
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iron oxide-promoted photochemical oxygen reduction to hydrogen peroxide (H2O2)†
Thomas
Freese
 a,
Jelmer T.
Meijer
a,
Maria B.
Brands
b,
Georgios
Alachouzos
a,
Marc C. A.
Stuart
c,
Rafael
Tarozo
a,
Dominic
Gerlach
d,
Joost
Smits
e,
Petra
Rudolf
d,
Joost N. H.
Reek
b and
Ben L.
Feringa
*a
a,
Jelmer T.
Meijer
a,
Maria B.
Brands
b,
Georgios
Alachouzos
a,
Marc C. A.
Stuart
c,
Rafael
Tarozo
a,
Dominic
Gerlach
d,
Joost
Smits
e,
Petra
Rudolf
d,
Joost N. H.
Reek
b and
Ben L.
Feringa
*a
aStratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: b.l.feringa@rug.nl
bvan’t Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands
cElectron Microscopy, Groningen Biomolecular Sciences and Biotechnology Institute, University of Groningen, Nijenborgh 7, 9747AG Groningen, The Netherlands
dZernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747AG Groningen, The Netherlands
eShell Global Solutions International BV, Grasweg 31, 1031 HW Amsterdam, The Netherlands
First published on 24th November 2023
Abstract
Hydrogen peroxide (H2O2) is a valuable green oxidant with a wide range of applications. Furthermore, it is recognized as a possible future energy carrier achieving safe operation, storage and transportation. The photochemical production of H2O2 serves as a promising alternative to the waste- and energy-intensive anthraquinone process. Following the 12 principles of Green Chemistry, we demonstrate a facile and general approach to sustainable catalyst development utilizing earth-abundant iron and biobased sources only. We developed several iron oxide (FeOx) nanoparticles (NPs) for successful photochemical oxygen reduction to H2O2 under visible light illumination (445 nm). Achieving a selectivity for H2O2 of >99%, the catalyst material could be recycled for up to four consecutive rounds. An apparent quantum yield (AQY) of 0.11% was achieved for the photochemical oxygen reduction to H2O2 with visible light (445 nm) at ambient temperatures and pressures (9.4–14.8 mmol g−1 L−1). Reaching productivities of H2O2 of at least 1.7 ± 0.3 mmol g−1 L−1 h−1, production of H2O2 was further possible via sunlight irradiation and in seawater. Finally, a detailed mechanism has been proposed on the basis of experimental investigation of the catalyst's properties and computational results.
Broader contextFacing the environmental crisis, the energy transition to renewables (wind and solar) increases global demand for future energy carriers. Next to its applications as an eco-friendly oxidant, H2O2 offers great potential as an energy carrier as it is fully soluble in water. Hence it offers an easy-to-handle liquid fuel alternative achieving safer operation, storage and transportation. The solar-driven reduction of molecular oxygen to produce H2O2 is an ecologically viable route, especially when sustainable and biobased photocatalysts are utilized. We established a strategy for the photochemical production of H2O2 catalysed by iron. The sustainable synthesis of iron oxide (FeOx) nanoparticles terminated with different surfactants was demonstrated, where specifically FeOx NPs with cis double bonds possess photoactivity for oxygen reduction to hydrogen peroxide. An apparent quantum yield (AQY) of 0.11% was achieved for the photochemical oxygen reduction to H2O2 with visible light (445 nm) at ambient temperatures and pressures (9.4–14.8 mmol g−1 L−1), corresponding to 1.7 ± 0.3 mmol g−1 L−1 h−1. The H2O2 yield could be increased by decreasing the pH, addition of cation exchangers and production in biphasic systems (heptane/DCM with Milli-Q water) (up to 19.5 ± 2.7 mmol g−1 L−1). The FeOx nanoparticles with oleic acid (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as a surfactant were successfully utilized in applications like wastewater treatment, polymerizations and in situ oxidations. Production of H2O2 was possible via sunlight irradiation and in seawater. Utilizing earth-abundant metals and biobased (co-)catalysts offers great potential for the photocatalytic production of hydrogen peroxide as a solar fuel. :1) as a surfactant were successfully utilized in applications like wastewater treatment, polymerizations and in situ oxidations. Production of H2O2 was possible via sunlight irradiation and in seawater. Utilizing earth-abundant metals and biobased (co-)catalysts offers great potential for the photocatalytic production of hydrogen peroxide as a solar fuel.
|
Introduction
Hydrogen peroxide (H2O2) is a versatile green oxidant with large scale applications in the chemical industry, pulp and paper bleaching, wastewater treatment and disinfectants.1–6 H2O2 is also utilized in fuel cells as an advantageous energy carrier over hydrogen (H2).7–14 Despite major advances in the generation of H2 from water, its storage and low energy density (per volume) are still a major bottleneck.15–18 Being far from practical with regards to the energy density of H2 per volume at atmospheric pressure, stationary fuel storage of H2 requires chemical transformation into transportable liquids (e.g. ammonia or formic acid).19–29 Pure H2 stored in a tank at 35 MPa (room temperature) delivers 2.8 MJ L−1 when operated in a fuel cell, which is very similar to the energy density of aqueous H2O2 (70 wt%) with 3.1 MJ L−1.30,31 Thus H2O2, being fully soluble in water, offers an easy-to-handle liquid fuel alternative achieving safer operation, storage and transportation.3,4 The global market for H2O2 is estimated to grow at a compound annual growth rate (CAGR) of 4.6% increasing to 5.7 million tons annual demand by 2028.32–34 Its characteristics as a high-energy fuel and as an ecological oxidant, generating water (H2O) and oxygen (O2) as the only by-products, constitute H2O2 being listed as one of the 100 most important chemicals on earth.35,36Currently more than 95% of H2O2 is produced via the anthraquinone process, comprising Pd-catalysed hydrogenation of an alkyl-anthraquinone and consecutive oxidation in organic solvents.1,37 However, this synthetic strategy involves high energy input and generates a substantial volume of wastewater and solid waste.1,36 With increasing demand for a sustainable alternative, the electrocatalytic oxygen reduction reaction (ORR) to form H2O2,38–40 as well as the direct synthesis of H2O2 from H2 and O2, offer apparent solutions to these problems.41–48 Nevertheless high energy consumption and inevitable high explosion risks of O2 and H2 gas mixtures hamper the industrial scale up of these systems.7,9
Consequently, utilizing green energy sources such as solar energy for the photochemical production of H2O2 directly from water serves as an alternative, cleaner method to meet the global demand for H2O2.4,9,33 By combining the photocatalytic two-electron reduction of O2 (ORR, +0.68 VNHE) and the catalytic four-electron oxidation of H2O (WOR, +1.23 VNHE), the overall photosynthesis of H2O2 from water can be achieved (eqn (1)).3,49
| 2H2O + O2 → 2H2O2 (ΔG° = 204 kJ mol−1) | (1) |
Despite these advances, the metal-based photocatalysts often rely on noble and scarce metals, the starting materials and solvents are not biobased and the catalyst synthesis requires special equipment or high temperatures.74 Thus, legitimate H2O2 production as a future energy carrier can only be achieved by a green photocatalyst complying with the Sustainable Development Goals and the 12 principles of Green Chemistry (Fig. 1A).75,76 Indeed, the development of sustainable materials such as catalysts,20,77–79 polymers,80,81 coatings82,83 and molecular motors84 from renewable and earth abundant sources has received high priority in the past, avoiding dependency on fossil resources. Considering element scarcity and climate change, iron is the fourth most abundant element in the earth's crust and the most abundant metal, and thus an ideal candidate as an environmental photocatalyst material.85,86
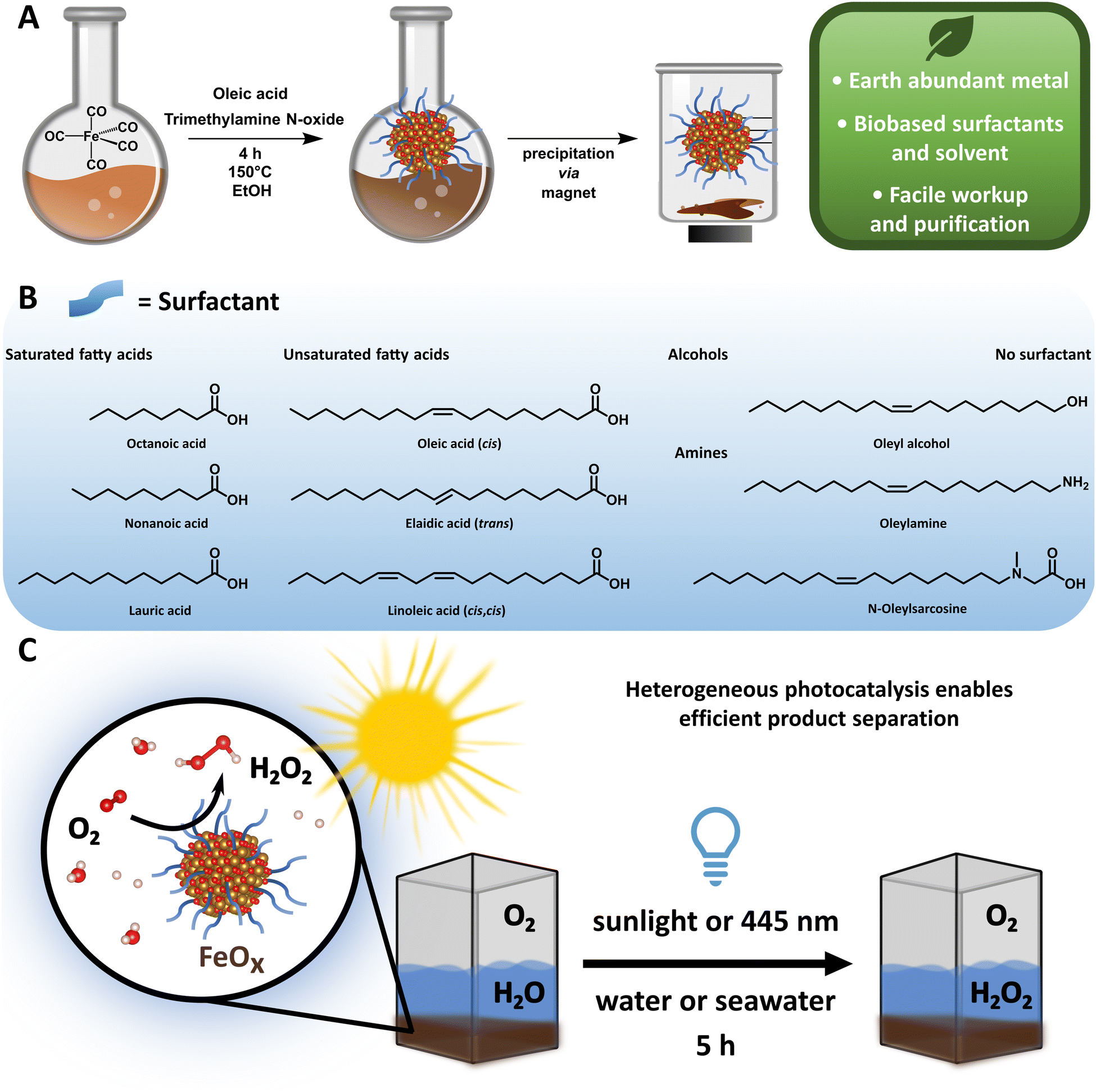 | ||
| Fig. 1 (A) Schematic representation of the synthesis of FeOx NPs in ethanol (EtOH), magnetic precipitation and its sustainability advantages. (B) Scope of different biobased surfactants assessed as ligands in FeOx NPs for H2O2 production. (C) Photochemical oxygen reduction to H2O2via FeOx NPs in water (1 mg mL−1) at 445 nm (5 h). | ||
Based on these considerations, we envisioned a possibility to adapt previously reported iron oxide (Fe3O4) nanoparticles (NPs) for our approach,87 as these particles, next to other iron oxides,88 already demonstrated promising electrocatalytic activity for the two-electron ORR to H2O2.89,90 We hypothesized that these might also have potential for the photocatalytic reduction of oxygen. Also, the proven electrochemical water splitting capability of iron oxide materials makes such catalysts interesting for photochemical H2O2 production including possible four-electron WOR.91,92 To the best of our knowledge, no pure iron oxide photocatalyst system has been reported to date, since iron oxide (Fe2O3) was only utilized as a support material before.55
Here we report an improved preparation method compared to the original synthesis of Fe3O4 NPs regarding solvent, energy consumption, workup and overall sustainability,93,94 and provide a scope of different nanoparticles, which can be synthesized and purified via a sustainable and facile route within six hours (Fig. 1A and B). The synthesized iron oxide nanoparticles (FeOx NPs) with oleic acid (OA, 2:1 ratio) as a surface ligand possess photochemical activity for oxygen reduction to selectively form H2O2 (up to 19.5 ± 2.7 mmol g−1 L−1, Fig. 1C) and could be recycled several times (up to four rounds). The production of H2O2 was achieved at ambient temperatures and pressures upon irradiation of the NPs with 445 nm light (5 h), and a detailed study of the oxygen reduction mechanism was conducted. Considering the high abundance of seawater and sunlight, the successful H2O2 production under these conditions via FeOx NPs provides indeed a basis for a sustainable solution in the H2O2 market.
Results and discussion
We investigated the ORR aiming at modifying the synthesis of Fe3O4 towards a more sustainable route and tuning the properties of the material towards a photocatalyst.87,95 This was especially of interest as similar iron-based materials possess electrochemical water splitting capability, which if tuned properly could lead to photochemical four-electron WOR and subsequent two-electron ORR properties.91The original solvent dioctyl ether (BP: 292 °C) was replaced with ethanol (BP: 78 °C), which not only allowed for less energy intensive reflux conditions, but also a greener solvent (Table 1).96–98 Instead of separation by centrifugation, utilizing the magnetic properties of the nanoparticles and precipitation on a magnet allowed for facile purification. An extensive investigation of the synthesis conditions was conducted yielding photoactive FeOx NP material eventually. As heterogeneous catalyst performance can strongly be impacted by changes in parameters such as temperature, particle size, pore dimensions and reactor configuration, we envisioned the material to be sensitive to changes in mass and heat transport.99 A thorough optimization of glassware (round bottom flask size, beaker size for magnetic precipitation), stirring bar size and shape, magnetic stirring vs. mechanical stirring, reflux temperature (78–150 °C), atmosphere (air, N2) and storage conditions was conducted (ESI-3.2 and ESI-3.3,†). The consistent synthesis of photoactive heterogeneous FeOx catalyst material could successfully be reproduced by several researchers in labs at different locations (ESI,† Fig. S110). As the reproducibility of heterogeneous catalyst materials is an often-overlooked aspect, achieving synthesis independent of iron(0) pentacarbonyl (FeCO5) suppliers (Sigma Aldrich, Acros Organics) from different Lot-numbers/continents was essential. The overall sustainability of the whole process has been assessed in Table 1.
| Principle | Justification |
|---|---|
| (1) Prevention of waste | As the synthesis of the Fe NPs is conducted in one step and purified via precipitation, stoichiometric amounts of waste could be minimized. Purification techniques such as column chromatography or centrifugation could be replaced by precipitation via magnetic properties of the NPs. As for the production of H2O2 no side products are obtained and the catalyst is not leached into the water. |
| (3) Less hazardous chemical synthesis | The synthetic method for the nanoparticles is designed for relatively low temperatures (150 °C), relies on biobased surfactants such as oleic acid, which can be obtained from olive oil, and utilizes non-harmful solvents such as ethanol in synthesis and workup.96,101 As for the metal, only earth-abundant iron (Fe) is utilized for the synthesis of the nanoparticles.85 In future optimizations the iron source (Fe(CO)5) will be replaced with other materials such as FeCl2 or FeCl3. The photochemical production of hydrogen peroxide is conducted with visible light in water, generating less harmful diluted H2O2 for application.9 |
| (5) Benign solvents & auxiliaries | Environmentally benign, biobased and only non-halogenated solvents (water, ethanol) were used throughout the synthesis and purification of the nanoparticles, as well as during the production of H2O2. Abundant seawater and lake water can also be utilized for the production.20 |
| (6) Energy efficiency | The original solvent dioctyl ether (BP: 292 °C) was replaced with ethanol (BP: 78 °C), which allowed for less energy intensive reflux conditions during catalyst synthesis. The photochemical production of H2O2 was fuelled by visible light (445 nm) and even by sunlight (September 2022) at room temperature. Thus, avoiding energy intensive high-pressure/temperature conditions. |
| (7) Renewable feedstocks | Considering element scarcity and CO2 footprint, iron is the fourth most abundant element in the earth's crust and the most abundant metal, and thus an ideal candidate as an environmental photocatalyst material.86,93 All surfactants utilized for the NP scope are biobased and renewable.100–102 |
| (8) No derivatives | Derivatization is avoided and the whole synthesis towards the catalyst material or H2O2 was conducted without the use of protecting groups. |
| (9) Catalysis | An environmentally benign heterogeneous photocatalyst has been developed for the production of H2O2 in water. Utilizing heterogeneous systems over homogeneous ones allows for facile product separation and purification. Replacing the Pd-catalysed anthraquinone process via heterogeneous photocatalysts is less energy intensive and generates less waste.3 |
| (10) Design for degradation | H2O2 as a product decomposes naturally on iron surfaces, which in fact can be used for degradation and wastewater treatment.111 The successful decomposition of an organic dye (methylene blue) via FeOx NPs has been confirmed (ESI-10.4). |
The general synthesis of FeOx NPs allowed for a wide variety of biobased capping agents (Fig. 1B), which were fully characterized (ESI-4,†).100–102 Analyses by transmission electron microscopy (TEM), scanning transmission electron microscopy (STEM), energy-dispersive X-ray spectroscopy (EDX), dynamic light scattering measurements (DLS), powder X-ray diffraction (XRD), UV-visible spectroscopy (UV-Vis) and elemental analysis were conducted to assess the properties for photochemical ORR activity. Specifically, FeOx with oleic acid and linoleic acid possessed photochemical activity for the production of H2O2. Additional immobilization of FeOx with oleic acid on graphene and activated carbon (C) was achieved (Fig. S47–S56, ESI†), where lower but still present activity for photochemical ORR was observed when FeOx@C was used. Through DLS measurements we correlated the particle size to the photochemical activity: FeOx NPs with oleic acid and linoleic acid were smaller (1.94 ± 0.34 nm and 1.54 ± 0.26 nm, respectively) than nanoparticles with other ligands (2.5–7.5 nm, Fig. S37, ESI†). While the synthesis offered great selectivity for consistent size below 10 nm, capping with oleyl alcohol or without surfactant led to larger particles (377 ± 166 nm and 1299 ± 100 nm, respectively, Table S1, ESI†).103 The monodisperse small nanoparticles were further visualized via TEM, STEM and EDX indicating successful incorporation of oxygen (O) and iron (Fe) (Fig. 2A–C). UV-Vis spectroscopy revealed that all catalyst materials possess similar absorption spectra (ESI,† Fig. S82). Interestingly, no significant difference between active FeOx NPs with oleic acid (cis)/linoleic acid (cis,cis) and inactive NPs with elaidic acid (trans) was observed (ESI,† Fig. S83). As FeOx NPs with oleic acid provided the highest photochemical production of H2O2 while also being the most abundant and cost-efficient surfactant,100,101 we analysed those in detail and will refer to those as standard when mentioning FeOx in upcoming sections.
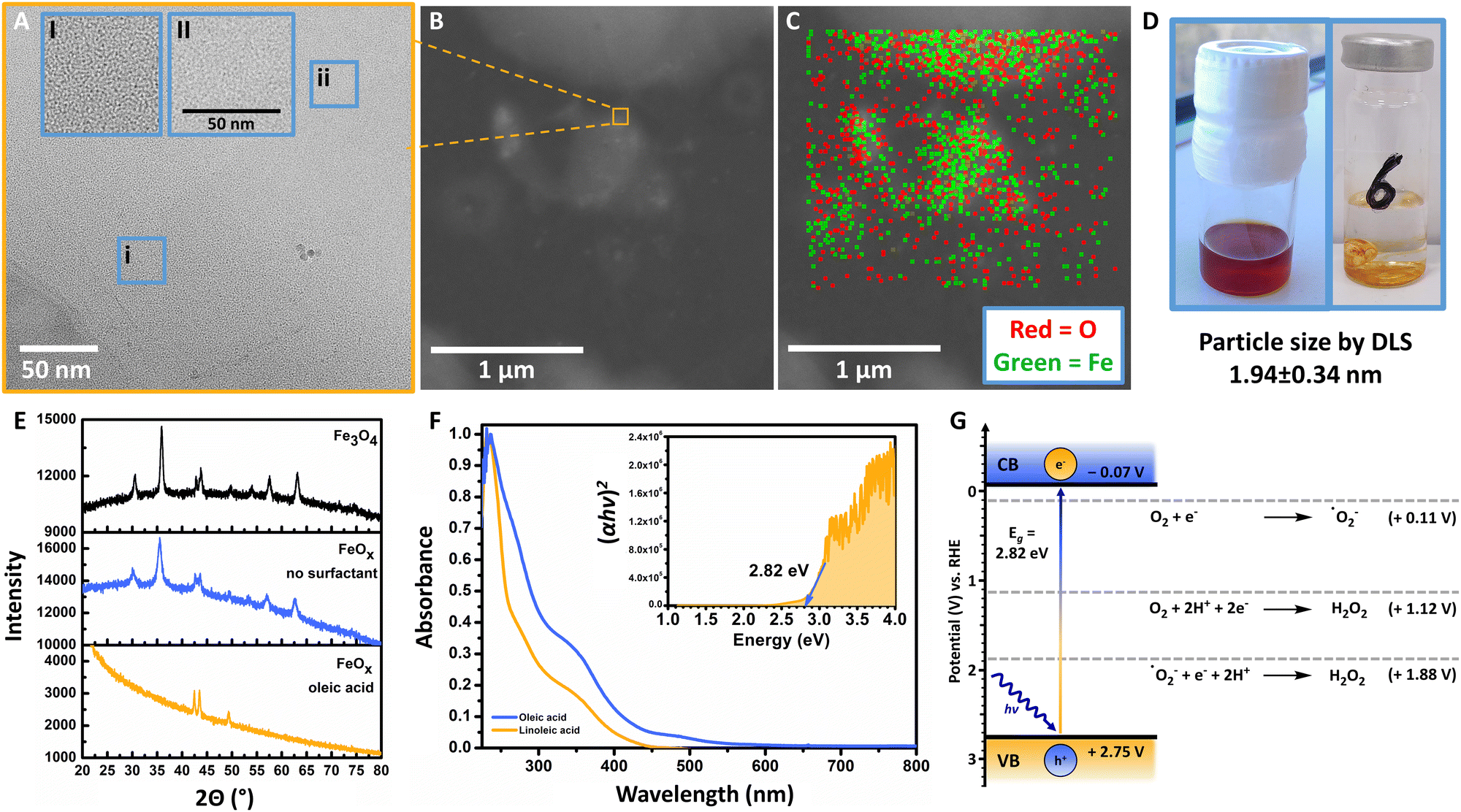 | ||
| Fig. 2 (A) Transmission electron microscopy (TEM) of FeOx (batch 131, oleic acid 2:1, Acros Organics, 1 mg mL−1 in THF), at a magnification of 100000× (inlet: zoomed); particle size by DLS 1.94 ± 0.34 nm. (B) Scanning transmission electron microscopy of FeOx (batch 131, oleic acid 2:1, 1 mg mL−1 in THF), inlet: zoom towards A. (C) EDX of FeOx (batch 131, oleic acid 2:1, 1 mg mL−1 in THF), drying spots of solvents contain more FeOx NPs and concentration decreases towards the edges of the droplets; oxygen is depicted in red – iron in green. (D) FeOx NPs stored in DCM and N2 atmosphere after synthesis (left); heterogeneous FeOx catalyst material after drying, insoluble in water for photoirradiation studies (1 mg mL−1). (E) XRD comparison between FeOx NPs with oleic acid (2:1, batch 135, Acros Organics) surfactant, without surfactant (batch 143, Acros Organics) and Fe3O4. The FeOx photocatalyst resembles Fe3O4 as indicated by the peaks in the region 40–55 degrees; peak broadening is observed due to small size and/or amorphous properties of the FeOx NP material. (F) Normalized UV-Vis absorption spectra for FeOx species possessing photochemical activity for oxygen reduction towards H2O2 (oleic acid: 0.125 mg mL−1, linoleic acid: 0.02 mg mL−1 in DCM); inlet: UV-Vis Tauc plot indicating the optical bandgap of the FeOx catalyst, where h = Planck's constant, ν = frequency of the radiation and α = absorption coefficient. The Tauc plots of different batches, solvents (THF and DCM) and heterogeneous deposition on the side of the cuvettes (AQY measurements) were compared. Conditions: batch 188 of FeOx NPs measured in DCM (c = 1 mg mL−1). (G) Energy band position of the FeOx catalyst material obtained via UV-Vis Tauc plot and electrochemical Mott-Schottky analysis via FeOx@FTO. | ||
Catalyst properties and performance
The synthesis of the FeOx NPs offered a consistent particle size of 1.94 ± 0.34 nm (DLS), which was also confirmed by TEM (Fig. 2A and B). Energy-dispersive X-ray spectroscopy allowed for the visualization of oxygen (O) and iron (Fe) on the images, indicating a higher concentration of NPs at the drying spots of the solvent (1 mg mL−1, THF, Fig. 2A and C). As previously confirmed by Xiao et al., comparing the FeOx NPs with Fe2O3 and Fe3O4via X-ray powder diffraction (XRD) revealed that the photocatalyst indeed resembles Fe3O4 (Fig. 2E). Possessing distinct peaks at 42.4°, 43.4° and 49.4° the FeOx does not resemble Fe2O3 but Fe3O4.104–106 Here, peak broadening was mainly correlated to the small size (1.94 nm) of the nanoparticles, but additionally could indicate the material being more amorphous than commercial Fe3O4 or FeOx synthesized without surfactant (Fig. 2E), which could also be observed via TEM.107,108 As depicted in Fig. 2D, the brown FeOx NPs are stable in hydrophobic solvents (DCM). This colour was retained when the heterogeneous catalyst was dried on glass for photoirradiation studies in water (1 mg mL−1, hydrophilic). Thus, the surfactant properties allow for facile catalyst and product separation from the product solution. The brown colour (absorbance up to 600 nm) of FeOx is also depicted in the UV-Vis spectrum in Fig. 2F. The Tauc plot analysis of the photocatalyst exhibits an adequate band gap in the visible-light region of 2.82 eV (Fig. 2F inlet). Mott-Schottky measurements (ESI-4.7,†) revealed that the flat-band potentials or conduction band (CB) of the FeOx NPs are positioned at −0.07 V vs. RHE. These results suggest that the catalyst material is able to facilitate indirect 2e− oxygen reduction (ORR: +0.11 V vs. RHE) via superoxide ˙O2− towards H2O2 without significant overpotential (Fig. 2G).Photoirradiation studies were generally conducted in a batch irradiation setup allowing for temperature controlled high-throughput screening of 24 conditions simultaneously (ESI-5,†).109 We opted for an Oslon SSL 80 royal blue LED (500 mW, λ = 445 nm, 180 mW cm−2) as the FeOx catalyst showed adequate absorbance in the visible-light region. After storage of the nanoparticles in DCM the catalyst was added to a vial (10 mL) to obtain a concentration of 4 mg per 4 mL solvent (generally H2O) after evaporation of DCM (Fig. 2D). The photoreactions were carried out in triplicate for 5 h in an oxygen atmosphere (20 °C) with irradiation from the bottom, where several blank reactions (including blanks in darkness) were performed in triplicate.
The general catalyst performance is depicted in Fig. 3, where successful photochemical production of H2O2 was achieved via the FeOx catalyst. Initially, an investigation of optimal catalyst loading was conducted, where 1 mg mL−1 offered the highest production of H2O2 (ESI,† Fig. S108) over 2 mg mL−1 and 0.5 mg mL−1. Thus, production is not limited to catalyst concentration, since more catalyst resulted in less production due to Fenton decomposition of hydrogen peroxide.110–112 The influence of the temperature on the photoactivity of the catalyst was investigated, where higher temperatures resulted in more production (ESI,† Fig. S113). Fig. 3A depicts the kinetics for the photochemical production of H2O2via FeOx NPs (1 h to 3 d), where production increased for the first 24 h and then stagnated for longer irradiation times (9.0 ± 0.4 mmol g−1 L−1 (5 h), 14.2 ± 1.1 mmol g−1 L−1 (20 h)). For future screenings, we opted for 5 h irradiation time at room temperature, as this yielded significant production of H2O2, which enabled quantification with peroxide test strips and titration. Furthermore, catalyst stability and recycling were also investigated, where the product solution was decanted off after each round of irradiation. We found that the catalyst could be recycled for up to four consecutive rounds (Fig. 3B, 20 h total). The catalyst material was stable for at least 6 months (including mixing of different batches) without loss of activity.
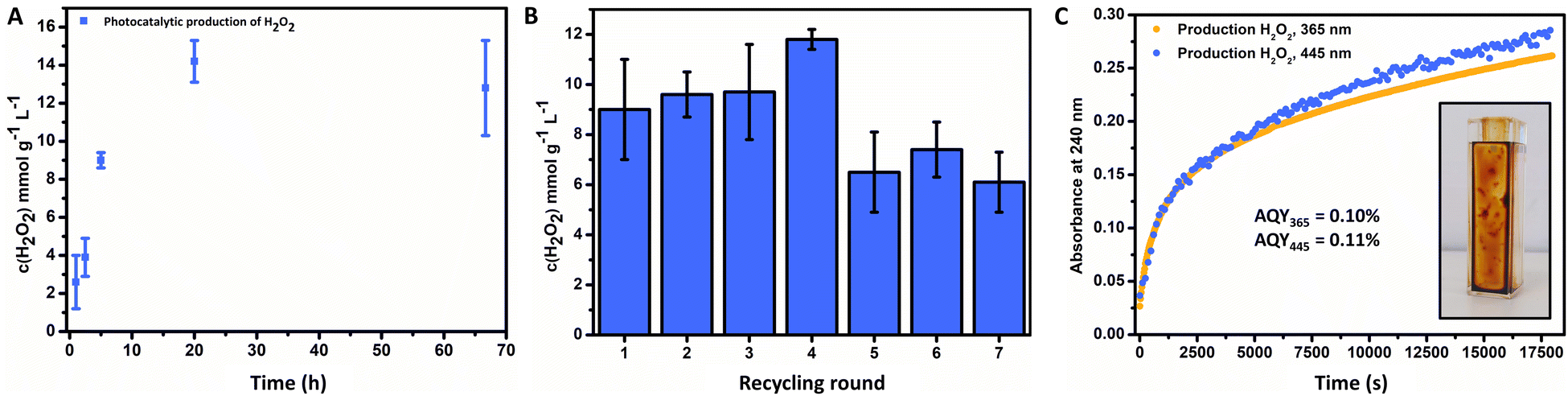 | ||
| Fig. 3 (A) Kinetics for the photochemical ORR of FeOx with oleic acid (2:1) surfactant (1 mg mL−1), obtained by irradiation with 445 nm in Milli-Q water at 20 °C. (B) Recycling of FeOx with oleic acid (2:1) surfactant (1 mg mL−1), obtained by irradiation for 5 h at 20 °C repeatedly with 445 nm in fresh Milli-Q water (30 min oxygenated) every round. (C) Production of hydrogen peroxide over time (∼5 h) upon irradiation with 445 nm and 365 nm, followed at 240 nm for apparent quantum yield (AQY) measurements; AQY365 = 0.10%, AQY445 = 0.11%. Inlet: immobilization. | ||
The apparent quantum yield (AQY) of the nanoparticles was measured via UV-Vis. After immobilization of the FeOx photocatalyst on the side of a cuvette (Fig. 3C, inlet), illumination with certain wavelengths was performed. The absorption of 240 nm was followed over time (5 h), corresponding to formed hydrogen peroxide (Fig. 3C) with an AQY365 = 0.10% and AQY445 = 0.11%. The catalyst stability towards hydrogen peroxide was explored (ESI,† Fig. S112), where subjection to a solution of H2O2 (1 mM) and irradiation (445 nm) for 5 h had no effect on the absorbance of the UV-Vis absorption spectrum, nor led to leaching of the material from the glass surface into solution. Hence the photocatalytic production of hydrogen peroxide or stronger oxidizing conditions do not have a deleterious effect on the photochemical catalyst properties. Previous studies suggested to analyse the catalyst stability for 24 h, as earlier decomposition greatly reduces practical applications, which as indicated is not the case for the FeOx NPs producing H2O2 for 3 d.68
Next, an extensive study on the quantification of H2O2 was conducted to cross-validate different quantification techniques,113–117 but also to confirm that solely H2O2 was formed, instead of other peroxides (e.g. autoxidation of unsaturated fatty acids). A method using high performance liquid chromatography-mass spectrometry (HPLC-MS) was developed, where H2O2 was found to leave the column at a retention time of 1.5 min, while amounts of oleic acid and its hydroperoxide could be separated and observed at 15–18 min (ESI-6.3,†). Utilizing cross-detection techniques of UV-DAD, MS and peroxide test strips, we confirmed a selectivity of >99% for the formation of H2O2. Thus, the formation of the allylic hydroperoxide of oleic acid and free (unligated) oleic acid was limited to trace amounts. Knowing that the photochemical oxygen reduction indeed led to H2O2 only, we further cross-validated UV-Vis iodometric quantification (ESI-6.4,†), UV-Vis of Ampliflu red in the presence of horseradish peroxidase towards resorufin (ESI-6.4,†), GC-MS (ESI-6.6,†), NMR (ESI-6.5,†), peroxide test strips (ESI-6.1,†) and iodometric titration (ESI-6.2,†).
Condition screening and mechanism studies
Only FeOx NPs with oleic acid and linoleic acid ligands were able to catalyse the photoproduction of H2O2 (Fig. 4A, 9.0 ± 0.4 mmol g−1 L−1 for oleic acid, 7.9 ± 1.9 mmol g−1 L−1 for linoleic acid, 5 h irradiation). Thus, the synthesis of a broad surfactant scope could indicate properties and allowed for predictions towards the reaction mechanism for oxygen reduction. Immobilization of the catalyst (FeOx NPs with oleic acid (2:1)) on different carbon materials (graphene, activated carbon) resulted in lowered activity, where accelerated recombination of charge carriers by enhanced conductivity could be an explanation. Utilizing oleyl alcohol or no surfactant resulted in microparticles instead of nanoparticles (ESI,† Fig. S36), indicating that small sizes of the nanoparticles (below 2 nm) enhance photoactivity through exciton transfer and quantum dot behavior.107,118–122 Nanoparticles with amines and alcohols were also found not to produce H2O2 (Fig. 4A). It is proposed that the NPs with amines or alcohols as a capping agent have different connectivity to the iron oxide surface than NPs with carboxylic acid123 and hence proper electron transfer to the active site is not warranted. Saturated fatty acids and unsaturated fatty acids with a trans double bond were all found to be inactive, suggesting a crucial role of the double bonds and their geometrical configuration (see oleic and linoleic acid) in the mechanism (Fig. 4A). No differences in the UV-Vis absorption spectrum between nanoparticles with trans-double bond elaidic and cis-double bond oleic acid could be observed, indicating no differences in its photochemical behaviour (ESI,† Fig. S83). However, elaidic acid NPs were not able to promote photocatalytic production towards hydrogen peroxide. Thus, the cis double bond of the nanoparticle bound surfactant (oleic acid and linoleic acid) is a crucial component to enable photocatalytic activity.
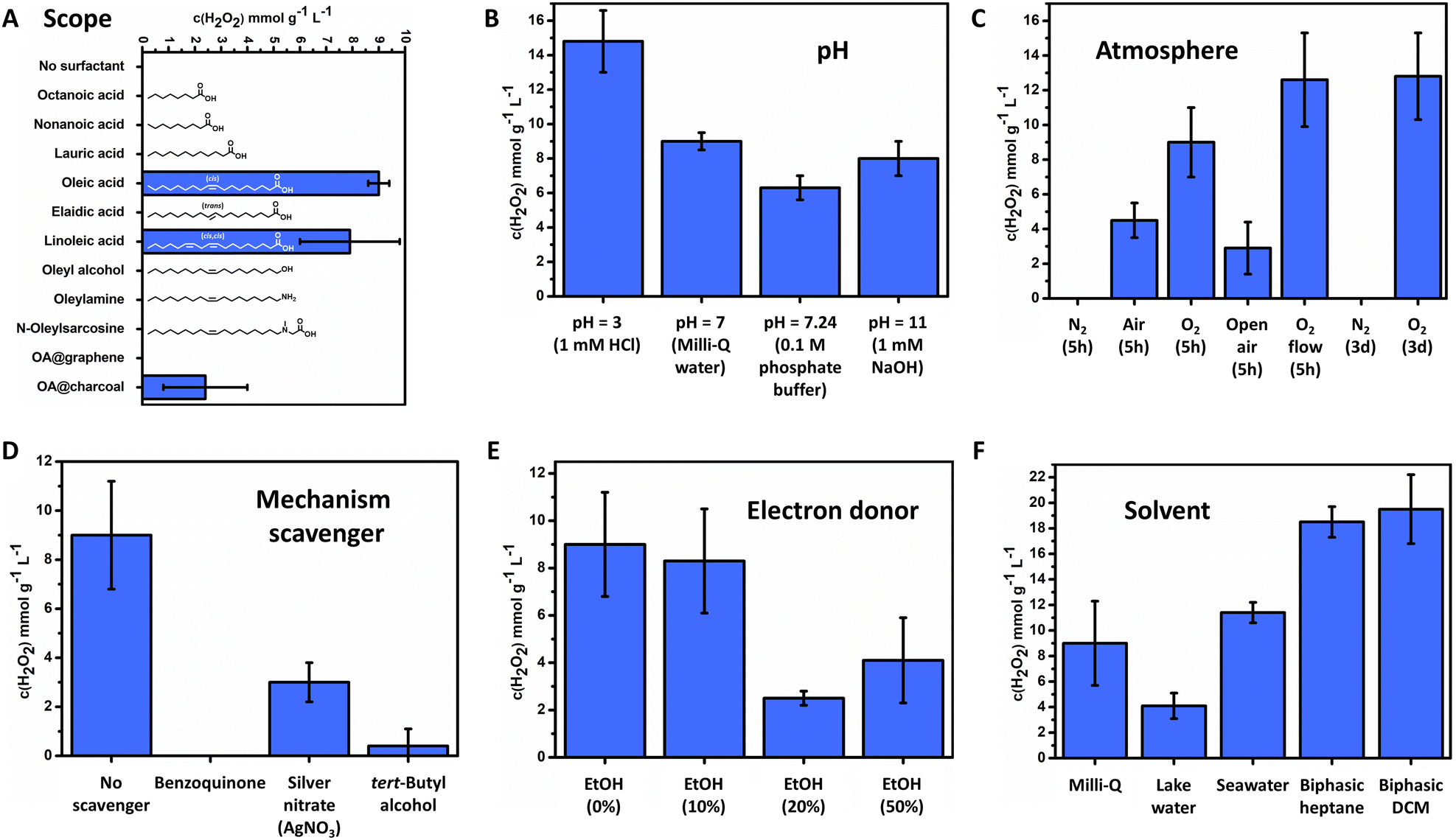 | ||
| Fig. 4 Production of H2O2 by FeOx NPs, depending on (A) surfactant type; (B) pH value; (C) different atmospheres; (D) presence of active species scavengers (12.5 mol L−1); (E) presence of electron donor ethanol; (F) solvent. General reactions conditions: FeOx NPs (1 mg mL−1), 4 mL solvent, O2 atmosphere (1 bar in 10 mL vial), LED OSRAM Oslon SSL 80 royal blue (500 mW, λ = 445 nm, 180 mW cm−2), 5 h, 293.15 K. H2O2 concentration values were obtained by iodometric redox titration. All yields are average values obtained from triplicate. | ||
Elucidating the influence of pH on the FeOx-catalyzed H2O2 photoproduction revealed that production in Milli-Q water, phosphate buffer and 1 mM NaOH resulted in similar amounts of H2O2 production (6.3–9.0 mmol g−1 L−1). Hence basic conditions or buffer solutions do not enhance or lower production. An increased production of H2O2 was obtained in an acidic environment (14.8 ± 1.8 mmol g−1 L−1 (pH = 3)). The low pH ensures more protons in solution, stabilizing H2O2 and decreasing the Fenton process, thus improving the balance between H2O2 formation and decomposition (Fig. 4B and 2G).124
The FeOx catalyst was then investigated in different atmospheres. As shown in Fig. 4C the H2O2 concentration decreased when switching from O2 (100%, 5 h, standard conditions) to air (21% O2). A nitrogen atmosphere (5 h and 3 d) resulted in no production of H2O2, which suggests that a water oxidation reaction (WOR) is not part of the FeOx catalyst system. Knowing that the FeOx material acts solely as an ORR catalyst, we envisioned open air and constant oxygen bubbling to increase the productivity. As only a slight increase in production was observed, the previous preparation method already ensured sufficient oxygen saturation and concentration. Long-term irradiation was conducted for three days, which only increased the production of H2O2 for about 1.4 times, while the irradiation time was about 13 times as long. This fact stresses the mismatch between the WOR and ORR, also suggesting that long-term irradiation studies do not necessarily lead to complete catalyst deactivation towards zero production of H2O2. The influence of stirring bars was examined by performing the reaction with Teflon and glass stirring bars added as well as without; no significant difference was observed as production was not affected by stirring or trace metals as new stirring bars were utilized (ESI,† Fig. S115). Active species trapping experiments of superoxide radicals (˙O2−), electrons (e−) and hydroxyl radicals (˙OH) by p-benzoquinone (BQ), silver nitrate (AgNO3) and tert-butyl alcohol, respectively, were conducted (Fig. 4D and ESI,† Schemes S2, S3)).70 These experiments should always be performed to check which active intermediates are participating in the H2O2 production mechanism. When AgNO3 and tert-butyl alcohol were added, H2O2 production dropped significantly, indicating the presence of electrons (e−) and hydroxyl radicals (˙OH) in the reaction pathway (3.0 ± 0.8 mmol g−1 L−1 (AgNO3), 0.4 ± 0.7 mmol g−1 L−1 (TBA)). Zero production of H2O2 was observed when BQ was present, stressing the importance of superoxide (˙O2−) and thus an indirect ORR pathway for the mechanism. These results confirm again the adequate band gap and its reaction pathway (Fig. 2G). Hence, superoxide radicals, electrons and hydroxyl radicals are all actively taking part in the mechanism of FeOx-catalysed photochemical H2O2 production.
In the literature, higher productions/productivities are often reported with the addition of sacrificial agents through filling of excess holes, produced by a photochemical mismatch in the WOR and ORR.98 Since the FeOx NPs nanoparticles were lacking the WOR, we opted for the addition of an electron donor (EtOH) to improve production and mismatch. However, as depicted in Fig. 4E, no significant increase was obtained. Other sacrificial agents investigated (methanol, isopropanol, benzyl alcohol) were also not compatible (ESI-8.2,†). An extensive evaluation of sacrificial agents was conducted by exposing sacrificial agents to air overnight, where it was found that benzyl alcohol and isopropanol produced peroxides.125–128 These findings are in line with previously reported non-innocent auto-photocatalytic oxidation of benzyl alcohol to benzaldehyde, where large quantities of H2O2 are produced upon irradiation.2,129,130 Therefore, we opted for only methanol and ethanol as trustworthy and biobased sacrificial agents. Methanol and ethanol did not inherently produce hydrogen peroxide in contact with oxygen by autoxidation.
A solvent screening was performed on biphasic systems and naturally occurring water resources, as shown in Fig. 4F. To our delight we obtained the production of H2O2 both in lake water and seawater, without any purification other than filtration (4.1 ± 1.0 mmol g−1 L−1 (lake water), 11.4 ± 0.8 mmol g−1 L−1 (seawater)). The FeOx NPs were thus able to perform the ORR in the presence of salts and other impurities present in lake and seawater.
We purposely decided against benzyl alcohol as a hole scavenger and ‘advantageous system’ for biphasic product (H2O2) separation from the active site of the catalyst, due to its inherent capability to absorb photons and undergo autoxidation (ESI-8.2,†). Heptane (0.6795 g cm−3) and DCM (1.3266 g cm−3) were chosen for biphasic systems as these do not undergo autoxidation and are immiscible with Milli-Q water (0.99705 g cm−3) (ESI-9.1,†). Significantly higher production was achieved (18.5 ± 1.2 mmol g−1 L−1 for heptane, 19.5 ± 2.7 mmol g−1 L−1 for DCM), the reason being circumvention of Fenton degradation through separation of the produced H2O2 and ˙O2− from the catalyst surface, as the catalyst dissolved in heptane and DCM while H2O2 migrated to the water layer.
Furthermore, the possibility of a cation-enhancement effect using various metals (Zn2+, Al3+, Ni2+, Fe2+, Fe3+) was investigated. Several iron salts and oxides (iron(II) sulfate heptahydrate, iron(II) chloride, iron(III) sulfate hydrate, iron(III) chloride and iron(III) nitrate nonahydrate and iron oxide (Fe2O3 and Fe3O4)) were investigated as additives resulting in zero production of H2O2 as Fenton chemistry was enhanced.131 The addition of zinc, aluminium and nickel salts to the reaction mixture was found to be tolerated for performing the ORR by the FeOx photocatalyst (5.2–8.3 mmol g−1 L−1). Interestingly, the addition of aluminium oxide (Al2O3) resulted in significantly increased production (15.5 ± 4.3 mmol g−1 L−1). Addition of pure sodium salts (in contrast to the salt mixtures in seawater) was found to significantly reduce production, probably through poisoning of active sites (ESI-9,†).132 In direct synthesis, these sodium salts are usually added in order to improve selectivity presumably by blocking of sites for O–O cleavage.133 This effect was however not found for the iron oxide nanoparticles.
Proposed photochemical oxygen reduction mechanism
After having established that the FeOx photocatalyst exclusively promotes oxygen reduction towards H2O2, we confirmed those findings further via Headspace GC-TCD (thermal conductivity detector). Neither O2 nor H2 formation was observed, indicating the absence of proton reduction as well as water oxidation for our catalyst system (ESI-8.1,†).XPS analysis revealed that the electrons for the photochemical oxygen reduction are provided by the FeOx catalyst material, as depicted in Table S10 (ESI†) and Fig. 5A. Fe2+ (2p photoemission line peaked at binding energy (BE) of 711.7 eV) species are oxidized to Fe3+ (BE = 714.3 eV) over time, transforming the Fe3O4 resembling catalyst (BE = 709.9 eV) into Fe2O3 (BE = 711.1 eV) (Fig. 5A).134–137 These observations are also corroborated by the loss of magnetism over consecutive rounds of catalyst recycling until deactivation (Fig. 3B). The cis double bond, playing a role as a co-catalyst, is also oxidized over time (C 1s peak at a BE of 286.5 eV, ESI,† Fig. S120). The general oxygen ratio is increasing due to oxidative conditions (ESI,† Fig. S119).134,138,139 Thus, the catalyst becomes inactive after four rounds of catalysis, i.e. ORR, as holes generated are not filled by WOR or sacrificial agents (Fig. 4C and E). Catalyst reactivation was attempted by reattaching new surfactant molecules but proved to be ineffective. Future research will focus on the implementation of the FeOx photocatalyst as a photoelectrodic material to not only achieve higher oxygen reduction rates to H2O2 but also avoid oxidation.89,140–142
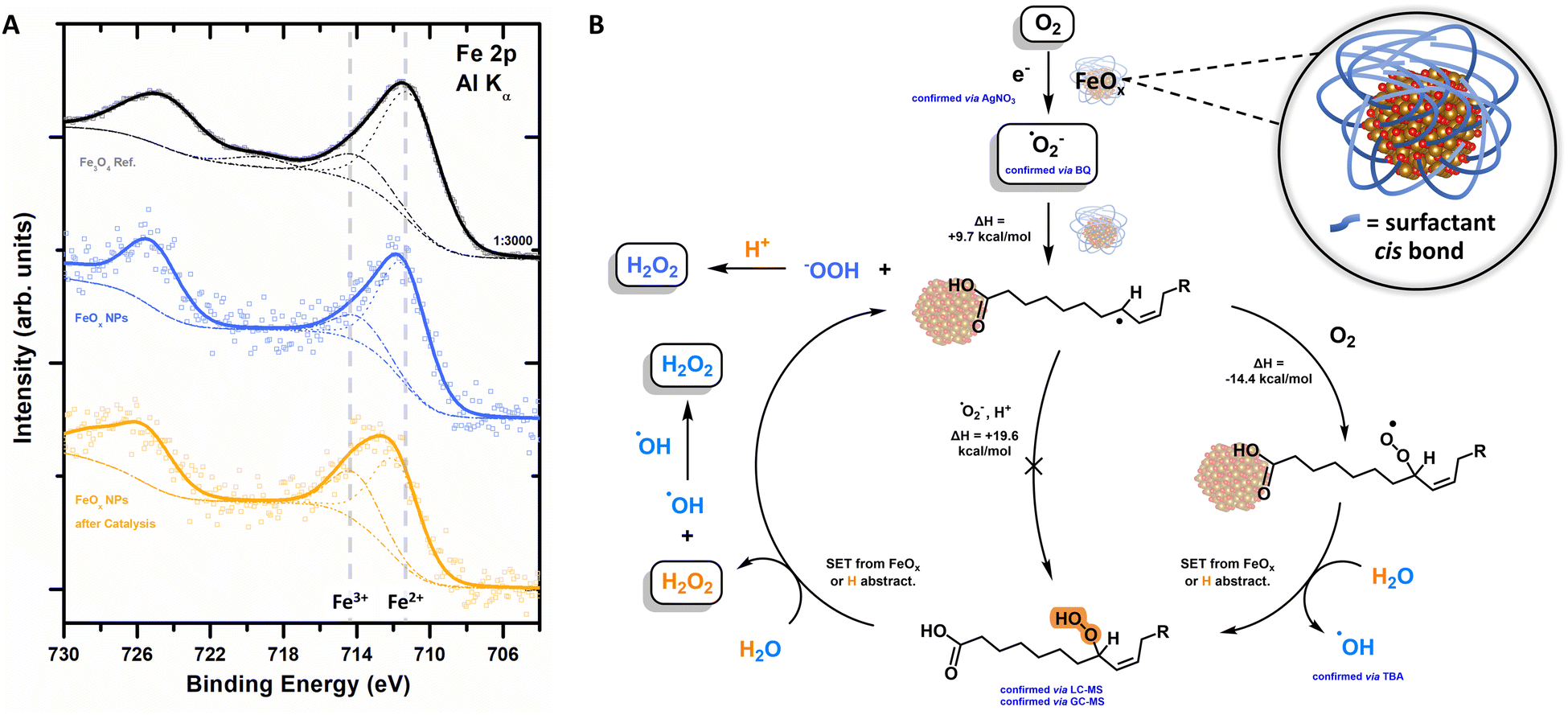 | ||
| Fig. 5 (A) XPS spectra of the Fe 2p core level region of the as-synthesised nanoparticles, black: FeOx without surfactant, blue: FeOx NPs before photochemical production of H2O2, orange: inactive FeOx NPs after 8 consecutive rounds of catalysis. (B) Proposed mechanism for FeOx promoted photochemical oxygen reduction to H2O2; inset: proposed active site. Energy barriers are calculated without the effect of the FeOx core. | ||
The extensive studies of the process led to the proposed mechanism depicted in Fig. 5B, which exhibits similar characteristics as the peroxidase and cyclooxygenase reaction of arachidonic acid (AA, cis) to Prostaglandin G2.143–146 H2O2 is produced photochemically with a selectivity of >99% (LC-MS) via the oxygen reduction reaction (ORR, atmosphere experiments).
Active species trapping experiments with AgNO3 indicate the presence e− (Fig. 4D), which are formed by photoexcitation of the photocatalyst (Fig. 2G). With these electrons, oxygen undergoes an indirect reduction mechanism towards superoxide ˙O2− (trapping experiments with BQ, Fig. 4D). The superoxide is able to attack the surfactant at the double bond forming first an allylic radical, followed by trapping of this allylic radical with molecular triplet oxygen to form an allylic peroxyl radical (Fig. 5B). A (i) subsequent single electron transfer (SET) event with either the FeOx core or with the solvated electrons; or (ii) subsequent hydrogen abstraction reaction of this peroxyl radical with water, both yield the hydroperoxide (intermediate trace amounts, confirmed by LC-MS and GC-MS) and hydroxyl radicals whose presence was confirmed by active species trapping with tert-butyl alcohol (Fig. 4D). The final cleavage of the hydroperoxide from the surfactant and regeneration of the catalytically active fatty acid allyl radical is also thought to proceed via either (i) SET event or (ii) hydrogen abstraction from the water solvent (calculated by density functional theory (DFT), ESI-8.5 and ESI-8.6,†). The presence of iron is crucial for the catalytic cycle proposed in Fig. 5B, indicating an active role of iron for adsorption and subsequent desorption from the catalyst surface.147,148 The active site (Fig. 5B inset) therefore seems to consist of iron oxide (Fe3O4, Feoct2+) connected to a carboxylic acid, which in proximity of the cis double bond forms a hydrophobic pocket favourable for oxygen affinity.87,149,150 Here, protons could possibly be supplied by (carboxylic) acids or water (Fig. 4B), while electron transfer is feasible from iron via its connectivity with the surfactant.151
From XRD and DLS measurements (ESI,† Table S1) it was found that particle sizes smaller than 2 nm (below the exciton Bohr radius) with a certain crystallinity were necessary for photoactivity through exciton transfer, while also TEM confirmed these small and round particles.118–120,152 This interplay between factors seems to be crucial for photochemical oxygen reduction activity towards hydrogen peroxide via FeOx.
Applications and in situ oxidations
After elucidating the catalytic properties, the oxygen reduction mechanism and the conditions for optimal photochemical production of hydrogen peroxide, we applied the FeOx NPs in several organic transformations and real-life conditions.Styrene is normally polymerized via free radical polymerization techniques initiated by the addition of a radical initiator (e.g. benzoyloxy peroxide).153 The O–O bond is cleaved at elevated temperatures when a peroxide initiator is used to form radicals for initiation (ESI-10.1,†). In situ polymerization of styrene (0.4 g, distilled) in methanol (2 mL) with iron oxide nanoparticles (4 mg) was possible after three days of irradiation by blue light (445 nm) (Scheme 1A) as indicated by the formation of a polystyrene solid in the reaction vial (ESI,† Scheme S5). This observation shows that hydrogen peroxide was formed and subsequently decomposed via photo-Fenton reactions towards radical species; these formed radical species were finally able to polymerize styrene. The polymerization reaction was slow as the polymer was only observed after three days. Oxygen present in the vial is known to inhibit free radical polymerization by reaction with active radicals.154 Other products formed were benzaldehyde, styrene oxide, benzene acetaldehyde and a dimethyl acetal as confirmed by GC-MS. A nucleophilic addition reaction was conducted on furfural (30 μL, 362 μmol) in oxygenated methanol (4 mL) by irradiation with 445 nm light catalysed by FeOx NPs (4 mg) (Scheme 1B). After 60 h, quantitative production of the dimethyl acetal of furfural was observed via1H-NMR (ESI,† Fig. S124). This reaction confirmed the absence of singlet oxygen in the mechanism as O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O would have led to the formation of hydroxybutenolide via [4+2] cycloaddition.82 Interestingly, with 17046 mmol g−1 L−1 of dimethyl acetal formed, a 1812× increase of H2O2 production was achieved (ESI-10.2,†), demonstrating that performing in situ reactions results in higher productivities because Fenton-decomposition is circumvented through direct reaction with organic substrates. An in situ oxidation reaction of α-terpinene (0.15 mL) in oxygenated isopropanol (5 mL) was catalysed by FeOx NPs (5 mg) when irradiating with 445 nm light (Scheme 1C). Also here, no singlet oxygen was formed, as there were no [4+2] cycloaddition products identified.80 The products of the in situ oxidations catalysed by FeOx NPs consisted of epoxides, ketones, ethers and alcohols. In total 7377 mmol g−1 L−1 of products were formed, which compared to previously investigated hydrogen peroxide production (5 h, 9.4 ± 1.3 mmol g−1 L−1) is a 784× increase concentration-wise. Again, in all these application reactions only benign solvents, room temperature and photoirradiation were utilized as sustainable conditions.155In situ oxidation of o-tolidene was also attempted as a quantification method via1H-NMR; however too little hydrogen peroxide was produced, not reaching the detection limit (ESI,† Fig. S107).
O would have led to the formation of hydroxybutenolide via [4+2] cycloaddition.82 Interestingly, with 17046 mmol g−1 L−1 of dimethyl acetal formed, a 1812× increase of H2O2 production was achieved (ESI-10.2,†), demonstrating that performing in situ reactions results in higher productivities because Fenton-decomposition is circumvented through direct reaction with organic substrates. An in situ oxidation reaction of α-terpinene (0.15 mL) in oxygenated isopropanol (5 mL) was catalysed by FeOx NPs (5 mg) when irradiating with 445 nm light (Scheme 1C). Also here, no singlet oxygen was formed, as there were no [4+2] cycloaddition products identified.80 The products of the in situ oxidations catalysed by FeOx NPs consisted of epoxides, ketones, ethers and alcohols. In total 7377 mmol g−1 L−1 of products were formed, which compared to previously investigated hydrogen peroxide production (5 h, 9.4 ± 1.3 mmol g−1 L−1) is a 784× increase concentration-wise. Again, in all these application reactions only benign solvents, room temperature and photoirradiation were utilized as sustainable conditions.155In situ oxidation of o-tolidene was also attempted as a quantification method via1H-NMR; however too little hydrogen peroxide was produced, not reaching the detection limit (ESI,† Fig. S107).
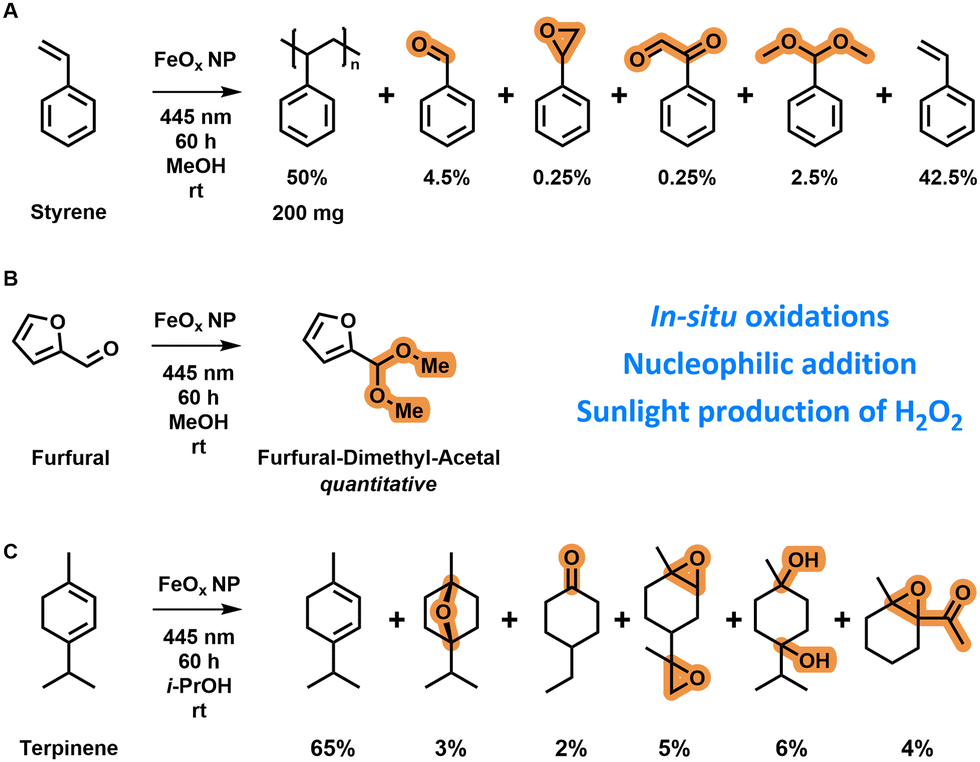 | ||
| Scheme 1 (A) Oxidation and polymerization of styrene (0.4 g, distilled) in methanol (2 mL) after 60 h of light irradiation (445 nm) in the presence of FeOx NPs (4 mg). (B) Nucleophilic addition reaction on furfural (30 μL, 362 μmol) to its dimethyl acetal after 60 h in oxygenated methanol (4 mL) by irradiation of FeOx NPs (4 mg) at 445 nm. (C) In situ oxidation of α-terpinene (0.15 mL) towards a mixture of products after 60 h in oxygenated isopropanol (5 mL) by irradiation of FeOx NPs (5 mg) at 445 nm. Product ratios based on GC-MS and 1H-NMR. | ||
A long-term irradiation experiment in real sunlight was conducted for 1 week. This was performed by putting a sample in a window, where it had approximately 8 h of sunlight daily (performed in September facing south in Groningen, NL, ESI-5.3,†). Photochemical production of H2O2 was also possible by 1 week irradiation by sunlight. Thus, ultimately a very sustainable and interesting process was achieved: iron oxide promoted photochemical oxygen reduction to hydrogen peroxide from sunlight in seawater.
Conclusion
Here, we established a strategy for the photochemical production of H2O2 catalysed by iron. Following the 12 principles of Green Chemistry we developed a sustainable synthesis for an entire scope of earth-abundant iron oxide nanoparticles terminated with biobased fatty acid surfactants. These materials were fully characterized and investigated for the photochemical production of H2O2. Specifically, FeOx NPs with fatty acid ligands possessing cis double bonds confer photoactivity for oxygen reduction to hydrogen peroxide. An extensive study for peroxide quantification revealed a selectivity of >99% for H2O2. Acting as an oxygen reduction material, the synthesized nanoparticles could be recycled for up to four consecutive rounds of 5 h irradiation. Through detailed experimental investigation of the catalyst properties and computational results, a mechanism and an active site were proposed for photochemical hydrogen peroxide production (Fig. 5). The H2O2 yield could be increased by decreasing the pH, addition of cation exchangers and by production in biphasic systems (heptane/DCM with Milli-Q water) (up to 19.5 ± 2.7 mmol g−1 L−1). Here a lower pH supplied protons, aluminium oxide facilitated electron transfer and biphasic systems circumvented Fenton-decomposition by separation of the product from the catalyst. The iron oxide nanoparticles with oleic acid (2:1) as a surfactant were also successfully utilized in other applications like wastewater treatment, polymerizations and in situ oxidations.
Crucially, the FeOx NPs could be synthesized from biobased and abundant materials. Oleic acid is the main component in olive oils, ethanol is obtained from biomass and iron is the most abundant metal in the earth's crust. An apparent quantum yield (AQY) of 0.11% was achieved for photochemical oxygen reduction to H2O2 at ambient temperatures and pressures upon irradiation of the FeOx catalyst with visible light (445 nm; 9.4–14.8 mmol g−1 L−1). Productivities to H2O2 of at least 1.7 ± 0.3 mmol g−1 L−1 h−1 were obtained. Production of H2O2 was possible via sunlight irradiation and in seawater.
This study demonstrates the importance of the development of sustainable catalyst materials if the replacement of outdated industrial processes is the ultimate goal. Utilizing earth-abundant metals and biobased (co-)catalysts offers great potential for the photocatalytic production of hydrogen peroxide as a solar fuel. The results presented here may open a new avenue for designing suitable and green photocatalysts for efficient H2O2 production from solar energy.
Author contributions
B. L. F. conceptualized the research project and coordinated it with the help of J. S.. T. F. and J. T. M. conducted the research and its validation. M. B. B., M. C. A. S., R. T. and D. G. conducted validation experiments with input from P. R. and J. N. H. R.. G. A. performed the DFT studies. T. F. and B. L. F. prepared the manuscript with input from J. T. M., M. B. B., G. A.. All authors reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank P. P. Pescarmona and M. Miola for critical assessment and discussions. Funding: this work is in collaboration with Shell and part of the Advanced Research Center for Chemical Building Blocks, ARC CBBC, which is cofounded and co-financed by the Netherlands Organization for Scientific Research (NWO, contract 736.000.000) and the Netherlands Ministry of Economic Affairs and Climate. We are grateful for the generous financial support to B. L. F. (the Ministry of Education, Culture and Science of the Netherlands Gravitation Programme no. 024.001.035), to G. A. (EMBO LTF232-2020 Postdoctoral Fellowship). Financial support from the Advanced Materials research programme of the Zernike National Research Centre under the Bonus Incentive Scheme of the Dutch Ministry for Education, Culture and Science is gratefully acknowledged.Notes and references
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- T. Freese, J. T. Meijer, B. L. Feringa and S. B. Beil, Nat. Catal., 2023, 6, 553–558 CrossRef CAS.
- X. Zeng, Y. Liu, X. Hu and X. Zhang, Green Chem., 2021, 23, 1466–1494 RSC.
- Y. Ding, S. Maitra, S. Halder, C. Wang, R. Zheng, T. Barakat, S. Roy, L. H. Chen and B. L. Su, Matter, 2022, 5, 2119–2167 CrossRef CAS.
- W. Yu, C. Hu, L. Bai, N. Tian, Y. Zhang and H. Huang, Nano Energy, 2022, 104, 107906 CrossRef CAS.
- Y. Guo, X. Tong and N. Yang, Nanomicro Lett., 2023, 15, 77 CAS.
- H. Hou, X. Zeng and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 17356–17376 CrossRef CAS PubMed.
- Y. Xue, Y. Wang, Z. Pan and K. Sayama, Angew. Chem., Int. Ed., 2021, 60, 10469–10480 CrossRef CAS PubMed.
- Y. Sun, L. Han and P. Strasser, Chem. Soc. Rev., 2020, 49, 6605–6631 RSC.
- S. Zaman, L. Huang, A. I. Douka, H. Yang, B. You and B. Y. Xia, Angew. Chem., Int. Ed., 2021, 60, 17832–17852 CrossRef CAS PubMed.
- S. Fukuzumi, Y. M. Lee and W. Nam, Chem. – Eur. J., 2018, 24, 5016–5031 CrossRef CAS PubMed.
- K. Mase, M. Yoneda, Y. Yamada and S. Fukuzumi, Nat. Commun., 2016, 7, 11470 CrossRef CAS PubMed.
- Y. Yamada, M. Yoneda and S. Fukuzumi, Energy Environ. Sci., 2015, 8, 1698–1701 RSC.
- C. J. McDonnell-Worth and D. R. MacFarlane, Aust. J. Chem., 2018, 71, 781–788 CrossRef CAS.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- P. Zhou, I. A. Navid, Y. Ma, Y. Xiao, P. Wang, Z. Ye, B. Zhou, K. Sun and Z. Mi, Nature, 2023, 613, 66–70 CrossRef CAS PubMed.
- I. Staffell, D. Scamman, A. Velazquez Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC.
- J. Andersson and S. Grönkvist, Int. J. Hydrogen Energy, 2019, 44, 11901–11919 CrossRef CAS.
- Y. Shiraishi, T. Takii, T. Hagi, S. Mori, Y. Kofuji, Y. Kitagawa, S. Tanaka, S. Ichikawa and T. Hirai, Nat. Mater., 2019, 18, 985–993 CrossRef CAS PubMed.
- A. Gopakumar, P. Ren, J. Chen, B. V. Manzolli Rodrigues, H. Y. Vincent Ching, A. Jaworski, S. Van Doorslaer, A. Rokicińska, P. Kuśtrowski, G. Barcaro, S. Monti, A. Slabon and S. Das, J. Am. Chem. Soc., 2022, 144, 2603–2613 CrossRef CAS PubMed.
- A. Klerke, C. H. Christensen, J. K. Nørskov and T. Vegge, J. Mater. Chem., 2008, 18, 2304–2310 RSC.
- J. Eppinger and K. W. Huang, ACS Energy Lett., 2017, 2, 188–195 CrossRef CAS.
- K. Sordakis, C. Tang, L. K. Vogt, H. Junge, P. J. Dyson, M. Beller and G. Laurenczy, Chem. Rev., 2018, 118, 372–433 CrossRef CAS PubMed.
- L. Piccirilli, B. Rabell, R. Padilla, A. Riisager, S. Das and M. Nielsen, J. Am. Chem. Soc., 2023, 145, 5655–5663 CrossRef CAS PubMed.
- X. Fu, J. B. Pedersen, Y. Zhou, M. Saccoccio, S. Li, R. Sažinas, K. Li, S. Z. Andersen, A. Xu, N. H. Deissler, J. Bjarke, V. Mygind, C. Wei, J. Kibsgaard, P. C. K. Vesborg, J. K. Nørskov and I. Chorkendorff, Science, 2023, 379, 707–712 CrossRef CAS PubMed.
- C. Ampelli, D. Giusi, M. Miceli, T. Merdzhanova, V. Smirnov, U. Chime, O. Astakhov, A. J. Martín, F. L. P. Veenstra, F. A. G. Pineda, J. González-Cobos, M. García-Tecedor, S. Giménez, W. Jaegermann, G. Centi, J. Pérez-Ramírez, J. R. Galán-Mascarós and S. Perathoner, Energy Environ. Sci., 2023, 16, 1644–1661 RSC.
- S. C. D’Angelo, A. J. Martín, S. Cobo, D. F. Ordóñez, G. Guillén-Gosálbez and J. Pérez-Ramírez, Energy Environ. Sci., 2023, 16, 3314–3330 RSC.
- Y. Chen, P. Ammari-Azar, H. Liu, J. Lee, Y. Xi, M. J. Castellano, S. Gu and W. Li, EES Catal., 2023, 1, 504–515 RSC.
- Y. Ni, Z. Han, Y. Chai, G. Wu and L. Li, EES Catal., 2023, 1, 459–494 RSC.
- R. S. Disselkamp, Int. J. Hydrogen Energy, 2010, 35, 1049–1053 CrossRef CAS.
- Y. Zhang, C. Pan, G. Bian, J. Xu, Y. Dong, Y. Zhang, Y. Lou, W. Liu and Y. Zhu, Nat. Energy, 2023, 8, 361–371 CrossRef CAS.
- AR6 Synthesis Report: Climate Change 2023, https://www.ipcc.ch/report/ar6/syr/, (accessed 21 March 2023).
- H. Cheng, J. Cheng, L. Wang and H. Xu, Chem. Mater., 2022, 34(10), 4259–4273 CrossRef CAS.
- Hydrogen Peroxide Market Size, Share, Demand, Analysis, Report 2023–2028, https://www.expertmarketresearch.com/reports/hydrogen-peroxide-market, (accessed 20 March 2023).
- R. L. Myers, The 100 most important chemical compounds: a reference guide, Greenwood Publishing Group: Westport, CT, 2007 Search PubMed.
- R. Ciriminna, L. Albanese, F. Meneguzzo and M. Pagliaro, ChemSusChem, 2016, 9, 3374–3381 CrossRef CAS PubMed.
- G. Goor, J. Glenneberg, S. Jacobi, J. Dadabhoy and E. Candido, Ullmann's Encyclopedia of Industrial Chemistry, Wiley, 2019, pp. 1–40 Search PubMed.
- S. C. Perry, D. Pangotra, L. Vieira, L. I. Csepei, V. Sieber, L. Wang, C. Ponce de León and F. C. Walsh, Nat. Rev. Chem., 2019, 3, 442–458 CrossRef CAS.
- D. Zhao, Z. Zhuang, X. Cao, C. Zhang, Q. Peng, C. Chen and Y. Li, Chem. Soc. Rev., 2020, 49, 2215–2264 RSC.
- K. Jiang, S. Back, A. J. Akey, C. Xia, Y. Hu, W. Liang, D. Schaak, E. Stavitski, J. K. Nørskov, S. Siahrostami and H. Wang, Nat. Commun., 2019, 10, 3997 CrossRef PubMed.
- J. S. Adams, M. L. Kromer, J. Rodríguez-López and D. W. Flaherty, J. Am. Chem. Soc., 2021, 143, 7940–7957 CrossRef CAS PubMed.
- N. M. Wilson, P. Priyadarshini, S. Kunz and D. W. Flaherty, J. Catal., 2018, 357, 163–175 CrossRef.
- D. W. Flaherty, ACS Catal., 2018, 8, 1520–1527 CrossRef CAS.
- J. K. Edwards, B. Solsona, N. Edwin Ntainjua, A. F. Carley, A. A. Herzing, C. J. Kiely and G. J. Hutchings, Science, 2009, 323, 1037–1041 CrossRef CAS PubMed.
- S. J. Freakley, Q. He, J. H. Harrhy, L. Lu, D. A. Crole, D. J. Morgan, E. N. Ntainjua, J. K. Edwards, A. F. Carley, A. Y. Borisevich, C. J. Kiely and G. J. Hutchings, Science, 2016, 351, 965–968 CrossRef CAS PubMed.
- J. K. Edwards, S. J. Freakley, R. J. Lewis, J. C. Pritchard and G. J. Hutchings, Catal. Today, 2015, 248, 3–9 CrossRef CAS.
- L. F. De Freitas, B. Puértolas, J. Zhang, B. Wang, A. S. Hoffman, S. R. Bare, J. Pérez-Ramírez, J. W. Medlin and E. Nikolla, ACS Catal., 2020, 10, 5202–5207 CrossRef.
- G. M. Lari, B. Puértolas, M. Shahrokhi, N. López and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2017, 56, 1775–1779 CrossRef CAS PubMed.
- F. E. Osterloh, ACS Energy Lett., 2017, 2, 445–453 CrossRef CAS.
- S. Kato, J. Jung, T. Suenobu and S. Fukuzumi, Energy Environ. Sci., 2013, 6, 3756–3764 RSC.
- Y. Wang, G. I. N. Waterhouse, L. Shang and T. Zhang, Adv. Energy Mater., 2021, 11, 2003323 CrossRef CAS.
- X. Wang, S. Blechert and M. Antonietti, ACS Catal., 2012, 2, 1596–1606 CrossRef CAS.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef CAS PubMed.
- T. Liu, Z. Pan, J. J. M. Vequizo, K. Kato, B. Wu, A. Yamakata, K. Katayama, B. Chen, C. Chu and K. Domen, Nat. Commun., 2022, 13, 1034 CrossRef CAS PubMed.
- Z. Zhang, T. Tsuchimochi, T. Ina, Y. Kumabe, S. Muto, K. Ohara, H. Yamada, S. L. Ten-no and T. Tachikawa, Nat. Commun., 2022, 13, 1499 CrossRef CAS PubMed.
- D. Tsukamoto, A. Shiro, Y. Shiraishi, Y. Sugano, S. Ichikawa, S. Tanaka and T. Hirai, ACS Catal., 2012, 2, 599–603 CrossRef CAS.
- H. Tada, Nanoscale Adv., 2019, 1, 4238–4245 RSC.
- C. Chu, D. Huang, Q. Zhu, E. Stavitski, J. A. Spies, Z. Pan, J. Mao, H. L. Xin, C. A. Schmuttenmaer, S. Hu and J. H. Kim, ACS Catal., 2019, 9, 626–631 CrossRef CAS.
- H. Tada, M. Teranishi and S. I. Naya, J. Phys. Chem. C, 2023, 127, 5199–5209 CrossRef CAS.
- Y. Isaka, Y. Kondo, Y. Kawase, Y. Kuwahara, K. Mori and H. Yamashita, Chem. Commun., 2018, 54, 9270–9273 RSC.
- Y. Kondo, Y. Kuwahara, K. Mori and H. Yamashita, Chem, 2022, 8, 2924–2938 CAS.
- Y. Kondo, K. Honda, Y. Kuwahara, K. Mori, H. Kobayashi and H. Yamashita, ACS Catal., 2022, 12, 14825–14835 CrossRef CAS.
- Q. Wu, J. Cao, X. Wang, Y. Liu, Y. Zhao, H. Wang, Y. Liu, H. Huang, F. Liao, M. Shao and Z. Kang, Nat. Commun., 2021, 12, 483 CrossRef CAS PubMed.
- Y. Zhang, C. Pan, G. Bian, J. Xu, Y. Dong, Y. Zhang, Y. Lou, W. Liu and Y. Zhu, Nat. Energy, 2023, 8, 361–371 CrossRef CAS.
- Z. Wei, M. Liu, Z. Zhang, W. Yao, H. Tan and Y. Zhu, Energy Environ. Sci., 2018, 11, 2581–2589 RSC.
- X. Zhang, P. Ma, C. Wang, L. Gan, X. Chen, P. Zhang, Y. Wang, H. Li, L. Wang, X. Zhou and K. Zheng, Energy Environ. Sci., 2022, 15, 830–842 RSC.
- Y. Shiraishi, M. Matsumoto, S. Ichikawa, S. Tanaka and T. Hirai, J. Am. Chem. Soc., 2021, 143, 12590–12599 CrossRef CAS PubMed.
- L. Liu, M. Y. Gao, H. Yang, X. Wang, X. Li and A. I. Cooper, J. Am. Chem. Soc., 2021, 143, 19287–19293 CrossRef CAS PubMed.
- X. Xu, R. Sa, W. Huang, Y. Sui, W. Chen, G. Zhou, X. Li, Y. Li and H. Zhong, ACS Catal., 2022, 12, 12954–12963 CrossRef CAS.
- W. Zhao, P. Yan, B. Li, M. Bahri, L. Liu, X. Zhou, R. Clowes, N. D. Browning, Y. Wu, J. W. Ward and A. I. Cooper, J. Am. Chem. Soc., 2022, 144, 9902–9909 CrossRef CAS PubMed.
- M. Kou, Y. Wang, Y. Xu, L. Ye, Y. Huang, B. Jia, H. Li, J. Ren, Y. Deng, J. Chen, Y. Zhou, K. Lei, L. Wang, W. Liu, H. Huang and T. Ma, Angew. Chem., Int. Ed., 2022, 61, e202200413 CrossRef CAS PubMed.
- S. Chai, X. Chen, X. Zhang, Y. Fang, R. S. Sprick and X. Chen, Environ. Sci.: Nano, 2022, 9, 2464–2469 RSC.
- C. Shao, Q. He, M. Zhang, L. Jia, Y. Ji, Y. Hu, Y. Li, W. Huang and Y. Li, Chin. J. Catal., 2023, 46, 28–35 CrossRef CAS.
- P.-I. Dassie, R. Haddad, M. Lenez, A. Chaumonnot, M. Boualleg, P. Legriel, A. Styskalik, B. Haye, M. Selmane, D. P. Debecker, C. Sanchez, C. Chaneac and C. Boissiere, Green Chem., 2023, 25, 2800–2814 RSC.
- THE 17 GOALS|Sustainable Development, https://sdgs.un.org/goals, (accessed 21 March 2023).
- J. B. Zimmerman, P. T. Anastas, H. C. Erythropel and W. Leitner, Science, 2020, 367, 397–400 CrossRef CAS PubMed.
- R. Shan, L. Lu, Y. Shi, H. Yuan and J. Shi, Energy Convers. Manage., 2018, 178, 277–289 CrossRef CAS.
- Y. Zhu, Z. Li and J. Chen, Green Energy Environ., 2019, 4, 210–244 CrossRef.
- D. Faust Akl, D. Poier, S. C. D’Angelo, T. P. Araújo, V. Tulus, O. V. Safonova, S. Mitchell, R. Marti, G. Guillén-Gosálbez and J. Pérez-Ramírez, Green Chem., 2022, 24, 6879–6888 RSC.
- J. G. H. Hermens, T. Freese, G. Alachouzos, M. L. Lepage, K. J. van den Berg, N. Elders and B. L. Feringa, Green Chem., 2022, 24, 9772–9780 RSC.
- J. G. H. Hermens, A. Jensma and B. L. Feringa, Angew. Chem., Int. Ed., 2022, 61, e202112618 CrossRef CAS PubMed.
- J. G. H. Hermens, T. Freese, K. J. Van Den Berg, R. Van Gemert and B. L. Feringa, Sci. Adv., 2020, 6, 26–42 Search PubMed.
- J. G. H. Hermens, M. L. Lepage, A. Kloekhorst, E. Keller, R. Bloem, M. Meijer and B. L. Feringa, React. Chem. Eng., 2022, 7, 2280–2284 RSC.
- T. Freese, B. Fridrich, S. Crespi, A. S. Lubbe, K. Barta and B. L. Feringa, Green Chem., 2022, 24, 3689–3696 RSC.
- Visualizing the Abundance of Elements in the Earth's Crust, https://www.visualcapitalist.com/visualizing-the-abundance-of-elements-in-the-earths-crust/, (accessed 21 March 2023).
- D. F. Akl, G. Giannakakis, A. Ruiz-Ferrando, M. Agrachev, J. D. Medrano-García, G. Guillén-Gosálbez, G. Jeschke, A. H. Clark, O. V. Safonova, S. Mitchell, N. López and J. Pérez-Ramírez, Adv. Mater., 2023, 2211464, DOI:10.1002/adma.202211464.
- Y. Xiao, J. Hong, X. Wang, T. Chen, T. Hyeon and W. Xu, J. Am. Chem. Soc., 2020, 142, 13201–13209 CrossRef CAS PubMed.
- W. R. P. Barros, Q. Wei, G. Zhang, S. Sun, M. R. V. Lanza and A. C. Tavares, Electrochim. Acta, 2015, 162, 263–270 CrossRef CAS.
- J. W. Jang, C. Du, Y. Ye, Y. Lin, X. Yao, J. Thorne, E. Liu, G. McMahon, J. Zhu, A. Javey, J. Guo and D. Wang, Nat. Commun., 2015, 6, 7447 CrossRef PubMed.
- B. H. R. Suryanto, Y. Wang, R. K. Hocking, W. Adamson and C. Zhao, Nat. Commun., 2019, 10, 5599 CrossRef CAS PubMed.
- M. Huck, L. Ring, K. Küpper, J. Klare, D. Daum and H. Schäfer, J. Mater. Chem. A, 2020, 8, 9896–9910 RSC.
- H. Liu, J. Yu, J. Lin, B. Feng, M. Sun, C. Qiu, K. Qian, Z. Si, B. Huang, J.-J. Delaunay, Y. Ikuhara and S. Yang, EES Catal., 2023, 1, 720–729 RSC.
- H. Duan, D. Wang and Y. Li, Chem. Soc. Rev., 2015, 44, 5778–5792 RSC.
- S. Iravani, RSC Sustainability, 2023, 1, 72–82 RSC.
- S. Ying, Z. Guan, P. C. Ofoegbu, P. Clubb, C. Rico, F. He and J. Hong, Environ. Technol. Innovation, 2022, 26, 102336 CrossRef CAS.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. Robert McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 7 CrossRef.
- L. Cseri, S. Kumar, P. Palchuber and G. Szekely, ACS Sustainable Chem. Eng., 2023, 11, 5696–5725 CrossRef CAS.
- J. S. Adams, A. Chemburkar, P. Priyadarshini, T. Ricciardulli, Y. Lu, V. Maliekkal, A. Sampath, S. Winikoff, A. M. Karim, M. Neurock and D. W. Flaherty, Science, 2021, 371, 626–632 CrossRef CAS PubMed.
- S. L. Scott, T. B. Gunnoe, P. Fornasiero and C. M. Crudden, ACS Catal., 2022, 12, 3644–3650 CrossRef CAS.
- J. Beare-Rogers, A. Dieffenbacher and J. V. Holm, Pure Appl. Chem., 2001, 73, 685–744 CrossRef CAS.
- D. J. Anneken, S. Both, R. Christoph, G. Fieg, U. Steinberner and A. Westfechtel, Fatty acids, Ullmann’s Encyclopedia of Industrial Chemistry, Wiley, 2006, 1, DOI:10.1002/14356007.A10_245.PUB2.
- J. Kubitschke, H. Lange and H. Strutz, Carboxylic acids, aliphatic, Ullmann's encyclopedia of industrial chemistry, Wiley, 2014 DOI:10.1002/14356007.a05_235.pub2.
- Z. Niu and Y. Li, Chem. Mater., 2014, 26, 72–83 CrossRef CAS.
- A. G. Roca, D. Niznansky, J. Poltierova-Vejpravova, B. Bittova, M. A. González-Fernández, C. J. Serna and M. P. Morales, J. Appl. Phys., 2009, 105, 114309 CrossRef.
- A. Kihal, G. Fillion, B. Bouzabata and B. Barbara, Phys. Status Solidi B, 2012, 249, 604–614 CrossRef CAS.
- S. Si, C. Li, X. Wang, D. Yu, Q. Peng and Y. Li, Cryst. Growth Des., 2005, 5, 391–393 CrossRef CAS.
- M. Inaba, A. Zana, J. Quinson, F. Bizzotto, C. Dosche, A. Dworzak, M. Oezaslan, S. B. Simonsen, L. T. Kuhn and M. Arenz, ACS Catal., 2021, 11, 7144–7153 CrossRef CAS.
- H. Yang, C. Li, T. Liu, T. Fellowes, S. Y. Chong, L. Catalano, M. Bahri, W. Zhang, Y. Xu, L. Liu, W. Zhao, A. M. Gardner, R. Clowes, N. D. Browning, X. Li, A. J. Cowan and A. I. Cooper, Nat. Nanotechnol., 2023, 18, 307–315 CrossRef CAS PubMed.
- G. S. Yedase, S. Kumar, J. Stahl, B. König and V. R. Yatham, Beilstein J. Org. Chem., 2021, 17, 1727–1732 CrossRef CAS PubMed.
- H. J. H. Fenton, J. Chem. Soc., Trans., 1894, 65, 899–910 RSC.
- A. Babuponnusami and K. Muthukumar, J. Environ. Chem. Eng., 2014, 2, 557–572 CrossRef CAS.
- G. Ruppert, R. Bauer and G. Heisler, J. Photochem. Photobiol., A, 1993, 73, 75–78 CrossRef CAS.
- O. Tantawi, A. Baalbaki, R. El Asmar and A. Ghauch, Sci. Total Environ., 2019, 654, 107–117 CrossRef CAS PubMed.
- T. M. Gill and X. Zheng, Chem. Mater., 2020, 32, 6285–6294 CrossRef CAS.
- Y. Wei, J. Zhang, Q. Zheng, J. Miao, P. J. J. Alvarez and M. Long, Chemosphere, 2021, 279, 130556 CrossRef CAS PubMed.
- J. E. Giaretta, H. Duan, F. Oveissi, S. Farajikhah, F. Dehghani and S. Naficy, ACS Appl. Mater. Interfaces, 2022, 14, 20491–20505 CrossRef CAS PubMed.
- C. Yang, M. Zhang, W. Wang, Y. Wang and J. Tang, Enzyme Microb. Technol., 2020, 140, 109620 CrossRef CAS PubMed.
- S. R. Ahmed, J. Cirone and A. Chen, ACS Appl. Nano Mater., 2019, 2, 2076–2085 CrossRef CAS.
- A. I. Galuza, A. B. Beznosov and V. V. Eremenko, Low Temp. Phys., 1998, 24, 726–729 CrossRef CAS.
- Y. P. He, Y. M. Miao, C. R. Li, S. Q. Wang, L. Cao, S. S. Xie, G. Z. Yang, B. S. Zou and C. Burda, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 125411, DOI:10.1103/PhysRevB.71.125411.
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706–8715 CrossRef CAS.
- L. E. Brus, J. Chem. Phys., 1984, 80, 4403–4409 CrossRef CAS.
- H. Sharifi Dehsari, R. A. Harris, A. H. Ribeiro, W. Tremel and K. Asadi, Langmuir, 2018, 34, 6582–6590 CrossRef CAS PubMed.
- Y. Pang, H. Xie, Y. Sun, M. M. Titirici and G. L. Chai, J. Mater. Chem. A, 2020, 8, 24996–25016 RSC.
- J. K. Lee, K. L. Walker, H. S. Han, J. Kang, F. B. Prinz, R. M. Waymouth, H. G. Nam and R. N. Zare, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 19294–19298 CrossRef CAS PubMed.
- J. Schneider and D. W. Bahnemann, J. Phys. Chem. Lett., 2013, 4, 3479–3483 CrossRef CAS.
- M. Sankar, E. Nowicka, E. Carter, D. M. Murphy, D. W. Knight, D. Bethell and G. J. Hutchings, Nat. Commun., 2014, 5, 3332 CrossRef PubMed.
- A. Bhattacharjee, A. R. LaVigne, S. M. Frazee, T. L. Herrera and T. M. McCormick, Chem. Commun., 2023, 59, 1090–1093 RSC.
- I. Krivtsov, A. Vazirani, D. Mitoraj and R. Beranek, ChemCatChem, 2023, 15, 1–8 CrossRef.
- B. C. Moon, B. Bayarkhuu, K. A. I. Zhang, D. K. Lee and J. Byun, Energy Environ. Sci., 2022, 15, 5082–5092 RSC.
- D. Meyerstein, Nat. Rev. Chem., 2021, 5, 595–597 CrossRef CAS PubMed.
- S. Li, C. Xiao, H. Jiang, Y. Li and C. Li, Sci. Bull., 2023, 68, 25–28 CrossRef CAS PubMed.
- P. Priyadarshini, T. Ricciardulli, J. S. Adams, Y. S. Yun and D. W. Flaherty, J. Catal., 2021, 399, 24–40 CrossRef CAS.
- A. Taufiq, R. E. Saputro, H. Susanto, N. Hidayat, S. Sunaryono, T. Amrillah, H. W. Wijaya, N. Mufti and F. M. Simanjuntak, Heliyon, 2020, 6, e05813 CrossRef CAS PubMed.
- T. Radu, A. Petran, D. Olteanu, I. Baldea, M. Potara and R. Turcu, Appl. Surf. Sci., 2020, 501, 144267 CrossRef CAS.
- NIST X-ray Photoelectron Spectroscopy (XPS) Database, Version 3.5, https://srdata.nist.gov/xps/, (accessed 2 June 2023).
- D. Tahir, S. Ilyas, B. Abdullah, B. Armynah and H. J. Kang, J. Electron Spectrosc. Relat. Phenom., 2018, 229, 47–51 CrossRef CAS.
- D. Wilson and M. A. Langell, Appl. Surf. Sci., 2014, 303, 6–13 CrossRef CAS.
- M. C. Biesinger, Appl. Surf. Sci., 2022, 597, 153681 CrossRef CAS.
- W. Zhu, Y. Wei, Z. Liu, Y. Zhang, H. He, S. Yang, Z. Li, Z. Zou and Y. Zhou, Catal. Sci. Technol., 2022, 12, 4372–4379 RSC.
- G. J. Meyer, J. M. Gardner, S. Kim and P. C. Searson, J. Nanomater., 2011, 46, 1–8, DOI:10.1155/2011/737812.
- T. R. Harris-Lee, F. Marken, C. L. Bentley, J. Zhang and A. L. Johnson, EES Catal., 2023, 1, 832 RSC.
- N. J. Skill, N. G. Theodorakis, Y. N. Wang, J. M. Wu, E. M. Redmond and J. V. Sitzmann, Am. J. Physiol.: Gastrointest. Liver Physiol., 2008, 295, 953–964 CrossRef PubMed.
- N. V. Chandrasekharan and D. L. Simmons, Genome Biol., 2004, 5, 241 CrossRef CAS PubMed.
- W. A. Van der Donk, A. L. Tsai and R. J. Kulmacz, Biochemistry, 2002, 41, 15451–15458 CrossRef CAS PubMed.
- A. K. Yadav, C. J. Reinhardt, A. S. Arango, H. C. Huff, L. Dong, M. G. Malkowski, A. Das, E. Tajkhorshid and J. Chan, Angew. Chem., Int. Ed., 2020, 59, 3307–3314 CrossRef CAS PubMed.
- H. Li, S. Kelly, D. Guevarra, Z. Wang, Y. Wang, J. A. Haber, M. Anand, G. T. K. K. Gunasooriya, C. S. Abraham, S. Vijay, J. M. Gregoire and J. K. Nørskov, Nat. Catal., 2021, 4, 463–468 CrossRef CAS.
- A. Cao and J. K. Nørskov, ACS Catal., 2023, 13, 3456–3462 CrossRef CAS.
- E. R. Vago and E. J. Calvo, J. Electroanal. Chem., 1992, 339, 41–67 CrossRef CAS.
- E. Calvo and D. Schiffrin, J. Electroanal. Chem. Interfacial Electrochem., 1988, 243, 171–185 CrossRef CAS.
- H. E. Rudel and J. B. Zimmerman, ACS Appl. Mater. Interfaces, 2023, 15, 34829–34837 CrossRef CAS PubMed.
- X. Zhou, H. Yang, C. Wang, X. Mao, Y. Wang, Y. Yang and G. Liu, J. Phys. Chem. C, 2010, 114, 17051–17061 CrossRef CAS.
- D. B. Priddy, Adv. Polym. Sci., 1994, 111, 66–114 CrossRef.
- R. Simič, J. Mandal, K. Zhang and N. D. Spencer, Soft Matter, 2021, 17, 6394–6403 RSC.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998, p. 30 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ey00256j |
| This journal is © The Royal Society of Chemistry 2024 |