
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemically recyclable fluorescent polyesters via the ring-opening copolymerization of epoxides and anhydrides†
Taylor B.
Young
a,
Owaen G.
Guppy
a,
Alysia J.
Draper
a,
Joshua M.
Whitington
a,
Benson M.
Kariuki
 a,
Alison
Paul
a,
Mark
Eaton
b,
Simon J. A.
Pope
*a and
Benjamin D.
Ward
*a
a,
Alison
Paul
a,
Mark
Eaton
b,
Simon J. A.
Pope
*a and
Benjamin D.
Ward
*a
aCardiff University, School of Chemistry, Main Building, Park Place, Cardiff CF10 3AT, UK. E-mail: WardBD@cardiff.ac.uk; PopeSJ@cardiff.ac.uk
bCardiff University, School of Engineering, Queen's Buildings, The Parade, Cardiff CF24 3AA, UK
First published on 17th April 2023
Abstract
Polymers rarely possess the desired properties for their intended application; additives (plasticizers, flame-retardants, stabilizers, colourants) modify their physical properties to those needed in the final product. Colourants are particularly detrimental to recycling technologies, since they cannot be removed by reprocessing; reprocessed plastic retains color from the original material. We have prepared single-component, coloured polymers by adding chromophore monomers to epoxide-anhydride ring-opening copolymerization reactions; chromophores need only be added at dopant levels to give highly coloured materials with little change in properties from the base polymer. We have depolymerized the polymers to the parent acid/alcohol and have remade colourless polymer from reformed epoxide and anhydride. Our approach paves the way to using dopant-level monomers to access recyclable polymers with dial-a-property tunability.
Introduction
Plastic pollution is one of the most publicized environmental concerns of recent years. Whilst plastics have conferred a great benefit to society, the difficulty of recycling them in an infinite series of make-unmake-remake cycles has led to a substantial environmental impact, with no realistic solution identified for many traditional plastics.1–5Epoxide-anhydride copolymers have significant structural diversity. This makes them adaptable to a wide range of applications, whilst the ester linkages make them amenable to recycling via depolymerization.6–8 These copolymers are prepared via the ring-opening copolymerization (ROCOP) of epoxides and cyclic anhydrides, which means that there are thousands of molecular identities (and therefore physical properties) that can, in principle, be accessed using a common preparation methodology.9–24
One of the greatest challenges to recycling plastic materials is the presence of additives, used to modify their properties.25 Examples typically include plasticizers and dyes; their complete removal is often impossible and thus renders recycled plastics sub-standard, either because they have inferior mechanical properties or because the reprocessed polymer is tainted with pigments from the original material, making it less attractive to consumers.26,27 The colouration of poly(lactic acid) (PLA) has been achieved by polymerizing lactide with chromophore-containing catalysts or initiators (the DyeCat process),28 which removes the need for environmentally-polluting dyeing processes. In contrast to PLA, the molecular identities of the monomers used in epoxide-anhydride ROCOP are much more diverse, and we therefore hypothesized that a chromophore-containing monomer might be added to the ROCOP reaction to colour the polymer, such that the dye becomes covalently bonded within the polymer chains (Fig. 1). This would afford a terpolymer consisting mainly of the two base monomers with only a small amount of chromophore necessary to invoke the desired colouration, in effect adding a dopant-level quantity of chromophore. The advantages of this approach are that the dye is held within the polymer by strong covalent bonds and is therefore expected to have the highest possible colour-fastness; the dye can then be recovered by depolymerization of the polymer since the chromophore would be liberated as a bis(alcohol) or bis(acid), depending on which monomer functionality is used. There is also no need to modify the catalyst used to prepare the coloured polymer since the functional group in the dopant is identical to that in the base monomer. Moreover, whilst we envisage that only a small quantity of chromophore will be required, the amount of added dopant is not a limitation for alternative applications (e.g. when employing the method introduced here to functionality other than colour) since a higher proportion of dopant can easily be substituted for one of the base monomers.
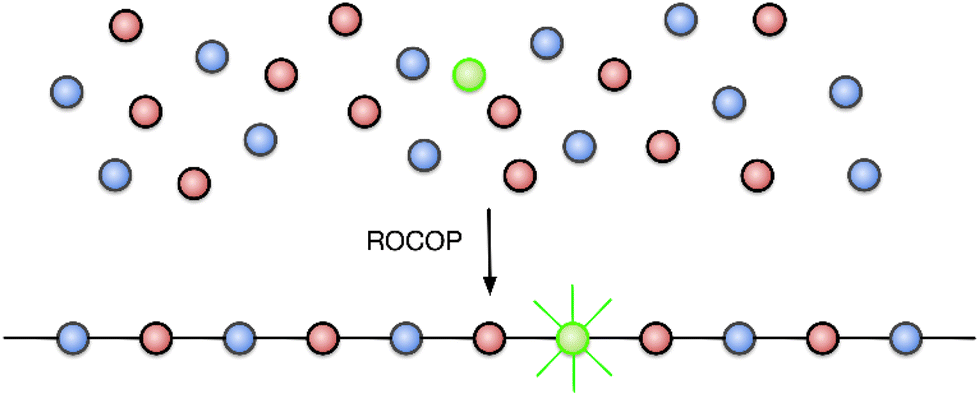 | ||
| Fig. 1 Chromophore-doped polymers via ring-opening copolymerization (ROCOP). | ||
Results and discussion
Synthesis of luminophore monomers and coloured polymers
Green and red luminescent monomers were prepared by functionalizing 4-piperidinyl-1,8-naphthalimide and 1-aminoanthraquinone with epichlorohydrin, to afford naphthalimide (Nap, 1) and aminoanthraquinone (AQ, 2) epoxides (Scheme 1).29,30 Their solid-state structures were determined by X-ray crystallography (S2.2 and S2.3).31–34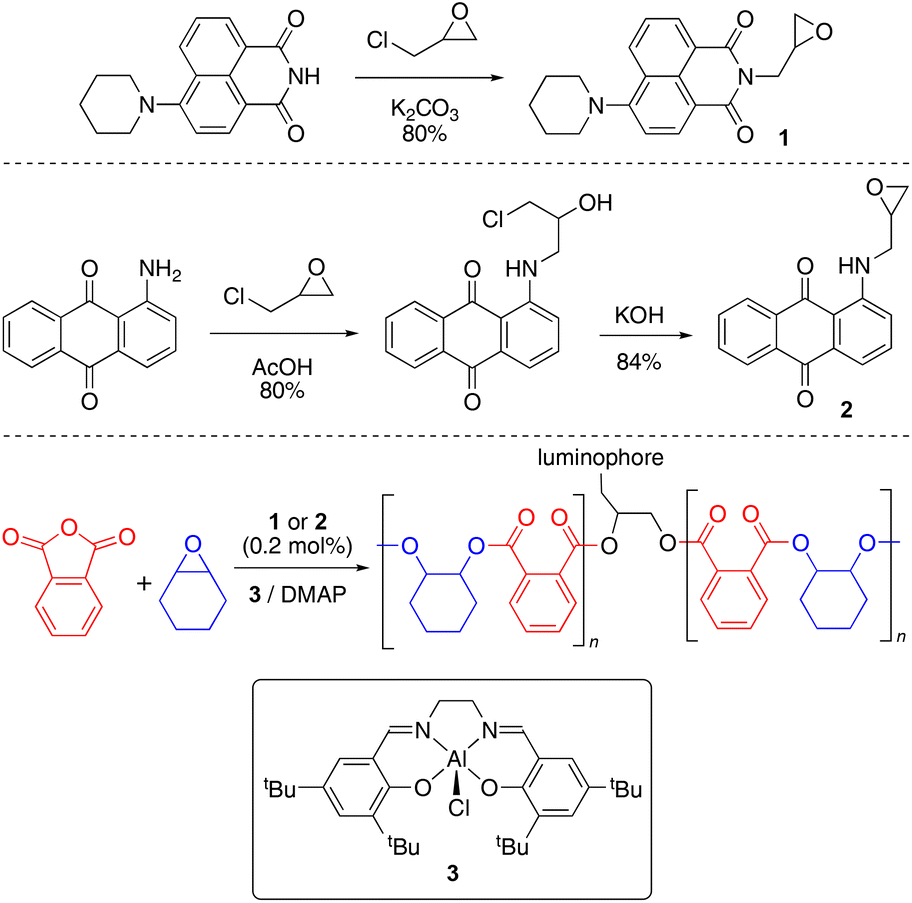 | ||
| Scheme 1 Synthesis of luminophore monomers and doped copolymers. | ||
The luminescent monomers were subsequently incorporated into copolymers of phthalic anhydride and cyclohexene oxide [poly(phthalic anhydride)-alt-(cyclohexene oxide), abbreviated to PA-CHO] via ring-opening copolymerization by [Al(tBu-salen)Cl] (3) and 4-dimethylaminopyridine (DMAP);35 polymer data are provided in Table 1. The polymerization reactions were performed in a 2 litre mechanically-stirred jacketed glass reactor (S1.3) on a 1.28 mol scale with 0.2 mol% of the appropriate dopant (and a control with no dopant) to afford 280–320 g of material (Table 1, entries 1, 2 and 6).‡ This scale is larger than usual in ROCOP research and the ability to prepare such quantities represents a significant step towards enabling materials research with anhydride-epoxide copolymers. The doped copolymers were obtained as intensely coloured powders (Fig. 2); AAQ-PA-CHO was orange and Nap-PA-CHO was bright yellow. The copolymers were easily melted and cast into simple shapes; the solid blocks thereby produced were brittle and would shatter easily on removal from the mould; cast AAQ-PA-CHO was produced as a deep red solid.
 | ||
| Fig. 2 ROCOP polymer samples; powdered products are shown below the corresponding melt-cast samples. From left to right, PA-CHO, AAQ-PA-CHO (0.2%), Nap-PA-CHO (0.2%). | ||
| Entry | Polymer | Dopant/mol% | Conv. % | T g/°C | M n /Da | % ester | Đ |
|---|---|---|---|---|---|---|---|
a Conditions: [Ep]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[Anh]0:[Al]0:[DMAP]0 = 400:400:1:2 in toluene; conversion based upon anhydride signals in 1H NMR spectra. Polymer was dried in a vacuum oven at 80 °C overnight.
b 3.16 mmol 3.
c 6.4 μmol 3.
d molecular weights measured by triple-detection GPC.
e No observable transition in the DSC data.
f Ester signals observed but ether signals obscured by chromophore and so %ester cannot be determined reliably.
g From the polymerization of PA and CHO obtained from the depolymerization of entry 2. :[Anh]0:[Al]0:[DMAP]0 = 400:400:1:2 in toluene; conversion based upon anhydride signals in 1H NMR spectra. Polymer was dried in a vacuum oven at 80 °C overnight.
b 3.16 mmol 3.
c 6.4 μmol 3.
d molecular weights measured by triple-detection GPC.
e No observable transition in the DSC data.
f Ester signals observed but ether signals obscured by chromophore and so %ester cannot be determined reliably.
g From the polymerization of PA and CHO obtained from the depolymerization of entry 2.
|
|||||||
| 1 | PA-CHOb | — | 96 | 140 | 12100 |
96 | 1.09 |
| 2 | Nap-PA-CHOb | 0.2 | 98 | 138 | 14600 |
94 | 1.06 |
| 3 | Nap-PA-CHOc | 12 | 82 | 128 | 8300 | 96 | 1.01 |
| 4 | Nap-PA-CHOc | 33 | 82 | 108 | 19000 |
89 | 1.01 |
| 5 | Nap-PA-CHOc | 63 | 85 | —e | 24600 |
—f | 1.36 |
| 6 | AAQ-PA-CHOb | 0.2 | 96 | 138 | 10200 |
94 | 1.05 |
| 7 | AAQ-PA-CHOc | 14 | 79 | 137 | 28700 |
97 | 1.20 |
| 8 | AAQ-PA-CHOc | 34 | 78 | 137 | 42300 |
96 | 1.14 |
| 9 | AAQ-PA-CHOc | 64 | 79 | 138 | 39300 |
95 | 1.23 |
| 10 | TCPA-CHOc | — | 91 | 172 | 56500 |
96 | 1.20 |
| 11 | PA-POc | — | 44 | 61 | 34000 |
94 | 1.01 |
| 12 | Nap-TCPA-CHOc | 0.2 | 96 | 157 | 24100 |
94 | 1.10 |
| 13 | Nap-PA-POc | 0.2 | 53 | 63 | 36000 |
98 | 1.47 |
| 14 | AAQ-TCPA-CHOc | 0.2 | 90 | 147 | 29100 |
94 | 1.24 |
| 15 | AAQ-PA-POc | 0.2 | 58 | 42 | 45600 |
95 | 1.06 |
| 16 | PA-CHOg | — | 51 | 135 | 11100 |
94 | 1.17 |
To minimize the effect of any residual solvent on the molecular weight determination by GPC, polymers were dried in vacuo at 80 °C for 24 hours prior to analysis. All of the molecular weight distributions were bi- or trimodal, as is routinely observed in ROCOP owing to the presence of trace amounts of diacid from the anhydride and/or adventitious water, each of which can act as chain transfer agents and add to the modality of the molecular weight distributions (see S3.1 for chromatograms).17 The microstructures of the polymers were probed by integration of the ester and ether regions of their 1H NMR spectra;36 the alternating selectivity was universally high and indicates that the ability to prepare well-defined alternating polyesters is unaffected by undertaking the reaction at >1 mole scale.
The flexibility of our approach to incorporating more dopant was demonstrated by substituting 10 mol%, 25 mol%, and 50 mol% of the CHO feedstock for each of the chromophores (Table 1, entries 3–5 and 7–9). In these cases, we were able to ascertain the amount of chromophore incorporated into the polymer using their 1H NMR spectra, giving 12%, 33%, and 63% for Nap and 14%, 34%, 64% for AAQ. Likewise, our approach is not limited to PA and CHO, since we substituted PA for tetrachlorophthalic anhydride (TCPA, entries 10, 12, 14) and CHO for propylene oxide (PO, entries 11, 13, 15).‡ In general, there was greater variation in molecular weight between dopant-free and 0.2% doped polymers with these alternative monomers; we note that PO is more volatile than CHO (34 °C vs. 130 °C in a reaction temperature of 80 °C) and TCPA has lower solubility under our reaction conditions; these factors may have a bearing on our observations. The variation in molecular weight likely explains the variation in Tg in these monomer combinations, since only 0.2% dopant is unlikely to have a measurable impact in this regard.
Although the polymer samples adopted the colour of the chromophore, our thesis relies on the dopants being integrated into the polymer chains, rather than co-existing as additives. Continual washing and re-precipitation of the doped polymer failed to remove the colour, unlike the undoped polymer with added chromophore-epoxide, from which the colour could be washed out with isopropanol. Moreover, we collected the waste stream into vials during GPC measurements and looked for the signature green fluorescence upon UV irradiation. For example, in the GPC analysis of Nap-PA-CHO we observed the fluorescent fractions co-eluting with the polymer (Fig. 3); a non-incorporated chromophore will elute at the end of the GPC experiment (small molecular size).
 | ||
| Fig. 3 Eluted GPC solvent showing the emission from the yellow (green-emissive) Nap-PA-CHO polymer in vials 5–7 (tR = 14–17 min; each vial = 1 min, i.e. 1 mL). | ||
Whilst the luminescent GPC vials in Fig. 3 are consistent with the dopant being incorporated onto the polymer backbone, it was impossible to obtain meaningful MALDI-ToF or DOSY NMR experiments owing to the very low amounts of chromophore, which are below the detection limits of these methods. Therefore, we studied the copolymers containing higher dopant loadings, for these materials have chromophore concentrations within a detectable range for DOSY NMR experiments (S3.2). DOSY data at all dopant concentrations (Table 1, entries 3–5 and 7–9) indicated that the chromophore signals have identical diffusion coefficients to those of the PA-CHO copolymer base. Taken together, these data are consistent with the chromophores being bonded within the same polymer chains as the base material.
During optimisation of the reaction time, we noted higher chromophore incorporation than expected for lower conversions. For example, with 10% AAQ feed into PA-CHO, 39% and 23% incorporation (relative to CHO) was observed for 29% and 48% conversion, respectively. These data are consistent with a bias towards the chromophore being incorporated at the start of the reaction. Addition of the AAQ monomer presumably has a lower activation energy than CHO in this case, and a bias towards early/late incorporation is expected if the activation energies are unequal,37 since epoxide opening is reported to be the rate-limiting step.38,39
Physical properties
DSC measurements were obtained for all polymers (S3.3), from which glass transition temperatures (Tg) were obtained. As expected for small dopant quantities and comparable molecular weights, the undoped and 0.2 mol% doped PA-CHO copolymers showed no significant variation in their Tg, lying between 138 and 140 °C. Changing the concentration of Nap led to a steady decrease in Tg from 138 °C to 108 °C as the proportion of Nap was increased from 0.2 to 10 and finally 25 mol%, with no visible transition being found for 50 mol%. Interestingly, increasing the quantity of AAQ had no effect on the Tg, with all values measured as 137–138 °C for all concentrations up to 50 mol%.Adding 0.2% chromophore to TCPA-CHO afforded a copolymer with substantially reduced molecular weight compared to the dopant-free material, and the Tg value showed a modest reduction from 172 °C to 147 °C and 157 °C for Nap and AAQ, respectively. Adding 0.2% Nap to PA-PO gave a comparable molecular weight (and Tg) whereas adding 0.2% AAQ gave a higher molecular weight material with a reduction in Tg from 61 °C to 42 °C.
Absorption and luminescence properties
The undoped polymer exhibited strong absorptions below 300 nm which are attributed to the π–π* transitions of the aromatic phthalate constituents; there are no absorption characteristics above 315 nm. Upon doping at 0.2% the appearance of the absorption spectra were modulated in the visible region in a way that was consistent with the incorporation of the different dopants. For example, the naphthalimide dopant exhibits a signature intramolecular charge transfer (ICT) absorption maximum at 410 nm, which is observed in the corresponding polymer (S4.1) and described via the movement of electron density from the piperidinyl group onto the aromatic and imide system (TD-DFT analyses in S5.1). The reduction in molar absorption coefficient presumably reflects the concentration of the naphthalimide dopant in the polymer. Similarly, addition of the anthraquinone dopant resulted in a new absorption band peaking at ca. 490 nm. Again, this can be attributed to the absorption features of the AAQ chromophore and are assigned to an absorption process with significant ICT character (amine to quinone).40 Assuming that the molar absorptivity coefficient of the luminophore is unaffected after polymerization, the absorption spectra give dopant concentrations of 0.24 mol% (Nap-PA-CHO) and 0.17 mol% (AAQ-PA-CHO), which are commensurate with the amount of dopant added to the polymerization reactions.Following elucidation of the absorption properties and confirmation of the contributions of the chromophoric dopants, the doped polymers were investigated for their luminescence properties. Firstly, upon irradiation with a benchtop UV lamp, the green emission of the naphthalimide doped Nap-PA-CHO could be clearly visualized (see Fig. 4), qualitatively indicating a material with green emissive character. Spectroscopic analysis of the solid-state Nap-PA-CHO samples revealed an apparent variation of emission wavelength upon the dopant concentration. Increasing levels of naphthalimide dopant (0.2% to 63%) led to a progressive bathochromic shift in the ICT emission maximum (note λem = 527 nm for 1 in the solid state) within the green region from 500 nm to 540 nm (Fig. 4). Given the well-known sensitivity of ICT excited state to the local environment, these data imply that the predominant locale of the naphthalimide fluorophore may vary with increasing dopant levels. Furthermore, time-resolved measurements on Nap-PA-CHO samples indicate, firstly, a fluorescence process (with lifetimes < 10 ns), but also a subtle variation according to dopant concentration. For example, increasing dopant level to 33–63% leads to a shortening of the fluorescence lifetime(s) and a decay profile that is more appropriately described by a dual exponential function (see Fig. 4, inset), and thus indicative of (at least) two different fluorophore environments. The modest quenching in lifetime may relate to the bathochromic shift in emission wavelength, and multiple components to the fitted decay may describe the different environments (for example, exposed surface sites versus those more embedded within the polymer). The emission spectra for the solid-state samples of the AAQ doped materials also showed a similar, but less pronounced, trend in emission maxima, but within the red region from 605–625 nm with increasing dopant level (note λem = 634 nm for 2 in the solid state). All lifetime measurements for AAQ-PA-CHO materials were obtained as <1 ns.
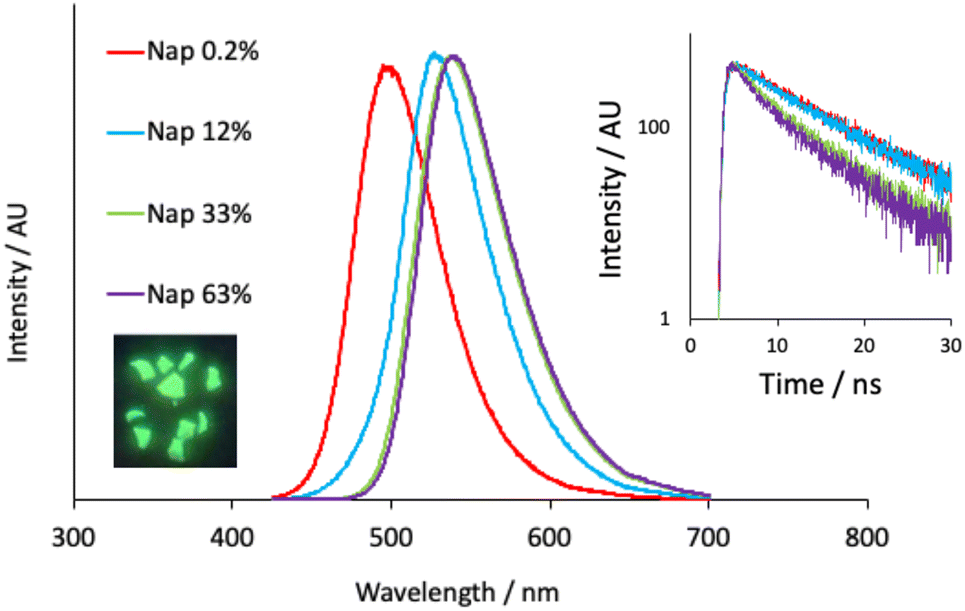 | ||
| Fig. 4 Solid state emission spectra of Nap-PA-CHO (λex = 410 nm) at different dopant levels. Inset: the corresponding decay profiles (λex = 295 nm) for the different materials (fits yielding lifetimes of 8.0, 7.9, 6.8 (80% weighting) and 2.6% (20% weighting), 7.3 (71% weighting) and 2.4 ns (29% weighting) for Nap dopant levels of 0.2%, 12%, 33% and 63%, respectively). | ||
Related measurements on dilute solutions of the 0.2% doped polymers were also recorded. Steady state emission measurements on Nap-PA-CHO gave an emission maximum at 510 nm (S4.7) and thus comparable to the solid-state characteristics. As seen with the absorption spectrum, the emission properties of the doped polymer are closely comparable to that for the native dopant, 2, with both polymer and monomer displaying similar quantum yield values of 45% and 47% respectively. The broad, featureless emission profile is again attributed to an emitting ICT excited state, and the corresponding emission lifetime (ca. 9 ns) confirmed the fluorescent nature of the emission. Similarly, for AAQ-PA-CHO the anthraquinone dopant displayed the expected ICT fluorescence (τ < 1 ns) band around 590 nm (consistent with the features of compound 3), and quantum yield values of ca. 4% and 5% respectively. In all cases it was therefore concluded that the doped polymers demonstrate fluorescence properties, in either solid or solution, that can be directly attributed to the characteristics of the fluorescent dopant providing both green and red emissive polymeric materials.
Finally, the parent undoped polymer was also investigated. Several excitation wavelengths were investigated and irradiation at 365 nm (i.e. where the undoped polymer did not display any significant absorption, S4.2) unexpectedly gave rise to an emission band at 480 nm. A comparison of the emission properties of undoped PA-CHO and the catalyst [Al(Salen)Cl] revealed a coincident emission feature to the spectra (S4.3). Therefore, it is likely that this observation may be attributed to some integration of the catalyst into the polymer. Previous work on the DyeCat process has deliberately exploited the integration of the catalyst; it remains bonded to the termini of the polymer chains, and in so doing, imparts chromophoric (introduced by ligand variants) character upon the polymer. While a similar observation cannot be excluded here, in the cases of Nap-PA-CHO and AAQ-PA-CHO, the emission characteristics are obviously dominated by the naphthalimide and anthraquinone fluorophores respectively, with no spectroscopic evidence of residual fluorescence from the catalyst.
Chemical recycling of Nap-PA-CHO
Epoxide-anhydride copolymers are known to depolymerize into the corresponding bis(carboxylic acid) and bis(alcohol). These can be used to remake copolymers via a dehydration step-growth polymerization process, thus rendering epoxide/anhydride copolymers circular from a recycling perspective. However, such an approach is beset with problems in that the remaking polymerization is inherently different to the initial process and may not produce an identical material, which could be undesirable in some settings. A more convincing circular economy of epoxide-anhydride copolymers would be obtained if the original monomers could be reformed for use in ROCOP, thus affording a perpetual make-degrade-remake recycling procedure.6,41 This approach has not yet been demonstrated, possibly since the formation of epoxide in particular is energetically expensive. However, it is possible to reform the anhydride and epoxide monomers from bis(acid) and bis(alcohol) respectively, which we demonstrate below.We sought to prove the viability of this approach and to demonstrate full circular economy of the doped polymers, to unambiguously demonstrate that circularity can be achieved, albeit in a synthetic laboratory setting. To this end, 0.2% Nap-PA-CHO was depolymerized by reacting a THF solution of polymer with 40% aqueous KOH (Scheme 2). This afforded a homogeneous liquid which separated into two fractions on cooling to 5 °C; 1H NMR spectra indicated only potassium phthalate in the aqueous fraction and trans-cyclohexane-1,2-diol in the organic fraction (Fig. 5). The residue from the organic fraction was yellow, indicating the presence of the naphthalimide chromophore; a mass spectrum of the organic-soluble residue indicated the presence of 4-piperidinyl-(N-propane-2,3-diol)-1,8-naphthalimide (S6.1). Considering that all the polymer was consumed, and that aromatic and aliphatic signals were detected in separate layers of the reaction mixture, this precludes the formation of oligomers and demonstrates complete depolymerization of the material.
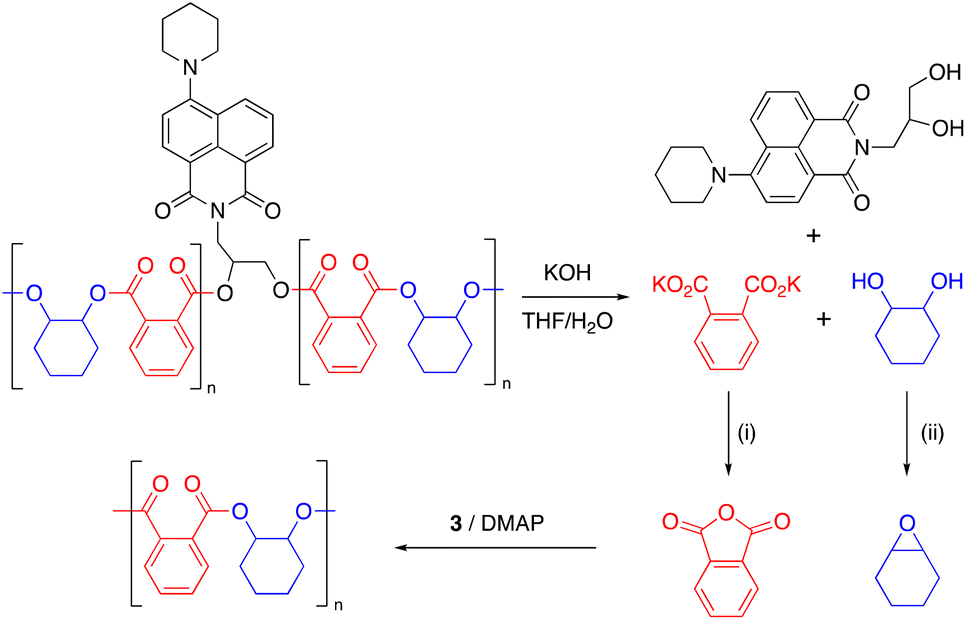 | ||
| Scheme 2 Chemical recycling of Nap-PA-CHO. Conditions: (i) HCl; acetic anhydride, reflux, 18 h. (ii) Ph3P, diethyl azodicarboxylate, rt, 18 h. | ||
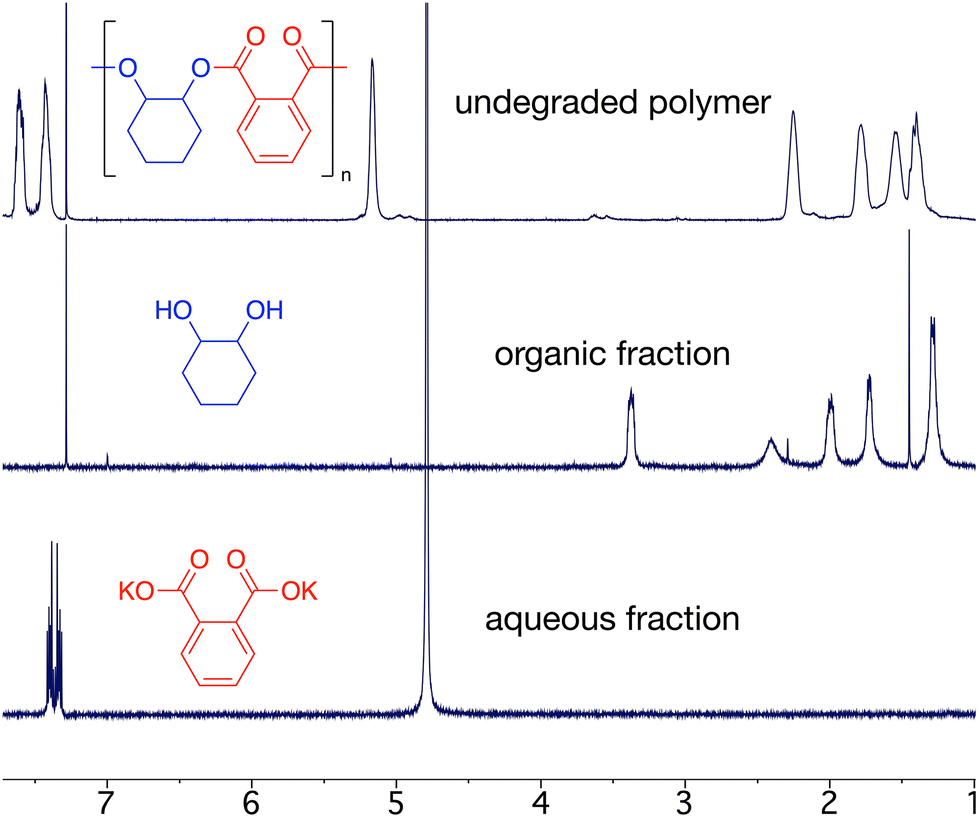 | ||
| Fig. 5 1H NMR spectra (300 MHz) of Nap-PA-CHO and crude organic and aqueous fractions after depolymerization with potassium hydroxide. No signals for the chromophore were observed in the spectra owing to the low concentration (0.2 mol%). | ||
Phthalic acid was isolated from the aqueous layer in 80% yield. Quantitative trans-cyclohexane-1,2-diol was obtained from the organic layer, which could be recrystallized from ethyl acetate to afford pure trans-cyclohexane-1,2-diol as a white crystalline solid in 46% isolated yield (S2.4); the yellow chromophore remained in the supernatant solution (Fig. 6). Analysis of the non-recrystallized diol mixture, the recovered trans-cyclohexane-1,2-diol, and the supernatant solution confirmed that the chromophore was entirely removed from the trans-cyclohexane-1,2-diol.
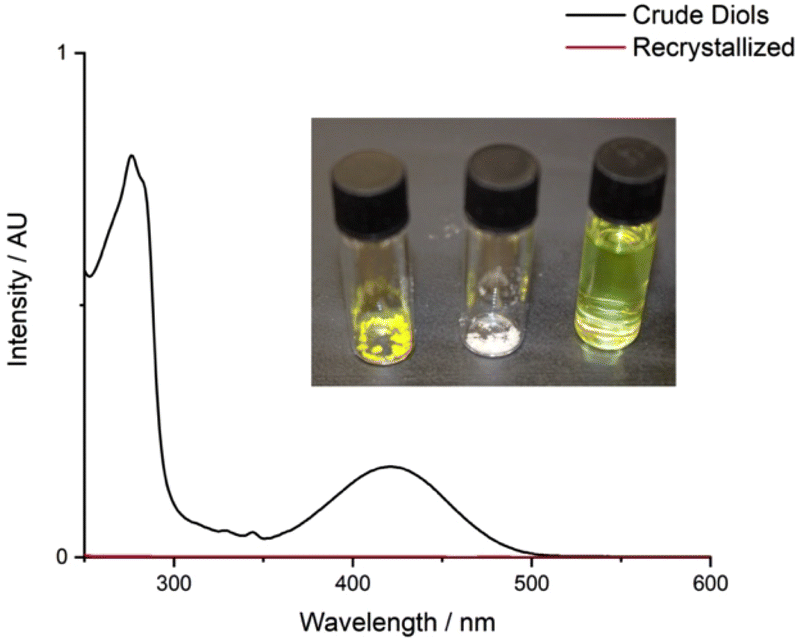 | ||
| Fig. 6 UV-vis absorption spectra of the crude and recrystallized diol mixture from depolymerization. Inset images from left to right show the crude mixture, the recrystallized trans-cyclohexane-1,2-diol, and the supernatant. | ||
The isolated phthalic acid was converted to phthalic anhydride by reaction with acetic anhydride,42 and trans-cyclohexane-1,2-diol was converted to cyclohexene oxide using the Mitsunobu reaction (S6.1.2).43 Although the Mitsunobu reaction suffers from poor atom economy, low yield, and the use of a highly reactive reagent, our data show that the depolymerization products can be re-formed into the original monomers, and we envisage future efforts finding more industrially-viable methods to achieve the same result that we report herein, with the certainty that the process is chemically viable.
The re-made monomers were used to prepare fresh white copolymer with 94% ester selectivity (compared to 96% for virgin PA-CHO) and comparable molecular weight and glass transition temperature to the original material (compare entries 1 and 16), thus demonstrating full circularity of these copolymers. A 1H NMR spectrum of the remade PA-CHO is shown in S6.5.
Conclusions
In conclusion, the addition of epoxide-functionalized chromophores to epoxide-anhydride copolymerization reactions leads to the incorporation of the chromophore into the polymer chains, inducing colouration and luminescence of the polymers. The doped polymers can be depolymerized, the chromophore removed, and the base monomers remade and repolymerized to give colour-free polymer in a circular economy of coloured polymers. The ability to take a coloured polymer and chemically recycle it to afford pure white polymer solves an important challenge in the plastics recycling industry; the scope of this approach is therefore profound. In addition, our strategy should be amenable to a wide range of dopants that can introduce different functionalities to impart bespoke physical and chemical properties upon the polymer. We therefore perceive a major expansion in the application of such doped polymers and are currently investigating a broad range of opportunities using these materials.Data availability
The data supporting the results presented in this article are freely available via the Cardiff University Data Catalogue at https://dx.doi.org/10.17035/d.2022.0152013843.Author contributions
Owaen G. Guppy undertook the initial proof of principle conceptual and experimental work, which was then expanded on by Taylor B. Young, Alysia J. Draper, and Joshua M. Whitington; Benson M. Kariuki collected the X-ray diffraction data, Alison Paul and Mark Eaton supervised the polymer materials testing and assisted with data analysis, Simon J. A. Pope and Benjamin D. Ward designed the experimental strategy and assisted with data analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by Cardiff University (studentship to OGG and research infrastructure grant) and the EPSRC (studentship to TBY and access to the National Crystallography Service). Access to the Advanced Research Computing facility at Cardiff University (ARCCA) is gratefully acknowledged.References
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- L. T. J. Korley, T. H. Epps, B. A. Helms and A. J. Ryan, Science, 2021, 373, 66–69 CrossRef CAS PubMed.
- M. MacLeod, H. P. H. Arp, M. B. Tekman and A. Jahnke, Science, 2021, 373, 61–65 CrossRef CAS PubMed.
- J. C. Worch and A. P. Dove, ACS Macro Lett., 2020, 1494–1506 CrossRef CAS PubMed.
- I. Vollmer, M. J. F. Jenks, M. C. P. Roelands, R. J. White, T. Harmelen, P. Wild, G. P. Laan, F. Meirer, J. T. F. Keurentjes and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2020, 59, 15402–15423 CrossRef CAS PubMed.
- G. L. Gregory, G. S. Sulley, L. P. Carrodeguas, T. T. D. Chen, A. Santmarti, N. J. Terrill, K.-Y. Lee and C. K. Williams, Chem. Sci., 2020, 11, 6567–6581 RSC.
- S. Kakadellis and G. Rosetto, Science, 2021, 373, 49–50 CrossRef CAS PubMed.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- W. Gruszka and J. A. Garden, Nat. Commun., 2021, 12, 3252 CrossRef CAS PubMed.
- S. Paul, Y. Zhu, C. Romain, R. Brooks, P. K. Saini and C. K. Williams, Chem. Commun., 2015, 51, 6459–6479 RSC.
- J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167–15197 CrossRef CAS PubMed.
- D. J. Darensbourg, R. R. Poland and C. Escobedo, Macromolecules, 2012, 45, 2242–2248 CrossRef CAS.
- R. C. Jeske, A. M. DiCiccio and G. W. Coates, J. Am. Chem. Soc., 2007, 129, 11330–11331 CrossRef CAS PubMed.
- M. J. Sanford, L. Peña Carrodeguas, N. J. Van Zee, A. W. Kleij and G. W. Coates, Macromolecules, 2016, 49, 6394–6400 CrossRef CAS.
- N. J. Van Zee and G. W. Coates, Angew. Chem., Int. Ed., 2015, 54, 2665–2668 CrossRef CAS PubMed.
- B. A. Abel, C. A. L. Lidston and G. W. Coates, J. Am. Chem. Soc., 2019, 141, 12760–12769 CrossRef CAS PubMed.
- M. J. Sanford, N. J. Van Zee and G. W. Coates, Chem. Sci., 2018, 9, 134–142 RSC.
- P. K. Saini, C. Romain, Y. Zhu and C. K. Williams, Polym. Chem., 2014, 5, 6068–6075 RSC.
- N. Yi, T. T. D. Chen, J. Unruangsri, Y. Zhu and C. K. Williams, Chem. Sci., 2019, 10, 9974–9980 RSC.
- C. Romain, Y. Zhu, P. Dingwall, S. Paul, H. S. Rzepa, A. Buchard and C. K. Williams, J. Am. Chem. Soc., 2016, 138, 4120–4131 CrossRef CAS PubMed.
- E. Hosseini Nejad, C. G. W. van Melis, T. J. Vermeer, C. E. Koning and R. Duchateau, Macromolecules, 2012, 45, 1770–1776 CrossRef.
- E. Hosseini Nejad, A. Paoniasari, C. G. W. van Melis, C. E. Koning and R. Duchateau, Macromolecules, 2013, 46, 631–637 CrossRef.
- S. Huijser, E. Hosseini Nejad, R. Sablong, C. de Jong, C. E. Koning and R. Duchateau, Macromolecules, 2011, 44, 1132–1139 CrossRef CAS.
- E. Hosseini Nejad, A. Paoniasari, C. E. Koning and R. Duchateau, Polym. Chem., 2012, 3, 1308 RSC.
- J. N. Hahladakis, C. A. Velis, R. Weber, E. Iacovidou and P. Purnell, J. Hazard. Mater., 2018, 344, 179–199 CrossRef CAS PubMed.
- G. Faraca and T. Astrup, Waste Manage., 2019, 95, 388–398 CrossRef CAS PubMed.
- Z. O. G. Schyns and M. P. Shaver, Macromol. Rapid Commun., 2021, 42, 2000415 CrossRef CAS PubMed.
- R. O. MacRae, C. M. Pask, L. K. Burdsall, R. S. Blackburn, C. M. Rayner and P. C. McGowan, Angew. Chem., Int. Ed., 2011, 50, 291–294 CrossRef CAS PubMed.
- P. Chi, C. Xie and J. Lin, Chin. J. Org. Chem., 2013, 33, 640–642 CrossRef CAS.
- V. Getautis, M. Dashkyavichene, I. Paulauskaite and A. Stanisauskaite, Chem. Heterocycl. Compd., 2005, 41, 426–436 CrossRef CAS.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683–689 RSC.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- M. Proverbio, N. G. Galotto, S. Losio, I. Tritto and L. Boggioni, Polymers, 2019, 11, 1222 CrossRef PubMed.
- B. Han, L. Zhang, B. Liu, X. Dong, I. Kim, Z. Duan and P. Theato, Macromolecules, 2015, 48, 3431–3437 CrossRef CAS.
- Z. A. Wood and M. E. Fieser, Polym. Chem., 2023, 14, 821–827 RSC.
- M. S. Shaw, M. R. Bates, M. D. Jones and B. D. Ward, Polym. Chem., 2022, 13, 3315–3324 RSC.
- M. E. Fieser, M. J. Sanford, L. A. Mitchell, C. R. Dunbar, M. Mandal, N. J. Van Zee, D. M. Urness, C. J. Cramer, G. W. Coates and W. B. Tolman, J. Am. Chem. Soc., 2017, 139, 15222–15231 CrossRef CAS PubMed.
- E. E. Langdon-Jones and S. J. A. Pope, Coord. Chem. Rev., 2014, 269, 32–53 CrossRef CAS.
- L. Feng, Y. Liu, J. Hao, X. Li, C. Xiong and X. Deng, Macromol. Chem. Phys., 2011, 212, 2626–2632 CrossRef CAS.
- R. Brown, K. Coulston, F. Eastwood and C. Vogel, Aust. J. Chem., 1988, 41, 1687 CrossRef CAS.
- O. Mitsunobu, J. Kimura, K. Iiizumi and N. Yanagida, Bull. Chem. Soc. Jpn., 1976, 49, 510–513 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental procedures and characterizing data, computational data and crystallographic data. CCDC 2094967–2094969. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3py00209h |
| ‡ Safety note. Large-scale reactions have the potential to give exothermic reactions from the homopolymerization of epoxide; these reactions were heated with an oil circulator with both heating and cooling capability and with the temperature controlled via a thermocouple inserted directly into the reaction medium, such that exotherms could be rapidly detected and the reaction cooled. PO reactions were undertaken above the boiling point. Screw cap vials were pressure tested prior to use and were shown to withstand a pressure of 1 bar without breakage; care should be taken in placing such reactions behind an appropriate screen to avoid injury. |
| This journal is © The Royal Society of Chemistry 2023 |