inhibitory concentration (IC50) of each compound was determined by treating cells for 48 h with different doses ranging from 0 to 40 μM. Untreated cells served as controls, and cell viability was assessed using the MTT assay. |
|||||
| 51a | Pr | H | C6H5 | >100 | >100 |
| 51b | C6H5 | H | C6H5 | 10.35 ± 1.55 | 17.01 ± 2.61 |
| 51c | C6H5 | H | 4-BrC6H4 | 2.61 ± 1.12 | 4.93 ± 1.13 |
| 51d | C6H5 | H | 4-ClC6H4 | 1.98 ± 1.10 | 4.50 ± 1.16 |
| 51e | 4-NO2C6H4 | H | C6H5 | 39.56 ± 1.05 | 40.5 ± 2.30 |
| 51f | 4-BrC6H4 | H | C6H5 | 6.35 ± 0.85 | 4.70 ± 1.38 |
| 51g | 4-FC6H4 | H | C6H5 | 9.51 ± 3.12 | 17.88 ± 3.36 |
| 51h | 4-MeOC6H4 | H | C6H5 | 42.25 ± 2.76 | 51.25 ± 3.06 |
| 51i | 2-MeOC6H4 | H | C6H5 | >100 | >100 |
| 51j | 4-CF3C6H4 | H | C6H5 | 5.98 ± 0.84 | 4.88 ± 1.73 |
| 51k | 2,3,4,5-(F)4C6H | H | C6H5 | 8.60 ± 0.59 | 9.9 ± 2.44 |
| 51l | 4-FC6H4 | H | 4-BrC6H4 | 2.67 ± 0.51 | 3.70 ± 0.34 |
| 51m | 4-BrC6H4 | H | 4-BrC6H4 | 2.73 ± 0.28 | 2.41 ± 0.57 |
| 51n | 2-FC6H4 | H | 4-BrC6H4 | 3.58 ± 0.88 | 10.22 ± 1.30 |
| 51o | 4-BrC6H4 | H | 4-CF3C6H4 | 3.37 ± 0.10 | 2.95 ± 0.18 |
| 51p | 4-BrC6H4 | H | 3,5-(Cl)2C6H3 | 5.56 ± 0.07 | 5.07 ± 1.18 |
| Afatinib | — | — | — | 8.46 ± 2.03 | 5.34 ± 0.94 |
| Gefitinib | — | — | — | 14.27 ± 4.20 | 13.86 ± 2.99 |
The same research group evaluated the anticancer activity of 1,3-diarylpyrazolones 51a–p against cutaneous melanoma (A375 and SKMEL-28) and non-melanoma skin cancer (A431 and SCC-12) cell lines using the MTT assay, with Celecoxib and Cisplatin as positive controls.61 Among them, compound 51c (R = C6H5, R1 = H, R2 = 4-BrC6H4) exhibited the highest potency against A375 and SKMEL-28 cells, with IC50 values of 14.3 ± 0.9 μM and 7.6 ± 0.6 μM, respectively. Compared to Celecoxib (IC50 = 26.8 ± 2.7 μM and 11.4 ± 2.4 μM, respectively), compound 51c was 1.9- and 1.5-fold more potent. However, in comparison to Cisplatin (IC50 = 1.49 ± 0.44 μM and 14.2 ± 0.24 μM, respectively), it was 9.6-fold less potent in A375 cells but 1.9-fold more potent in SKMEL-28 cells. Similarly, compound 51c exhibited the highest potency against A431 and SCC-12 cells, with IC50 values of 3.7 ± 0.5 μM and 12.2 ± 0.6 μM, respectively. Compared to Celecoxib (IC50 = 7.4 ± 0.6 μM and 44.1 ± 1.1 μM, respectively), compound 51c was 2.0- and 3.6-fold more potent. However, in comparison to Cisplatin (IC50 = 7.7 ± 0.3 μM and 4.4 ± 0.2 μM, respectively), it was 2.1-fold more potent in A431 cells but 2.8-fold less potent in SCC-12 cells. Treatment of A431 and SK-MEL-28 cells with compound 51c induced a pronounced, concentration-dependent induction of apoptosis, as evidenced by a significant increase in the levels of activated caspase-3, caspase-9, and cleaved PARP compared to untreated controls (Fig. 7).61 These findings suggest that compound 51c induces apoptosis in A431 and SKMEL-28 cells via the intrinsic mitochondrial apoptotic pathways, involving PARP activation.
 | ||
| Fig. 7 Concentration-dependent effects (0, ½IC50, IC50, and 1½IC50) of compound 51c on the expression levels of apoptotic markers (caspase-3, caspase-9, Bax, Bcl2, and cleaved PARP) in SK-Mel-28 and A431 cancer cells after 48 h of treatment. β-actin was used as the loading control. Statistical significance was determined using Bonferroni's tests, with *p < 0.05 and **p < 0.01 considered significant. This is an open-access article distributed under the terms of the Creative Commons CC BY license from ref. 61. | ||
Mótyán et al. described the nucleophilic addition of the amine group from various arylhydrazines 2 to the carbonyl group of dehydroepiandrosterone derivatives 52 using a microwave reactor set at 120 °C for 10–20 min, resulting in the formation of D-ring-fused steroidal 5-amino-1-arylpyrazoles 53 with yields ranging from 80% to 94% (Scheme 13).62 The anticancer activity of the synthesized compounds 53a–h was evaluated against breast (MCF-7), prostate (PC-3), lung (A549), cervical (HeLa), and osteosarcoma (U2Os) cell lines using the MTT assay, with Cisplatin as the positive control. Among them, compound 53g (R = 4-CNC6H4) exhibited the highest potency with IC50 values of 6.2 ± 1.0 μM in MCF-7 cells, 4.9 ± 1.1 μM in PC-3 cells, 7.9 ± 1.0 μM in A549 cells, and 4.0 ± 1.0 μM in HeLa cells, corresponding to approximately 54-, 184-, 47-, and 64-fold greater potency, respectively, compared to Cisplatin (IC50 = 338.5 ± 1.1 μM, 902 ± 1.6 μM, 369.8 ± 1.2 μM, and 254.5 ± 1.2 μM, respectively).
 | ||
| Scheme 13 Synthesis and anticancer evaluation of D-ring-fused steroidal 5-amino-1-arylpyrazoles 53. | ||
Ashok et al. reported a condensation reaction between 1-phenyl-1H-pyrazole-4-carbaldehydes 54 and o-phenylene diamine 55 in DMF using a microwave reactor set at 100 °C and 300 W for 7–9 min, resulting in the formation of pyrazole-based benzo[d]imidazoles 56 with yields ranging from 77% to 89% (Table 4).63 The synthesized compounds 56a–g were evaluated for their anticancer activity against brain (C6) and breast (MCF-7) cancer cell lines using the MTT assay, with Cisplatin as a positive control. Among them, compound 56b (R = MeO) exhibited IC50 values of 0.102 μM and 0.110 μM against C6 and MCF-7 cells, respectively, demonstrating 1.2- and 5.4-fold higher potency compared to Cisplatin (IC50 = 0.122 μM and 0.596 μM, respectively).
|
||||
|---|---|---|---|---|
| Compound | R | Yield | IC50b (μM) | |
| C6 | MCF-7 | |||
| a Reaction conditions: 1-phenyl-1H-pyrazole-4-carbaldehydes 54 and o-phenylene diamine 55 in DMF using a microwave reactor set at 100 °C and 300 W for 7–9 min.b The half-maximal inhibitory concentration (IC50) of each compound was determined by treating cells for 24 h with different doses (0.5, 0.25, 0.125, 0.0625, 0.0312, and 0.0156 mg mL−1). Untreated cells served as controls, and cell viability was assessed using the MTT assay. | ||||
| 56a | H | 83 | 0.564 | 0.253 |
| 56b | MeO | 84 | 0.102 | 0.110 |
| 56c | F | 81 | 0.243 | 0.144 |
| 56d | Cl | 89 | 3.220 | 0.185 |
| 56e | Br | 87 | 0.219 | 0.450 |
| 56f | Me | 85 | 0.120 | 0.303 |
| 56g | NO2 | 77 | 0.560 | 0.452 |
| Cisplatin | — | — | 0.122 | 0.596 |
Liao et al. reported the synthesis of 3,5-bis(styryl)pyrazoles 58 in acceptable yields through a cyclocondensation reaction of various curcuminoids 57 with hydrazine hydrate 4 in a 1:1 mixture of N,N-dimethylformamide and acetic acid, using a microwave reactor set at 80 °C for 5 min (Scheme 14).64 The synthesized compounds 58a–l were evaluated for their antiproliferative activity against the prostate cancer cell line (PC-3) using the MTT assay, with Methotrexate as the reference drug. Among them, compounds 58a (R = R3 = R4 = H, R1 = MeO, R2 = OH) and 58l (R = R4 = Cl, R1 = R2 = R3 = H) exhibited the highest potency against PC-3 cells with GI50 values of 0.85 ± 0.34 μM and 2.21 ± 0.33 μM, respectively. However, compounds 58a and 58l exhibited significantly lower potency than Methotrexate (GI50 = 0.012 ± 0.008 μM), being 71- and 184-fold less potent, respectively. Cell cycle analysis of PC-3 cells treated with bis(styryl)pyrazoles 58a and 58l for 72 h revealed a significant increase in the percentage of cells arrested in the G2/M phase, reaching 89.6% and 89.3%, respectively. This G2/M arrest inhibited cell division, leading to a reduced cell population in the G0/G1 phase (8.0% and 4.4%) and the S phase (2.2% and 6.3%). Furthermore, compound 58a (Kd = 4.6 ± 1.1 μM) exhibited a 12-fold lower binding affinity for tubulin compared to compound 58l (Kd = 0.4 ± 0.1 μM).
 | ||
| Scheme 14 Synthesis and anticancer evaluation of 3,5-bis(styryl)pyrazoles 58. | ||
Molecular docking analysis was conducted to examine the interactions between compound 58l and the tubulin-combretastatin A4 complex (PDB ID: 5LYJ). The binding energy of 58l with tubulin was determined to be −8.4 kcal mol−1. Remarkably, compound 58l forms two hydrogen bonds within the paclitaxel binding site, interacting with the peptide backbone of residues T273 and P271 at distances of 2.2 Å and 2.6 Å, respectively (Fig. 8).64
 | ||
| Fig. 8 3D representation of interactions between compound 58l and the tubulin-combretastatin A4 complex (PDB ID: 5LYJ). This is an open-access article distributed under the terms of the Creative Commons CC BY license from ref. 64. | ||
Al-Wahaibi et al. reported a time-efficient microwave-assisted synthesis of 3,5-diaminopyrazole derivatives 60 in high yields through a cyclization reaction between arylhydrazines 2 and 2-((3-nitrophenyl)diazenyl)malononitrile 59 in ethanol (Scheme 15).65 This method was successfully extended to thiosemicarbazide 18 and carbohydrazide derivatives 35 under similar reaction conditions. The synthesized compounds 60a–f were evaluated for their anticancer activity against the breast cancer cell lines MCF-7 and MDA-MB-231 using the MTT assay, with Doxorubicin as the reference drug. Among them, compound 60a (R = C6H5) exhibited an IC50 value of 6.20 ± 0.40 μM against MCF-7 cells, demonstrating 5.4-fold greater potency than Doxorubicin (IC50 = 33.20 ± 3.50 μM). Although compound 60c (R = CONH2) showed the highest activity against MDA-MB-231 cells with an IC50 value of 14.50 ± 1.10 μM, it was 4.5-fold less potent than Doxorubicin (IC50 = 3.20 ± 0.10 μM).
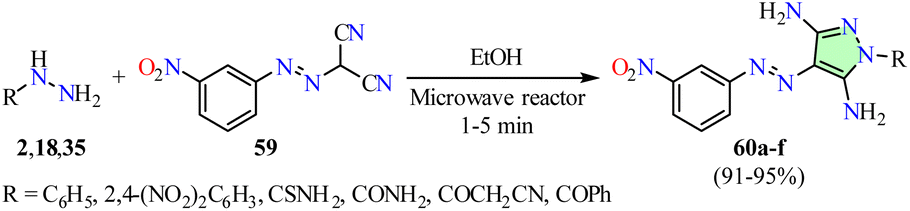 | ||
| Scheme 15 Synthesis and anticancer evaluation of 3,5-diaminopyrazole derivatives 60. | ||
Sanad et al. reported the synthesis of pyrazole-linked pyrimidinones 62 in 72–85% yields through the reaction of 3-aryl-1H-pyrazol-5-amines 33 with thieno[2,3-b]pyridine-based enamines 61 in dioxane, using a microwave reactor set at 100 °C and 300 W for 30–45 min (Table 5).66 The synthesized compounds 62a–e were evaluated for their anticancer activity against breast (MCF-7), colon (Caco2), and liver (HEPG2) cancer cell lines using the MTT assay, with 5-fluorouracil (5-Fu) as the reference drug. Among them, compound 62c (R = NO2) exhibited IC50 values of 4.20 ± 0.33 μM in MCF-7 cells, 7.62 ± 0.51 μM in Caco2 cells, and 3.65 ± 0.29 in HEPG2 cells, demonstrating 1.9-, 1.6-, and 1.7-fold higher potency than 5-fluorouracil, respectively.
|
|||||
|---|---|---|---|---|---|
| Compound | R | Yield | IC50b (μM) | ||
| MCF-7 | Caco2 | HEPG2 | |||
| a Reaction conditions: 3-aryl-1H-pyrazol-5-amines 33 and thieno[2,3-b]pyridine-based enamines 61 in dioxane using a microwave reactor set at 100 °C and 300 W for 30–45 min.b The half-maximal inhibitory concentration (IC50) of each compound was determined by treating cells for 24 h with different doses (1, 2.5, 5, 10, 20, and 40 μg mL−1). Untreated cells served as controls, and cell viability was assessed using the MTT assay. | |||||
| 62a | H | 81 | 17.12 ± 1.38 | 23.93 ± 1.11 | 13.05 ± 0.72 |
| 62b | Cl | 77 | 11.35 ± 0.92 | 13.38 ± 0.74 | 6.59 ± 0.48 |
| 62c | NO2 | 72 | 4.20 ± 0.33 | 7.62 ± 0.51 | 3.65 ± 0.29 |
| 62d | Me | 83 | 33.45 ± 1.25 | 44.72 ± 1.82 | 25.37 ± 0.97 |
| 62e | MeO | 85 | 35.61 ± 1.85 | 47.55 ± 1.98 | 27.19 ± 1.15 |
| 5-FU | — | — | 8.02 ± 0.77 | 12.20 ± 1.14 | 6.12 ± 0.52 |

Molecular docking analysis was performed to investigate the interactions between compound 62c and thymidylate synthase (PDB ID: 6QXG) (Fig. 9). The binding energy of 62c with thymidylate synthase was determined to be −12.0 kcal mol−1. Docking studies revealed hydrogen-bond interactions between the nitrogen atom and carbonyl oxygen of the pyrimidinone moiety with Arg215 and Tyr258, respectively. Additionally, compound 62c formed hydrogen-bond interactions between the nitrogen atom of the pyrazole ring and Asp218, as well as between its nitro group and His196. Furthermore, a π–H stacking interaction was observed between the pyridine ring and Arg50, along with a π–cation stacking interaction involving the pyrimidinone ring and Arg50.66
 | ||
| Fig. 9 2D representation of the interactions between compound 62c and thymidylate synthase (PDB ID: 6QXG). Reproduced with permission from ref. 66. Copyright Elsevier Inc., 2024. | ||
Anwer et al. reported a time-efficient synthesis of pyrazolone derivatives 64 in high yields through a cyclization reaction between arylhydrazines 2 and ethyl 2-cyano-2-{[4-(phenyldiazenyl)phenyl]diazenyl} acetate 63 in ethanol, using a microwave reactor for 0.5–3 min (Scheme 16).67 This method was successfully extended to hydrazine hydrate 4 and carbohydrazide derivatives 35 under similar reaction conditions. The synthesized compounds 64a–d were evaluated for their anticancer activity against liver (HEPG2), colon (HCT-116), breast (MCF-7), and lung (A549) cancer cell lines using the MTT assay, with Sorafenib and Erlotinib as reference drugs. Among them, compound 64b (R = C6H5) exhibited the highest activity with IC50 values of 7.80 ± 0.70 μM in HEPG2 cells, 8.12 ± 0.90 μM in HCT-116 cells, 6.98 ± 1.10 μM in MCF-7 cells, and 6.50 ± 1.50 μM in A549 cells. However, it was 2.0-, 1.6-, 1.2-, and 1.6-fold less potent than Sorafenib, which exhibited IC50 values of 4.00 ± 0.33 μM, 5.05 ± 0.50 μM, 5.58 ± 0.55 μM, and 4.04 ± 0.33 μM, respectively. In contrast, compound 64b exhibited comparable potency to Erlotinib (IC50 = 7.73 ± 0.67 μM, 13.91 ± 1.30 μM, 8.20 ± 0.34 μM, and 5.49 ± 0.45 μM, respectively) against HEPG2 and A549 cells, while exhibiting 1.7- and 1.2-fold greater potency against HCT-116 and MCF-7 cells, respectively. Moreover, compounds 64a–d were further evaluated for their dual inhibitory effects on VEGFR-2 and EGFRT790M. Among them, compound 64b exhibited the strongest inhibition of VEGFR-2 with an IC50 value of 1.25 ± 0.50 μM, demonstrating 1.5-fold lower potency than Sorafenib (IC50 = 0.84 ± 0.04 μM). Additionally, compounds 64b and 64c (R = 2,4-(NO2)2C6H3) exhibited the highest inhibitory effects on EGFRT790M with IC50 values of 0.40 ± 0.35 μM and 0.35 ± 0.15 μM, respectively, showing 1.7- and 1.4-fold lower potency compared to Erlotinib (IC50 = 0.24 ± 0.22 μM).
 | ||
| Scheme 16 Synthesis and anticancer evaluation of pyrazolone derivatives 64. | ||
3. Ultrasound-assisted synthesis of pyrazole derivatives
Ultrasound-assisted organic synthesis (UAOS) has become a crucial tool in sustainable chemistry, offering significant advantages over traditional methods. Its effectiveness is primarily attributed to the phenomenon of acoustic cavitation – the formation, growth, and implosive collapse of bubbles within a liquid medium – which generates localized high temperatures and pressures.68,69 These “hot spots” greatly enhance chemical reactivity, facilitating a wide range of chemical transformations. UAOS enables reactions to proceed with greater efficiency, achieving higher yields, and faster reaction rates at lower temperatures compared to conventional and microwave heating methods, thereby establishing itself as a cornerstone in the progress of sustainable chemistry.70,71In this context, Ahmed et al. reported a cyclocondensation reaction of diketones 36 with 2-(2,7-dimethyl-1,8-naphthyridin-4-yloxy) acetohydrazide 65 in ethanol, using an ultrasonic bath for 10 min. This process resulted in the formation of pyrazole-based 1,8-naphthyridines 66a and 66b in 98% and 97% yields, respectively (Scheme 17).72 The synthesized compounds 66a,b were evaluated for their anticancer activity against the liver cancer cell line (HEPG2) using the MTT assay, with Doxorubicin as the reference drug. Among them, compounds 66a (R = Me) and 66b (R = C6H5) exhibited the highest potency against HEPG2 cells with IC50 values of 0.071 μM and 0.064 μM, respectively. However, compounds 66a and 66b exhibited lower potency than Doxorubicin (IC50 = 0.04 μM), making them 1.8- and 1.6-fold less potent, respectively.
 | ||
| Scheme 17 Synthesis and anticancer evaluation of pyrazole-based 1,8-naphthyridines 66. | ||
Suresh et al. reported a three-component synthesis of pyrazolo[1,5-a]pyrimidines 68 in 37–88% yields through the reaction of 5-aminopyrazoles 33, aromatic aldehydes 12, and terminal alkynes 67 in acetic acid, using an ultrasonic bath set at 35 kHz and 60 °C for 30 min (Scheme 18).73 To maintain a stable bath temperature of 60 °C during prolonged ultrasound irradiation, cold water was periodically added for temperature control. Selected compounds 68 were evaluated for their anticancer activity against breast cancer cell lines (MCF-7 and MDA-MB-231) using the SRB assay, with Gemcitabine as the reference drug. Inhibition percentages were recorded after 72 h of treatment with selected compounds 68 at a concentration of 10 μM. Among them, compound 68b (R = CO2Et, R1 = R2 = 4-MeC6H4) exhibited the highest inhibition percentage (25.12 ± 2.43%) against MCF-7 cells, being 2.0-fold more potent than Gemcitabine (49.56 ± 6.71%). Similarly, compound 68e (R = CO2Et, R1 = C6H5, R2 = 4-(n-Pent)C6H4) showed the highest inhibition percentage (62.16 ± 6.89%) against MDA-MB-231 cells, being 1.6-fold more potent than Gemcitabine (100.01 ± 1.13%).
 | ||
| Scheme 18 Synthesis and anticancer evaluation of pyrazolo[1,5-a]pyrimidines 68. | ||
Bhatt et al. described an efficient synthesis of pyrazole-based isoxazol-5(4H)-ones 71 in 82–96% yields through a three-component reaction involving 4-formylpyrazoles 6, β-ketoester 69, hydroxylamine hydrochloride 70, and pyridine in a 1:1 ethanol–water mixture, using an ultrasonic bath set at 50 °C for 30–45 min (Scheme 19).74 The National Cancer Institute (NCI) evaluated the anticancer activity of compounds 71a–i across a panel of 60 human cancer cell lines using the SRB assay at a concentration of 10 μM. However, data for the reference drug were not available. Notably, compounds 71d (R = 4-BrC6H4) and 71e (R = 4-MeC6H4) exhibited the highest growth inhibition percentages, with values of −73.60% and −98.00%, respectively, against the LOX-IMVI melanoma cell line.
 | ||
| Scheme 19 Synthesis and anticancer evaluation of pyrazole-based isoxazol-5(4H)-ones 71. | ||
Nitulescu et al. reported the synthesis of pyrazole derivatives 73 in 67–77% yields through a one-pot reaction between 5-aminopyrazoles 33 and benzoyl isothiocyanates 72, which were generated in situ from benzoyl chlorides and ammonium thiocyanate in acetonitrile, using an ultrasonic bath for 15–30 min (Scheme 20).75 The anticancer activity of compounds 73a–f was evaluated against colon (HT-29) and acute monocytic leukemia (THP-1) cell lines after 24 h of exposure at concentrations of 6.25, 12.5, 25, and 50 μg mL−1, using the MTT assay. However, data for the reference drug were not available. THP-1 cells displayed greater sensitivity to the synthesized compounds than HT-29 cells. Notably, compound 73e (R = Br, R1 = Cl) exhibited an IC50 value of 40.34 μg mL−1, while the other compounds showed IC50 values ranging from 42.97 μg mL−1 to 48.96 μg mL−1. Apoptosis and necrosis were assessed in HT-29 cells treated with 50 μg mL−1 of compounds 73a–f for 24 h using flow cytometry and the Annexin V-PI assay. Compound 73e showed the highest necrosis-inducing effect, with 84.7% viable cells, 5.69% early apoptotic cells, 1.16% late apoptotic cells, and 8.43% necrotic cells. Furthermore, cell cycle analysis of HT-29 cells exposed to 50 μg mL−1 of compound 73e for 24 h revealed a significant increase in the percentage of cells arrested in the G2/M phase (80.28%), leading to a reduced cell population in the G0/G1 (4.17%) and S (11.32%) phases compared to the control (G0/G1 = 65.81%, S = 22.49%, and G2/M = 10.03%). The G2/M phase arrest induced by compound 73e was associated with the upregulation of cyclin A, cyclin B, CDK1, and CDC20 gene expression.75
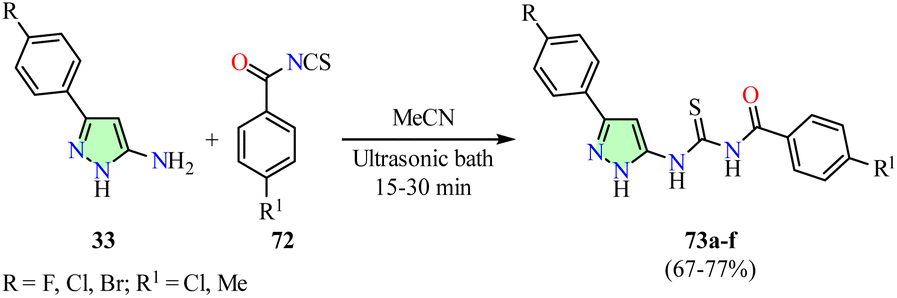 | ||
| Scheme 20 Synthesis and anticancer evaluation of pyrazole derivatives 73. | ||
Nagasundaram et al. reported a catalyst-free synthesis of 1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitriles 75 in 82–93% yields through a four-component reaction involving hydrazine hydrate 4, malononitrile 48, β-ketoester 49, and arylazo-salicylaldehydes 74 in a 1:1 ethanol–water mixture, using an ultrasonic reactor set at 40 kHz and 60 °C for 16–23 min (Scheme 21).76 The anticancer activity of compounds 75h (R = 4-NO2) and 75i (R = 3-NO2) was evaluated against the HeLa cervical cancer cell line after 24 h of exposure at concentrations of 0.39, 0.78, 1.56, 3.12, 6.25, 12.5, 25, 50, and 100 μg mL−1, using the MTT assay with Doxorubicin as the reference drug. Among them, compound 75i exhibited the highest potency against HeLa cells with an IC50 value of 5.75 μg mL−1, which is comparable to that of Doxorubicin (IC50 = 6.03 μg mL−1).
 | ||
| Scheme 21 Synthesis and anticancer evaluation of 1,4-dihydropyrano[2,3-c]pyrazole-5-carbonitriles 75. | ||
Molecular docking analysis was performed to investigate the interactions between compound 75i and the epidermal growth factor receptor (EGFR) tyrosine kinase domain (PDB ID: 1M17) (Fig. 10). The binding energy of 75i with EGFR was determined to be −11.7 kcal mol−1. Docking studies revealed seven hydrogen-bond interactions involving residues Lys721, Arg817, Asn818, Ala698, Asp813, and Gly833. Additionally, three electrostatic interactions were identified with Lys721, Glu738, and Asp831, along with one π–π stacking hydrophobic interaction with Phe699.76
 | ||
| Fig. 10 3D representation of the interactions between compound 75i and the epidermal growth factor receptor (EGFR) tyrosine kinase domain (PDB ID: 1M17). Reproduced with permission from ref. 76. Copyright Elsevier Inc., 2024. | ||
Dofe et al. reported the synthesis of tetrazole-based pyrazole derivatives 77 in 93–98% yields through a cyclization reaction between hydrazine hydrate 4 and tetrazole-based chalcones 76 in ethanol, using an ultrasonic bath for 10–14 min (Table 6).77 The compounds 77a–h were evaluated for their anticancer activity against breast (MCF-7), lung (A549), and liver (HEPG2) cancer cell lines using the MTT assay, with Combretastatin A-4 (CA-4) as a positive control. Among them, compound 77c (R = H, R1 = F) exhibited the highest potency with IC50 values of 0.92 μM in MCF-7 cells, 0.94 μM in A549 cells, and 0.85 μM in HEPG2 cells. However, compound 77c exhibited 23-, 19-, and 94-fold lower potency against MCF-7, A549, and HEPG2 cells, respectively, compared to Combretastatin A-4 (IC50 = 0.04 μM, 0.05 μM, and 0.009 μM, respectively). Moreover, compound 77c displayed significant inhibition of tubulin polymerization (IC50 = 2.16 μM), being 1.3-fold less potent than Combretastatin A-4 (IC50 = 1.62 μM).
|
||||||
|---|---|---|---|---|---|---|
| Compound | R | R1 | Yield | IC50b (μM) | ||
| MCF-7 | A549 | HEPG2 | ||||
| a Reaction conditions: hydrazine hydrate 4 and tetrazole-based chalcones 76 in ethanol using an ultrasonic bath for 10–14 min.b The half-maximal inhibitory concentration (IC50) of each compound was determined by treating cells for 72 h with different doses (200, 100, 50, 25, 12.5, 6.5, and 3.125 mg/100 μL). Untreated cells served as controls, and cell viability was assessed using the MTT assay. | ||||||
| 77a | H | H | 98 | 3.16 | 2.98 | 3.15 |
| 77b | H | Cl | 97 | 1.04 | 1.11 | 0.96 |
| 77c | H | F | 96 | 0.92 | 0.94 | 0.85 |
| 77d | MeO | MeO | 95 | 2.94 | 2.82 | 3.06 |
| 77e | OH | H | 96 | 2.16 | 2.24 | 2.88 |
| 77f | H | MeO | 95 | 3.10 | 3.12 | 2.98 |
| 77g | H | Me | 93 | 1.76 | 0.99 | 1.08 |
| 77h | H | OH | 94 | 1.94 | 1.16 | 2.02 |
| CA-4 | — | — | — | 0.04 | 0.05 | 0.009 |

Molecular docking analysis was performed to investigate the interactions between compound 77c and the colchicine binding site of α,β-tubulin (PDB ID: 1SA0) (Fig. 11). Docking studies revealed that the tetrazole ring aligns coplanarly with the Val238 residue, enabling it to act as a hydrogen bond donor. The 2-methoxy group on the central phenyl ring functions as a hydrogen bond acceptor for the thiol proton of Cys241. Additionally, the pyrazoline and substituted phenyl groups are positioned within the hydrophobic pocket of tubulin, interacting with residues Asn258, Met259, Val315, Ala316, Val318, Asn350, Val351, and Lys352.77
 | ||
| Fig. 11 3D representation of the interactions of compound 77c with α,β-tubulin at the colchicine binding site (PDB ID: 1SA0). Reproduced with permission from ref. 77. Copyright Elsevier Inc., 2024. | ||
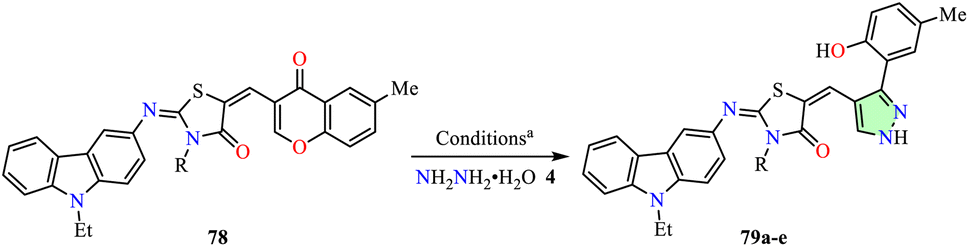
Hassanin et al. reported the synthesis of carbazolyl–thiazolyl–pyrazole hybrids 79 in 88–92% yields through the cyclization of hydrazine hydrate 4 with carbazole–thiazolidinone–chromone hybrids 78 in ethanol, using an ultrasonic bath at 50 °C for 15 min (Table 7).78 The anticancer activity of these compounds was evaluated against colon (HCT-116), prostate (PC-3), and liver (HEPG2) cancer cell lines using the SRB assay, with Doxorubicin as the reference drug. Among them, compound 79b (R = allyl) exhibited the highest potency against HCT-116 and PC-3 cell lines, with IC50 values of 28.5 ± 1.5 μM and 4.9 ± 0.8 μM, respectively. Although compound 79b was 4.1-fold less potent against HCT-116 cells, it exhibited 1.3-fold greater potency against PC-3 cells compared to Doxorubicin (IC50 = 6.9 ± 0.5 μM and 6.2 ± 0.9 μM, respectively). Additionally, compound 79e (R = 4-ClC6H4) exhibited the highest anticancer activity against the HEPG2 cell line with an IC50 value of 9.1 ± 0.5 μM, making it 1.2-fold less potent than Doxorubicin (IC50 = 7.9 ± 1.3 μM).
|
|||||
|---|---|---|---|---|---|
| Compound | R | Yield | IC50b (μM) | ||
| HCT-116 | PC-3 | HEPG2 | |||
| a Reaction conditions: hydrazine hydrate 4 and carbazole–thiazolidinone–chromone hybrids 78 in ethanol using an ultrasonic bath at 50 °C for 15 min.b The half-maximal inhibitory concentration (IC50) of each compound was determined by treating cells for 72 h with different doses (0.01, 0.1, 1, 10, 100, and 1000 mg mL−1). Untreated cells served as controls, and cell viability was assessed using the SRB assay. | |||||
| 79a | Me | 88 | 69.5 ± 1.5 | 12.9 ± 3.8 | 20.1 ± 0.2 |
| 79b | Allyl | 88 | 28.5 ± 1.5 | 4.9 ± 0.8 | 18.5 ± 0.4 |
| 79c | Adamantan-1-yl | 89 | 30.9 ± 2.3 | 37.4 ± 1.6 | 25.3 ± 1.4 |
| 79d | C6H5 | 92 | >100 | >100 | >100 |
| 79e | 4-ClC6H4 | 90 | 39.9 ± 0.1 | 9.2 ± 0.4 | 9.1 ± 0.5 |
| Doxorubicin | — | — | 6.9 ± 0.5 | 6.2 ± 0.9 | 7.9 ± 1.3 |
Molecular docking analysis was performed to investigate the interactions between compound 79b and the vascular endothelial growth factor receptor 2 (VEGFR2) kinase domain (PDB ID: 3EWH) (Fig. 12). The binding energy of 79b with VEGFR2 was determined to be −11.2 kcal mol−1. Docking studies revealed that the NH group of the pyrazole ring forms a hydrogen bond with Glu917, while the CO group of the thiazole ring interacts with Thr916. Additionally, the thiazole ring engages in a π–cation interaction with Lys868, and the phenyl ring of the carbazole moiety participates in a π–anion interaction with Asp1046. Furthermore, π–alkyl interactions are observed between the carbazole or thiazole moieties and the residues Val848 and Leu889.78
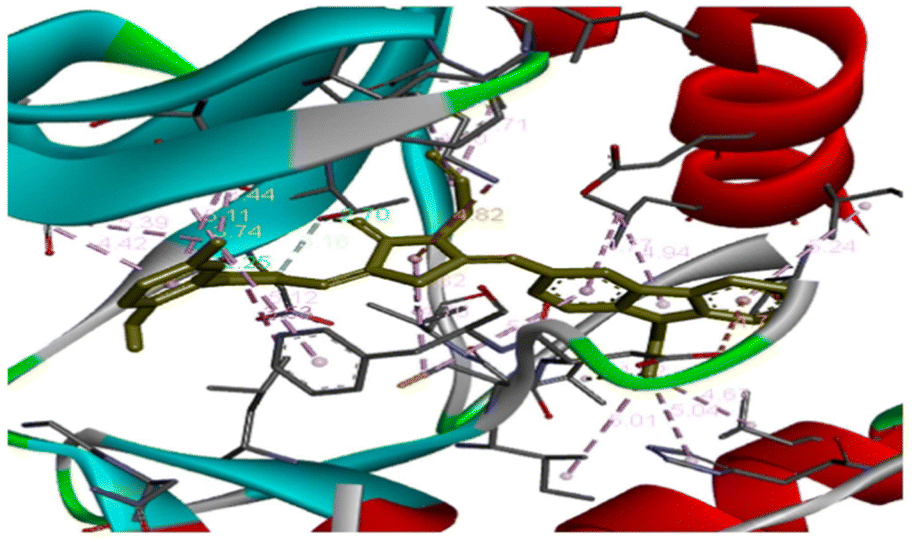 | ||
| Fig. 12 3D representation of the interactions between compound 79b and the vascular endothelial growth factor receptor 2 (VEGFR2) kinase domain (PDB ID: 3EWH). This is an open-access article distributed under the terms of the Creative Commons CC BY license from ref. 78. | ||
4. Mechanochemical-assisted synthesis of pyrazole derivatives
Mechanochemistry utilizes mechanical energy generated through compression, shear, or friction to facilitate chemical transformations. This method is predominantly utilized under solvent-free conditions, offering notable advantages such as accelerated reaction rates, reduced reaction times, and enhanced control over product selectivity.79,80 These benefits stem from the absence of solvation effects and the high reagent concentrations inherent to solvent-free systems.79 Mechanochemical activation is typically achieved through three techniques: (a) grinding with a mortar and pestle, (b) planetary ball milling, and (c) high-speed vibration milling in a mixer mill.81,82In this context, Al-Wahaibi et al. reported a solvent-free synthesis of 3,5-diaminopyrazole derivatives 60 in 77–79% yields by grinding arylhydrazines 2 with 2-((3-nitrophenyl)diazenyl)malononitrile 59 at room temperature for 10–18 min (Scheme 22).65 This methodology was successfully extended to thiosemicarbazide 18 and carbohydrazide derivatives 35 under similar reaction conditions. The anticancer activity of compounds 60a–f was previously discussed in the section on microwave-assisted synthesis of pyrazole derivatives (Scheme 15).
 | ||
| Scheme 22 Solventless synthesis of 3,5-diaminopyrazole derivatives 60. | ||
Rashdan et al. reported a solvent-free synthesis of pyrazolo[1,2-b]phthalazinediones 83 in 73–92% yields through a three-component reaction involving 1,2,3-triazolyl-pyrazolecarbaldehydes 80, 6-nitrophthalhydrazide 81, active methylene compounds such as malononitrile 48 or ethyl cyanoacetate 82, and sodium hydroxide. The reaction was performed by grinding the components at room temperature for 20–30 min (Table 8).83 The anticancer activity of these compounds was evaluated against HEPG2 liver cancer cells and BALAB/3T3 normal cells using the MTT assay, with Doxorubicin as the reference drug. Among them, compound 83f (R = C6H5, R1 = CO2Et) exhibited the highest potency against the HEPG2 cell line with an IC50 value of 3.01 ± 0.21 μg mL−1, making it 1.2-fold more potent than Doxorubicin (IC50 = 3.56 ± 0.46 μg mL−1). Notably, compound 83f showed no cytotoxicity toward the normal BALAB/3T3 cell line, demonstrating its selective anticancer activity.
|
|||||
|---|---|---|---|---|---|
| Compound | R | R1 | Yield | IC50b (μg mL−1) | |
| HEPG2 | BALAB/3T3 | ||||
| a Reaction conditions: 1,2,3-triazolyl-pyrazolecarbaldehydes 80, 6-nitrophthalhydrazide 81, active methylene compounds 48 or 82, and sodium hydroxide under grinding at room temperature for 20–30 min.b The half-maximal inhibitory concentration (IC50) of each compound was determined by treating cells for 72 h with doses ranging from 0.1 to 100 μg mL−1. Untreated cells served as controls, and cell viability was assessed using the MTT assay. ND: not detected at the concentrations tested. | |||||
| 83a | C6H5 | CN | 88 | 13.51 ± 4.48 | 18.24 ± 3.61 |
| 83b | 4-ClC6H4 | CN | 82 | ND | ND |
| 83c | 4-MeOC6H4 | CN | 73 | 22.73 ± 5.36 | 28.34 ± 7.61 |
| 83d | 4-BrC6H4 | CN | 87 | ND | ND |
| 83e | 4-NO2C6H4 | CN | 86 | ND | ND |
| 83f | C6H5 | CO2Et | 82 | 3.01 ± 0.21 | ND |
| 83g | 4-ClC6H4 | CO2Et | 82 | ND | ND |
| 83h | 4-MeOC6H4 | CO2Et | 82 | ND | ND |
| 83i | 4-BrC6H4 | CO2Et | 92 | 26.37 ± 6.17 | 30.04 ± 8.52 |
| 83j | 4-NO2C6H4 | CO2Et | 91 | ND | ND |
| Doxorubicin | — | — | — | 3.56 ± 0.46 | ND |

Edrees et al. reported the cyclization of pyrazoline 84 with various hydrazonoyl chlorides 85 and 87 in the presence of 1,4-diazabicyclo[2.2.2]octane (DABCO), resulting in the formation of pyrazolylthiazoles 86 and 88 in 67–75% and 67–74%, respectively (Scheme 23).84 Similarly, reacting α-bromoacetophenones 89 under identical conditions gave pyrazolylthiazoles 90 in 71–82% yields. In all cases, the reaction mixtures were ground for 10–20 min and subsequently poured into water. The resulting products were filtered and recrystallized from appropriate solvents to obtain the corresponding pyrazolylthiazoles. The anticancer activity of these compounds was evaluated against the HEPG2 liver cancer cell line using the MTT assay, with Cisplatin as the positive control. Among the tested compounds, pyrazolylthiazoles 90f (R = 4-FC6H4) and 90d (R = 4-ClC6H4) exhibited the highest potency against HEPG2 cells with IC50 values of 1.70 μM and 2.98 μM, respectively. However, compounds 90f and 90d were 1.9- and 3.3-fold less potent than Cisplatin (IC50 = 0.90 μM), respectively.
 | ||
| Scheme 23 Synthesis and anticancer evaluation of pyrazolylthiazoles 86, 88, and 90. | ||
5. The structure activity relationship (SAR)
The biological activity of pyrazole derivatives is strongly influenced by the nature and position of substituents on the pyrazole core, as these modifications significantly affect their interactions with pharmacophoric targets, thereby modulating cytotoxicity and enzymatic inhibition. Structure–activity relationship (SAR) analysis demonstrates that specific substitutions at positions 1, 3, 4, and 5, along with structural fusions, are critical for enhancing anticancer activity. This enhancement is attributed to precise interactions with biological targets, including the induction of cell cycle arrest and the inhibition of key enzymes or proteins involved in cancer progression (Fig. 13). Moreover, Table 9 summarizes the structure–activity relationship (SAR) trends observed in this study, emphasizing the influence of specific substitutions on the pyrazole ring in anticancer activity. It highlights key substitution positions and molecular docking interactions with pharmacophoric targets. This comprehensive overview provides valuable insights into the structural features that contribute to enhanced anticancer activity.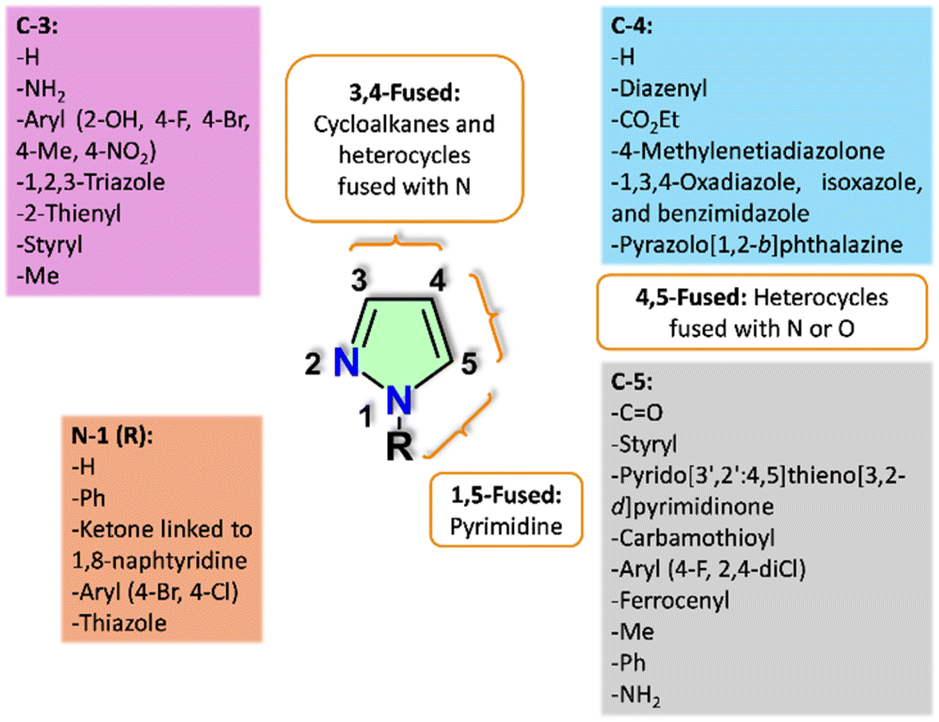 | ||
| Fig. 13 SAR analysis of pyrazole derivatives with anticancer activity. | ||
| Substituted positionsa | Key substitutions for enhanced activity | Compound | Anticancer activity or pharmacophoric interactions |
|---|---|---|---|
| a The position numbers and types of substitutions on the pyrazole core are specified to categorize compounds with similar modifications, facilitating the SAR analysis of their effects on anticancer activity. | |||
| 3,4 | 3-Amine and 4-diazenyl groups | 30b,52 31,52 45b,57 64b67 |
Exhibits enhanced activity against MCF-7, HepG2, and/or HCT-116 cancer cell lines |
| 1,5 | Pyrimidine fusion in pyrazolo[1,5-a]pyrimidine with an amino group at position 3 and a diazenyl group at position 4 commonly observed | 30b,52 31,52 34d,53 45b,57 68b,73 68e73 |
Exhibits enhanced activity in MCF-7 and/or HCT-116 cancer cell lines |
| 3,4 and 4,5 | Fusion at positions 3,4 and 4,5 with cycloalkanes, as well as nitrogen- and oxygen-containing heterocycles | 22g,49 24e,50 43m,56 50h,59 53g,62 75i76 |
Exhibits enhanced activity against MCF-7 and HeLa cancer cell lines, with significant interactions observed in the epidermal growth factor receptor (EGFR) tyrosine kinase domain (PDB ID: 1M17) or through mTOR inhibition (PDB ID: 4JT5) |
| 1,3,4 | 1-NH, 3-aryl, and 4-methylenetiadiazolone, with aryl groups containing OH and Me substitutions | 79b78 |
Docking interactions with the vascular endothelial growth factor receptor 2 (VEGFR2) kinase domain (PDB ID: 3EWH) |
| 1,3,4 | 1,3-Diaryl, 1-aryl-3-(2-tienyl), and 4-hetaryl (oxadiazole, isoxazole, and benzimidazole) groups directly attached to the pyrazole core or linked via methylene bridges | 13a,45 13d,45 17a,48 17b,48 56b,63 71d,74 71e,74 83f83 |
Enhances activity against Hela, A549, MCF-7, LOX-IMVI, and HEPG2 cancer cell lines |
| 1,3,5 | 1-NH, 3,5-bis(styryl), 3,5-diaryl and 3-aryl-5-carbamothioyl moieties within a pyrazole or pyrazoline scaffold | 58a,64 58l,64 62c,66 73e,75 77c77 |
Exhibits docking interactions with the tubulin-combretastatin A4 complex (PDB ID: 5LYJ) and the colchicine-binding site of α,β-tubulin (PDB ID: 1SA0), as well as interactions with thymidylate synthase (PDB ID: 6QXG). Additionally, it induces G2/M phase arrest in acute monocytic leukemia (THP-1) and PC-3 cancer cell lines |
| 1,3,5 | Substituted at position 1 with a ketone linked to 1,8-naphthyridine, an aryl, or a thiazole, while positions 3 and 5 are substituted with alkyl, aryl, or hetaryl groups, as well as ferrocene and carbonyl within a pyrazole or pyrazoline scaffold | 47c,58 51c,60 51d,60 66a,72 66b,72 90d,84 90f84 |
Exhibits docking interactions with the tyrosine kinase domain (PDB ID: 1M17) and the human IDH1 mutant (R132H), along with significant activity against the HEPG2 liver cancer cell line |
| 1,3,4,5 | Substituted at position 1 with an aryl or amide group (CONH2), along with 3,5-diamine and 4-diazenyl groups | 60a,65 60c65 |
Enhances activity against MCF-7 and MDA-MB-231 breast cancer cell lines |
The aryl and heteroaryl substituents at position 1 play a crucial role in determining the binding affinity and biological activity of pyrazole derivatives. For instance, pyrazole derivatives substituted at position 1 with a ketone linked to 1,8-naphthyridine, aryl, or thiazole, in combination with 3,5-alkyl/aryl or hetaryl groups, exhibit significant biological activity. Docking studies have revealed strong interactions with the tyrosine kinase domain and the human IDH1 mutant, demonstrating potent activity against HEPG2 liver cancer cells, as confirmed by the MTT assay, with inhibitory effects that induce apoptosis and disrupt the cell cycle.58,60,72,84 Additionally, substituents at positions 3 and 4, such as the 3-amino and 4-diazenyl groups, have demonstrated notable cytotoxic activity against MCF-7, HepG2, and HCT-116 cancer cell lines. These functional groups are essential for enhancing molecular interactions with cellular targets, thereby significantly improving cytotoxic efficacy.52,57,67
Pyrazolo[1,5-a]pyrimidine-fused derivatives substituted at positions 1 and 5 exhibit significant anticancer activity, particularly against MCF-7 and HCT-116 cell lines. The presence of an amino group at position 3 and a diazenyl group at position 4 enhances interactions with active sites, thereby improving anticancer efficacy.52,53,57,73 Additionally, cyclofused derivatives at positions 3,4 and 4,5 containing cycloalkanes or nitrogen/oxygen-based heterocycles, have demonstrated notable activity against MCF-7 and HeLa cell lines. These derivatives inhibit key proteins involved in cancer progression, such as the epidermal growth factor receptor (EGFR) tyrosine kinase domain (e.g., Lys721 and Asn818) and mTOR.49,50,56,59,62,76
In 1,3,4-substituted derivatives, the presence of 1-NH, 3-aryl, and 4-methylenetiadiazolone moieties facilitates strong docking interactions with the vascular endothelial growth factor receptor 2 (VEGFR2) kinase domain, further validating their anticancer activity.78 Furthermore, 1,3,4-diaryl and hetaryl derivatives, including oxadiazole, isoxazole, and benzimidazole, exhibit broad-spectrum anticancer activity against HeLa, A549, MCF-7, LOX-IMVI, and HEPG2 cell lines, which can be attributed to their ability to form π–π interactions and hydrogen bonds with active site residues.45,48,63,74,83
At positions 1, 3, and 5, pyrazole and pyrazoline derivatives containing 1-NH, 3,5-bis(styryl), and 3-aryl/5-carbamothioyl substituents exhibit strong docking interactions with the tubulin-combretastatin A4 complex. These interactions disrupt microtubule polymerization, induce G2/M phase arrest, and effectively inhibit cell division. Additionally, these derivatives interact with thymidylate synthase, forming hydrogen bonds with key residues such as Asp218, which enhance their activity against acute monocytic leukemia (THP-1).64,66,75,77
In 1,3,4,5-substituted derivatives, the presence of 1-aryl or CONH2 groups, combined with 3,5-diamine and 4-diazenyl moieties, exhibits significant activity against MCF-7 and MDA-MB-231 cell lines. Their biological activity is attributed to interactions within key protein domains and mechanisms that induce G2/M cell cycle arrest, effectively inhibiting cancer cell proliferation.65
6. Conclusions and perspectives
This review highlights the diverse methodologies – microwave, ultrasound, and mechanochemical – employed in the synthesis of pyrazole derivatives with notable anticancer properties, emphasizing the strategic importance of these approaches in medicinal chemistry and drug discovery. The SAR analysis clearly demonstrates that targeted substitutions and structural modifications at positions 1, 3, 4, and 5 significantly enhance the anticancer activity of pyrazole derivatives by enabling precise interactions with key active site residues, including Lys721 (EGFR), Asn818 (EGFR), and Cys241 (tubulin), through hydrogen bonding, π–π interactions, and hydrophobic stabilization. These interactions drive critical mechanisms, including G2/M phase arrest, apoptosis induction, and enzymatic inhibition, highlighting the potential of pyrazole derivatives as promising candidates for targeted anticancer therapies and future drug discovery initiatives.Our analysis reveals that microwave irradiation is the most widely used method, representing 68% of the reviewed studies, with reaction times ranging from 0.5 to 80 minutes and temperatures between 80 and 150 °C. Among microwave-assisted methodologies, dedicated microwave reactors are employed in 86% of cases, while conventional microwave ovens are employed in 14%. Ultrasound irradiation ranks as the second most applied technique, accounting for 22% of cases, with reaction times between 10 and 45 minutes and temperatures ranging from 50 to 60 °C. Within ultrasound-assisted methodologies, ultrasonic baths are utilized in 86% of cases, whereas ultrasonic reactors are applied in 14%. Mechanochemical activation is the least frequently employed method, representing 10% of cases, with reaction times ranging from 10 to 30 minutes at room temperature. Notably, all mechanochemical-assisted methodologies rely on grinding with a mortar and pestle. The integration of ultrasonic reactors and ball mills into the synthesis of anticancer pyrazole derivative presents significant potential for enhancing atom economy and step efficiency. Importantly, the lower market cost of ultrasonic reactors and ball mills compared to microwave reactors enables the development of more sustainable and cost-effective synthetic processes at reduced temperatures.
Data availability
No primary research results have been included and no new data were generated or analysed as part of this review.Author contributions
The authors have made significant contributions to the development of this review, with no involvement from any other individuals. The specific contribution of each author are as follows: Dr Diana Becerra. Literature investigation, writing, manuscript review and editing, and conceptualization. Dr Juan-Carlos Castillo. Literature investigation, writing, manuscript review and editing, conceptualization, and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J.-C. C. acknowledges the partial financial support provided by the Dirección de Investigaciones at the Universidad Pedagógica y Tecnológica de Colombia (Project SGI-3724). Additionally, D. B. and J.-C. C. express their sincere gratitude to Dr Irina Morales Castaño, Dean of the Facultad de Ciencias, and Dr Sandra Chaparro Acuña, Coordinator of the Doctorado en Ciencias Químicas, for their invaluable financial assistance.Notes and references
- J.-C. Castillo, B. Castro Agudelo, J. Gálvez, Y. Carissan, J. Rodríguez and Y. Coquerel, Periselectivity in the aza-Diels–Alder cycloaddition between α-oxoketenes and N-(5-pyrazolyl)imines: A combined experimental and theoretical study, J. Org. Chem., 2020, 85, 7368–7377 CrossRef CAS PubMed.
- Y. Zhang, C. Wu, N. Zhang, R. Fan, Y. Ye and J. Xu, Recent advances in the development of pyrazole derivatives as anticancer agents, Int. J. Mol. Sci., 2023, 24, 12724 CrossRef CAS PubMed.
- G. Li, Y. Cheng, C. Han, C. Song, N. Huang and Y. Du, Pyrazole-containing pharmaceuticals: Target, pharmacological activity, and their SAR studies, RSC Med. Chem., 2022, 13, 1300–1321 RSC.
- Q. Fu, P.-P. Cai, L. Cheng, L.-K. Zhong, C.-X. Tan, Z.-H. Shen, L. Han, T.-M. Xu and X.-H. Liu, Synthesis and herbicidal activity of novel pyrazole aromatic ketone analogs as HPPD inhibitor, Pest Manage. Sci., 2020, 76, 868–879 CrossRef CAS PubMed.
- J.-C. Castillo, N.-F. Bravo, L.-V. Tamayo, P.-D. Mestizo, J. Hurtado, M. Macías and J. Portilla, Water-compatible synthesis of 1,2,3-triazoles under ultrasonic conditions by a Cu(I) complex-mediated click reaction, ACS Omega, 2020, 5, 30148–30159 CrossRef CAS PubMed.
- A. Tigreros, J.-C. Castillo and J. Portilla, Cyanide chemosensors based on 3-dicyanovinylpyrazolo[1,5-a]pyrimidines: Effects of peripheral 4-anisyl group substitution on the photophysical properties, Talanta, 2020, 215, 120905 CrossRef CAS PubMed.
- E. Moreno-Suárez, R. Avila-Acosta, K. Sánchez-Ramírez, J.-C. Castillo and M. A. Macías, Crystallographic, spectroscopic and thermal studies of 1-(4-bromophenyl)-5-(2,5-dimethyl-1H-pyrrol-1-yl)-3-methyl-1H-pyrazole, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2023, 79, 472–479 CrossRef PubMed.
- D. Fonseca, S. M. Leal-Pinto, M. V. Roa-Cordero, J. D. Vargas, E. M. Moreno-Moreno, M. A. Macías, L. Suescun, Á. Muñoz-Castro and J. J. Hurtado, Inhibition of C. albicans dimorphic switch by cobalt(II) complexes with ligands derived from pyrazoles and dinitrobenzoate: Synthesis, characterization and biological activity, Int. J. Mol. Sci., 2019, 20, 3237 CrossRef CAS PubMed.
- A. Kurt and M. Koca, Synthesis, characterization and thermal degradation kinetics of a new pyrazole derived methacrylate polymer, poly(1,3-diphenyl-1H-pyrazol-5-yl methacrylate), Acta Chim. Slov., 2022, 69, 466–477 CrossRef CAS PubMed.
- J. Zhang, W. Yang, M. He, Z. Peng and G. Wang, Development of novel pyrazole-1,2,4-triazole derivatives as tyrosinase inhibitors: Design, preparation, mechanism of action and anti-browning application, Food Chem., 2024, 460, 140722 CrossRef CAS PubMed.
- M. Guin, R. A. Roopa, P. Jain and N. B. Singh, Heterocyclic surfactants and their applications in cosmetics, ChemistrySelect, 2022, 7, e202103989 CrossRef CAS.
- T.-F. Wang, S. R. Kosuru, S.-C. Yu, Y.-C. Chang, H.-Y. Lai, Y.-L. Chang, K.-H. Wu, S. Ding and H.-Y. Chen, Use of pyrazoles as ligands greatly enhances the catalytic activity of titanium iso-propoxide for the ring-opening polymerization of L-lactide: A cooperation effect, RSC Adv., 2020, 10, 40690–40696 RSC.
- M. J. Naim, O. Alam, F. Nawaz, J. Alam and P. Alam, Current status of pyrazole and its biological activities, J. Pharm. BioAllied Sci., 2016, 8, 2–17 CrossRef CAS PubMed.
- B. Insuasty, A. Montoya, D. Becerra, J. Quiroga, R. Abonía, S. Robledo, I. D. Vélez, Y. Upegui, M. Nogueras and J. Cobo, Synthesis of novel analogs of 2-pyrazoline obtained from [(7-chloroquinolin-4-yl)amino]chalcones and hydrazine as potential antitumor and antimalarial agents, Eur. J. Med. Chem., 2013, 67, 252–262 CrossRef CAS PubMed.
- K. Karrouchi, S. Radi, Y. Ramli, Y. N. Mabkhot, J. Taoufik, F. A. Al-aizari and M. Ansar, Synthesis and pharmacological activities of pyrazole derivatives: A review, Molecules, 2018, 23, 134 CrossRef PubMed.
- M. Faisal, A. Saeed, S. Hussain, P. Dar and F. A. Larik, Recent developments in synthetic chemistry and biological activities of pyrazole derivatives, J. Chem. Sci., 2019, 131, 70 CrossRef.
- O. Ebenezer, M. Shapi and J. A. Tuszynski, A review of the recent development in the synthesis and biological evaluations of pyrazole derivatives, Biomedicines, 2022, 10, 1124 Search PubMed.
- D. Becerra, R. Abonía and J.-C. Castillo, Recent applications of the multicomponent synthesis for bioactive pyrazole derivatives, Molecules, 2022, 27, 4723 CrossRef CAS PubMed.
- J. J. Cui, M. Tran-Dubé, H. Shen, M. Nambu, P.-P. Kung, M. Pairish, L. Jia, J. Meng, L. Funk, I. Botrous, M. McTigue, N. Grodsky, K. Ryan, E. Padrique, G. Alton, S. Timofeevski, S. Yamazaki, Q. Li, H. Zou, J. Christensen, B. Mroczkowski, S. Bender, R. S. Kania and M. P. Edwards, Structure based drug design of Crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal–epithelial transition factor (c-MET) kinase and anaplastic lymphoma Kinase (ALK), J. Med. Chem., 2011, 54, 6342–6363 CrossRef CAS PubMed.
- V. Subbiah, P. A. Cassier, S. Siena, E. Garralda, L. Paz-Ares, P. Garrido, E. Nadal, J. Vuky, G. Lopes, G. P. Kalemkerian, D. W. Bowles, M. Seetharam, J. Chang, H. Zhang, J. Green, A. Zalutskaya, M. Schuler, Y. Fan and G. Curigliano, Pan-cancer efficacy of pralsetinib in patients with RET fusion–positive solid tumors from the phase 1/2 ARROW trial, Nat. Med., 2022, 28, 1640–1645 CrossRef CAS PubMed.
- M. C. Heinrich, R. L. Jones, M. Mehren, P. Schöffski, C. Serrano, Y.-K. Kang, P. A. Cassier, O. Mir, F. Eskens, W. D. Tap, P. Rutkowski, P. S. P. Chawla, J. Trent, M. Tugnait, E. K. Evans, T. Lauz, T. Zhou, M. Roche, B. B. Wolf, S. Bauer and S. George, Avapritinib in advanced PDGFRA D842V-mutant gastrointestinal stromal tumour (NAVIGATOR): A multicentre, open-label, phase 1 trial, Lancet Oncol., 2020, 21, 935–946 CrossRef CAS PubMed.
- J. Schoepfer, W. Jahnke, G. Berellini, S. Buonamici, S. Cotesta, S. W. Cowan-Jacob, S. Dodd, P. Drueckes, D. Fabbro, T. Gabriel, J.-M. Groell, R. M. Grotzfeld, A. Q. Hassan, C. Henry, V. Iyer, D. Jones, F. Lombardo, A. Loo, P. W. Manley, X. Pellé, G. Rummel, B. Salem, M. Warmuth, A. A. Wylie, T. Zoller, A. L. Marzinzik and P. Furet, Discovery of asciminib (ABL001), an allosteric inhibitor of the tyrosine kinase activity of BCR-ABL1C, J. Med. Chem., 2018, 61, 8120–8135 CrossRef CAS PubMed.
- A. S. Harney, G. S. Karagiannis, J. Pignatelli, B. D. Smith, E. Kadioglu, S. C. Wise, M. M. Hood, M. D. Kaufman, C. B. Leary, W.-P. Lu, G. Al-Ani, X. Chen, D. Entenberg, M. H. Oktay, Y. Wang, L. Chun, M. Palma, J. G. Jones, D. L. Flynn and J. S. Condeelis, The selective Tie2 inhibitor rebastinib blocks recruitment and function of Tie2Hi macrophages in breast cancer and pancreatic neuroendocrine tumors, Mol. Cancer Ther., 2017, 16, 2486–2501 CrossRef CAS PubMed.
- M. A. Alam, Pyrazole: An emerging privileged scaffold in drug discovery, Future Med. Chem., 2023, 15, 2011–2023 CrossRef CAS PubMed.
- A. A. Gaber, A. M. El-Morsy, F. F. Sherbiny, A. H. Bayoumi, K. M. El-Gamal, K. El-Adl, A. A. Al-Karmalawy, R. R. Ezz Eldin, M. A. Saleh and H. S. Abulkhair, Pharmacophore-linked pyrazolo[3,4-d]pyrimidines as EGFR-TK inhibitors: Synthesis, anticancer evaluation, pharmacokinetics, and in silico mechanistic studies, Arch. Pharm., 2021, e2100258 CrossRef PubMed.
- M. F. Harras and R. Sabour, Design, synthesis and biological evaluation of novel 1,3,4-trisubstituted pyrazole derivatives as potential chemotherapeutic agents for hepatocellular carcinoma, Bioorg. Chem., 2018, 78, 149–157 CrossRef CAS PubMed.
- R. P. Paitandi, V. Sharma, V. D. Singh, B. K. Dwivedi, S. M. Mobin and D. S. Pandey, Pyrazole appended quinoline-BODIPY based arene ruthenium complexes: Their anticancer activity and potential applications in cellular imaging, Dalton Trans., 2018, 47, 17500–17514 RSC.
- L. Gomez, M. D. Kack, J. Wu, J. J. M. Wiener, H. Venkatesan, A. Santillán, D. J. Pippel, N. Mani, B. J. Morrow, S. T. Motley, K. J. Shaw, R. Wolin, C. A. Grice and T. K. Jones, Novel pyrazole derivatives as potent inhibitors of type II topoisomerases. Part 1: Synthesis and preliminary SAR analysis, Bioorg. Med. Chem. Lett., 2007, 17, 2723–2727 CrossRef CAS PubMed.
- C. Yamali, H. I. Gul, A. Ece, S. Bua, A. Angeli, H. Sakagami, E. Sahin and C. T. Supuran, Synthesis, biological evaluation and in silico modelling studies of 1,3,5-trisubstituted pyrazoles carrying benzenesulfonamide as potential anticancer agents and selective cancer-associated hCA IX isoenzyme inhibitors, Bioorg. Chem., 2019, 92, 103222 CrossRef CAS PubMed.
- J. Zhou, Q. Zhou and J.-P. Wan, Recent advances in the multicomponent synthesis of pyrazoles, Org. Biomol. Chem., 2024, 22, 8065–8077 RSC.
- S. Sharma, P. Sharma, B. Budhalakoti, A. Kumar, K. Singh, R. K. Kamboj and K. Bhatrola, Exploring the impact of ionic liquids on pyrazole derivatives synthesis: A critical review, ChemistrySelect, 2024, 9, e202401925 CrossRef CAS.
- S. Sharma, V. Singh, Vaishali, R. Kumar, R. Jamra, N. Banyal and Jyoti, From 2011 to 2022: The development of pyrazole derivatives through the α,β-unsaturated carbonyl compounds, J. Heterocycl. Chem., 2024, 61, 232–284 CrossRef CAS.
- R. Singh, R. Kaur, P. Ahlawat, P. Kaushik and K. Singh, Green methods for the synthesis of pyrazoles: A review, Org. Prep. Proced. Int., 2021, 53, 317–351 CrossRef CAS.
- S. P. Chandrasekharan, A. Dhami, S. Kumar and K. Mohanan, Recent advances in pyrazole synthesis employing diazo compounds and synthetic analogues, Org. Biomol. Chem., 2022, 20, 8787–8817 RSC.
- P. K. Mykhailiuk, Fluorinated pyrazoles: From synthesis to applications, Chem. Rev., 2021, 121, 1670–1715 CrossRef CAS PubMed.
- B. Nehra, V. Chawla, P. A. Chawla and M. Kumar, Recent advancements in microwave assisted synthesis of pyrazole analogues: An ecological synthetic approach, Polycyclic Aromat. Compd., 2024, 2024, 1–47 Search PubMed.
- F. Tok and B. Koçyiğit-Kaymakçıoğlu, Recent advances in the microwave and ultrasound-assisted synthesis of pyrazole scaffolds, Curr. Org. Chem., 2023, 27, 1053–1071 CrossRef CAS.
- S. Singh, S. Yadav, M. Minakshi and R. Pundeer, Green synthesis of pyrazoles: Recent developments in aqueous methods, SynOpen, 2023, 7, 297–312 Search PubMed.
- D. Dallinger and C. O. Kappe, Microwave-assisted synthesis in water as solvent, Chem. Rev., 2007, 107, 2563–2591 CrossRef CAS PubMed.
- A. Hoz, Á. Díaz-Ortiz and A. Moreno, Microwaves in organic synthesis. Thermal and non-thermal microwave effects, Chem. Soc. Rev., 2005, 34, 164–178 RSC.
- C. O. Kappe, Microwave dielectric heating in synthetic organic chemistry, Chem. Soc. Rev., 2008, 37, 1127–1139 RSC.
- K. Martina, G. Cravotto and R. S. Varma, Impact of microwaves on organic synthesis and strategies toward flow processes and scaling, J. Org. Chem., 2021, 86, 13857–13872 CrossRef CAS PubMed.
- M. Sankaran, C. Uvarani, K. Chandraprakash, M. U. Maheswari and P. S. Mohan, An expedient approach for the synthesis of bioactive pyrazole, isoxazole and benzodiazepine-substituted quinolin-2(1H)-one derivatives, J. Heterocycl. Chem., 2015, 52, 1082–1092 CrossRef CAS.
- D. Ashok, K. Padmavati, B. V. Lakshmi and M. Sarasija, Microwave-assisted one-pot synthesis of pyrazolyl-substituted benzochroman-4-one derivatives and evaluation of their anticancer activity, Chem. Heterocycl. Compd., 2016, 52, 15–20 CrossRef CAS.
- N. C. Desai, G. M. Kotadiya, A. R. Trivedi, V. M. Khedkar and P. C. Jha, Design, synthesis, and biological evaluation of novel fluorinated pyrazole encompassing pyridyl 1,3,4-oxadiazole motifs, Med. Chem. Res., 2016, 25, 2698–2717 CrossRef CAS.
- D. Insuasty, J. Castillo, D. Becerra, H. Rojas and R. Abonía, Synthesis of biologically active molecules through multicomponent reactions, Molecules, 2020, 25, 505 CrossRef PubMed.
- D. Hurtado-Rodríguez, A. Salinas-Torres, H. Rojas, D. Becerra and J.-C. Castillo, Bioactive 2-pyridone-containing heterocycle syntheses using multicomponent reactions, RSC Adv., 2022, 12, 35158–35176 RSC.
- S. M. Gomha, M. M. Edrees, R. A. M. Faty, Z. A. Muhammad and Y. N. Mabkhot, Microwave-assisted one-pot three-component synthesis of some novel pyrazole scaffolds as potent anticancer agents, Chem. Cent. J., 2017, 11, 37 CrossRef PubMed.
- T. Arasakumar, S. Mathusalini, S. Gopalan, S. Shyamsivappan, A. Ata and P. S. Mohan, Biologically active perspective synthesis of heteroannulated 8-nitroquinolines with green chemistry approach, Bioorg. Med. Chem. Lett., 2017, 27, 1538–1546 CrossRef CAS PubMed.
- G. Mótyán, M. K. Gopisetty, R. E. Kiss-Faludy, A. Kulmány, I. Zupkó, É. Frank and M. Kiricsi, Anti-cancer activity of novel dihydrotestosterone-derived ring A-condensed pyrazoles on androgen non-responsive prostate cancer cell lines, Int. J. Mol. Sci., 2019, 20, 2170 CrossRef PubMed.
- A. Ballesteros-Casallas, M. Paulino, P. Vidossich, C. Melo, E. Jiménez, J.-C. Castillo, J. Portilla and G. Pietro Miscione, Synthesis of 2,7-diarylpyrazolo[1,5-a]pyrimidine derivatives with antitumor activity. Theoretical identification of targets, Eur. J. Med. Chem. Rep., 2022, 4, 100028 CAS.
- A. M. Fouda, H.-A. S. Abbas, E. H. Ahmed, A. A. Shati, M. Y. Alfaifi and S. E. I. Elbehairi, Synthesis, in vitro antimicrobial and cytotoxic activities of some new pyrazolo[1,5-a]pyrimidine derivatives, Molecules, 2019, 24, 1080 CrossRef PubMed.
- K. Shekarrao, P. P. Kaishap, V. Saddanapu, A. Addlagatta, S. Gogoi and R. C. Boruah, Microwave-assisted palladium mediated efficient synthesis of pyrazolo[3,4-b]pyridines, pyrazolo[3,4-b]quinolines, pyrazolo[1,5-a]pyrimidines and pyrazolo[1,5-a]quinazolines, RSC Adv., 2014, 4, 24001 RSC.
- S. Aydın, N. Kaushik-Basu, S. Özba-Turan, J. Akbuga, P. Mega Tiber, O. Orun, K. R. Gurukumar, A. Basu and G. Küçükgüzel, Synthesis of 1-aroyl-3,5-dimethyl-1H-pyrazoles as anti-HCV and anticancer agents, Lett. Drug Des. Discovery, 2014, 11, 121–131 CrossRef.
- A. Thalassitis, A.-M. Katsori, K. Dimas, D. J. Hadjipavlou-Litina, F. Pyleris, N. Sakellaridis and K. E. Litinas, Synthesis and biological evaluation of modified purine homo-N-nucleosides containing pyrazole or 2-pyrazoline moiety, J. Enzyme Inhib. Med. Chem., 2014, 29, 109–117 CrossRef CAS PubMed.
- G. L. Reddy, S. K. Guru, M. Srinivas, A. S. Pathania, P. Mahajan, A. Nargotra, S. Bhushan and R. A. Vishwakarma, Synthesis of 5-substituted-1H-pyrazolo[4,3-d]pyrimidin-7(6H)-one analogs and their biological evaluation as anticancer agents: mTOR inhibitors, Eur. J. Med. Chem., 2014, 80, 201–208 CrossRef CAS PubMed.
- A. M. A. Alnaja, T. A. Farghaly, H. S. A. El-zahabi and M. R. Shaaban, Cytotoxicity, docking study of new fluorinated fused pyrimidine scaffold: Thermal and microwave irradiation synthesis, Med. Chem., 2021, 17, 501–518 CAS.
- E. V. Filho, J. W. S. Pina, M. K. Antoniazi, L. B. Loureiro, M. A. Ribeiro, C. B. Pinheiro, C. J. Guimaraes, F. C. E. de Oliveira, C. Pessoa, A. G. Taranto and S. J. Greco, Synthesis, docking, machine learning and antiproliferative activity of the 6-ferrocene/heterocycle-2-aminopyrimidine and 5-ferrocene-1H-pyrazole derivatives obtained by microwave-assisted Atwal reaction as potential anticancer agents, Bioorg. Med. Chem. Lett., 2021, 48, 128240 CrossRef CAS PubMed.
- P. H. Parikh, J. B. Timaniya, M. J. Patel and K. P. Patel, Microwave-assisted synthesis of pyrano[2,3-c]pyrazole derivatives and their anti-microbial, anti-malarial, anti-tubercular, and anti-cancer activities, J. Mol. Struct., 2022, 1249, 131605 CrossRef CAS.
- A. Dahal, M. Lo, S. Singh, H. Vo, D. ElHage, S. D. Jois and S. Murru, 1,3-Diarylpyrazolones as potential anticancer agents for non-small cell lung cancer: Synthesis and antiproliferative activity evaluation, Chem. Biol. Drug Des., 2022, 99, 620–633 CrossRef CAS PubMed.
- S. T. Boatenga, T. Roya, K. Torrey, U. Owunna, S. Banang-Mbeumi, D. Basnet, E. Niedda, A. D. Alexander, D. El Hage, S. Atchimnaidu, B. M. Nagalo, D. Aryal, A. Findley, N. P. Seeram, T. Efimova, M. Sechi, R. A. Hill, H. Ma, J. C. Chamcheu and S. Murru, Synthesis, in silico modelling, and in vitro biological evaluation of substituted pyrazole derivatives as potential anti-skin cancer, anti-tyrosinase, and antioxidant agents, J. Enzyme Inhib. Med. Chem., 2023, 38, 2205042 CrossRef PubMed.
- G. Mótyán, Á. Baji, M. A. Marc, M. K. Gopisetty, D. I. Adamecz, M. Kiricsi, É. A. Enyedy and É. Frank, Microwave-assisted synthesis, proton dissociation processes, and anticancer evaluation of novel D-ring-fused steroidal 5-amino-1-arylpyrazoles, Appl. Sci., 2020, 10, 229 CrossRef.
- D. Ashok, M. R. Reddy, N. Nagaraju, R. Dharavath, K. Ramakrishna, S. Gundu, P. Shravani and M. Sarasija, Microwave-assisted synthesis and in vitro antiproliferative activity of some novel 1,2,3-triazole-based pyrazole aldehydes and their benzimidazole derivatives, Med. Chem. Res., 2020, 29, 699–706 CrossRef CAS.
- V. W. Y. Liao, A. Kumari, R. Narlawar, S. Vignarajan, D. E. Hibbs, D. Panda and P. W. Groundwater, Tubulin-binding 3,5-bis(styryl)pyrazoles as lead compounds for the treatment of castration-resistant prostate cancer, Mol. Pharmacol., 2020, 97, 409–422 CrossRef CAS PubMed.
- L. H. Al-Wahaibi, M. A. I. Elbastawesy, N. E. Abodya, B. G. M. Youssif, S. Bräse, S. N. Shabaan, G. H. Sayed and K. E. Answer, New pyrazole/pyrimidine-based scaffolds as inhibitors of heat shock protein 90 endowed with apoptotic anti-breast cancer activity, Pharmaceuticals, 2024, 17, 1284 CrossRef CAS PubMed.
- S. M. H. Sanad and A. E. M. Mekky, New thieno[2,3-b]pyridine-fused pyrimidin-4(3H)-ones as potential thymidylate synthase inhibitors: Synthesis, SAR, in vitro and in silico study, J. Mol. Struct., 2023, 1282, 135236 CrossRef CAS.
- K. E. Anwer, S. S. A. El-Hddad, N. E. A. A. El-Sattar, A. El-morsy, F. Khedr, S. Mohamady, D. E. Keshek, S. A. Salama, K. El-Adl and N. S. Hanafy, Five and six membered heterocyclic rings endowed with azobenzene as dual EGFRT790M and VEGFR-2 inhibitors: Design, synthesis, in silico ADMET profile, molecular docking, dynamic simulation and anticancer evaluations, RSC Adv., 2023, 13, 35321–35338 RSC.
- T. J. Mason, Ultrasound in synthetic organic chemistry, Chem. Soc. Rev., 1997, 26, 443–451 RSC.
- G. Cravotto and P. Cintas, Power ultrasound in organic synthesis: Moving cavitational chemistry from academia to innovative and large-scale applications, Chem. Soc. Rev., 2006, 35, 180–196 RSC.
- S. V. Sancheti and P. R. Gogate, A review of engineering aspects of intensification of chemical synthesis using ultrasound, Ultrason. Sonochem., 2017, 36, 527–543 Search PubMed.
- M. Draye, G. Chatel and R. Duwald, Ultrasound for drug synthesis: A green approach, Pharmaceuticals, 2020, 13, 23 CrossRef CAS PubMed.
- N. S. Ahmed, K. O. AlFooty and S. S. Khalifah, Synthesis of 1,8-naphthyridine derivatives under ultrasound irradiation and cytotoxic activity against HepG2 cell lines, J. Chem., 2014, 2014, 1–8 Search PubMed.
- N. Suresh, B. V. Durgarao, A. Ratnakar, S. K. Kolli, M. A. Ashfaq, M. V. B. Rao and M. Pal, Ultrasound-Assisted 3-component reaction in acetic acid alone: Catalyst/promoter/ligand free synthesis of bioactive pyrazolo[1,5-a]pyrimidines, Lett. Drug Des. Discovery, 2017, 14, 1176–1183 CAS.
- T. D. Bhatt, D. G. Gojiya, P. L. Kalavadiya and H. S. Joshi, Rapid, greener and ultrasound irradiated one-pot synthesis of 4-(substituted-1H-pyrazol-4-yl)methylene)-3-isopropylisoxazol-5(4H)-ones and their in vitro anticancer activity, ChemistrySelect, 2019, 4, 11125–11129 CrossRef CAS.
- G. M. Nitulescu, L. Matei, I. M. Aldea, C. Draghici, O. T. Olaru and C. Bleotu, Ultrasound-assisted synthesis and anticancer evaluation of new pyrazole derivatives as cell cycle inhibitors, Arabian J. Chem., 2019, 12, 816–824 CrossRef CAS.
- N. Nagasundaram, K. Padmasree, S. Santhosh, N. Vinoth, N. Sedhu and A. Lalitha, Ultrasound promoted synthesis of new azo fused dihydropyrano[2,3-c]pyrazole derivatives: In vitro antimicrobial, anticancer, DFT, in silico ADMET and molecular docking studies, J. Mol. Struct., 2022, 1263, 133091 CrossRef CAS.
- V. S. Dofe, A. P. Sarkate, S. V. Tiwari, D. K. Lokwani, K. S. Karnik, I. A. Kale, S. Dodamani, S. S. Jalalpure and P. V. L. S. Burra, Ultrasound assisted synthesis of tetrazole based pyrazolines and isoxazolines as potent anticancer agents via inhibition of tubulin polymerization, Bioorg. Med. Chem. Lett., 2020, 30, 127592 CrossRef CAS PubMed.
- N. M. Hassanin, T. E. Ali, M. A. Assiri and S. M. Abdel-Kariem, Novel carbazolyl–thiazolyl–chromone and carbazolyl–thiazolyl–pyrazole hybrids: Synthesis, cytotoxicity evaluation and molecular docking studies, RSC Adv., 2024, 14, 17245 RSC.
- M. Leonardi, M. Villacampa and J. C. Menéndez, Multicomponent mechanochemical synthesis, Chem. Sci., 2018, 9, 2042–2064 RSC.
- J.-L. Do and T. Friščić, Mechanochemistry: A force of synthesis, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed.
- T. K. Achar, A. Bose and P. Mal, Mechanochemical synthesis of small organic molecules, Beilstein J. Org. Chem., 2017, 13, 1907–1931 CrossRef CAS PubMed.
- V. Martinez, T. Stolar, B. Karadeniz, I. Brekalo and K. Užarević, Advancing mechanochemical synthesis by combining milling with different energy sources, Nat. Rev. Chem., 2023, 7, 51–65 CrossRef CAS PubMed.
- H. R. M. Rashdan, S. M. Gomha, M. S. El-Gendey, M. A. El-Hashash and A. M. M. Soliman, Eco-friendly one-pot synthesis of some new pyrazolo[1,2-b]phthalazinediones with antiproliferative efficacy on human hepatic cancer cell lines, Green Chem. Lett. Rev., 2018, 11, 264–274 CrossRef CAS.
- M. M. Edrees, S. Abu-Melha, A. M. Saad, N. A. Kheder, S. M. Gomha and Z. A. Muhammad, Eco-Friendly synthesis, characterization and biological evaluation of some novel pyrazolines containing thiazole moiety as potential anticancer and antimicrobial agents, Molecules, 2018, 23, 2970 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2025 |