
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreferential N–H⋯:C![[double bond splayed right]](https://www.rsc.org/images/entities/h2_char_e00a.gif) hydrogen bonding involving ditopic NH-containing systems and N-heterocyclic carbenes†
hydrogen bonding involving ditopic NH-containing systems and N-heterocyclic carbenes†
Zacharias J. Kinney a,
Arnold L. Rheingoldb and
John D. Protasiewicz*a
a,
Arnold L. Rheingoldb and
John D. Protasiewicz*a
aDepartment of Chemistry, Case Western Reserve University, Cleveland, Ohio 44106, USA. E-mail: protasiewicz@case.edu
bDepartment of Chemistry and Biochemistry, University of California, La Jolla, San Diego, California 92093, USA
First published on 20th November 2020
Abstract
Hydrogen bonding plays a critical role in maintaining order and structure in complex biological and synthetic systems. N-heterocyclic carbenes (NHCs) represent one of the most versatile tools in the synthetic chemistry toolbox, yet their potential as neutral carbon hydrogen bond acceptors remains underexplored. This report investigates this capability in a strategic manner, wherein carbene-based hydrogen bonding can be assessed by use of ditopic NH-containing molecules. N–H bonds are unique as there are three established reaction modes with carbenes: non-traditional hydrogen bonding adducts (X–H⋯:C![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) ), salts arising from proton transfer ([H–C]+[X]−), or amines from insertion of the carbene into the N–H bond. Yet, there are no established rules to predict product distributions or the strength of these associations. Here we seek to correlate the hydrogen bond strength of symmetric and asymmetric ditopic secondary amines with 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene (IPr, a representative NHC). In symmetric and asymmetric ditopic amine adducts both the solid-state (hydrogen bond lengths, NHC interior angles) and solution-state (1H Δδ of NH signals, 13C signals of carbenic carbon) can be related to the pKa of the parent amine.
), salts arising from proton transfer ([H–C]+[X]−), or amines from insertion of the carbene into the N–H bond. Yet, there are no established rules to predict product distributions or the strength of these associations. Here we seek to correlate the hydrogen bond strength of symmetric and asymmetric ditopic secondary amines with 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene (IPr, a representative NHC). In symmetric and asymmetric ditopic amine adducts both the solid-state (hydrogen bond lengths, NHC interior angles) and solution-state (1H Δδ of NH signals, 13C signals of carbenic carbon) can be related to the pKa of the parent amine.
Introduction
Hydrogen bonding interactions play a vital role throughout nature and are central to our understanding of complex biological systems.1 The biological examples of such systems are incalculable, but prominent examples such as DNA, RNA, and enzymatic processes deserve mention.2–4 Non-biological examples are equally diverse, ranging from controlling the reactivity of small molecules,5 being central to the unique properties of water,6,7 to being a key mechanism for generating complex folded structures.8,9 While the role of hydrogen bonding in complex systems is indisputable, the development of methods to predict and assess the strengths of these interactions are vital to rationally designing novel systems.10,11Hydrogen bond adducts X–H⋯A involve three-center four-electron bonds formed by interaction of lone pair electrons on a hydrogen bond acceptor (A) with a σ* orbital of a hydrogen bond donor (X–H).12 In traditional hydrogen bonding, X is an electronegative atom (such as N, O, S, or F) which sufficiently polarizes the X–H bond leading to a reduction of the σ* orbitals energy; and A is generally an electronegative atom (N, O, or halogens) having accessible lone pair electrons.12–14 Non-traditional hydrogen bonds involving carbon centers are known but relatively rare, with association strengths that are generally weaker than conventional hydrogen bonds.15 Most often these non-traditional species are mediated via formally charged carbon centers (such as isonitriles and carbanions) as the hydrogen bond acceptor.16,17
Since the first isolation of singlet carbenes their proliferation to all corners of chemistry are readily apparent.18–22 Though a variety of singlet carbenes are now synthetically accessible,23,24 N-heterocyclic carbenes (NHCs) based on an imidazole core remain one of the most well-studied and utilized class of carbenes.21,22,25–27 Shortly after the discovery of isolable NHCs, it was postulated that these compounds could serve as strong hydrogen bond acceptors: with their divalent carbon bearing a lone pair (σ-donor) to interact with a myriad of Brønsted acid X–H species.28,29 Arduengo described the first example of a carbene-centered hydrogen bond between 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ium hexafluorophosphate and 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene (IMes).30 Clyburne and Davidson later expanded these findings to amine-based non-traditional hydrogen bonding between diphenylamine (DPA) and IMes (DPA⋯IMes, Fig. 1a), showing these species can be structurally authenticated.31 Movassaghi and Schmidt also identified a MeOH⋯IMes adduct in their efforts to catalyze amidation reactions with carbenes, authenticating the adduct both in solid- and solution-state (Fig. 1a).32
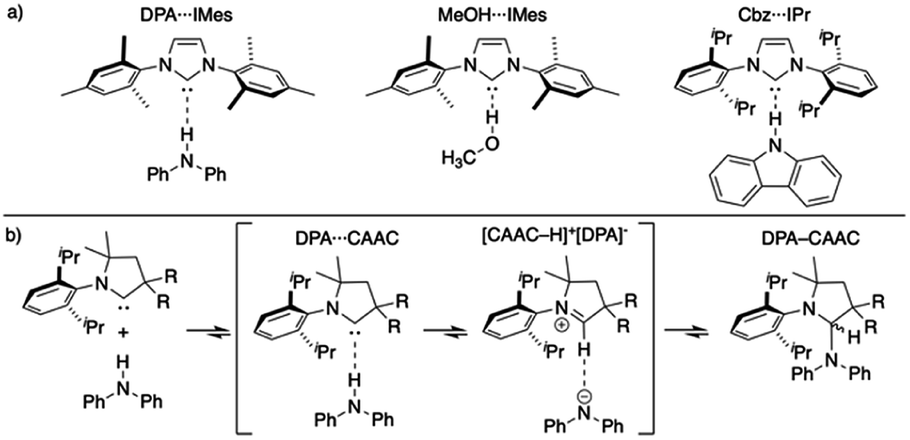 | ||
| Fig. 1 (a) Representative authenticated X–H⋯:C systems;31–33 (b) general reaction progression elucidated by Bertrand of CAAC inserting into a N–H bond.34 | ||
In addition to these reports, other computational35–39 and experimental33,40–50 studies on non-traditional X–H⋯:C hydrogen bonding have emerged (:C is neutral carbene species). The balance between non-traditional hydrogen bonding and proton transfer relies on the weak nucleophilicity but high Lewis basicity of NHCs, which can be further modulated via variation of the substituents on the imidazole-core.51,52 Comparison of the available solid-state structures of X–H⋯:C hydrogen bonding reveal significant differences can occur. An example of this can be realized in comparing the hydrogen bond lengths (H⋯:C distance) in secondary amine-based adducts of carbazole (Cbz) and DPA with common NHCs 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene (IPr) and IMes. The hydrogen bond lengths of Cbz⋯IPr (2.07(3) Å)33 and DPA⋯IMes (2.30(2) Å)31 suggest a stronger interaction for the carbazole NH than for the DPA NH when employing similar NHCs.53 The interior N–C–N angles of the imidazole-2-ylidene units are slightly distorted as well, where Cbz⋯IPr = 102.2(2)° and DPA⋯IMes = 101.7(1)°.
These differences might be attributed to the differing steric profiles of IMes and IPr (2,4,6-trimethylphenyl, Mes versus 2,6-diisopropylphenyl, Dipp), though more probably these differences are due to the acidities of DPA (pKa ≈ 25.0 in DMSO)54 versus Cbz (pKa ≈ 19.9 in DMSO).55 Consistent with the latter hypothesis, MeOH⋯IMes adduct (Fig. 1a) displays a shorter hydrogen bond length (1.96(2) Å) and a more pronounced N–C–N angle of 102.5(1)° (MeOH pKa ∼ 29.0 in DMSO)56,57 than the amine-based species depicted in Fig. 1a. Although making comparisons between the reactivities of different NHCs can be troublesome, relatively minor differences in the basicity of IPr and IMes can be assumed, for the pKa of their parent imidazoliums' are within error (pKa IPr·HCl = 19.29 ± 0.07, pKa IMes·HCl = 19.40 ± 0.12).58,59
Though the isolation and authentication of hydrogen bonding adducts of carbenes has thus far been limited to imidazolylidene-based carbenes, hydrogen bonding adducts role in catalysis60–63 and X–H insertion chemistry has been postulated for some time. Bertrand recently provided computational evidence for these non-traditional hydrogen bonded species as key intermediates for the reversible insertion of cyclic (alkyl)(amino)(carbene)s (CAACs) into the N–H bond of DPA (Fig. 1b).34 The calculations suggested that the process of a CAAC inserting into an N–H bond proceeds via non-traditional hydrogen bonded and proton transfer intermediates.
Our recent work has established that Cbz and IPr form a hydrogen bonding adduct (Cbz⋯IPr, Fig. 1a) that has a strength of association in solution (C6D6) of −2.8 kcal mol−1.33 Previous studies of non-traditional X–H⋯:C hydrogen bonding (X = N (ref. 31) or O (ref. 32, 47, 48 and 64)) did not quantitate the strengths of interaction. Interestingly, the corresponding reaction of Cbz with CAAC from this study is reversible and yields the product of insertion of the carbene into the N–H bond of Cbz. The use of more strongly acidic N–H species with CAACs however results in proton transfer to forms salts.33,65 Collectively, these experimental efforts help “visualize” the proposed intermediates for insertion reactions of CAACs into X–H bonds.
While these efforts have proven fruitful in identifying new species with unique hydrogen bonds, there are no steadfast rules to predict reactivity or product distribution. Crystallographic information on H⋯:C hydrogen bonds shed some insight into the strength of these associations, but there are no obvious ways to correlate these metrics to solution-state measurements. Computational efforts have been made to determine the hydrogen bond basicity of carbenes, but thus far these measures have been purely theoretical.66,67
It is of interest therefore to gain further insights into the factors that determine when hydrogen bonding occurs in preference to proton transfer or N–H insertion for reactions between isolable carbenes and NH-containing molecules. An important part of this effort involves delineating the strengths of non-conventional hydrogen bonding. In order to probe this latter question in more detail, a series of symmetric and asymmetric ditopic NH-containing systems consisting of Cbz and DPA units (Chart 1; 3,3′-bicarbazole,68 BC; N,N′-diphenylbenzidine, DPB; N,N′-diphenyl-p-phenylenediamine, DPPD, 4-(carbazol-3-yl)-N-phenylaniline, CD), which are structurally similar with differing acidities, will be reacted with a single NHC (IPr). These unusual platforms will allow for the gradual tuning of a single component (X–H) while having the carbene (A) remain consistent. This array of hydrogen bonded adducts will be used to assess how a NHC participates in non-traditional hydrogen bonding when presented with multiple reactive sites within single molecules.
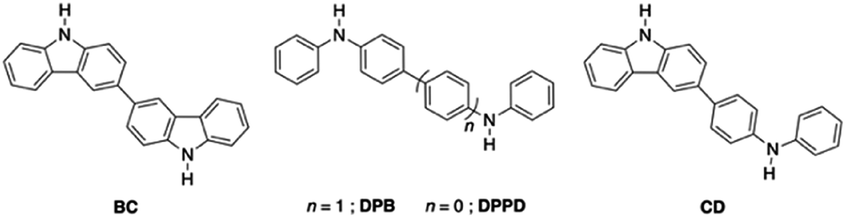 | ||
| Chart 1 Ditopic secondary amines. | ||
Results and discussion
BC⋯2IPr adduct
Previous studies on a Cbz⋯IPr hydrogen bonded adduct suggested that IPr could form a di-hydrogen bonded adduct with BC, and that this adduct should be labile and display concentration dependent behavior. 1H NMR analysis of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 mixture of BC and IPr in THF-d8 reveals that significant shifts (Δδ > 0.05 ppm) occur for all proton resonances of the individual components, as highlighted in Fig. 2. In particular, the drastic downfield shift of Hα (Δδ ≈ +1.7 ppm) is indicative of hydrogen bonding, and is similar to the corresponding shift of Hα during formation of Cbz⋯IPr. The number of resonances indicate a symmetric structure for the BC⋯2IPr di-hydrogen bonded adduct. Consistent with this formulation, signals 1b and 2b show a substantial upfield shift (Δδ ≈ −0.9 ppm), since upon complexation they are positioned directly into the face of the Dipp groups on IPr. Inspection of the 13C{1H} NMR spectrum of BC⋯2IPr reveals a single carbenic resonance (δ 215.2 ppm) that is noticeably upfield relative to the carbenic resonance of IPr (δ 221.1 ppm in THF-d8).
:2 mixture of BC and IPr in THF-d8 reveals that significant shifts (Δδ > 0.05 ppm) occur for all proton resonances of the individual components, as highlighted in Fig. 2. In particular, the drastic downfield shift of Hα (Δδ ≈ +1.7 ppm) is indicative of hydrogen bonding, and is similar to the corresponding shift of Hα during formation of Cbz⋯IPr. The number of resonances indicate a symmetric structure for the BC⋯2IPr di-hydrogen bonded adduct. Consistent with this formulation, signals 1b and 2b show a substantial upfield shift (Δδ ≈ −0.9 ppm), since upon complexation they are positioned directly into the face of the Dipp groups on IPr. Inspection of the 13C{1H} NMR spectrum of BC⋯2IPr reveals a single carbenic resonance (δ 215.2 ppm) that is noticeably upfield relative to the carbenic resonance of IPr (δ 221.1 ppm in THF-d8).
 | ||
| Fig. 2 Partial 1H NMR spectra (500 MHz, THF-d8) of BC (50 mM) and BC⋯2IPr. Dashed lines indicate chemical shift changes upon hydrogen bonding to IPr. | ||
Vapor diffusion of hexanes into a solution of BC⋯2IPr in THF yielded pale yellow crystals suitable for X-ray diffraction. Crystallographic analysis of one such crystal of BC⋯2IPr is portrayed in Fig. 3 (see ESI† for full details). The structure confirms a two-fold hydrogen bonding adduct, with an average H1⋯C13 distance of 2.10 Å and a N2–C12–N3 angle of 102.2°, which are nearly identical to corresponding parameters for the mono-functional counterpart Cbz⋯IPr. Interestingly, the imidazole-2-ylidene C3N2 and Cbz ring systems are nearly coplanar as indicated by ϕ N2–N3–C12–C1 = 13.5°, which is in line with the monotopic Cbz⋯IPr system (ϕ = 11.0°).
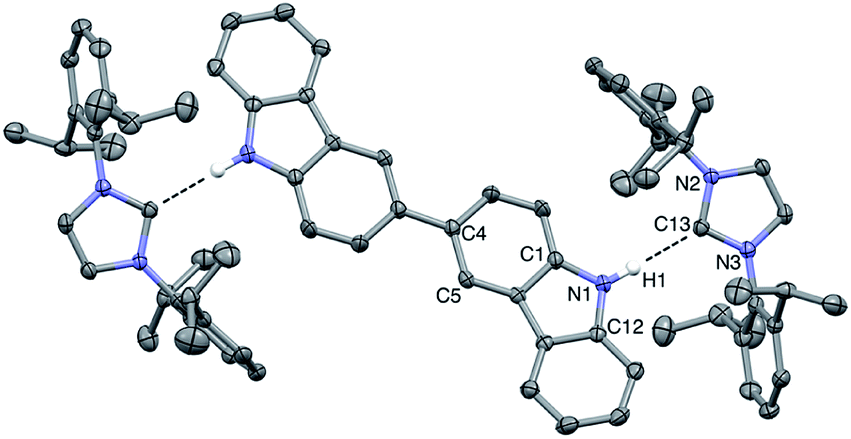 | ||
| Fig. 3 ORTEP diagram (ellipsoids drawn at 50% probability) of BC⋯2IPr. Non-hydrogen bonding hydrogens and co-crystallized THF are omitted for clarity. Average select bond lengths (Å) and angles (°): H1⋯C13, 2.10; N1⋯C13, 2.96; N2–C13–N3, 102.2; N2–N3–C12–C1, 13.5. | ||
Model DPA⋯IPr adduct
Structural data for a DPA⋯IMes hydrogen bonded adduct were previously reported,31 but for the present study related details on the corresponding DPA⋯IPr adduct are necessary so that closer comparisons can be made. This material was synthesized by the addition of IPr to a THF solution of DPA. Removal of solvent, followed by recrystallization from toluene at −35 °C affords pure DPA⋯IPr as white crystals. Hydrogen bond formation is clearly indicted in C6D6 by an upfield shift of the carbenic carbon resonance (218.2 ppm) compared to parent IPr (220.6 ppm in C6D6).69 Preliminary 1H NMR analysis of this material showed concentration dependent behavior. To ascertain the strength of this association a 1H NMR concentration dependent study of DPA⋯IPr was performed in C6D6 over a broad concentration range (75–1 mM). Benzene is an ideal solvent in such an analysis as its impact on hydrogen bonding is minimal,70 while also allowing for comparison to our previous measurements on Cbz⋯IPr (C6D6, concentration range of 50–1 mM).33 This analysis revealed the association strength of DPA⋯IPr to be substantially weaker than Cbz⋯IPr (Kd = 105 ± 3 M−1; ΔG ≈ −2.8 kcal mol−1), with only the NH resonance being significantly influenced upon complexation (Kd = 0.85 ± 1.05 M−1 and ΔG ≈ −0.10 kcal mol−1, see ESI† for full details).71 This drastic preference for the more acidic amine of Cbz was also verified via an exchange reaction (eqn (1)):| DPA⋯IPr + Cbz ⇌ Cbz⋯IPr + DPA | (1) |
Ditopic DPB⋯2IPr and DPPD⋯2IPr adducts
Both ditopic DPB and DPPD yield 1:2 adducts DPB⋯2IPr and DPPD⋯2IPr, respectively, upon reaction with two equivalents of IPr (Fig. 4, top) in THF. As previously observed, DPB⋯2IPr and DPPD⋯2IPr are in equilibrium with their individual components, and at higher NMR concentrations these solutions exist predominately as the di-hydrogen bonded species. 1H NMR analyses (DPB⋯2IPr, DPPD⋯2IPr), as shown in Fig. 4, highlights the subtle chemical shift changes (Δδ < 0.20 ppm) for resonances compared to BC⋯2IPr (Δδ > 0.15 ppm). Though these shifts in proton resonances are smaller, the overall directionality matches that observed for BC⋯2IPr. This is best realized in the NH signal, Hβ, which shifts by Δδ = 0.20 ppm (DPB⋯2IPr) and Δδ = 0.15 ppm (DPPD⋯2IPr), indicative of a significantly weaker hydrogen bond.
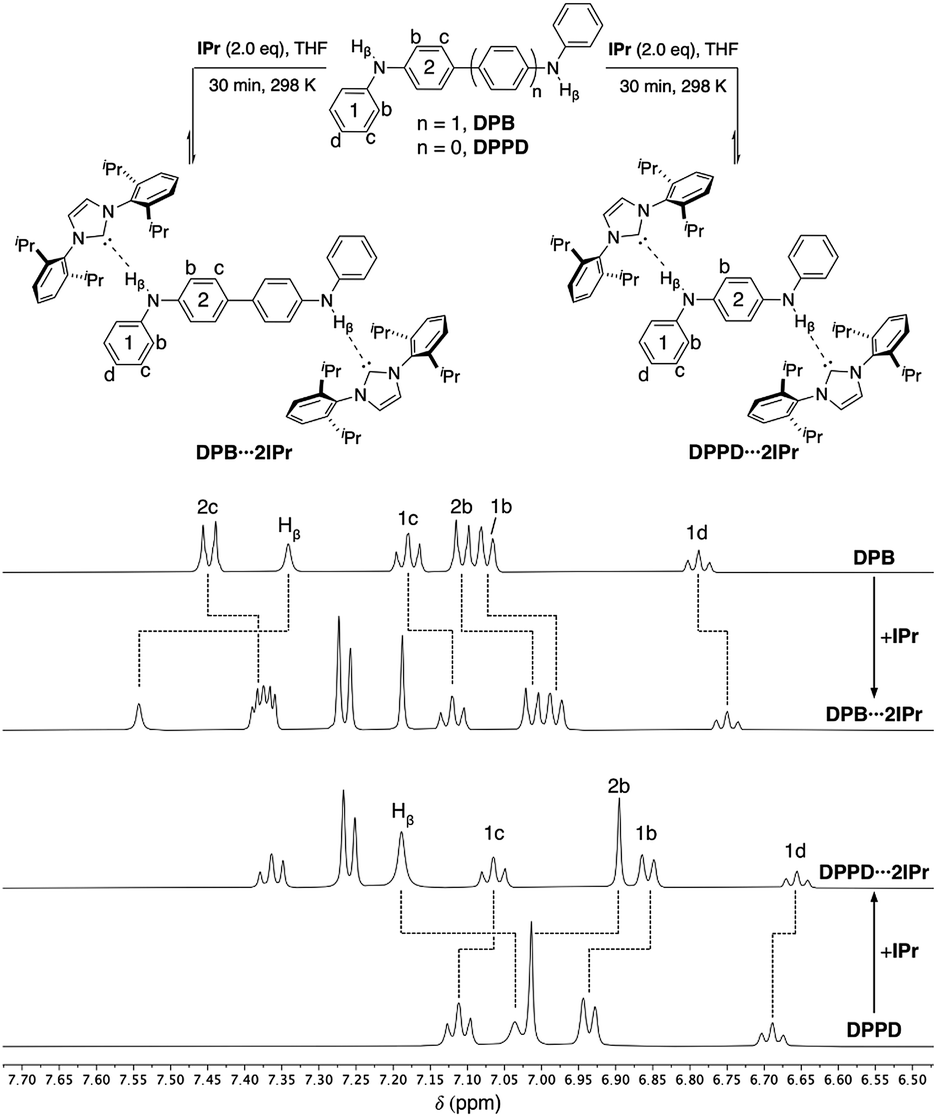 | ||
| Fig. 4 Partial 1H NMR spectra (500 MHz, THF-d8) of DPB (50 mM), DPB⋯2IPr, DPPD⋯2IPr, and DPPD (50 mM). Dashed lines highlight the chemical shift changes upon hydrogen bonding to IPr. | ||
Crystals of DPPD⋯2IPr suitable for a X-ray diffraction studies were obtained by chilling a solution of DPPD⋯2IPr in a 3:2 hexanes:THF solvent mixture at −35 °C for several days. The resulting solid-state structure of DPPD⋯2IPr is portrayed in Fig. 5. The H1⋯C24 distance of 2.30(2) Å matches that found for the DPA⋯IMes adduct, as is the interior N2–C13–N3 angle of 101.9(1)°. The dihedral between the pseudo-planes of DPPD⋯2IPr (ϕ N2–N3–C2–C4 = 17°) are in agreement with BC⋯2IPr, and slightly less pronounced than DPA⋯IMes (ϕ = 38°). The N1–H1⋯C24 bond angle is nearly linear (171°), albeit quite bent relative to DPA⋯IMes (N–H⋯:C = 179°). The hydrogen bond distances are within the sum of the van der Waal radii, and display closer contacts than the hydrogen bond adducts of the Ong group, where carbenes with pendant secondary amines form dimers in the solid-state.44,45 By contrast, all attempts to crystallize DPB⋯2IPr resulted in dissociation of the adduct into individual components. It appears that the poor solubility of DPB leads to preferential precipitation/crystallization of DPB, which is in facile equilibrium with IPr and DPB⋯2IPr (vide infra).
 | ||
| Fig. 5 ORTEP diagram (ellipsoids drawn at 50% probability) of DPPD⋯2IPr. Non-hydrogen bonding hydrogens are omitted for clarity. Select bond lengths (Å) and angles (°): H1⋯C24, 2.30(2); N1⋯C24, 3.174(2); N2–C13–N3, 101.9(1); N2–N3–C4–C2, −17.41(7). | ||
Analysis of the metric data obtained for the symmetric ditopic NH-bearing adducts in both solid- and solution-state reveals several interesting insights. In the solid-state the N–C–N angles within IPr (101.5°)69 relax upon formation of hydrogen bonded species DPPD⋯2IPr (101.9°) and BC⋯2IPr (102.2°). A greater distortion of the N–C–N angle is thus associate with greater interaction, and if taken to an extreme would result in proton transfer (where imidazolium salts typically feature N–C–N angles of ∼108°).30 The length of the hydrogen bonds (H⋯:C distance; DPPD⋯2IPr, 2.30 Å; BC⋯2IPr, 2.10 Å) also correlate well with the presumed strength of association based on measurements on the monofunctional amine counterparts (vide supra).
While 1H NMR spectra show that the changes in the chemical shifts for protons involved in hydrogen bonding can be predictive of association strengths, the shifts observed in the 13C{1H} NMR spectra for the carbenic carbon are also quite sensitive to these external interactions. As the strength of the hydrogen bond increases the carbenic carbon shifts upfield.30
Specifically, the 13C{1H} NMR spectra of BC⋯2IPr (215.2 ppm) shows a greater Δδ (free IPr δ 221.1 ppm) than either DPB⋯2IPr (δ 220.1 ppm) and DPPD⋯2IPr (δ 220.3 ppm). 13C spectroscopy might thus serve as a new tool for assessing the strength of the hydrogen bond in non-traditional systems, adding to other experimental measures used to assess the properties of carbenes as ligands (i.e. measuring π-acidity via 31P or 77Se NMR spectroscopy72 and carbene-metal ligand strength assessed via 13C spectroscopy73).
Internal competitive hydrogen bonding
To probe the competitive nature of these hydrogen bonding adducts mixed carbazole-diphenylamine species CD was synthesized via Suzuki–Miyaura cross-coupling in modest yield (55% isolated, see ESI† for details). Reactions of CD with one and two equivalents of IPr were thus examined (Fig. 6, upper). As expected, formation of the mono- and di-carbene adducts CD⋯IPr and CD⋯2IPr is observable by NMR spectroscopy at elevated concentrations. Fig. 6 compares partial 1H NMR spectra of these products contrasted against CD, highlighting the proton resonances impacted by NHC association. In alignment with the findings above, preferential interaction of the NHC occurs with the carbazole unit of CD upon addition of one equivalent of IPr (black dotted lines in Fig. 6). This assessment is confirmed by the signals for carbazole fragment of CD shifting in the same fashion as the symmetric BC⋯2IPr, with the caveat that shifts are only ∼65% the overall Δδ of their symmetric counterpart. This may reflect the fact that IPr equilibrates between both carbazole and diphenylamine NH residues to some extent. The perturbations of the proton resonances on rings 3 and 4 (orange dotted lines) are reminiscent of DPB⋯2IPr and DPPD⋯2IPr, albeit only minor in comparison to rings 1 and 2.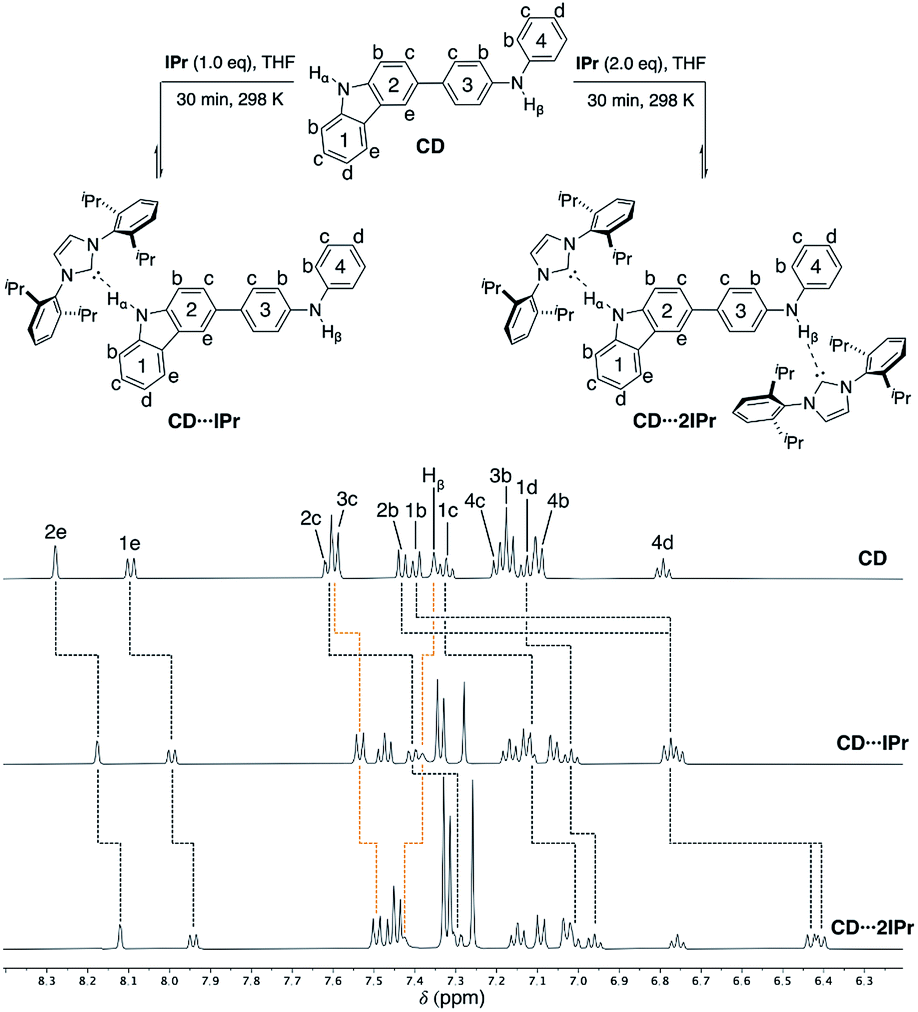 | ||
| Fig. 6 Partial 1H NMR spectra (500 MHz, THF-d8) of CD (50 mM), CD⋯IPr, and CD⋯2IPr, highlighting the chemical shift changes upon hydrogen bonding to IPr. CD Hα (10.25 ppm), CD⋯IPr Hα (11.30 ppm), and CD⋯2IPr Hα (12.40 ppm) are broadened due to exchange, see ESI† (rings 1 and 2 = black dotted lines, rings 3 and 4 = orange dotted lines). | ||
Upon addition of two equivalents of IPr to CD, the 1H NMR chemical shifts of CD⋯2IPr are a near match to the symmetric diamine counterparts, BC⋯2IPr and DPB⋯2IPr respectively. The realignment of the 1H spectra to mirror the symmetric analogues confirms formation of CD⋯2IPr. Due to the rapid exchange of IPr between both NH sites on CD the 13C{1H} NMR spectra yield a singular shift for the carbenic resonance. The observed shifts for asymmetric CD⋯2IPr (δ 217.4 ppm) and CD⋯IPr (δ 216.4 ppm) fall between their respective symmetric counterparts (BC⋯2IPr, δ 215.2 ppm, DPB⋯2IPr, δ 220.1 ppm). Unsurprisingly the 13C{1H} NMR signal in CD⋯IPr resides closer to BC⋯2IPr, verifying our 1H measurements which show that IPr preferentially binds to the carbazole side of the ditopic system. When both NH sites are satisfied in CD⋯2IPr the carbenic carbon resonance is a near perfect average of the observed resonances for the symmetric species (BC⋯2IPr and DPB⋯2IPr average resonance carbenic resonance ≈ 217.6 ppm).
Unfortunately attempts to obtain single crystals of either CD⋯IPr and CD⋯2IPr were thwarted by the preferential crystallization of CD from solutions of these materials (see ESI† for crystallographic details). While this may be due to the reduced solubility of CD relative to IPr, the probable cause is the labile nature of the N–H⋯:C bond not being conducive to solid-state analysis.
Conclusion
Through a series of symmetric and asymmetric ditopic secondary amines the strength of a X–H⋯:C hydrogen bond can be directly assessed via several solid- and solution-state factors, summarized in Table 1. Paramount in predicting the strength of the association is the pKa of the parent X–H species, which in these examples correlates well with both solid- and solution-state metrics. In the solid-state limiting the degrees of freedom in the parent molecule is vital, wherein systems with even weak hydrogen bonds can be crystallized in their adduct form (DPPD⋯2IPr). In BC, DPB, and DPPD systems solution-state analysis is simplified due their high degree of symmetry and rapid equilibrium of the N–H⋯IPr interaction. In each of these species both the Δδ of the proton participating in hydrogen bonding and the 13C resonance of the carbenic carbon are indicators of hydrogen bond strength. 1H NMR spectra show that upon hydrogen bonding with IPr the monomers are rigidified, with proton resonances far removed from area of interaction changing significantly in comparison to the unhindered molecules. When starving the asymmetric CD system of IPr, the carbene primarily interacts with the more acidic carbazole residue as evidenced by both 1H and 13C spectroscopy. When enough IPr is supplied to satisfy both NH sites the carbenic carbon resonance falls directly between the symmetric 13C residues, indicating a system that is in rapid equilibrium on the NMR timescale. The reversible nature of these systems and the ability to predict hydrogen bond strength through both solid- and solution-state metrics could lead to further developments in systems chemistry. Efforts to explore higher functionality NH-containing systems and carbenes are currently underway.
| N–C–N (°) | 13C δ (ppm) | 1H Δδ (ppm) | pKa | |
|---|---|---|---|---|
| a 1H Δδ is calculated relative to each adducts parent species at 50 mM in THF-d8.b See ref. 69.c pKa of the conjugate acid (IPr·HCl).58,59d Average values. | ||||
| IPr | 101.5b | 221.1 | — | 19.3c |
| BC⋯2IPr | 102.2 | 215.2 | 1.66 | 19.9 |
| DPB⋯2IPr | — | 220.1 | 0.20 | 25.0 |
| DPPD⋯2IPr | 101.9 | 220.3 | 0.15 | 25.0 |
| CD⋯2IPr | — | 217.4 | 0.99d | 22.5d |
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We thank the National Science Foundation (CHE-1464855 and CHE-1955845) for their support of this work.Notes and references
- G. A. Jeffrey and W. Saenger, Hydrogen Bonding in Biological Systems, Springer, Berlin, 1991 Search PubMed.
- L. McFail-Isom, C. C. Sines and L. D. Williams, DNA Structure: Cations in Charge?, Curr. Opin. Struct. Biol., 1999, 9, 298–304 CrossRef CAS.
- J. A. Subirana and M. Soler-López, Cations as Hydrogen Bond Donors: A View of Electrostatic Interactions in DNA, Annu. Rev. Biophys. Biomol. Struct., 2003, 32, 27–45 CrossRef CAS.
- D. Meyer, P. Neumann, R. Ficner and K. Tittmann, Observation of a Stable Carbene at the Active Site of a Thiamin Enzyme, Nat. Chem. Biol., 2013, 9, 488–490 CrossRef CAS.
- A. G. Doyle and E. N. Jacobsen, Small-Molecule H-Bond Donors in Asymmetric Catalysis, Chem. Rev., 2007, 107, 5713–5743 CrossRef CAS.
- P. Gallo, K. Amann-Winkel, C. A. Angell, M. A. Anisimov, F. Caupin, C. Chakravarty, E. Lascaris, T. Loerting, A. Z. Panagiotopoulos, J. Russo, J. A. Sellberg, H. E. Stanley, H. Tanaka, C. Vega, L. Xu and L. G. M. Pettersson, Water: A Tale of Two Liquids, Chem. Rev., 2016, 116, 7463–7500 CrossRef CAS.
- F. Cipcigan, V. Sokhan, G. Martyna and J. Crain, Structure and Hydrogen Bonding at the Limits of Liquid Water Stability, Sci. Rep., 2018, 8, 1–8 CrossRef CAS.
- S. H. Gellman, Foldamers: A Manifesto, Acc. Chem. Res., 1998, 31, 173–180 CrossRef CAS.
- D. Núñez-Villanueva, G. Iadevaia, A. E. Stross, M. A. Jinks, J. A. Swain and C. A. Hunter, H-Bond Self-Assembly: Folding versus Duplex Formation, J. Am. Chem. Soc., 2017, 139, 6654–6662 CrossRef.
- H. Szatyłowicz, Structural Aspects of the Intermolecular Hydrogen Bond Strength: H-Bonded Complexes of Aniline, Phenol and Pyridine Derivatives, J. Phys. Org. Chem., 2008, 21, 897–914 CrossRef.
- S. J. Pike, E. Lavagnini, L. M. Varley, J. L. Cook and C. A. Hunter, H-Bond Donor Parameters for Cations, Chem. Sci., 2019, 10, 5943–5951 RSC.
- M. D. Joesten, Hydrogen Bonding and Proton Transfer, J. Chem. Educ., 1982, 59, 362–366 CrossRef CAS.
- J. Emsley, Very Strong Hydrogen Bonding, Chem. Soc. Rev., 1968, 9, 91–124 RSC.
- R. H. Crabtree, Hypervalency Secondary Bonding and Hydrogen Bonding: Siblings under the Skin, Chem. Soc. Rev., 2017, 46, 1720–1729 RSC.
- G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond: In Structural Chemistry and Biology, 2001 Search PubMed.
- J. A. Platts, S. T. Howard and K. Woźniak, Quantum Chemical Evidence for C–H⋯C Hydrogen Bonding, Chem. Commun., 1996, 1, 63–64 RSC.
- A. S. Batsanov, M. G. Davidson, J. A. K. Howard, S. Lamb and C. Lustig, Phosphonium Ylides as Hydrogen Bond Acceptors: Inter Molecular C–H⋯C Interactions in the Crystal Structure of Triphenylphosphonium Benzylide, Chem. Commun., 1996, 15, 1791–1792 RSC.
- A. J. Arduengo, R. L. Harlow and M. Kline, A Stable Crystalline Carbene, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, An Overview of N-Heterocyclic Carbenes, Nature, 2014, 510, 485–496 CrossRef CAS.
- J. Chen and Y. Huang, Asymmetric Catalysis with N-Heterocyclic Carbenes as Non-Covalent Chiral Templates, Nat. Commun., 2014, 5, 1–8 CAS.
- V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, NHCs in Main Group Chemistry, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS.
- E. Peris, Smart N-Heterocyclic Carbene Ligands in Catalysis, Chem. Rev., 2018, 118, 9988–10031 CrossRef CAS.
- M. Soleilhavoup and G. Bertrand, Cyclic (Alkyl)(Amino)Carbenes (CAACs): Stable Carbenes on the Rise, Acc. Chem. Res., 2015, 48, 256–266 CrossRef CAS.
- M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Cyclic (Alkyl)(Amino)Carbenes (CAACs): Recent Developments, Angew. Chem., Int. Ed., 2017, 56, 10046–10068 CrossRef CAS.
- A. J. Arduengo, Looking for Stable Carbenes: The Difficulty in Starting Anew, Acc. Chem. Res., 1999, 32, 913–921 CrossRef CAS.
- R. Visbal and M. C. Gimeno, N-Heterocyclic Carbene Metal Complexes: Photoluminescence and Applications, Chem. Soc. Rev., 2014, 43, 3551–3574 RSC.
- A. Doddi, M. Peters and M. Tamm, N-Heterocyclic Carbene Adducts of Main Group Elements and Their Use as Ligands in Transition Metal Chemistry, Chem. Rev., 2019, 119, 6994–7112 CrossRef CAS.
- I. Alkorta, I. Rozas and J. Elguero, Non-Conventional Hydrogen Bonds, Chem. Soc. Rev., 1998, 27, 163–170 RSC.
- J. K. W. Chui, T. Ramnial and J. A. C. Clyburne, N-Heterocyclic Carbenes: Exotic Molecules as Precursors to Unusual Hydrogen Bonds, Comments Inorg. Chem., 2003, 24, 165–187 CrossRef CAS.
- A. J. I. Arduengo, S. F. Gamper, M. Tamm, J. C. Calabrese, F. Davidson and H. A. Craig, A Bis(Carbene)—Proton Complex: Structure of a C—H—C Hydrogen Bond, J. Am. Chem. Soc., 1995, 117, 572–573 CrossRef CAS.
- J. A. Cowan, J. A. C. Clyburne, M. G. Davidson, R. L. W. Harris, J. A. K. Howard, P. Küpper, M. A. Leech and S. P. Richards, On the Interaction between N-Heterocyclic Carbenes and Organic Acids: Structural Authentication of the First N—H⋯C Hydrogen Bond and Remarkably Short C—H···O Interactions, Angew. Chem., Int. Ed., 2002, 41, 1432–1434 CrossRef CAS.
- M. Movassaghi and M. A. Schmidt, N-Heterocyclic Carbene-Catalyzed Amidation of Unactivated Esters with Amino Alcohols, Org. Lett., 2005, 7, 2453–2456 CrossRef CAS.
- J. M. Kieser, Z. J. Kinney, J. R. Gaffen, S. Evariste, A. M. Harrison, A. L. Rheingold and J. D. Protasiewicz, Three Ways Isolable Carbenes Can Modulate Emission of NH-Containing Fluorophores, J. Am. Chem. Soc., 2019, 141, 12055–12063 CrossRef CAS.
- D. Tolentino, S. Neale, C. Isaac, S. A. Macgregor, M. K. Whittlesey, R. Jazzar and G. Bertrand, Reductive Elimination at Carbon under Steric Control, J. Am. Chem. Soc., 2019, 141, 9823–9826 CrossRef CAS.
- I. Alkorta and J. Elguero, Carbenes and Silylenes as Hydrogen Bond Acceptors, J. Phys. Chem., 1996, 100, 19367–19370 CrossRef CAS.
- A. M. Magill, K. J. Cavell and B. F. Yates, Basicity of Nucleophilic Carbenes in Aqueous and Nonaqueous Solvents—Theoretical Predictions, J. Am. Chem. Soc., 2004, 126, 8717–8724 CrossRef CAS.
- J.-L. Mieusset and U. H. Brinker, The Carbene Reactivity Surface: A Classification, J. Org. Chem., 2008, 73, 1553–1558 CrossRef CAS.
- O. Hollóczki, Unveiling the Peculiar Hydrogen Bonding Behavior of Solvated N-Heterocyclic Carbenes, Phys. Chem. Chem. Phys., 2016, 18, 126–140 RSC.
- J. E. Del Bene, I. Alkorta and J. Elguero, Hydrogen-Bonded Complexes with Carbenes as Electron-Pair Donors, Chem. Phys. Lett., 2017, 675, 46–50 CrossRef CAS.
- L. A. Leites, G. I. Magdanurov, S. S. Bukalov and R. West, Intermolecular =C–H···:C Hydrogen Bond in a Crystalline Unsaturated Arduengo-Type Carbene, Mendeleev Commun., 2008, 18, 14–15 CrossRef CAS.
- M. Kapitein, M. Balmer and C. von Hanisch, A Sterically Encumbered 13/15 Cycle and Its Cleavage with N-Heterocyclic Carbenes and Other Lewis Bases, Z. Anorg. Allg. Chem., 2016, 642, 1275–1281 CrossRef CAS.
- P. A. Petrov, T. S. Sukhikh and M. N. Sokolov, NHC Adducts of Tantalum Amidohalides: The First Example of NHC Abnormally Coordinated to an Early Transition Metal, Dalton Trans., 2017, 46, 4902–4906 RSC.
- H. Jong, B. O. Patrick and M. D. Fryzuk, Amine-Tethered N-Heterocyclic Carbene Complexes of Rhodium(I), Can. J. Chem., 2008, 86, 803–810 CrossRef CAS.
- W. C. Shih, C. H. Wang, Y. T. Chang, G. P. A. Yap and T. G. Ong, Synthesis and Structure of an Amino-Linked N-Heterocyclic Carbene and the Reactivity of Its Aluminum Adduct, Organometallics, 2009, 28, 1060–1067 CrossRef CAS.
- C.-Y. Y. Li, Y.-Y. Y. Kuo, J.-H. H. Tsai, G. P. A. A. Yap and T.-G. G. Ong, Amine-Linked N-Heterocyclic Carbenes: The Importance of an Pendant Free-Amine Auxiliary in Assisting the Catalytic Reaction, Chem.–Asian J., 2011, 6, 1520–1524 CrossRef CAS.
- H. Braunschweig, W. C. Ewing, K. Geetharani and M. Schäfer, The Reactivities of Iminoboranes with Carbenes: BN Isosteres of Carbene-Alkyne Adducts, Angew. Chem., Int. Ed., 2015, 54, 1662–1665 CrossRef CAS.
- M. A. Schmidt, P. Müller and M. Movassaghi, On the Interactions of N,N′-Bismesitylimidazolin-2-Yl and Alcohols, Tetrahedron Lett., 2008, 49, 4316–4318 CrossRef CAS.
- N. A. Giffin, M. Makramalla, A. D. Hendsbee, K. N. Robertson, C. Sherren, C. C. Pye, J. D. Masuda and J. A. C. Clyburne, Anhydrous TEMPO-H: Reactions of a Good Hydrogen Atom Donor with Low-Valent Carbon Centres, Org. Biomol. Chem., 2011, 9, 3672–3680 RSC.
- A. H. Raut, G. Karir and K. S. Viswanathan, Matrix Isolation Infrared and Ab Initio Study of the Interaction of N-Heterocyclic Carbene with Water and Methanol: A Case Study of a Strong Hydrogen Bond, J. Phys. Chem. A, 2016, 120, 9390–9400 CrossRef CAS.
- R. Srivastava, R. Moneuse, J. Petit, P. A. Pavard, V. Dardun, M. Rivat, P. Schiltz, M. Solari, E. Jeanneau, L. Veyre, C. Thieuleux, E. A. Quadrelli and C. Camp, Early/Late Heterobimetallic Tantalum/Rhodium Species Assembled Through a Novel Bifunctional NHC-OH Ligand, Chem.–Eur. J., 2018, 24, 4361–4370 CrossRef CAS.
- B. Maji, M. Breugst and H. Mayr, N-Heterocyclic Carbenes: Organocatalysts with Moderate Nucleophilicity but Extraordinarily High Lewis Basicity, Angew. Chem., Int. Ed., 2011, 50, 6915–6919 CrossRef CAS.
- A. Levens, F. An, M. Breugst, H. Mayr and D. W. Lupton, Influence of the N-Substituents on the Nucleophilicity and Lewis Basicity of N-Heterocyclic Carbenes, Org. Lett., 2016, 18, 3566–3569 CrossRef CAS.
- T. Steiner, The Hydrogen Bond in the Solid State, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- F. G. Bordwell, J. C. Branca, D. L. Hughes and W. N. Olmstead, Equilibriums Involving Organic Anions in Dimethyl Sulfoxide and N-Methylpyrrolidin-2-One: Acidities, Ion Pairing, and Hydrogen Bonding, J. Org. Chem., 1980, 45, 3305–3313 CrossRef CAS.
- F. G. Bordwell, G. E. Drucker and H. E. Fried, Acidities of Carbon and Nitrogen Acids: The Aromaticity of the Cyclopentadienyl Anion, J. Org. Chem., 1981, 46, 632–635 CrossRef CAS.
- P. Ballinger and F. A. Long, Acid Ionization Constants of Alcohols. II. Acidities of Some Substituted Methanols and Related Compounds, J. Am. Chem. Soc., 1960, 82, 795–798 CrossRef CAS.
- W. N. Olmstead, Z. Margolin and F. G. Bordwell, Acidities of Water and Simple Alcohols in Dimethyl Sulfoxide Solution, J. Org. Chem., 1980, 45, 3295–3299 CrossRef CAS.
- M. H. Dunn, N. Konstandaras, M. L. Cole and J. B. Harper, Targeted and Systematic Approach to the Study of pKa Values of Imidazolium Salts in Dimethyl Sulfoxide, J. Org. Chem., 2017, 82, 7324–7331 CrossRef CAS.
- Z. Wang, X. S. Xue, Y. Fu and P. Ji, Comprehensive Basicity Scales for N-Heterocyclic Carbenes in DMSO: Implications on the Stabilities of N-Heterocyclic Carbene and CO2 Adducts, Chem.–Asian J., 2020, 15, 169–181 CrossRef CAS.
- S. Santra, A. Porey, B. Jana and J. Guin, N-Heterocyclic Carbenes as Chiral Brønsted Base Catalysts: A Highly Diastereo- and Enantioselective 1,6-Addition Reaction, Chem. Sci., 2018, 9, 6446–6450 RSC.
- S. Gehrke and O. Hollóczki, Hydrogen Bonding of N-Heterocyclic Carbenes in Solution: Mechanisms of Solvent Reorganization, Chem.–Eur. J., 2018, 24, 11594–11604 CrossRef CAS.
- O. Hollóczki, The Mechanism of N-Heterocyclic Carbene Organocatalysis through a Magnifying Glass, Chem.–Eur. J., 2020, 26, 4885–4894 CrossRef.
- S. Santra, U. Maji and J. Guin, Enantioselective α-Amination of Acyclic 1,3-Dicarbonyls Catalyzed by N-Heterocyclic Carbene, Org. Lett., 2020, 22, 468–473 CrossRef CAS.
- R. Guo, X. Huang, M. Zhao, Y. Lei, Z. Ke and L. Kong, Bifurcated Hydrogen-Bond-Stabilized Boron Analogues of Carboxylic Acids, Inorg. Chem., 2019, 58, 13370–13375 CrossRef CAS.
- C. Pi, X. Yu and W. Zheng, Imidazolium 1,3-Benzazaphospholide Ion Pairs with Strong C–H⋯N Hydrogen Bonds — Synthesis, Structures, and Reactivity, Eur. J. Inorg. Chem., 2015, 2015, 1804–1810 CrossRef CAS.
- J. A. Platts, Theoretical Prediction of Hydrogen Bond Donor Capacity, Phys. Chem. Chem. Phys., 2000, 2, 973–980 RSC.
- M. H. Abraham, J. Elguero and I. Alkorta, The Hydrogen-Bond Basicity of Carbenes, Croat. Chem. Acta, 2018, 91, 121–124 CrossRef.
- S. Mallick, S. Maddala, K. Kollimalayan and P. Venkatakrishnan, Oxidative Coupling of Carbazoles: A Substituent-Governed Regioselectivity Profile, J. Org. Chem., 2019, 84, 73–93 CrossRef CAS.
- A. J. Arduengo, H. A. Craig, J. R. Goerlich, W. J. Marshall and M. Unverzagt, Imidazolylidenes, Imidazolinylidenes and Imidazolidines, Tetrahedron, 1999, 55, 14523–14534 CrossRef CAS.
- J. L. Cook, C. A. Hunter, C. M. R. Low, A. Perez-Velasco and J. G. Vinter, Solvent Effects on Hydrogen Bonding, Angew. Chem., Int. Ed., 2007, 46, 3706–3709 CrossRef CAS.
- M. Chu, A. N. Scioneaux and C. S. Hartley, Solution-Phase Dimerization of an Oblong Shape-Persistent Macrocycle, J. Org. Chem., 2014, 79, 9009–9017 CrossRef CAS.
- D. Munz, Pushing Electrons — Which Carbene Ligand for Which Application?, Organometallics, 2018, 37, 275–289 CrossRef CAS.
- D. Tapu, D. A. Dixon and C. Roe, 13C NMR Spectroscopy of “Arduengo-Type” Carbenes and Their Derivatives, Chem. Rev., 2009, 109, 3385–3407 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, supplemental figures and analysis, NMR spectra, and crystallographic results. CCDC 2011390, 2011391 and 2011392. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra08490e |
| This journal is © The Royal Society of Chemistry 2020 |