
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen bonded polymer complex thin films for highly stretchable gas barriers†
Sarah G.
Fisher
 a,
Hsu-Cheng
Chiang
a,
Ethan T.
Iverson
a,
Edward
Chang
b and
Jaime C.
Grunlan
*abc
a,
Hsu-Cheng
Chiang
a,
Ethan T.
Iverson
a,
Edward
Chang
b and
Jaime C.
Grunlan
*abc
aDepartment of Chemistry, Texas A&M University, College Station, TX 77843, USA. E-mail: jgrunlan@tamu.edu
bDepartment of Mechanical Engineering, Texas A&M University, College Station, TX 77843, USA
cDepartment of Materials Science & Engineering, Texas A&M University, College Station, TX 77843, USA
First published on 8th January 2024
Abstract
Clay-containing polyelectrolyte complexes have been investigated as simple and effective gas barrier coatings for a variety of substrates. Some applications for barrier coatings, such as stretchable electronics, require films that exhibit high extensibility as well as low oxygen transmission rate. This combination of properties is difficult to achieve, as barrier tends to go hand-in-hand with film density. In this work, a stretchable thin film with low oxygen transmission rate (OTR) was prepared through the addition of montmorillonite (MMT) clay into a hydrogen bonded complex of poly(ethylene oxide) (PEO) and poly(acrylic acid) (PAA). Interpolymer hydrogen bonding and MMT clay distribution were found to improve with an increasing ratio of PEO to PAA in the complex, leading to the creation of a highly stretchable coating with a high gas barrier. This elastomeric system, deposited in three steps on 760 μm thick natural rubber, reduces the OTR by two orders of magnitude with a thickness of 1.8 μm. This film maintains a >90% reduction in OTR even after it is subjected to 50% strain. This is believed to be the first gas barrier coating deposited as a buffer-cured, hydrogen bonded polymer complex.
Introduction
Gas barrier films are desirable for applications such as food and drug preservation, electronic device encapsulation, and fuel cell membranes.1–5 While barrier tends to improve with increased stiffness,6,7 highly stretchable gas barrier films are required for applications in next-generation stretchable electronics, such as deformable batteries and flexible OLEDs.8,9 Gas barrier films are often constructed of polymeric materials due to their light weight, low cost, and easily processable nature.10 Barrier properties of polymer films can be significantly improved via the incorporation of inorganic nanomaterials, even in low concentrations.11,12 These impermeable nanoparticles, most often nanoplatelets, decrease gas transmission in polymeric thin films by reducing the area available for gas diffusion and increasing the tortuosity of the diffusion pathway.13 While polymer nanocomposites have been prepared with a wide variety of inorganic materials, including carbon nanotubes and graphene oxide, clays are widely used due to their availability, low cost, high aspect ratio, platelet geometry, and straightforward processing.14–17 Several factors, including aspect ratio, loading fraction, intercalation, and orientation of nanofillers, can impact the tortuosity of the barrier formed.18A number of high gas barrier nanocomposite films have been prepared via layer-by-layer deposition (LbL).19–21 In LbL assembly, highly ordered films are built up in layers via alternating exposure to species with relatively strong intermolecular interactions (ionic, hydrogen bonding, pi stacking, etc.).19,22 For barrier films, these interactions are most commonly ionic, utilizing polyelectrolytes in combination with nanomaterials to create “nanobrick wall” structures that exhibit high barrier due to high nanoparticle alignment.19,23,24 Ionically assembled LbL films tend to be stiff and brittle, especially at high filler loadings, making them impractical for applications requiring stretchability.25 One approach to combine stretchability and low gas permeability is the preparation of LbL films through hydrogen bonding interactions.26–28
One of the most widely-studied hydrogen bonded systems, consisting of PEO and PAA, was synthesized as a freestanding LbL film that exhibited rubbery behavior when stretched up to 3× its original length.26 A major drawback of hydrogen bonded LbL films is their higher gas permeability, as they are less dense than ionic LbL films due to lower bonding energy.26,29 Even so, when nanoparticles are incorporated, hydrogen bonded LbL films have been shown to exhibit good barrier properties with some retained stretchability.18,30 For example, a PEO/PAA LbL thin film containing MMT platelets reduced the oxygen transmission rate (OTR) of polyurethane rubber by two orders of magnitude (and retained a 54× reduction in OTR after exposure to a 20% strain).28 The primary drawback with the LbL procedure is the number of processing steps and the time required to deposit such coatings,22 which is not ideal from an industrial standpoint.
Buffer-cured polyelectrolyte complex (PEC) coatings represent an alternative way that gas barrier films can be prepared. PEC coatings exploit interpolymer interactions that are similar to the LbL process, but instead of dipping a substrate into alternating solutions, both polymers coexist in one solution with the pH and/or salt content manipulated to minimize intermolecular interaction. After this coating solution is applied to the desired substrate, it can then be buffer cured, changing the pH to induce complexation. Ionically assembled PEC films have already demonstrated excellent gas barrier.6,31,32 For example, a solution of polyethyleneimine (PEI) and PAA was deposited on a poly(ethylene terephthalate) substrate at pH 8, then cured with a pH 3 citric acid buffer to result in a coating with an undetectable OTR (<0.005 cc (m2 day)−1) at 90% relative humidity.6 Despite exhibiting high barrier, these high modulus films are not expected to be stretchable without cracking.
In this work, a buffer-cured, hydrogen bonded polymer complex (HBPC) coating consisting of PEO, PAA, and MMT was prepared to combine barrier with stretchability. Polymer ratios were manipulated to maximize interpolymer hydrogen bonding interactions for elastomeric behavior. Simultaneously, increased PEO content was found to improve MMT platelet dispersion to create a stronger barrier against gas molecules. The resulting 1.8 μm thick HBPC film provides a 99% reduction in the OTR of the natural rubber substrate. Deposited in three steps, this coating also exhibits high stretchability, maintaining a 93% reduction in OTR after being subjected to 50% strain. This stretchable gas barrier coating exhibits higher flexibility and greater OTR reduction relative to a previously reported LbL analogue, while being deposited with significantly fewer processing steps.28 This system represents a simple and effective barrier coating for improved protection of flexible electronics and pressure retention in various types of inflatable devices.
Experimental
Materials
Polyethylene oxide (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 g mol−1) was purchased from Polysciences (Warrington, PA). Poly(acrylic acid) (250000 g mol−1, 35 wt% aqueous solution), branched polyethyleneimine (25000 g mol−1), and citric acid (CA) (98%) were purchased from Sigma-Aldrich (Milwaukee, WI). Natural sodium montmorillonite clay was purchased from Southern Clay products, Inc. (Gonzales, TX). All solutions were prepared using 18 MΩ deionized (DI) water.
000000 g mol−1) was purchased from Polysciences (Warrington, PA). Poly(acrylic acid) (250000 g mol−1, 35 wt% aqueous solution), branched polyethyleneimine (25000 g mol−1), and citric acid (CA) (98%) were purchased from Sigma-Aldrich (Milwaukee, WI). Natural sodium montmorillonite clay was purchased from Southern Clay products, Inc. (Gonzales, TX). All solutions were prepared using 18 MΩ deionized (DI) water.
Solution preparation
Solutions of PEI were prepared with DI water and adjusted to pH 11 with 5 M NaOH. Solutions of PEO were prepared with DI water and adjusted to pH 12 using 5 M NaOH. PAA was dissolved in DI water, then MMT was added and the solution was stirred overnight. The PAA + MMT solution was adjusted to pH 12 using 5 M NaOH. Equal masses of the PEO and PAA + MMT solutions were combined and mixed overnight to form the coating solution. All coating solutions were 0.5 wt% MMT and the total polymer content was 1 wt%. Coating solutions were prepared in three polymer mass ratios: 2:1 (0.67 wt% PEO + 0.33 wt% PAA), 1:1 (0.5 wt% PEO + 0.5 wt% PAA), and 1:2 (0.33 wt% PEO + 0.67 wt% PAA). The recipes are denoted PPM 2:1, PPM 1:1, and PPM 1:2 respectively (Table 1).
| Sample name | Contents | PEO:PAA mass ratio |
PEO:PAA molar ratio |
|---|---|---|---|
| PPM 1:2 |
0.33 wt% PEO + 0.67 wt% PAA + 0.5 wt% MMT | 1:2 |
0.8:1 |
| PPM 1:1 |
0.5 wt% PEO + 0.5 wt% PAA + 0.5 wt% MMT | 1:1 |
1.6:1 |
| PPM 2:1 |
0.67 wt% PEO + 0.33 wt% PAA + 0.5 wt% MMT | 2:1 |
3.2:1 |
Substrates
Single-side polished, 500-μm-thick Si wafers were purchased from University Wafer (South Boston, MA) and used as substrates for profilometry, atomic force microscopy (AFM), nanoindentation, Fourier-transform infrared spectroscopy (FTIR), differential scanning calorimetry (DSC), and thermogravimetric analysis (TGA). X-ray diffraction (XRD) samples were prepared on indium-tin-oxide (ITO)-coated glass slides, also purchased from University Wafer. 7 mil polyethylene terephthalate (PET) film was purchased from Tekra (New Berlin, WI) and used as the substrate for coat weight measurements. 760 μm thick natural rubber was purchased from McMaster-Carr (Elmhurst, IL) and used as the substrate for oxygen transmission rate (OTR) testing and scanning electron microscopy (SEM). All substrates were rinsed with water, then methanol, then water again, dried with compressed air, and plasma treated for five minutes before priming to improve primer adhesion.Coating deposition
Plasma-treated substrates were primed by dipping in 0.1 wt% PEI at pH 11 for 5 minutes, then dried at 70 °C for 5 minutes. This was done to improve coating adhesion to the substrate. Next, substrates were dipped in a given coating solution for 5 minutes, then dried at 70 °C for 15 minutes. Dried substrates were cured by dipping in a 200 mM citric acid buffer at pH 2.5 for 15 minutes, then dip-rinsed in 3 separate portions of pH 2.5 DI water for 20 seconds each to remove excess material without disturbing the hydrogen bonded network. Cured samples were dried at 70 °C for 5 minutes, then stored in a drybox prior to characterization. Samples for FTIR, DSC, and TGA were scraped off the silicon wafer substrate with a razor before testing. Fig. 1a summarizes the coating process.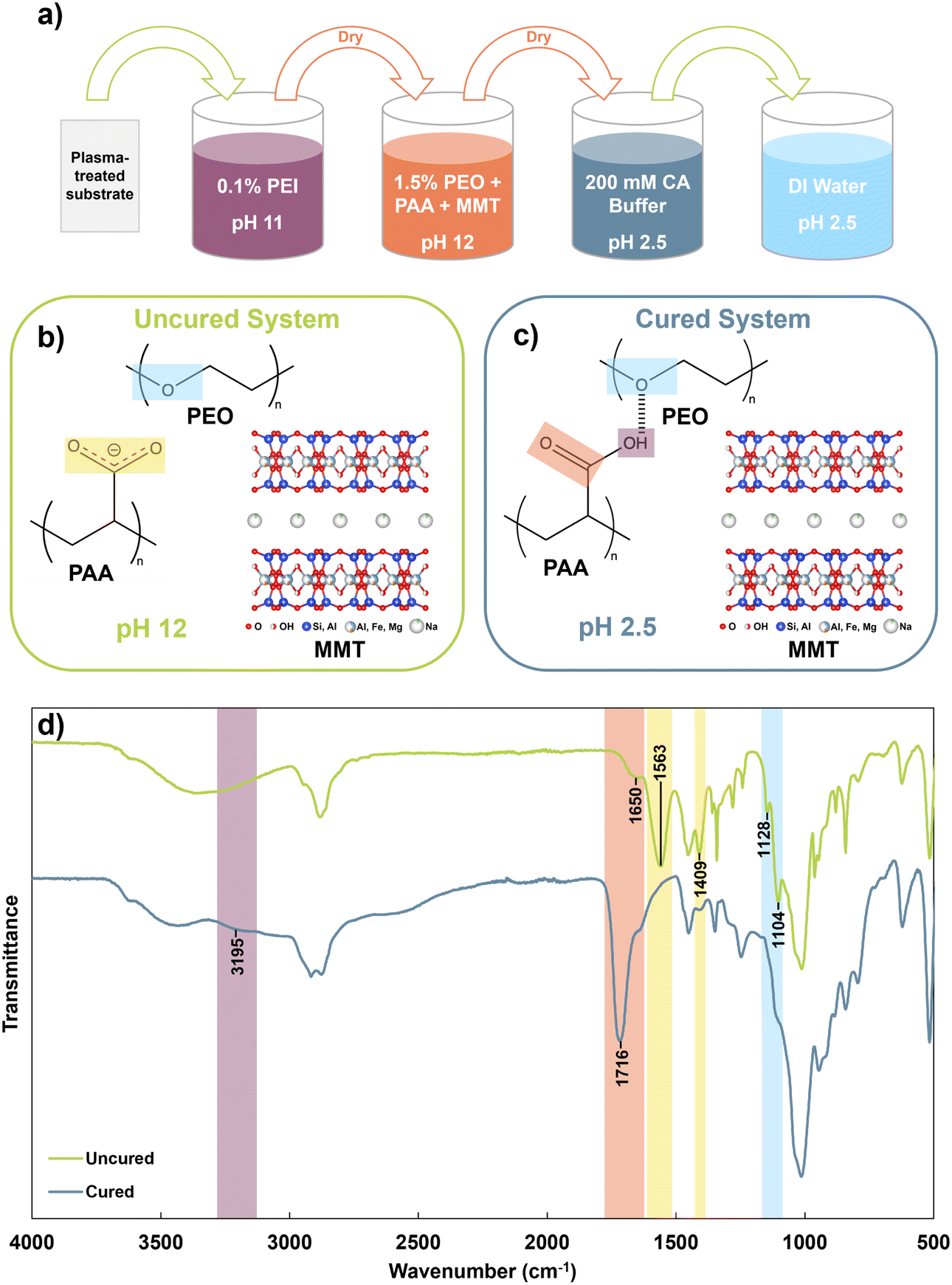 | ||
| Fig. 1 (a) Schematic of the HBPC coating process, (b) chemical structures of materials in coating solution, (c) chemical structures of materials in the cured coating, and (d) FTIR spectra of cured and uncured PPM 2:1 coatings, with key peaks labelled. | ||
Characterization
Film thickness was measured in triplicate using a KLA-Tencor P-6 Stylus Profilometer (Milpitas, CA). FTIR spectroscopy was performed using an Alpha Platinum-ATR FTIR spectrometer (Bruker Optics Inc., Billerica, MA), with air as a background. A minimum of 64 scans were signal averaged for each sample and the scan resolution was 2 cm−1. The thermal properties of samples were measured using a TA Instruments DSC 2500 (New Castle, DE). 3–5 mg of dry polymer or PPM complex was placed in an aluminum pan and heated from −70 to 150 °C at a ramp rate of 5 °C min−1, with a 1-minute isotherm at the beginning and ending of each ramp. The second heating cycle was used for analysis. TGA was performed using a Q-50 thermogravimetric analyzer (TA Instruments, New Castle, DE, USA). Samples were heated isothermally at 100 °C for 20 minutes to remove any residual water, after which the temperature was increased by 10 °C min−1 up to 900 °C under a 60 mL s−1 air flow. XRD was performed using a diffractometer (Bruker AXS model D8 Discover) with copper K-alpha radiation equipped with a Vantec 500 2D detector. Samples were analyzed at a maximum of 40 kV and 40 mA. Individual polymer films were prepared by drop casting polymer solutions on ITO-coated glass. All samples were run as films on ITO-coated glass except MMT, which was run as a powder. An Instron model 4411 tensile tester (Norwood, MA) was used to apply different strains to coated and uncoated rubber for 2 minutes. Scanning electron microscope images of uncoated and coated rubber before and after application of various strains were obtained using a Model JSM-7500F FE-SEM (JEOL, Tokyo, Japan) after sputter coating with 4 nm of a platinum/palladium alloy. Film surface morphology was analyzed via atomic force microscopy using a Bruker Dimension Icon AFM (Billerica, MA). Nanoindentation was performed to determine the reduced modulus and hardness of the films before and after curing using a TI 950 Triboindenter (Hysitron, Inc., Minneapolis, MN). The loading force was 200 μN to ensure indentation depth of ≈10% and mitigate the substrate effect. The loading profile consisted of ten seconds of loading, five seconds at a stationary position, and two seconds of unloading. The coat weight was determined by coating each recipe on a 7 mil PET substrate, then punching 1.9 cm diameter circles of uncoated and coated PET and measuring the mass on an analytical balance. Oxygen transmission rate testing was performed by Ametek MOCON, Inc. (Minneapolis, MN) using an Oxtran 2/10 instrument in accordance with ASTM F2622. Oxygen was utilized as a test gas at 23 °C, with a relative humidity (RH) of 0%.Results and discussion
Structure and bonding in complexed films
Fig. 1b and c illustrate the components of the uncured and cured system, respectively. At pH 12, the carboxyl groups of PAA are expected to be largely deprotonated, as the pKa of PAA is 4.5.33 At this elevated pH, PAA and PEO can coexist in solution without significant hydrogen bonding interactions. After curing at pH 2.5, PAA is expected to become highly protonated and act as a hydrogen bond donor to PEO, forming an insoluble but flexible complex.34 This complexation behavior is demonstrated through the FTIR spectra of the uncured and cured PPM 2:1 system (Fig. 1d). The FTIR spectra of each individual component of the system are shown in Fig. S1.†
In the uncured complex, the ether stretching peak of PEO is observed as a maximum peak at 1104 cm−1, with a smaller peak at 1128 cm−1. In reality, this is a triplet peak, but the righthand shoulder is overshadowed by the large MMT peak centered at 1000 cm−1. In the cured spectrum, the major peak broadens, indicating increased participation in hydrogen bonding, and the side peak decreases in intensity, suggesting a loss in PEO crystallinity that can be attributed to hydrogen bonding.35 The uncured spectrum shows strong carboxylate peaks at 1409 and 1563 cm−1, corresponding to symmetric and asymmetric stretching, respectively.36 These peaks are significantly decreased in the cured system, indicating that most of the carboxyl groups have been protonated. Protonation is further confirmed by the shift of the minor carbonyl stretch at 1650 cm−1 in the uncured material to a strong peak at 1716 cm−1 in the cured material. The increase in peak intensity indicates that the PAA becomes largely protonated, while the shift to a higher wavenumber suggests that PAA participates in intermolecular hydrogen bonding in the cured system rather than only dimerization with itself.26,37 Finally, the appearance of an OH stretch at ∼3200 cm−1 in the cured system can be attributed to the increased presence of hydrogen bonded hydroxyl groups in the cured material. While “free” hydroxyls typically appear in the 3400–3600 cm−1 region, hydrogen bonded hydroxyls show a lower stretching region of 3100–3200 cm−1.38
FTIR spectra of cured PPM 2:1, PPM 1:1 and PPM 1:2 were also compared to evaluate the relative levels of PAA-PEO hydrogen bonding in the different recipes, as shown in Fig. 2. The carbonyl stretching peak at ∼1700 cm−1 consists of multiple distinct peaks. The peak at 1710 cm−1 corresponds to PAA dimerization, while the peak at 1725 cm−1 is attributed to PAA–PEO hydrogen bonding interactions.39,40 The presence of a minor stretching peak at 1650 cm−1 is attributed to some uncured material in the film. The 1850 to 1550 cm−1 region was deconvoluted using a Gaussian model, a common technique for many types of experimental spectra, including FTIR.41–43 Comparing the deconvoluted spectra of PPM 1:2 (Fig. 2a), PPM 1:1 (Fig. 2b), and PPM 2:1 (Fig. 2c), it is apparent that PAA–PEO hydrogen bonding (at 1725 cm−1) increases relative to PAA dimerization (at 1710 cm−1) as the PEO:PAA ratio increases. Li et al. observed a similar phenomenon.44
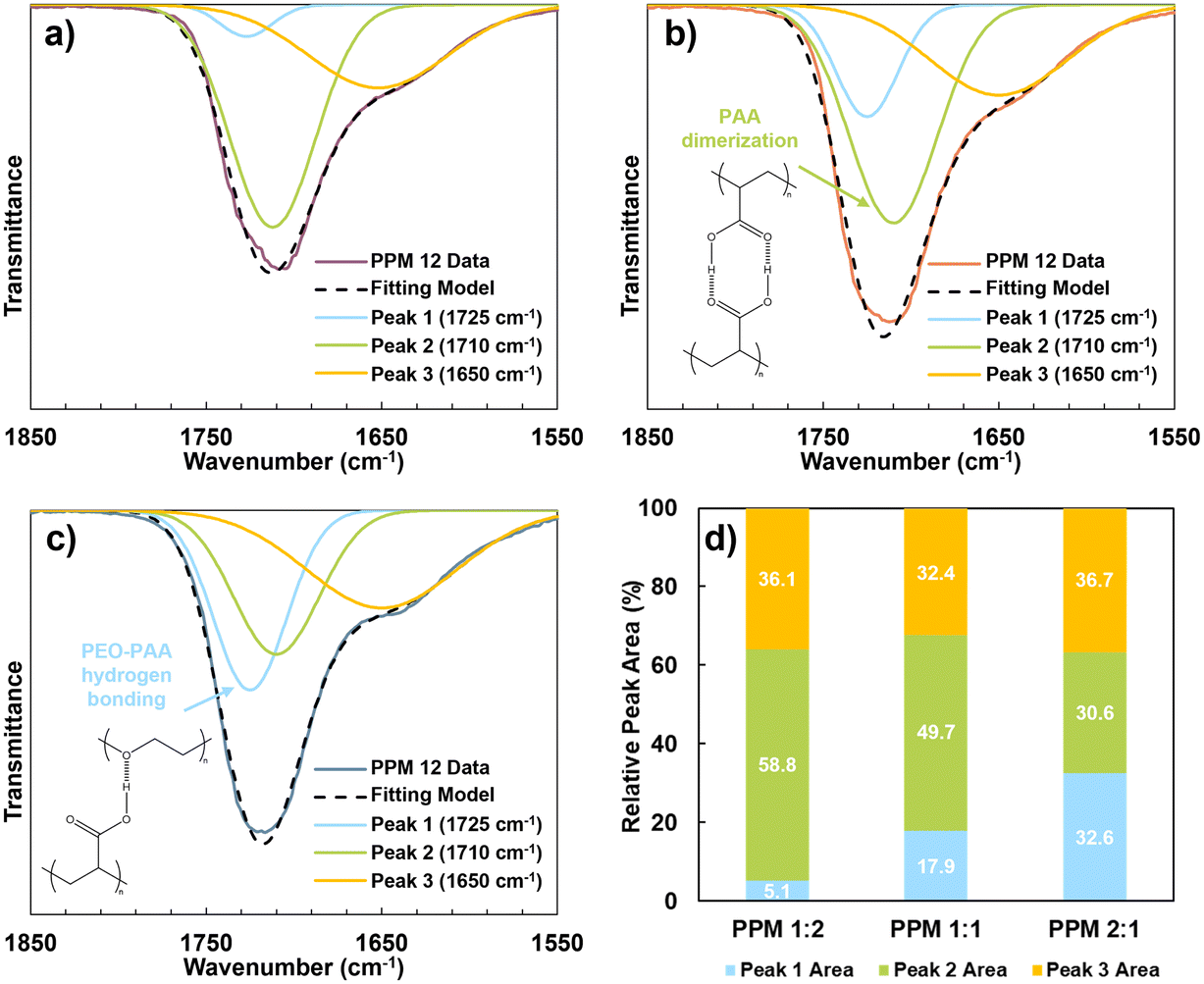 | ||
| Fig. 2 FTIR spectral deconvolution of (a) cured PPM 1:2, (b) cured PPM 1:1, and (c) cured PPM 2:1 from 1850 to 1550 cm−1. (d) Tabulated relative peak areas for each spectrum. Schemes of (b) PAA dimerization and (c) PEO–PAA hydrogen bonding are included. | ||
Intimate mixing of the two polymers is evidenced by DSC and XRD analyses. In the XRD spectra for all materials except MMT, which was analyzed as a powder, ITO peaks can be observed due to the ITO-coated glass substrate used for film analysis. These peaks, represented by black triangles in Fig. 3a, are in agreement with reported values in the literature.45,46 Although the basal (001) peak of montmorillonite is not observed, other characteristic peaks, such as (003) and (110), are observed in the MMT spectrum as well as the three PPM spectra, confirming the presence of MMT clay in each coating.47,48 These peaks are represented by pink circles in Fig. 3a. It should be noted that the small shoulders on the (110) and (005) MMT peaks have been observed in the literature, and are attributed to quartz impurities in the clay sample.49,50
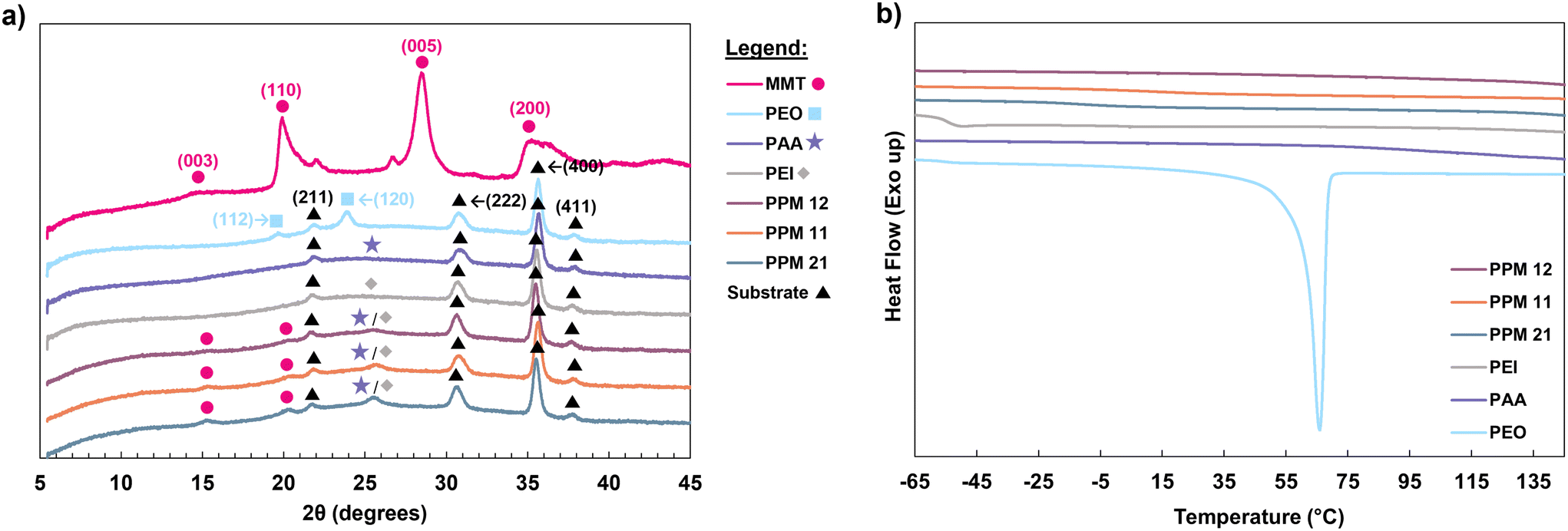 | ||
| Fig. 3 (a) XRD spectra of coating components and PPM recipes and (b) DSC thermograms of coating components and PPM complexes. | ||
The XRD spectra of PAA and PEI both exhibit broad peaks around 25°, marked with a purple star and a gray diamond in Fig. 3a, indicating that these polymer films are amorphous.51 Similar broad peaks appear in the PPM complexes, indicating the presence of amorphous PEI and PAA in the coatings. Most importantly, the (112) and (120) characteristic peaks of semicrystalline PEO are observed in the pure PEO spectrum at 19 and 23°, respectively (represented by blue squares).52,53 These same PEO peaks are not observed in any of the PPM complexes, suggesting that PEO forms minimal crystalline domains in the PPM films. Furthermore, in DSC, semicrystalline PEO exhibits a melting point at ∼66 °C (Fig. 3b). None of the PPM complexes exhibit a melting transition in this region (Fig. 3b), indicating that there are no regions of crystallized PEO within the coatings. In addition, the Tgs of PEO and PAA (−53 and 105 °C, respectively) are not observed the PPM coatings (Fig. 3b).44 This lack of Tg signatures in DSC suggests that the two polymers form a single continuous phase within the coating, which is typical of polymer complexes.54
Oxygen barrier of hydrogen bonded complexes
PPM 1:2, 1:1, and 2:1 were coated on 760 μm thick natural rubber substrates for OTR measurement. As shown in Table 2, film barrier properties improve significantly as the PEO content increases relative to PAA. PPM 1:2 provides a modest 1.8× reduction in OTR compared to uncoated rubber, while PPM 1:1 reduces the OTR of rubber by an order of magnitude. PPM 2:1 reduces the OTR of natural rubber by two orders of magnitude at a similar thickness (∼1.8 μm) to PPM 1:2 and 1:1. A more complete set of barrier values, at various strains, along with coat weights, are provided in Table S1.† This improvement cannot be attributed to the quantity of MMT in the PPM 2:1 coating, as the total clay content of all three systems was confirmed to be the same (∼40%) via thermogravimetric analysis (Fig. S2†). The improved barrier of PPM 2:1 is attributed to the greater relative amount of PEO in the system, leading to improved clay distribution, despite the total clay content of each system being the same. It has been observed that MMT is more fully intercalated and more uniformly distributed in a PEO solution as the PEO content increases.55,56 Although MMT is expected to be fully exfoliated when dispersed in water,57 it is likely that increased interactions with PEO will lead to more uniform nanosheet distribution within the complex, leading to an increase in tortuosity for gas molecules.
| Sample name | OTR (cc (m2 day)−1) | Film thickness (μm) | Total permeability (×10−14 cc cm (cm2 s Pa)−1) | Film permeabilitya (×10−16 cc cm (cm2 s Pa)−1) |
|---|---|---|---|---|
| a Film permeability was decoupled from the substrate using a previously described method.58 | ||||
| Substrate | 2110 | — | 184 | — |
| PPM 1:2 |
1150 | 1.07 | 100 | 61.8 |
| PPM 1:1 |
262 | 1.63 | 22.9 | 11.2 |
| PPM 2:1 |
21.4 | 1.79 | 1.87 | 0.9 |
Improved distribution of MMT platelets with increasing PEO content can be observed via FTIR. As shown in Fig. 4, pure PEO exhibits a broad asymmetric CH2 stretching peak between 2940 and 2840 cm−1, as well as two narrow, weak bands at 2740 and 2693 cm−1. In contrast, when PEO is associated with MMT, these peaks should shift to become two defined peaks at 2918 and 2880 cm−1.55 These peaks, while present in the FTIR spectra for all three PPM recipes, grow in sharpness and intensity as PEO content increases (Fig. 4). These data support the hypothesis that increased PEO content results in increased polymer interactions with MMT platelets in the PPM complex. The improved OTR of PPM 2:1 relative to PPM 1:1 and 1:2 can also be partially attributed to the richer hydrogen bonding network, which increases the cohesive energy.4 It has previously been demonstrated that increasing interpolymer hydrogen bonding interactions within a PEO–PAA film decreases oxygen permeability, while increasing intramolecular interactions between PAA repeat units does little to improve barrier properties.25,27 As PEO content increases in the coating, interactions are maximized between PEO and each of the other coating components (PAA and MMT) leading to a greatly improved oxygen barrier.
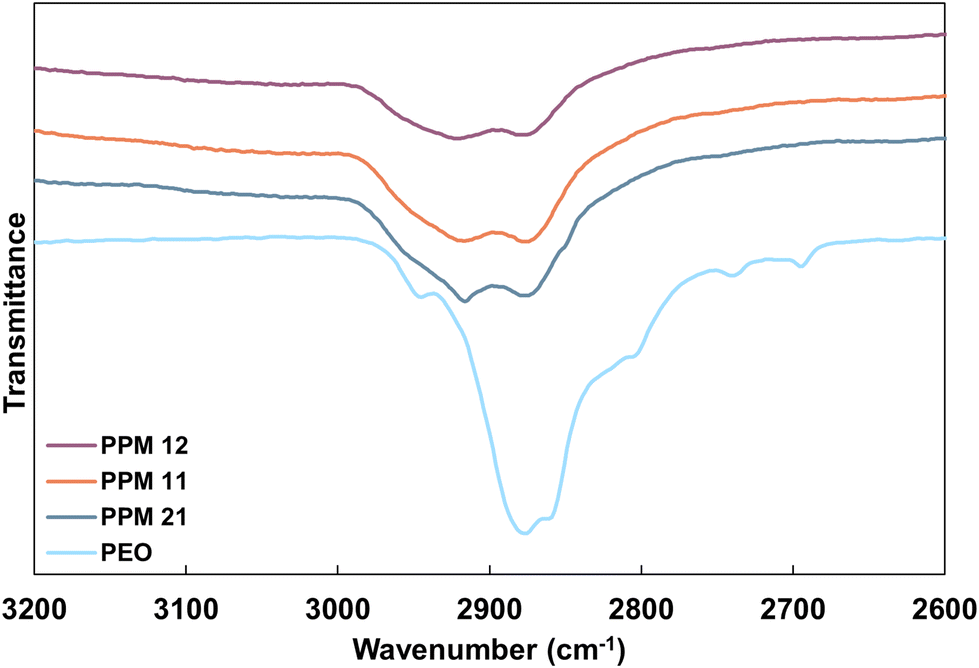 | ||
| Fig. 4 FTIR spectra of cured PPM 1:2, 1:1, and 2:1 complexes and pure PEO from 3200 to 2600 cm−1. | ||
AFM data supports the assertion that increased PEO content leads to improved coating uniformity. As shown in Fig. 5, film roughness significantly decreases as the PEO:PAA ratio increases, with PPM 2:1 being about twice as smooth as PPM 1:2. Roughness may be attributed to increased PAA dimerization (at the expense of PEO–PAA hydrogen bonding) that leads to larger individual polymer domains within the film, decreasing homogeneity and increasing the roughness.27 Additionally, PPM 2:1 exhibits increased dispersion of MMT platelets due to higher PEO content, which could lead to a smoother film surface. This assertion is further demonstrated via phase mapping of the same AFM images (Fig. 5d–f). When compared to the other films, the PPM 2:1 coating appears to show more clearly dispersed clay particles throughout the darker polymer matrix. MMT is less evenly distributed in the PPM 1:2 and 1:1 coatings compared to in PPM 2:1, leading to increased permeability and surface roughness.
 | ||
| Fig. 5 AFM height images (a–c) and phase images (d–f) of PPM 1:2 (a and d), PPM 1:1 (b and e), and PPM 2:1 (c and f), including average roughness (Ra) and root-mean-square roughness (Rq) values. | ||
Mechanical behavior of complexes
The PEO:PAA ratio also plays a major role in determining the film's mechanical properties. Neither PEO nor PAA are elastomeric on their own, but hydrogen bonded complexes of the two demonstrate incredibly elastic behavior.44,59 The low Tg, highly mobile PEO in the complex allows it to be stretched, while the hydrogen bonds between PEO and PAA act as physical crosslinks that allow the complex to restore its shape after stress is removed.26,44 The elasticity of the coating improves with increasing relative PEO content, because the PEO in the system is well above its Tg of −53 °C and is prevented from crystallizing by complexation with PAA.38,51,54 As a result, these highly mobile PEO chains have a greater ability to relax when strain is applied. Due to its higher relative PEO content, the PPM 2:1 complex is expected to behave more elastically than the PPM 1:1 or 1:2 complexes.44,59,60 Even small changes in polymer ratios can have a considerable impact on mechanical behavior. As shown in Fig. 6a, the indentation moduli of PPM 1:2, 1:1, and 2:1 were measured to be 19.2 ± 2.8, 7.1 ± 0.8, and 3.1 ± 0.2 GPa, respectively. PPM 2:1 was chosen for analysis as a stretchable gas barrier because it has the lowest OTR and the greatest elasticity.
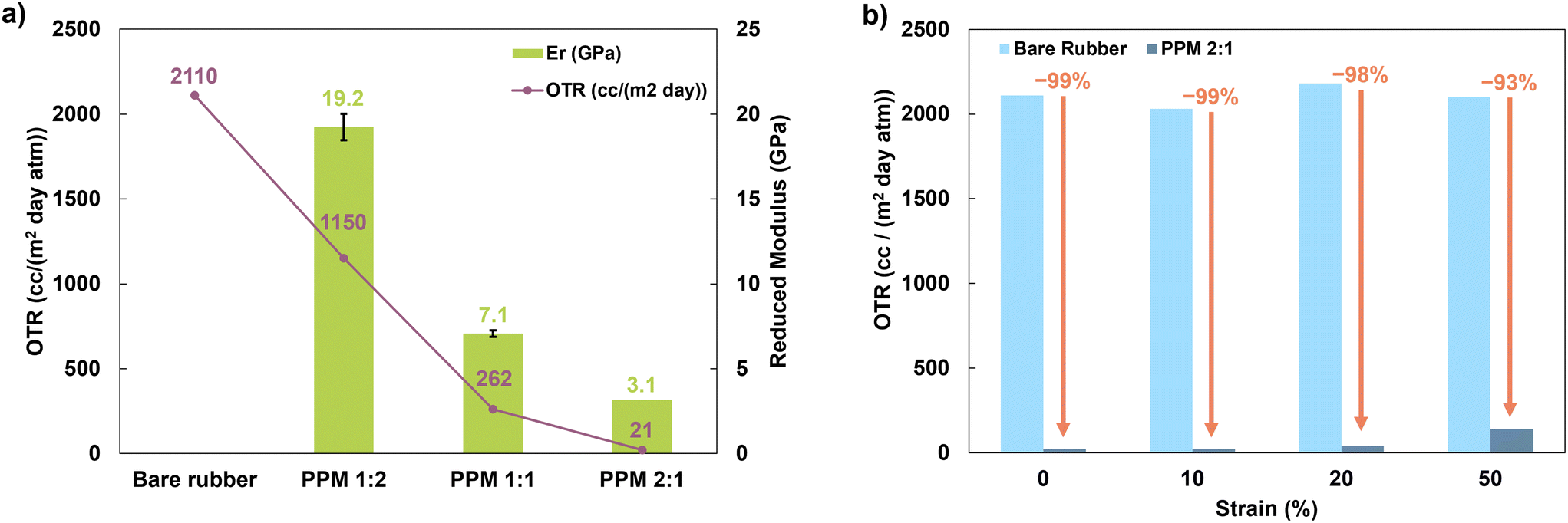 | ||
| Fig. 6 (a) OTR of uncoated and coated rubber with reduced moduli (from nanoindentation) of PPM coatings, and (b) OTR of uncoated and PPM 2:1-coated rubber as a function of strain applied before OTR testing. | ||
Influence of strain on gas barrier
The PPM 2:1 coating demonstrates excellent retention of barrier properties upon stretching. When subjected to no strain, the coating decreases the OTR of natural rubber by 99%. After 10% strain, the OTR is still reduced by 99%, and after 20% strain, the OTR is reduced by 98%. SEM images of coated and uncoated substrates after being subjected to 0, 10, 20 and 50% strain reveal that the coating does not begin to show signs of damage until 50% strain (Fig. S4†). Even after 50% strain, the PPM 2:1 coating still reduces the OTR by 93%, as shown in Fig. 6. The retention of barrier properties even after stretching is attributed to the highly elastomeric nature of the PPM 2:1 film.
The reduction in OTR and number of processing steps are considerably improved compared to a previously published PEO/PAA + MMT LbL system.28 The previous 10-bilayer system, which required a total of 21 individual dipping steps and 61 rinsing steps, reduced the OTR of the polyurethane substrate by 80×, and after 20% strain (the maximum strain applied to the coated substrate), reduced the OTR by 46×. In contrast, the coating presented here requires a total of three deposition steps and three rinsing steps, reduces the OTR of the substrate by 99×, and maintains a 54× reduction in OTR after 20% strain. Furthermore, this coating is able to withstand 50% strain, while still providing a 15× reduction in OTR.
It is interesting to note that an inverse relationship between barrier and ductility of polymer complex films is expected based on previous work,6 but the present study exhibits the opposite situation. Indeed, when low permeability is largely ascribed to high cohesive energy density, barrier properties are expected to correlate to film stiffness.7 In this work, however, the barrier must be attributed primarily to the high level of MMT dispersion obtained by manipulating the PEO content of the system. Independently, the PEO:PAA ratio in the system is tuned to obtain high elasticity. In this way, the manipulation of PEO content allows for the creation of a coating that maximizes stretchability, while minimizing oxygen transmission.
It is likely that a maximum PEO:PAA ratio exists (above PPM 2:1) that would result in the best possible combination of high stretchability and low OTR. This ratio would be wherever the critical number of hydrogen bond crosslinks required to maintain stretchability and prevent PEO crystallization occurs. At this “critical ratio”, enough crosslinks would be present to maintain elastomeric behavior, while MMT exfoliation would simultaneously be maximized due to high PEO content. Above this ratio, stretchability would likely decrease and OTR increase due to an insufficient number of hydrogen bonding interactions. Excess PEO in the HBPC would be expected to crystallize, resulting in crystalline peaks in XRD and a melting peak in DSC, neither of which were observed for any of the ratios investigated in this work.
Conclusions
An effective and highly elastic gas barrier coating was realized through the development of a hydrogen bonded polymer complex of poly(acrylic acid), poly(ethylene oxide), and montmorillonite clay. FTIR verifies the protonation of PAA, and resulting hydrogen bonding interactions, upon changing the pH of the coating with a buffer step. XRD and DSC analyses indicate intimate blending of the polymers at all ratios investigated, as no polymer crystallinity was observed for any of the ratios studied. FTIR analysis further demonstrates that increasing PEO content relative to PAA increases interpolymer hydrogen bonds and strengthens interactions between PEO and MMT clay. As a result, the HBPC with the highest PEO:PAA ratio exhibits the lowest roughness, the lowest oxygen permeability, and the greatest elasticity of the complexes. This HBPC coating decreases the oxygen transmission rate of the natural rubber substrate by two orders of magnitude. Even after exposure to 50% strain, the best-performing coating retains a 93% reduction in OTR as compared to the uncoated substrate. Deposited in three steps, with water-based processing, this hydrogen bonded complex represents a promising development for the facile construction of stretchable gas barrier films.
Author contributions
Sarah G. Fisher: investigation, formal analysis, validation, writing – original draft, writing – review & editing, visualization. Hsu-Cheng Chiang: conceptualization, methodology, investigation. Ethan T. Iverson: methodology, investigation, formal analysis, writing – review & editing. Edward Chang: investigation. Jaime C. Grunlan: supervision, resources, writing – review & editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the Texas A&M Materials Characterization Facility (RRID: SCR_022202) for assistance obtaining SEM data and the Texas A&M University Soft Matter Facility (RRID: SCR_022482) for assistance obtaining DSC data.References
- A. Behrendt, C. Friedenberger, T. Gahlmann, S. Trost, T. Becker, K. Zilberberg, A. Polywka, P. Görrn and T. Riedl, Adv. Mater., 2015, 27, 5961–5967 CrossRef CAS PubMed.
- Y. Su, V. G. Kravets, S. L. Wong, J. Waters, A. K. Geim and R. R. Nair, Nat. Commun., 2014, 5, 4843 CrossRef CAS PubMed.
- Y. Dou, T. Pan, S. Xu, H. Yan, J. Han, M. Wei, D. G. Evans and X. Duan, Angew. Chem., Int. Ed., 2015, 54, 9673–9678 CrossRef CAS PubMed.
- K. Shi, X. Xu, S. Dong, B. Li and J. Han, AIChE J., 2021, 67, e17373 CrossRef CAS.
- S. Gupta, S. Sindhu, K. A. Varman, P. C. Ramamurthy and G. Madras, RSC Adv., 2012, 2, 11536–11543 RSC.
- H.-C. Chiang, E. T. Iverson, K. Schmieg, D. L. Stevens and J. C. Grunlan, J. Appl. Polym. Sci., 2023, 140, e53473 CrossRef CAS.
- D. Benczédi, Trends Food Sci. Technol., 1999, 10, 21–24 CrossRef.
- N. Ochirkhuyag, Y. Nishitai, S. Mizuguchi, Y. Isano, S. Ni, K. Murakami, M. Shimamura, H. Iida, K. Ueno and H. Ota, ACS Appl. Mater. Interfaces, 2022, 14, 48123–48132 CrossRef CAS PubMed.
- S. Shin, H. W. Yoon, Y. Jang and M. Hong, Appl. Phys. Lett., 2021, 118, 181901 CrossRef CAS.
- T. V. Duncan, J. Colloid Interface Sci., 2011, 363, 1–24 CrossRef CAS PubMed.
- A. Idris, A. Muntean and B. Mesic, J. Coat. Technol. Res., 2022, 19, 699–716 CrossRef CAS.
- G. H. Fredrickson and J. Bicerano, J. Chem. Phys., 1999, 110, 2181–2188 CrossRef CAS.
- E. L. Cussler, S. E. Hughes, W. J. Ward and R. Aris, J. Membr. Sci., 1988, 38, 161–174 CrossRef CAS.
- Y. Cui, S. Kumar, B. R. Kona and D. van Houcke, RSC Adv., 2015, 5, 63669–63690 RSC.
- J.-T. Chen, Y.-J. Fu, Q.-F. An, S.-C. Lo, S.-H. Huang, W.-S. Hung, C.-C. Hu, K.-R. Lee and J.-Y. Lai, Nanoscale, 2013, 5, 9081–9088 RSC.
- D. Feldman, J. Macromol. Sci., Part A: Pure Appl.Chem., 2013, 50, 441–448 CrossRef CAS.
- H. M. C. de Azeredo, Food Res. Int., 2009, 42, 1240–1253 CrossRef CAS.
- F. B. Dhieb, E. Jalali Dil, S. H. Tabatabaei, F. Mighri and A. Ajji, RSC Adv., 2019, 9, 1632–1641 RSC.
- M. A. Priolo, K. M. Holder, T. Guin and J. C. Grunlan, Macromol. Rapid Commun., 2015, 36, 866–879 CrossRef CAS PubMed.
- L. Wågberg and J. Erlandsson, Adv. Mater., 2021, 33, 2001474 CrossRef PubMed.
- Y. Li, Z. Tang, W. Wang, X. Huang, Y. Lv, F. Qian, Y. Cheng and H. Wang, Carbohydr. Polym., 2021, 253, 117227 CrossRef CAS PubMed.
- Multilayer Thin Films, eds G. Decher and J. B. Schlenoff, Wiley-VCH Verlag & Co., Weinheim, 2nd edn, 2012 Search PubMed.
- D. Han, Y. Luo, Q. Ju, X. Xiao, M. Xiao, N. Xiao, S. Chen, X. Peng, S. Wang and Y. Meng, Polymers, 2018, 10, 1082 CrossRef PubMed.
- B. Palen, T. J. Kolibaba, J. T. Brehm, R. Shen, Y. Quan, Q. Wang and J. C. Grunlan, ACS Omega, 2021, 6, 8016–8020 CrossRef CAS PubMed.
- F. Xiang, S. M. Ward, T. M. Givens and J. C. Grunlan, ACS Macro Lett., 2014, 3, 1055–1058 CrossRef CAS PubMed.
- J. L. Lutkenhaus, K. D. Hrabak, K. McEnnis and P. T. Hammond, J. Am. Chem. Soc., 2005, 127, 17228–17234 CrossRef CAS PubMed.
- F. Xiang, S. M. Ward, T. M. Givens and J. C. Grunlan, Soft Matter, 2015, 11, 1001–1007 RSC.
- S. Qin, Y. Song, M. E. Floto and J. C. Grunlan, ACS Appl. Mater. Interfaces, 2017, 9, 7903–7907 CrossRef CAS PubMed.
- M. Humood, S. Chowdhury, Y. Song, P. Tzeng, J. C. Grunlan and A. A. Polycarpou, ACS Appl. Mater. Interfaces, 2016, 8, 11128–11138 CrossRef CAS PubMed.
- C. Cho, Y. Song, R. Allen, K. L. Wallace and J. C. Grunlan, J. Mater. Chem. C, 2018, 6, 2095–2104 RSC.
- R. J. Smith, C. T. Long and J. C. Grunlan, Langmuir, 2018, 34, 11086–11091 CrossRef CAS PubMed.
- J. Li, G. van Ewijk, D. J. van Dijken, J. van der Gucht and W. M. de Vos, ACS Appl. Mater. Interfaces, 2021, 13, 21844–21853 CrossRef CAS PubMed.
- D. F. Anghel, V. Alderson, F. M. Winnik, M. Mizusaki and Y. Morishima, Polymer, 1998, 39, 3035–3044 CrossRef CAS.
- J. Choi and M. F. Rubner, Macromolecules, 2005, 38, 116–124 CrossRef CAS.
- A. M. Rocco, R. P. Pereira and M. I. Felisberti, Polymer, 2001, 42, 5199–5205 CrossRef CAS.
- B. C. Smith, Spectroscopy, 2018, 33, 20–23 Search PubMed.
- H. Kaczmarek and A. Szalla, J. Photochem. Photobiol., A, 2006, 180, 46–53 CrossRef CAS.
- M. M. Coleman, P. C. Painter and J. F. Graf, Specific Interactions and the Miscibility of Polymer Blends, CRC Press, 1995 Search PubMed.
- J. L. Lutkenhaus, K. McEnnis and P. T. Hammond, Macromolecules, 2007, 40, 8367–8373 CrossRef CAS.
- M. M. Coleman, J. Y. Lee, C. J. Serman, Z. Wang and P. C. Painter, Polymer, 1989, 30, 1298–1307 CrossRef CAS.
- P. Laurson, P. Raudsepp, H. Kaldmäe, A. Kikas and U. Mäeorg, AIP Adv., 2020, 10, 085214 CrossRef CAS.
- A. R. M. Tones, E. Eyng, C. L. Zeferino, S. O. Ferreira, A. A. A. Alves, M. R. Fagundes-Klen and E. Sehn, Sci. Total Environ., 2020, 713, 136301 CrossRef CAS PubMed.
- C.-M. Popescu, G. Singurel, M.-C. Popescu, C. Vasile, D. S. Argyopoulos and S. Willför, Carbohydr. Polym., 2009, 77, 851–857 CrossRef CAS.
- J. Li, Z. Wang, L. Wen, J. Nie, S. Yang, J. Xu and S. Z. D. Cheng, ACS Macro Lett., 2016, 5, 814–818 CrossRef CAS PubMed.
- B. W. N. H. Hemasiri, J.-K. Kim and J.-M. Lee, Sci. Rep., 2017, 7, 17868 CrossRef PubMed.
- M. Thirumoorthi and J. Thomas Joseph Prakash, J. Asian Ceram. Soc., 2016, 4, 124–132 CrossRef.
- G. Damian, F. Damian, Z. Szakács, G. Iepure and D. Aştefanei, Minerals, 2021, 11, 938 CrossRef CAS.
- X. Zhang, H. Yi, H. Bai, Y. Zhao, F. Min and S. Song, RSC Adv., 2017, 7, 41471–41478 RSC.
- N. Tahiri, L. Khouchaf, M. Elaatmani, G. Louarn, A. Zegzouti and M. Daoud, IOP Conf. Ser.: Mater. Sci. Eng., 2014, 62, 012002 CrossRef CAS.
- I. Jeon and K. Nam, Sci. Rep., 2019, 9, 9878 CrossRef PubMed.
- Y. Deng, Y. Xu, X. Ni, W. LiYu, Y. Wang and Z. Yang, J. Mater. Eng. Perform., 2023, 32, 6123–6132 CrossRef CAS.
- K. K. Kumar, M. Ravi, Y. Pavani, S. Bhavani, A. K. Sharma and V. V. R. N. Rao, J. Non-Cryst. Solids, 2012, 358, 3205–3211 CrossRef.
- A. M. Abdelghany, M. O. Farea and A. H. Oraby, J. Mater. Sci.: Mater. Electron., 2021, 32, 6538–6549 CrossRef CAS.
- R. F. Shamoun, H. H. Hariri, R. A. Ghostine and J. B. Schlenoff, Macromolecules, 2012, 45, 9759–9767 CrossRef CAS.
- Z. Shen, G. P. Simon and Y.-B. Cheng, Polymer, 2002, 43, 4251–4260 CrossRef CAS.
- J. Wu and M. M. Lerner, Chem. Mater., 1993, 5, 835–838 CrossRef CAS.
- Y. Zhao, H. Yi, F. Jia, H. Li, C. Peng and S. Song, Powder Technol., 2017, 306, 74–79 CrossRef CAS.
- A. P. Roberts, B. M. Henry, A. P. Sutton, C. R. M. Grovenor, G. A. D. Briggs, T. Miyamoto, M. Kano, Y. Tsukahara and M. Yanaka, J. Membr. Sci., 2002, 208, 75–88 CrossRef CAS.
- Y. Wang, X. Liu, S. Li, T. Li, Y. Song, Z. Li, W. Zhang and J. Sun, ACS Appl. Mater. Interfaces, 2017, 9, 29120–29129 CrossRef CAS PubMed.
- Y. Hu, D. Shao, Y. Chen, J. Peng, S. Dai, M. Huang, Z.-H. Guo, X. Luo and K. Yue, ACS Appl. Energy Mater., 2021, 4, 10886–10895 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: FTIR spectra of PEO, PAA, and MMT; TGA thermograms of PPM complexes under air; SEM images of uncoated and coated substrate; SEM images of coated and uncoated substrate before and after strain application. See DOI: https://doi.org/10.1039/d3lp00194f |
| This journal is © The Royal Society of Chemistry 2024 |