
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA review: advancing organic electronics through the lens of ionic liquids and polymerized ionic liquids
Swati
Arora†
 *ab and
Nagendra
Verma‡
c
*ab and
Nagendra
Verma‡
c
aDepartment of Chemistry, University of Pittsburgh, Pittsburgh, PA, USA
bPatheon/Thermo Fisher Scientific, Florence, South Carolina, USA. E-mail: swa10@pitt.edu; swati.arora2@thermofisher.com
cDepartment of Biomedical Sciences, Cedars Sinai Medical Center, Los Angeles, CA, USA. E-mail: Nagendra.verma@cshs.org; nagendraverma516@gmail.com
First published on 15th April 2024
Abstract
The technological world has undergone significant progress over the last 60 years through advances in electronics; nonetheless, a lot needs to be accomplished for constructing devices that can toggle between desired functions. This review provides a brief look at recent developments in innovative chemistries involving fine-tuning the molecular properties of polymerized ionic liquids to create new kinds of responsive organic electronic devices, thus highlighting the versatility of polymerized ionic liquids in promising a bright future for advanced organic electronics. This review also reinforces the need to customize materials for contemporary applications, as doing so can improve the functionality of sophisticated devices. In addition, the review sheds light on a novel class of materials referred to as “doubly polymerized ionic liquids”. This class of materials features negligible ionic conductivity, providing a solid groundwork for constructing ion-locked devices that can be used to attain novel technological applications. We anticipate this review will close research gaps and promote discoveries in the fields of organic electronics, polymer chemistry, and polymer physics.
 Swati Arora | Dr Swati Arora received her Bachelor's in Chemistry from St Stephen's College, Delhi University, India, in 2009. She further pursued her Master's in Organic Chemistry from St Stephen's College in 2011. She then moved to the USA in 2012 where she began her career in research in the Department of Chemistry at Purdue University, Indiana, where she worked at the interface of Organic Chemistry and Chemical Biology. During her Master's at Purdue University, she synthesized intermediates for Medicinal Chemistry applications using Organoborane and Fluororganic Chemistry. She further investigated light-toggled gene expression in mammalian cells, which triggered her interest in polymers. This led her to continue her Ph.D. research in the Department of Chemistry at the University of Pittsburgh where she worked at the interface of Organic Chemistry and Polymer Chemistry and designed materials like doubly-polymerized ionic liquids, mechanophores, and resorcinarene-based polymeric membranes for specific applications. After graduating with her Ph.D., Dr Arora is currently working as a Scientist-III, Process Research and Development at Thermo Fisher Scientific, where she is developing robust and scalable chemical processes for the commercial production of active pharmaceutical ingredients for medicines. She has contributed to numerous research papers, including reviews and editorials, and published in national and international peer-reviewed journals. |
 Nagendra Verma | Dr Nagendra Verma is currently a Postdoctoral Scientist in the Department of Regenerative Medicine at Cedar Sinai Medical Centre in Los Angeles, California, USA. Previously, he worked as a Postdoctoral scholar in the Cancer Centre at Cedar Sinai Medical Centre in Los Angeles, USA. He obtained his doctoral degree from the Department of Molecular Medicine at Sapienza University of Rome in Rome, Italy. He also holds an M.Sc. degree from the School of Life Sciences, Department of Biotechnology, Central University of Rajasthan in Ajmer, Rajasthan, India. Dr Verma's research focuses on regenerative medicine and its applications in a variety of conditions, including cancer and cornea wound healing. Specifically, he investigates the use of stem cells and extracellular vesicles to discover drugs and biomarkers for disease profiling at the biomolecular level. He is interested in profiling the bioactive content of extracellular vesicles for biomedical and healthcare purposes, and he aims to translate his findings from basic research into practical clinical applications and diagnostics. He has contributed to numerous research papers, including reviews and editorials, and published in national and international peer-reviewed journals. |
1 Introduction
Ionic liquids are salts that are composed of weakly bound charged cations and anions.1,2 As the name indicates, these salts are liquid at room temperature. Generally, the charged cations and anions constituting the ionic liquids are bulky organic species. Their low volatilities and substantially high ionic conductivities at temperatures relevant to applications make them suitable for a wide range of uses, including flexible electronics and fuel cell electrolytes.3–8 In the last few years, a related class of materials known as singly polymerized ionic liquids (SPILs) has been illuminated.9–11 As suggested by the name, one of the two ionic species present in these materials is mobile, and the other is covalently bonded to a polymer backbone. This results in facile ion transport with high charge mobility in these mechanically strong materials.Researchers have made various attempts to enhance the ionic conductivity of SPILs. These include altering the ionic groups’ chemical makeup,12 how far apart they are from the polymer backbone,13 and how they are spaced out along the chains.13,14 It is interesting to note that under specific experimental conditions, these methods are unable to modify the mobile ion content of polymerized ionic liquids (PILs). In order to overcome this challenge, scientists have created a brand-new class of PILs known as “triggerable polymerized ionic liquids” that react to a variety of chemical and physical stimuli.15 This exemplifies a novel approach for “on-command” change in ionic conductivity of PILs and has led to significant advancement in designing organic electronics.
However, developing a deeper molecular-scale understanding of ion motion and charge transport in these materials is critical for practical applications with additional capabilities in response to simple chemical and physical triggers. Broadband dielectric spectroscopy (BDS) is one such promising technique and is widely used in the polymers community for investigating the dynamics of ion motion in SPILs, considering spectroscopic techniques like nuclear magnetic resonance (NMR) and infrared (IR) spectroscopy are not substantiated for probing the coulombic interactions among the ions.16 This technique has provided a plethora of knowledge regarding the charge transport mechanism in these SPILs, providing exciting avenues for measuring the energetics and ion motion timescales in these cutting-edge organic electronics.17–20
In the scientific community, to date, the majority of researchers have conducted comprehensive investigations for designing PILs with enhanced ionic mobility without sacrificing mechanical properties. Therefore, designing PILs with a significant decrease in ionic mobility would be counterintuitive. However, to realize certain emerging applications, like electrical double-layer locking, on application-relevant timescales in advanced organic electronics, restricting ion motion would be desirable. To achieve this goal, researchers have examined doubly polymerized ionic liquids (DPILs) as a viable platform for ion-locking applications.21,22
Unlike SPILs, where only one ionic species is bound to the polymer backbone, DPILs have both ionic species attached to the polymer chain.12,22–26 Chemically locking the ions in place in DPILs drastically decreases the ionic conductivity. This is not only fundamentally important, but also has the potential to stimulate the development of exciting classes of organic electronics. Developing devices based on DPIL requires a far deeper understanding of the parameters that govern the electrical double layer (EDL) retention time, which is the duration for which the ions can be held in place. Inspecting these factors will further assist in the design of precise and controllable DPIL-based devices. Because DPILs respond to dielectric relaxation measurements, scientists have recently gained more insight into the mechanisms underlying the ion transport blockage in these materials.22 These rigorous studies illuminated the timescales for the functioning of devices dependent on ion locking.
Over the years, several researchers have enlightened us on the emerging advances in the design, synthesis, and applications of polymerized ionic liquids. However, in this comprehensive review, we present a novel perspective to envisage PILs for fabricating stimuli-responsive and ion-locked devices. A significant rigorous understanding of ion transport and ion-locking mechanisms in PILs is critical to building a solid foundation for creating these organic electronics. We hope to push the envelope of what we can achieve through DPILs and their potential applications in advanced organic electronics (Fig. 1).
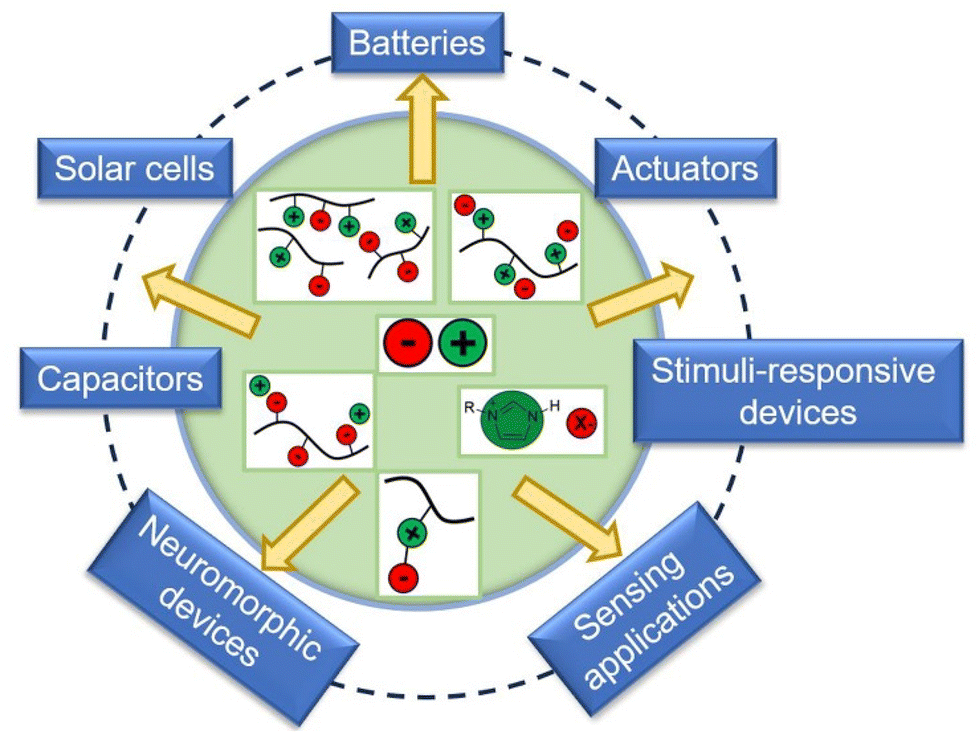 | ||
| Fig. 1 Schematic illustration describing the applications of different ionic liquids to energy storage and devices. | ||
2 Ionic liquids
The polymer community has shown a great deal of interest in ionic liquids during the past ten years. These are low melting point salts made up of an inorganic anion and an organic cation. With the cation being asymmetric, the crystallization of the salt is impeded, resulting in a substantially lower melting point than that of a common salt.27 Their remarkable properties including negligible vapor pressure, nonflammability, excellent electrochemical stability, good ion mobility, and miscibility with a wide range of organic materials make them enticing for a variety of applications.1,28 Furthermore, with different possible combinations of anions and cations, their physiochemical and electrochemical properties can be easily tailored.292.1 Synthetic route to ionic liquids
Ionic liquids (ILs) have been generally synthesized in a two-fold process. In the first step, a quaternization reaction takes place in which a haloalkane alkylates a base followed by an anion exchange in the metathesis reaction in the second step.30 This process works efficiently with water as a reaction solvent for a metathesis reaction where hydrophobic ILs are readily isolated from the reaction media. However, the synthesis of water-miscible ionic liquids becomes challenging by this route. Researchers have addressed this challenge by conducting the metathesis reaction at the melting temperature of the amine halide salts, with amine acting as a solvent.31 This synthetic strategy was not only cost-effective but also scalable and environmentally friendly.2.2 Electrochemical stability of ionic liquids
When choosing an ionic liquid for a particular application, the electrochemical stability of the liquid is one of its physiochemical properties that is most important to consider.32 It is typically expressed as the voltage differential between the ionic liquid's oxidation and reduction potentials, and is independent of the reference utilized for the measurements.29 Impurity and water content in ionic liquids significantly influence their electrochemical stability.33 Furthermore, electrochemical stability has also been shown to be greatly influenced by anion structure, and typically highly fluorinated anions such as Tf2N− and BF4− are more electrochemically inert to redox reactions.34 Notably, with the cathodic stability of cations, the following trend has been observed: piperidinium ≈ pyrrolidinium ≈ ammonium > imidazolium > pyridinium.35 Interestingly, many ionic liquids like morpholinium, pyrrolidinium, and piperidinium (m)ethyl sulfate have displayed electrochemical windows wider than those of imidazolium-based ionic liquids by at least 4.5 V.36Extensive studies on cationic degradation mechanisms have shown that the C2 proton in the imidazole ring accounts for the low stability of imidazolium salts in electrochemical applications.37 This is accompanied by the loss in aromaticity and the formation of the first neutral radical. The radical finally gets converted into carbene with the evolution of hydrogen gas (Fig. 2a). However, as shown by Witkamp et al., the formed C2 radical might undergo dimer formation through the coupling of two unpaired radicals or disproportionation to form neutral species (Fig. 2b).38
 | ||
| Fig. 2 Reduction of [C4mim] cation 1 through radical and carbene formation followed by dimer formation or disproportionation, or cage-like structure formation of the [C4mim] radical. Reproduced from ref. 37 with permission from ACS, Copyright [2005]. | ||
Similarly, the most commonly and widely used bis(trifluoromethylsulfonylimide)imide anion is not very stable in some of the applications. MacFarlane et al., while conducting the electrocyclic experiment, observed the reduction of bistriflimide to [NSO2CF3]2−, which they attributed to the presence of two strong electron-withdrawing groups on the nitrogen atom.39 Furthermore, in the reduced species, [NSO2CF3]2−, the LUMO is centered mainly on the S atom. Consequently, the electron will be added there, leading to the weakening of the S–N bond. Subsequently, the anion will be cleaved into a nitrogen-centered radical and an anion, each of which can then further undergo reduction (Fig. 3).
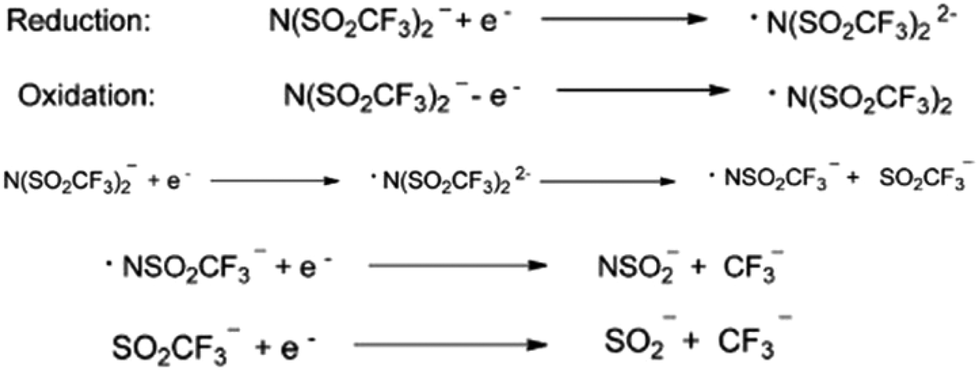 | ||
| Fig. 3 Reduction and oxidation of the bistriflimide anion. Reproduced from ref. 32 with permission from Wiley-VCH, Copyright [2014]. | ||
Researchers have also extensively used dicyanamide anion [N(CN)2]− to achieve low-viscosity ionic liquids. Deacon et al. reported the first electrochemical degradation of this type of anion owing to the dimerization of dicyanamide anion.40
Overall, with electrochemical stability windows in the range between 4 and 7 V, ionic liquids have been broadly used in organic electronics as gate dielectrics for field-effect transistors5,6 and electrolytes for ion transport membranes,7,8 batteries, and flexible electronics.2–4
2.3 Charge transport in ionic liquids
This technique involves assembling the sample in the form of a parallel plate capacitor and applying an oscillating voltage across the sample cell's electrodes (Fig. 4).43 It measures the current response that results. Roughly speaking, the in-phase portion of the response provides information about the sample's resistance, which allows us to determine conductivity, and the out-of-phase portion furnishes information about the sample's capacitance, which allows us to determine the material's dielectric permittivity. By carrying out this measurement at various frequencies, we are able to obtain a comprehensive understanding of the material's response across a broad range of timescales. It is typically convenient to Fourier-transform the measured current and use it to calculate the frequency-dependent complex permittivity and complex conductivity (Fig. 4).16,44
 | ||
| Fig. 4 Characterization of ionic liquids using dielectric relaxation spectroscopy and further fitting the dielectric response to a combination of a power law representing the electrode polarization response, and a Havriliak–Negami (HN) relaxation function. | ||
The dielectric function being responsive to dipolar species furnishes timescales of dielectric relaxation in the material that stems from segmental motion, side-chain orientation, or ion transport in ionic liquids.45–47 Primarily, it is useful to look at the dielectric derivative spectra, which helps remove contributions from ionic conductivity and allows the molecular relaxation to be resolved better.48,49
To extract quantitative information about the timescales of the ionic rearrangement, we fit the dielectric derivative spectra to the combination of a power law that exemplifies the electrode polarization response50 and a Havriliak–Negami (HN) relaxation function,16,44 which is a helpful empirical relaxation function that can catch both symmetric and asymmetric broadening of the relaxation around a centre frequency (Fig. 4). With this fit, we are able to extract the relaxation strength, which is indicative of how powerfully the material can reciprocate on that timescale, as well as the particular frequency of each mode, which is reciprocal of relaxation time.
Rössler and coworkers probed the mechanism of charge transport in BMIM-based room-temperature ionic liquids (RTILs) with varied counter anions by gauging the complex dielectric function over a wide temperature range.51 In their study, they found that RTIL displays an ionic conductor behavior above the glass transition temperature (Tg) and a glassy behavior below it.
Kremer and coworkers have investigated mass and charge transport in ionic liquids based on the imidazolium cation.52 They extracted important molecular parameters including the mobilities and number density of the charge carriers and showed how a similar temperature dependence of mobility gave rise to the Vogel–Fulcher–Tammann (VFT) behavior of conductivity while the number density of charge carriers possessed Arrhenius-type thermal activation.
Furthermore, they uncovered how charge transport and glassy dynamics in imidazole-based ionic liquids are impacted by alkyl side chains.53 Their principal finding was that the structural relaxation rate declines with chain length. They attributed this to increased viscosity in longer chains, so the molecules required more time to relax structurally. Additionally, they observed a two-decade longer delay in structural relaxation than charge transport, due to the proton-conducting nature of imidazole.
2.4 Ionic liquids in action
Ionic liquid-based gels have shown significant advancement in the design of electrochemical and electronic devices such as FET,54 actuators,55,56 batteries,57–59 sensors,60 supercapacitors,61 and solar batteries.62,63Ionic liquids have been successfully used as electron transporters for dye-sensitized solar cells (DSSCs). The I−/I3− redox couple is frequently utilized as an electron carrier in these cells and therefore it is desirable to employ ionic liquids to screen the coulombic repulsion of I−/I3− in ILs.64,65 Grätzel et al. demonstrated the role played by a Grotthuss-like mechanism in the additional accelerated charge transport of an I−/I3− redox couple that arises due to an exchange reaction between the I− and I3− in an IL electrolyte (Fig. 5).65,66 This magnifies the performance of DSSCs containing ILs.67,68
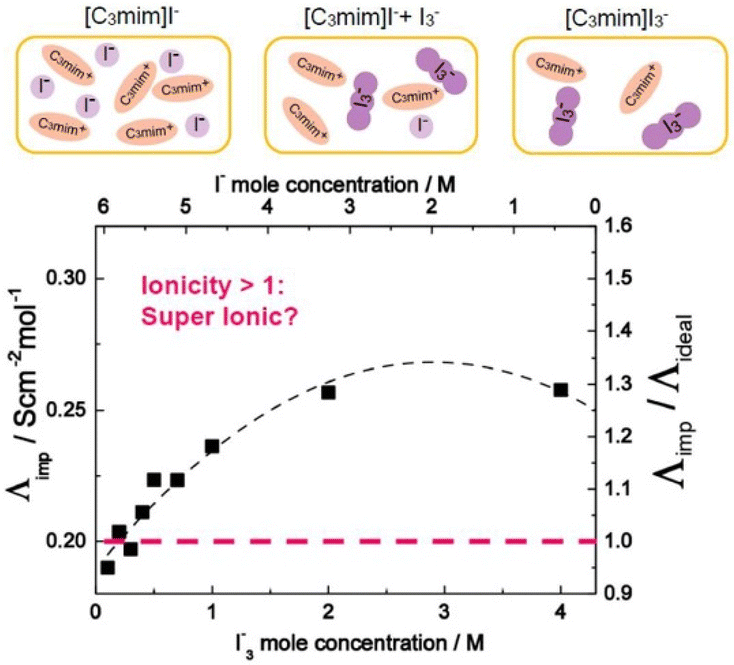 | ||
| Fig. 5 Molar conductivity (Λimp) and ionicity (Λimp/Λideal) of [C3mim]I− with added I2 as a function of I3− (I−) concentration at iso-viscosity state of 0.2 Pa s. Reproduced from ref. 65 with permission from RSC, Copyright [2018]. | ||
Furthermore, it was shown that the DSSC containing an IL electrolyte generated photocurrent that was greater than 80% of that of the cell using a standard organic electrolyte solution.67
Later researchers demonstrated the successful application of polymerized ionic liquid electrolyte for solid-state DSSCs. Interestingly, it was hypothesized that since the charge transport based on exchange reactions requires minimal translational motion of the I−/I3− redox couple, it can take place even in the polymeric phase.69
Ionic liquids have also been studied for use in conjugated polymer electrochemical devices. Li et al. demonstrated that when π-conjugated polymers like polyaniline, polythiophene etc. are electrochemically cycled in imidazolium-based ionic liquids like 1-butyl-3-methylimidazole with BF4− or PF6− anions, they exhibit an enhanced lifetime of close to a million cycles and rapid cycle switching speeds (Fig. 6).70 The ionic liquid was further utilized for the fabrication and functioning of highly stable electrochromic devices and conjugated polymeric mechanical actuators, each of which showed remarkable performance.
 | ||
| Fig. 6 (A) Electrochemical reactions of polyaniline and polypyrrole in the presence of ionic liquid; [BMIM]+[PF6]−. (B) Demonstration of these polymers in electrochemical devices; the transmittance change obtained at 560 nm for a pixel of the same numeric display upon voltage switching between −1.5 V and +1.5 V with the pulse width of 1 s. (C) The charge versus time (dashed line) and ΔT versus time (solid line) curves for a display pixel upon voltage switching between −1.5 and 1.5 V with a pulse width of 1 s. Reproduced from ref. 70 with permission from Science, Copyright [2002]. | ||
This demonstration, highlighting the application of environmentally stable, room-temperature ionic liquids in π-conjugated polymer electrochemical devices, significantly stimulated the further investigation of ionic liquids in electronic devices.
Lee et al. for the first time reported the application of an ionic liquid-based gel in an organic film transistor.71 The gel network, bearing special structural and chemical properties, was a tri-block copolymer, poly(styrene-block-ethylene oxide block-styrene)(SOS) mixed with [BMIM][PF6] (Fig. 7).
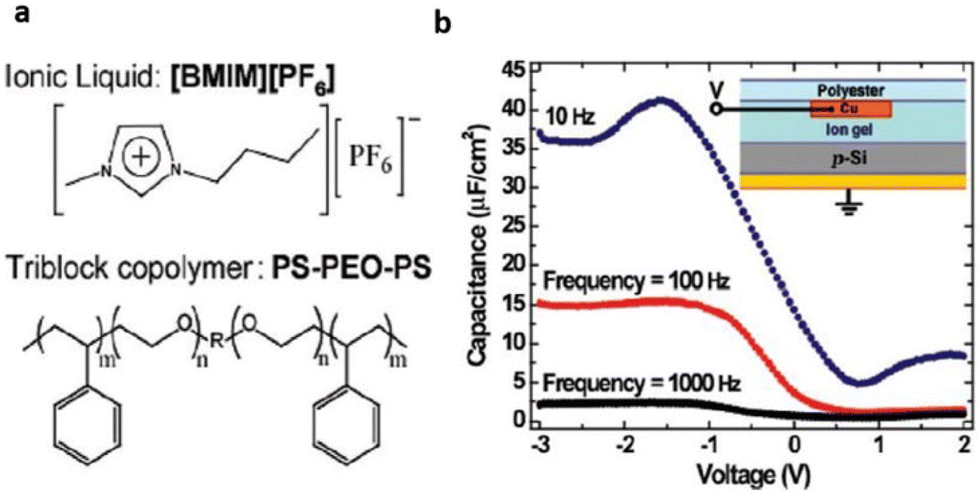 | ||
| Fig. 7 (a) Structure of the ionic liquid and triblock copolymer ion gel components (b) C–V characteristics of a p-Si/ion gel/Cu test structure (see inset) at three frequencies (right). The C–V curves indicate large frequency-dependent hole accumulation in the Si at negative bias on the top contact. Reproduced from ref. 71 with permission from ACS, Copyright [2007]. | ||
The designed ABA-tri-block polymer was successfully employed for constructing various transistors with different requirements. This illustrates the ionic-gel material's good compatibility with organic FETs and, as a result, further clarifies the boundaries of flexible electronic technology with reference to dielectric materials.
Cho et al. combined a photocurable mixture material with a chemically stable ionic liquid [EMIM] [TFSI] to create patterned graphene transistors.72 The designed transistor displayed outstanding polarization response speed, surface capacitance, and stability over time.
However, ILs did not turn out to be safe for in vivo applications. Taking this into consideration, Tang et al. designed biocompatible ion-gate transistor (IGT) constituted of poly(ε-decalactone)-b-poly(DL-lactide)-b-poly(ε-decalactone) (DLD) and [P14][TFSI].73 This composition enabled a minimum capacitance of 2 μF cm−2 with 20% ionic gel, and furthermore the system could perform under low voltage (Fig. 8).
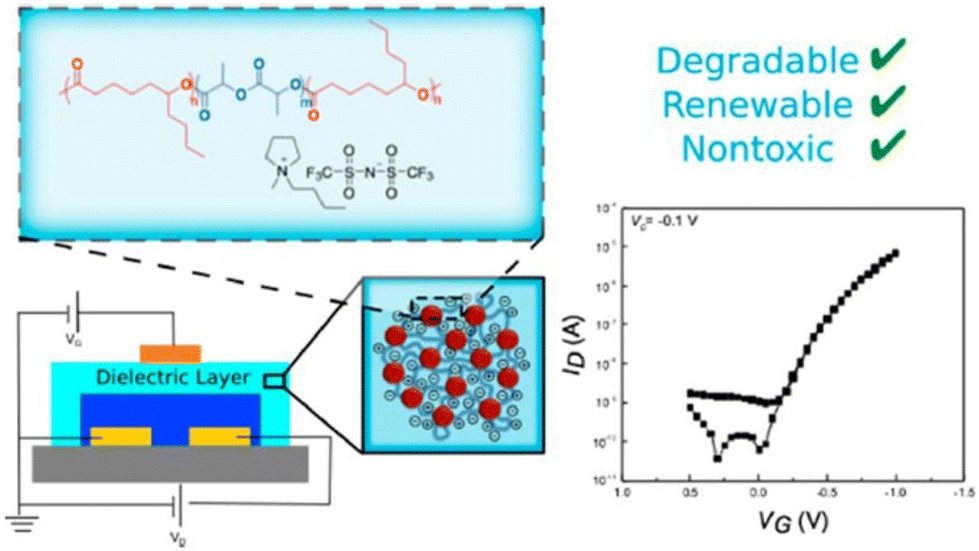 | ||
| Fig. 8 Schematic representation of a degradable and biocompatible ion gels from a renewable ABA triblock polyester and ionic liquid. Reproduced from ref. 73 with permission from ACS, Copyright [2017]. | ||
A mobility of 1 cm2 V−1 s−1, an Ion/Ioff ratio of 105, and low hysteresis could be obtained by using P3HT as the channel layer in the composed IGT, which makes it promising for printed electronic applications. These IGTs have been further used for fabricating neuromorphic devices, especially synaptic transistors.74,75
The use of organic electrolytes in lithium batteries is restricted by their high temperature volatility, decomposition, and propensity to crystallize.76,77 However, researchers have demonstrated the use of additives in electrolytes to overcome this issue. Wang et al. employed 1% 1-ethyl-3-methylimidazolium tetrafluoroborate (EMIBF4) in nanomaterial-based lithium batteries as a low-temperature electrolyte additive that increased the capacity retention from 82% to 93% at −10 °C after 150 cycles (Fig. 9).78
 | ||
| Fig. 9 Electrochemical performance characteristics of the NCM523/graphite full cells with different amounts of the EMI-BF4 additive. (a) dQ/dV curves of the NCM523/graphite full cells. (b) Cycle stability and coulombic efficiency of the NCM523/graphite full cell at a voltage range of 2.75–4.2 V at room temperature. (c) Discharge curves of the NCM523/graphite full cells at the temperatures of −30 °C, −20 °C, and −25 °C at 0.5C. (d) The cycle stability and coulombic efficiency of the NCM523/graphite full cells at a voltage range of 2.75–4.2 V at −10 °C. Reproduced from ref. 78 with permission from Elsevier, Copyright [2019]. | ||
Interestingly, the presence of BF4− on the surfaces of graphite anodes and nanomaterial cathodes aided in the development of low-impedance interfacial films, which improved the batteries’ performance at low temperatures.
Because ILs have medium conductivity and are highly viscous at room temperature, they have a negative effect on battery cycling in lithium batteries that use them as electrolytes. To alleviate this issue, researchers have combined ILs with thermally stable organic solvents. Dong et al. designed an E60 electrolyte using 0.5 M LiDFOB in PP14TFSI and sulfolane (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 wt) (Fig. 10). This electrolyte performed significantly better than LiPF6 in carbonate electrolyte, exhibiting a wider electrochemical potential window in the temperature range of 55 °C to 70 °C.79
:2 wt) (Fig. 10). This electrolyte performed significantly better than LiPF6 in carbonate electrolyte, exhibiting a wider electrochemical potential window in the temperature range of 55 °C to 70 °C.79
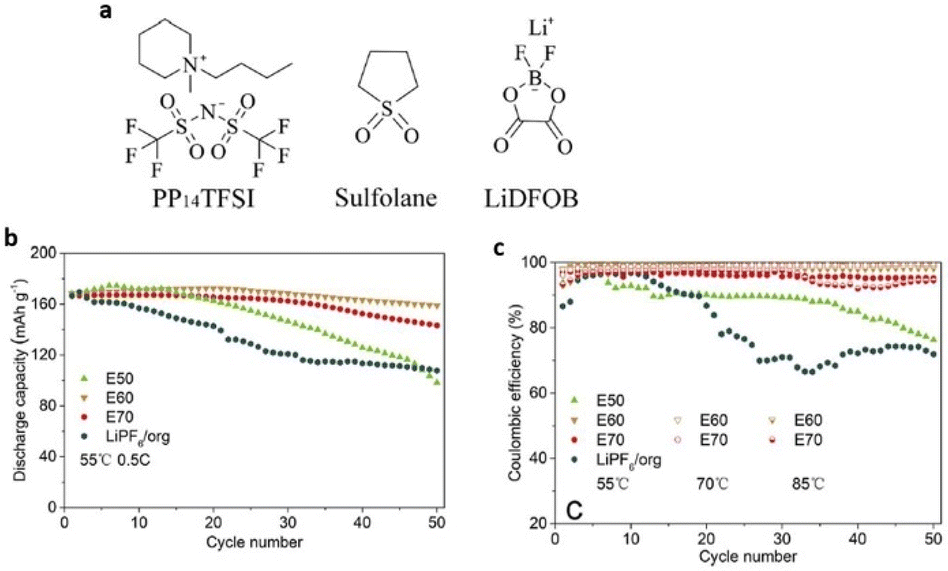 | ||
| Fig. 10 (a) Chemical structures of 1-methyl-1-butylpiperidinium bis(trifluoromethanesulfonyl)-imide (PP14TFSI), sulfolane and lithium difluoro(oxalato)borate (LiDFOB). (b and c) Cycling stability and coulombic efficiency and rate performance of Li/Li-rich half cells at 55 and 70 °C at 0.5C and 85 °C at 1C under a voltage range of 2.0–4.6 V. Reproduced from ref. 79 with permission from Elsevier, Copyright [2018]. | ||
Additionally, the combined E60 electrolyte creates a solid cathode electrode–electrolyte interface that is extremely thermally stable, provides reduced resistance to safeguard the cathode material structure, and preserves cycling stability at high voltage and high temperature.
To address the poor ion-transport properties of ionogels resulting from the formation of ion pairs in lithium-secondary batteries, researchers have modified the surface of a polymeric matrix with a zwitterionic surfactant to enable stabilized ionogels. The characteristic feature of these ionogels is the alignment of the surfactant dipole structure towards the IL-based electrolyte, promoting effective dipole interactions and thus facilitating lithium salt dissociation in IL-based electrolytes. Yoo et al. synthesized such an ionogel by photocuring a polymerizable monomer, 1,12-dodecanethiol dimethacrylate, DDDA, an imidazolium-based electrolyte, [EMIM][TFSI] with LiTFSI, and a zwitterionic surfactant composed of a long alkyl chain, a negatively charged perfluoroalkylsulfonylimide group, and a positively charged imidazolium group (Fig. 11).80 Notably, the designed zwitterionic surfactant-stabilized ionogel exhibited significant enhancement in lithium-ion conductivity. As a result, there was an improvement in the lithium plating/stripping behavior, with the best rate capability of 116.8 mA h g−1 at 1C and cycling performance with a capacity fading rate of 0.0669% per cycle and an initial discharge capacity of 148.9 mA h g−1.
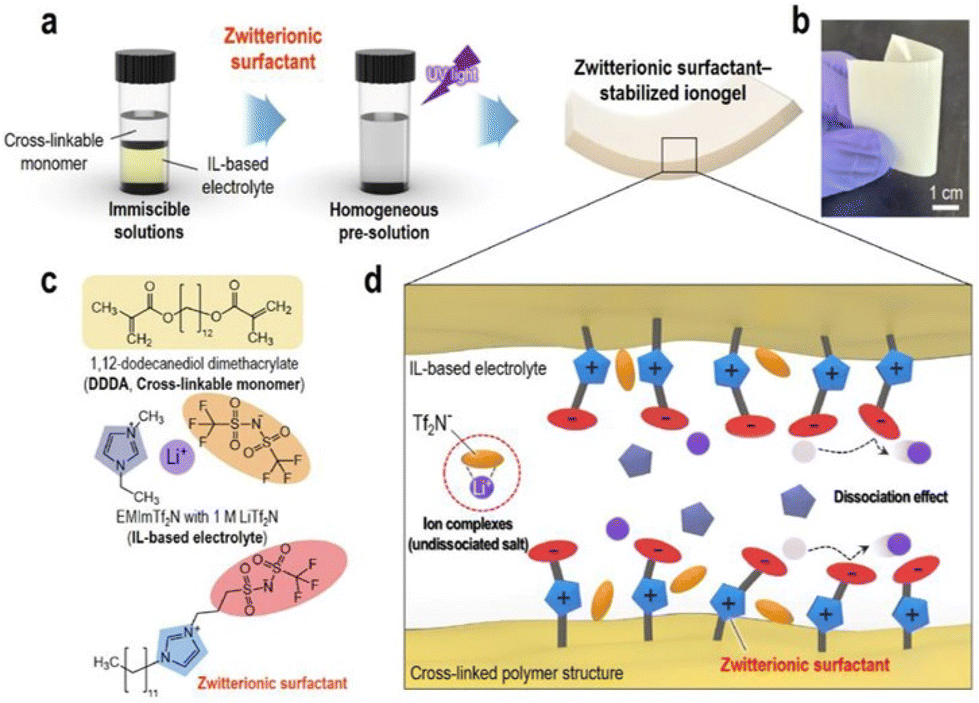 | ||
| Fig. 11 (a) Schematic preparation and (b) representative photograph of zwitterionic surfactant-stabilized ionogels. (c) Chemical structures of pre-solution components. (d) Schematic of ion-dipole interactions within zwitterionic surfactant-stabilized ionogel electrolytes. Reproduced from ref. 80 with permission from Elsevier, Copyright [2023]. | ||
Ionic liquids have also been used for fabricating supercapacitors that are energy reservoirs. Redox reactions or electric double-layer formation at the electrode surface enables supercapacitors to store energy.81–85 Supercapacitors have conveniently replaced batteries as an alternative renewable energy source in electronics, laptops, mobiles, etc.86,87
Using LiCl in EMIMBF4 ionic liquid hybrid as the electrolyte and N-doped micropore-dominant carbon (N-MPC) electrode as the electrode, Gao et al. recently designed a dual-ion battery-supercapacitor hybrid device (DIB-SCHD) (Fig. 12).88 A mass capacitance of 374 Fg−1 at a high voltage of 3.5 V was made possible by this synergism.
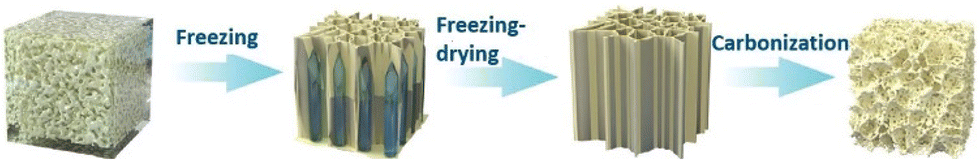 | ||
| Fig. 12 Schematic illustration of the fabrication of N-MPC. Reproduced from ref. 88 with permission from Wiley-VCH, Copyright [2021]. | ||
A remarkable specific power of 22834 W kg−1 at 77 W h kg−1 and a further significantly high specific energy of 208 W h kg−1 at 1144 W kg−1 were achieved. These values exceeded the current state of the art. Mechanistic studies highlighted the contribution of the anions BF4− and cations EMIM+ to the capacitor-type charge storage via surface accumulation on porous carbon electrodes. In contrast, Li+ and Cl− intercalated into the graphitic carbon layers, executing a battery-type charge storage mechanism. This innovation might serve as a promoter of novel-type battery-supercapacitor hybrid devices.
Ionic liquids have been successfully used for crafting actuators. In ionic polymer actuators, the movement of ions in the electrolyte membranes occurs upon voltage application, and typically low voltage under 5 V displays large deformation.89,90 These electrolyte membranes are mostly hydrated polyelectrolyte membranes. Owing to the solvent evaporation from the electrolyte membranes, the durability of conventional ionic polymer actuators is low in an open atmosphere. This obstructs the function of the actuator since the solvent is critical for the movement of ions. To alleviate this issue, researchers have developed ionic polymer actuators employing ILs that can perform in an open atmosphere as well as under vacuum.91,92 Therefore, polymers have been blended with ILs and further used as additives for effective ion conduction in ionic polymer actuators.
In efforts towards achieving high ionic conductivity, researchers have successfully incorporated ionic liquids into a bifunctional polymer for fabricating solid-state polymer electrolytes. Park et al. recently designed an innovative solid-polymer electrolyte established on a bifunctional poly(3-hydroxy-4-sulfonated styrene) (PS-3H4S) polymer for its successful application in soft actuators and lithium–metal batteries (Fig. 13).93 The incorporation of the ionic liquid led to the formation of interconnected rod-like channels within the glassy polymer matrix. This facilitated proton hopping through the glassy polymer matrix resulting from the efficient decoupling of ion relaxation from polymer relaxation. To enable such a solid-polymer electrolyte, PS-3H4S polymer was doped with an imidazolium-based ionic liquid and bis(trifluoromethanesulfonyl)imide (HTFSI). This weakened the ion-clustering behavior and doubled the proton dissociation by suppressing electrostatic interactions of glassy polymer matrix with embedded ionic liquid.
 | ||
| Fig. 13 (a) Schematic representation of PS-3H4S polymer used in the study. (b) Schematic illustration depicting bending deformation of ionic polymer actuators under an applied voltage. (c) Conductivity–modulus relationship of PS-3H4S-2.0, PS-3M4S-2.0, and PS-4S-2.0. (d) Bending displacements of PS-3H4S actuator at ±0.5, ±1 and ±2 V, compared with PS-4S counterparts. Reproduced from ref. 93 with permission from Wiley-VCH, Copyright [2022]. | ||
The exceptional actuation performance of the polymer/ionic liquid composite electrolyte was demonstrated by its high storage modulus of roughly 100 MPa and ionic conductivity of 10−3 S cm−1 at ambient temperatures.
Therefore, a number of critical factors, such as the kind of ILs, the quantity of IL loadings, and the kind of polymers, have a substantial impact on the actuators’ performances.94 The incorporation of ILs into the ionic polymer layer of electro-active actuators reduces Young's modulus, facilitates ion transport, and enhances the electrochemical stability of the polymer electrolytes.70,95 Consequently, this significantly improves the strain, response time, and lifetime of actuators, thus improving their performance.
2.5 Ionic liquid crystals
Ionic liquid crystals have been the subject of scrutiny for numerous years.96 They are characterized as a subclass of liquid crystals bearing ionic groups in their molecular structures and possess oriental order.97–99 Investigations have been conducted on ionic liquid crystals exhibiting diverse molecular formations, encompassing those with rod-like,100 wedge-like,101 and disc-like structures.102 Interestingly, these ionic liquid crystals display structure-dependent functions and exhibit properties that are unachievable by regular ionic liquids with disordered structures.103 These have been meticulously generated and recorded in previous evaluations.104 Nevertheless, this present study delves into only a limited number of these ionic liquid crystals.Researchers have incorporated liquid crystal properties into zwitterionic systems, with both cations and anions linked together through the covalent bonds, to create anisotropic matrices capable of selectively conducting target ions.105 With the goal of arranging zwitterion/HTf2N systems into a one-dimensional nanochannel framework, researchers have developed phosphonium-based zwitterions (3n) that possess three long alkyl chains (n = 6 and 8). These zwitterions form uniform adducts when combined with equimolar amounts of HTf2N (Fig. 14). The formation of these specific adducts is attributed to the favorable interactions between the soft phosphonium cation of zwitterions 3n and the soft Tf2N anion. In the presence of water, this adduct displays lyotropic columnar liquid-crystalline phases.106
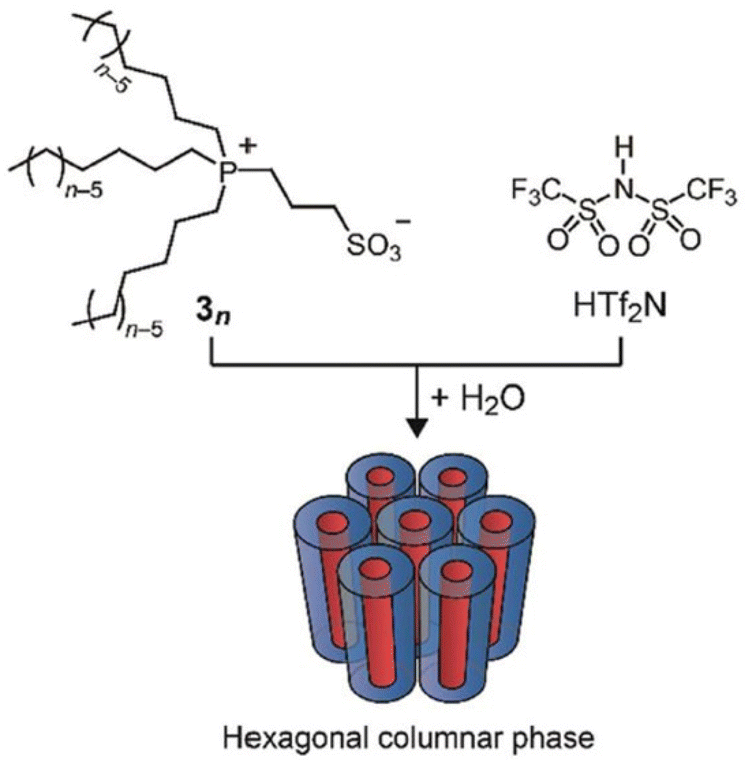 | ||
| Fig. 14 Zwitterionic compounds 3n consisting of a phosphonium cation and a sulfonate anion. An equimolar mixture of 3n and HTf2N forms a homogeneous complex that exhibits lyotropic columnar liquid crystalline phases in the presence of water. Reproduced from ref. 104 with permission from RSC, Copyright [2011]. | ||
These materials possess the intriguing capability of arranging their columnar axis in parallel to the direction of an exerted mechanical force, thereby enabling the execution of 1D anisotropic ion conduction measurements. Interestingly, the ascertained ionic conductivity was found to be 7.9 × 10−2 S cm−1 at a temperature of 25 °C. This numerical value is approximately 40-fold greater than that exhibited by columnar liquid crystal samples in a polydomain state. Consequently, researchers deduced that protons have the capacity to traverse the 1D water nanochannels with ease.
Researchers have integrated ionic liquid crystals into conjugated polymer systems to fabricate molecular materials constituting aligned thin films of mixed electron-ion conducting polymers. Such macromolecular architectures are envisaged to be of great potential in electrochemical devices.
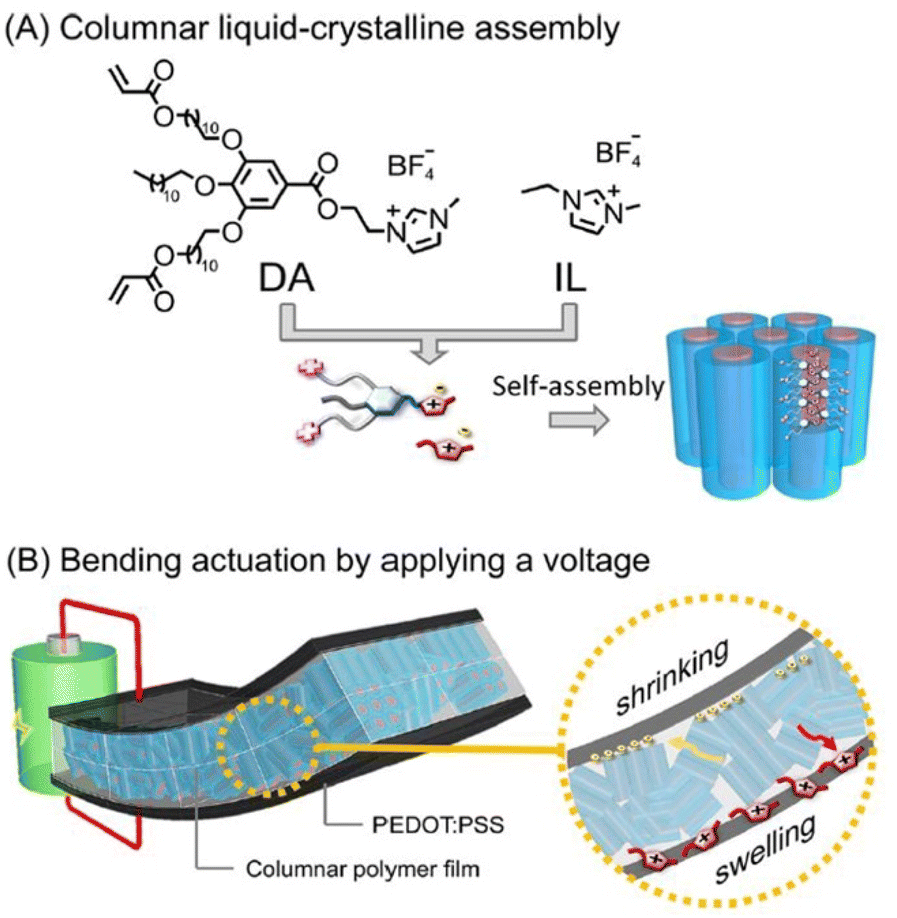 | ||
| Fig. 15 (A) Molecular structures of the wedge-shaped imidazolium compound with diacrylate groups (DA) and ionic liquid [EMIM][BF4] (IL) and their self-assembly into the columnar liquid-crystalline phase forming 1D ion channels. (B) Illustration of the electric-field-driven bending actuation of the three-layer membranes composed of the columnar liquid crystalline polymer electrolyte and PEDOT:PSS electrodes. Reproduced from ref. 107 with permission from ACS, Copyright [2022]. | ||
The designed nanostructured polymer membrane actuator displayed significant bending displacement rapidly and large force generation in contrast to the non-ordered actuator fabricated using the amorphous membrane that was generated by photopreservation of diacrylate in the isotropic liquid phase.
Recently, Osuji and coworkers incorporated biphenyl-containing 1-alkyl-3-methylimidazolium salts with bromide counter ions into conjugated sulfonated poly(alkyl thiophenes) backbones (Fig. 16).108 The ionic liquid crystals of imidazolium salts functioned as ionic conduction elements in the side chains through ionic bonding in these thermotropic liquid crystalline supramolecular conjugates.
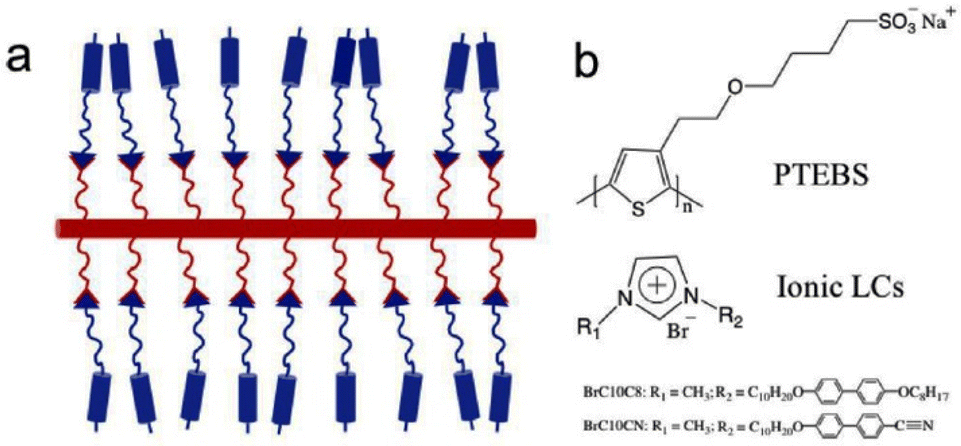 | ||
| Fig. 16 (a) Schematic of supramolecular complex of conjugated polyelectrolytes coupling with ionic LCs, and (b) Chemical structures of conjugated polyelectrolytes and ionic LCs. The red and blue rods represent a conjugated backbone and mesogen unit, respectively; the red and blue curves represent hydrocarbons; the red and blue triangles represent ionic groups. Reproduced from ref. 108 with permission from Elsevier, Copyright [2022]. | ||
Furthermore, the amorphous polythiophene exhibited a semicrystalline behavior upon coordination with ionic liquid crystals. Under appropriate conditions, these supramolecular complexes exhibited bulk alignment under mechanical shear and application of magnetic field. Thus, in emerging electrochemical devices, this study furnishes a facile way through supramolecular complexation for regulating the structural alignment of mixed electron-ion conducting polymers.
Recently, researchers have demonstrated lyotropic liquid crystals (LLC) as potential ion conductors for application in energy devices. Tan and coworkers synthesized lamellar nanostructured quasi-solid LLC electrolytes by mixing an alkylimidazolium compound with an appropriate liquid electrolyte (Fig. 17).109 The liquid electrolyte comprised tetra methylammonium 5-mercapto-1-methyltetrazole, di-5-(1-methyltetrazole)disulfide, 4-tert-butylpyridine, and LiClO4. The layered regulation of liquids in these LLCs not only significantly improved the device stability but also enhanced the charge transport and the ion conduction, with the observed conductivity value of 4.2 × 10−3 S cm−1 at 20 °C and device efficiency of 6.1% at 25 °C by merely consuming 40% liquid electrolyte at ambient temperature. This work signifies the potential of LLCs to overtake the state-of-the-art liquid electrolytes for sustainable energy devices.
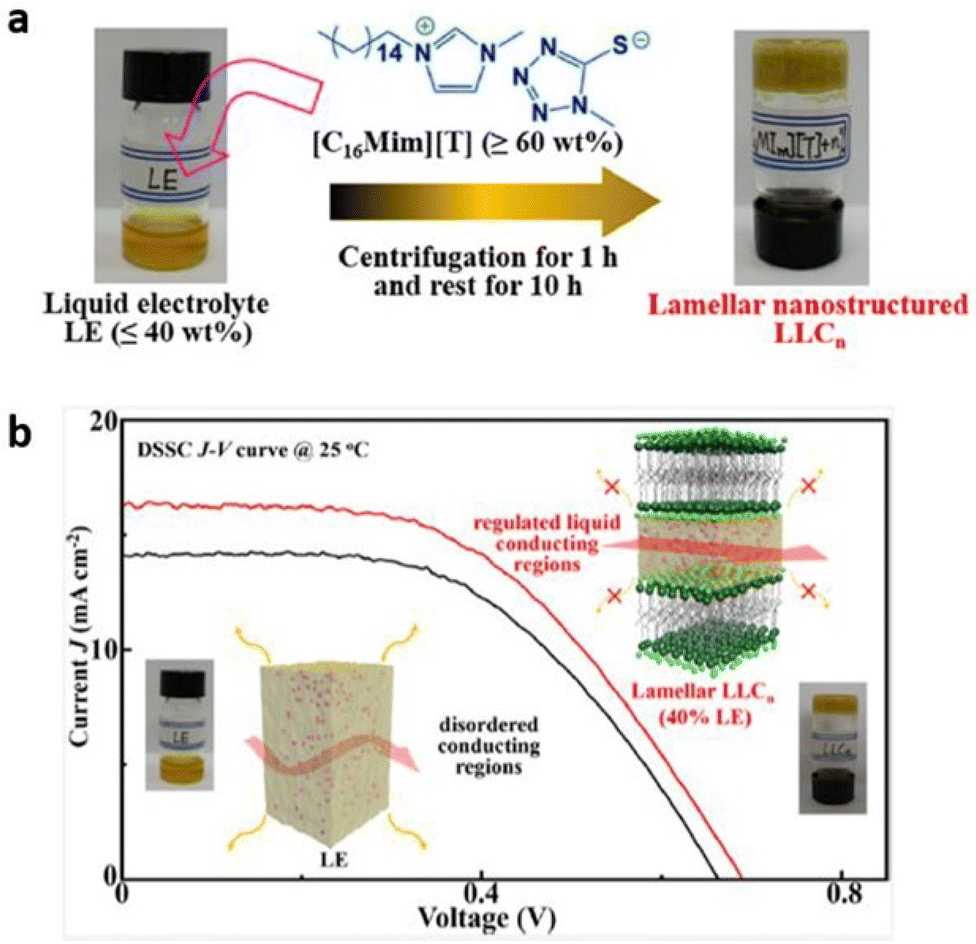 | ||
| Fig. 17 (a) Preparation of LLCn from organic LE and [C16Mim][T]. (b) Schematic representation of lyotropic liquid crystals inducing liquid–fluid lamellar conducting highways. Reproduced from ref. 109 with permission from ACS, Copyright [2022]. | ||
Researchers have designed novel nanostructured columnar ionic liquid crystal/polymer composite actuators that have exhibited significantly fast response at low voltages. Recently Yoshio et al. reported such an actuator possessing a free-standing stretchable membrane electrolyte composed by blending a zwitterionic rod-like molecule, an ionic liquid, and poly(vinyl alcohol) (Fig. 18).110 The obtained ionic polymer membrane displayed significant enhancement in ionic conductivity, which is a consequence of the free movement of dissociated ions via a continuous 3D ionophilic matrix. Interestingly, its capability to demonstrate fast response at low voltages arises owing to its stable contact formation with conducting polymer electrodes of poly(3,4-ethylenedioxythiophene) doped with poly(styrenesulfonate) (PEDOT:PSS). Furthermore, this actuator based on supramolecular self-assembly was demonstrated to be promising for advanced applications in optical devices by laminating it with an aluminium-coated flexible polymer film.
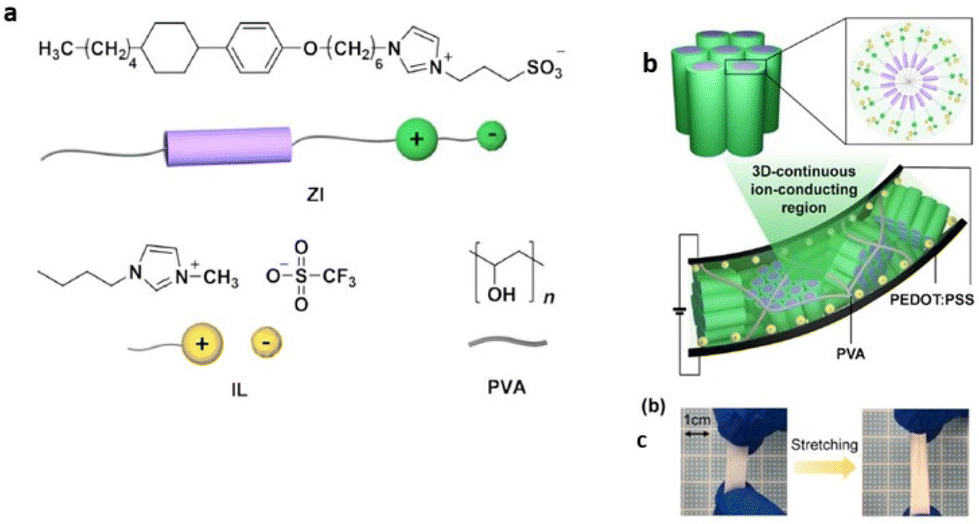 | ||
| Fig. 18 (a) Chemical structures and illustrations of the zwitterionic liquid-crystalline compound (ZI), ionic liquid (IL), and poly(vinyl alcohol) (PVA). (b and c) Schematic illustration of the deformed ionic electroactive polymer actuator composed of the hexagonal columnar liquid-crystalline polymer composite electrolyte membrane of ZI/IL/PVA sandwiched between two PEDOT:PSS electrode layers under an applied DC electric field at 70% RH. Reproduced from ref. 110 with permission from ACS, Copyright [2022]. | ||
Interestingly, the formation of phase-separated morphologies in IL-embedded polymers has cleared the roadblocks for developing advanced actuators using block copolymer electrolytes by elevating the ion transport rate using nanometre-sized ionic channels.56,111,112
Recently, researchers have successfully employed the approach of supramolecular columnar LC self-assembly of amphiphilic monomers and the IL and in situ photopolymerization. Yoshio and coworkers constructed a liquid-crystalline polymer electrolyte membrane using wedge-shaped amphiphilic molecules bearing two ionic heads and a lipophilic tail that was further blended with low amounts of ionic liquid (Fig. 19).113 The crafted membrane was mechanically tough and contained three-dimensionally interconnected ionic pathways, enabling its potential as an electrolyte layer in soft robotic actuators.
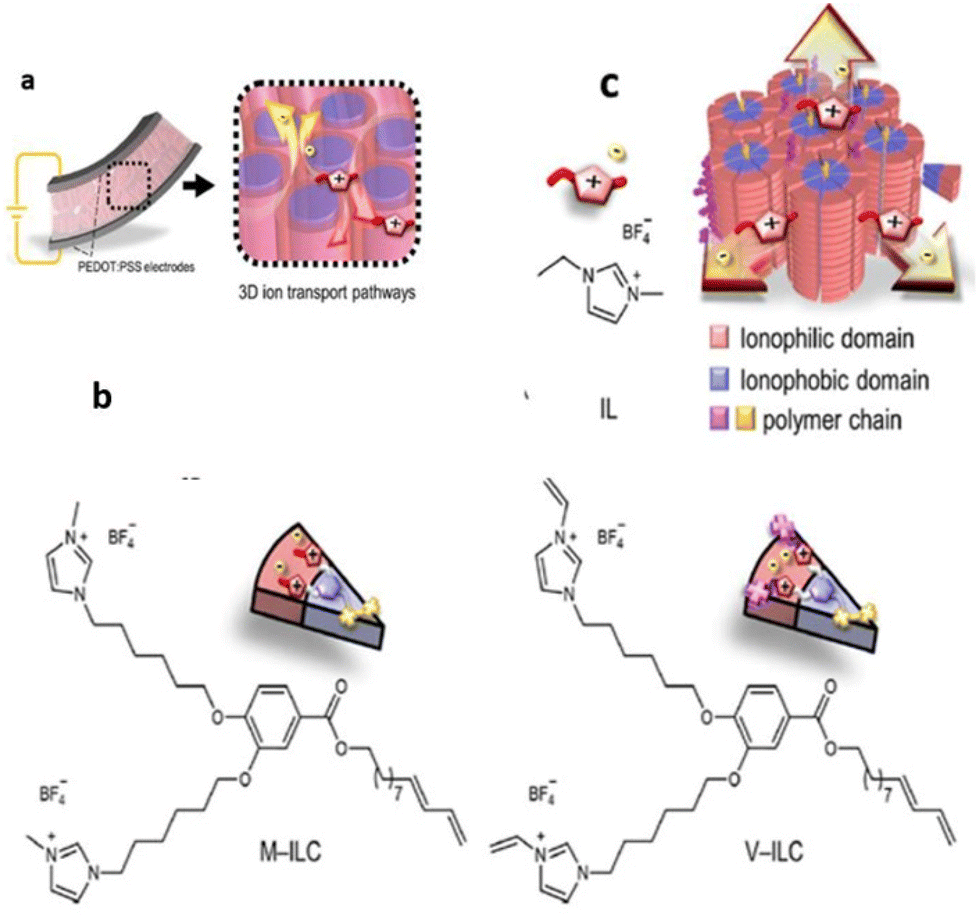 | ||
| Fig. 19 (a) 3D ion conductive Colh LC polymer membrane electrolyte with interconnected ionic paths, which is sandwiched between a pair of PEDOT/PSS electrodes to fabricate electromechanical actuators. (b) New polymerizable Colh ionic LC molecules: the methyl imidazolium salt MILC and the vinyl imidazolium salt V-ILC. Structure of an IL, 1-ethyl-3-methylimidazolium tetrafluoroborate (IL). (c) Schematic illustration of 3D continuous ion transport in a nanosegregated columnar LC structure (red: ionic parts and blue: insulating parts) formed by self-assembly of the wedge-shaped ionic amphiphiles and the IL. Reproduced from ref. 113 with permission from ACS, Copyright [2023]. | ||
To realize such a membrane, methyl-imidazolium salt M-ILC and its analogous vinyl imidazolium salt V-ILC were used, and further only 5.6 wt% of 1-ethyl-3-methylimidazolium tetrafluoroborate IL was chosen as a source of mobile ions for the 3D interconnected ionic pathways. This membrane displayed efficient actuation performance with a high bending strain of 0.8% at 2 V and 0.01 Hz and a wide frequency response of 0.01–20 Hz. It is noteworthy that this LC membrane exhibited a long-term stability of >16000 cycles, and even without the operation of the bias voltage, its shape memory effect sustained the blocking force.
2.6 Protic ionic liquids
A subset of ionic liquids known as protic ionic liquids are easily produced by mixing a Brønsted acid with a Brønsted base.114 This combination is accompanied by a neutralization reaction. There exists a substantial multitude of possible combinations of acids and bases that can be employed to fabricate novel protic ionic liquids. The proton transfer from the acid to the base, which creates proton-donor and acceptor sites that can be used to create a hydrogen-bonded network, is one of the characteristics that set protic ionic liquids apart from others. Several reviews have encompassed the established scope of protic ionic liquids, documenting their physiochemical attributes and contexts in which they have been employed.104,115 In the present review, however, only a few of the protic ionic liquids are discussed.Protic ILs containing TFSI anion, alkylammonium cations, and imidazolium cations have been found to possess high thermal stability with decomposition temperatures >200 °C.116,117 The ones composed of carboxylate anion, especially formate, display lower thermal stability due to a plausible condensation reaction leading to amide bond formation.118
Protic ionic liquids containing cations other than that of imidazolium and pyridinium cations are also known. These cations are assessed based on the ionic conductivity and electrochemical activity of protic ionic liquids for their effectiveness in fuel cells.119 Extensive research has been undertaken to identify protic ILs that fulfill the aforementioned requirements, resulting in the discovery of diethylmethylammonium trifluoromethanesulfonate ([dema][TfO]) as an exceptional protic IL (Fig. 20).120
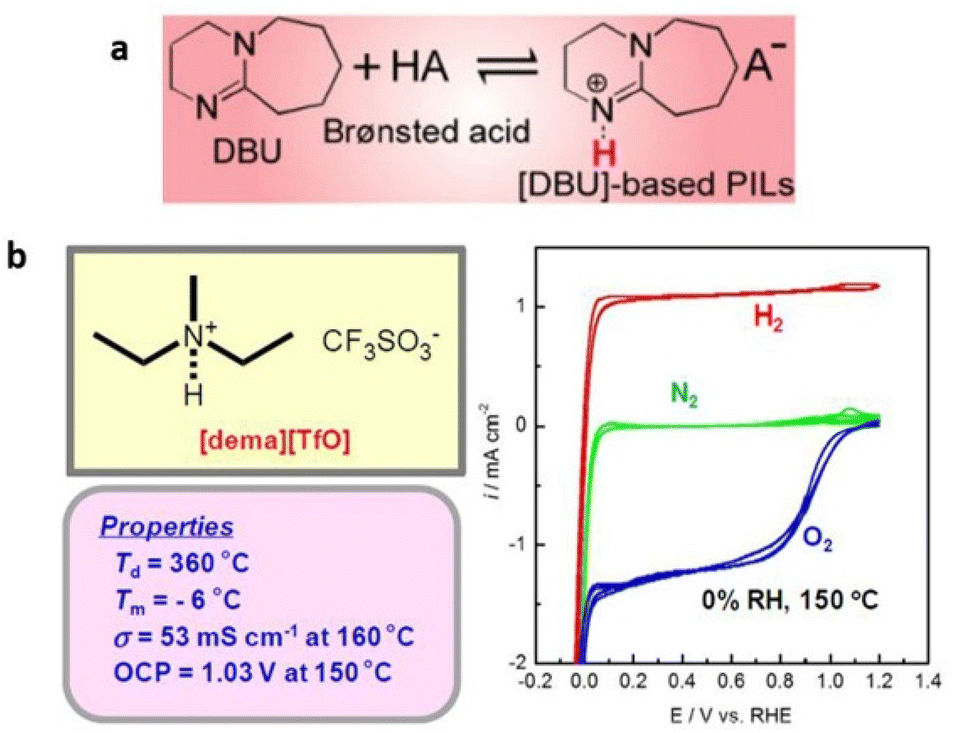 | ||
| Fig. 20 (a) Thermal stability and ionicity for different protic ILs based on a super-strong base, DBU, as a function of ΔpKa. Reproduced from ref. 121 with permission from RSC, Copyright [2019]. (b) Bulk and electrochemical properties of [dema][TfO]. The cyclic voltammograms (right) are recorded under N2, H2, O2 bubbling conditions at a Pt wire working electrode equipped with a platinized Pt wire. A reference electrode, RHE, is a Pt wire in H2 bubbling conditions, placed close to the working electrode through a Luggin capillary. Reproduced from ref. 120 with permission from RSC, Copyright [2007]. | ||
This particular IL exhibits a remarkably high decomposition temperature, a low melting point (Tm), substantial ionic conductivity through the migration and diffusion of the [dema]+ by vehicular mechanism, and notable electrochemical activity.
Protic ILs containing a super-strong base, 1,8-diazabicyclo-[5,4,0]-undec-7-ene (DBU), have also been synthesized by its neutralization reaction with various Brønsted acids (Fig. 20).121 The synthesized protic ILs typically had melting temperatures below 100 °C. Furthermore, these protic ILs displayed remarkable thermal stability up to 450 °C under inert conditions. However, they displayed lower iconicity when compared with aprotic ILs, and this was attributed to the hydrogen-bonding interactions among the NH protons in [BH]+ and the anions, as well as the increased specificity of these interactions.
Due to the presence of these hydrogen-bonding interactions, the interaction energies of the stable ion pairs in a few protic ionic liquids exceed that of their aprotic ILs counterparts with the same structural isomers as the protic ILs by approximately 40 kJ mol−1. Furthermore, the specificity of these interactions also increases in the case of the protic ILs.
Protic ionic liquids with low lithium coordination numbers furnish all the natural advantages of aprotic ionic liquids for lithium-ion batteries.122,123 Furthermore, with dissociable protons, these are of great potential application as electrolytes in fuel cells,116 enabling the use of anhydrous conditions and thus the operation temperature can exceed 100 °C.124,125 Some of the applications of protic ionic liquids are discussed in the following section.
Using protic ionic liquids, researchers have designed polymeric membranes analogous to Nafion. Tigelaar et al. designed such a polymer membrane composed of sulfonated aromatic backbones that were interconnected with diamine-terminated PEO oligomers.127 This membrane was designed to increase the amount of ionic liquid in the membrane, in contrast to Nafion where the perfluorinated nature was decreasing the uptake. Imidazolium-CF3SO3H was used to achieve a significantly high ionic conductivity of 50 mS cm−1 at high temperatures by soaking the polymer membranes in a 1:1 mixture of protic ionic liquid and water. Researchers have further successfully demonstrated the application of protic ionic liquid-based crosslinked polymer electrolytes to enable solid electrical double-layer capacitors and solid lithium–metal batteries capable of exhibiting great performance at room temperature. Gerbaldi and coworkers prepared such a polymer electrolyte encompassing photocrosslinked PEO, 1-butylpyrrolidinium bis(trifluoromethanesulfonyl)imide, and lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) (Fig. 21).128
 | ||
| Fig. 21 (A) Schematics of materials, polymer electrolyte preparation, and crosslinking at different magnifications with indication of the temperature range of stability. (B) Voltage profile versus specific capacity during CC cycling of a Li/PEO_HPyr-VC/LFP cell at C/20 and C/10 rate and RT. (C) Specific capacity and coulombic efficiency versus cycle number upon CC cycling of the proof-of-concept lab-scale solid like Li/PEO_HPyr-VC/LFP Li–metal cell in which the surface of the lithium–metal electrode was wetted with few drops of a solution of PyrH4TFSI and LiTFSI (having a PIL:salt molar ratio equal to 4:1) added with 10% by weight of VC prior to coming into contact with the crosslinked PEO_HPyr polymer electrolyte. Reproduced from ref. 128 with permission from Wiley-VCH, Copyright [2020]. | ||
With its broad thermal and electrochemical stability and high ionic conductivity, this protic ionic liquid-based crosslinked polymer electrolyte was appealing for energy storage devices. Utilizing this solid electrolyte in a laboratory-scale lithium metal cell equipped with lithium iron phosphate cathodes resulted in a noteworthy 150 mA h g−1 capacity at C/20 and allowed for reversible cycling at various current rates in ambient conditions.
3 Journey from ionic liquids to polymerized ionic liquids
3.1 Limitations of ionic liquids for device applications
Ionic liquids have limited applicability in many practical systems because both their liquids and solids suffer from issues of high viscosity and leakage of dissociated ions.129–131 For applications of ILs in organic electronic devices, film-like ion conductive materials are typically favoured from the standpoint of processing, treatment, and packaging.130,131 To address this issue, researchers have incorporated polymerizable groups into ILs and subsequently polymerized these groups, forming polymerized ionic liquids (PILs).14,23 This alleviates the stability and leakage issues in electronic devices. Further immobilizing the ions by covalently linking them to a polymeric backbone enables the transference number of the corresponding ion to approach unity.132 PILs bring a broad range of new possibilities into consideration for the design of a new generation of advanced organic electronics with entirely new sets of features and expectations.3.2 Polymerized ionic liquids
000 g mol−1.136 Polymerized ionic liquids are made by a variety of radical polymerizations, like atom transfer radical polymerization,137,138 nitrous oxide-mediated polymerization,139 and reversible addition–fragmentation transfer polymerization.140–143 Bouchet et al. synthesized a triblock copolymer PSTFSI-Li-b-PEO-PSTFSI-Li by nitroxide-mediated polymerization using a PEO precursor, and meticulously maintained an optimum balance of PSTFSI-Li and PEO to achieve a desirable Li-ion conductivity.144
PILs have also been extensively synthesized by chemical modification of the chain via a post-polymerization processing technique.145 In this approach, the starting point is the uncharged neutral polymer in contrast to the corresponding IL monomer. Consequently, the obtained PIL possesses the structure of the starting polymer and significant flexibility to undergo post-polymerization modification to achieve a specific reactivity. Researchers have also attempted to synthesize PILs with a high specific surface area that is desirable for certain applications by crosslinking the PIL chain with another polymer.146 In general, the synthesis of cationic PILs has been less challenging in contrast to the anionic PILs, thus cationic PILs have been widely investigated.
Unlike commercially available membranes based on ionomers like Nafion that show a drastic drop in ionic conductivity at 90 °C, PILs exhibit high ionic conductivity of 90 mS cm−1 at higher temperatures of 100 °C.149 Researchers have also applied PILs for stabilizing another sister class of conducting polymers or their dispersions over graphene structures to boost their electrochemical properties.150–153 This strategy has been employed to facilitate the interaction of conductive polymers and carbon nanotubes.154 This is extremely beneficial for solar cell materials, as significantly high efficacy, as great as 8%, for energy conversion can be achieved.155–159
PILs have been used to functionalize graphene for improved electrochemical performance.160 This results from the enhanced electrochemical accessibility of the electroactive material, as the electrochemical reactivity of carbon atoms is reduced owing to the flat structure of graphene.161,162 Additionally, this can improve the electrode wettability and electrode/electrolyte interface structure for superior affinity with the electrolyte.
PILs with a soft polymer matrix can be blended with various materials to improve the optical properties of the devices. Researchers have fabricated efficient luminescent materials with a broad range of optical properties by facilitating the bond formation of the light-emitting precursors with the PILs.163–166 PILs with both hydrophobic and hydrophilic behavior aid in regulating the growth mechanism for the formation of nanoparticles. This is of specific significance for optical materials due to the large active surface area, and furthermore the optical properties are polished because of the ion interactions.167–170
PILs can also serve as good hosts for electroactive materials due to their ion-rich medium and electrochemically accessible matrix.157–159 PILs have therefore been used in catalytic systems for sensing applications for the detection of various species.171–176
However, the low ionic conductivity of polymerized ionic liquids has been a concern for their application in electronic devices.177–179 Therefore, over the years, researchers have made rigorous efforts to enhance the mobile ion content and improve the ionic conductivity of PILs. Thus, scrutinizing the mechanism of ion transportation in PILs is critical for uncovering the factors that can be manipulated to amplify the ionic conductivity in PILs.
3.3 Ion transport in polymerized ionic liquids
The low ionic conductivity of polymerized ionic liquids due to the small fraction of mobile ions and sluggish local dynamics of polymer chains limits their application in electrochemical devices.180,181 Several questions regarding the specific interactions that influence the properties of these ion-containing polymers remain unanswered. Therefore, it is essential to comprehend these particular interactions that affect molecular configurations and ion transport in polymerized ionic liquids in order to develop these materials for use in devices. Analogous to ILs, researchers have also rigorously investigated PILs by BDS to illuminate factors affecting ionic conductivity and elucidate the mechanism of ion transport in PILs.16 The ionic conductivity is a measure of the bulk ion transport in the polymerized ionic liquid. The BDS technique quantifies the complex conductivity function, σ*(σ′ + iσ′′) and as shown in Fig. 22, there exists a direct relationship between the real part of ionic conductivity and frequency.16,20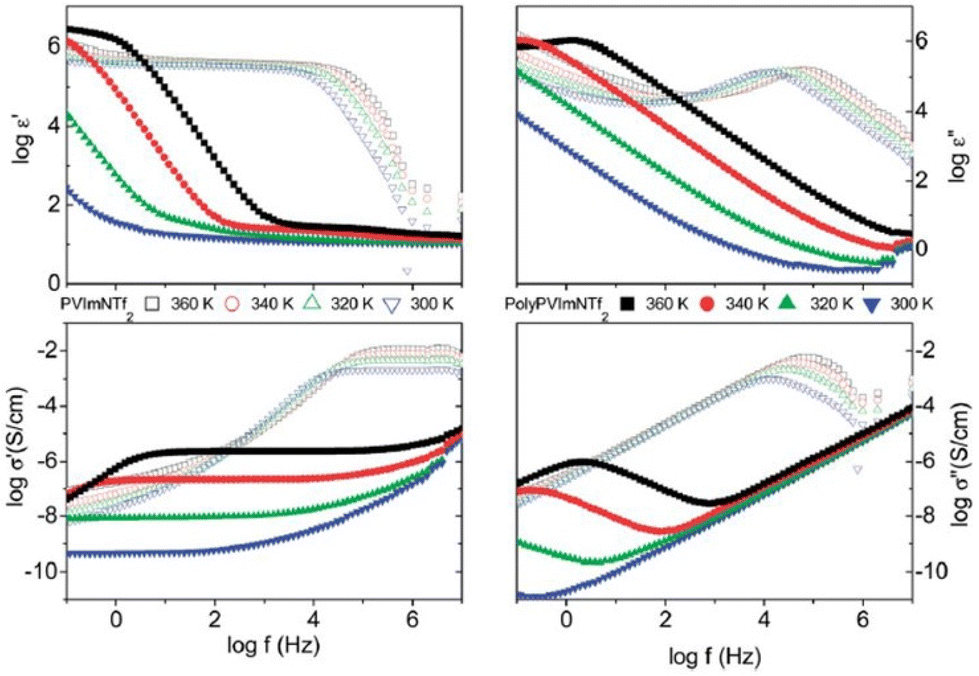 | ||
| Fig. 22 The complex dielectric function ε*(ε′ − iε′′) and the complex conductivity function σ*(σ′ + iσ′′) versus frequency at different temperatures as indicated for the polymerized ionic liquid (poly(PVIM) NTf2) (closed symbols) and the corresponding low molecular weight ionic liquid (open symbols) at selected temperatures. Reproduced from ref. 20 with permission from RSC, Copyright [2014]. | ||
Therefore, in an ionic conductor one observes an increase in σ′ with increasing frequency. Furthermore, there exists a distinct plateau at higher temperature that corresponds to DC conductivity, which is attributed to a frequency-independent response resulting from the long-distance movements of species responsible for ionic conduction.182 At frequencies lower than this plateau a reduction in σ′ can be detected, and this phenomenon is primarily ascribed to electrode polarization, which signifies an accumulation of mobile ions at the electrodes.183 This, in turn, results in a diminished electric field experienced by ions within the bulk of the substance, ultimately leading to a decrease in its observed conductivity.
The DC conductivity of the polymerized ionic liquid demonstrates a Vogel–Fulcher–Tammann-type of thermal activation above the glass transition temperature, and shifts to an Arrhenius-type of temperature dependence below the glass transition temperature (Fig. 23). Below the glass transition temperature, the segmental dynamics freeze, leading to the dominant contribution of the counterion hopping within the glassy polymeric matrix to the ionic conductivity.20
 | ||
| Fig. 23 The dc conductivity, σo (black symbols), as well as the inverse shear viscosity 1/ηo (blue symbols) versus Tg/T for the polymerized ionic liquid (poly(PVIM) NTf2 – full symbols) and the low molecular weight ionic liquid PVIM NTf2 (open symbols). Inset: (a) BNN plot; dc conductivity, σo, versus the characteristic frequency, ωc, for the materials under study as indicated, and, (b) product of the σo and the zero-shear viscosity normalized with respect to the high temperature values for the materials investigated versus Tg/T. Reproduced from ref. 20 with permission from RSC, Copyright [2014]. | ||
Consequently, when plotting the ionic conductivity against the ratio of Tg to the temperature (T), the discrepancies in Tg between the PILs are mitigated, while the variations in conductivity arising from the differences in molecular volume or size of the counterions are accentuated.
Considering the polymeric chain is composed of cations implies that the movement of cations is exceedingly sluggish, rendering it insufficient to significantly contribute to the DC conductivity observed.20 Instead, it is the contribution of anions that dominates. The mobility of anions ought to be considerably greater than that of cations. This outcome aligns with the relatively high ionic conductivity at the glass transition temperature (Tg) in polyIL. At this temperature, the mobility of cations is essentially immobilized within the confines of the experimental timescale.
For the polyILs, it is postulated that the interconnection of the cations’ chains leads to an augmentation of the accessible free space owing to the repulsive interaction among the adjacent covalently bonded cations.20 It can be anticipated that even below Tg, when the movement of polymer segments is effectively halted on the pertinent experimental timescales, the anions continue to exhibit significant mobility due to their diminutive dimensions and the presence of available free space. Ion transportation in this particular scenario bears resemblance to the transportation found in inorganic glasses, exhibiting the conventional temperature dependence described by the Arrhenius equation below Tg.185
The successful application of BDS to a wide range of polymerized ionic liquids has provided immense scientific knowledge that has been a blueprint for fine-tuning ionic conductivity in PILs and fabricating advanced organic electronics with desired properties. Thus far, numerous factors have been examined in order to facilitate the creation of polyILs.
3.4 Fine-tuning ionic conductivity in polymerized ionic liquids
An alternative technique involves increasing the decoupling of ion transportation from segmental dynamics, which potentially could be accomplished through the confinement of morphological properties and/or adjustment of specific interactions within PILs.188
The copolymer composition of the PIL has been shown to significantly affect its glass transition temperature. Elabd et al. observed an increase in ionic conductivity by more than a factor of ten after adding hexyl methacrylate to methacrylate-based imidazolium PILs, thereby lowering their glass transition temperature.129
Research has indicated that polyILs containing NH4+ exhibit greater DC ionic conductivity compared with their imidazolium counterparts at their Tg, as evidenced by the findings from quantitative BDS, WAXS, etc. measurements.18,197 PILs with imidazolium cations and increased spacer (n = 3) are more electrically conducting in contrast to PILs with another onium ion such as pyrrolidinium cation.130 However, imidazolium cation is electrochemically less stable when compared with piperidinium cation and undergoes reduction, making it less versatile for battery electrolytes. PolyILs containing electron-rich trisaminocyclopropenium ions with a highly delocalized charge have exhibited a 4–6 order-of-magnitude increase in ionic conductivity due to enhanced ion-hopping.198
Segalman and coworkers investigated the influence of polymer structure and packing on ionic conductivity in PILs.199 They probed the role of ion placement in imidazolium bis(trifluoromethane)sulfonimide PILs composed of pendant charges and charges in the backbone, and observed significantly higher conductivity in backbone PIL systems.
The BDS technique has also successfully demonstrated the significant influence of these materials’ nanoscale structure on the dynamics of ion motion. Depending on the polymer chemistry, these materials segregate into polar and non-polar domains. The presence of mesoscale aggregates with alkyl spacers larger than butyl in imidazolium ILs was demonstrated by Sangoro et al., who also shed light on the relationship between the dynamics of nanoscale hydrophobic aggregates and slow sub-alpha relaxations.18,202 Similarly, Frenzel et al. ascribed the lower conductivity of polyisobutylene-based ionic liquids in contrast to neat ILs to the microphase segregation of the polymeric part and the IL-like moieties.203 Using BDS, Sokolov and colleagues separated the temperature dependence of ionic conductivity in these polymeric materials’ structural relaxation and demonstrated a strong correlation between the decoupling strength and the materials’ fragility.20,42
These studies elucidate certain fundamental approaches for amplifying the ionic conductivity of PILs and thereby establish a robust framework for the development of cutting-edge devices.
3.5 Mechanical properties of PILs
Significant progress has been made in the field of synthesizing and characterizing polymerized ionic liquids. However, there are still opportunities to be explored in the molecular design of PILs, with the goal of achieving desired ionic conductivities and mechanical strengths simultaneously.Mechanical strength and ionic conductivity are the two antagonistic properties that are both critical to enable PILs in device applications.4 There exists a trade-off relationship between the ionic conductivity and mechanical robustness. This trade-off arises from the strong association between ion motion and polymer chain motion. Consequently, ionomers with elevated levels of ionic conductivity tend to sacrifice their elastic modulus. This necessitates a careful selection of the composition of PIL.43
Mechanical properties are typically evaluated based on the stress–strain curve acquired through dynamic mechanical analyzer (DMA) measurements. The minimum stress concentration observed upon straining PILs is the hallmark of great mechanical strength in these materials.4 The property of mechanical strength indeed holds significant importance among polymer electrolytes that have been designed for implementation as polymer actuators.204
Analogous to ionic conductivity, the mechanical properties of PIL can also be fine-tuned. Considerable efforts to enhance the mechanical characteristics have been documented, including the incorporation of inorganic additives,205 alteration of the polymer composition,206 and introduction of homogeneity into the polymer network structure.207 Notably, with a homogeneous PEG network scaffold, researchers have accomplished enhanced ion transport and superior mechanical properties with an elastic modulus of 171 kPa and fracture energy of 1890 kJ m−3 (Fig. 24).204
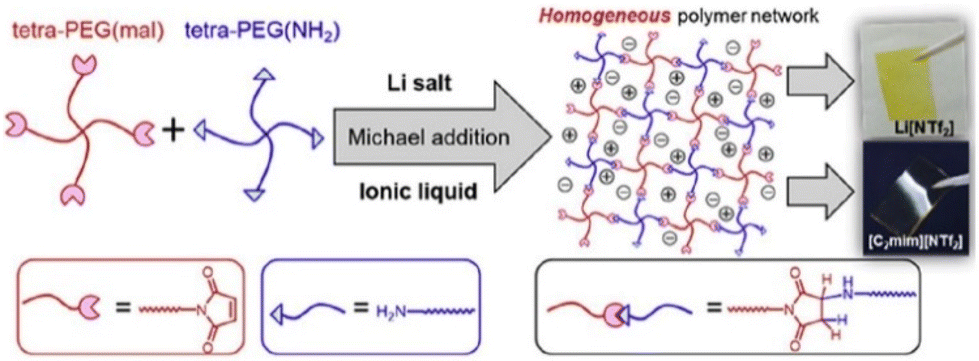 | ||
| Fig. 24 Formation of polymer electrolytes with a homogeneous PEG network via Michael addition reaction and containing either Li[NTf2] or [C2mim][NTf2]. Reproduced from ref. 204 with permission from Elsevier, Copyright [2019]. | ||
Controlled radical polymerization presents a powerful strategy to create novel block copolymers with distinct transport and mechanical properties. Using RAFT polymerization, Long et al. synthesized a A-BC-A triblock copolymer, poly[Sty-b-(Sty-Tf2N-Li-co-DEGMEMA)-b-Sty] with the central block constituting anionic –SO2-N−-SO2-CF3 group and DEGMEMA with low Tg, whereas the external polystyrene block A conferred mechanical strength (Fig. 25).208
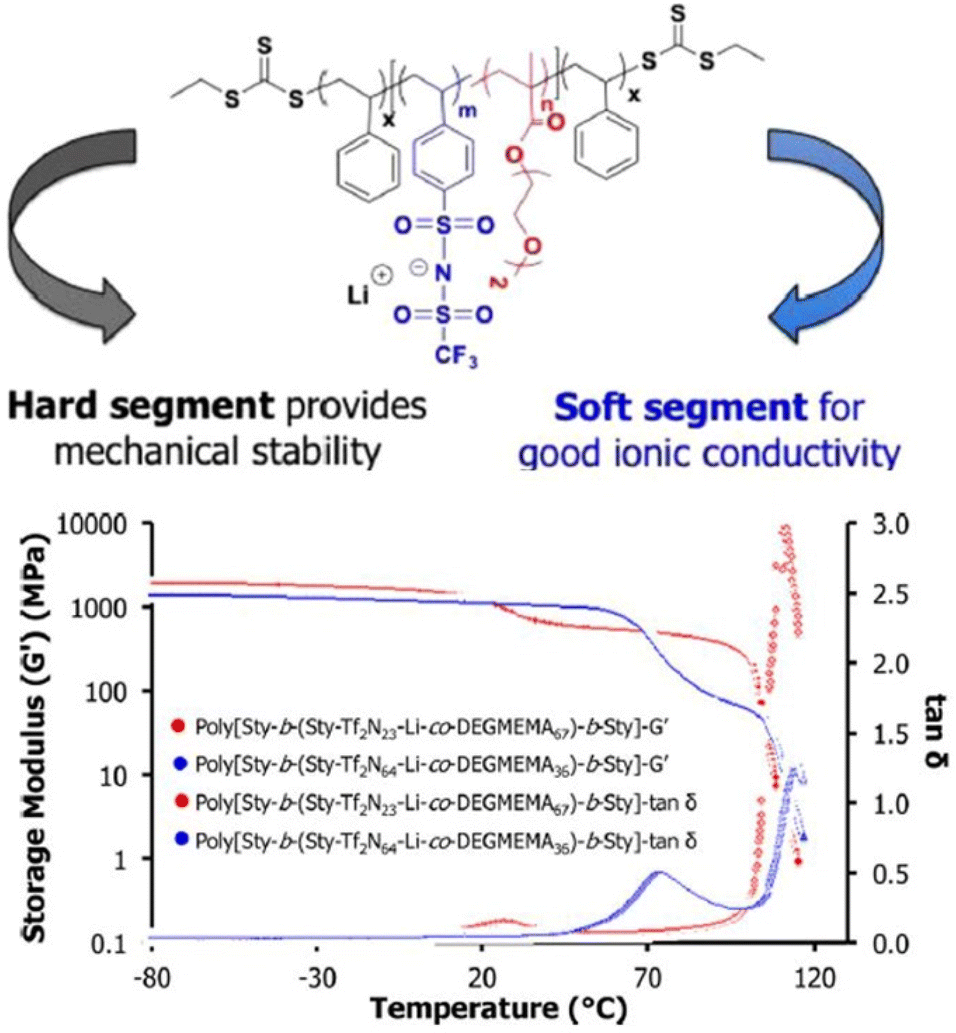 | ||
| Fig. 25 Schematic representation of the sulfonimide-containing triblock copolymers for improved conductivity and mechanical performance. Reproduced from ref. 208 with permission from ACS, Copyright [2015]. | ||
These ion-containing triblock copolymers, A-BC-A, demonstrated exceptional ion-transport properties with ionic conductivity of ∼10−1 S cm−1 and high mechanical strength with storage moduli G′ > 108 Pa at room temperature, rendering them suitable for utilization in energy storage apparatus.
As mentioned above, careful selection of the composition of PIL is critical to achieving high mechanical strength in PILs. Moon and coworkers successfully synthesized a gel-type electrolyte, P[S-r-VBMI][PF6], through the judicious selection of P[S-r-VBMI][PF6] and [EMI][TFSI] composition (Fig. 26).4
 | ||
| Fig. 26 (a) Schematic representation of the random copolymer-based ionic conductive gel. (b) Resistance (Z′) versus frequency (Hz) plots and (c) stress–strain curves for the ion gels composed of 40 wt% P[S-r-VBMI][PF6] or P[VBMI][PF6] and 60 wt% [EMI][TFSI]. The inset of (b) depicts the electrode/ion gel/electrode cell structure used in electrochemical impedance spectroscopy (EIS) experiments. Reproduced from ref. 4 with permission from Wiley-VCH, Copyright [2018]. | ||
The synthesized ion gel displayed both excellent mechanical robustness with an elastic modulus of 0.105 MPa and enhanced ionic conductivity of approximately 1.15 mS cm−1.
Given that the attainment of a heightened mechanical modulus generally transpires below the glass transition temperature, it becomes intriguing to explore approaches that enhance ionic conductivity while circumventing the dependency on Tg.
Inoue and colleagues reported an additional approach for augmenting the mechanical characteristics of polymerized ionic liquids while simultaneously ensuring a relatively high level of ionic conductivity (Fig. 27).43 In this work, they examined the linear viscoelastic response of three imidazolium-based PILs, poly(1-butyl-3-vinylimidazolium) ionic liquid. These PILs were composed of imide-based counteranions with varied perfluoroalkyl chain lengths, namely bis(trifluoromethanesulfonyl) (TFSI−), perfluorosulfonimide (PFSI−), and bis(heptafluoropropanesulfonyl)imide (HFSI−). The aim of this study was to analyse the impact of counteranions with larger ionic volume compared with the side-chain cation on the polymer dynamics of poly(1-butyl-3-vinylimidazolium) ionic liquids.
 | ||
| Fig. 27 Elastic modulus of imidazolium-based polymerized ionic liquids upon introducing large counteranions. Reproduced from ref. 43 with permission from ACS, Copyright [2018]. | ||
They demonstrated that the elastic modulus is significantly increased at temperatures greater than the glass transition temperature, specifically when the ionic volume of the counteranion surpasses that of the side-chain cation.
Sokolov and coworkers further demonstrated the dependence of mechanical properties of acrylate-based tetralkylammonium polymerized ionic liquids on the pendant group structure in PIL.42 They correlated the fragility of these PILs with mechanical measurements and concluded a higher level of packing efficiency in flexible polymers. This results in obstruction in ion-diffusion in a densely packed polymeric matrix and consequently necessitates a greater reorganization of polymer segments. Conversely, in a loosely packed polymer matrix, ion diffusion can still occur upon frozen segmental dynamics. Overall, they found that a reduction in the fragility of the polymer (due to enhanced packing efficiency) corresponds to a decrease in the extent to which ion transport and segmental dynamics are decoupled.
These studies emphasize various approaches for achieving high ionic conductivity and mechanical robustness in polymerized ionic liquids (PILs) with the aim of potentially utilizing them in the field of organic electronics.
3.6 Polymerized ionic liquids in action
Researchers have conducted exhaustive investigations and toggled a variety of parameters to enable the application of PILs in devices. Some of these applications are illuminated in the following subsections.To enable such a device application, Mecerreyes and coworkers mixed vinyl-imidazolium based-PIL with corresponding IL bearing the same counterion (Fig. 28).179 They observed a drastic increase in ionic conductivity of these tailor-made PILs with increasing ionic liquid content. Such tailor-made PILs when used in electrochromic devices showed an enhanced cycle life of up to 70000 cycles, which is comparable to previously used poly(ethylene oxide) electrolytes.
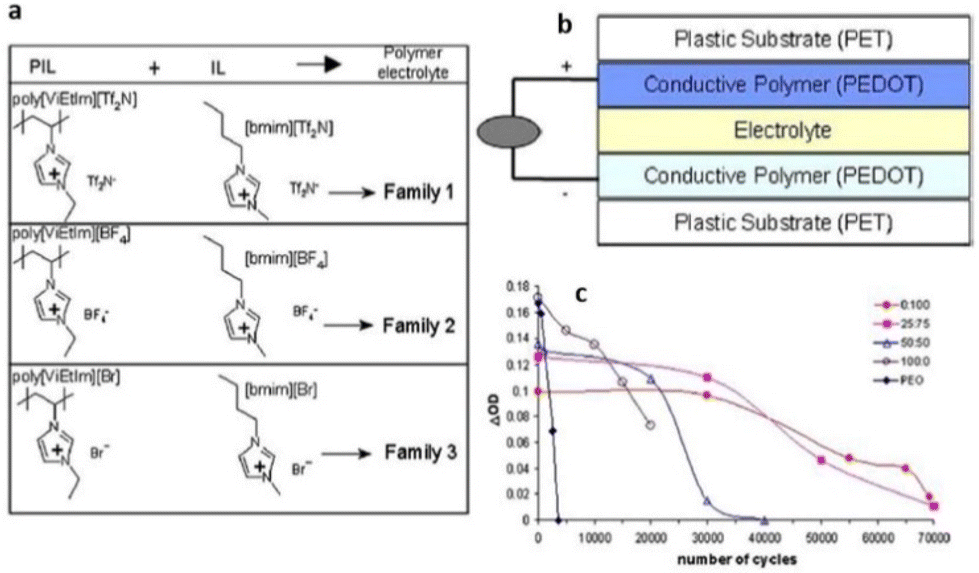 | ||
| Fig. 28 (a) Composition of the polymer electrolyte families. (b) All-polymeric five-layer electrochromic device configuration. (c) Variation of DOD as a function of the number of cycles for ECDs using polymer electrolytes with different ratios [EMIM][Br]: poly[ViEtIm][Br] and using PEO/lithium triflate electrolyte. Reproduced from ref. 179 with permission from Elsevier, Copyright [2006]. | ||
Although the aforementioned observation highlighted that the ionic conductivity of the electrolyte is one of the contributing factors to impact the performance of ECDs, this study did not fully comprehend/elucidate the intricate electrochemical processes occurring between the ionic liquids and conducting polymer.
For the first time, Elabd and associates showed that a PIL block copolymer with a high ionic conductivity of >1 mS cm−1 could be used as separators and solid-state electrolytes in lithium batteries to achieve high mechanical strength and ion conduction (Fig. 29).209
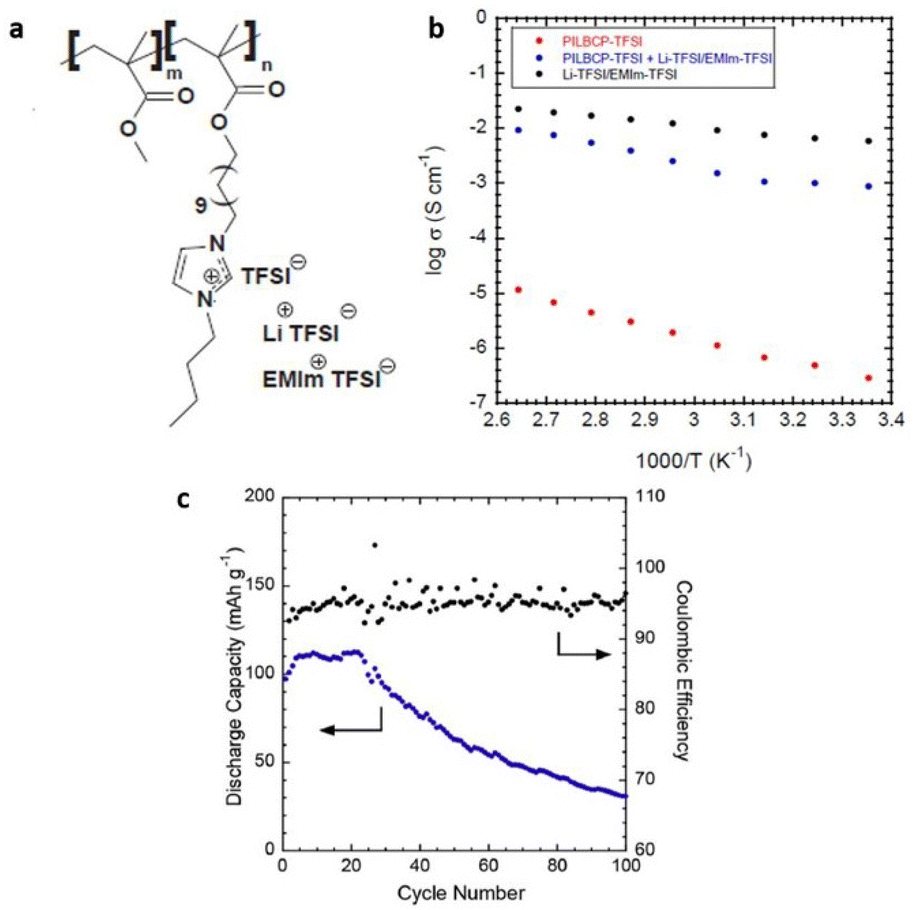 | ||
| Fig. 29 (a) Schematic representation of the PILBCP-TFSI polymer. (b) Temperature-dependent dry ionic conductivity of PILBCP-TFSI (red), PILBCPTFSI + Li-TFSI/EMIm-TFSI (blue), and Li-TFSI/EMIm-TFSI (black). (c) Discharge capacity and coulombic efficiency as a function of cycle number and (b) rate is 0.1 C. Reproduced from ref. 209 with permission from Elsevier, Copyright [2016]. | ||
The ionic liquid monomer, (1-[(2-methacryloyloxy)undecyl]-3-butylimidazolium bis(trifluoromethane)sulfonamide) (MUBIm-TFSI) and the non-ionic monomer methyl methacrylate (MMA) made up the polymerized diblock copolymer poly(MMA-b-MUBIm-TFSI). It is noteworthy that coulombic efficiency of >94% and a discharge capacity of 112 mA h g−1 at 0.1 C was maintained over 100 cycles.
However, the low electrochemical stability of the designed PIL-block copolymer of 2.7 V demanded further significant improvements, considering that high electrochemical stability is a prerequisite for a PIL to function in batteries.
This electrochemical stability challenge has been elegantly addressed by Gerbaldi and coworkers (Fig. 30).210 They designed an ionic liquid-like copolymer that comprised poly(ethylene glycol) methyl ether methacrylate blocks with a fixed length and a second block of poly(lithium 1-[3-(methacryloyloxy)-propylsulfonyl]-1-(trifluoromethylsulfonyl)imide), the length of which was varied. A thorough investigation of these copolymers resulted in polyelectrolytes with significantly lower Tg and high ionic conductivity. Prototypes of lithium batteries with great specific capacities and high charging/discharging efficiencies were made possible by these polyelectrolytes.
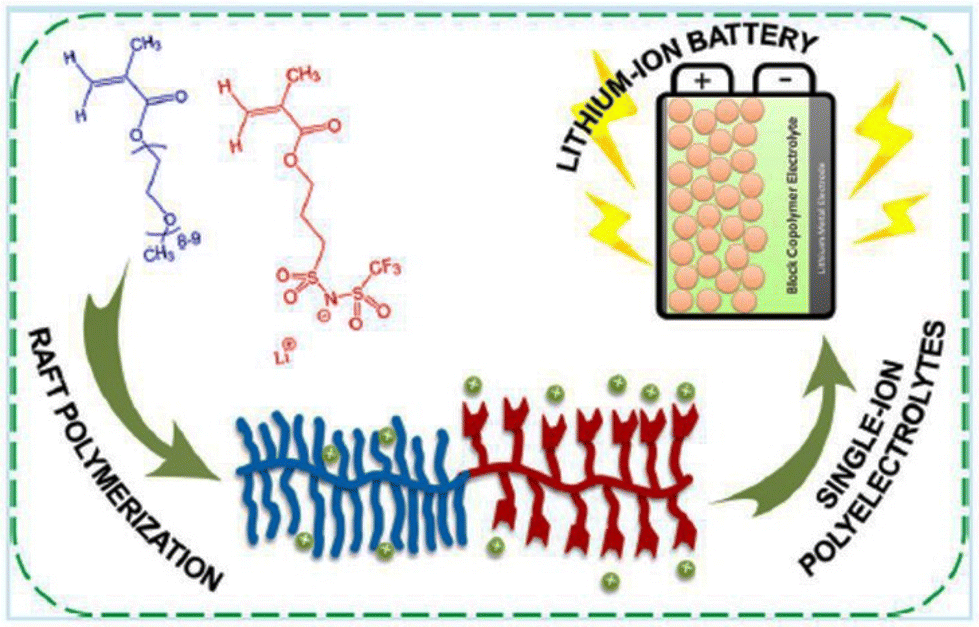 | ||
| Fig. 30 Schematic representation of the single-ion block copoly(ionic liquid)s as electrolyte for Li ion battery. Reproduced from ref. 210 with permission from ACS, Copyright [2016]. | ||
However, the inherent solid-state characteristic of this polymer electrolyte renders it considerably inadequate for exceptionally demanding applications, such as plug-in vehicles which are progressively aspiring to greater heights with each passing year.
Therefore, the flexible chemistry of polyurethanes has been leveraged to modulate the composition and chemical structure of single-ion polymer electrolyte as desired. Researchers have constructed polyurethane-based single-ion conducting poly (ionic liquid)s with significantly lower Tg and appreciable ionic conductivity (Fig. 31).211
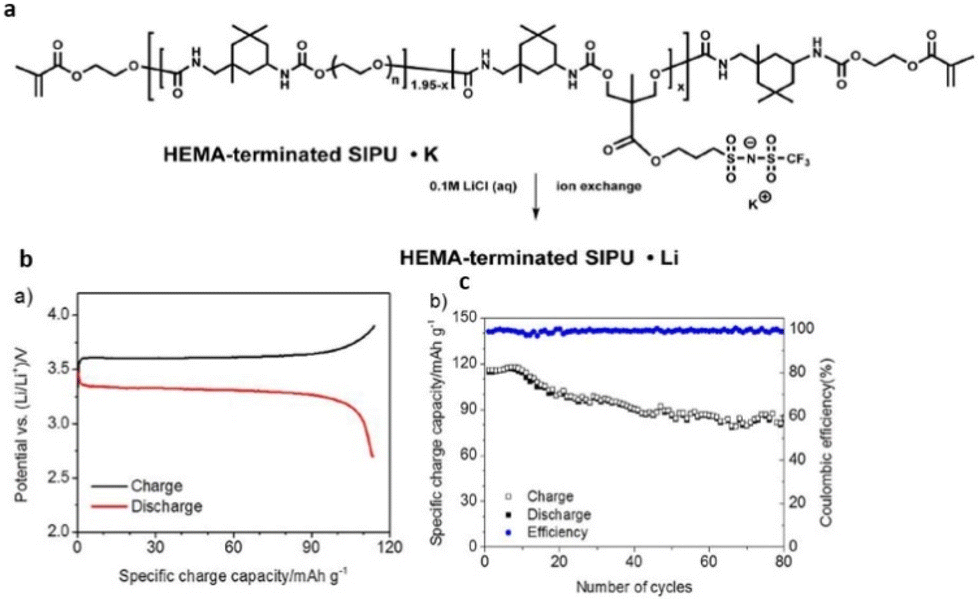 | ||
| Fig. 31 (a) Schematic representation of the polyurethane-based polymer investigated in the current study. (b) Lithium metal/XSIPU-PC/LiFePO4 cell potential versus specific discharge capacity at C/10 and ambient temperature; (c) specific capacity versus cycle number. Reproduced from ref. 211 with permission from Elsevier, Copyright [2017]. | ||
These materials when further tested in lithium metal cells exhibited nearly 100% coulombic efficiency with capacity retention exceeding 72.8% after 80 cycles at C/10. The polyurethane skeleton imparts good segmental motion of the polymer chains, facilitating a favorable pathway for ion conduction, thus making polyurethane-based poly(ionic liquid)s viable for application in the field of battery science.
However, the propensity of the acidic protons in the urethane linkage to react with lithium has been acknowledged for a considerable time as a disadvantage of polymer electrolytes based on polyurethanes. This reactivity accounts for the undesirable decline in capacity during cycling.
Interestingly, ortho-quinones redox-active polymers (RAPs) have recently gained attention to resolve this issue; ortho-quinone RAPs are appealing due to their enhanced life cycle (which can reach 3400 cycles while retaining 98% of their capacity). Researchers have therefore integrated multi-walled carbon nanotubes and redox-active catechol moieties into a poly(N-vinylimidazolium) backbone with TFSI counterion (Fig. 32).212 Li+ and TFSI− mobility in the electrode's bulk and at the electrode–electrolyte interface were both enhanced by this configuration, resulting in improved performance metrics. A high reversible capacity of 247 mA h g−1 at 0.2 C and a remarkable capacity was 134 mA h g−1 at 60 C could be achieved.
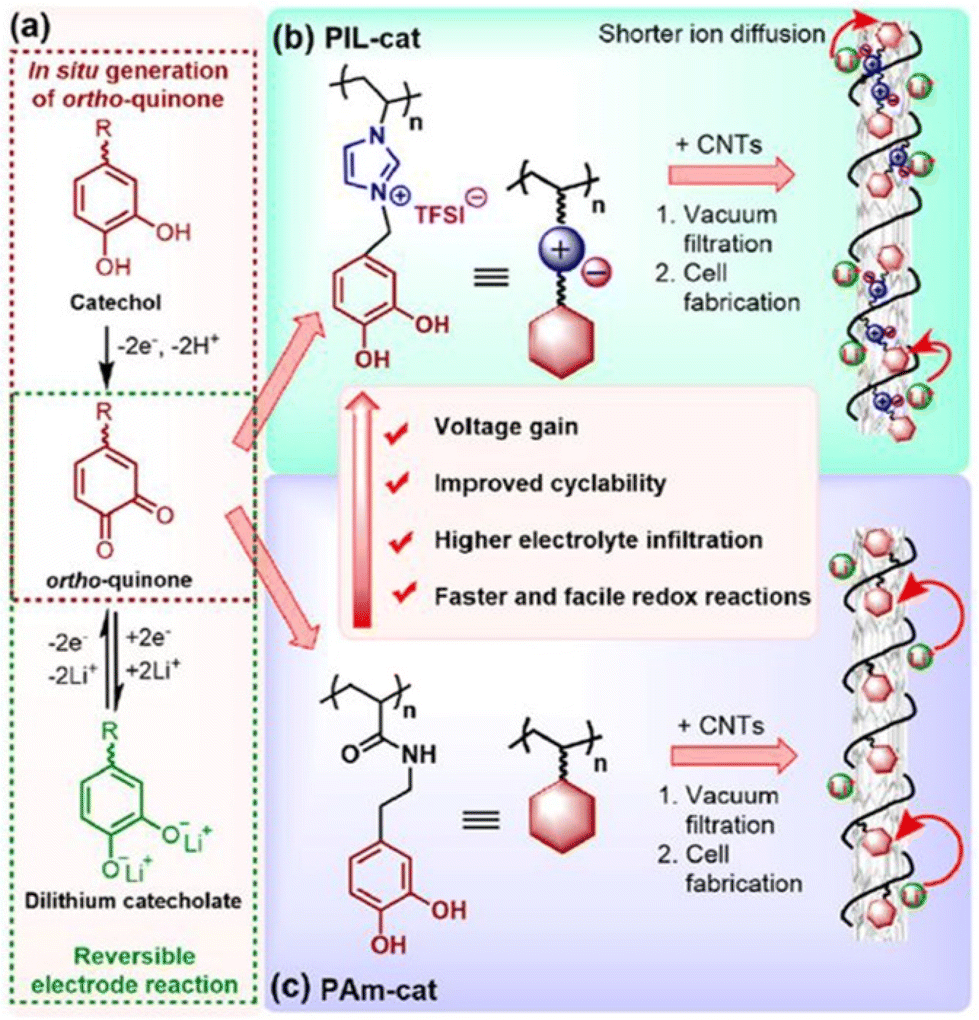 | ||
| Fig. 32 Construction of catechol-based PIL/CNTs buckypaper for LIBs. (a) ortho-Quinone/dilithium catecholate redox reaction. (b and c) Enhancement in the electrochemical performance of cationic PIL-cat (b) compared to its neutral counterpart PAm-cat (c). Reproduced from ref. 212 with permission from ACS, Copyright [2018]. | ||
It is beyond doubt that this methodology facilitates the development of sustainable and high-functioning electrochemical energy storage technologies; however, future investigations focused on streamlining the polymer backbone might further enhance the capacities.
Excellent rate capacity, however, has been achieved by combining PIL frameworks with organic molecules. This has facilitated Li+ transport and minimized IL leakage and leaching issues. Such a system is a solid–liquid hybrid where the entire volume is covered by the entire phase volume. This enables substantial thermal stability and ionic conductivity of the electrolyte. Cheng et al. demonstrated that in contrast to the polyolefin-based separators, the PIL-based separator furnishes enhanced sites for Li+ transport in the electrolyte (Fig. 33).213 This is a consequence of high porosity and enhanced thermal dimensional stability achieved upon combining PILs with organic electrolytes.
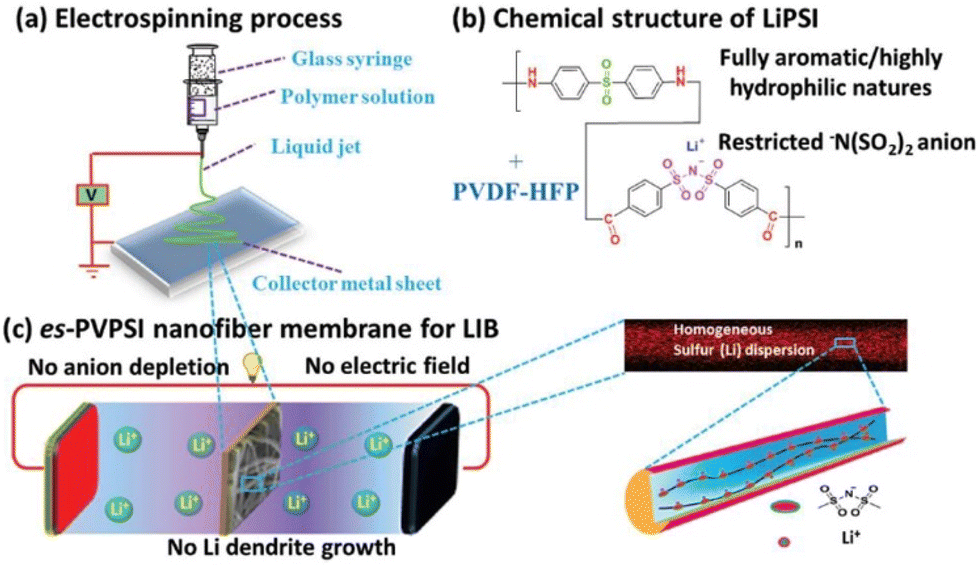 | ||
| Fig. 33 Schematic illustration of the fabrication (a), composition (b), and operation (c) of the es-PVPSI nanofiber-based membrane, acting as single-ion conducting polymer electrolyte in LIBs. Reproduced from ref. 213 with permission from Wiley-VCH, Copyright [2019]. | ||
This combination inhibits the growth of lithium dendrites, a crucial characteristic for the application of PILs in high-energy Li metal batteries. However, this study lacks computational investigations that could yield profound comprehension of the alterations in microstructure upon coordination between PIL and organic electrolytes and their corresponding effects on Li+ motion.
Lithium batteries are extensively utilized in the realm of commercial compact electronic gadgets. However, the incorporation of these batteries into electric vehicles (EVs) and airborne transportation modalities like drones necessitates substantial enhancements in terms of both safety and energy density. Therefore, Forsyth and coworkers exerted their efforts in the direction of achieving safe and high-energy-density Li–metal batteries (Fig. 34).214 They designed a solvent-free polymer solid electrolyte composed of PDADMAFSI and LiTFSI in high concentration. The solid polymer electrolyte showed a significantly higher Li+ transference number and ionic conductivity at a molar ratio of 1.5 of PIL to LiTFSI. It is interesting to note that polycation (N+) and FSI− interaction leads to enhanced Li+ transport number and facilitates the dissociation of Li-FSI to Li+ and FSI−.
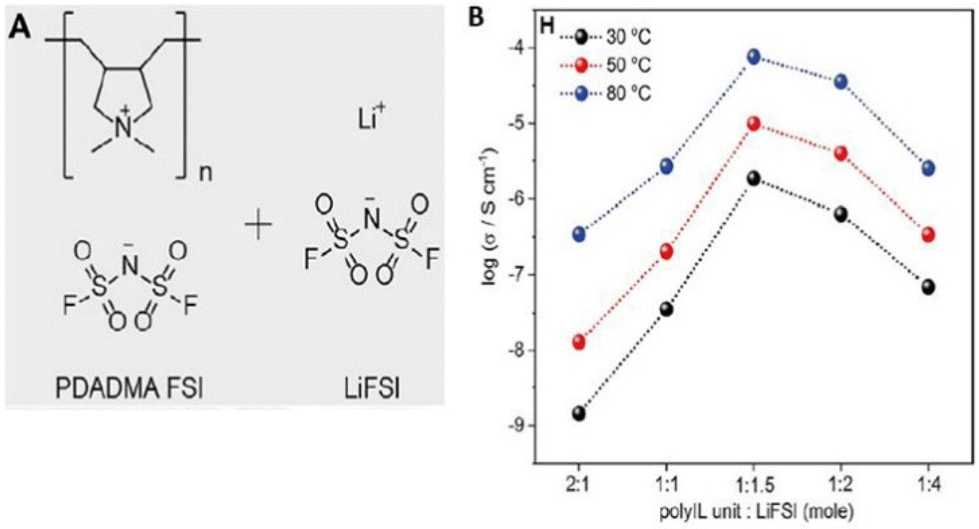 | ||
| Fig. 34 (A) The chemical structures of poly(diallyldimethylammonium) bis(fluorosulfonyl)imide (PDADMA FSI) and Li bis(fluorosulfonyl)imide (LiFSI) salt. (B) The ion conductivities of different polyIL/salt systems at 30 °C, 50 °C, and 80 °C, respectively. The highest conductivity of 0.7 × 104 S cm−1 is achieved for the 1:1.5 system at 80 °C. Reproduced from ref. 214 with permission from Elsevier, Copyright [2019]. | ||
However, this study needs further refinement in the chemistry of polyILs and or Li/salt to inhibit crystallization and enhance the efficiency of Li transport in polyIL-based solid electrolytes.
To address the challenges associated with Li+ transport, researchers have utilized side-chain chemistry to effectively regulate the conductivity in semiconducting polymers and achieve precise transport in Li+ batteries. Recently, Segalman and coworkers demonstrated a significant role played by diffused imidazolium cation side chains for Li+ conduction in electrochemical applications (Fig. 35).215 Substitution of an ether-functionalized side chain in poly(3-(methoxyethoxymethyl)-thiophene) (P3MEEMT) with an imidazolium ionic liquid side chain in polythiophene significantly improved ionic conductivity and lithium transference number.
 | ||
| Fig. 35 Chemical structures for poly-(3-(methoxyethoxyethoxymethyl)thiophene) (P3MEEMT) and poly(3-(6′-(N-methylimidazolium)hexyl)thiophene) (P3HT-Im + TFSI−) used in the study. Reproduced from ref. 215 with permission from ACS, Copyright [2022]. | ||
At 80 °C, the ionic conductivity of poly(3-(6′-(N-methylimidazolium)hexyl)thiophene TFSI−) (P3HT-Im+TFSI−) was ∼10−3 S cm−1 with a Li+ transference number (tLi+) of 0.36, while P3MEEMT, which had a conductivity of 10−5 S cm−1, showed that tLi+ was ≈0 at the same temperature. Coordination of TFSI anion with side chain imidazolium cation weakens the Li-TFSI interaction, enabling facile movement of Li+ throughout the polymer network and accounting for the observed transference number.
This study provides a thorough understanding of the application-specific chemical design to regulate the motion of Li+ with respect to its counterion in mixed conducting polymers for advanced organic electronics. However, this study lacks the systematic investigation of varying the chemical nature of each of the charged units that would have illuminated how these factors would have affected the ionic conductivity and a Li+ transference number.
The lithium-ion transference number of polymeric electrolytes has been further improved for their application in Li+ batteries. A 3-D gel polymer electrolyte with a Li+ transport number of 0.622 and a high ionic conductivity of 2.74 × 10−5 S cm−1 at room temperature was created by Xu et al. (Fig. 36).216
 | ||
| Fig. 36 (a) The images of the precursor solution and SLIMs after in situ crosslinking, with the corresponding change of structures. (b) Preparation process of SLIMs by in situ crosslinking reaction. Reproduced from ref. 216 with permission from Elsevier, Copyright [2020]. | ||
To enable this 3D structured gel, polyethylene glycol dimethacrylates (PEGDMA) were crosslinked with LiTFSI and saturated with commercialized electrolytes. The high ionic conductivity and high Li+ transference number was attributed to the effective Li+ migration sites provided by the anionic PIL backbone.
Furthermore, apart from possessing a substantial degree of ionic conductivity and a notable Li+ transfer number, it is greatly advantageous to encompass a broad operational temperature range in batteries. This particular attribute has been meticulously exemplified by the work of Hu and coworkers, in which they synthesized a monolithic ionogel electrolyte that constituted PIL poly(diallyldimethylammonium) bis(trifluoromethanesulfonyl)imide (PDADMATFSI) nanofibers (NF), an IL solvent, N-methoxyethyl-N-methylpyrrolidinium bis(trifluoromethanesulfonyl)imide (Pyr1201TFSI) and cross-linked poly(2,2,2-trifluoroethyl methacrylate) (PTFEMA) (Fig. 37).217
 | ||
| Fig. 37 Schematic illustration of the structure and composition of the IGEM. Reproduced from ref. 217 with permission from Wiley-VCH, Copyright [2021]. | ||
The presence of PTFEMA in ionogel electrolytes facilitates the achievement of both high ionic conductivity and a high Li+ transference number with a wide operational temperature range from 0 °C to 90 °C. This is attributed to the collaborative effect of PTFEMA in reducing the impedance at the electrode/electrolyte interface and PIL-NF in providing the ionogel with sufficient strength and thermal stability.
The performance of Li+ batteries is also significantly influenced by the type of binder used and it is critical to ensure its non-resistive nature. Researchers have therefore implemented stable binders for application in high-energy batteries. Key to their approach was the stabilization of a blend of a charged conjugated polymer with an oppositely charged polyelectrolyte using electrostatics. Segalman et al. complexed a cationic polymeric ionic liquid, poly[(3-methyl-1-propylimidazolylacrylamide)-co-3-methyl-1-(propylacrylamide)] to an anionic conjugated polyelectrolyte, poly[6-(thiophen-3-yl)hexane-1-sulfonate-co-3-(hexylthiophene)] (PTHS:P3HT) to compose a CPE-PIL complex (CPC) (Fig. 38).218 This synergism enabled tremendous cycling stability with significant capacity retention of 93% and 63% after 100 and 400 cycles at C/2 when used in LiFePO4 (LFP) composite cathodes.
 | ||
| Fig. 38 (a) Conventional lithium-ion batteries composed of a composite anode, a composite cathode, and a separator soaked with a liquid electrolyte that allows the flow of ions while preventing the passage of electronic current. (b) Cathode composite composition and interactions. The crystalline active material (black spheres) is responsible for redox activity. Conductive additives (black) improve electron transport, while a binder (purple) holds the active material + conductive additive system together. Current technologies use resistive plastics for the latter, such as PVDF. During the charge/discharge process, ion transport occurs between the electrodes via the electrolyte, requiring good ionic diffusion at the electrolyte/active material interface, and electrons flow between the electrodes via the external circuit, requiring fast electron transport from the conductive additive to the current collector and vice versa. Resistive binders create barriers to the transport of both of these charged species, reducing overall performance. (c) The polymer complex serves as a multifunctional binder, enabling ion and electron transport, as well as binding. Reproduced from ref. 218 with permission from ACS, Copyright [2023]. | ||
These values demonstrate drastic improvement over the existing state-of-the-art PVDF binder, reinforcing this electrostatically stabilized complexation strategy as a promising strategy to realize a binder with high ionic and electronic conductivity for high-power demanding applications. The authors envisage the potential application of their designed binder in electric vehicles; however, the study lacks the investigation of the binder efficiency under environmentally relevant temperature conditions.
Rigorous efforts have been exerted to further improve the operational efficiency of Li+ batteries. Researchers have successfully designed an effective approach for promoting simultaneous electron and ion conduction that is critical to the operation of electrochemical devices. To enable this approach, Segalman et al. covalently tethered the imidazolium ionic liquid group to a polythiophene-based conjugated polymer backbone.219 This modelled conjugated polymeric ionic liquid, poly{3-[6′-(N-methylimidazolium)hexyl]thiophene}BF4− (P3HT-IM) was oxidized and treated with salt (Fig. 39).
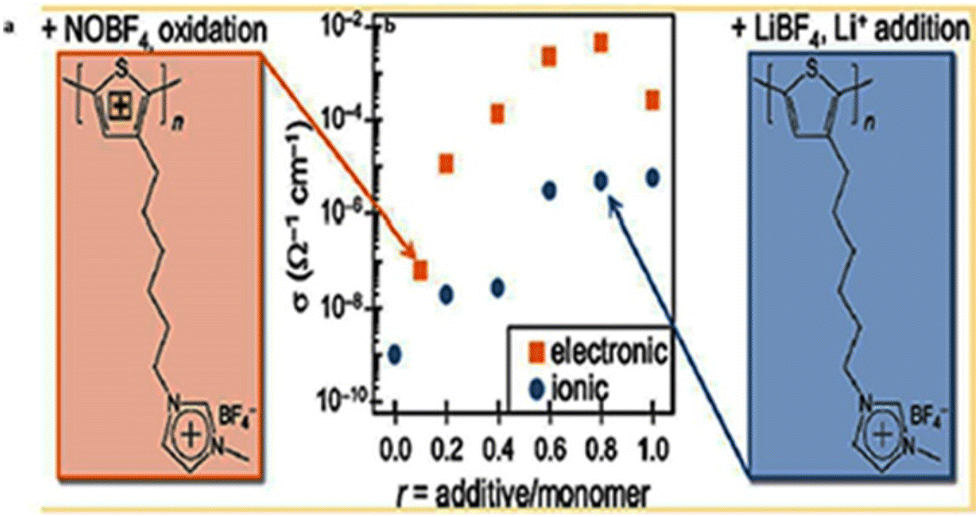 | ||
| Fig. 39 (a) Schematic structure of the thiophene-based conjugated polymeric ionic liquid used in this study. (b) Electronic conductivity as a function of NOBF4 oxidant addition. Reproduced from ref. 219 with permission from ACS, Copyright [2021]. | ||
This was accompanied by an increase in electronic and ionic conductivity owing to a monotonic increase in mobile ionic and electronic carriers. Notably, this first study investigating both the electronic and ionic conductivity of a conjugated polymer functionalized with an ionic liquid holds profound significance for lithium-ion batteries.
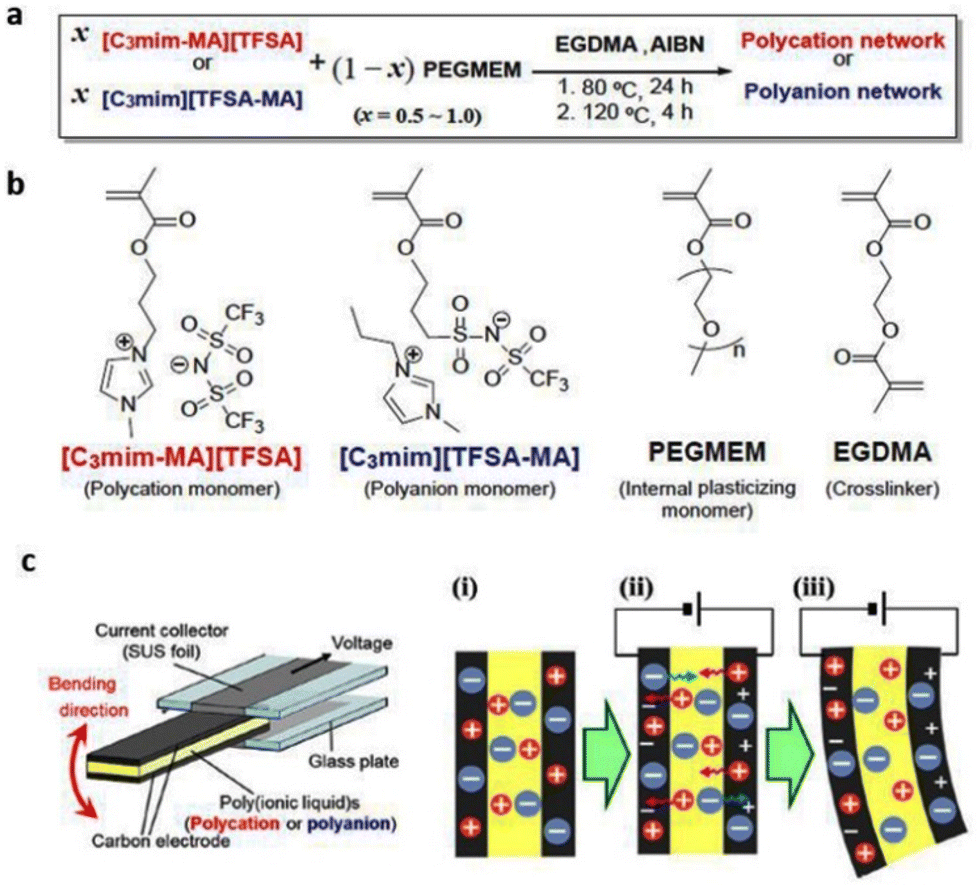 | ||
| Fig. 40 (a) Scheme for preparation of polyIL networks and the chemical structures of the monomers, internal plasticizing monomer, and cross-linking agent used in this study; (b) structure of polymer actuator in this study and (c) driving mechanism of the polymer actuator using ionic liquids or ionic salts as an electrolyte in the case of (t+ > t−); (i) before applying voltage, (ii) after applying voltage, ion migration to both EDL associated with charging, (iii) deformation toward positive electrode due to the increase in the volume of negative electrode. Reproduced from ref. 132 with permission from Elsevier, Copyright [2018]. | ||
This reflects there is a necessity to enhance the ionic conductivity of the polymer electrolyte to ameliorate the speed of response. Going forward, it is imperative to undertake further refinement in low ionic conductivity of the polymer electrolyte in order to optimize the longevity of the actuator's operational cycle, facilitate greater displacement and generating force, and enhance actuation speed.
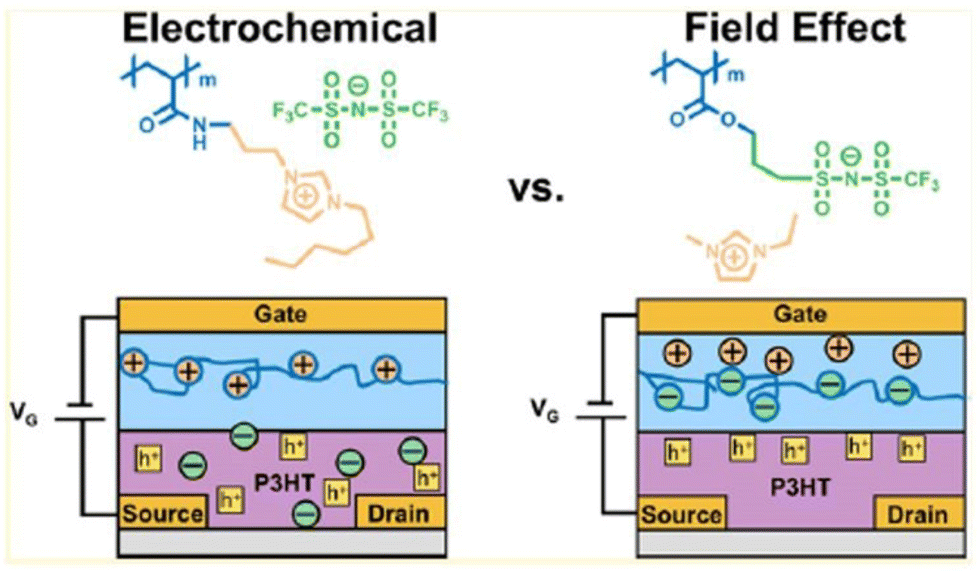 | ||
| Fig. 41 p-type semiconductor (P3HT) with the cation-tethered PIL and the anion-tethered PIL used as gate dielectrics in the study. Reproduced from ref. 220 with permission from ACS, Copyright [2019]. | ||
These devices with p-type semiconducting polymer P3HT disclosed that electrical double layer (EDL) doping facilitated an enhanced uniform even distribution of carriers between the ordered and amorphous regions of the film in contrast to electrochemical (EC) doping. This reflects that for EDL-based devices with anion-tethered PIL higher mobilities can be achieved at low doping levels in P3HT with lower onset voltages. On the contrary, for applications demanding high currents electrochemical doping may be adapted, enabling a higher charge capacity for a given semiconductor. However, this study lacks the comprehensive investigation on the intricate process involving ion interaction and electron transport that could further accelerate the exploration of these EGTs in biosensor or bioelectronic devices.
The photoconversion of PILs into a readily processable material has also enabled their application in field-effect transistors. Segalman and coworkers reported an intriguing strategy for transforming a highly viscous PIL into a flexible and soft solid via a pericyclic reaction involving [4π + 4π] cycloaddition reaction of anthracene upon UV radiation (Fig. 42).221 This approach enabled the designing of PILs with enhanced mechanical properties, resulting from modeling the polymer to a crosslinked polymer network without compromising ionic conductivity, owing to negligible changes in parameters like ion concentration, ion mobility, and polymer segmental dynamics upon cross-linking. The presented molecular design enabled facile processability of the material for attractive applications like patterning substrates for gating semiconductors in a thin-film geometry.
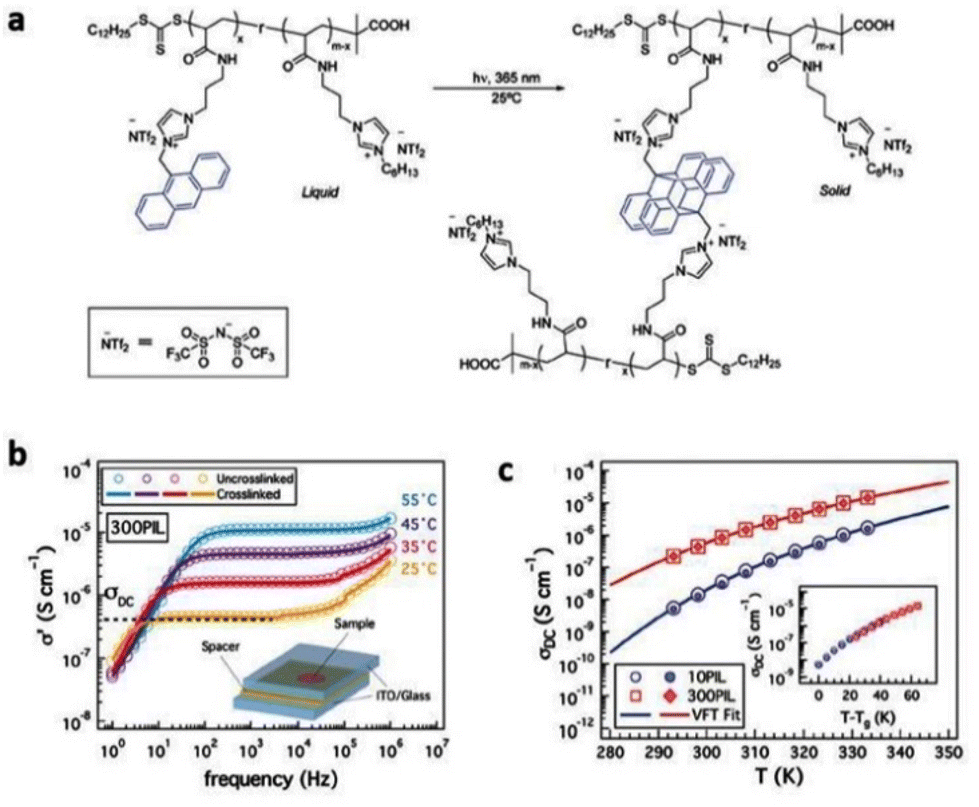 | ||
| Fig. 42 (a) Dimerization of anthracene enables conversion of a PIL from a polymer liquid to a soft solid. (b) Ionic conductivity of anthracene-functionalized PILs before and after crosslinking: real part of complex conductivity as a function of frequency and temperature. The plateau from the frequency-independent part of the spectrum was taken as the DC conductivity. The open circles and solid lines respectively correspond to the measurements performed on uncrosslinked and crosslinked (60 min exposure at 298 K) PIL. The inset illustrates the schematic of the in situ conductivity measurement cell comprising the polymer sandwiched between two ITO/glass transparent electrodes, and separated by a 150 mm Kapton spacer. (c) Temperature-dependent ionic conductivity for the PILs. The open and closed symbols are the data for uncrosslinked and crosslinked PILs, respectively, while the solid lines are the VFT fits. The inset illustrates the Tg-normalized conductivity data for both PILs that fall onto a single master curve. Reproduced from ref. 221 with permission from RSC, Copyright [2018]. | ||
Although the presented molecular design enabled facile processability of the material for applications like patterning substrates for gating semiconductors in a thin film geometry, it lacks the demonstration of the reversible phenomenon with light as external stimulus.
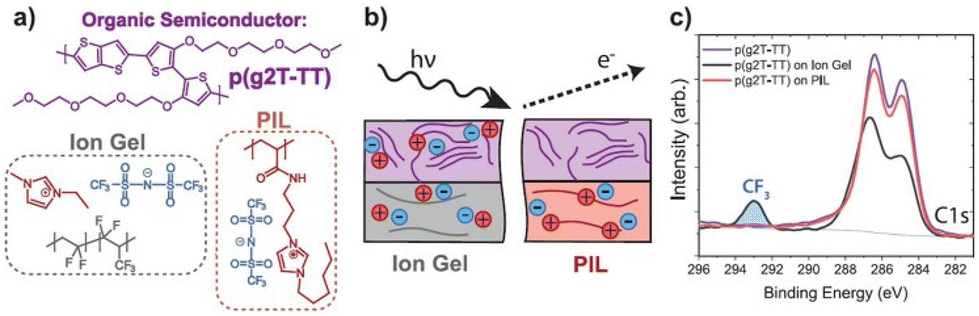 | ||
| Fig. 43 (a) Chemical structures of the organic semiconductor and electrolytes used in this work. The EMIM:TFSI ion gel has both anions and cations mobile whereas the PIL only has mobile TFSI anions. (b) Schematic illustrating XPS samples and measurement geometry. (c) XPS spectra of pristine p(g2T-TT) (purple) and p(g2T-TT) films spun on ion gel (black) and PIL (red) electrolytes. The spectra show significant signal corresponding to the ionic liquid ions that have entered the p(g2T-TT) film, whereas the PIL spectra resembles that of the pristine polymer. Peak fits corresponding to the EMIM cation (red) and TFSI anion (blue) of the are shown for the p(g2T-TT) on ion gel spectra. Reproduced from ref. 222 with permission from Wiley-VCH, Copyright [2021]. | ||
These investigation studies involved comparing the devices composed of imidazolium polymerized ionic liquid with mobile TFSI anion with the corresponding devices made of PVDF-HFP gel permeated with ionic liquids constituting mobile TFSI anion and mobile EMIM cation.
Their XPS studies confirmed passive ionic liquid uptake within the bulk of OMIEC films introduced by ion gel electrolytes did not affect the charge transport pathways of the semiconductor. In contrast, no ion pair uptake was observed in the device with PIL owing to the covalent attachment of the cation to the polymeric backbone.
This work effectively demonstrated the significance of assessing the cationic hole compensation in ion-gel-based organic electrochemical devices, a strategy for realizing high-performance devices but at the cost of device speed, thereby creating an opportunity for further scope for improvement.
3.7 Polymerized ionic liquids for constructing stimuli-responsive devices
In addition to increasing the ionic conductivity of PILs, it is also imperative to control their mobile ion content for advanced organic electronics. This demands the incorporation of stimuli-responsive handles in PILs to develop triggerable PILs. Researchers have made significant progress in this direction to enable new classes of technological applications.Guo et al. synthesized ionic liquids constituting imidazolium-based cation and light-responsive anions of trans-ortho-methoxycinnamic acid ([OMCA]) to achieve highly efficient irreversible conductivity modulation in aqueous solution (Fig. 44).223
 | ||
| Fig. 44 (a) Photoisomerism of [Cnmim][OMCA] ionic liquids. Effects of the concentration of the ILs, the alkyl chain length of the cations and light intensity on the light-conversion efficiency of [Cnmim][OMCA] in aqueous solutions. (b) Concentration effects of [C10mim][OMCA], 0.1 mol kg−1, 0.01 mol kg−1, 0.001 mol kg−1. (c) Alkyl chain length effects of cation at the IL concentration of 0.001 mol kg−1 [C4mim][OMCA], [C10mim][OMCA], [C16mim][OMCA]. Reproduced from ref. 223 with permission from RSC, Copyright [2014]. | ||
Upon UV exposure the anion undergoes irreversible photoisomerization from trans to cis, resulting in conductivity modulation with a maximum increase of ∼130% under 3 h irradiation in aqueous solutions.
Yan and colleagues used acrylic acid (AA), N-isopropylacrylamide (NIPAM), and acrylonitrile to photocopolymerize imidazolium type IL monomers to create a thermo- and pH-responsive poly(ionic liquid) (Fig. 45).224
 | ||
| Fig. 45 (a) Reaction scheme for the preparation of PIL-based membranes via in situ photocrosslinking (DVB as the cross-linking agent). (b) Mechanism for thermo-and pH-responsive behaviors. Reproduced from ref. 224 with permission from RSC, Copyright [2016]. | ||
Temperature-responsive characteristics are imparted to the resulting PIL membranes by the temperature-sensitive nature of PNIPAM segments. The association and dissociation of the hydrogen-bonding interactions with acrylic acid segments account for the pH-responsive behavior of the PIL membranes.
The produced dual-responsive PIL membranes have improved mechanical properties, making them suitable for use in functional materials for information inscription and sophisticated sensors.
Yuan et al. electrochemically controlled the sol–gel transition behavior of the IL gemini guests containing ferrocene (Fc) and bis(trifluoromethyl-sulfonyl)imide as the α- and ω-guest groups (Fig. 46).225 This IL along with the host polymer containing β-cyclodextrin (CD) constituted the electroactive supramolecular gels through the host–guest interactions.
 | ||
| Fig. 46 (a) Preparation of ionic liquid gemini guest cross-linked electroactive supramolecular gels and (b−e) their assembly and disassembly induced by multistimuli. Reproduced from ref. 225 with permission from ACS, Copyright [2014]. | ||
In addition to reversible sol–gel transitions in response to multiple stimuli like temperature, electrochemical, and chemical redox reactions, this supramolecular gel shows conductivity switching synchronously with such stimuli. The authors envisage these multi-stimuli-responsive supramolecular gels to be of great potential application in advanced sensor and semisolid state electrolyte materials.
Along the same lines, Zheng and colleagues used host–guest interaction between β-CD and zwitterionic liquid (ZIL) containing an azobenzene and bis(triflurosulfonylmethane imide) in place of a ferrocene moiety to fabricate a photo- and thermoresponsive supramolecular hydrogel (Fig. 47).226 Upon UV irradiation, the hydrogel generates a fluid sol state with trans-to-cis isomerization of the azo moiety. Subsequent exposure to visible light or heat treatment reverts the cis moiety to the trans form.
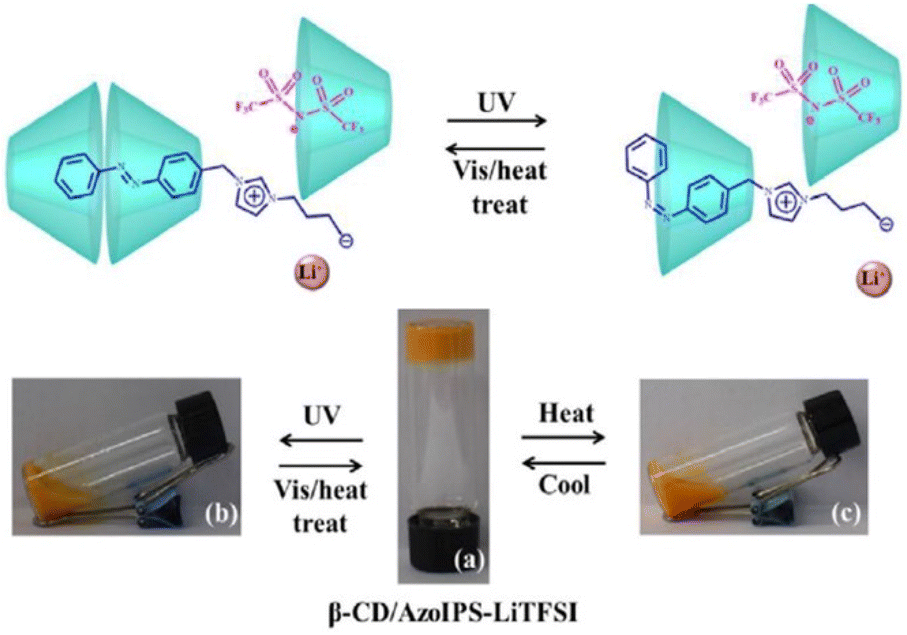 | ||
| Fig. 47 (a) Mode of inclusion between b-CD and AzoIPS-LiTFSI before and after UV irradiation. (b) Preparation of the supramolecular hydrogel: (a) b-CD/AzoIPS-LiTFSI (3:1), and its disassembly induced by: (b) UV light, and (c) temperature. Reproduced from ref. 226 with permission from Wiley-VCH, Copyright [2018]. | ||
Owing to the sol–gel phase transition accompanied by reversible switching of the conductivity, these supramolecular hydrogels demonstrate a novel strategy for laying the foundation of multi-responsive electrical sensing materials.
Using azobenzene moiety, researchers have also designed an intriguing multiwavelength organic photodetector (Fig. 48).227 This photodetector, which is based on an azobenzene polymeric ionic liquid, can detect light at several wavelengths that correspond to the azobenzene's absorption spectrum. Bates and coworkers forged this light-detecting device by blending a light-responsive azobenzene-based polymeric ionic liquid as a gate layer with a semiconducting p-type polymer poly[(4,4-dihexadecyl-4H-cyclopenta[1,2-b:5,4-b′]dithiophene-2,6-diyl)-alt-(2,3-difluoro-1,4-phenylene)](PhF-2,3). This device showed a significant amplification in current when exposed to UV and visible light at a constant voltage. Once the light source is turned off, the current decays rapidly.
 | ||
| Fig. 48 Multiwavelength stimulated increase in photocurrent driven by absorption of azobenzene in Azo-PIL. Reproduced from ref. 227 with permission from ACS, Copyright [2021]. | ||
The charge transfer processes in the semiconducting polymer and photoexcited azobenzene polymeric ionic liquid account for the increased light-responsive current. Despite multiple ON/OFF cycles, it is interesting to note that the performance of this light-mediated system is fatigue-resistant and adjustable with wavelengths well beyond visible wavelengths (660 to 700 nm).
Sumitani et al. designed a Ru-containing PIL comprising a Ru sandwich complex [Ru(C5H5)−{C6H3(OC6H12CN)3}]+ and a polymeric anion[–CH2-CH(SO2N−SO2CF3)–]n− (Fig. 49).228,229 The ionic conductivity of this PIL was reversibly controlled by the application of heat and light. Upon UV irradiation, the cations in the PIL formed a coordination network that remodelled the PIL into an amorphous hybrid coordination polymer, gradually decreasing the ionic conductivity. Subsequently heating the coordination polymer regenerated the PIL and increased the conductivity.
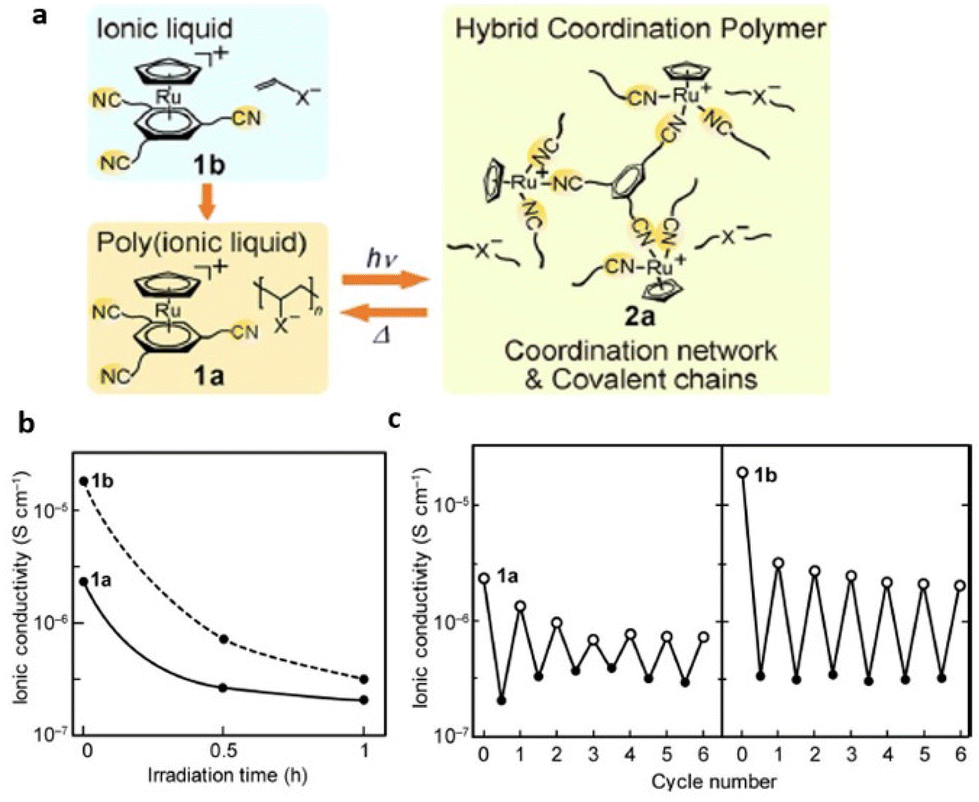 | ||
| Fig. 49 (a) Schematic representation of metal-containing poly(ionic liquid) exhibiting photogeneration of coordination network. (b) Ionic conductivity of PIL 1a and IL 1b at 25 °C. (c) Ionic conductivity changes for alternating cycles of UV photoirradiation for 1 h (filled circles) and heating at 120 °C for 20 min (open circles). Reproduced from ref. 228 with permission from ACS, Copyright [2020]. | ||
This demonstrated the modulation in ionic conductivity by thermal and photochemical reactions resulting in a coordinated structure transformation. The authors anticipate these PILs to be advantageous for constructing photocontrollable electronic and photoresist materials.
Zhang and coworkers constructed a phase-transformable ionogel (PTIG) by incorporating a phase-transformable IL, 1,3-dimethyl imidazolium bis(triflurosulfonylmethane imide) into a polymer network, polybutyl acrylate-co-polyfluorinated butyl acrylate (Fig. 50).230 Within an insulating polymer network, the IL provides a continuous conductive phase. However, upon cooling to the phase-transition temperature of −15 °C a significant drop in ionic conductivity (∼107 times) is observed.
 | ||
| Fig. 50 (a) Chemical structures of PTIG precursor materials. (b) Schematic illustration of the PTIG concept with a reversible ICIT. The PTIG is a gel with a phase-transformable IL immobilized within a soft-polymer matrix, which is homogeneous and transparent at room temperature. The IL forms a continuous conductive phase within an insulating polymer network, providing good conductivity. A sharp conductor–insulator transition occurs when cooled to the phase-transition temperature due to the first-order solid–liquid transition of the IL, which is reversed after warming. Reproduced from ref. 230 with permission from Wiley-VCH, Copyright [2020]. | ||
The ionic conductivity is regained upon warming the PTIG. Thus, PTIG exhibited a reversible conductor–insulator transition. Furthermore, it displayed high transmittance (∼97%) with good stability and stretchability (up to 84% strain), demonstrating its great potential for stretchable iontronic applications.
Recently Alaniz and coworkers synthesized light-responsive PILS using photochromic diarylethenes (DAEs) tethered to imidazolium cation, which is covalently attached to a poly(ethylene oxide) backbone (Fig. 51).231
 | ||
| Fig. 51 (a) Photochromism of imidazolium-bearing DAE; (b) scheme of PIL with photoresponsive cation anion interaction; (c) solid-state PIL film with tunable, photoresponsive ionic conductivity. Reproduced from ref. 231 with permission from Wiley-VCH, Copyright [2020]. | ||
The cation character of the imidazolium bridging unit was modulated upon UV exposure led to a 70% drop in conductivity, and upon subsequent irradiation with visible light the PIL regained its ionic conductivity, thus demonstrating reversible control over ionic conductivity. To enable such photoresponsive PIL, the binding between mobile anions and imidazolium containing DAE cations was controlled where the ring-open isomer (DAE-O) possessed a delocalized cation charge due to the aromatic imidazolium ring, whereas the ring-closed version (DAE-C) contained more localized charge.
This method opens the door for the development of organic electronics, where an external stimulus like light can be used to control ionic conductivity.
Different to light, Laaser et al. created a heat-responsive polymerized ionic liquid made up of temperature-sensitive furan-maleimide Diels–Alder linkages between the methacrylate backbone and the charged imidazolium backbone (Fig. 52).15 Heating the PIL above the retro-Diels–Alder temperature of 90 °C enabled thermally triggered ion release. Concrete proof that the retro-Diels–Alder reaction is responsible for the increased mobile ion content and, consequently, the corresponding increase in ionic conductivity, was provided by electrical characterization studies conducted in conjunction with the temperature dependence of the capacitor response during the heating cycle.
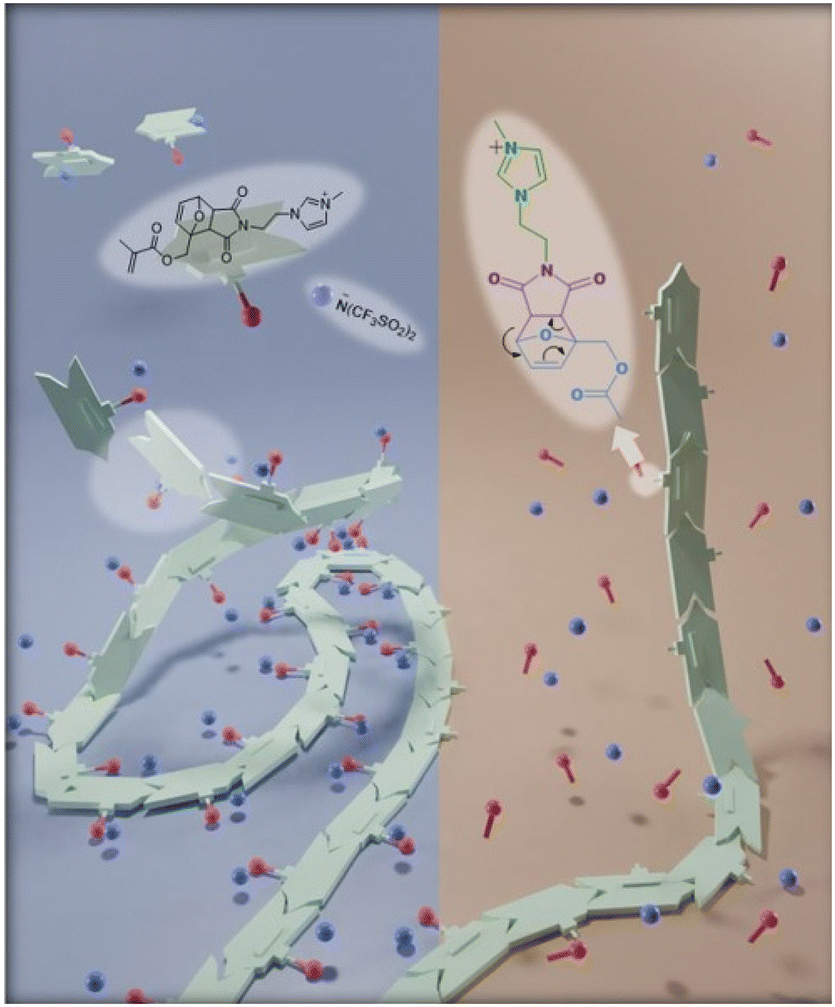 | ||
| Fig. 52 Schematic illustration of temperature-dependent ion release in polymerized ion liquids containing thermally labile Diels–Alder linkages. Reproduced from ref. 15 with permission from ACS, Copyright [2020]. | ||
By adding chemical triggers to release ions “on-demand”, this work demonstrated a novel method for varying the mobile ion content in polymeric material. This interesting capability of changing the mobile ion content is currently missing among the ion-containing polymers. This is usually because variables affecting ionic conductivity, such as ion concentration, ionic group composition, ionic group spacing along polymer chains, and ionic group separation from the polymer backbone, are usually fixed under specific experimental conditions and cannot be changed while the material is being used. While it is possible to raise the PIL's ionic conductivity above its Tg for a short while,20,232 once the PIL cools, its ionic conductivity eventually returns to its original level. Consequently, ionic conductivity is essentially fixed at a particular temperature. Nonetheless, this work beautifully demonstrates the ability to alter the mobile ion content and conductivity at a specific temperature.
Chabinyc and coworkers enabled flexible redox active polymers with a low Tg by exploiting two weakly interacting ionic groups in a polymeric ionic liquid (Fig. 53).233 Phenothiazine (PTZ) with N-substituted (2-(2-ethoxyethoxy)ethoxy)ethyl (EE) was used as a redox-active group and attached closer to the polymer backbone to facilitate efficient charge hopping between the PTZ groups. Partial doping with TFSI enhanced charge conduction, elucidating pPTZ-EE as an enticing material for deformable electronics. pPTZ-EE displayed mechanical softness upon mild heating that could be further enhanced via cross-linking.
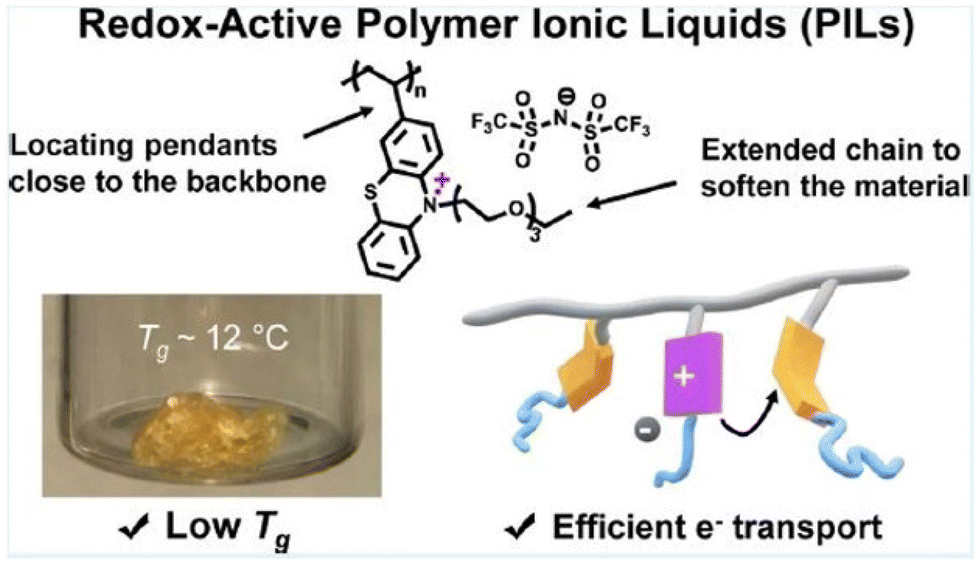 | ||
| Fig. 53 Schematic structure of redox polymers with tethered chains inserted in between the backbone and pendants to target for low Tg charge conducting polymers. Reproduced from ref. 233 with permission from ACS, Copyright [2021]. | ||
Thus, clearly pPTZ-EE, within one polymeric system demonstrating dual properties of soft texture and charge conducting properties, holds considerable promise for the development of wearable electronics.
Furthermore, Chabinyc and coworkers incorporated redox-active croconate dianions in poly(3-hexylthiophene) semiconducting polymer having tetraethylene glycol side chains, leading to double doping that alters electrostatic interactions between the carriers and counterions (Fig. 54).234 The resulting P(g42T-T): croconate2− possesses lower activation energy for electrical conductivity, maintaining the crystalline order in P(g42T-T).
 | ||
| Fig. 54 (a) Structure of the P(g42T-T) with croconate−2. Thermoelectric parameters of chemically doped and ion exchange P(g42T-T) films. (b) Seeback coefficients (S) and (c) power factors (PF) of P(g42T-T):PF6−, P(g42T-T):PF6−, low, and P(g42T-T): croconate−2 thin films Reproduced from ref. 234 with permission from Wiley-VCH, Copyright [2023]. | ||
This strategy exemplifies a novel approach for enhancing charge transport and optimizing the thermoelectric performance of the semiconductor polymer by fine-tuning the doping levels of croconate counterions. The sufficient stability of the croconate dianion makes it applicable for energy harvesting with wearable electronics.
Recently, researchers have installed coumarin light-sensitive groups to PILs for fabricating smart PIL-based electronic devices. Zhang and coworkers synthesized a novel photoresponsive PIL-based solid electrolyte by merging coumarin-containing PIL with polyvinylidene fluoride hexafluoropropylene (Fig. 55).235 Interestingly, the ionic conductivity of the designed PIL could be reversibly modulated with a maximum modulation of 95% up to at least 7 times upon exposure to alternative light irradiation of 365 and 254 nm.
 | ||
| Fig. 55 (a) Reversible dimerization and cleavage of coumarin groups upon exposure to 365/254 nm light. (b) Schematic diagram of crosslinking network formation and dissociation. Reproduced from ref. 235 with permission from BME-PT, Copyright [2023]. | ||
Upon exposure to UV irradiation of 365 nm, coumarin derivatives undergo [2 + 2] cycloaddition forming a cyclobutane structure resulting in decreased ionic conductivity. Furthermore, this cyclobutane structure is cleaved on exposure to UV light of 254 nm and is accompanied by an increase in ionic conductivity. This photodimerization and photocleavage of coumarin components significantly alters the polymer segmental motion and thus justifies the observed reversible control over ionic conductivity.
This wide range of potential applications makes polymerized ionic liquids a prime candidate for further development, making them viable in organic electronics.
3.8 Challenges and future perspective: PILs and stimuli-responsive PILs
Several significant, yet unanswered inquiries obstruct the advancement of more sophisticated polymerized ionic liquids for device applications, as shown in Fig. 56. | ||
| Fig. 56 Schematic illustration highlighting the scientific gaps that need to be filled to enable PILs for advanced organic electronics. | ||
These inquiries circle understanding and creating a balance among the specific interactions encompassing ion-dipole interactions, electrostatic interactions, hydrogen-bonding, and acid–base interactions that govern the dynamics of ion transport and the selectivity of ion absorption in these materials. The fundamental insights derived from these inquiries will facilitate the identification of robust design rules to stimulate innovative device applications.
Although technological advancements offer potent and resilient instruments like BDS,16 SAOS, SAXS, WAXS,236etc. for revealing pivotal insights regarding these materials, it is evident that modeling and simulations are indispensably required to supplement the discovery of experimental outcomes and steer additional experimental avenues to augment our comprehension of the mechanisms of ion transportation, as well as the progression of enhanced materials for device applications.
Over several decades of extensive research on PILs, a significant amount of crucial knowledge on the relationship between structure and properties has been uncovered. Nonetheless, there are still numerous challenges that need to be addressed. These challenges consist of achieving both mechanical stability and rapid ion transport in a synergistic manner, facilitating the separation of ion transport from segmental motion, and organizing attached ionic groups in a way that promotes efficient ion transport.
Finding solutions to these problems would represent a significant advancement in the development of PILs as highly promising materials to guide the design of cutting-edge organic electronics. Discerning the phenomenon of nanophase separation, specifically the properties of the hydrophobic region in relation to the ion-conducting phase, will be paramount in the rational development of PILs that exhibit high conductivity and maintain mechanical and chemical steadiness in a variety of operational scenarios.
Furthermore, there are still considerable challenges that remain unresolved with regard to the processing and preparation of PILs for large-scale industrialization for their rapid, sustainable, and low-cost application in advanced organic electronic devices.
The application of PILs for constructing fuel cells such that the fuel cell membranes can function without the need for humidification and at significantly higher temperatures necessitates further investigation.
Although significant interest has been directed towards the advancement of neuromorphic devices, present advancements are currently confined to a limited number of nerve or synaptic devices, all of which are focused on basic biological nerve functions.74,75 Subsequently, it would be beneficial to conduct an in-depth investigation of PIL-based materials for the construction of high-performing neuromorphic devices.
Very few studies have been conducted thus far to progress the technology in the direction of achieving rapidly responsive and low-voltage-driven electro-active actuators. Consequently, it is highly desirable for researchers to focus on the development of such electro-active actuators. It is possible to anticipate a significant advancement in the progress of electro-active actuators operated at low voltages through the achievement of efficient enhancement in both ion transport characteristics and mechanical properties of ionic polymer actuators, which are built upon polymer electrolytes and ILs, and are designed optimally.
Studies are still lacking in the comprehensive analysis of the design, synthesis, function and implementation of PILs as biodegradable materials in contemporary organic devices. It is critical to investigate the distinctive characteristics of these materials, which enable the creation of devices that are both biodegradable and environmentally friendly, thereby promoting sustainability.237 Moreover, biodegradable materials would be valuable for fabricating transient electronic devices that are pivotal for security and military operations and disintegrate into biologically harmless smaller components after fulfilling their intended purpose.238
However, in order to enhance the operational lifetimes of transient organic electronic devices, a deeper comprehension of the degradation kinetics of such PIL-based materials is necessary for their implementation in practical applications. To do so, it would be interesting to investigate how the degradation rate in different environments is contingent upon the chemical composition of the PIL, the influence of electron-donating/withdrawing groups and the extent of branching.
Consequently, critically assessing the role of the biodegradable PIL component in the electronic device, as well as the distinctive properties that enable the devices to be employed in specific biodegradable applications, is needed. These studies will make a valuable contribution in delineating the fundamental approaches for mitigating environmental pollution and attaining sustainable development. Furthermore, it will facilitate the future advancement of this burgeoning field: biodegradable “green” electronics.
We posit that a significant progression would entail the formulation of methodologies for the design of mechanically stable PIL materials that yield not only high-performing devices, but also contribute to the enhancement of their environmental durability. We maintain that such persistent endeavors will undoubtedly direct the implementation of PILs in transient electronics. Hence, forthcoming investigations must prioritize the enhancement of electronic device functionality by leveraging the design of biodegradable PILs.
The development of triggerable PILs with heat and light-controllable conductivity, which integrates advantageous characteristics from both materials classes, has surprisingly fallen behind other classes of ion-conducting materials. Cutting-edge applications that significantly rely on photo- and thermally conductive properties would benefit greatly from progress in tuneable PILs, and would also control the operational lifetime of transient electronics.
Furthermore, the non-intrusive characteristic of the photomodulation process, which circumvents the utilization of supplementary chemical components to regulate ionic conductivity, introduces an unparalleled potential for subsequent mechanistic investigations that is critical to achieving a wide array of stimuli-responsive PILs for device applications.
One intriguing constraint of existing PILs is that their levels of mobile ions are typically fixed under specific experimental conditions. However, the prospect of novel device categories could materialize if the mobile ion content of PILs could be altered in the presence of a straightforward external chemical or physical stimulus. For instance, this type of material could be utilized to conceive electronic devices that can be permanently activated or deactivated upon command. Hence, the attainment of “triggered” ion release PILs poses a significant challenge in order to facilitate the emergence of new technological applications.
The acquisition of more quantitative information regarding the change in ionic conductivity in triggerable PIL materials could be achieved through impedance measurements, and is more accurate; however, the execution and interpretation of such measurements is challenging. Conducting these measurements in the lateral geometry required for UV-polymerizable materials is not a straightforward task.
Furthermore, interpreting the data in terms of absolute mobile ion concentrations would still pose difficulties. The application of models to dielectric/impedance data and the extraction of relevant mobile ion concentrations are hindered by various factors, including substrate effects. Thus, it is necessary to develop BDS models that can precisely measure the mobile ion concentration in these triggerable materials. This is vital for the design of devices that can be permanently activated or deactivated upon using external stimuli.
4 Zwitterionic PolyILs
Progress towards achieving long-lasting and high-energy lithium-ion batteries has been impeded by instabilities occurring at the interfaces between the electrolyte and electrode. These instabilities result in a lack of cycling stability, and there are also safety concerns related to the use of lithium metal anodes, which have a high energy density.239 To address these issues, solid polymeric electrolytes (SPEs) with their high stability and mechanical robustness have been proposed as a potential solution.240 However, the effectiveness of SPEs is limited by the slow dynamics of the polymer segments, which hampers their conductivity.241 Recently, zwitterionic polyILs have been extensively investigated to mitigate these challenges.A zwitterion refers to a form of substance that is electrically neutral, while simultaneously being composed of covalently bonded anion and cation entities. The coexistence of bonded positive and negative charges in zwitterions can induce a dipole moment. This dipole moment serves to diminish the electrostatic attraction between cations and anions through dipole rotation, which is known as the zwitterion effect.242
In a zwitterionic polymer, the interaction between lithium and framework ions is significantly weakened and there exists ample free volume for the superionic transport of Li+. Consequently, the Li+ conduction is decoupled from segmental relaxation and the glass transition temperature, which are typically relied upon by traditional polyethylene oxide-based solid polymer electrolytes.243
Furthermore, the zwitterionic functional groups have the capability to create dipole–dipole physically cross-linked scaffolds amongst themselves when integrated into a polymer matrix. This phenomenon enables a significant improvement in the mechanical rigidity of gel polymer electrolytes.244
Researchers have successfully administered zwitterionic PIL-based polymer electrolytes in Li–metal batteries. To facilitate this, Zheng et al. used propylene carbonate as the solvent of the gel polymer electrolyte to photopolymerize imidazolium-type zwitterion 3-(1-vinyl-3-imidazolio)propanesulfonate (VIPS) and LiTFSI (Fig. 57).245
 | ||
| Fig. 57 PIL organogel after crosslinking in the presence of PC. Reproduced from ref. 245 with permission from ACS, Copyright [2017]. | ||
The polymerized matrix of [VIPS][LiTFSI] furnished Li+-rich channels to promote Li+ transportation, consequently resulting in high ionic conductivity of 10−3 S cm−1. Furthermore, the half-cell's cycling performance was markedly improved by the assembly of the PIL-based polymer electrolyte.
Moving forward, Panzer and colleagues conducted a study in which they described a physically crosslinked polymer-supported gel electrolyte that contained a lithium salt/ionic liquid solution (Fig. 58).246
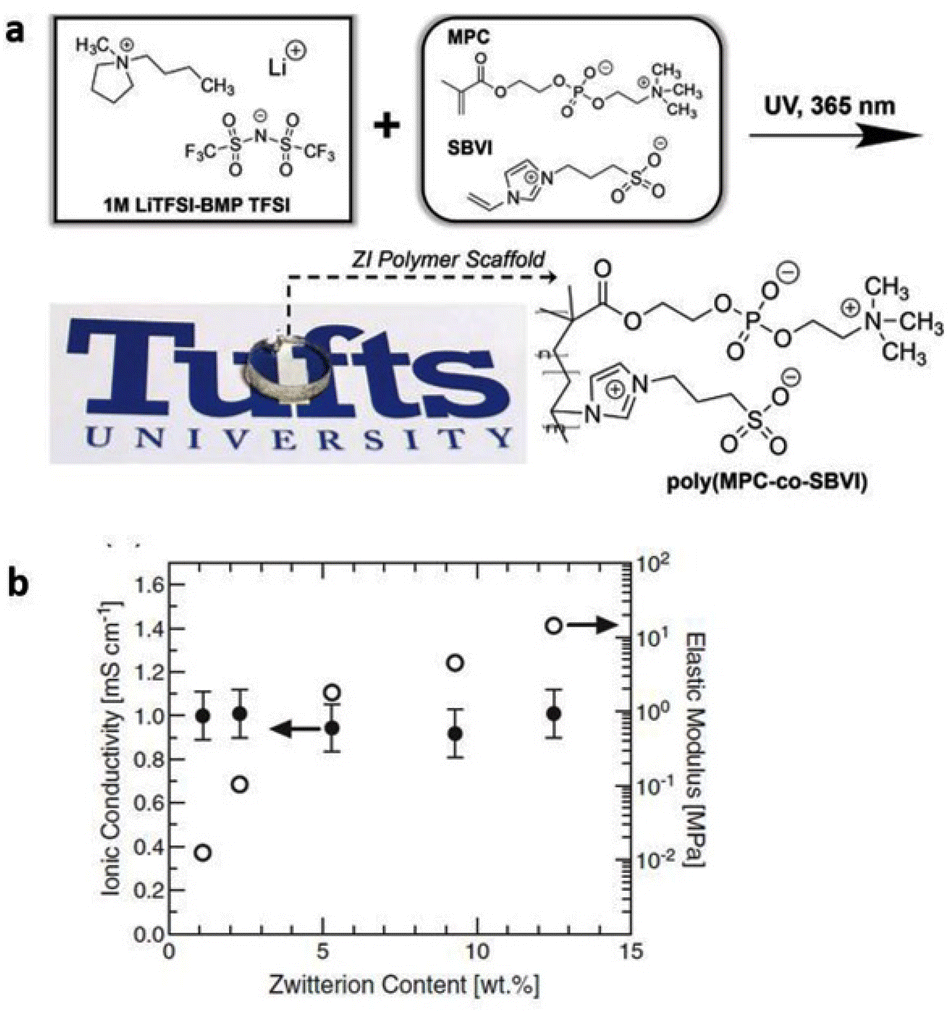 | ||
| Fig. 58 (a) Schematic diagram showing the molecular structures of ions present in the base liquid electrolyte, LiTFSI dissolved in the ionic liquid N-butyl-N-methylpyrrolidinium bis(trifluoromethylsulfonyl)imide (BMP TFSI), to which are added two zwitterionic (ZI) comonomers, 2-methacryloyloxyethyl phosphorylcholine (MPC) and sulfobetaine vinylimidazole (SBVI), resulting in the creation of a physically cross-linked ZI gel electrolyte via in situ UV-initiated free radical copolymerization. (b) Room-temperature ionic conductivity and compressive elastic modulus values for ZI gel electrolytes plotted as a function of zwitterion content. Reproduced from ref. 246 with permission from Wiley-VCH, Copyright [2018]. | ||
This gel electrolyte incorporates a fully zwitterionic (ZI) copolymeric network. The inclusion of zwitterionic functional groups in the polymer network contributed to the mechanical strength of the gel polymer electrolyte (GPE), reaching a remarkable value of 14.3 MPa. Additionally, the presence of amphoteric ions in the polymer allows for stronger interactions with the ILs and lithium salts in the GPE. As a result, the coordination shell of Li+ in the electrolyte undergoes changes, leading to an enhancement in the electrochemical performance of the GPE electrolyte. The ZI copolymer-supported gels exhibit comparable conductivity to the base liquid electrolyte, with a value of approximately 1 mS cm−1 at room temperature. Being compatible with lithium metal anode, these materials hold great potential for the advancement of lithium-based electrochemical energy storage devices in the future.
Furthermore, zwitterionic solid polymeric electrolytes capable of self-assembling into superionically conductive domains have also been designed and fabricated (Fig. 59).243
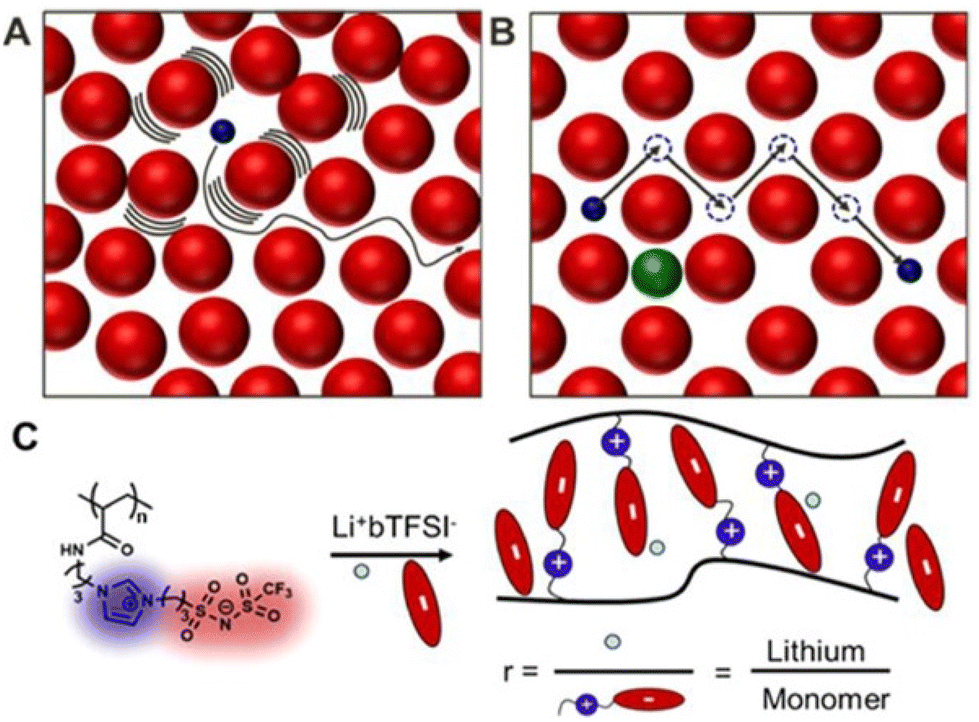 | ||
| Fig. 59 Schematic representation of the path of an ion through (A) a typical solid polymer electrolyte, in which ion transport is coupled to the timescale of local rearrangements, and (B) an ordered solid with sufficient free volume to enable superionic transport. The atoms of a crystal are typically confined to specific lattice positions, allowing sufficiently small ions with a low charge (blue spheres) to diffuse though the matrix (red spheres) via successive discrete hops involving vacant lattice sites or interstitial sites, while the motion of larger or highly charged ions (green sphere) is excluded. (C) The zwitterionic polymer studied herein comprises IL-inspired ions tethered to the backbone. Mobile ions are doped in through the addition of a Li+TFSI− salt at select molar ratios. Reproduced from ref. 243. | ||
These polymeric electrolytes are easily processable and possess unparalleled lithium-ion conduction and selectivity. As a pendant to the polymer group, bulky imidazolium-trifluoromethanesulfonamide was selected to produce weaker coulombic interactions and facilitate Li+ movement by sufficient void space in the formed crystal.
The self-assembly design allowed the separation of Li+ transport from the polymeric matrix's fluidity. As a result, the decoupling facilitated labile ion–ion interactions and demonstrated notably high selectivity of t+ ≈ 0.6–0.8 and Li+ conductivity of 1.6 mS cm−1, both of which were significantly higher than the state-of-the-art PEO currently in use (t+ ≈ 0.2). These findings show that polymeric zwitterionic liquids have the potential to serve as a novel solid polymer electrolyte support structure with superionic conduction, improving the efficiency of electrochemical cells.
Recently, researchers have designed a new and an intriguing kind of zwitterionic single-ion conducting polymer electrolyte that displays high dielectric constant by minimizing the electrostatic attraction force and facilitating ion dissociation. To enable such a zwitterionic polyIL for efficient Li+ conduction, Cheng and coworkers chemically co-grafted two propanesulfonyl(trifluoromethanesulfonyl)imide groups to the nitrogen atom of poly(biphenyl piperdinium), PBP-g-Li(PSI)2 (Fig. 60).247
 | ||
| Fig. 60 (a) Schematic representation of grafted poly(biphenyl piperidinium)s including PBP-g-Li(PSI)2 (b) dielectric spectra of PBP-g-Li(PSI)2 and PBP-g-LiPSI (c) ionic conductivity of PBP-g-LiPSI and PBP-g-Li(PSI)2 gel blend polymer electrolyte membranes. Reproduced from ref. 247 with permission from Elsevier, Copyright [2022]. | ||
This assembly establishes an ion-dipole adjacent to the lithium-ion carrier. Consequently, the zwitterionic, PBP-g-Li(PSI)2 electrolyte exhibits 3.5 times higher dielectric constant of 22.3 at 2 MHz and twice the ionic conductivity of 2.41 × 10−4 S cm−1 at 25 °C in contrast to the control PBP-g-Li(PSI) gel polymer electrolyte membrane, in which only one propanesulfonyl(trifluoromethanesulfonyl)imide group is grafted to poly(biphenyl piperidinium). Furthermore, the designed zwitterionic, PBP-g-Li(PSI)2 electrolyte was not only able to significantly resist galvanostatic cycling in a lithium symmetric cell at ±0.5 mA cm−2@ 2 mA h cm−2 for 2000 h, but could also conduct steadily in lithium metal secondary battery for extended duration.
These studies clearly showcase the remarkable potential of polymeric zwitterionic liquids for the rapid and selective Li+ conduction, although additional research is necessary to demonstrate the in operando durability and stability of electrochemical cells constructed upon these design principles.
5 Doubly polymerized ionic liquids (DPILs)
The characteristics, synthesis, uses, and mechanism of ion transport in ionic liquids and polymerized ionic liquids have all been thoroughly studied by researchers to this point. In addition, rigorous efforts have been made to enhance the ionic conductivity of PILs without compromising the mechanical strength. Significant investigations have been conducted to develop stimuli-responsive polymerized ionic liquids for fabricating advanced organic electronics. However, the doubly polymerized ionic liquid (DPIL), a novel class of materials, has gained attention only very recently.12,22 As the name reflects, in these materials both the cation and the anion are tethered to the polymeric backbone, unlike in singly polymerized ionic liquid where either the cation or the anion is covalently attached to the polymer backbone. In fact, the cationic and anionic monomers in these materials form a 1:1 copolymer.
At initial observation, DPILs exhibit a structural resemblance to zwitterionic polyILs due to the presence of covalently bonded cation and anion in both types of polymerized ionic liquids. Nevertheless, notable differences exist between these two categories. In zwitterionic polymeric electrolytes, the cation is directly covalently attached to the polymer chain, while the anion is not. Instead, the anion is attached to the cation through a spacer, typically consisting of 2 to 3 carbon atoms, as reported to date. In contrast, in DPILs both the positively and negatively charged species are directly covalently bonded to the polymer chain (Fig. 61).
 | ||
| Fig. 61 Schematic representation of the structural difference between (a) Zwitterionic PIL and (b) DPIL. | ||
These types of polymerized ionic liquids for the very first time were investigated by Yoshizawa et al., and constituted of vinylimidazole and vinylsulfonic acid (Fig. 62).23
 | ||
| Fig. 62 (a) Structure of ionic liquid monomers obtained by neutralization. (b) Effect of LiTFSI concentration on the ionic conductivity of corresponding polymers P1 and P2. Reproduced from ref. 23 with permission from Wiley-VCH, Copyright [2002]. | ||
The oppositely charged ionic species were copolymerized in the bulk ionic liquid, which resulted in a sharp decrease in conductivity that was measured to be on the order of 10−9 Siemens per cm. A small amount of non-polymerizable LiTFSI was added in order to highlight the crucial part that free ions play in the ionic conductivity of these materials. This led to 4–5 orders of magnitude increase in conductivity due to the generation of carrier ions in the ionic liquid copolymers.
Shaplov and coworkers copolymerized pyrrolidinium or imidazolium-based cationic monomer with sulfonate-based anionic monomer and further completely removed the mobile counterions (Fig. 63).12
 | ||
| Fig. 63 Schematic representation of the copolymerization of oppositely charged monomers. Reproduced from ref. 12 with permission from ACS, Copyright [2011]. | ||
Ionic conductivity drastically decreased as a result, to the order of 10−12 Siemens per cm. These copolymers were found to be highly thermally stable and also exhibited higher glass transition temperatures.
Recently, Mezzenga and coworkers copolymerized cations and anions each containing a vinyl polymerizable group via RAFT polymerization (Fig. 64).25 Acrylic acid, vinylphosphonic acid, vinylbenzoic acid, and bis[2-(methacryloyloxy)ethyl]phosphate constituted a series of polymerizable anions, while vinyl-1,2,4-triazole was selected as the polymerizable cation.
 | ||
| Fig. 64 (a) Protic ionic liquids investigated in the study (b) copolyampholytes produced from RAFT polymerization of protic ILs. (c) Solid-state 2D NMR for the identification of the molecular arrangement of the polymer backbone. (d) Representation of the mechanical properties of the synthesized copolyampholytes. Reproduced from ref. 25 with permission from ACS, Copyright [2017]. | ||
The solid-state NMR analysis illuminated the molecular arrangement of the polymeric backbone and indicated the presence of copolymers with both random and alternating copolymer chains. Essentially these polyampholytic copolymers were found to be glassy and amorphous, having Young's moduli in the range of 1.4–2.2 GPa which is a typical range for polymers in a glassy state (Fig. 64).
Laaser and coworkers recently developed a model DPIL system similar to the commonly used and commercially available [EMIM][TFSI] ionic liquid.22 As shown in Fig. 66, the designed DPIL consisted of an imidazolium cation and a triflurosulfonylimide anion with methacrylate as a polymerizable group on both ionic species. Each ionic species in the DPIL had its methacrylate group efficiently polymerized, producing a statistical copolymer with both ionic species covalently bonded to the polymer chains. The designed DPIL displayed negligible ionic conductivity with ions locked in place. This property is in marked contrast to zwitterionic polyILs that display high ionic conductivity, to which the key is decoupling of ion transport from segmental motion.243 Furthermore, the dielectric behavior of DPILs was significantly different in contrast to singly polymerized ionic liquids (SPILs).
Designing DPILs with negligible ionic conductivity exemplifies a counter-intuitive approach in the polymer community, where tremendous efforts have been made to enhance ionic conductivity without sacrificing mechanical properties for constructing advanced organic electronics. However, sometimes certain specific applications call for an overall decrease in the mobile ion content and ionic conductivity rather than an increase, as discussed below.
5.1 DPILs in action
Ion-locking has been widely used in the semiconductor community to study the characteristics of two-dimensional semiconductors and interfaces.248,249 Researchers have mostly used PEO as the polymer matrix for EDL-locking experiments. However, the need to conduct experiments at cryogenic temperatures considering its low glass transition temperature (approx. −70 °C in the pure polymer, or −40 °C to −10 °C with added salt) minimizes its potential for room-temperature applications.250 Furthermore, its incompatibility with accustomed semiconductor processing methods makes its amalgamation into existing device manufacturing lines arduous.251In efforts to address this issue, researchers have recently used a PVA/LiClO4 system to achieve EDL locking at room temperatures (Fig. 65).251 Using the PVA/LiClO4 system, an EDL retention time of approximately 4 hours was achieved, demonstrating a significant improvement of six orders of magnitude enhancement in EDL retention time than the standard PEO-based electrolytes.
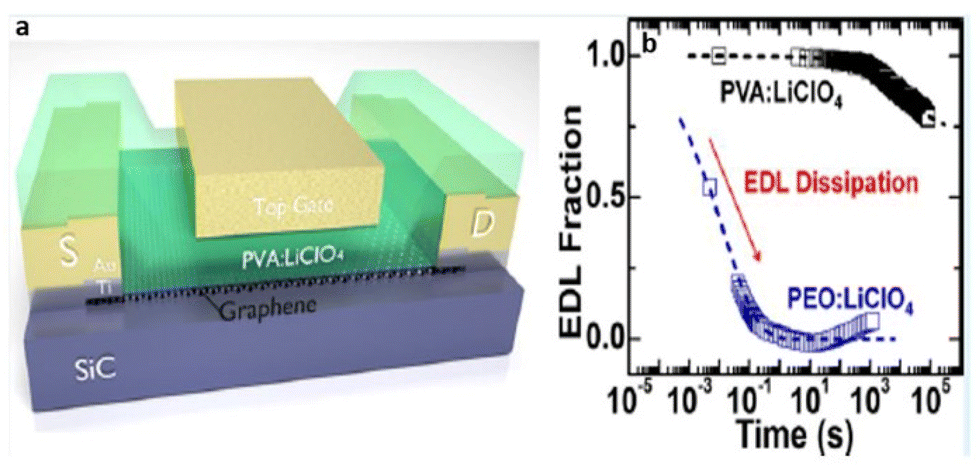 | ||
| Fig. 65 (a) Schematic representation of the graphene FET fabricated; (b) enhancing the room-temperature electric double layer retention time in 2-D crystal FETs. Reproduced from ref. 251 with permission from ACS, Copyright [2017]. | ||
However, practical applications entail appreciable refinements to attain EDL retention times substantially greater than 4 hours and controllable enough to precisely modulate the ion locking of EDL-locked materials. Recently, Liang et al. for the first time demonstrated the application of the ion-locking phenomenon in [EMIM][TFSI]-modelled DPIL for constructing stable p–n junctions (Fig. 66).21
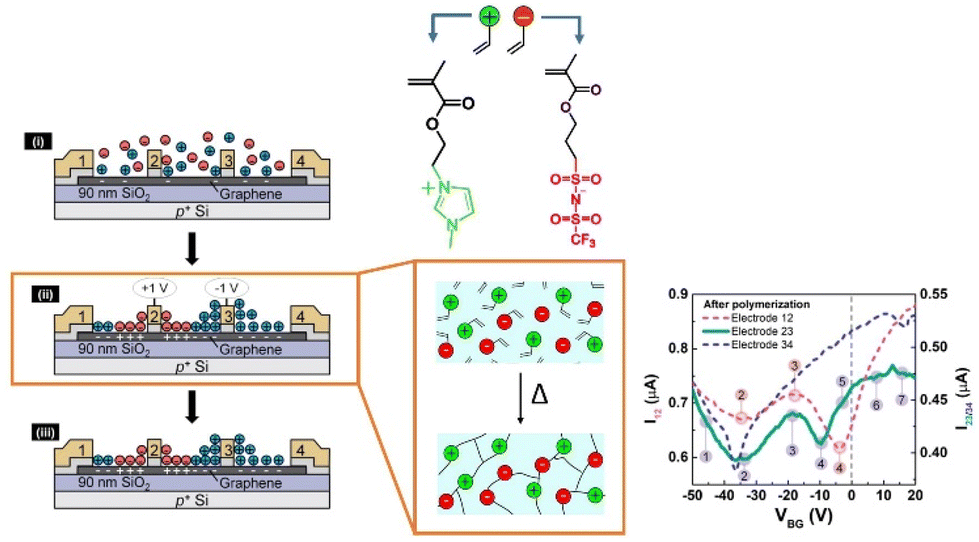 | ||
| Fig. 66 Schematic representation of ion-locking phenomenon in [EMIM][TFSI]-modeled DPIL to create a stable p–n junction in a two-dimensional semiconductor. The resulting transfer characteristics of a representative device are shown here; the presence of two current minima with variation in the back gate voltage are the tell-tale signature of successful formation of a p–n junction. Reproduced from ref. 21. | ||
In this work, DPIL was deposited on a 2-D semiconductor graphene. Applying the appropriate voltages across the electrodes caused the ions to drift to the interface, where they caused a p–n junction doping pattern in the semiconductor. In addition to locking the ions in place after the polymerization of DPIL in this state, the researchers were also able to create a stable p–n junction that held up even after the programming voltages were turned off. This is interesting because, unlike conventional electrolytes, this approach does not require the constant application of a gate voltage to set the doping state and hold the ions in place. These studies clearly demonstrate the criticality of developing a profound understanding of the mechanism of ion locking in DPIL materials and further describe their conduction and relaxation timescale that will govern the functional lifetimes of ion-locked electronic devices.
5.2 BDS for understanding ion locking in DPILs
For many years, scientists in the polymer community have been delving deeply into the mechanism of ion transport in singly polymerized ionic liquids.17,194,195,252 Nevertheless, there is presently no thorough analysis that sheds light on the dynamics of ion motion in DPIL materials. The use of DPIL systems inside organic thin-film transistors or field-effect transistors requires a detailed study of the dynamics of ion motion in these materials. In a recent step forward in this direction, Laaser and colleagues compared and examined the dielectric response and ion transport of DPIL with singly polymerized ionic liquids (Fig. 67).22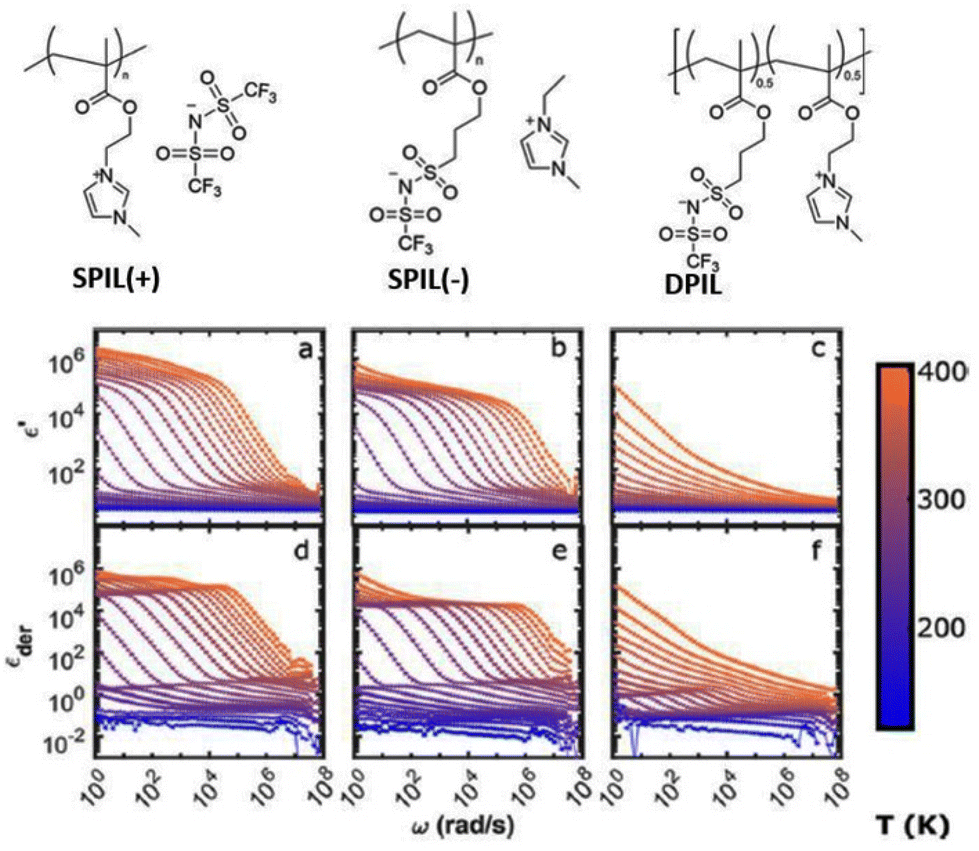 | ||
| Fig. 67 Schematic representation of the polymers investigated in the study. Frequency dependence of (a–c) the dielectric permittivity and (d–f) the dielectric loss derivative function εder for (a and d) SPIL(+), (b and e) SPIL(−), and (c and f) DPIL across the range of temperatures investigated in the study. Reproduced from ref. 22 with permission from ACS, Copyright [2021]. | ||
Their BDS investigations on DPIL materials provide unambiguous evidence of the presence of robust physical crosslinks created by ion pairs between polymer chains as a result of the two ionic species’ copolymerization. These strong ionic interactions between the polymer chains are homologous to the one present in polyampholyte gels, and account for the obstruction of ion transport in these materials.253
The idea that ionic interactions between the polymer chains in the DPIL material predominate was further supported by the notably reduced DC conductivity of DPIL compared with its SPIL counterparts. The incapability of VFT dynamics to explain the temperature dependence of DC conductivity in DPIL systems reinforces the decoupling of bulk ion transport in DPIL to the available free volume.184
Multiple ion pairs would therefore need to relax simultaneously in order to facilitate ion transport in DPIL material that is extensively physically cross-linked. These inter-chain crosslinks will probably need to be broken and reformed significantly in order to facilitate any ion transport. These disturbances require a lot of energy and happen slowly (Fig. 68). This is in marked contrast to zwitterionic PILs that display a superionic mechanism even in the presence of physical cross-links.
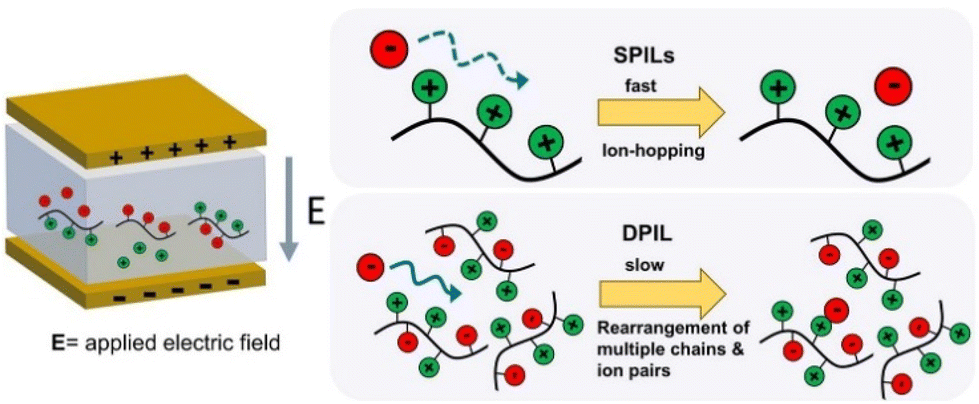 | ||
| Fig. 68 Schematic illustration of the bulk ion transport in DPILs vs. SPILs. In SPILs, individual ions can move relatively independently of other charged units. However, in DPILs movement of a single ion requires rearrangement of multiple chains and ion pairs. Reproduced from ref. 22 with permission from ACS, Copyright [2021]. | ||
Taken together, their BDS findings explicitly suggest that polymerizing both the ionic species results in the formation of extensive cross-links between the polymer chains that slows down the molecular relaxations holding the ions longer in place, indicating ionic cross-linking is a key driver to the “ion-locking” phenomenon in DPILs.
Analogous to modulation in ionic conductivity in polymerized ionic liquids in response to external stimuli, it would be interesting to design DPIL-based stimuli-responsive materials. This would eventually lead to “on-command” ion locking and unlocking in response to simple chemical and physical triggers and enable tuneable functional devices with pre-determined EDL-retention time. To advance these applications, however, will require a thorough comprehension of the mechanism underlying ion locking in these materials as well as an understanding of how these mechanisms impact the creation of design guidelines for tuneable functional devices.
6 Challenges and future motivations
This review provides an overview of both ionic liquids and polymerized ionic liquids and their applications in electronic devices. Considering some applications in the polymer community demand not only increasing the ionic conductivity but also decreasing the ionic conductivity,21 this review presents a new outlook to researchers for implementing polymerized ionic liquids with restricted ionic conductivity like DPILs in device applications.However, a lot of work still needs to be done before significant success can be achieved in fabricating devices using DPILs (Fig. 69). Undoubtedly, we are at the proof-of-concept juncture, and shortly, DPILs will have to be thoroughly and rigorously investigated to address critical questions at the intersection of polymer chemistry and organic electronics.
 | ||
| Fig. 69 Schematic illustration highlighting the scientific gaps that need to be filled to enable DPIL-based materials for advanced organic electronics. | ||
As illustrated by Laaser and coworkers, the failure of VFT dynamics to describe conductivity dependence on temperature in DPIL-based materials, even above the material's Tg, explicitly demands new models analogous to VFT and Arrhenius models that can explain this conductivity behavior.22,254 Furthermore, the mobile ion content that has been previously evaluated by researchers in polymerized ionic liquids using electrode polarization analysis encompassing the MacDonald model cannot be extrapolated to DPIL-based materials.50 Thus, this accounts for the need to develop models that can accurately quantify the mobile ion content in these materials to enable the quantification of dependence of EDL retention times on the free ion content, which is critical for designing devices that require significant restriction of ion transport in these materials.
Ganesan and co-workers have successfully scrutinized the computational modelling and simulation aspects associated with the mechanism of ion transport in polymerized ionic liquids.8,255 However, this rigorous investigation is still missing for DPIL-based materials due to the lack of accurate and reliable force fields to model the ion transport mechanisms in these materials. The nanoscale structure of singly polymerized ionic liquids has a significant impact on ion transport.200,256,257 This emphasizes the need to incorporate the influence of nanoscale segregation into polar and nonpolar domains in DPIL-based materials in computational simulations to thoroughly comprehend how the nanostructure of these materials affects the dynamics of ion motion and further EDL retention times for their application in devices.
The significant restriction of ion transport in DPIL-based materials for device application entails the development of physically motivated design rules involving the exploration of several chemical and physical factors to further aid the design of these materials, providing detailed insights into their molecular mechanisms. Thus, it is time for researchers to reimagine polymerized ionic liquids with negligible ionic conductivity like DPILs in contrast to singly polymerized ionic liquids with relatively higher ionic conductivity for advanced organic electronics.
We anticipate this review will not only prove riveting and educational to readers of the polymer community who are engrossed in scrutinizing the fundamental properties of ion-containing polymers, but also to those involved in advancing new device applications.
7 Conclusions
In this review, we take the readers on a journey in the polymer community through the sequential advances made in ionic liquids to polymerized ionic liquids and then further doubly polymerized ionic liquids. We further briefly touch upon the rigorous efforts made by researchers over the years to increase the ionic conductivity of polymerized ionic liquids without sacrificing the mechanical properties of the PILs. We shed light on the use of broadband dielectric spectroscopy to gain a thorough understanding of the variables influencing the ionic conductivity of polymerized ionic liquids and illuminate the ion transport mechanism in these ion-containing polymers. In addition, we provide experimental findings, where researchers have incorporated stimuli-responsive handles to toggle the ionic conductivity and the mobile ion content on-command.Given that the polymerized ionic liquid community has placed a great deal of emphasis on improvising ion transport thus far, this review provides additional insight into an intriguing class of materials known as doubly polymerized ionic liquids, which represent a counterintuitive class of materials where ion motion is intentionally restricted. These materials introduce us to an interesting “ion-locking” phenomenon that results from extensive physical crosslinks in these materials. Further rigorous investigation of the chemical and physical factors governing this phenomenon should aid in the composition of DPIL materials for applications demanding considerable restriction of ion transport in ion-containing polymers.
Significant research on polymerized ionic liquids has already been completed, yet they are still lagging in practical application. The fundamental knowledge of polymerized ionic electrolytes as smart, tailorable materials needs to be increased for the development of advanced organic electronics for versatile applications. We believe this review will revitalize development across polymer chemistry, polymer physics, and organic electronics. We anticipate it will be of broad interest to readers inquisitive about novel chemistries for regulating the ion content of polymeric materials and stimulating the advancement of polymerized ionic liquids and doubly polymerized ionic liquids in electronic devices.
Author contributions
This manuscript was primarily written by Swati Arora. The author would like to thank Nagendra Verma's contribution to reviewing, editing, and producing pictures. Both the authors read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.References
- R. Brooke, M. Fabretto, M. Krasowska, P. Talemi, S. Pering, P. Murphy and D. Evans, J. Mater. Chem. C, 2016, 4, 1550–1556 RSC.
- H. C. Moon, C. Kim, T. P. Lodge and C. D. Frisbie, ACS Appl. Mater. Interfaces, 2016, 8, 6252–6260 CrossRef CAS PubMed.
- M. Watanabe, M. L. Thomas, S. Zhang, K. Ueno, T. Yasuda and K. Dokko, Chem. Rev., 2017, 117, 7190–7239 CrossRef CAS PubMed.
- D. G. Seo and H. C. Moon, Adv. Funct. Mater., 2018, 28, 1706948 CrossRef.
- R. Misra, M. A. McCarthy and A. F. Hebard, Appl. Phys. Lett., 2007, 90, 052905 CrossRef.
- T. Fujimoto and K. Awaga, Phys. Chem. Chem. Phys., 2013, 15, 8983 RSC.
- M. L. Hoarfrost and R. A. Segalman, Macromolecules, 2011, 44, 5281–5288 CrossRef CAS.
- S. Mogurampelly, O. Borodin and V. Ganesan, Annu. Rev. Chem. Biomol. Eng., 2016, 7, 349–371 CrossRef PubMed.
- J. Yuan, D. Mecerreyes and M. Antonietti, Prog. Polym. Sci., 2013, 38, 1009–1036 CrossRef CAS.
- A. S. Shaplov, D. O. Ponkratov and Y. S. Vygodskii, Polym. Sci., Ser. B, 2016, 58, 73–142 CrossRef CAS.
- W. Qian, J. Texter and F. Yan, Chem. Soc. Rev., 2017, 46, 1124–1159 RSC.
- A. S. Shaplov, P. S. Vlasov, E. I. Lozinskaya, D. O. Ponkratov, I. A. Malyshkina, F. Vidal, O. V. Okatova, G. G. Pavlov, C. Wandrey, A. Bhide, M. Schönhoff and Y. S. Vygodskii, Macromolecules, 2011, 44, 9792–9803 CrossRef CAS.
- H. Ohno, M. Yoshizawa and W. Ogihara, Electrochim. Acta, 2004, 50, 255–261 CrossRef CAS.
- M. Yoshizawa and H. Ohno, Electrochim. Acta, 2001, 46, 1723–1728 CrossRef CAS.
- S. Arora, J. Liang, S. K. Fullerton-Shirey and J. E. Laaser, ACS Mater. Lett., 2020, 2, 331–335 CrossRef CAS.
- F. Kremer and A. Schönhals, Broadband dielectric spectroscopy, 2003 Search PubMed.
- U. H. Choi, A. Mittal, T. L. Price, H. W. Gibson, J. Runt and R. H. Colby, Macromolecules, 2013, 46, 1175–1186 CrossRef CAS.
- C. Iacob, A. Matsumoto, M. Brennan, H. Liu, S. J. Paddison, O. Urakawa, T. Inoue, J. Sangoro and J. Runt, ACS Macro Lett., 2017, 6, 941–946 CrossRef CAS PubMed.
- K. Nakamura, T. Saiwaki and K. Fukao, Macromolecules, 2010, 43, 6092–6098 CrossRef CAS.
- J. Sangoro, C. Iacob, A. L. Agapov, Y. Wang, S. Berdzinski, H. Rexhausen, V. Strehmel, C. Friedrich, A. P. Sokolov and F. Kremer, Soft Matter, 2014, 10, 3536–3540 RSC.
- J. Liang, K. Xu, S. Arora, J. E. Laaser and S. K. Fullerton-Shirey, Materials, 2020, 13, 1089 CrossRef CAS PubMed.
- S. Arora, J. Rozon and J. E. Laaser, Macromolecules, 2021, 54, 6466–6476 CrossRef CAS.
- M. Yoshizawa, W. Ogihara and H. Ohno, Polym. Adv. Technol., 2002, 13, 589–594 CrossRef CAS.
- H. Gu, F. Yan and J. Texter, Macromol. Rapid Commun., 2016, 37, 1218–1225 CrossRef CAS PubMed.
- C. C. J. Fouillet, T. L. Greaves, J. F. Quinn, T. P. Davis, J. Adamčík, M. Sani, F. Separovic, C. J. Drummond and R. Mezzenga, Macromolecules, 2017, 50, 8965–8978 CrossRef CAS.
- Y. Wang, R. Ma, W. Nie, X. Zhao and J. Yin, Langmuir, 2023, 39, 14006–14014 CrossRef CAS PubMed.
- M. D. Fayer, Chem. Phys. Lett., 2014, 616–617, 259–274 CrossRef CAS.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS PubMed.
- Z. Xue, L. Qin, J. Jiang, T. Mu and G. Gao, Phys. Chem. Chem. Phys., 2018, 20, 8382–8402 RSC.
- K. Nakamura and T. Shikata, ChemPhysChem, 2010, 11, 285–294 CrossRef CAS PubMed.
- H. Srour, H. Rouault, C. C. Santini and Y. Chauvin, Green Chem., 2013, 15, 1341 RSC.
- N. De Vos, C. Maton and C. V. Stevens, ChemElectroChem, 2014, 1, 1258–1270 CrossRef CAS.
- Y. Cao, Y. Chen, X. Sun, Z.-M. Zhang and T. Mu, Phys. Chem. Chem. Phys., 2012, 14, 12252 RSC.
- S. Kazemiabnavi, Z. Zhang, K. Thornton and S. Banerjee, J. Phys. Chem. B, 2016, 120, 5691–5702 CrossRef CAS PubMed.
- K. Li, Y. Zhou, H. Han, S. Zhou, W. Feng, J. Nie, H. Li, X. Huang, M. Armand and Z. Zhou, Electrochim. Acta, 2010, 55, 7145–7151 CrossRef.
- T. Y. Wu, S. G. Su, S. T. Gung, M. Lin, Y. Lin, C. A. Lai and I. Sun, Electrochim. Acta, 2010, 55, 4475–4482 CrossRef CAS.
- S. T. Handy and M. Okello, J. Org. Chem., 2005, 70, 1915–1918 CrossRef CAS PubMed.
- M. C. Kroon, W. Buijs, C. J. Peters and G. Witkamp, Green Chem., 2006, 8, 241–245 RSC.
- P. C. Howlett, E. I. Izgorodina, M. Forsyth and D. R. MacFarlane, Zeitschrift für Physikalische Chemie, 2006, 220, 1483–1498 CrossRef CAS.
- D. R. MacFarlane, J. J. Golding, S. A. Forsyth, M. Forsyth and G. B. Deacon, Chem. Commun., 2001, 1430–1431 RSC.
- M. Chen, J. W. Dugger, X. Li, Y. Wang, R. Kumar, K. M. Meek, D. Uhrig, J. F. Browning, L. A. Madsen, T. E. Long and B. S. Lokitz, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1346–1357 CrossRef CAS.
- F. Fan, Y. Wang, T. Hong, M. Heres, T. Saito and A. P. Sokolov, Macromolecules, 2015, 48, 4461–4470 CrossRef CAS.
- A. Matsumoto, C. Iacob, T. Noda, O. Urakawa, J. Runt and T. Inoue, Macromolecules, 2018, 51, 4129–4142 CrossRef CAS.
- G. G. Raju, Dielectrics in Electric Fields, CRC Press, 2nd edn, 2016, pp. 83–136 Search PubMed.
- A. Rivera and E. A. Rössler, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73 DOI:10.1103/physrevb.73.212201.
- P. J. Griffin, A. P. Holt, K. Tsunashima, J. Sangoro, F. Kremer and A. P. Sokolov, J. Chem. Phys., 2015, 142, 084501 CrossRef PubMed.
- P. Sippel, P. Lunkenheimer, S. Krohns, E. Thoms and A. Loidl, Sci. Rep., 2015, 5, 13922 CrossRef CAS PubMed.
- J. Van Turnhout and M. Wübbenhorst, J. Non-Cryst. Solids, 2002, 305, 50–58 CrossRef CAS.
- M. Wübbenhorst and J. Van Turnhout, J. Non-Cryst. Solids, 2002, 305, 40–49 CrossRef.
- R. J. Klein, S. Zhang, S. Dou, B. H. Jones, R. H. Colby and J. Runt, J. Chem. Phys., 2006, 124, 144903 CrossRef PubMed.
- A. Rivera, A. Brodin, A. M. Pugachev and E. A. Rössler, J. Chem. Phys., 2007, 126, 114503 CrossRef CAS PubMed.
- J. Sangoro, A. Serghei, C. B. Haymob, P. Galvosas, J. Kärger, C. Wespe, F. Bordusa and F. Kremer, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2008, 77, 051202–051204 CrossRef CAS PubMed.
- C. Iacob, J. Sangoro, A. Serghei, C. B. Haymob, Y. Korth, J. Kärger, C. Friedrich and F. Kremer, J. Chem. Phys., 2008, 129, 234511 CrossRef CAS PubMed.
- T.-G. Kim, U. J. Kim, H. B. Son and J. Hur, Sci. Adv. Mater., 2017, 9, 1589–1594 CrossRef CAS.
- X. Ma, X. Lan, L. Wu, L. Wang, Q. Gu, Y. Shi, X. Gu and Z. Luo, Eur. Polym. J., 2020, 123, 109446 CrossRef CAS.
- S. Imaizumi, H. Kokubo and M. Watanabe, Macromolecules, 2011, 45, 401–409 CrossRef.
- D. Kim, X. Liu, B. Yu, S. Mateti, L. A. O’Dell, Q. Rong and Y. Chen, Adv. Funct. Mater., 2020, 30, 1910813 CrossRef CAS.
- L. Chen, J. Fu, Q. Lü, L. Shi, M. Li, L. Dong, Y. Xu and R. Jia, Chem. Eng. J., 2019, 378, 122245 CrossRef CAS.
- M. Forsyth, L. Porcarelli, X. Wang, N. Goujon and D. Mecerreyes, Acc. Chem. Res., 2019, 52, 686–694 CrossRef CAS PubMed.
- B. J. Park, S. Oh, F. S. Kim and S. T. Chang, J. Mater. Chem. C, 2019, 7, 10264–10272 RSC.
- J. A. Sirviö, Y. Yang, C. Liu, W. Chen, C. Ling, H. Lian and H. Liimatainen, J. Mater. Chem. C, 2020, 8, 550–560 RSC.
- C. Qian, J. Sun, J. Yang and Y. Gao, RSC Adv., 2015, 5, 14567–14574 RSC.
- J. Yoon, D. K. Kang, J. Won, J.-Y. Park and Y. S. Kang, J. Power Sources, 2012, 201, 395–401 CrossRef CAS.
- B. C. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- A. Andrew, et al. , Faraday Discuss., 2018, 206, 113–129 RSC.
- N. Papageorgiou, Y. Athanassov, M. Armand, P. Bonhôte, H. Pettersson, A. Azam and M. Grätzel, J. Electrochem. Soc., 1996, 143, 3099–3108 CrossRef CAS.
- R. Kawano, H. Matsui, C. Matsuyama, A. Sato, Md. A. B. H. Susan, N. Tanabe and M. Watanabe, J. Photochem. Photobiol., A, 2004, 164, 87–92 CrossRef CAS.
- H. Matsui, K. Okada, T. Kawashima, T. Ezure, N. Tanabe, R. Kawano and M. Watanabe, J. Photochem. Photobiol., A, 2004, 164, 129–135 CrossRef CAS.
- R. Kawano, T. Katakabe, H. Shimosawa, Md. K. Nazeeruddin, M. Grätzel, H. Matsui, T. Kitamura, N. Tanabe and M. Watanabe, Phys. Chem. Chem. Phys., 2010, 12, 1916 RSC.
- W. Li, A. G. Fadeev, B. Qi, E. Smela, B. R. Mattes, J. Ding, G. M. Spinks, J. Mazurkiewicz, D. Zhou, G. G. Wallace, D. R. MacFarlane, S. A. Forsyth and M. Forsyth, Science, 2002, 297, 983–987 CrossRef PubMed.
- J. Lee, M. J. Panzer, Y. He, T. P. Lodge and C. D. Frisbie, J. Am. Chem. Soc., 2007, 129, 4532–4533 CrossRef CAS PubMed.
- Q. Sun, W. Seung, B. J. Kim, S. Seo, S. W. Kim and J. H. Cho, Adv. Mater., 2015, 27, 3411–3417 CrossRef CAS PubMed.
- B. Tang, D. K. Schneiderman, F. Z. Bidoky, C. D. Frisbie and T. P. Lodge, ACS Macro Lett., 2017, 6, 1083–1088 CrossRef CAS PubMed.
- Y. LeCun, Y. Bengio and G. E. Hinton, Nature, 2015, 521, 436–444 CrossRef CAS PubMed.
- D. Wang, L. Wang, W. Ran, S. Zhao, R. Yin, Y. Yan, K. Jiang, Z. Lou and G. Shen, Nano Energy, 2020, 76, 105109 CrossRef CAS.
- J. Hou, M. Yang, D. Wang and J. Zhang, Adv. Energy Mater., 2020, 10, 1904152 CrossRef CAS.
- Q. Zhao, X. Liu, J. Zheng, Y. Deng, A. Warren, Q. Zhang and L. A. Archer, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 26053–26060 CrossRef CAS PubMed.
- W. Wang, T. Yang, S. Li, W. Fan, Z. Xin, C. Fan, L. Yu, S. Zhou, X. Zuo, R. Zeng and J. Nan, Electrochim. Acta, 2019, 317, 146–154 CrossRef CAS.
- D. Liang, F. Liang, D. Wang, C. Zhu, J. Liu, D. Gui and C. Li, Electrochim. Acta, 2018, 270, 426–433 CrossRef.
- J. H. Lee, J. C. Shin, J. Kim, J. Ho, W. J. Cho, M. J. Park, G. Yi, M. Lee and P. J. Yoo, J. Power Sources, 2023, 557, 232565 CrossRef CAS.
- Y. Du, Y. Mao and Y. Chen, Materials, 2021, 14, 1908 CrossRef CAS PubMed.
- A. Ray, D. Korkut and B. Saruhan, Nanomaterials, 2020, 10, 1768 CrossRef CAS PubMed.
- F. Ivol, M. Porcher, A. Ghosh, J. Jacquemin and F. Ghamouss, Molecules, 2020, 25, 2697 CrossRef CAS PubMed.
- H. M. Yadav, J. D. Park, H. C. Kang, J. Kim and J. J. Lee, Nanomaterials, 2021, 11, 395 CrossRef CAS PubMed.
- G. Chen, X. Zhang, Y. Ma, H. Song, C. Pi, Y. Zheng, B. Gao, J. Fu and P. K. Chu, Molecules, 2020, 25, 3218 CrossRef CAS PubMed.
- C. Yin, C. Yang, M. Jiang, C. Deng, L. Yang, J. Li and D. Qian, ACS Appl. Mater. Interfaces, 2016, 8, 2741–2752 CrossRef CAS PubMed.
- Z. Huang, C. Qin, J. Wang, L. Cao, Z. Ma, Q. Yuan, Z. Lin and P. Zhang, Materials, 2021, 14, 1714 CrossRef CAS PubMed.
- Z. Xiong, P. Guo, Y. Yang, S. Yuan, N. Shang, C. Wang, Y. Zhang, H. Wang and Y. Gao, Adv. Energy Mater., 2021, 12, 2103226 CrossRef.
- K. Asaka, K. Oguro, Y. Nishimura, M. Mizuhata and H. Takenaka, Polym. J., 1995, 27, 436–440 CrossRef CAS.
- R. H. Baughman, C. Cui, A. A. Zakhidov, Z. Iqbal, J. N. Barisci, G. M. Spinks, G. G. Wallace, A. Mazzoldi, D. De Rossi, A. G. Rinzler, O. Jaschinski, S. Roth and M. Kertész, Science, 1999, 284, 1340–1344 CrossRef CAS PubMed.
- M. D. Bennett and D. J. Leo, Sens. Actuators, A, 2004, 115, 79–90 CrossRef CAS.
- T. Fukushima, K. Asaka, A. Kosaka and T. Aida, Angew. Chem., Int. Ed., 2005, 44, 2410–2413 CrossRef CAS PubMed.
- R. Wang, S. Jeong, H. Ham, J. Kim, H. Lee, C. Y. Son and M. J. Park, Adv. Mater., 2023, 35, 2203413 CrossRef CAS PubMed.
- J. Li, W. Ma, S. Li, Z. Niu, L. Cai, Q. Zeng, X. Zhang, H. Dong, D. Zhao, W. Zhou and S. Xie, Nano Lett., 2011, 11, 4636–4641 CrossRef CAS PubMed.
- K. Mukai, K. Asaka, T. Sugino, K. Kiyohara, I. Takeuchi, N. Terasawa, D. N. Futaba, K. Hata, T. Fukushima and T. Aida, Adv. Mater., 2009, 21, 1582–1585 CrossRef CAS.
- S. Ujiie and K. Iimura, Macromolecules, 1992, 25, 3174–3178 CrossRef CAS.
- K. Binnemans, Chem. Rev., 2005, 105, 4148–4204 CrossRef CAS PubMed.
- K. Goossens, K. Lava, C. W. Bielawski and K. Binnemans, Chem. Rev., 2016, 116, 4643–4807 CrossRef CAS PubMed.
- K. V. Axenov and S. Laschat, Materials, 2011, 4, 206–259 CrossRef CAS PubMed.
- P. H. J. Kouwer and T. M. Swager, J. Am. Chem. Soc., 2007, 129, 14042–14052 CrossRef CAS PubMed.
- G. Park, K. Goossens, T. J. Shin and C. W. Bielawski, Chem. – Eur. J., 2018, 24, 6399–6411 CrossRef CAS PubMed.
- Y. Gao, J. M. Slattery and D. W. Bruce, New J. Chem., 2011, 35, 2910 RSC.
- T. Kato, M. Yoshio, T. Ichikawa, B. Soberats, H. Ohno and M. Funahashi, Nat. Rev. Mater., 2017, 2, 17001 CrossRef.
- T. Ichikawa, T. Kato and H. Ohno, Chem. Commun., 2019, 55, 8205–8214 RSC.
- M. Yoshizawa and H. Ohno, Chem. Commun., 2004, 1828 RSC.
- S. Ueda, J. Kagimoto, T. Ichikawa, T. Kato and H. Ohno, Adv. Mater., 2011, 23, 3071–3074 CrossRef CAS PubMed.
- C.-H. Wu, W. Meng and M. Yoshio, ACS Mater. Lett., 2022, 4, 153–158 CrossRef CAS.
- S. Zhang, E. S. Beach, P. T. Anastas, L. D. Pfefferle and C. O. Osuji, Giant, 2022, 9, 100088 CrossRef CAS.
- C. Wang, J. Zhou, L. Ji, K. Lü, H. Ruan, Q. Zhao, J. Ji, Y. Wu and S. Tan, Ind. Eng. Chem. Res., 2022, 61, 17930–17936 CrossRef CAS.
- S. Cao, J. Aimi and M. Yoshio, ACS Appl. Mater. Interfaces, 2022, 14, 43701–43710 CrossRef CAS PubMed.
- R. Gao, D. Wang, J. R. Heflin and T. E. Long, J. Mater. Chem., 2012, 22, 13473 RSC.
- P. H. Vargantwar, K. E. Roskov, T. K. Ghosh and R. J. Spontak, Macromol. Rapid Commun., 2011, 33, 61–68 CrossRef PubMed.
- C.-H. Wu, W. Meng, K. Iakoubovskii and M. Yoshio, ACS Appl. Mater. Interfaces, 2023, 15, 4495–4504 CrossRef CAS PubMed.
- M. Hirao, H. Sugimoto and H. Ohno, J. Electrochem. Soc., 2000, 147, 4168 CrossRef CAS.
- M. Watanabe, Bull. Chem. Soc. Jpn., 2021, 94, 2739–2769 CrossRef CAS.
- Md. A. B. H. Susan, A. Noda, S. Mitsushima and M. Watanabe, Chem. Commun., 2003, 938 RSC.
- H. Ohno, Solid State Ionics, 2002, 154–155, 303–309 CrossRef CAS.
- T. L. Greaves, A. Weerawardena, C. Fong, I. Krodkiewska and C. J. Drummond, J. Phys. Chem. B, 2006, 110, 22479–22487 CrossRef CAS PubMed.
- T. Yasuda and M. Watanabe, MRS Bull., 2013, 38, 560–566 CrossRef CAS.
- H. Nakamoto and M. Watanabe, Chem. Commun., 2007, 2539–2541 RSC.
- M. S. Miran, M. Hoque, T. Yasuda, S. Tsuzuki, K. Ueno and M. Watanabe, Phys. Chem. Chem. Phys., 2019, 21, 418–426 RSC.
- S. Menne, J. Pires, M. Anouti and A. Balducci, Electrochem. Commun., 2013, 31, 39–41 CrossRef CAS.
- S. Menne, T. Vogl and A. Balducci, Phys. Chem. Chem. Phys., 2014, 16, 5485 RSC.
- S. M. Haile, Acta Mater., 2003, 51, 5981–6000 CrossRef CAS.
- S. Litster and G. F. McLean, J. Power Sources, 2004, 130, 61–76 CrossRef CAS.
- T. Mukai, M. Yoshio, T. Kato, M. Yoshizawa-Fujita and H. Ohno, Electrochemistry, 2005, 73, 623–626 CrossRef CAS.
- D. M. Tigelaar, J. Waldecker, K. M. Peplowski and J. D. Kinder, Polymer, 2006, 47, 4269–4275 CrossRef CAS.
- T. Stettner, G. Lingua, M. Falco, A. Balducci and C. Gerbaldi, Energy Technol., 2020, 8, 2000742 CrossRef CAS.
- H. Chen, J. Choi, D. S. La Cruz, K. I. Winey and Y. A. Elabd, Macromolecules, 2009, 42, 4809–4816 CrossRef CAS.
- W. Ogihara, S. Washiro, H. Nakajima and H. Ohno, Electrochim. Acta, 2006, 51, 2614–2619 CrossRef CAS.
- H. Ohno, Macromol. Symp., 2007, 249–250, 551–556 CrossRef.
- H. Kokubo, R. Sano, K. Murai, S. Ishii and M. Watanabe, Eur. Polym. J., 2018, 106, 266–272 CrossRef CAS.
- J. Illescas, M. Casu, V. Alzari, D. Nuvoli, M. A. Scorciapino, R. Sanna, V. Sanna and A. Mariani, J. Polym. Sci., Part A: Polym. Chem., 2014, 3521–3532 CAS.
- F. Joubert, R. Yeo, G. J. Sharples, O. M. Musa, D. Hodgson and N. R. Cameron, Biomacromolecules, 2015, 16, 3970–3979 CrossRef CAS PubMed.
- W. Xu, P. A. Ledin, V. V. Shevchenko and V. V. Tsukruk, ACS Appl. Mater. Interfaces, 2015, 7, 12570–12596 CrossRef CAS PubMed.
- M. D. Green, D. S. La Cruz, Y. Ye, J. M. Layman, Y. A. Elabd, K. I. Winey and T. E. Long, Macromol. Chem. Phys., 2011, 212, 2522–2528 CrossRef CAS.
- R. Sood, M. M. Obadia, B. P. Mudraboyina, B. Zhang, A. Serghei, J. Bernard and É. Drockenmuller, Polymer, 2014, 55, 3314–3319 CrossRef CAS.
- H. He, D. R. Luebke, H. Nulwala and K. Matyjaszewski, Macromolecules, 2014, 47, 6601–6609 CrossRef CAS.
- S.-J. Long, F. Wan, W. Yang, H. Guo, X. He, J. Ren and J. Gao, J. Appl. Polym. Sci., 2012, 128, 2687–2693 CrossRef.
- J. Yuan, H. Schlaad, C. Giordano and M. Antonietti, Eur. Polym. J., 2011, 47, 772–781 CrossRef CAS.
- K. Vijayakrishna, S. K. Jewrajka, A. Ruiz, R. Marcilla, J. A. Pomposo, D. Mecerreyes, D. Taton and Y. Gnanou, Macromolecules, 2008, 41, 6299–6308 CrossRef CAS.
- K. Vijayakrishna, D. Mecerreyes, Y. Gnanou and D. Taton, Macromolecules, 2009, 42, 5167–5174 CrossRef CAS.
- Y. Ye, J. Choi, K. I. Winey and Y. A. Elabd, Macromolecules, 2012, 45, 7027–7035 CrossRef CAS.
- R. Bouchet, S. Maria, R. Meziane, A. Aboulaich, L. Liénafa, J.-P. Bonnet, T. N. T. Phan, D. Bertin, D. Gigmès, D. Devaux, R. Denoyel and M. Armand, Nat. Mater., 2013, 12, 452–457 CrossRef CAS PubMed.
- M. M. Obadia, B. P. Mudraboyina, A. Serghei, T. N. T. Phan, D. Gigmès and É. Drockenmuller, ACS Macro Lett., 2014, 3, 658–662 CrossRef CAS PubMed.
- D. Kuźmicz, P. Coupillaud, Y. Men, J. Vignolle, G. Vendraminetto, M. Ambrogi, D. Taton and J. Yuan, Polymer, 2014, 55, 3423–3430 CrossRef.
- A. Roy, A. Ray, S. Saha, M. Ghosh, T. Das, M. Nandi, G. Lal and S. Das, Int. J. Energy Res., 2021, 45, 16908–16921 CrossRef CAS.
- D. Deb, P. Bose and S. Bhattacharya, Ionics, 2020, 27, 123–136 CrossRef.
- Y.-T. Kim, M. Song, K. Kim, S. Park, S.-K. Min and H. Rhee, Electrochim. Acta, 2004, 50, 645–648 CrossRef CAS.
- T. H. Lee, T. Y. Kim, H. T. T. Duong, J. Kim and K. S. Suh, Synth. Met., 2009, 159, 2453–2457 CrossRef CAS.
- T. T. Tùng, T. Y. Kim, J. P. Shim, W. S. Yang, H. Kim and K. S. Suh, Org. Electron., 2011, 12, 2215–2224 CrossRef.
- H. Mao, J. Liang, H. Zhang, Q. Pei, D. Liu, S. Wu, Y. Zhang and X. Song, Biosens. Bioelectron., 2015, 70, 289–298 CrossRef CAS PubMed.
- T. Y. Kim, H. Lee, M. D. Stoller, D. R. Dreyer, C. W. Bielawski, R. S. Ruoff and K. S. Suh, ACS Nano, 2010, 5, 436–442 CrossRef PubMed.
- T. Y. Kim, T. T. Tùng, T. H. Lee, J. Kim and K. S. Suh, Chem. – Asian J., 2010, 5, 256–260 CrossRef CAS PubMed.
- X. Chen, J. Zhao, J. Zhang, L. Qiu, D. Xu, H. Zhang, X. Han, B. Sun, G. Fu, Y. Zhang and F. Yan, J. Mater. Chem., 2012, 22, 18018 RSC.
- J. Guo, L. Qiu, Z. Deng and F. Yan, Polym. Chem., 2013, 4, 1309 RSC.
- Y. Rong, Z. Ku, M. Xu, L. Liu, M. Hu, Y. Yang, J. Chen, A. Mei, T. Liu and H. Han, RSC Adv., 2014, 4, 9271 RSC.
- B. Lin, T. Feng, F. Chu, S. Zhang, N. Yuan, G. Qiao and J. Ding, RSC Adv., 2015, 5, 57216–57222 RSC.
- N. Jeon, D. K. Hwang, Y. S. Kang, S. S. Im and D.-W. Kim, Electrochem. Commun., 2013, 34, 1–4 CrossRef CAS.
- J. P. C. Trigueiro, R. L. Lavall and G. G. Silva, J. Power Sources, 2014, 256, 264–273 CrossRef CAS.
- A. Eftekhari and H. García, Mater. Today Chem., 2017, 4, 1–16 CrossRef.
- A. Eftekhari and P. Jafarkhani, J. Phys. Chem. C, 2013, 117, 25845–25851 CrossRef CAS.
- Z. Li, J. Wang, M. Chen and Y. Wang, Chem. – Asian J., 2016, 11, 745–749 CrossRef CAS PubMed.
- Q. Ru, Z. Xue, Y. Wang, Y. Liu and H. Li, Eur. J. Inorg. Chem., 2013, 2014, 469–474 CrossRef.
- T. Zhou, L. Yuan, H. Zhang, P. Zhang, C. Yan, Z. Zheng, Y. Chen and Y. Yu, ACS Appl. Mater. Interfaces, 2016, 8, 23431–23436 CrossRef CAS PubMed.
- S. Ohisa, Y. Pu and J. Kido, J. Mater. Chem. C, 2016, 4, 6713–6719 RSC.
- K. Cui, X. Lü, W. Cui, J. Wu, X. Chen and Q. Lu, Chem. Commun., 2011, 47, 920–922 RSC.
- N. Şahiner, A. O. Yasar and N. Aktaş, Int. J. Hydrogen Energy, 2016, 41, 20562–20572 CrossRef.
- N. Patil, D. Cordella, A. Aqil, A. Debuigne, S. Admassie, C. Jérôme and C. Detrembleur, Macromolecules, 2016, 49, 7676–7691 CrossRef CAS.
- V. Bui-Thi-Tuyet, G. Trippé-Allard, J. Ghilane and H. Randriamahazaka, ACS Appl. Mater. Interfaces, 2016, 8, 28316–28324 CrossRef CAS PubMed.
- A. Dani, V. Crocellà, L. Maddalena, C. Barolo, S. Bordiga and E. Groppo, J. Phys. Chem. C, 2016, 120, 1683–1692 CrossRef CAS.
- Y. Gu, I. Favier, C. Pradel, D. L. Gin, J. Lahitte, R. D. Noble, M. Gómez and J.-C. Rémigy, J. Membr. Sci., 2015, 492, 331–339 CrossRef CAS.
- M. R. Nabid, Y. Bide and Z. Habibi, RSC Adv., 2015, 5, 2258–2265 RSC.
- Y. Yu, C. Yu, T. Yin, S. Ou, X. Sun, X. Wen, L. Zhang, D. Tang and X. Yin, Biosens. Bioelectron., 2017, 87, 278–284 CrossRef CAS PubMed.
- J. R. Nykaza, Y. Li, Y. A. Elabd and J. Snyder, J. Electroanal. Chem., 2016, 783, 182–187 CrossRef CAS.
- F. Fathirad, A. Mostafavi and D. Afzali, Anal. Chim. Acta, 2016, 940, 65–72 CrossRef CAS PubMed.
- T. Sato, T. Morinaga, S. Marukane, T. Narutomi, T. Igarashi, Y. Kawano, K. Ohno, T. Fukuda and Y. Tsujii, Adv. Mater., 2011, 23, 4868–4872 CrossRef CAS PubMed.
- N. Matsumi, K. Sugai, M. Miyake and H. Ohno, Macromolecules, 2006, 39, 6924–6927 CrossRef CAS.
- R. Marcilla, F. Alcaide, H. Sardón, J. A. Pomposo, C. Pozo-Gonzalo and D. Mecerreyes, Electrochem. Commun., 2006, 8, 482–488 CrossRef CAS.
- V. Bocharova, Ż. Wojnarowska, P. Cao, Y. Fu, R. Kumar, B. Li, V. N. Novikov, S. Zhao, A. Kisliuk, T. Saito, J. W. Mays, B. G. Sumpter and A. P. Sokolov, J. Phys. Chem. B, 2017, 121, 11511–11519 CrossRef CAS PubMed.
- C. Y. Son and Z. Wang, J. Chem. Phys., 2020, 153, 100903 CrossRef CAS PubMed.
- H. Jain and S. Krishnaswami, Solid State Ionics, 1998, 105, 129–137 CrossRef CAS.
- A. Serghei, M. Treß, J. Sangoro and F. Kremer, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 184301 CrossRef.
- M. Watanabe, K. Sanui, N. Ogata, T. Kobayashi and Z. Ohtaki, J. Appl. Phys., 1985, 57, 123–128 CrossRef CAS.
- J. C. Dyre, P. Maass, B. Roling and D. L. Sidebottom, Rep. Prog. Phys., 2009, 72, 046501 CrossRef.
- H. Ohno and K. Ito, Chem. Lett., 1998, 27, 751–752 CrossRef.
- D. Mecerreyes, Prog. Polym. Sci., 2011, 36, 1629–1648 CrossRef CAS.
- Ż. Wojnarowska, H. Feng, Y. Fu, S. Cheng, B. Carroll, R. Kumar, V. N. Novikov, A. Kisliuk, T. Saito, N. Kang, J. W. Mays, A. P. Sokolov and V. Bocharova, Macromolecules, 2017, 50, 6710–6721 CrossRef.
- Y. Ye and Y. A. Elabd, Polymer, 2011, 52, 1309–1317 CrossRef CAS.
- C. Jangu, A. Schultz, C. E. Wall, A. R. Esker and T. E. Long, Macromol. Rapid Commun., 2016, 37, 1212–1217 CrossRef CAS PubMed.
- J. R. Keith, N. Rebello, B. Cowen and V. Ganesan, ACS Macro Lett., 2019, 8, 387–392 CrossRef CAS PubMed.
- A. Wang, H. Xu, X. Liu, R. Gao, S. Wang, Q. Zhou, J. Chen, X. Liu and L. Zhang, Polym. Chem., 2017, 8, 3177–3185 RSC.
- M. Lee, U. H. Choi, R. H. Colby and H. W. Gibson, Chem. Mater., 2010, 22, 5814–5822 CrossRef CAS.
- U. H. Choi, M. Lee, S. Wang, W. Liu, K. I. Winey, H. W. Gibson and R. H. Colby, Macromolecules, 2012, 45, 3974–3985 CrossRef CAS.
- U. H. Choi, Y. Ye, D. S. La Cruz, W. Liu, K. I. Winey, Y. A. Elabd, J. Runt and R. H. Colby, Macromolecules, 2014, 47, 777–790 CrossRef CAS.
- A. S. Shaplov, D. O. Ponkratov, P. S. Vlasov, E. I. Lozinskaya, L. I. Komarova, I. A. Malyshkina, F. Vidal, G. T. M. Nguyen, M. Armand, C. Wandrey and Y. S. Vygodskii, Polym. Sci., Ser. B, 2013, 55, 122–138 CrossRef CAS.
- M. Heres, T. Cosby, E. U. Mapesa, H. Liu, S. Berdzinski, V. Strehmel, M. Dadmun, S. J. Paddison and J. Sangoro, Macromolecules, 2018, 52, 88–95 CrossRef.
- P. J. Griffin, J. L. Freyer, N. Han, N. Geller, X. Yin, C. D. Gheewala, T. H. Lambert, L. M. Campos and K. I. Winey, Macromolecules, 2018, 51, 1681–1687 CrossRef CAS.
- C. M. Evans, C. R. Bridges, G. E. Sanoja, J. Bartels and R. A. Segalman, ACS Macro Lett., 2016, 5, 925–930 CrossRef CAS PubMed.
- F. Frenzel, P. Borchert, A. M. Anton, V. Strehmel and F. Kremer, Soft Matter, 2019, 15, 1605–1618 RSC.
- J. Sangoro, C. Iacob, A. Serghei, C. B. Haymob, P. Galvosas, J. Kärger, C. Wespe, F. Bordusa, A. Stoppa, J. Hunger, R. Buchner and F. Kremer, J. Chem. Phys., 2008, 128, 214509 CrossRef CAS PubMed.
- T. Cosby, Z. Vicars, Y. Wang and J. Sangoro, J. Phys. Chem. Lett., 2017, 8, 3544–3548 CrossRef CAS PubMed.
- F. Frenzel, M. Y. Folikumah, M. Schulz, A. M. Anton, W. H. Binder and F. Kremer, Macromolecules, 2016, 49, 2868–2875 CrossRef CAS.
- R. Shioiri, H. Kokubo, T. Horii, Y. Kobayashi, K. Hashimoto, K. Ueno and M. Watanabe, Electrochim. Acta, 2019, 298, 866–873 CrossRef CAS.
- H. Ji, W. Wang, H. Peng, M. Guo, Y. Feng and Z. Xue, Macromolecules, 2017, 50, 1970–1980 CrossRef.
- H. B. Youcef, O. García-Calvo, N. Lago, D. Shanmukaraj and M. Armand, Electrochim. Acta, 2016, 220, 587–594 CrossRef.
- S. Ishii, H. Kokubo, K. Hashimoto, S. Imaizumi and M. Watanabe, Macromolecules, 2017, 50, 2906–2915 CrossRef CAS.
- C. Jangu, A. M. Savage, Z. Zhang, A. Schultz, L. A. Madsen, F. L. Beyer and T. E. Long, Macromolecules, 2015, 48, 4520–4528 CrossRef CAS.
- J. R. Nykaza, A. M. Savage, Q. Pan, S. Wang, F. L. Beyer, M. H. Tang, C. Y. Li and Y. A. Elabd, Polymer, 2016, 101, 311–318 CrossRef CAS.
- L. Porcarelli, A. S. Shaplov, M. Salsamendi, J. R. Nair, Y. S. Vygodskii, D. Mecerreyes and C. Gerbaldi, ACS Appl. Mater. Interfaces, 2016, 8, 10350–10359 CrossRef CAS PubMed.
- L. Porcarelli, K. Manojkumar, H. Sardón, O. Llorente, A. S. Shaplov, K. Vijayakrishna, C. Gerbaldi and D. Mecerreyes, Electrochim. Acta, 2017, 241, 526–534 CrossRef CAS.
- N. Patil, M. Aoudia, A. Aqil, F. Ouhib, R. Marcilla, A. Minoia, R. Lazzaroni, C. Jérôme and C. Detrembleur, Chem. Mater., 2018, 30, 5831–5835 CrossRef CAS.
- C. Li, B. Qin, Y. Zhang, A. Varzi, S. Passerini, J. Wang, J. Dong, D. Zeng, Z. Liu and H. Cheng, Adv. Energy Mater., 2019, 9, 1803422 CrossRef.
- X. Wang, F. F. Chen, G. M. A. Girard, H. Zhu, D. R. MacFarlane, D. Mecerreyes, M. Armand, P. C. Howlett and M. Forsyth, Joule, 2019, 3, 2687–2702 CrossRef CAS.
- G. Pace, O. Nordness, K. Asham, R. J. Clément and R. A. Segalman, Chem. Mater., 2022, 34, 4672–4681 CrossRef CAS.
- X. Guan, Q. Wu, X. Zhang, X. Guo, C. Li and J. Xu, Chem. Eng. J., 2020, 382, 122935 CrossRef CAS.
- L. Yu, Q. Liu, M. Tao, S. Wang and X. Hu, Adv. Funct. Mater., 2021, 32, 2110653 CrossRef.
- G. Pace, M. L. Le, R. J. Clément and R. A. Segalman, ACS Energy Lett., 2023, 8, 2781–2788 CrossRef CAS.
- D. Rawlings, D.-W. Lee, J. Kim, I.-B. Magdău, G. Pace, P. M. Richardson, E. M. Thomas, S. P. O. Danielsen, S. H. Tolbert, T. F. Miller, R. Seshadri and R. A. Segalman, Chem. Mater., 2021, 33, 6464–6474 CrossRef CAS.
- D. Rawlings, E. M. Thomas, R. A. Segalman and M. L. Chabinyc, Chem. Mater., 2019, 31, 8820–8829 CrossRef CAS.
- B. C. Popere, G. E. Sanoja, E. M. Thomas, N. S. Schauser, S. D. Jones, J. Bartels, M. E. Helgeson, M. L. Chabinyc and R. A. Segalman, J. Mater. Chem. C, 2018, 6, 8762–8769 RSC.
- T. J. Quill, G. LeCroy, A. Melianas, D. Rawlings, Q. Thiburce, R. Sheelamanthula, C. Cheng, Y. Tuchman, S. T. Keene, I. McCulloch, R. A. Segalman, M. L. Chabinyc and A. Salleo, Adv. Funct. Mater., 2021, 31, 2104301 CrossRef CAS.
- J. Yang, J. Wang, Y. Zhang and Z. Guo, Chem. Commun., 2014, 50, 14979–14982 RSC.
- F. Chen, J. Guo, D. Xu and F. Yan, Polym. Chem., 2016, 7, 1330–1336 RSC.
- C. Yuan, J. Guo, M. Tan, M. Guo, L. Qiu and F. Yan, ACS Macro Lett., 2014, 3, 271–275 CrossRef CAS PubMed.
- A. Wu, P. Sun, N. Sun and L. Zheng, Chem. – Eur. J., 2018, 24, 10452–10459 CrossRef CAS PubMed.
- A. M. Scheuermann, H. Wakidi, A. T. Lill, S. Oh, L. C. Llanes, C. D'Ambra, S. Antoine, M. Wang, M. L. Chabinyc, T. Nguyen, J. R. De Alaniz and C. M. Bates, ACS Appl. Polym. Mater., 2021, 3, 5125–5133 CrossRef CAS.
- R. Sumitani and T. Mochida, Macromolecules, 2020, 53, 6968–6974 CrossRef CAS.
- R. Sumitani, H. Yoshikawa and T. Mochida, Chem. Commun., 2020, 56, 6189–6192 RSC.
- X. Ming, L. Shi, H. Zhu and Q. Zhang, Adv. Funct. Mater., 2020, 30, 2005079 CrossRef CAS.
- H. Nie, N. S. Schauser, N. D. Dolinski, J. Hu, C. J. Hawker, R. A. Segalman and J. R. De Alaniz, Angew. Chem., Int. Ed., 2020, 59, 5123–5128 CrossRef CAS PubMed.
- Y. Wang, F. Fan, A. L. Agapov, T. Saito, J. Yang, X. Yu, K. Hong, J. W. Mays and A. P. Sokolov, Polymer, 2014, 55, 4067–4076 CrossRef CAS.
- S. Oh, A. F. Nikolaev, K. Tagami, T. V. Tran, D.-W. Lee, S. Mukherjee, R. A. Segalman, S. Han, J. R. De Alaniz and M. L. Chabinyc, ACS Appl. Mater. Interfaces, 2021, 13, 5319–5326 CrossRef CAS PubMed.
- D. Yuan, E. Plunkett, P. H. Nguyen, D. Rawlings, M. L. Le, R. Kroon, C. Müller, R. A. Segalman and M. L. Chabinyc, Adv. Funct. Mater., 2023, 33, 2300934 CrossRef CAS.
- W. Xü, M. Zhang, Y. Chen, Q. Tian, X. Zhou, L. Zhang, X. Wang and W. Zhang, eXPRESS Polym. Lett., 2023, 17, 406–416 CrossRef.
- G. Gebel and O. Diat, Fuel Cells, 2005, 5, 261–276 CrossRef CAS.
- J. H. Lee, J. S. Heo, Y. Kim, J. Eom, H. J. Jung, J. Kim, I.-S. Kim, H.-H. Park, H. S. Mo, Y. Kim and S. K. Park, Adv. Mater., 2020, 32, 2000969 CrossRef CAS PubMed.
- Y. Gao, Y. Zhang, W. Xu, K. Sim, J. Liu, J. Chen, X. Feng, H. Xu and C. Yu, Sci. Adv., 2017, 3, e1701222 CrossRef PubMed.
- Y. Kang, H. J. Kim, E. Kim, B. Oh and J. H. Cho, J. Power Sources, 2001, 92, 255–259 CrossRef CAS.
- D. T. Hallinan and N. P. Balsara, Annu. Rev. Mater. Res., 2013, 43, 503–525 CrossRef CAS.
- K. I. S. Mongcopa, M. Tyagi, J. P. Mailoa, G. G. Samsonidze, B. Kozinsky, S. A. Mullin, D. A. Gribble, H. Watanabe and N. P. Balsara, ACS Macro Lett., 2018, 7, 504–508 CrossRef CAS PubMed.
- N. Byrne, P. C. Howlett, D. R. MacFarlane and M. Forsyth, Adv. Mater., 2005, 17, 2497–2501 CrossRef CAS.
- S. D. Jones, H. Nguyen, P. M. Richardson, Y. Chen, K. E. Wyckoff, C. J. Hawker, R. J. Clément, G. H. Fredrickson and R. A. Segalman, ACS Cent. Sci., 2022, 8, 169–175 CrossRef CAS PubMed.
- F. Lind, L. Rebollar, P. Bengani-Lutz, A. Asatekin and M. J. Panzer, Chem. Mater., 2016, 28, 8480–8483 CrossRef CAS.
- F. Lü, X. Gao, A. Wu, N. Sun, L. Shi and L. Zheng, J. Phys. Chem. C, 2017, 121, 17756–17763 CrossRef.
- A. J. D’Angelo and M. J. Panzer, Adv. Energy Mater., 2018, 8, 1801646 CrossRef.
- X. He, L. Huang, W. Li, S. Gu, D. Zeng, Y. Zhang, Y. Sun and H. Cheng, J. Membr. Sci., 2022, 651, 120452 CrossRef.
- H. Xu, S. Fathipour, E. Kinder, A. Seabaugh and S. K. Fullerton-Shirey, ACS Nano, 2015, 9, 4900–4910 CrossRef CAS PubMed.
- H. M. Li, K. Xu, B. Bourdon, H. Lu, Y. C. Lin, J. A. Robinson, A. Seabaugh and S. K. Fullerton-Shirey, J. Phys. Chem. C, 2017, 121, 16996–17004 CrossRef CAS.
- S. K. Fullerton-Shirey and J. K. Maranas, Macromolecules, 2009, 42, 2142–2156 CrossRef CAS.
- E. Kinder, A. M. Fuller, Y. C. Lin, J. A. Robinson and S. K. Fullerton-Shirey, ACS Appl. Mater. Interfaces, 2017, 9, 25006–25013 CrossRef CAS PubMed.
- E. W. Stacy, C. Gainaru, M. Gobet, Ż. Wojnarowska, V. Bocharova, S. Greenbaum and A. P. Sokolov, Macromolecules, 2018, 51, 8637–8645 CrossRef CAS.
- F. Luo, T. L. Sun, T. Nakajima, D. R. King, T. Kurokawa, Y. Zhao, A. B. Ihsan, X. Li, H. Guo and J. P. Gong, Macromolecules, 2016, 49, 2750–2760 CrossRef CAS.
- S. Arora, PhD dissertation, University of Pittsburgh, 2022.
- V. Ganesan, Mol. Syst. Des. Eng., 2019, 4, 280–293 RSC.
- M. Kofu, M. S. Tyagi, Y. Inamura, K. Miyazaki and O. Yamamuro, J. Chem. Phys., 2015, 143, 234502 CrossRef PubMed.
- S. Mogurampelly, J. R. Keith and V. Ganesan, J. Am. Chem. Soc., 2017, 139, 9511–9514 CrossRef CAS PubMed.
Footnotes |
| † Dr Swati Arora: Scientist-III: Process Research & Development. |
| ‡ Dr Nagendra Verma: Research Scientist. |
| This journal is © The Royal Society of Chemistry 2024 |