DOI:
10.1039/D3RA00650F
(Paper)
RSC Adv., 2023,
13, 8190-8201
α-Glucosidase inhibitory activities of flavonoid derivatives isolated from Bouea macrophylla: in vitro and in silico studies†
Received
31st January 2023
, Accepted 24th February 2023
First published on 13th March 2023
Abstract
In continuation of our search for bioactive compounds from the Bouea macrophylla (B. macrophylla) plant, we describe herein eight flavonoid-type compounds including mearsetin (1), mearnsitrin (2), kampferol (3), afzelin (4), quercetin (5), quercitrin (6), myricitin (7), and naringenin (8) with the aim of investigating their antidiabetic properties. Compounds 3 and 5 were selected for aromatic bromination to provide two new products 3a and 5a, respectively. All compounds showed promising α-glucosidase inhibition, with IC50 values ranging from 9.2 to 266 μM apart from compound (2). Remarkably, compound 5a, 8-bromoquercetin, showed the highest inhibition activity, and it was thirty-seven times better than the standard drug acarbose. Pose 261/compound 5a interacted well with enzyme 3TOP in silico docking, and the complex of pose 261 and target enzyme proved its stability in MD. Compound 5a, pose 261 was predicted to be safe and seemed to have good absorption, distribution, metabolism, and excretion properties as assessed via the ADMET model in silico. Our findings revealed the α-glucosidase inhibitory potential of the flavonoids isolated from the leaves of B. macrophylla with a predictive pharmacokinetics profile, which may be helpful in their development as potential drugs.
1. Introduction
In 2021, the International Diabetes Federation (IDF) reported that at least 500 million individuals are affected with type 2 diabetes mellitus (T2DM) and suffering from its complications, causing >6.7 million deaths. Noticeably, this figure is predicted to rise to 643 million by 2030 and 783 million by 2045, causing one of the world's most serious public health challenges. Currently, besides changing to a healthy lifestyle (food, exercise), some specific drugs and insulin work together to manage T2DM.1 Despite there being some available therapies such as SGLT2 inhibitors and GLP-1 RAs to treat T2DM, managing glycemic control remains a major challenge and searching some alternative treatment options for obtaining a better glycemic control without complications is a continuous effort. Recently, alpha-glucosidase (AGs) inhibitors are among the class of oral hypoglycemic agents that have widely proved to be significantly effective and more reliable in reducing hyperglycemia.
Bouea macrophylla Griff. (B. macrophylla) is a medicinal plant (known as marian plum or plum mango), which has been widely grown in tropical countries,2 including Vietnam. In traditional medicine, various parts of the plant are used to treat or prevent some illness.3 Previously, antioxidant, anticancer, and antibacterial activities were observed in this plan, suggesting some great potential for drug development.3–5 Interestingly, our previous studies have initially confirmed that B. macrophylla was a prolific source of novel bioactive compounds such as botulinic acid and methyl gallatic that exhibited α-glucosidase inhibition significantly (IC50 = 1.4–143.3 μM).4
In continuation of our interest in the isolation of AGs inhibitors, herein, we report some more AG inhibitors from B. macrophylla. The chemical structures of eight compounds were clearly elucidated in comparison to those available in the literature (Fig. 1). Also, two new brominated products were synthesized from quercetin and kaempferol. The eight was evaluated for anti α-glucosidase and the mechanisms involved were elucidated through molecular docking studies.
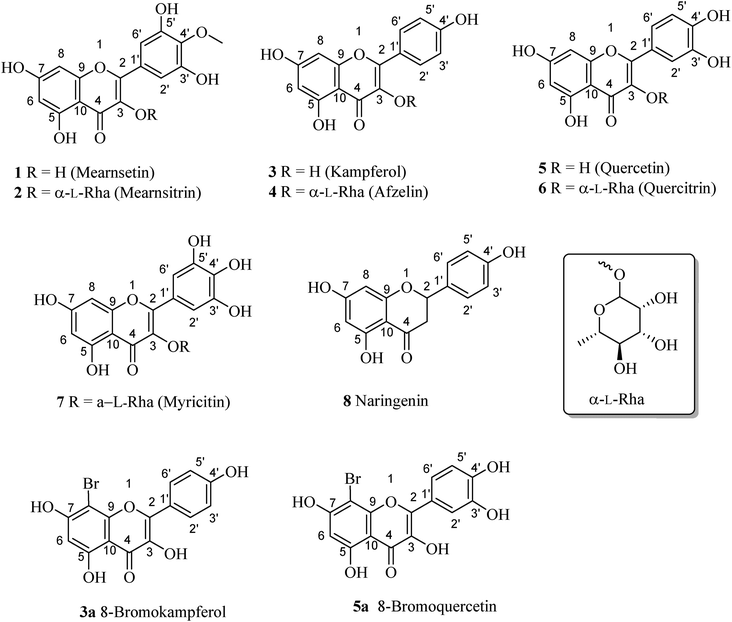 |
| | Fig. 1 Chemical structures of isolated compounds. | |
2. Experimental
2.1. General experimental procedures
NMR spectra were recorded on a Bruker Avance III spectrometer (500 MHz for 1H-NMR and 125 MHz for 13C-NMR) using deuterated solvents: acetone-d6 and chloroform-d. Aluminum TLC plate, silica gel plates (normal phase and C18 reverse phase, Merck), and spots were visualized using vanillin/H2SO4 solution. Silica gel column chromatography used silica gel 60 powder (Himedia, India).
2.2. Source of plant material
About 27 kg fresh leaves of B. macrophylla were harvested from Vinh Long, Vietnam between May and July 2020. All plant material was clearly identified by Prof. Luan Dinh and voucher herbarium specimens (UP-019) deposited at the VNUH.
2.3. Screening of α-glucosidase inhibitory activity
The α-glucosidase enzyme was derived from Saccharomyces cerevisiae. Acarbose and 4-nitrophenyl β-D-glucopyranoside (pNPG) were purchased from Sigma-Aldrich Co, Saint Louis, MI, USA. The α-glucosidase inhibition assay was performed using a slight modification of a published method.6 The IC50 values of the inhibitors were determined using five serial dilutions of α-glucosidase inhibitor. The results were recorded as the concentration of the inhibitor at which the α-glucosidase activity was inhibited by 50%.
2.4. α-Glucosidase inhibition by 5a
The type of inhibition of 5a was determined by Lineweaver–Burk plots using modified methods like those reported previously.7 Compound 5a (0, 1.98, 3.97, and 7.93 μM) was prepared and tested in three replicates to get its IC50 values and the inhibition constants.
2.5. Molecular docking, MD simulations, and like-drugness pharmacokinetic parameters
The molecular docking model was made based on the general procedure in Scheme S1.† The highly active compound in vitro α-glucosidase enzyme inhibition was additionally performed using the molecular docking model with an enzyme 3TOP: protein data bank code.8 The central active on the enzyme were determined at coordinates of (X = −41.386, Y = 12.243, Z = −16.271) on side chain A, which correspond to the center of the grid box. The grid parameters in dock.gpf file was set up by the numbers of the grid points in X, Y, Z as 60, 60, 60, respectively, and spacing of 0.5 Å. The input and output of docking parameters applied by Genetic Algorithm parameters (number of GA runs = 400, population size = 400, and maximum number of evals = 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000) and Lamarckian GA methods. The validations of the molecular docking model were calculated by the PyMOL package.9 The molecular dynamics (MD) simulation of the best docked pose of the protein–ligand (pose 120/400) was executed using the GROMOS96 43a1 force field.10,11 PRODRG tool12 was used to get the compound (5g or pose 120) topology. For the best docked pose of the protein–ligand complex, the simple point charge (SPC) was chosen as a solvent model (triclinical water box size 50 × 75 × 70 Å). The system set for MD simulation was neutralized with sodium or chlorine ions based on the total charges. The steepest descent algorithm (5000 steps) was used to minimize the system before conducting the MD simulation. The MD simulations were then run with 0.15 M NaCl in the presence of a constant temperature (300 K) and pressure (1.0 bar). The number of frames in each simulation was estimated to be about 5000. 100 ns MD simulation was chosen as the simulation time. The simulation trajectories obtained from MD simulation were used to calculate several parameters such as the root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), hydrogen bonds, solvent-accessible surface area (SASA), and radius of gyration (Rg) values to achieve better understanding about the stability of the protein–ligand complex. The pharmacokinetic and like drug-likeness were determined by ADMETlab 2.0.13
000000) and Lamarckian GA methods. The validations of the molecular docking model were calculated by the PyMOL package.9 The molecular dynamics (MD) simulation of the best docked pose of the protein–ligand (pose 120/400) was executed using the GROMOS96 43a1 force field.10,11 PRODRG tool12 was used to get the compound (5g or pose 120) topology. For the best docked pose of the protein–ligand complex, the simple point charge (SPC) was chosen as a solvent model (triclinical water box size 50 × 75 × 70 Å). The system set for MD simulation was neutralized with sodium or chlorine ions based on the total charges. The steepest descent algorithm (5000 steps) was used to minimize the system before conducting the MD simulation. The MD simulations were then run with 0.15 M NaCl in the presence of a constant temperature (300 K) and pressure (1.0 bar). The number of frames in each simulation was estimated to be about 5000. 100 ns MD simulation was chosen as the simulation time. The simulation trajectories obtained from MD simulation were used to calculate several parameters such as the root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), hydrogen bonds, solvent-accessible surface area (SASA), and radius of gyration (Rg) values to achieve better understanding about the stability of the protein–ligand complex. The pharmacokinetic and like drug-likeness were determined by ADMETlab 2.0.13
2.6. Extraction and isolation
Leaves of B. macrophylla (27 kg) were washed, then ground to a powder (20 kg). This was macerated with MeOH (3 × 30 L) at room temperature, followed by filtration and evaporation under reduced pressure. The crude MeOH residue (790 g) was yielded and applied to liquid–liquid extraction using different solvents, such as n-hexane, n-hexane-EtOAc (1:1, v/v), and EtOAc, to provide n-hexane (H, 219 g), n-hexane-EtOAc (HEA, 30.6 g), and EtOAc (EA, 390 g) extracts, respectively. The EA extract was applied to CC with n-hexane:EtOAc (1:1, v/v) to get EA1–5 fractions.
Fraction EA3 (63 g) was applied to silica gel CC with the eluent n-hexane:EtOAc:acetone (4:2:1, v/v) to obtain fractions EA3.1–3.4. Fraction EA3.1 (18 g) was then subjected to silica gel CC under n-hexane:EtOAc:CHCl3:MeOH (8:2:3:1, v/v) to give fractions EA3.1.1–EA3.1.4. Compound 8 (8.3 mg) was obtained from fraction EA3.1.3 (7.2 g) using silica gel CC with the mobile phase of n-hexane:EtOAc:CHCl3:MeOH (4:2:3:1, v/v).
Fraction EA4 (48 g) was subjected to Sephadex LH-20 and washed with MeOH to release fractions EA4.1–4.3. Fraction EA4.3 (15 g) was then applied to silica gel CC and washed with n-hexane:CHCl3:EtOAc:MeOH (2:5:2:2, v/v/v/v) to obtain fractions EA4.3.1–EA4.3.5. Fraction EA4.3.2 (5.2 g) was subjected to silica gel CC and eluted with n-hexane:EtOAc:MeOH (1:1:1, v/v/v/v) to give compounds 3 (121 mg) and 5 (72 mg). The purification of fraction EA4.3.3 (2.8 g) by CC with n-hexane:CHCl3:EtOAc:MeOH (4:10:3:3, v/v/v/v) as an eluent provided compound 2 (6.4 mg), compound 6 (4.7 mg), and compound 1 (7.8 mg). Fraction EA4.3.4 (1.2 g) was chromatographed by CC and eluted with CHCl3:EtOAc:MeOH (3:2:1, v/v/v) to afford compound 7 (7.54 mg) and compound 4 (5.2 mg).
2.7. General procedure to synthesize compounds 3a and 5a
Acetic acid and DMSO (1:1, v/v), kaempferol (3) (50.0 mg, 0.175 mmol), and sodium bromide (89.8 mg, 0.873 mol) were dissolved at RT. 0.18 mL (1.750 mmol) 30% hydrogen peroxide was added to the reaction flask and the reaction was conducted in 1 h. The resultant mixture was neutralized with saturated sodium hydrogen carbonate and extracted with ethyl acetate–water (1:1, v/v). The organic layer was washed with brine and dried over anhydrous Na2SO4, followed by filtration to produce the residue. This residue was further subjected to silica gel column chromatography and eluted with the gradient system of n-hexane:ethyl acetate:acetic acid (27:9:1, v/v/v) to afford 3a (10.9 mg). A similar procedure was applied to quercetin (5) (20.0 mg, 0.0662 mmol) to afford 5a (22.3 mg).
3. Results
3.1. Phytochemical identification of isolated compounds 1–8
Eight flavonoid-type compounds were isolated from the leaves of Bouea macrophylla. Their chemical structures were elucidated as mearnsetin (1),14 mearnsitrin (2),15 kampferol (3),16 afzelin (4),16 quercetin (5),16 quercitrin (6),16 myricitin (7),17 and naringenin (8).18
3.1.1 Mearnsetin (1). 1H-NMR (500 MHz, acetone-d6) δ: 12.10 (1H, s, 5-OH), 9.86 (1H, s, 7-OH), 8.39 (2H, s, 3′-OH, 5′-OH), 8.13 (1H, s, 3-OH), 7.39 (2H, s, H-2′, 6′), 6.53 (1H, d, J = 2.0 Hz, H-8), 6.28 (1H, d, J = 2.0 Hz, H-6), 3.90 (3H, s, 4′-OMe). 13C-NMR (125 MHz, acetone-d6) δ: 176.8 (C-4), 165.3 (C-7), 162.4 (C-5), 157.9 (C-9), 151.4 (C-3′, 5′), 146.3 (C-2), 137.6 (C-3, 4′), 127.4 (C-1′), 108.4 (C-2′, 6′), 104.3 (C-10), 99.3 (C-6), 94.5 (C-8), 60.8 (4′-OMe). See Fig. S1 and S2.†
3.1.2 Mearnsitrin (2). 1H-NMR (500 MHz, acetone-d6) δ: 12.65 (1H, s, 5-OH), 7.015 (2H, s, H-2′,6′), 6.47 (1H, d, J = 2 Hz, H-8), 6.26 (1H, d, J = 2 Hz, H-6), 5.45 (1H, s, H-1′′), 4.22 (1H, s, H-3′′), 3.88 (1H, s, H-4′-OMe), 3.73 (1H, s, H-4′′), 3.34 (3H, s, 5′′), 0.92 (3H, d, J = 5.95 Hz, H-6′′). 13C-NMR (125 MHz, acetone-d6) δ: 179.4 (C-4), 165.4 (C-7), 163.2 (C-5), 158.2 (C-2), 158.1 (C-9), 151.2 (C-3′,5′), 138.7 (C-4′), 136.3 (C-3), 126.6 (C-1′), 109.5 (C-2′,6′), 105.8 (C-10), 102.9 (C-1′′), 99.7 (C-6), 94.6 (C-8), 73.0 (C-4′′), 72.1 (C-3′′), 71.5 (C-5′′), 71.3 (C-2′′), 60.8 (4′-OMe), 17.8 (C-6′′). See Fig. S3 and S4.†
3.1.3 Kaempferol (3). 1H-NMR (500 MHz, acetone-d6) δ: 12.20 (1H, s, 5-OH), 8.16 (1H, d, J = 9.2 Hz, H-2′,6′), 7.02 (1H, d, J = 9.2 Hz, H-3′,5′), 6.54 (1H, d, J = 2.0 Hz, H-8), 6.26 (1H, d, J = 2.0 Hz, H-6). See Fig. S5.†
3.1.4 Afzelin (4). 1H-NMR (500 MHz, acetone-d6) δ: 7.85 (2H, d, J = 4 Hz, H-3′, 5′), 7.01 (2H, dd, J = 6.5 Hz, 2 Hz, H-2′, 6′), 6.46 (1H, d, J = 2 Hz, H-8), 6.26 (1H, d, J = 2 Hz, H-6), 5.54 (1H, d, J = 1.5 Hz, H-1′′), 4.21 (1H, dd, J = 3.5 Hz, 1.5 Hz, H-2′′), 3.68 (1H, dd, J = 9 Hz, 3.5 Hz, H-5′′), 3.60 (1H, s, H-3′′), 3.31 (1H, m, H-4′′), 0.90 (3H, d, J = 6 Hz, H-6′′). 13C-NMR (125 MHz, acetone-d6) δ: 172.7 (C-4), 161.4 (C-7), 160.0 (C-4′), 158.2 (C-5), 152.4 (C-9), 147.2 (C-2), 137.7 (C-3), 131.7 (C-2′, 6′), 122.6 (C-1′), 116.3 (C-3′, 5′), 102.7 (C-10), 102.0 (C-1′′), 99.7 (C-6), 94.4 (C-8), 72.9 (C-4′′), 72.1 (C-3′′), 71.3 (C-2′′), 70.9 (C-5′′), 17.9 (C-6′′). See Fig. S6 and S7.†
3.1.5 Quercetin (5). 1H-NMR (500 MHz, acetone-d6) δ: 13.03 (1H, s, 5-OH), 7.96 (2H, d, J = 8.5 Hz, H-6), 7.04 (2H, d, J = 9.0 Hz, H-5′), 6.65 (1H, s, H-2′), 6.56 (1H, d, J = 2.0 Hz, H-8), 6.27 (1H, d, J = 2.0 Hz, H-6). 13C-NMR (125 MHz, acetone-d6) δ: 176.3 (s, C-4), 164.3 (s, C-7), 161.1 (s, C-5), 156.6 (s, C-9), 148.1 (s, C-4′) 147.3 (s, C-2), 145.5 (s, C-3′), 136.1 (s, C-3), 122.4 (s, C-1′), 120.5 (s, C-6′), 116.1 (s, C-5′), 115.5 (s, C-2′), 103.5 (s, C-10), 98.7 (s, C-6), 93.9 (s, C-8). See Fig. S8.†
3.1.6 Quercitrin (6). 1H-NMR (500 MHz, acetone-d6) δ: 7.50 (1H, d, J = 2 Hz, H-2′), 7.39 (1H, dd, J = 3.5 Hz, 2 Hz, H-6′), 6.99 (1H, d, J = 8 Hz, H-5′), 6.46 (1H, d, J = 1.5 Hz, H-8), 6.26 (1H, d, J = 2, H-6′), 5.52 (1H, s, H-1′′), 4.18 (1H, d, J = 1.5 Hz, H-2′′), 3.73 (1H, dd, J = 3.8 Hz, 2.8 Hz, H-5′′), 3.61 (1H, s, H-3′′), 3.38 (1H, m, H-4′′), 0.91 (1H, d, J = 6 Hz, H-6′′). 13C-NMR (125 MHz, acetone-d6) δ: 179.3 (C-4), 165.0 (C-7), 163.2 (C-9), 158.4 (C-5), 158.0 (C-2), 149.0 (C-4′), 145.8 (C-3′), 135.9 (C-3), 122.9 (C-1′), 122.6 (C-6′), 116.8 (C-5′), 116.1 (C-2′), 105.8 (C-10), 102.8 (C-1′′), 99.3 (C-6), 94.7 (C-8), 73.0 (C-4′′), 72.1 (C-3′′), 71.5 (C-2′′), 71.3 (C-5′′), 17.8 (C-6′′). See Fig. S9 and S10.†
3.1.7 Myricitrin (7). 1H-NMR (500 MHz, acetone-d6) δ: 7.10 (2H, s, H-2′,6′), 6.46 (1H, d, J = 2 Hz, H-8), 6.25 (1H, d, J = 2.5 Hz, H-6), 5.48 (1H, s, H-1′′), 4.22 (1H, dd, J = 3.5 Hz, 1.5 Hz, H-2′′), 3.78 (1H, dd, J = 9.5 Hz, 3.5 Hz, H-3′′), 3.53 (1H, m, H-5′′), 3.36 (1H, m, H-4′′), 0.93 (3H, d, J = 6, H-6′′). 13C-NMR (125 MHz, acetone-d6) δ: 179.4 (C-4), 165.0 (C-7), 163.2 (C-5), 158.5 (C-2), 157.9 (C-9), 146.4 (C-3′,5′), 137.1 (C-4′), 135.9 (C-3), 121.9 (C-1′), 109.4 (C-2′,6′), 105.8 (C-10), 102.8 (C-1′′), 99.5 (C-6), 94.5 (C-8), 73.1 (C-4′′), 72.1 (C-3′′), 71.5 (C-5′′), 71.3 (C-2′′), 17.8 (C-6′′). See Fig. S11 and S12.†
3.1.8 Naringenin (8). 1H-NMR (500 MHz, acetone-d6) δ: 12.18 (1H, s, 5-OH), 7.39 (2H, d, J = 8.5 Hz, H-2′,6′), 6.90 (2H, d, J = 8.5 Hz, H-3′,5′), 5.95 (1H, d, J = 3 Hz, H-6,8), 5.46 (1H, dd, J = 12.5 Hz, 3 Hz, H-2), 3.18 (1H, dd, J = 17.1 Hz, 12.8 Hz, H-3a), 2.73 (1H, dd, J = 17.1 Hz, 3.1 Hz, H-3b). See Fig. S13.†
3.2. Bromination of 3 and 5
Compounds 3 and 5 were elected for bromination for making new flavones (Fig. 2) using hydrogen peroxide and potassium bromide in acetic acid following the previous report with modifications (Fig. 2).19 As a result, compounds 3a and 5a were prepared with isolated yields of 93% and 88%, respectively. The 1H NMR spectra of 3a and 5a were highly like those of their mother compounds 3 and 5, respectively. The only difference was the A-ring with the disappearance of one aromatic proton (H-8), indicating that this position was brominated. The data are described in the figure, which was consistent with the similar compounds reported by Dao and co-workers (2021).19
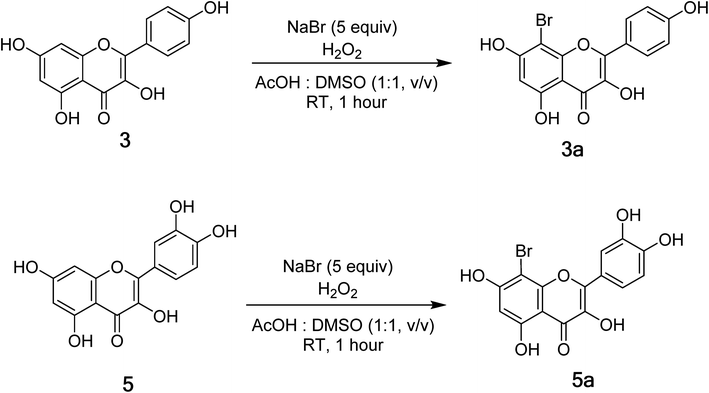 |
| | Fig. 2 General synthesis route toward analogues 3a and 5a. | |
8-Bromokaempferol (3a) is a light-yellow powder with 93% yield. 1H-NMR (500 MHz, acetone-d6) δ: 12.19 (1H, s, 5-OH), 8.32 (2H, d, J = 10.0 Hz, H-2′,6′), 7.08 (2H, d, J = 10.0 Hz, H-3′,5′), 6.55 (1H, s, H-6). 13C-NMR (125 MHz, acetone-d6) δ: 196.3 (C-4), 161.5 (C-7), 161.1 (C-5), 159.5 (C-4′), 154.0 (C-2), 151.4 (C-9), 135.8 (C-3), 129.6 (C-2′,6′), 120.6 (C-1′), 115.6 (C-3′,5′), 107.6 (C-10), 98.9 (C-6), 87.0 (C-8). HR-ESI-MS m/z: 362.9516 [M − H]− (Calcd for C15H8BrO6: 362.9504). See Fig. S14 and S15.†
8-Bromoquercetin (5a), isolated yield 88%, light yellow powder. 1H-NMR (500 MHz, acetone-d6) δ: 12.20 (1H, s, 5-OH), 7.88 (1H, d, J = 8.5 Hz, H-6′), 7.03 (1H, d, J = 8.5 Hz, H-5′), 6.49 (1H, s, H-2′), 6.27 (1H, s, H-6).13C-NMR (125 MHz, Acetone-d6) δ: 175.6 (C-4), 163.8 (C-7), 160.7 (C-5), 152.8 (C-9), 149.2 (C-4′), 147.8 (C-2), 145.1 (C-3′), 136.1 (C-3), 122.8 (C-1′), 120.9 (C-6′), 119.8 (C-5′), 115.5 (C-2′), 104 (C-10), 98.4 (C-6), 86.3 (C-8). HR-ESI-MS m/z: 378.9465 [M − H] (Calcd for C15H9BrO7: 378.9465). See Fig. S17 and S19.†
3.3. Alpha-glucosidase inhibition of 1–8, 3a, and 5a
All compounds revealed strong inhibitory activity toward glucosidase present with IC50 values between 9.2 and 266 μM, except for compound (2), which showed no activity (Table 1).
Table 1 IC50 of pure compounds for α-glucosidase inhibitory activities
| Compound |
IC50 (μM) |
Compound |
IC50 (μM) |
| 1 |
205.3 ± 5.6 |
5 |
117 ± 1.9 |
| 2 |
Negative |
5a |
9.2 ± 0.72 |
| 3 |
230 ± 2.7 |
6 |
255 ± 3.31 |
| 3a |
173 ± 2.35 |
7 |
266 ± 2.97 |
| 4 |
118 ± 0.68 |
Acarbose |
332 ± 3.9 |
3.4. Alpha-glucosidase inhibition type and inhibition constants of 5a
To investigate the inhibition type of 5a, different concentrations of 5a (0, 1.98, 3.97, and 7.93 μM) were investigated for their activity. As a result, a group of lines with different slopes and intercepts intersected the y-axis in the second quadrant in the Lineweaver–Burk plots (Fig. 3A), indicating that 5a acted as a mixed mode inhibitor. The inhibition constants of 5a binding with the free enzyme (Ki) and with the enzyme–substrate complex (K′i) were determined to be 5.58 ± 0.35 μM and 10.33 ± 0.85 μM, respectively (Fig. 3B and C).
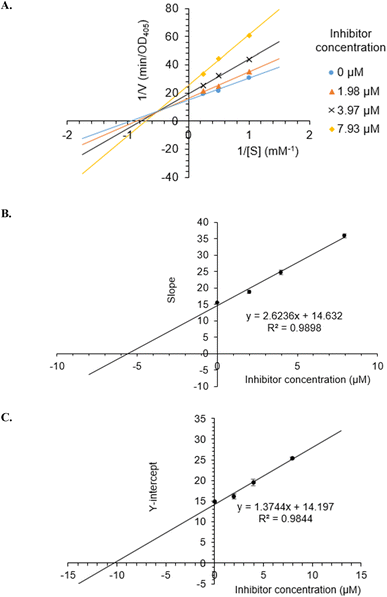 |
| | Fig. 3 Lineweaver–Burk plot (A) for α-glucosidase inhibition by 5a and the secondary plots of slope and Y-intercept vs. inhibitor concentration (B and C). | |
3.5. In silico modeling and molecular docking of compound 5a
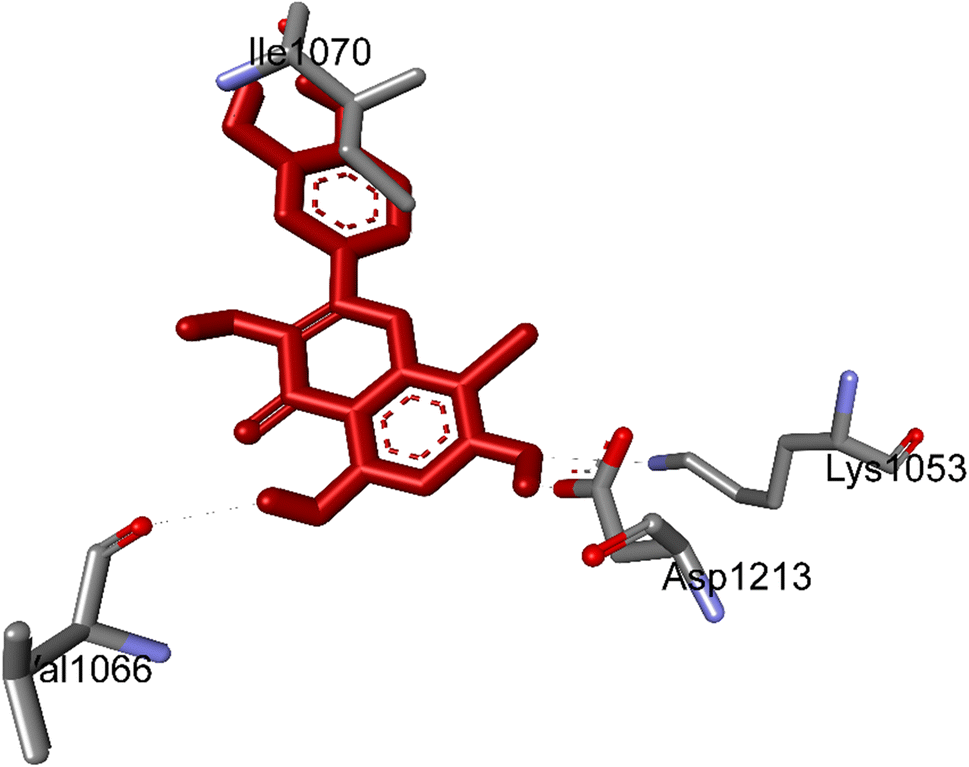
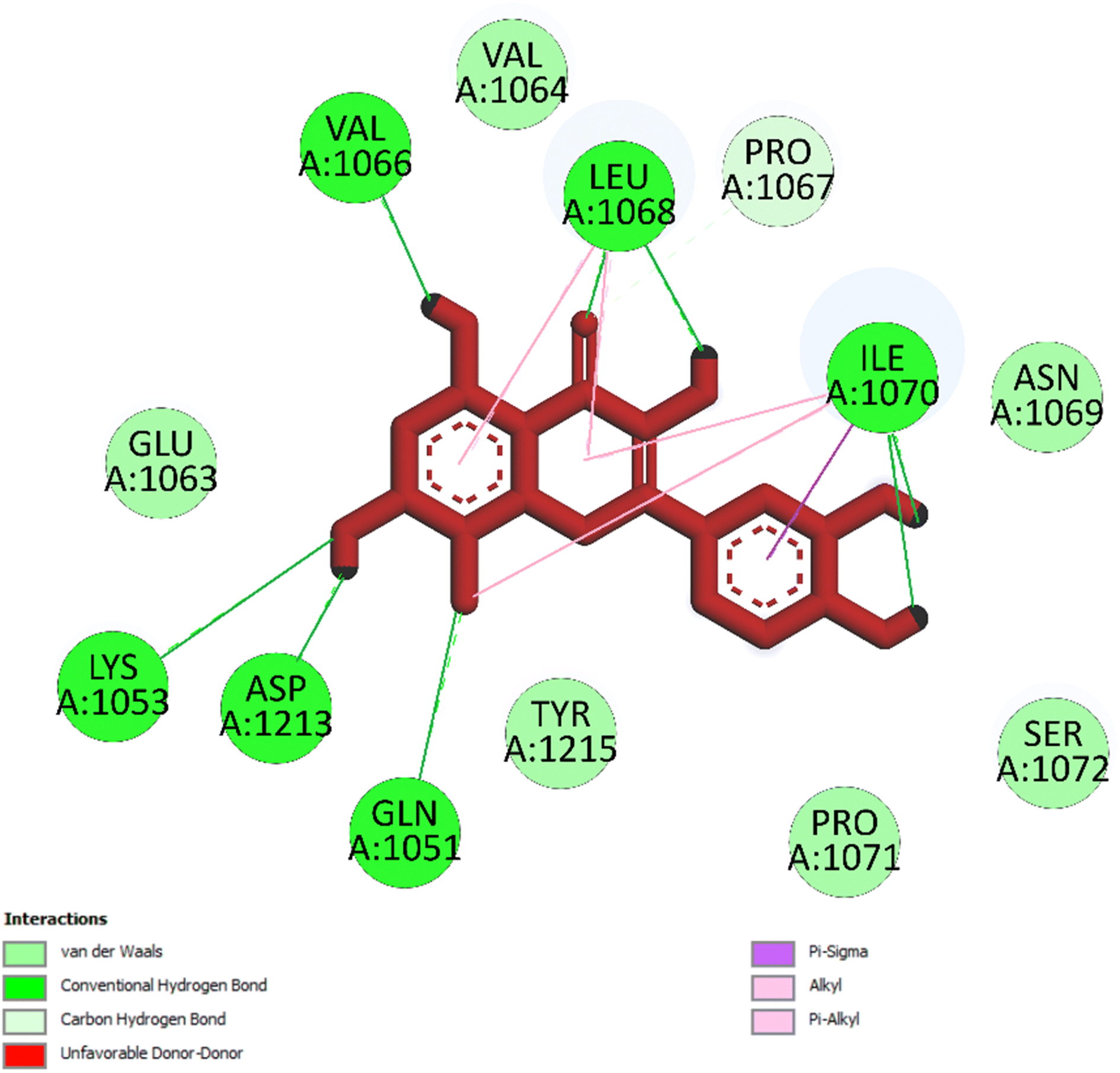
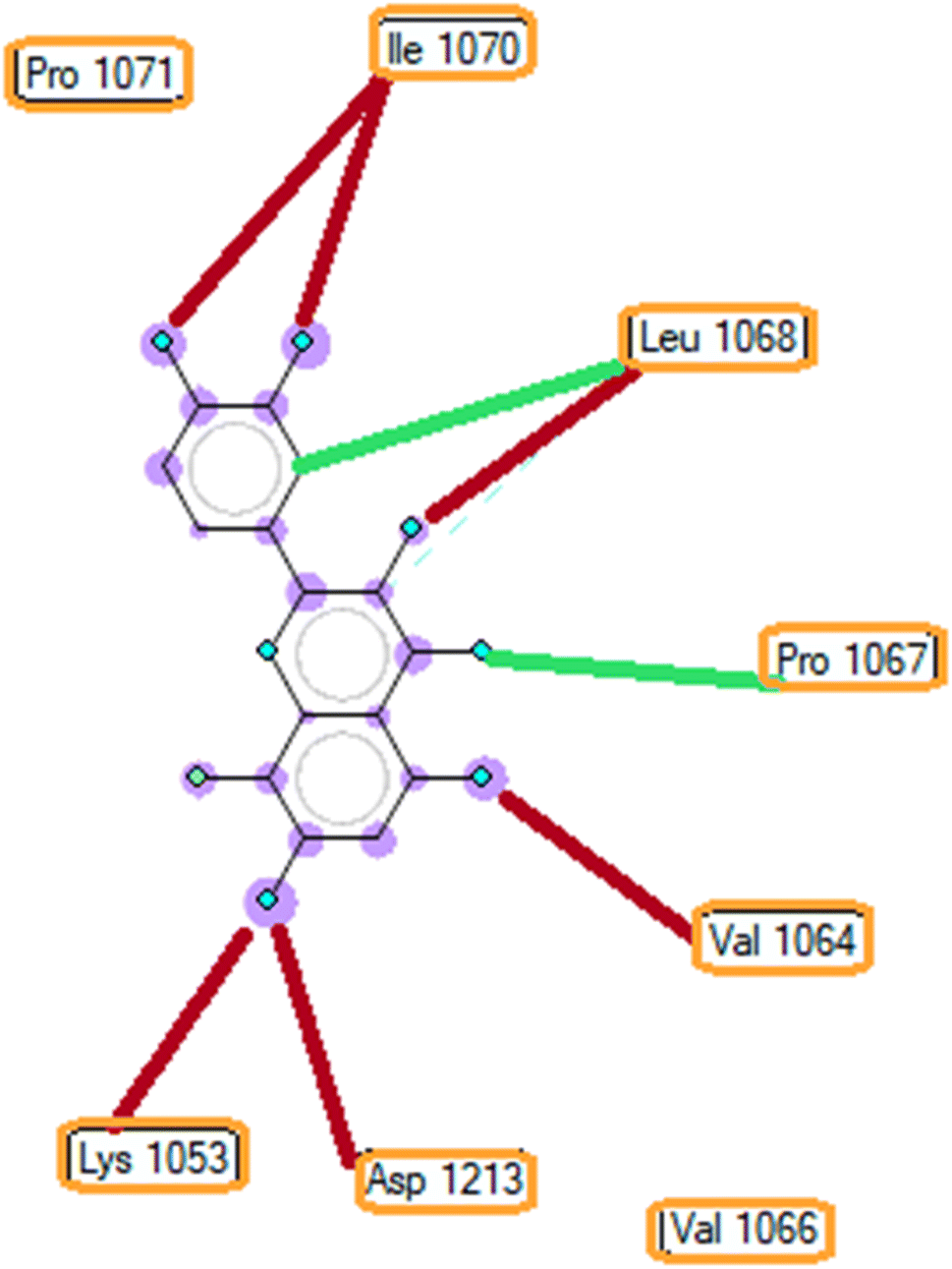
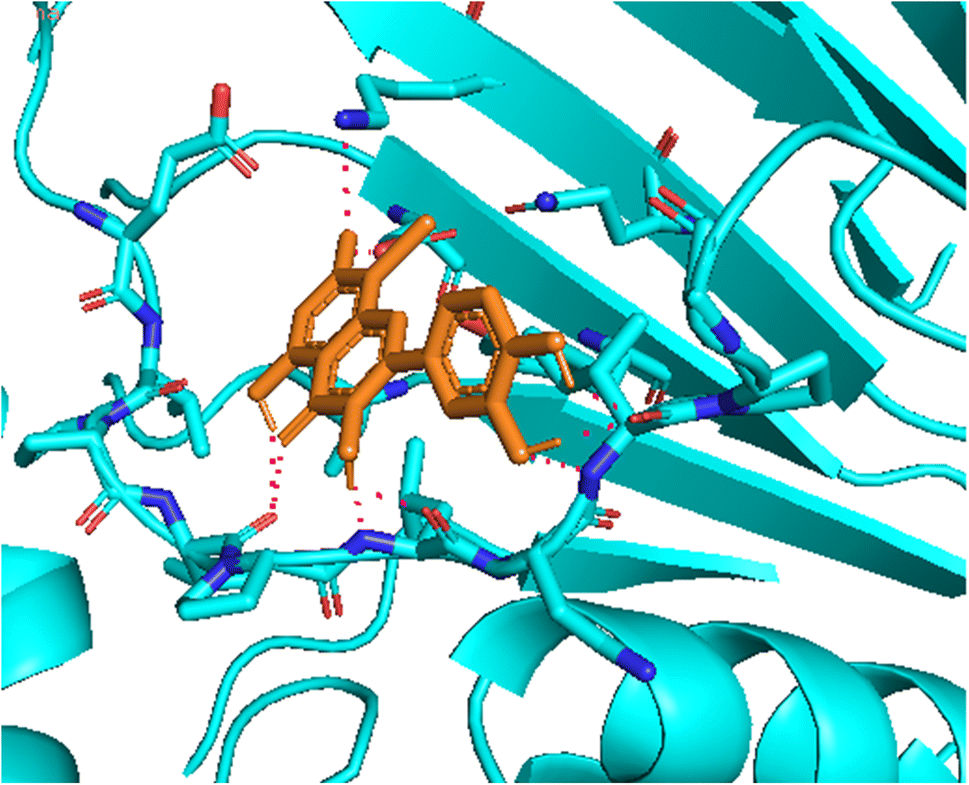
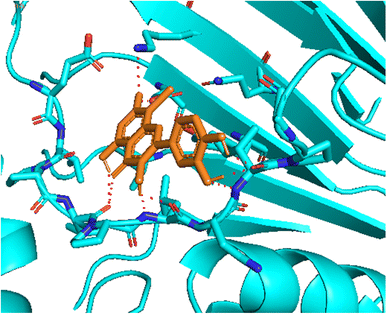
3.5.1 Pose 261/compound 5a. The significant calulation in silico results of compound 5a are presented in Table 2, Fig. 4 to 7. The compound 5a or ranked pose 261/400 docked to 3TOP α-glucosidase enzyme, one enzyme leads to the hydrolysis of carbohydrate to glucose in blood in silico molecular docking model with free energy of binding and inhibition constant values of −6.42 and 19.85 μM, respectively, which were calculated from ADT, as shown in Table 2. Pose 261 is the best docking pose, which was selected among 400 poses and is the most stable conformation ligand of compound 5a. As shown in Fig. 4 and Table 2, the ranked pose 261 formed seven hydrogen bindings with enzyme 3TOP with residual amino acid, which are relative to hydrophilic interactions such as Lys 1053, Asp 1213, Lys 1053, Leu 1068, and Ile 1070. Among hydrophilic interactions, one strongest hydrogen bonding linked from H in pose 261 to Asp 1213 with one bond length of 1.71 Å. Also, as shown in Fig. 5, significant ligand interactions between 3TOP and pose 261 were presented in one 2D diagram, which proved that pose 261 interacted well with enzyme because this pose has 3 parts, which include the capping groups (identification of protein via identification of the aromatic ring/heterocyclic ring)–hydrophobic interactions, linker part/connecting unit (CU)-identification aliphatic chain, or aromatic–hydrophobic interactions, and functional group (detection of functional group via hydrophilic interactions or hydrogen bond). One ligand or pose like this is considered a good interaction in the ligand interactions model.1 The functional group was detected by hydrogen bonding from Lys 1053, Val 1066, Leu 1068, Ile 1070, and Glu 1051 to oxygen atoms and hydrogen atom on pose 261. The capping groups was detected by alkyl or pi-alkyl from Leu 1068 and Ile 1070 to the aromatic ring, carbonyl ring, and brom atom on pose 261. The CU was identified via one pi–sigma interaction from Ile 1070 to the system of pi electrons. The ligand map showed secondary interactions that formed around pose 261, as indicated in Fig. 6. They included hydrogen bonding (red lines) from Ile 1070, Leu 1068, Val 1064, Asp 1213, and Lys 1053, steric interactions such as Leu 1068 and Pro 1067 (green lines), and overlap interactions (violet circles). They proved that there are not many interactions forming pose 261. As shown in Fig. 7, hydrogen bonding was formed between pose 261 and residual amino acids on 3TOP enzyme at the active site around this pose in the 3D diagram obtained using PyMOL software.
Table 2 The significant result of the best docking poses to enzyme 3TOP: PDB, α-glucosidase inhibition enzyme
| Entry |
Pose |
Free energy of binding |
Ki |
The number of hydrogen bonds |
The property and bond length |
| 5 |
250/400 |
−5.76 |
59.80 |
2 |
A: Arg 1098: N – 5: O (2.85 Å) |
| A: Arg 1098: N – 5: O (2.95 Å) |
| 5a |
261/400 |
−6.42 |
19.85 |
|
5a: H – A: Asp 1213: O (1.79 Å) |
| A: Lys 1053: N – 5a: O (3.15 Å) |
| 5a: H – A: Val 1066: O (2.33 Å) |
| A: Leu 1068: N – 5a: O (3.13 Å) |
| 5a: H – A: Leu 1068: O (1.99 Å) |
| A: Ile 1070: N – 5a: O (2.84 Å) |
| 5a: H – A: Ile 1070: O (2.06 Å) |
| Small ligand (in 3TOP) |
260/400 |
−3.85 |
1500 |
7 |
A: Arg 1235: N – small ligand: O (3.01 Å) |
| Small ligand: H – A: Glu 1495: O (2.08 Å) |
| A: Tyr 1761: O – small ligand: O (2.64 Å) |
| Small ligand: H – A: Tyr 1761: O (1.91 Å) |
| Small ligand: H – A: Thr 1628: O (1.71 Å) |
| Small ligand: H – A: Thr 1628: O (2.42 Å) |
| Small ligand: H – A: Asp 1759: O (1.86 Å) |
| Small ligand: H – A: Asp 1759: O (1.78 Å) |
| Small ligand: H – A: Asp 1759: O (2.18 Å) |
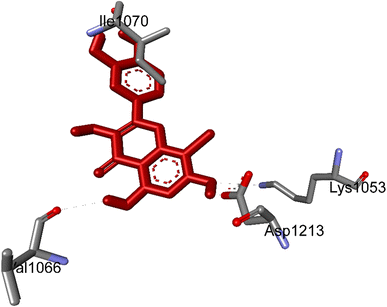 |
| | Fig. 4 The hydrogen bonding formed from active atoms on best docking 261/compound 5a to residual amino acids on enzyme 3TOP: PDB: A chain, which inhibited α-glucosidase enzyme. | |
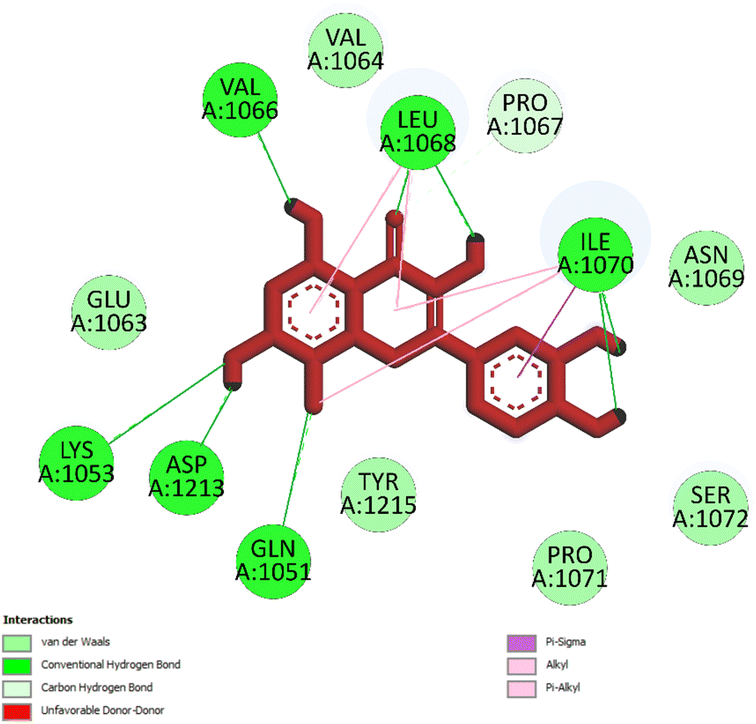 |
| | Fig. 5 One 2D diagram shows the significant ligand interactions between the ranked pose 261/compound 5a and enzyme 3TOP: A chain. | |
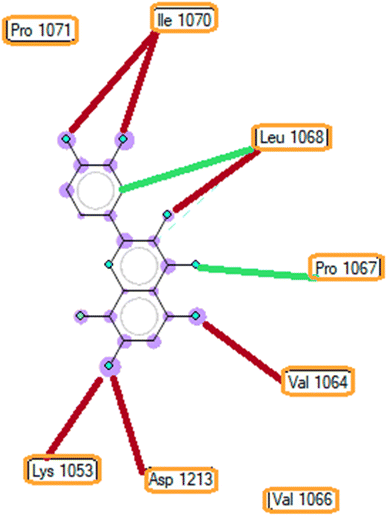 |
| | Fig. 6 The ligand map indicates the secondary interactions between the ranked pose 261/compound 5a and enzyme 3TOP. | |
 |
| | Fig. 7 The one 2D diagram shows the hydrogen bonding between pose 261/compound 5a and 3TOP enzyme by the PyMOL software. | |
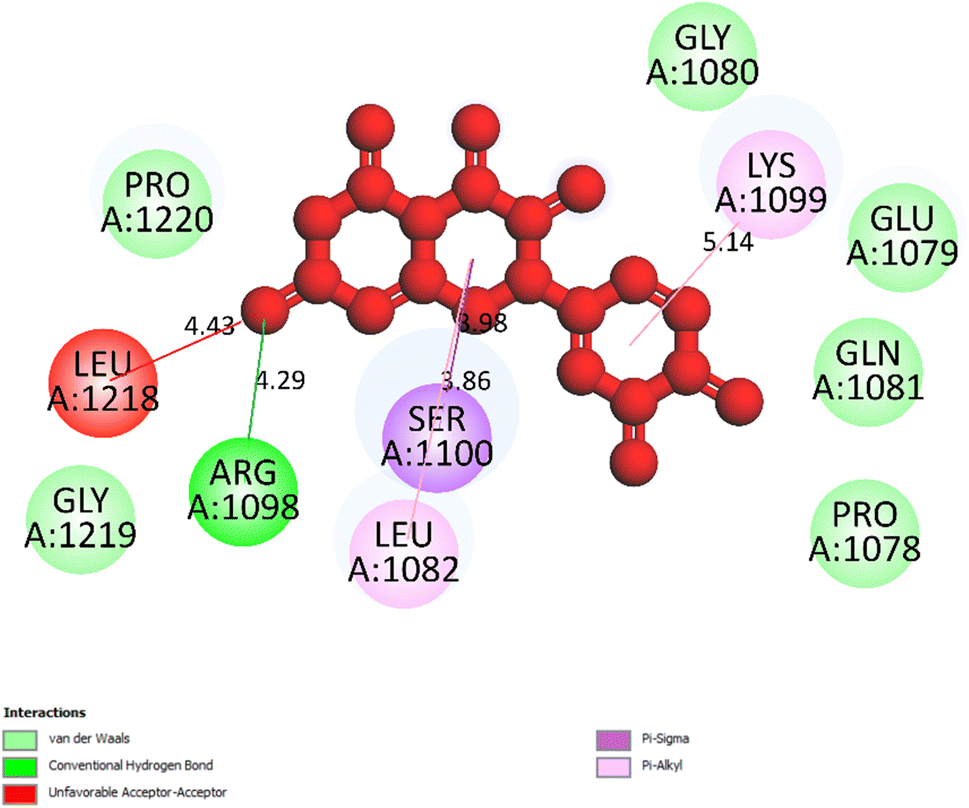
3.5.2 Pose 250/compound 5. Pose 250, the best docking pose of compound 5 was docked to 3TOP enzyme with the free Gibbs energy of binding and inhibition constant of −5.76 kcal mol−1 and 59.80, respectively, as shown in Table 2. At thermodynamic, pose 261 interacted with the enzyme better than pose 250 did because its free energy of binding is lower than that of pose 250. As shown in Fig. 8, pose 250 did not interact well with enzyme because one unfavorable acceptor–acceptor formed from Leu 1218 to oxygen atom of carbonyl group on pose 250.
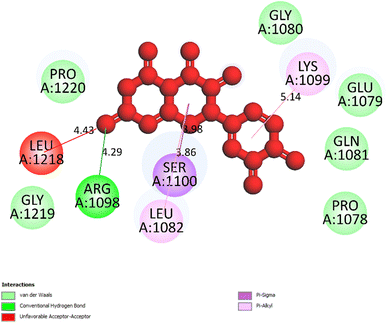 |
| | Fig. 8 The one 2D diagram shows the significant ligand interactions between pose 250/compound 5 and 3TOP enzyme, A: chain. | |
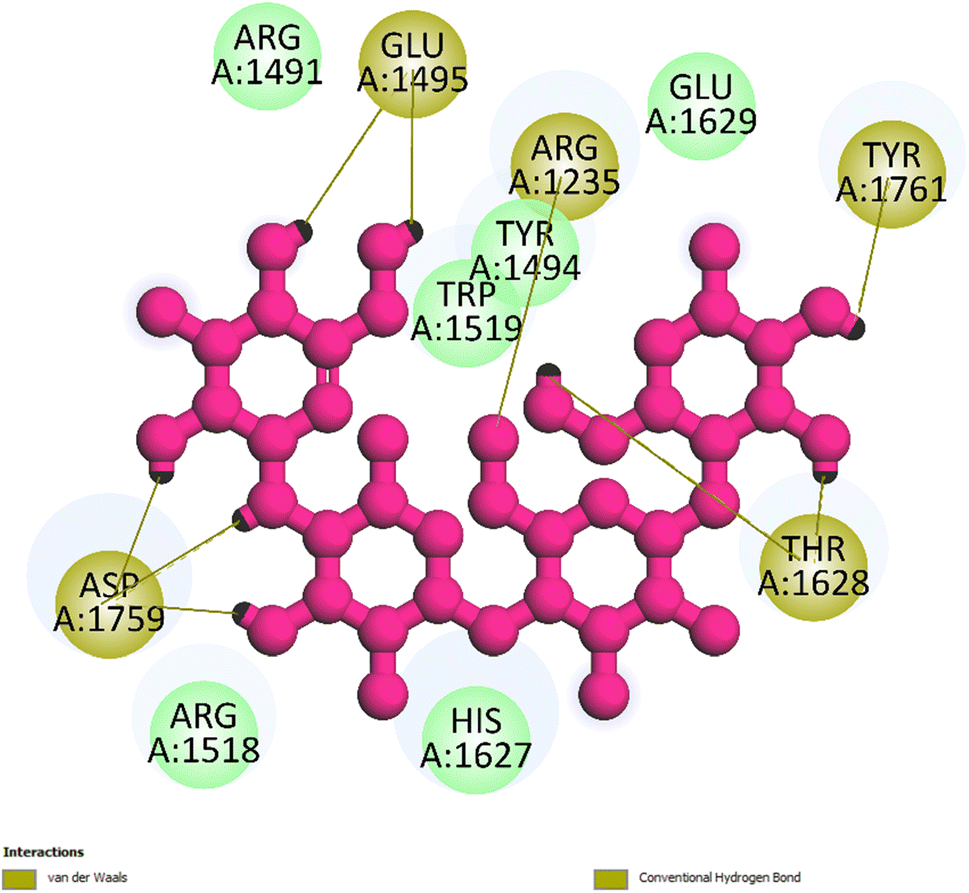
3.5.3 Pose 260. The best docking pose of small ligand in 3TOP enzyme was used to redock to this enzyme and anchored to 3TOP with the free energy of binding and inhibition constant values of −3.85 kcal mol−1 and 1500 μM, respectively, as shown in Table 2. As shown in Fig. 9, the most important ligand interactions indicated that this pose interacted with the enzyme by polar interactions or hydrophilic interactions, and pose 260 did not link well to this enzyme. In the ligand interaction model, the ability of pose was ranked by pose 261/compound 5a > pose 260/small ligand > pose 250/compound 5.
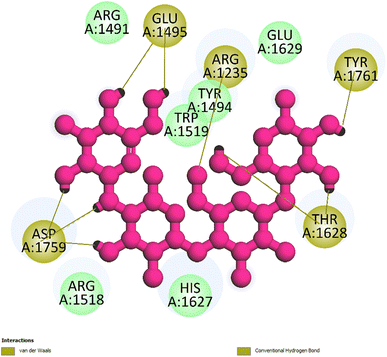 |
| | Fig. 9 The one 2D diagram shows the significant ligand interactions between pose 260/small ligand, which is available in 3TOP and 3TOP enzyme (redocking). | |
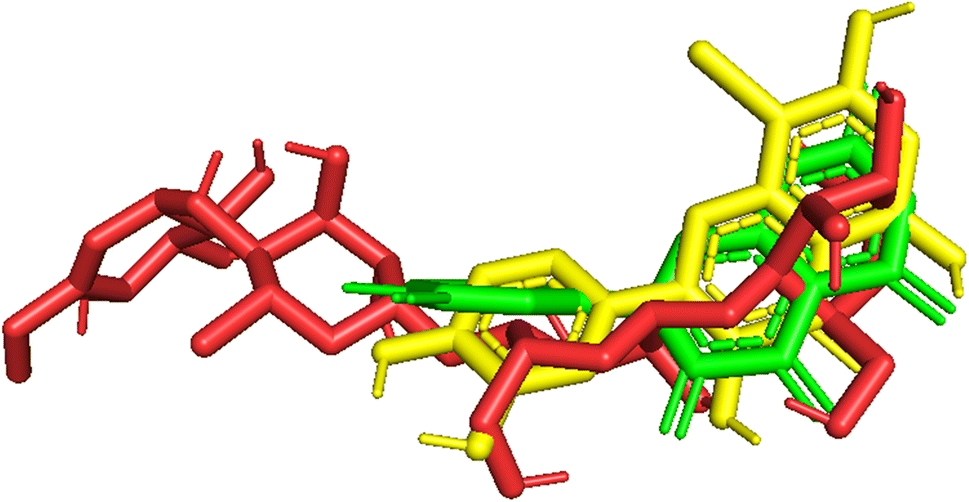
3.5.4 The validation of the model. As indicated in Fig. 10 and Table 3, pose 261 and pose 250 were aligned to pose 260, and the values of RMSD of pair poses were calculated by (pose 250, pose 260) and (pose 261, pose 260), which were 3.010 and 3.842 Å, respectively. The redocking of pose 250, pose 261 to pose 260 proved the validations of the model used in the method, orientational docking, docking parameters, and model validation with an interesting compound. As the values of RMSD are higher than 2.0 Å, the ligand interactions between pose 261 and 3TOP obtained via molecular dynamics (MD) simulation evaluated the stable ligand complex (pose 261-3TOP complex).
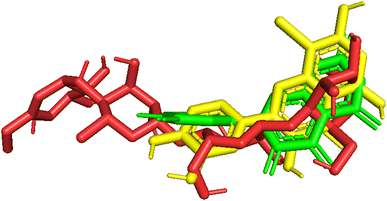 |
| | Fig. 10 Pose_250 or compound 5, pose_261 or compound 5a were aligned to pose_260, one small ligand that is available in the 3TOP enzyme for the values of RMSD calculation. | |
Table 3 The values of RMSD of pair poses, pose 260, one small ligand in 3TOP enzyme
| |
Pose_250, compound 5 |
Pose_261, compound 5a |
Pose_260, reference pose, small ligand in 3TOP: PDB |
| Pose_250, compound 5 |
0 |
— |
3.010 |
| Pose_261, compound 5a |
— |
0 |
3.842 |
| Pose_260, reference pose, small ligand in 3TOP: PDB |
— |
— |
0 |
3.6. MD simulation
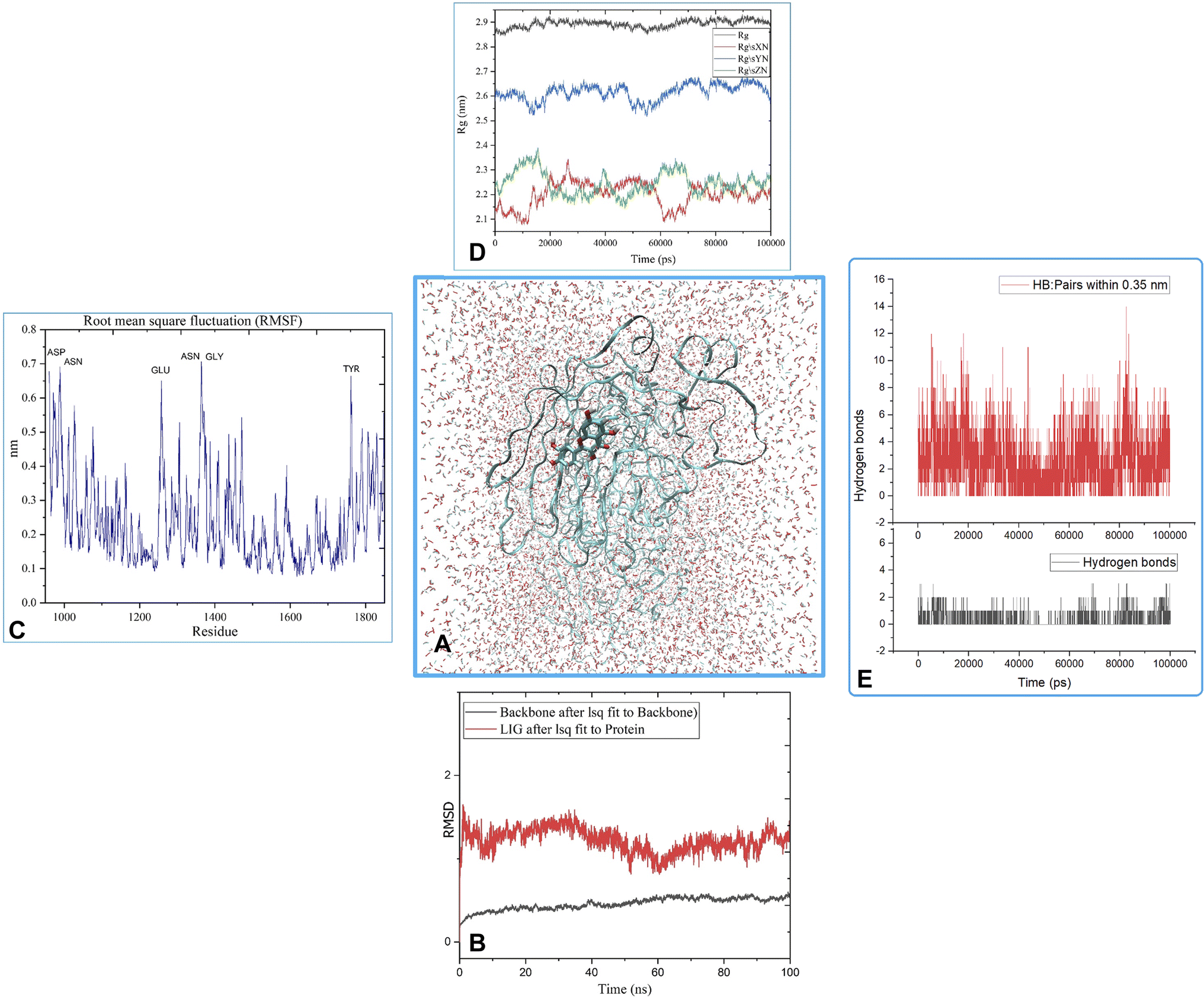
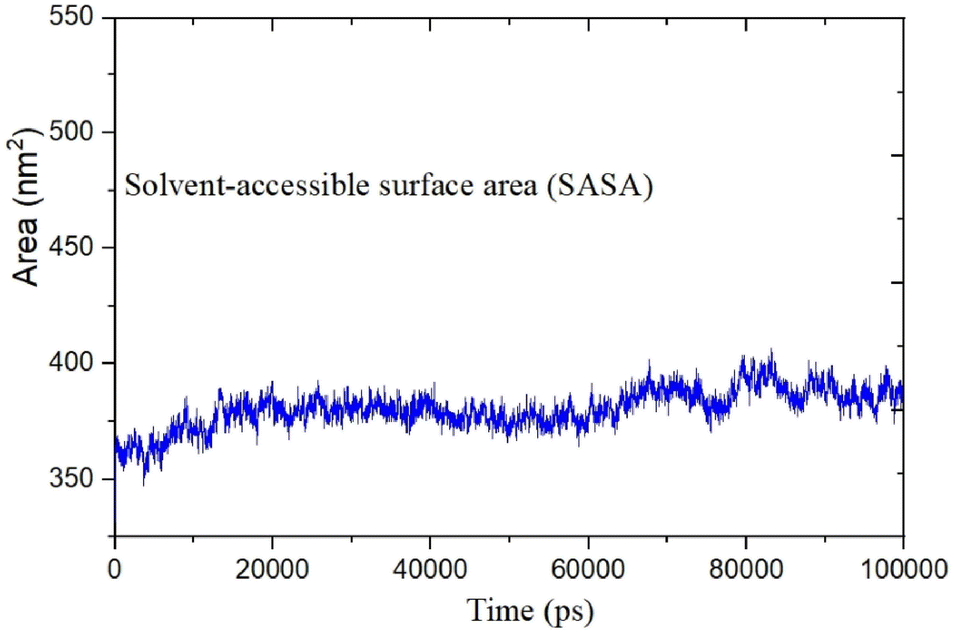
MD was utilized to examine the ligand's binding effectiveness toward the protein over time at an atomic level. Several characteristics, including RMSD, RMSF, radius of gyration, volume, density, and hydrogen bonds, play a major role in providing information about the binding pattern. As a result, a 100 ns MD simulation study was done to compare with the crystal structure of the C-terminal subunit of Human Maltase-Glucoamylase complexed with Acarbose to evaluate the overall stability and binding effectiveness of compound 5a or pose 120/400 with Human Maltase-Glucoamylase, as shown in Fig. 11A. RMSD parameter analysis provides detailed structural information for understanding the conformational stability of the system. Thus, RMSD studies for the protein (3TOP.pdb) in the presence of compound 5a were performed. The analysis revealed that the protein-compound 5a or pose 261/400 complex is highly equilibrated and stable; yet, before the 40 ns simulation, as seen in Fig. 11B, the ligand-complex was slightly fluctuating within its limit, indicating that the protein-compound 5 or pose 261/400 complex is stable overall. The root mean square fluctuation (RMSF) method examines the flexibility of the protein residues in the presence of compound 5a. As shown in Fig. 11C, the protein-compound (5a) complex exhibited a nearly identical fluctuation pattern, confirming constrained motions over the 100 ns simulation. However, some residues, namely, Asp, Asn, Glu, Asn, Gly, and Tyr, that were not directly involved in the catalytic site and had a high level of mobility with fluctuations ranging from 0.2 to 0.7 nm were non-significant, whereas residues involved in the catalytic site to form the best pose with compound 5a had significant dynamic behavior that led to a stable protein-compound 5a complex. In addition, the complex-ligand radius of gyration (Rg) was measured, as shown in Fig. 11D. The radius of gyration indicates the protein's compactness with protein folding and unfolding structures via thermodynamic effect during the 100 ns MD simulation. Fig. 11D depicts total Rg (Rx, Ry and Rz) values in nm for protein–ligand complexes. Under MD simulation, the Rg value of the best docking pose started at 2.86 nm, and the structure gradually raised the Rg value until it reached 2.90 nm at 20 ns (20000 ps). The Rg value was steady at 2.90 nm, with equilibrium at 100 ns and a slight decline at 99 ns, showing that the receptor-ligand 5a or pose 120/400 was stable and tightly packed, as indicated in Fig. 11D. To count the number of H-bonds involved in the MD simulation, the Gromacs g_hbond utility was used. The number of H bonds formed between the ligand 5a or pose 120 and the protein were determined using 100 ns simulation trajectories, as illustrated in Fig. 11E. Compound 5a or pose 261 demonstrated a continuous maximum of 1–3 H-bonds; hence, this output assists the ligand to be stabilized with no changes in the RMSD value during the simulation period. The hydrogen bond with pairs within 0.35 nm has a maximum of 12–13 H-bonds; however, this is rare in the system in Fig. 11E, showing red color. Aside from these findings, the solvent accessible surface area (SASA) was calculated for two complex systems to examine the interaction between the protein–ligand complexes and solvents. SASA was utilized to anticipate how the protein will change its shape because of binding, as shown in Fig. 11E. The solvation effects, such as the desolation of the protein cavity, ligand, and solvent molecule rearrangement, are linked to ligand–protein binding affinity. Based on the simulation results as seen in Fig. 12, the protein–ligand complex was shown to be stable, with a modest fluctuation in area between 370 and 400 nm2 (area) and small conformational changes of the protein during simulation.
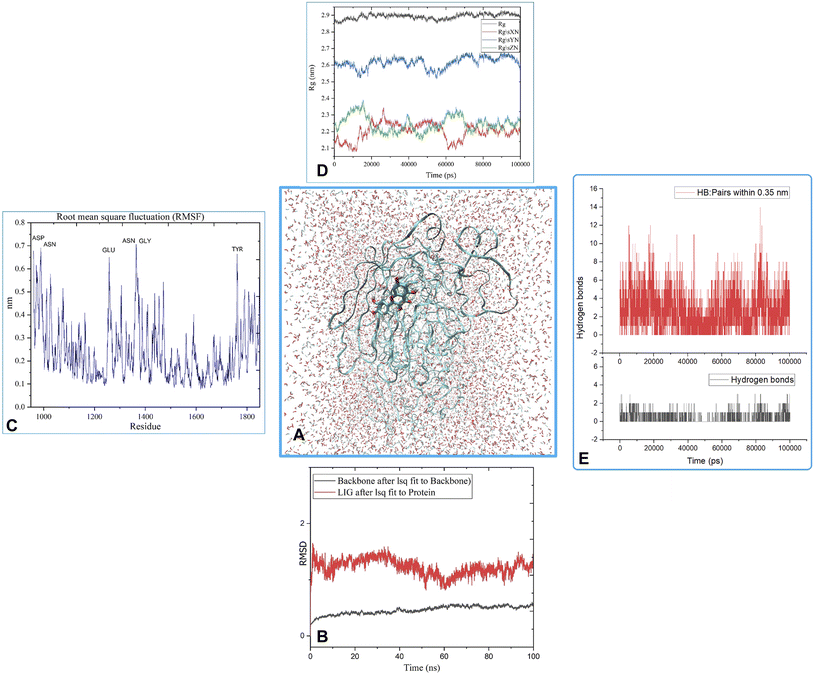 |
| | Fig. 11 (A) MD simulation analysis of the best pose 120/400 to receptor, 3TOP: A chain, (B) the root means square deviation (RMSD) of (5a) and protein backbone 3TOP calculated using 100 ns MD simulation, (C) root mean square fluctuations (RMSF), (D) radius of gyration (Rg) plot, (E) the number of hydrogen bonds. | |
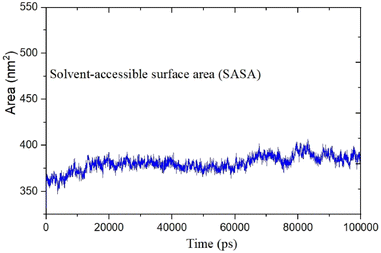 |
| | Fig. 12 The calculated solvent accessible surface area of the protein-compound (5a) complex. | |
3.7. Physicochemical, medicinal chemistry, and pharmacokinetic properties
The physicochemical, medicinal, and pharmacokinetic properties of compound 5a were calculated and are presented in Table S1–S10.† As shown in Table S1,† the physiochemistry properties exposed that these values are in the standard range. As revealed in Table S2,† an the important parameters of medicinal chemistry followed the standard exception for the values such as QED, Fsp,3 and MEC-18. The absorption properties like-drug of compound 5a are in the permissible range. As seen in Table S3,† the value of Caco-2 permeability is near the optimal value or little higher than that of the optimal value. As shown in Table S4,† the drug distribution of compound 5a exposed that the Plasma Protein Binding (PPB) parameter is higher than the optimal value of about 9%, and most of them were determined to be in this range. The properties of the drug metabolism of compound 5a were detected in the optimal values (Table S5†). The properties of excretion and toxicity of drug is depicted in Table S6 and S7,† respectively, and most of them followed the standard values. The properties of clearance and T1/2 indicated that the drug is clear and has a long half-life, respectively, in seen in Table S6.† As shown in Table S8,† the environmental toxicities of drug (5a) is in the acceptable range. The properties of the Tox21 pathway of compound 5a, as seen in Table S9,† are out of ranges and its toxicophoric rules are in the optimal ranges, as seen in Table S10.†
4. Discussion
Since the pathogenesis of diabetes is really complex and involves many mechanisms, increased research on the discovery of targets, pathways, and treatments has been reported.20 Among all the enzymes involved in the digestion process, α-glucosidase is the most important in digesting the uptake of carbohydrates and postprandial hyperglycemia. Hence, various natural products, new plants, and their extract have been well-known to exhibit their inhibitory activities against α-glucosidase and have been increasingly investigated. To further identify potential α-glucosidase inhibitory agents from B. macrophylla, we have reported some active constituents of the plant, which showed the most potent α-glucosidase inhibitory activity in this screening. All these compounds were tested for their α-glucosidase inhibitory activity to identify candidates for the development of drugs.
Assessments were performed regarding the α-glucosidase-inhibiting effects of isolated compounds 1–7. Most compounds revealed significant α-glucosidase inhibitory activity with IC50 values in the range of 9.2–266.3 μM (Table 1). Among the antidiabetic potential, the novel compound 8-bromoquercetin was found to have a significant active (IC50 ∼ 9.2 ± 0.72 μM) than acarbose, the first approved drug in α-glucosidase inhibitor (IC50 ∼ 332.4 μM) as previously described,21 whereas compound 2 has no activity.
The known compound 3 was identified as kaempferol, which showed a potent inhibitory activity on α-glucosidase with an IC50 value of 230 ± 2.7 μM. The compound is well-known to exhibit much stronger α-glucosidase inhibitory activity than acarbose ∼95.1% inhibition. Compound 3 can interact with some amino acid residues located within the active site of α-glucosidase regarding the catalytic center of α-glucosidase to refrain from the p-nitrophenyl-α-D-glucopyranoside entering and resulting in enzyme inactivity.22 Remarkably, to our knowledge, this is the first report on the inhibition of kaempferol on α-glucosidase from B. macrophylla.
In addition, the present results also revealed that the two compounds 4 and 6, Afzelin and Quercitrin, might be potent α-glucosidase inhibitors with IC50 values of the two compounds for inhibiting α-glucosidase of 118 ± 0.68 μM, and 255 ± 3.31 μM, respectively, which were lower than that of acarbose (a clinically applied α-glucosidase inhibitor, IC50 value was 332 ± 3.9 μM). The α-glucosidase inhibitory effects of the flavonoids in this study were similar to that of a previous study by in Zhang et al.'s research whose IC50 value was 0.231 ± 0.033 mg mL−1 and 0.215 ± 0.004 mg mL−1.23 The enhanced α-glucosidase inhibitory capacity of the identified flavonoids in this study may result from the different substituent group at the C-3 position of ring C, indicating that the two compounds might be potent α-glucosidase inhibitors.
Compounds 5 and 7, quercetin and myricetin, exhibited more potent activities, with IC50 values ranging from 117 ± 1.9 to 266 ± 2.97 μM, than the positive control acarbose (IC50, 332 ± 3.9 μM). It has been well-known that quercetin is an α-glucosidase inhibitor that was previously isolated from Forsythia suspensa (Thunb) Vahl,24 Matricaria recutita L.,25 Eucommia ulmoides,26 and Salicornia herbacea.27 Interestingly, as a flavonoid, myricetin is abundant in most plants, which was able to cut down the fasting blood glucose level through STZ-induced diabetic rats28 and in mice fed a high fat and high sucrose diet.29 Also, this compound was found to inhibit yeast α-glucosidase with an IC50 of 5 μg mL−1 in vitro.30 Thus, the hypoglycemic effect of myricetin could also be due to α-glucosidase inhibition. However, the α-glucosidase inhibitory effect of myricetin from B. macrophylla was not reported previously.
Interestingly, brominated substitution using sodium bromide and hydroperoxide was conducted on 3 and 5 to provide products 3a and 5a. This reaction was selected based on enhancing the alpha-glucosidase inhibition of brominated derivatives from the previous reports. Particularly, the bromination of flavonoids, kamatakenin, and ayanin dramatically increased their alpha-glucosidase inhibition.19 Further, brominated lichen metabolites showed more potent activity than their mother depsidones.31 Pose 120 is considered to have good ligand interactions with enzyme 3TOP or good enzyme inhibition in this model due to 3 parts of the ligand such as capping unit, connecting unit, and functional group of the ligand, which exposed fully ligand interactions.32 The capping group of this pose 120 detected the protein of enzyme 3TOP via alkyl or pi-alkyl from Lys 1089 of A chain to aromatic ring, alkyl, or pi-alkyl from Leu 1086: A chain to aromatic ring, and pi–sigma interaction from Lys 1086 to aromatic ring. The linker part of ligand is identified via alkyl or pi-alkyl from Lys 1088 to aromatic ring via one pi–sigma interaction. Finally, the functional group of pose 120 revealed hydrophilic interactions or hydrogen bonds from Gln 1109, Asn 1090, Ile 1087, and Tyr 1010 to hydrogen atoms of hydroxyl phenolic rings and oxygen atom of the carbonyl group of the ketone ring. Another ligand interaction, halogen interaction, is linked from the bromo atom on the pose to Lys 1089 on enzyme 3TOP. The secondary interactions is revealed in Fig. 4 between residual amino acids of A chain and active atom on pose 120 such as hydrogen bonding interactions (brown lines), which are involved in Tyr 1010, Asn 1090, Gln 1109, and Ile 1087 and active atoms on this pose. The steric interactions (light green lines) are exposed from Lys 1089, Ile 1087, Lys 1038, and Asn 1080 to active atoms on pose 120. The overlap interactions are indicated by violet circles on the pose. The sizes of violet circles are larger, and the overlap interactions are stronger. The ligand map indicated the strengths of ligand interactions in the processing of conformation. As shown in Fig. 6, the steric interactions are not strong because few amino acids can make around this pose. The value of RMSD of the pair pose (pose 120, compound 5a) are 3.598, which proved a small change of conformation of compound 5a after completing the docking of compound 5a.33 The values of RMSD are in the permissible range. It confirmed the validations of the model in redocking, orientational docking, conformation, active center, and interactions.34 Compound 5a also evaluated like-drugless via physiochemistry and ADMET model13 considering it as a candidate drug in in silico pharmacokinetic properties.
5. Conclusion
The current study employed combined in vitro and in silico approaches to evaluate the α-glucosidase inhibitory activity of isolated compounds from B. macrophylla. The results of the docking studies indicated that out of the studied compounds, 8-bromokaempferol (3a) and 8-bromoquercetin (5a) compounds displayed potent α-glucosidase inhibitory effect with an IC50 value of 173 ± 2.35 μM and 9.2 ± 0.72 μM, which were much better than that of acarbose (IC50: 1332 ± 3.9 μM). Meanwhile, molecular docking was employed to investigate the probable mechanism for α-glucosidase–compounds 3a/5a interactions. The findings were expected to additionally provide the research and development of stronger α-glucosidase inhibitors, where the flavonoid may act as the lead medicinal chemical compound, suggesting some lead compounds for the development of novel α-glucosidase inhibitor from B. macrophylla.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research is funded by Vietnam National University Ho Chi Minh City (VNU-HCM) under grant number C2022-44-04.
References
- J. Upadhyay, S. A. Polyzos, N. Perakakis, B. Thakkar, S. A. Paschou, N. Katsiki, P. Underwood, K. H. Park, J. Seufert, E. S. Kang, E. Sternthal, A. Karagiannis and C. S. Mantzoros, Metab. Clin. Exp., 2018, 78, 13–42 CrossRef CAS PubMed.
- N. Dechsupa, J. Kantapan, M. Tungjai and S. Intorasoot, Heliyon, 2019, 5, e02052 CrossRef PubMed.
- N. H. Nguyen, T. T. Nguyen, P. C. Ma, Q. T. H. Ta, T.-H. Duong and V. G. Vo, Molecules, 2020, 25, 1996 CrossRef CAS PubMed.
- K.-N. Ha, T.-V.-A. Nguyen, D.-T. Mai, N.-M.-A. Tran, N.-H. Nguyen, G. V. Vo, T.-H. Duong and H. Truong Nguyen, Saudi J. Biol. Sci., 2022, 29, 1029–1042 CrossRef CAS PubMed.
- G. Van Vo, P. C. Guest and N. H. Nguyen, Methods mol. biol., 2022, 2343, 215–228 CrossRef CAS PubMed.
- A. P. Devi, T. H. Duong, S. Ferron, M. A. Beniddir, M. H. Dinh, V. K. Nguyen, N. K. Pham, D. H. Mac, J. Boustie, W. Chavasiri and P. L. Pogam, Planta Med., 2020, 86, 1216–1224 CrossRef CAS PubMed.
- C. L. Tran, T. B. Dao, T. N. Tran, D. T. Mai, T. M. Tran, N. M. Tran, V. S. Dang, T. X. Vo, T. H. Duong and J. Sichaem, Molecules, 2021, 26, 2257 CrossRef CAS PubMed.
- G. Singh, A. Singh, V. Singh, R. K. Verma, J. S. Tomar and R. Mall, Med. Chem. Res., 2020, 1–21 Search PubMed.
- E. W. Bell and Y. Zhang, J. Cheminformatics, 2019, 11, 40 CrossRef PubMed.
- S. W. Chiu, S. A. Pandit, H. L. Scott and E. Jakobsson, J. Phys. Chem. B, 2009, 113, 2748–2763 CrossRef CAS PubMed.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. Berendsen, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- A. W. Schüttelkopf and D. M. van Aalten, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 1355–1363 CrossRef PubMed.
- G. Xiong, Z. Wu, J. Yi, L. Fu, Z. Yang, C. Hsieh, M. Yin, X. Zeng, C. Wu, A. Lu, X. Chen, T. Hou and D. Cao, Nucleic Acids Res., 2021, 49, W5–w14 CrossRef CAS PubMed.
- M. Jay, A. Hasan, B. Voirin, J. Favre-Bonvin and M. R. Viricel, Phytochemistry, 1978, 17, 1196–1198 CrossRef CAS.
- V. Seidel, F. Bailleul and P. G. Waterman, Phytochemistry, 2000, 55, 439–446 CrossRef CAS PubMed.
- F. Utari, A. Itam, S. Syafrizayanti, W. H. Putri, M. Ninomiya, M. Koketsu, K. Tanaka and M. Efdi, J. App. Pharm. Sci., 2019, 9(8), 53–65 CrossRef CAS.
- I.-W. Hwang and S.-K. Chung, Prev. Nutr. Food Sci., 2018, 23, 341–346 CrossRef PubMed.
- J. Rencoret, M. J. Rosado, H. Kim, V. I. Timokhin, A. Gutiérrez, F. Bausch, T. Rosenau, A. Potthast, J. Ralph and J. C. Del Río, Plant Physiol., 2022, 188, 208–219 CrossRef CAS PubMed.
- T.-B.-N. Dao, T.-M.-T. Nguyen, V.-Q. Nguyen, T.-M.-D. Tran, N.-M.-A. Tran, C. H. Nguyen, T.-H.-T. Nguyen, H.-H. Nguyen, J. Sichaem, C.-L. Tran and T.-H. Duong, Molecules, 2021, 26, 2531 CrossRef CAS PubMed.
- H. Y. Hung, K. Qian, S. L. Morris-Natschke, C. S. Hsu and K. H. Lee, Nat. Prod. Rep., 2012, 29, 580–606 RSC.
- A. E. Martin and P. A. Montgomery, Am. J. Health-Syst. Pharm., 1996, 53, 2277–2290 CrossRef CAS PubMed.
- X. Peng, G. Zhang, Y. Liao and D. Gong, Food Chem., 2016, 190, 207–215 CrossRef CAS PubMed.
- X. Zhang, B. Cheng, X. Liu, Y. Li, J. Hou, S. Chen, J. Chen and S. Li, ChemistrySelect, 2020, 5, 8440–8446 CrossRef CAS.
- W. Kang, J. Wang and L. Zhang, Zhongguo Zhongyao Zazhi, 2010, 35, 1156–1159 CAS.
- H. Ichiki, O. Takeda, I. Sakakibara, S. Terabayashi, S. Takeda and H. Sasaki, J. Nat. Med., 2007, 61, 146–153 CrossRef CAS.
- J. Watanabe, J. Kawabata, H. Kurihara and R. Niki, Biosci. Biotechnol. Biochem., 1997, 61, 177–178 CrossRef CAS PubMed.
- D. Lee, J. Y. Park, S. Lee and K. S. Kang, Processes, 2021, 9, 483 CrossRef CAS.
- K. C. Ong and H. E. Khoo, Life Sci., 2000, 67, 1695–1705 CrossRef CAS PubMed.
- H. N. Choi, M. J. Kang, S. J. Lee and J. I. Kim, Nutr. Res. Pract., 2014, 8, 544–549 CrossRef CAS PubMed.
- K. Tadera, Y. Minami, K. Takamatsu and T. Matsuoka, J. Nutr. Sci. Vitaminol., 2006, 52, 149–153 CrossRef CAS PubMed.
- N.-K.-T. Pham, N.-M.-A. Tran, H. Truong Nguyen, D.-D. Pham, T.-Q.-T. Nguyen, T.-H.-A. Nguyen, H.-T. Nguyen, T.-H. Do, N.-H. Nguyen and T.-H. Duong, Arab. J. Chem., 2022, 15, 103535 CrossRef CAS.
- G. Giannini, W. Cabri, C. Fattorusso and M. Rodriquez, Future Med. Chem., 2012, 4, 1439–1460 CrossRef CAS PubMed.
- M. N. Sibanyoni, S. K. Chaudhary, W. Chen, H.-R. Adhami, S. Combrinck, V. Maharaj, D. Schuster and A. Viljoen, Fitoterapia, 2020, 146, 104650 CrossRef CAS PubMed.
- S. Hidalgo-Figueroa, A. Rodríguez-Luévano, J. C. Almanza-Pérez, A. Giacoman-Martínez, R. Ortiz-Andrade, I. León-Rivera and G. Navarrete-Vázquez, Eur. J. Pharmacol., 2021, 907, 174244 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Thuc-Huy Duong*c and
Giau Van Vo
b,
Thuc-Huy Duong*c and
Giau Van Vo