
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modified Cu–Zn–Al mixed oxide dual function materials enable reactive carbon capture to methanol†
Chae
Jeong-Potter
 ,
Martha A.
Arellano-Treviño
,
W. Wilson
McNeary
,
Alexander J.
Hill
,
Daniel A.
Ruddy
* and
Anh T.
To
*
,
Martha A.
Arellano-Treviño
,
W. Wilson
McNeary
,
Alexander J.
Hill
,
Daniel A.
Ruddy
* and
Anh T.
To
*
National Renewable Energy Laboratory, Catalytic Carbon Transformation and Scale-Up Center, Golden, Colorado 80401, USA. E-mail: anh.to@nrel.gov; dan.ruddy@nrel.gov
First published on 8th November 2023
Abstract
Reactive carbon capture (RCC), an integrated CO2 capture and conversion process that does not require generating a purified CO2 stream, is an attractive carbon management strategy that can reduce costs and energy requirements associated with traditionally separate capture and conversion processes. Dual function materials (DFMs) comprised of co-supported sorbent sites and catalytic sites have emerged as a promising material design to enable RCC. DFMs have been extensively studied for methane production, but the noncompetitive economics of methane necessitates the development of DFMs to target more valuable, useful, and versatile products, like methanol. Herein, we report the development of modified Cu–Zn–Al mixed oxide (Alk/CZA, Alk = K, Ca) DFMs for combined capture and conversion of CO2 to methanol. CO2 chemisorption, in situ DRIFTS characterization, and co-fed hydrogenation performance revealed that K and Ca have different effects on the CO2 capture and catalytic behavior of the parent CZA. K-modification resulted in the greatest promotional effect on capture capacity but the most detrimental effect on co-fed hydrogenation catalytic activity. Interestingly, when used in a cyclic temperature-and-pressure-swing RCC operation, K/CZA exhibited a greater conversion of adsorbed CO2 (94.4%) with high methanol selectivity (46%), leading to greater methanol production (59.0 μmol gDFM−1) than the parent CZA or Ca/CZA (13.2 and 18.9 μmol gDFM−1, respectively). This study presents the foundational methodology for the design and evaluation of novel DFMs to target renewable methanol synthesis, highlighted by a critical learning that co-fed CO2 hydrogenation performance is not an effective indicator of RCC performance.
Broader contextWith the rise of atmospheric CO2 concentrations from anthropogenic emissions, the development of carbon capture and utilization technologies is increasingly necessary to access renewable fuels and chemicals. Reactive carbon capture (RCC) processes, where CO2 capture and conversion occur in a single reactor, can be energetically and economically attractive by avoiding the need to purify, compress, and transport the captured CO2. To realize these potential advantages, there is a need to discover and develop dual function materials (DFMs) that enable RCC to useful and versatile C1 products, with a focus on methanol here. The accompanying process development is also needed to maximize carbon efficiency to target product and advance the technology towards commercialization. Here we present the discovery of DFMs based on a commercial methanol synthesis catalyst and provide foundational learnings in the design and testing of these DFMs. |
1. Introduction
As atmospheric CO2 concentrations continue to reach historically high levels (420 ppm as of June 2022) and as predictions forecast the prolonged use of carbon-derived and carbon-emitting fuels and chemicals, the implementation of carbon management technologies is necessary to mitigate the negative effects of associated climate change.1 While CO2 removal strategies such as point-source carbon capture and storage (CCS) and direct air carbon capture and storage (DACCS) are required to achieve net-zero and net-negative emission goals, wide-spread deployment is still limited due to high capture costs, lack of efficient transportation infrastructure, and low intrinsic value of CO2.2Carbon capture and utilization (CCU) overcomes these disadvantages by providing a revenue stream to offset capture costs by converting CO2 to more valuable chemicals and fuels. However, traditional CCU pathways are limited by the energy penalty for desorption of the captured CO2 during sorbent regeneration (60–100 kJ mol−1) and the associated purification, transport, and pressurization of CO2 from dilute sources.2–4 To this end, a reactive carbon capture (RCC) process, where CO2 capture and conversion are integrated in a stepwise approach in a single reactor (or a series of reactors), has been more recently investigated.5–11 A recent report highlights how this approach can eliminate the need for separate CO2 desorption and downstream processes using methanol (MeOH) as a target product, thereby providing a route to reduced cost and reduced energy input.10 A promising RCC technology platform is based on dual function materials (DFMs), which are solid-phase materials composed of sorbents and catalysts co-dispersed on the same high surface area carrier. As shown in Fig. 1, the sorbent component allows for selective capture of CO2 from a gas stream (R2) and the catalyst component subsequently performs the conversion of the adsorbed CO2 upon introduction of a reactive gas (typically H2, R1).5,7,12 Catalytic conversion of adsorbed CO2 with H2 simultaneously regenerates the solid adsorbent and releases products without the need for the energy-intensive separation steps.
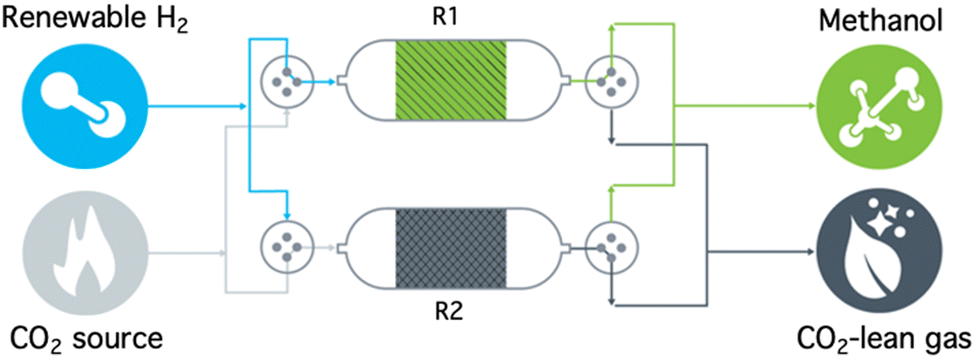 | ||
| Fig. 1 Conceptualized process flow diagram of RCC-to-methanol. | ||
The most well-developed DFM is comprised of Ru and/or Ni with an alkaline sorbent for methanation of CO2.13,14 While renewable methane would be an excellent transition fuel, fossil methane is inexpensive (averaging $6.45 per MMBTU in 2022 in the US15) and the economics of renewable methane utilization are noncompetitive (generally >$10 per MMBTU16). This requires the design and development of DFMs that enable RCC to more valuable and more useful C1 products. MeOH is a particularly attractive target molecule, being a versatile building block in both the chemicals industry (e.g., formaldehyde, acetic acid) and fuels industry via direct use in the shipping industry or upgrading to energy dense synthetic fuels in difficult-to-decarbonize sectors (e.g., heavy-duty vehicles, aviation).17,18 The value of MeOH is reflected in its comparatively higher average price of $32.20 per MMBTU in 2022 in the US.19 Despite the increasing demand for MeOH for fuel and chemical production in recent decades, renewable MeOH represents only 0.2% of current annual production.20 Reducing the cost and energy requirements of CO2 to MeOH are needed to decarbonize and defossilize MeOH for the fuel and chemical industries.21 To this end, RCC to MeOH has been reported to have the potential to reduce capital costs and energy requirements by about 50% when compared to separate CO2 capture and conversion.10 The key results of that case study are highly dependent on the development of materials and processes that have CO2 capture capacities and MeOH yields that are comparable and competitive to a separate capture and conversion system.
Integrated capture and conversion approaches to target MeOH production have been demonstrated in a variety of ways. The most reported approach involves capturing CO2 using a liquid-phase solvent (e.g., amine8,22,23 or hydroxide solutions24–26). The CO2 loaded capture media is then passed over a solid catalyst bed23 or mixed with a homogeneous catalyst8,22,25,26 to convert the absorbed CO2. While favorable results were reported using this approach (>90% MeOH yield25), it requires the use of two reactors (one for the capture solvent and one for the catalyst) and subsequent separation of MeOH product from the solvent, which would not see the same cost reduction potentials as a solid-phase system that allows the use of a single material in a single reactor. However, design of solid-phase DFMs and their accompanying RCC systems for MeOH synthesis, as shown in Fig. 1, has been attempted just a few times thus far.27–29 The reported attempts have exhibited low overall conversion of initially captured CO2 (i.e., low working capacity) and low MeOH yield. For example, in a recently reported solid-phase Pd-amine DFM, less than 5% of the captured CO2 was converted to MeOH for a gravimetric activity (i.e., productivity) of ca. 20 μmol g−1, leaving much room for improvement in both DFM and process design.28 In an alternative approach using a stacked bed of sorbent and catalyst, a similarly low productivity of 12 μmol g−1 was reported.29
Herein, we report an investigation of a novel DFM and RCC to MeOH process. We propose a DFM design that modifies the industrial MeOH synthesis catalyst (mixed oxide of Cu–Zn–Al, CZA) with alkali and alkaline metal sorbents (termed Alk/CZA, Alk = K, Ca). K and Ca were chosen as initial representatives of the group 1 and 2 metal oxide sorbents that have demonstrated good performance in methanation DFMs. The DFMs and the benchmark, parent CZA were first characterized for their CO2 capture behavior and catalytic activity in co-fed steady-state CO2 hydrogenation to MeOH. We then evaluated the DFM for cyclic capture and conversion using a robust methodology for a temperature-and-pressure swing RCC process. This approach resulted in an exceptional methanol productivity exhibited by K/CZA of 59 μmol g−1, 4.5× greater than unmodified CZA.
2. Results and discussion
2.1. Preparation and characterization of Alk/CZA DFMs
Alk/CZA DFMs were prepared through incipient wetness impregnation of commercial CZA using aqueous solutions of K2CO3 and Ca(NO3)2 salts targeting 5 wt% loading of metal oxide, which is typically investigated in other DFMs studies.30,31 Elemental analysis provided weight loadings of 3.71% for K and 2.97% for Ca, corresponding to 4.5% and 4.2% for K2O and CaO, respectively. As an initial structural characterization, X-ray diffraction (XRD) analysis was performed. The addition of Alk species did not change the CZA reflections for CuO and ZnO, suggesting that there are no major disruptions to the parent structure (Fig. S1, ESI†). Detailed investigations into the exact location of these species, especially under RCC conditions, is the subject of future investigation, while the focus here is to investigate the ability of these Alk metals to improve CO2 capture and hydrogenation to MeOH when dispersed on CZA.Strongly chemisorbed CO2 was measured over a range of temperatures to probe the effects of Alk-modification of CZA on CO2 capture capacity (Fig. 2(a) and Table S1, ESI†). The materials were reduced at 250 °C in H2 prior to analysis, as informed by H2-TPR (Fig. S2, ESI†). As the analysis temperature increased from 50 to 250 °C, the total CO2 uptake decreased markedly for CZA (132 to 42 μmol g−1), moderately for Ca/CZA (197 to 105 μmol g−1), and only minorly for K/CZA (265 to 206 μmol g−1). At low temperatures (50 °C), modifying CZA with Ca increased strong adsorption capacity by 50% (Fig. 2(b)). At moderate temperatures (100, 150 °C), the strong adsorption capacity increase on Ca/CZA was less pronounced at 35% and 27%, respectively. At high temperatures relevant to CO2 conversion (200, 250 °C), the strong CO2 adsorption capacities of Ca/CZA (144, 105 μmol g−1) were much greater than those of unmodified CZA (74, 42 μmol g−1), equating to 95% and 148% increase in capacity, respectively. K-modification greatly enhanced strong CO2 adsorption capacity at all temperatures when compared to both Ca/CZA and unmodified CZA. Even at lower temperatures (50–150 °C), the strong adsorption capacity of K/CZA (265 μmol g−1) was about double that of the unmodified CZA (132 μmol g−1). The adsorption enhancement of K-modification was greatest at 250 °C, accounting for a 390% increase in capacity (206 vs. 42 μmol g−1). K/CZA also exhibited the smallest change in capacity as a function of analysis temperature, and retained the highest capacity at relevant conversion temperatures above 200 °C. These data demonstrate that the addition of sorbent sites can greatly increase CO2 uptake capacity on CZA and a significant amount of CO2 remains strongly adsorbed on the DFMs at high temperatures (above 100 °C), which is an important factor for reactive desorption, as discussed later.
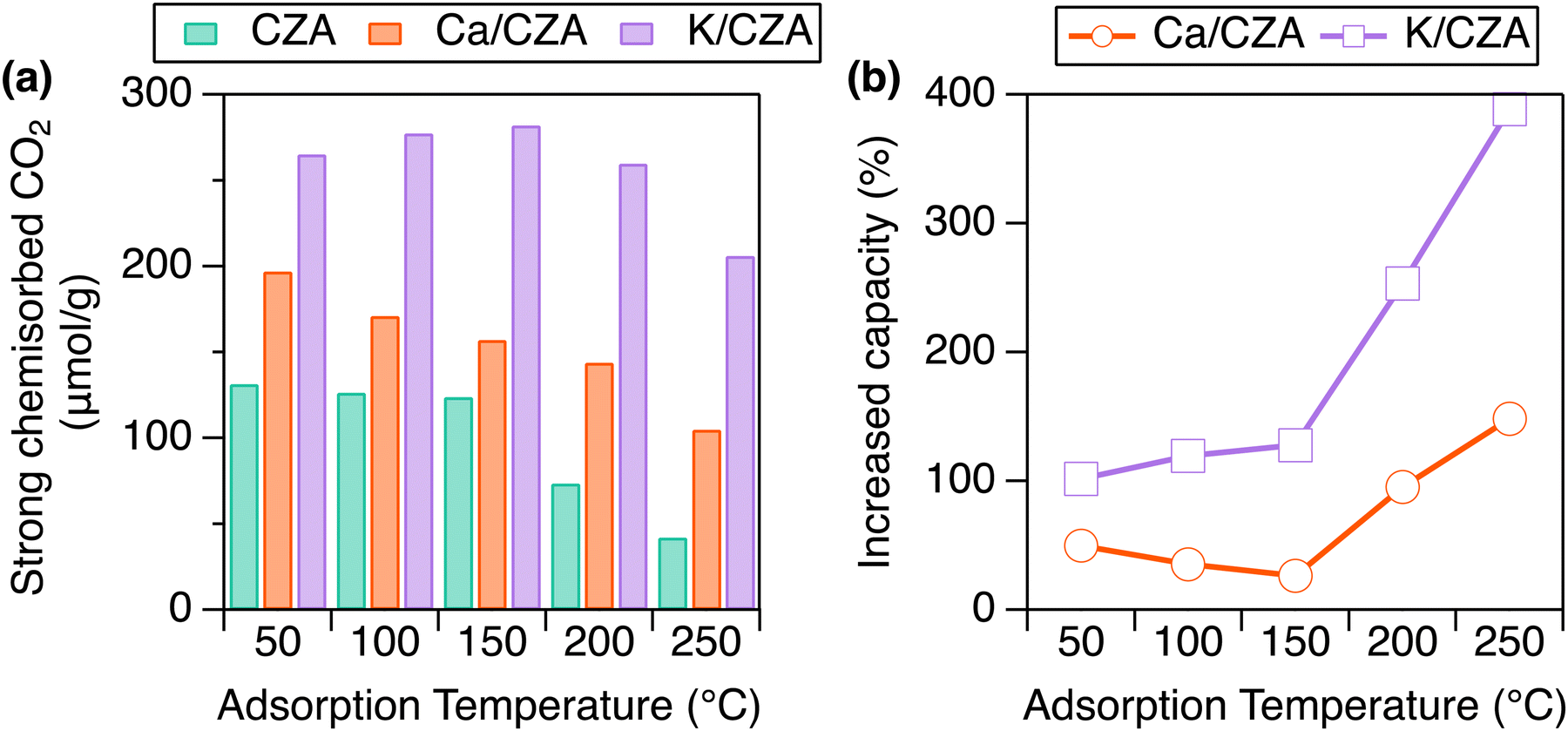 | ||
| Fig. 2 (a) Strong CO2 chemisorption values for CZA (teal), Ca/CZA (orange), and K/CZA (purple) at specified temperatures. (b) Increase in strong CO2 adsorption capacity of the DFMs as compared to parent CZA. Samples were reduced in H2 at 250 °C for 8 h, followed by an 8 h evacuation before analysis. | ||
In situ DRIFTS was utilized to investigate the binding geometry of strongly adsorbed CO2 on these DFMs. The spectra after a H2 reduction, 30 min of exposure to 5% CO2/He at 100 °C, and a 1 h purge at 100 °C to remove weakly bound CO2 are displayed in Fig. 3(a). Observed surface carbonate species (CO3*) and their respective peak positions are summarized in Fig. 3(b), (c), and Table S2 (ESI†). It has been reported that CO2 binds to CZA above ambient temperature as bridged bidentate carbonates.26 Peaks indicating this adsorption geometry were observed on the unmodified CZA at 1604 and 1379 cm−1. Polydentate carbonates have also been reported and were observed here (1537 cm−1). These two surface species have been related to different binding strengths, with the polydentate being more strongly bound.32 The spectrum for Ca/CZA was not remarkably different from unmodified CZA. The surface carbonate features of alumina-supported Ca species have been reported at 1630 cm−1 and 1340 cm−1 associated with the monodentate geometry, which are in close proximity to the peaks associated with the bidentate geometry on CZA.33 Though it appears there was a slight broadening of peaks in this region, it is difficult to distinguish CO2 binding on Ca sites from CO2 binding on CZA sites. Given the similar spectra for CZA and Ca/CZA observed here, we attribute the dominant surface species on Ca/CZA to the same bridged bidentate and polydentate carbonates as observed on unmodified CZA, with possible contributions from monodentate geometry on Ca sites at coincident peak positions.
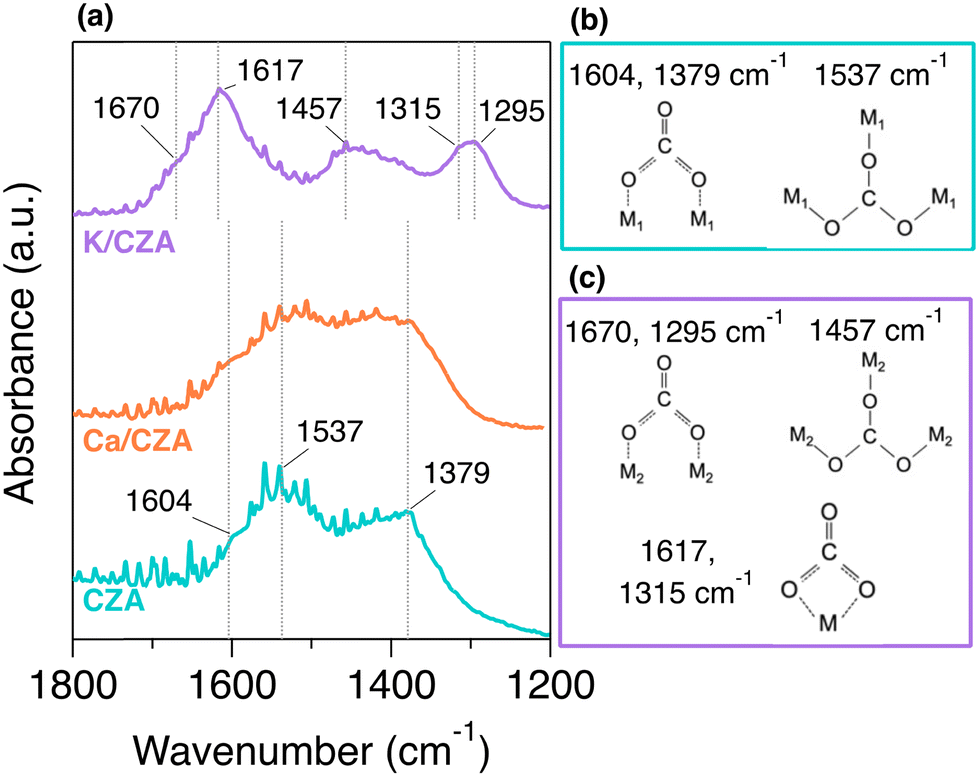 | ||
| Fig. 3 (a) In situ DRIFTS spectra of CZA (bottom, teal), Ca/CZA (middle, orange), and K/CZA (top, purple) after reduction with 50% H2/He at 250 °C for 8 h, 30 min of exposure to 5% CO2/He at 100 °C, and a 1 h purge with inert gas. Geometry of surface species observed on (b) CZA and Ca/CZA, and (c) those observed on K/CZA. Peak position assignments labelled here were aided by the spectra of the samples prior to the inert purge, which can be found in Fig. S3 (ESI†). | ||
In contrast, the spectrum for K/CZA was dramatically different than those of CZA and Ca/CZA. Strong absorbances were observed in the regions of 1700–1600 cm−1, 1500–1400 cm−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1, and 1350–1250 cm−1, which are all distinct from the peaks observed on the CZA and Ca/CZA samples. It has been reported that CO2 chemisorbs onto K-containing materials in diverse CO3* arrangements, such as different types of bicarbonates, bidentate, and polydentate carbonates,31,34,35 and the peaks observed here can be attributed to three CO3* surface species (Fig. 3(c)). A broad shoulder at 1670 cm−1 and peak centered at 1295 cm−1 correlate well with bridged bidentate carbonates on K species.34 Although similar in structure to those observed on CZA and Ca/CZA, the different peak position indicates a different binding strength at K sites. The splitting Δν3 associated with this geometry on K-sites is 375 cm−1, which is greater than splitting Δν3 = 225 cm−1 associated with the CZA-bidentate, indicating a less stable (and possibly more reactive) carbonate. The peak centered at 1617 cm−1 has a companion peak at 1315 cm−1, and these can be assigned to a chelating bidentate carbonate, which is a geometry distinct from CO3* on Ca or CZA sites.31,35 Lastly, the broad peak centered around 1457 cm−1 correlates best with a polydentate carbonate associated with a K site, again a similar geometry with a different strength than on CZA and Ca/CZA.34 While carbonate geometries arising from the parent CZA are likely also present, adsorption at K sites dominates the spectrum for this material.
1, and 1350–1250 cm−1, which are all distinct from the peaks observed on the CZA and Ca/CZA samples. It has been reported that CO2 chemisorbs onto K-containing materials in diverse CO3* arrangements, such as different types of bicarbonates, bidentate, and polydentate carbonates,31,34,35 and the peaks observed here can be attributed to three CO3* surface species (Fig. 3(c)). A broad shoulder at 1670 cm−1 and peak centered at 1295 cm−1 correlate well with bridged bidentate carbonates on K species.34 Although similar in structure to those observed on CZA and Ca/CZA, the different peak position indicates a different binding strength at K sites. The splitting Δν3 associated with this geometry on K-sites is 375 cm−1, which is greater than splitting Δν3 = 225 cm−1 associated with the CZA-bidentate, indicating a less stable (and possibly more reactive) carbonate. The peak centered at 1617 cm−1 has a companion peak at 1315 cm−1, and these can be assigned to a chelating bidentate carbonate, which is a geometry distinct from CO3* on Ca or CZA sites.31,35 Lastly, the broad peak centered around 1457 cm−1 correlates best with a polydentate carbonate associated with a K site, again a similar geometry with a different strength than on CZA and Ca/CZA.34 While carbonate geometries arising from the parent CZA are likely also present, adsorption at K sites dominates the spectrum for this material.
Based on chemisorption and DRIFTS analysis, modification with K has a greater effect on the overall adsorption performance – increasing capacity at relevant reaction temperatures – and a greater effect on adsorption geometry – different binding strengths and a unique geometry of a chelating bidentate carbonate. The effects on capacity from Ca are more moderate and more apparent at higher temperatures. This characterization of the DFMs shows that the CO2 adsorption capacity, strength, and geometry are a function of Alk identity. With this knowledge, it is important to then consider how these parameters subsequently affect the catalytic performance of these DFMs under co-fed steady state CO2 hydrogenation and adsorption–reactive desorption steps during an RCC cycle.
2.2. Performance of DFMs in co-fed CO2 hydrogenation experiments
The DFMs and parent CZA were tested for CO2 hydrogenation activity as an initial assessment of Alk-modification on catalytic performance, especially MeOH synthesis activity. The materials were tested under continuous flow of H2 and CO2 (molar ratio of H2:CO2 = 3:1) over a range of pressures (1–3 MPa) and temperatures (150–250 °C). The resulting conversion and product selectivities are presented in Fig. 4 and Table S3 (ESI†).
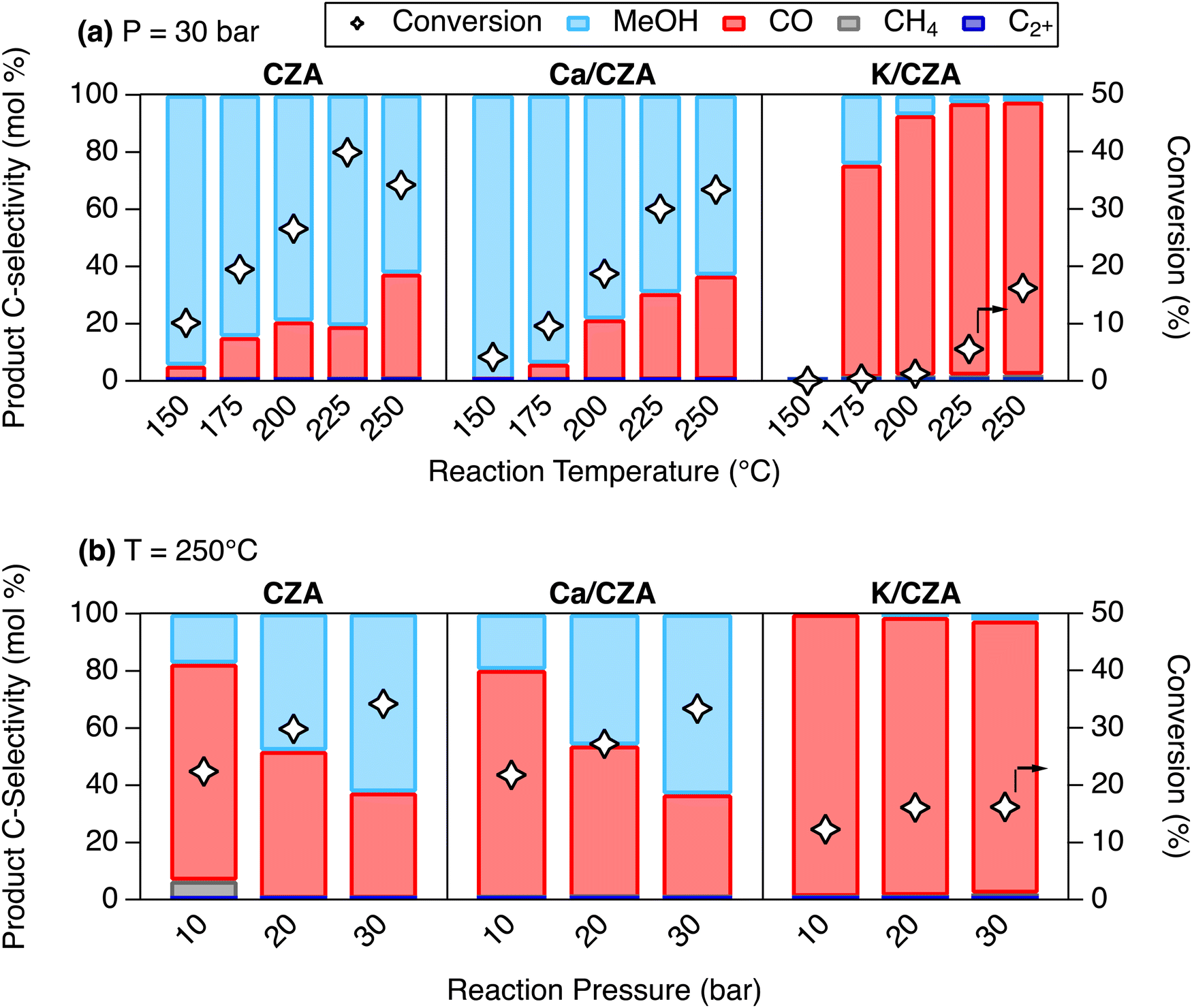 | ||
| Fig. 4 CO2 conversion (markers) and product selectivity (bars) of CZA, Ca/CZA, and K/CZA in the co-fed CO2 hydrogenation reaction at (a) T = 150–250 °C at 30 bar and (b) P = 10–30 bar at 250 °C. Reaction conditions: WHSV of 0.5 gCO2 gcat−1 h−1 and H2:CO2 molar ratio of 3:1. Materials were pre-reduced in 95% H2/Ar for 16 h at 250 °C. | ||
When the reaction temperature was increased at a constant pressure of 30 bar (Fig. 4(a)), CZA exhibited increasing CO2 conversion (10.1–34.2%) and decreasing MeOH selectivity (95.6–62.6%). Ca/CZA demonstrated slightly lower conversion throughout most of the temperature range but reached a similar conversion at 250 °C (33.4% vs. 34.2% for CZA). On the other hand, K/CZA did not show any significant conversion (< 1%) until 200 °C and only achieved a maximum conversion of 16.2% at 250 °C. At lower temperatures (150, 175 °C), Ca/CZA gave slightly higher selectivity to MeOH (99.9%, 93.9%) when compared to unmodified CZA (94.6%, 84.6%). At higher reaction temperatures (≥ 200 °C), MeOH selectivity was comparable between CZA and Ca/CZA, ranging from 79.1–62.6% for CZA and 78.4–63.1% for Ca/CZA between 200–250 °C. In contrast, modification with K resulted in preferential formation of CO at all temperatures. MeOH selectivity with K/CZA was highest at 200 °C at just 2.2%.
At a reaction temperature of 250 °C and pressures between 10 and 30 bar (Fig. 4(b)), CZA and Ca/CZA exhibited similar conversion and MeOH selectivity. Both conversion and MeOH selectivity increased for these two materials as pressure increased from 10 to 30 bar. In stark contrast, increased pressure did not enhance conversion or MeOH production on K/CZA. Overall, it can be concluded that modification with K was significantly detrimental to overall hydrogenation activity, especially methanol yield. Indeed, this result aligns well with previous reports that impregnated alkali species can inhibit the activity of CZA catalysts.36 Given that Ca-modification provided a moderate promotional effect to adsorption capacity, with minor effects on CO2 binding geometries and catalytic performance, one would expect that Ca/CZA would be the best candidate for combined capture and conversion to MeOH. The next section exemplifies a key learning that we present in the development of DFMs for RCC processes: CO2 hydrogenation evaluation of a DFM in a typical co-fed experiment is not always a reliable indicator for performance in cyclic capture and conversion operation.
2.3. Performance of DFMs in CO2 adsorption–reactive desorption cycles
The steps used for cyclic RCC performance evaluation are described below and graphically represented in Fig. S4 (ESI†). A 1% CO2 stream was used during the CO2 adsorption step for ease of method development and performance evaluation. CO2 adsorption was conducted at 100 °C and atmospheric pressure for 1 h. Upon introduction of flowing CO2, reaction of CO2 with the freshly reduced CZA surface was evidenced by CO formation, but CO concentration gradually decreased with adsorption time (Fig. S5, ESI†). This reaction was suppressed over Ca/CZA and K/CZA (Table S4, ESI†), which is consistent with the lower hydrogenation activity observed in the CO2 hydrogenation, especially for K/CZA. An inert purge was performed after adsorption to evacuate the reactor of excess CO2, remove weakly bound CO2, and ensure that any products detected in the subsequent conversion step evolved from the surface-bound CO2 on the DFM (i.e., strongly bound CO2), and not as a result of reactions from residual gaseous species. This type of purge will also be important in future investigations of this and similar RCC technology when O2 is present in the CO2 stream to prevent the mixing of H2 and O2 for safety considerations. The CO2 that remains on the DFM surface after the inert purge is defined as “strong CO2 adsorption”, which is the reactant for the subsequent reactive desorption stage (i.e., hydrogenation reaction). The presence of strongly bound CO2 are consistent with the in situ DRIFTS study above. Importantly, strong CO2 adsorption neglects the portion of CO2 consumed to produce CO during the adsorption step as mentioned earlier. After purging, the reactor was pressurized to 30 bar with H2 before ramping the temperature to 250 °C for a 2 h hydrogenation. MeOH and CO were the major products at this stage, while DME, methane, and desorbed CO2 were also observed (Fig. S5 and Table S5, ESI†). The pressure was then lowered back to atmospheric pressure and held for 1 h to release residual products and reduce the catalyst for the next cycle. During this release step, CO was the major product observed (Fig. S5 and Table S5, ESI†). CZA and each DFM were tested for 5 RCC cycles (Fig. S6, ESI†), and the performance was compared by averaging results from the last 3 cycles – the first two cycles were disregarded to avoid any “start-up” effects, which would not be representative of cyclic performance (Fig. 5 and Tables S4, S5, ESI†).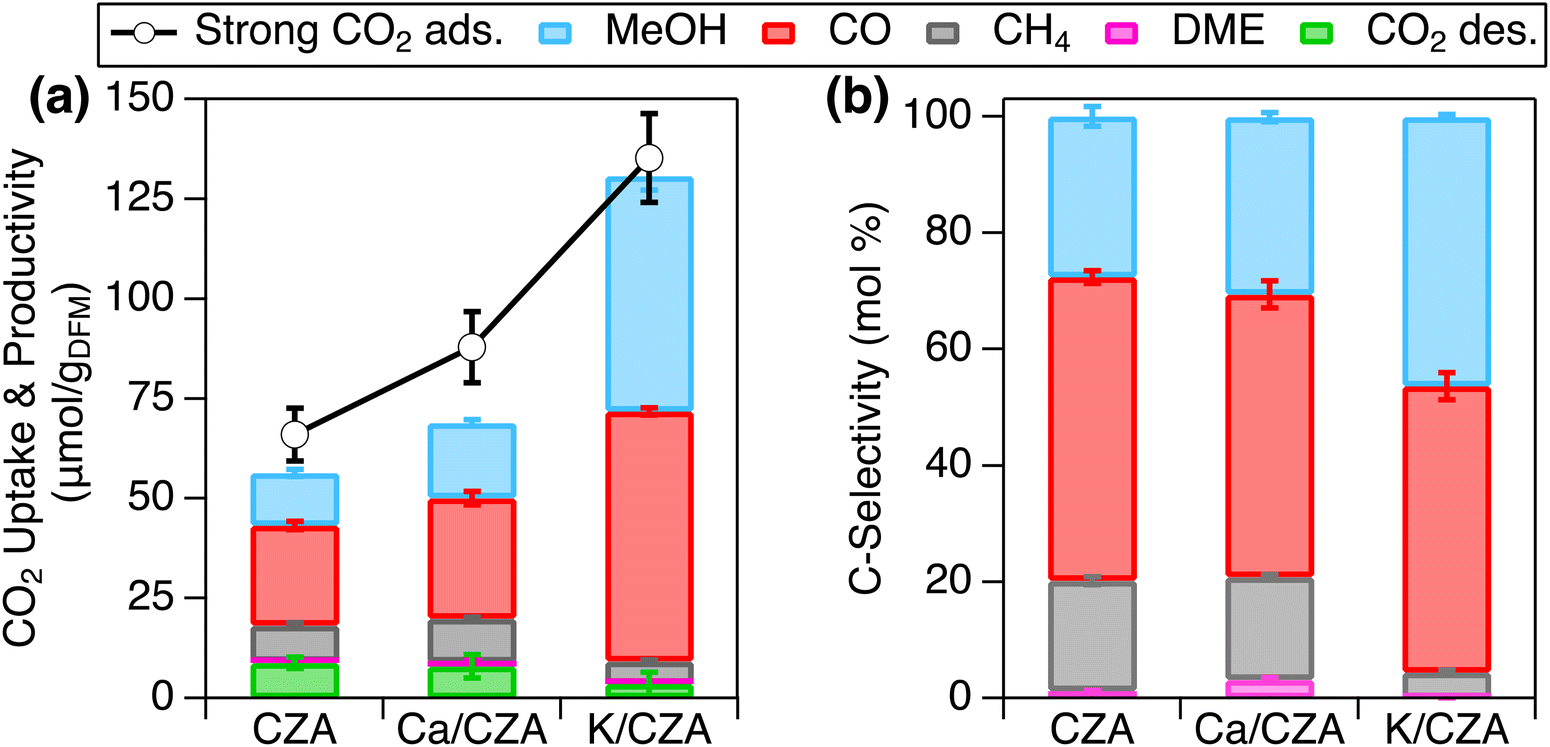 | ||
| Fig. 5 (a) Strong CO2 adsorption (markers) and productivity (stacked bars), and (b) C-selectivity of all products during reactive desorption over CZA, Ca/CZA and K/CZA. Data are averages of the last 3 cycles with standard deviations. CO2 adsorption followed by inert purge was performed at 100 °C and 0.8 bar pressure; reactive desorption was performed in pure H2 at 250 °C and 30 bar for 2 h followed by pressure release and purge at 0.8 bar pressure for 1 h. Additional experimental details can be found in ESI.† | ||
In accordance with the chemisorption results in Fig. 2, the introduction of Ca and K increased the strong CO2 capture capacity of the DFMs in an increasing order of CZA < Ca/CZA < K/CZA (Fig. 5(a) and Table S4, ESI†). In addition, the amount of desorbed CO2 during the hydrogenation step also decreased in the same order, from 8.8 μmol g−1 over CZA to just 3.5 μmol g−1 over K/CZA (Table S5, ESI†). Comparing Ca/CZA with the parent CZA, greater strong CO2 adsorption was observed with Ca/CZA (66.0 vs. 87.8 μmol g−1, Table S4, ESI†), consistent with the CO2 chemisorption data presented above. A slight decrease in desorbed, unreacted CO2 was also exhibited by Ca/CZA (8.8 vs. 7.9 μmol g−1). Overall conversion of adsorbed CO2 to products were similar between Ca/CZA (71.2%) and CZA (73.1%). Comparable product selectivities to CO, MeOH, and CH4 were also observed for CZA and Ca/CZA, with Ca/CZA having slightly greater MeOH selectivity at 30.5% versus 27.6% for CZA (Fig. 5(b) and Table S5, ESI†). Combining the comparable MeOH selectivity with enhanced adsorption capacity, Ca-modification results in a moderate improvement in RCC performance, highlighted by a 43% increase in MeOH productivity (i.e., specific activity) from 13.2 μmol g−1 for CZA to 18.9 μmol g−1 for Ca/CZA (Fig. 5(a)). Comparing K/CZA to both CZA and Ca/CZA, K/CZA exhibited the highest strong CO2 adsorption (135 μmol g−1) and the lowest amount of CO2 lost to desorption during reactive desorption (3.6 μmol g−1). In addition, the conversion of adsorbed CO2 to hydrogenated products was greatest over K/CZA (94% vs. 71–73% for Ca/CZA and CZA), leading to the greatest productivity to total hydrogenated products (average of 127 μmol g−1). This activity is comparable to top-performing methanation DFMs (150–200 μmol g−1).13,37 The selectivity to MeOH was greatly enhanced over K/CZA (46% vs. 28–30% for CZA and Ca/CZA), and importantly, the selectivity to CH4 was greatly suppressed (4.4% vs. 18–19%). Increased CO2 capture capacity, conversion of adsorbed CO2, and selectivity to MeOH resulted in a MeOH productivity of 59.0 μmol g−1, which is 3× greater than Ca/CZA and 4.5× greater than parent CZA.
There have been three recent reports targeting the production of MeOH from CO2 in a capture-conversion approach. As alluded to above, Pd-amine functionalized silica DFMs were tested in a cyclic, temperature swing process.28 These materials exhibited high CO2 adsorption (up to 1.2 mmol g−1) and achieved highly selective conversion to MeOH at mild reaction conditions (140 °C, 1 atm); however, ca. 70% of the captured CO2 was desorbed (unreacted) during the inert purge and temperature ramp steps of the cycles, resulting in a 20 μmol g−1 MeOH productivity. In a related approach that is not directly relevant, but is worth noting here, an integrated capture and conversion approach was employed with a liquid amine capture agent, followed by conversion of the carbamate to MeOH over a Pt/TiO2 catalyst.23 This approach reported high MeOH selectivity (51.5%) and productivity (150 μmol g−1), but CH4 selectivity was considerable at 27%. Most recently, a stacked bed approach was investigated, where a bed of Na/Al2O3 was layered on top of a bed of commercial MeOH synthesis catalyst (i.e., CZA).29 While this configuration was able to capture ca. 150 μmol g−1 of CO2, the MeOH productivity was considerably lower at 12 μmol g−1 due to low conversion of 20% and MeOH selectivity of 22%. Although this approach used a similar combination of alkali sorbent and CZA, we hypothesize that the chemistry resulting from a stacked bed of two separate materials (i.e., alkali sorbent and CZA) is different than what occurs when CZA and the sorbent are co-located on the same surface by impregnation (this report). With the separate bed configuration, CO2 was proposed to desorb from the sorbent bed during the initial temperature ramp and then subsequently convert as a gas phase reactant on the downstream bed.29 However, on the Alk/CZA DFMs employed here, we hypothesize that the co-location of the sorbent and catalyst sites offer a synergistic mechanism, in which adsorbed and activated CO2 reacts with nearby hydrogenation sites through a surface migration or spillover effect. Similar mechanisms have been proposed for many solid-phase DFMs such as methanation DFMs38 and the Pd-amine DFMs.27,28 Detailed mechanistic studies, including in situ or operando DRIFTS under relevant reactive desorption conditions coupled with computational analysis, are necessary to precisely define the mechanism, which will be the topic of future reports. The K/CZA DFM presented in this work marks a great advancement in RCC to MeOH, exhibiting a combination of high CO2 adsorption capacity, high conversion of adsorbed CO2 in the reactive desorption stage, and high MeOH selectivity with low selectivity to CH4, ultimately leading to a 3× increase in MeOH production when compared to a similar report using solid-phase DFMs in a cyclic approach.23
In the context of developing an accompanying RCC process, it is important to note that CO is a major by-product from this RCC process, especially during the low-pressure reactive desorption step when the reactor pressure was released and the catalyst was reduced for the next cycle. In contrast, the majority of MeOH was produced in the high-pressure step of reactive desorption (Fig. S5 and Table S5, ESI†), which is consistent with the thermodynamic preference for MeOH synthesis from CO2 hydrogenation reactions.39–41 The high selectivity to CO during the low-pressure step, coupled with low CH4 selectivity during the entire reactive desorption stage, could be beneficial for downstream separation and may enable a partial recycle of the product stream to improve overall, integrated MeOH yield through a series of RCC reactors. This is illustrated in the conceptualized process flow diagram in Fig. S7 (ESI†).
As noted in the previous section, the evaluation of CO2 hydrogenation activity of these novel DFMs in a co-fed experiment (with a H2:CO2 ratio of 3:1) did not accurately predict their RCC performance. Although K/CZA exhibited low conversion and low selectivity towards MeOH in a co-fed hydrogenation experiment, it exhibited the highest CO2 uptake, conversion of adsorbed CO2, and MeOH selectivity in RCC evaluation. We hypothesize that this inconsistency may arise from different H2:CO2 surface coverages when co-fed at ratio of 3 versus delivered in a cyclic fashion where the H2:CO2 ratio is substantially greater. Based on the strong CO2 adsorption value for K/CZA (ca. 135 μmol g−1) and our catalyst bed volume (ca. 1 mL), we estimate a H2:CO2 ratio of 7 at 30 bar and 100 °C at the start of reactive desorption. This ratio is expected to increase as the reaction progresses and surface-bound CO2 is consumed. Another hypothesis is that the most abundant surface intermediates may be different in the co-fed CO2 hydrogenation environment versus the reactive desorption step in the RCC cycles. DRIFTS characterization representing the adsorption step of the RCC cycles revealed that the strongly adsorbed CO2 was bound to CZA and DFMs as various carbonate species (CO3*). Therefore, the following reactive desorption step may involve the reaction between dissociated H2 (H*) with these various CO3* species. In contrast, under co-fed conditions over CZA, the CO2 hydrogenation reaction has been proposed to proceed via a formate intermediate, which is mainly formed by the reaction between molecularly adsorbed CO2 (CO2*) and H*.42,43 A more in-depth mechanistic study is required for a precise explanation for this difference in performance, which will be the topic of a separate report, and may necessitate a combination of chemical computations and operando characterization.
Similarly, when comparing the performance in RCC cycles, we rationalize the observed similar activity and product selectivity for CZA and Ca/CZA based on DRIFTS data that indicated similar binding strengths and geometries of the surface CO3*. In contrast, dramatically different binding strengths and geometries were observed on K/CZA compared to CZA and Ca/CZA. The high overall CO2 conversion and MeOH selectivity observed over K/CZA may be due to the reaction pathway proceeding through these unique K–CO3* species (i.e., the K-bound carbonates being inherently activated for greater MeOH production based on geometry and proximity to hydrogenation sites). Thus, as a second learning in the design of DFMs for MeOH synthesis via RCC, an analysis of the CO2 binding geometry may be more instructive to anticipate differences between the parent material and novel DFMs.
3. Conclusion
Motivated by the potential for energy and cost reductions for renewable MeOH synthesis via an RCC approach, we synthesized, characterized, and tested two modified CZA DFMs. CO2 chemisorption analysis of the materials revealed that total capture capacity and strong CO2 uptake increased from CZA to Ca/CZA and K/CZA, with K-modification having the greatest promotional effect. We identified the dominant CO3* binding geometries on these materials with in situ DRIFTS characterization, and the binding modes and strengths on K/CZA were drastically different when compared to CZA and Ca/CZA. These DFMs were tested for CO2 hydrogenation activity in a co-fed experiment, during which K-modification exhibited a detrimental effect on catalytic activity. In stark contrast, K/CZA provided the best performance in RCC to MeOH, exhibiting the highest CO2 conversion, MeOH selectivity, and MeOH productivity. Ca/CZA, while offering an increased capture capacity, did not significantly improve hydrogenation activity compared to CZA during both the co-fed experiments and RCC cycles. These results highlight the importance of careful methodology in screening novel DFMs, and a consideration of surface coverage and surface structure of intermediates.This report provides a strong foundation for subsequent, continued research to understand and improve both the DFM composition and the RCC-to-MeOH process design and operation. This report leverages CZA as a robust commercial catalyst that is produced and available at scale, and we demonstrate the feasibility of RCC to MeOH utilizing Alk/CZA materials. For DFM development, materials that offer high CO2 capacity and even higher MeOH selectivity should be pursued, taking note of the principles presented here. Additional mechanistic studies are also of interest, to precisely describe the reactive desorption mechanism and explain the difference between RCC behavior and co-fed hydrogenation behavior. For RCC process development, the cycling conditions presented here have not been optimized to maximize capture and conversion efficiency, warranting a further detailed parametric study to improve process performance (e.g., improve MeOH productivity, reduce cycle time). Evaluation at higher cycle numbers, and the assessment of oxygen/water stability is of particular importance. Such contaminants can have a major impact in the efficiency of RCC cycles, especially on catalyst stability, CO2 capture capacity, and hydrogen demand, affecting the resulting process design and techno-economic analysis of this approach.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was authored by the National Renewable Energy Laboratory, operated by Alliance for Sustainable Energy, LLC, for the U.S. Department of Energy (DOE) under contract no. DE-AC36-08GO28308. Funding was provided by U.S. Department of Energy, Office of Fossil Energy and Carbon Management. The views expressed in this article do not necessarily represent the views of the DOE or the U.S. Government. The U.S. Government retains and the publisher, by accepting the article for publication, acknowledges that the U.S. Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this work, or allow others to do so, for U.S. Government purposes.References
- IPCC, Global Warming of 1.5 °C: IPCC Special Report on Impacts of Global Warming of 1.5 °C above Pre-industrial Levels in Context of Strengthening Response to Climate Change, Sustainable Development, and Efforts to Eradicate Poverty, Cambridge University Press, 1st edn, 2022 Search PubMed.
- R. G. Grim, Z. Huang, M. T. Guarnieri, J. R. Ferrell, L. Tao and J. A. Schaidle, Transforming the carbon economy: challenges and opportunities in the convergence of low-cost electricity and reductive CO2 utilization, Energy Environ. Sci., 2020, 13, 472–494 RSC.
- M. Fasihi, O. Efimova and C. Breyer, Techno-economic assessment of CO2 direct air capture plants, J. Cleaner Prod., 2019, 224, 957–980 CrossRef CAS.
- A. Kätelhön, R. Meys, S. Deutz, S. Suh and A. Bardow, Climate change mitigation potential of carbon capture and utilization in the chemical industry, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 11187–11194 CrossRef PubMed.
- M. S. Duyar, M. A. A. Treviño and R. J. Farrauto, Dual function materials for CO2 capture and conversion using renewable H2, Appl. Catal., B, 2015, 168–169, 370–376 CrossRef CAS.
- C. Jeong-Potter, M. Abdallah, C. Sanderson, M. Goldman, R. Gupta and R. Farrauto, Dual function materials (Ru + Na2O/Al2O3) for direct air capture of CO2 and in situ catalytic methanation: The impact of realistic ambient conditions, Appl. Catal., B, 2022, 307, 120990 CrossRef CAS.
- I. S. Omodolor, H. O. Otor, J. A. Andonegui, B. J. Allen and A. C. Alba-Rubio, Dual-Function Materials for CO2 Capture and Conversion: A Review, Ind. Eng. Chem. Res., 2020, 59, 17612–17631 CrossRef CAS.
- S. Kar, R. Sen, A. Goeppert and G. K. S. Prakash, Integrative CO2 Capture and Hydrogenation to Methanol with Reusable Catalyst and Amine: Toward a Carbon Neutral Methanol Economy, J. Am. Chem. Soc., 2018, 140, 1580–1583 CrossRef CAS PubMed.
- I. Ioannou, J. Javaloyes-Antón, J. A. Caballero and G. Guillén-Gosálbez, Economic and Environmental Performance of an Integrated CO2 Refinery, ACS Sustainable Chem. Eng., 2023, 11, 1949–1961 CrossRef CAS PubMed.
- M. C. Freyman, Z. Huang, D. Ravikumar, E. B. Duoss, Y. Li, S. E. Baker, S. H. Pang and J. A. Schaidle, Reactive CO2 capture: A path forward for process integration in carbon management, Joule, 2023, 7, 631–651 CrossRef CAS.
- R. E. Siegel, S. Pattanayak and L. A. Berben, Reactive Capture of CO2: Opportunities and Challenges, ACS Catal., 2023, 13, 766–784 CrossRef CAS.
- M. S. Duyar, S. Wang, M. A. Arellano-Treviño and R. J. Farrauto, CO2 utilization with a novel dual function material (DFM) for capture and catalytic conversion to synthetic natural gas: An update, J. CO2 Util., 2016, 15, 65–71 CrossRef CAS.
- S. Wang, R. J. Farrauto, S. Karp, J. H. Jeon and E. T. Schrunk, Parametric, cyclic aging and characterization studies for CO2 capture from flue gas and catalytic conversion to synthetic natural gas using a dual functional material (DFM), J. CO2 Util., 2018, 27, 390–397 CrossRef CAS.
- M. A. Arellano-Treviño, N. Kanani, C. W. Jeong-Potter and R. J. Farrauto, Bimetallic catalysts for CO2 capture and hydrogenation at simulated flue gas conditions, Chem. Eng. J., 2019, 375, 121953 CrossRef.
- Natural Gas: Henry Hub Natural Gas Spot Price, https://www.eia.gov/dnav/ng/hist/rngwhhdA.htm, (accessed 1 July 2023).
- J. Gorre, F. Ortloff and C. Van Leeuwen, Production costs for synthetic methane in 2030 and 2050 of an optimized Power-to-Gas plant with intermediate hydrogen storage, Appl. Energy, 2019, 253, 113594 CrossRef CAS.
- M. Svanberg, J. Ellis, J. Lundgren and I. Landälv, Renewable methanol as a fuel for the shipping industry, Renewable Sustainable Energy Rev., 2018, 94, 1217–1228 CrossRef CAS.
- R. G. Grim, A. T. To, C. A. Farberow, J. E. Hensley, D. A. Ruddy and J. A. Schaidle, Growing the Bioeconomy through Catalysis: A Review of Recent Advancements in the Production of Fuels and Chemicals from Syngas-Derived Oxygenates, ACS Catal., 2019, 9, 4145–4172 CrossRef CAS.
- About Methanol: Pricing, https://www.methanex.com/about-methanol/pricing/, (accessed 1 July 2023).
- S. Kang, Innovation outlook: renewable methanol, International Renewable Energy Agency, Abu Dhabi, 2021 Search PubMed.
- S. Sollai, A. Porcu, V. Tola, F. Ferrara and A. Pettinau, Renewable methanol production from green hydrogen and captured CO2: A techno-economic assessment, J. CO2 Util., 2023, 68, 102345 CrossRef CAS.
- R. Sen, A. Goeppert and G. K. S. Prakash, Integrated carbon capture and utilization to methanol with epoxide-functionalized polyamines under homogeneous catalytic conditions, J. Organomet. Chem., 2022, 965–966, 122331 CrossRef CAS.
- J. Kothandaraman, J. S. Lopez, Y. Jiang, E. D. Walter, S. D. Burton, R. A. Dagle and D. J. Heldebrant, Integrated Capture and Conversion of CO2 to Methanol in a Post-Combustion Capture Solvent: Heterogeneous Catalysts for Selective C–N Bond Cleavage, Adv. Energy Mater., 2022, 12, 2202369 CrossRef CAS.
- S. Kar, A. Goeppert, V. Galvan, R. Chowdhury, J. Olah and G. K. S. Prakash, A Carbon-Neutral CO2 Capture, Conversion, and Utilization Cycle with Low-Temperature Regeneration of Sodium Hydroxide, J. Am. Chem. Soc., 2018, 140, 16873–16876 CrossRef CAS PubMed.
- R. Sen, C. J. Koch, V. Galvan, N. Entesari, A. Goeppert and G. K. S. Prakash, Glycol assisted efficient conversion of CO2 captured from air to methanol with a heterogeneous Cu/ZnO/Al2O3 catalyst, J. CO2 Util., 2021, 54, 101762 CrossRef CAS.
- S. Kar, A. Goeppert and G. K. S. Prakash, Integrated CO2 Capture and Conversion to Formate and Methanol: Connecting Two Threads, Acc. Chem. Res., 2019, 52, 2892–2903 CrossRef CAS PubMed.
- J. Pazdera, E. Berger, J. A. Lercher and A. Jentys, Conversion of CO2 to methanol over bifunctional basic-metallic catalysts, Catal. Commun., 2021, 159, 106347 CrossRef CAS.
- J. Pazdera, D. Issayeva, J. Titus, R. Gläser, O. Deutschmann and A. Jentys, Impact of the Local Environment of Amines on the Activity for CO2 Hydrogenation over Bifunctional Basic – Metallic Catalysts, ChemCatChem, 2022, 14, e202200620 CrossRef CAS.
- L. C. Wirner, F. Kosaka, T. Sasayama, Y. Liu, A. Urakawa and K. Kuramoto, Combined capture and reduction of CO2 to methanol using a dual-bed packed reactor, Chem. Eng. J., 2023, 470, 144227 CrossRef CAS.
- M. A. Arellano-Treviño, Z. He, M. C. Libby and R. J. Farrauto, Catalysts and adsorbents for CO2 capture and conversion with dual function materials: Limitations of Ni-containing DFMs for flue gas applications, J. CO2 Util., 2019, 31, 143–151 CrossRef.
- A. Porta, R. Matarrese, C. G. Visconti, L. Castoldi and L. Lietti, Storage Material Effects on the Performance of Ru-Based CO2 Capture and Methanation Dual Functioning Materials, Ind. Eng. Chem. Res., 2021, 60, 6706–6718 CrossRef CAS.
- M. Smyrnioti, C. Tampaxis, T. Steriotis and T. Ioannides, Study of CO2 adsorption on a commercial CuO/ZnO/Al2O3 catalyst, Catal. Today, 2020, 357, 495–502 CrossRef CAS.
- P. Gruene, A. G. Belova, T. M. Yegulalp, R. J. Farrauto and M. J. Castaldi, Dispersed Calcium Oxide as a Reversible and Efficient CO2-Sorbent at Intermediate Temperatures, Ind. Eng. Chem. Res., 2011, 50, 4042–4049 CrossRef CAS.
- T. Numpilai, N. Chanlek, Y. Poo-Arporn, C. K. Cheng, N. Siri-Nguan, T. Sornchamni, M. Chareonpanich, P. Kongkachuichay, N. Yigit, G. Rupprechter, J. Limtrakul and T. Witoon, Tuning Interactions of Surface-adsorbed Species over Fe–Co/K–Al2O3 Catalyst by Different K Contents: Selective CO2 Hydrogenation to Light Olefins, ChemCatChem, 2020, 12, 3306–3320 CrossRef CAS.
- F. Prinetto, M. Manzoli, S. Morandi, F. Frola, G. Ghiotti, L. Castoldi, L. Lietti and P. Forzatti, Pt–K/Al2O3 NSR Catalysts: Characterization of Morphological, Structural and Surface Properties, J. Phys. Chem. C, 2010, 114, 1127–1138 CrossRef CAS.
- P. Kowalik, W. Próchniak and T. Borowiecki, The effect of alkali metals doping on properties of Cu/ZnO/Al2O3 catalyst for water gas shift, Catal. Today, 2011, 176, 144–148 CrossRef CAS.
- C. Jeong-Potter, A. Porta, R. Matarrese, C. G. Visconti, L. Lietti and R. Farrauto, Aging study of low Ru loading dual function materials (DFM) for combined power plant effluent CO2 capture and methanation, Appl. Catal., B, 2022, 310, 121294 CrossRef CAS.
- L. Proaño, E. Tello, M. A. Arellano-Trevino, S. Wang, R. J. Farrauto and M. Cobo, In situ DRIFTS study of two-step CO2 capture and catalytic methanation over Ru,“Na2O”/Al2O3 Dual Functional Material, Appl. Surf. Sci., 2019, 479, 25–30 CrossRef.
- G. H. Graaf, P. J. J. M. Sijtsema, E. J. Stamhuis and G. E. H. Joosten, Chemical equilibria in methanol synthesis, Chem. Eng. Sci., 1986, 41, 2883–2890 CrossRef CAS.
- A. T. To, M. A. Arellano-Treviño, C. P. Nash and D. A. Ruddy, Direct synthesis of branched hydrocarbons from CO2 over composite catalysts in a single reactor, J. CO2 Util., 2022, 66, 102261 CrossRef CAS.
- G. Leonzio, E. Zondervan and P. U. Foscolo, Methanol production by CO2 hydrogenation: Analysis and simulation of reactor performance, Int. J. Hydrogen Energy, 2019, 44, 7915–7933 CrossRef CAS.
- F. Studt, M. Behrens, E. L. Kunkes, N. Thomas, S. Zander, A. Tarasov, J. Schumann, E. Frei, J. B. Varley, F. Abild-Pedersen, J. K. Nørskov and R. Schlögl, The Mechanism of CO and CO2 Hydrogenation to Methanol over Cu-Based Catalysts, ChemCatChem, 2015, 7, 1105–1111 CrossRef CAS.
- X.-K. Wu, G.-J. Xia, Z. Huang, D. K. Rai, H. Zhao, J. Zhang, J. Yun and Y.-G. Wang, Mechanistic insight into the catalytically active phase of CO2 hydrogenation on Cu/ZnO catalyst, Appl. Surf. Sci., 2020, 525, 146481 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ey00254c |
| This journal is © The Royal Society of Chemistry 2024 |