
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Linear not cyclic – unravelling an anionic initiation pathway for Lewis pair polymerization of lactones†
Vincent
Nieboer
 a,
Noé
Fanjul-Mosteirín
b,
Peter
Olsén
*b and
Karin
Odelius
*b
a,
Noé
Fanjul-Mosteirín
b,
Peter
Olsén
*b and
Karin
Odelius
*b
aDepartment of Fibre and Polymer Technology, KTH Royal Institute of Technology, SE-100 44, Stockholm, Sweden
bWallenberg Wood Science Center, WWSC, Department of Fibre and Polymer Technology, KTH Royal Institute of Technology, SE-100 44, Stockholm, Sweden. E-mail: polsen@kth.se; hoem@kth.se
First published on 26th April 2023
Abstract
Zwitterionic Lewis pair (LP) catalysis is potent towards the polymerization of lactone monomers to form cyclic polymers. In pursuit of faster polymerization kinetics, the use of weaker Lewis acids, such as diethylzinc (ZnEt2), has hitherto been suggested. However, the strong Brønsted base character of ZnEt2 brings the question of the actual initiation mechanism. Here, the ZnEt2-1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) LP was studied as catalyst for the initiation and polymerization reactions of ω-pentadecalactone (PDL), ε-caprolactone, δ-valerolactone, and γ-butyrolactone. Collective MALDI-ToF MS, NMR, FT-IR, and Ubbelhode viscometry studies revealed a polymerization mechanism proceeding through deprotonation of the α-protons on the lactone and not zwitterionic ring-opening, yielding an anionic propagation mechanism and linear polymers. The polymerization kinetics display an initiation period that correlates to ethyl decomposition on ZnEt2 and the initiation period is shortened by increasing the reaction temperature, Lewis base equivalents, and the lactones, e.g. ε-caprolactone, δ-valerolactone, and γ-butyrolactone in the system.
Introduction
Lewis pair (LP) polymerization combines the respective and unique properties of Lewis acids and Lewis bases to achieve advantages over standard metal, acid or basic catalysts, such as readily available catalysts, a wide assortment of polymerizable monomers, defined polymer topology, sequence architecture, and good control over molecular weight and dispersity.1,2 An interesting aspect of LP catalysis of lactones, is the ability to perform ring-opening zwitterionically to yield cyclic polymers.3,4 Zwitterionic ring-opening polymerization of lactones by LPs, is achieved through a Lewis base bound to the ester carbonyl group and is stabilized by a Lewis acid at the alkoxide propagating chain end.3–11 Due to the differences in interacting nature between a LP and the monomer, LPs exhibit large variations in activity, where some LPs show high activity for some monomers, but at the same time are unable to achieve any conversion of others.12–18An example of a monomer considered ‘challenging to polymerize’ is the macrolactone ω-pentadecalactone (PDL), as it has low ring strain and the polymerization thermodynamics are entropy driven.19–21 Yet, PDL is of high commercial and scientific interest, since the properties of poly(ω-pentadecalactone) PPDL resemble those of low density polyethylene (LDPE).22–24 The polymerization of PDL has found great benefit from the fast reaction kinetics associated LP polymerization.4,13,25 Nevertheless, polymerization of PDL generally requires high reaction temperatures and long reaction times to achieve high conversions,4 decreasing the environmental benefit of its use.
In pursuit of faster LP polymerization kinetics, several Lewis acids have been evaluated and an increased reactivity is found with decreasing Lewis acidity from ZnEt2 > Zn(C6H5)2 > Zn(C6F5)2 in combination with 4-(dimethylamino)pyridine (DMAP) towards the zwitterionic ring-opening polymerization of ε-caprolactone (εCL) and lactide.5 This observation was attributed to an enhanced initiation rate, since low Lewis acidity promotes dissociation and activation of the LP.5 This makes zwitterionic Lewis pair polymerization kinetically very similar to anionic polymerization, where the initiation by the Lewis base and the strength of the Lewis acid (counter ion) mainly dictates the rate and control over the polymerization.9
Although a weaker ZnEt2 Lewis acid accelerates the reaction rate, ZnEt2 is inherently more prone to side reactions due to a higher basicity of the ethyl anions compared to e.g. the pentafluoro benzene anion. Furthermore, self-initiated polymerization of εCL by Zn(C6F5)2 or Al(C6F5)3 have been reported to yield low number average molecular weight polymers attributed to uncontrolled side reactions.3,6,7 It should be noted that ZnEt2 has very diverse chemical features and has been used for the addition of alcohols to carbodiimides,26 reductions of imines,27 reduction and nucleophilic addition of and to ketones and aldehydes,28–31 reduction of acylsilanes,32 and deprotonation of β-keto esters,33 clearly testifying to its high and diverse reactivity. In addition, when we tried to employ ZnEt2 to synthesise cyclic poly(εCL), ZnEt2 was found proficient in the self-initiated ring-opening polymerization of εCL without requiring any additional initiator (Table S1,† entries 1 and 2) which is in agreement with literature.34
During anionic polymerization, side reactions between the initiator and the lactone monomer may occur to yield a deprotonated monomer functioning as an initiator.35–38 Moreover, the well-known class of Claisen condensation coupling reactions may employ strong Brønsted bases (e.g. alkyl-O–K/Na or NaH) to form β-keto esters from two esters through a deprotonation mechanism.39 We hypothesize that these observations explain the self-initiation ability of some organo-metal Lewis acids including ZnEt2 towards εCL. This would suggest that LP polymerization with basic metal ligands carry more anionic character than they are credited for.
The importance of understanding the underlying mechanism of LP polymerization with respect to previously reported literature, prompted us to investigate the polymer structure and initiation reaction of γ-butyrolactone (γBL), δ-valerolactone (δVL), εCL, and PDL as model monomers for the ZnEt2-DBU catalytic LP polymerization. Special attention is put on demystify the non-linearity in conversion during the initial polymerization stage. Our results show that under the conditions investigated, initiation occurred via α-proton abstraction (Scheme 1), generating linear polymers via an anionic propagation mechanism.
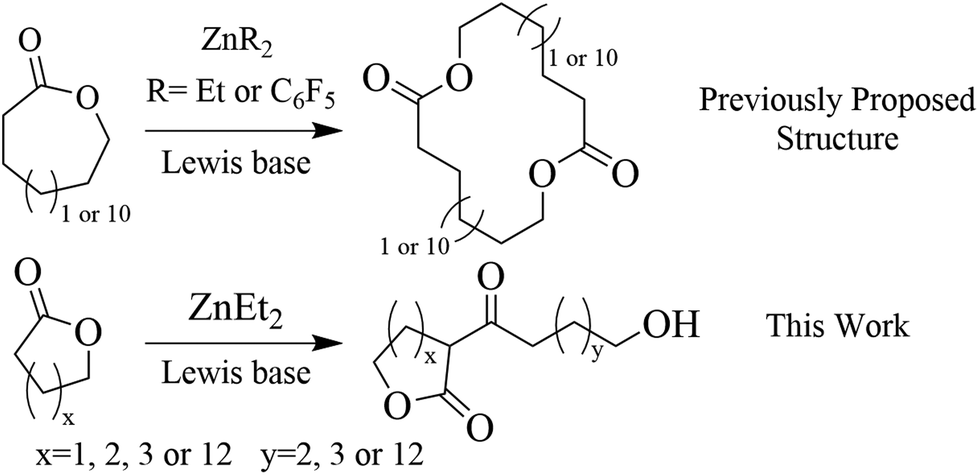 | ||
| Scheme 1 Polymerization of lactones PDL and εCL catalyzed by organo zinc-based Lewis pairs. | ||
Results and discussion
The polymerization of lactones using ZnEt2 as Lewis acid in LP polymerization is intriguing, due to the high activity and non-linearity of the polymerization rate. However, these features also bring questions about the actual initiation mechanism during LP polymerization with ZnEt2. This work addresses the mechanism in the ZnEt2-DBU LP catalyzed polymerization of PDL in detail, by investigating the polymer topology, initiation mode, and polymerization kinetics.Initiation reaction mechanism
As εCL can be polymerized by ZnEt2 alone, the first step was to evaluate if a LP was necessary to polymerize PDL or if a Lewis acid or base alone was sufficient. Hence, DBU or ZnEt2 were evaluated as catalysts to polymerize PDL. At 120 °C with toluene as solvent, no reaction of PDL proceeds solely with ZnEt2 nor DBU within 4 hours (Table S1,† entries 3 and 5). This suggests that DBU either acts as;(i) an initiator to generate a zwitterionic structure through an acyl transfer of the carbonyl to DBU.
(ii) a ligand coordinating to the zinc centre and that initiation occurs by ZnEt2 deprotonating the α-CH2 to the PDL ester.
At present, scenario (i) is accepted as the general route for most Lewis acids,3–11 and is capable of yielding cyclic polymers.3 However, if scenario (ii) is correct cyclic polymers will not be achieved and an initiation mechanism resembling that of Claisen condensation will occur.
To get insights into the polymeric structure formed, we used matrix assisted laser desorption/ionization time of flight mass spectrometry (MALDI-ToF MS) analysis (Fig. 1a) of PPDL synthesized by ZnEt2-DBU catalyzed polymerization ([PDL] = 1 M in toluene at 90 °C). The results showed that the formed PPDL consists only of one molecular weight population with different number of replications of the PDL repeating units (m/z = 240.21 × n + 23). Although this could be interpreted as proof of cyclic polymer formation, we need to keep in mind that linear polymers generated by the deprotonation of and subsequent initiation from PDL would generate the same number average molecular weight (Fig. 1b). MALDI-ToF MS is therefore not a reliable technique to determine the polymer topology. In this context, it should also be highlighted that end-groups of high number average molecular weight polymers are difficult to detect in 1H or 13C NMR (Fig. S1†), and the absence of end groups in NMR can be interpreted as proof of the formation of cyclic polymers. Based on these observations, ZnEt2 has been proposed as a part of a catalyst system to generate cyclic polymers.5 Yet contrary, εCL can efficiently be polymerized in the sole presence of ZnEt2 (Table S1,† entries 1 and 2) to yield a MALDI-ToF MS spectra based only on the molecular weight of only εCL repeating units (Fig. S2†), in parallel to what is observed for PDL. This contradicts the concept that zwitterionic structures derived from LP cooperation, scenario (i), are responsible for cyclic polymers and suggests that initiation is reliant on the ZnEt2 and not DBU.
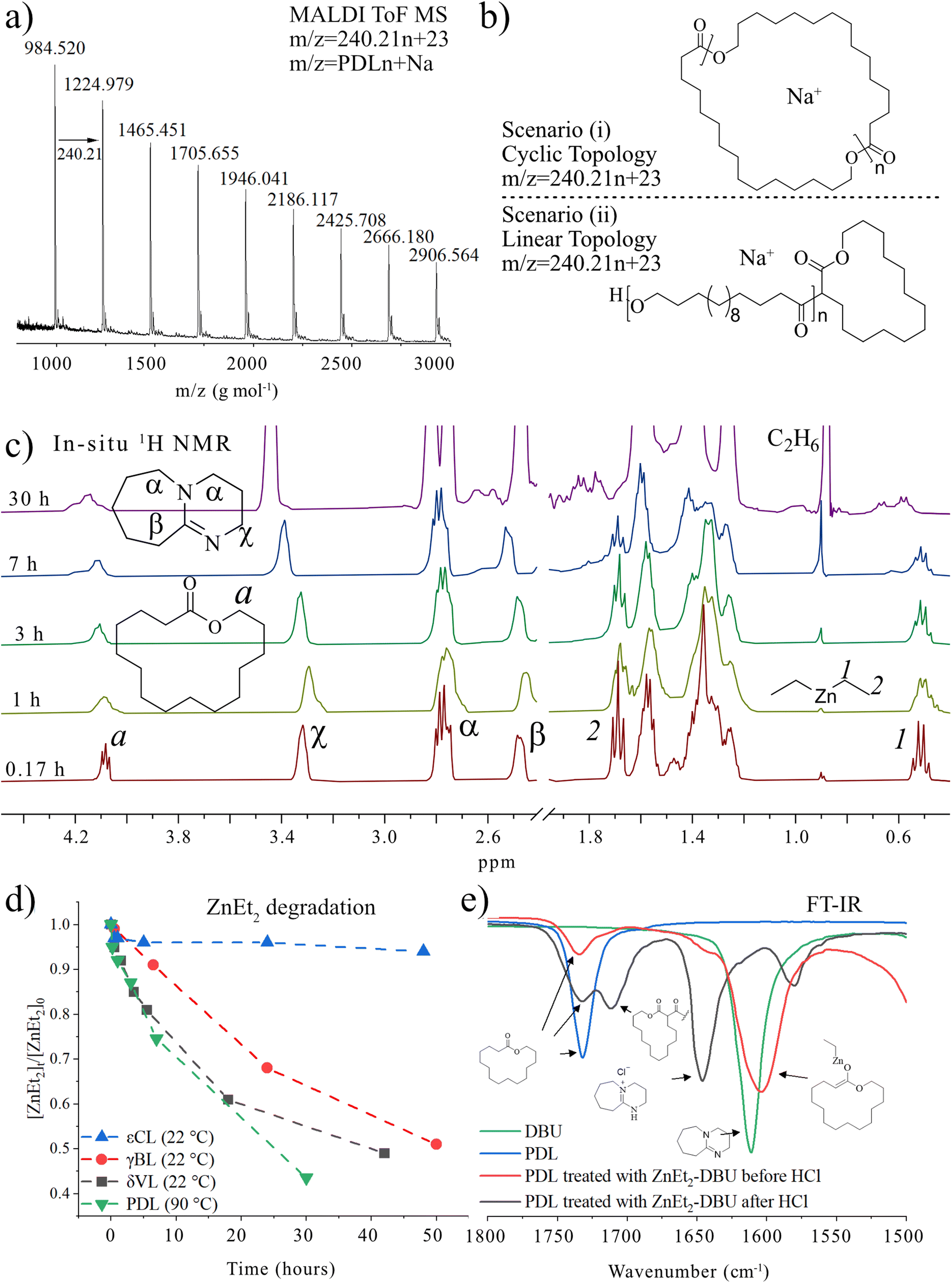 | ||
| Fig. 1 Analysis of low number average molecular weight PPDL synthesized by [PDL]/[ZnEt2]/[DBU] = 20/1/2 at 90 °C in toluene (Mn,SEC = 22.6 kg mol−1, Đ = 1.85) (a) MALDI-ToF MS analysis and (b) description of the molecular weights of that would be found for linear and cyclic PPDL. (c) 1H NMR of [PDL]0 = 0.29 M in toluene with [PDL]/[DBU]/[ZnEt2] = 1/2/1 at different heating times. Spectra was recorded in inert atmosphere in non-deuterated toluene containing a capillary with CDCl3 as shown in Fig. S3.† (d) ZnEt2 concentration overtime derived from integration of (c) (PDL), S4 (δVL),† S5 (γBL),† and S6 (εCL).† (e) FT-IR spectra of PDL, DBU, and reaction product of PDL treated as [PDL]/[DBU]/[ZnEt2] = 1/1/1 at 120 °C for 2 hours in bulk. | ||
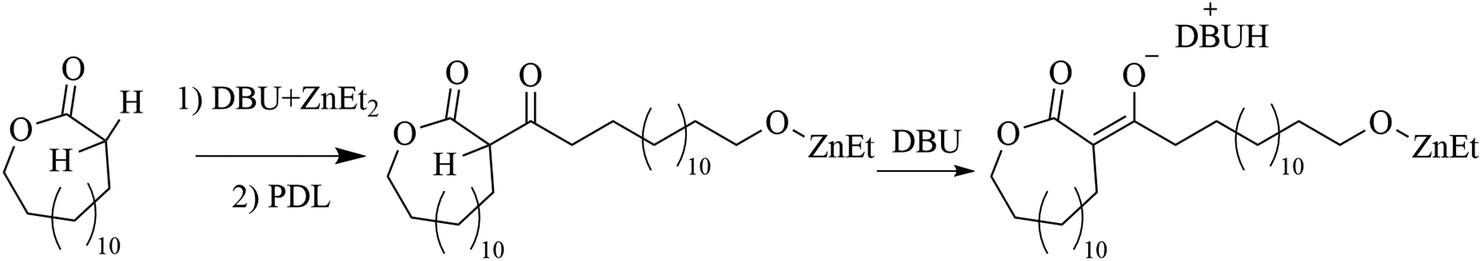
In an attempt to clarify the polymerization mechanism and to determine which of the scenarios (i) or (ii) is most probable, the initiation was investigated in detail. A solution of [PDL]/[ZnEt2]/[DBU] = 1/1/2 in toluene was heated to 120 °C and 1H NMR spectra of the crude solutions were recorded over time (Fig. 1c). The most notable observations are the splitting of the signals belonging to the ZnEt2, at CH3CH2 = 1.69 ppm and CH3CH2 = 0.52 ppm, as well as splitting of the PDL ω-CH2 (4.10 ppm) signal. This experiment was repeated for δVL (Fig. S4†), γBL (Fig. S5†), and εCL (Fig. S6†), at room temperature for these strained lactones. When a solution of PDL, ZnEt2, and DBU in toluene was set at room temperature, no reaction was observed (Table S1†). Room temperature In situ1H NMR revealed that the formation of new peaks and decomposition of ZnEt2 for δVL and γBL occurred in an almost identical way as for PDL with ZnEt2-DBU. εCL showed only minor reaction with ZnEt2-DBU, which is likely the results of a competitive polymerization reaction that consumed all monomeric εCL before it could react with the LP, which was feasible due to the high ceiling temperature of this monomer.40,41 On that note, δVL and γBL both have low thermodynamic ceiling temperatures, which prevents competitive monomer consumption through polymerization reaction.40–42 For all runs, a signal formed at ∼0.85 ppm with increased intensity as a function of time, which was interpreted as ZnEt2 decomposition products in the form of ethane, as the shift matches with literature values of ethane dissolved in toluene.43 Comparing the integration of the ethyl peaks on the ZnEt2 to the DBU peaks for γBL, δVL, εCL, and PDL in their reaction with the ZnEt2-DBU LP, revealed a decrease in the amount of ZnEt2 over time (Fig. 1d), suggesting that the ethyl groups are involved in the reaction generating ethane gas by deprotonation of acidic protons. FT-IR analysis suggests that deprotonation of the α-CH2 (to the carbonyl) is the most likely reaction, as demonstrated by the shift to a lower wavenumber, caused by an increase in electron density of the carbonyl group of PDL that has been treated with ZnEt2-DBU in the same manner (Fig. 1e and Fig. S7a†). The addition of anhydrous HCl to the ZnEt2 treated PDL (Scheme 2), generated two carbonyl peaks at lower wavenumbers, showing peaks which would be expected for β-keto ester structures, strongly suggesting a deprotonation mechanism (Fig. 1e and Fig. S7b†).
 | ||
| Scheme 2 Hypothesized mechanism of deprotonation of PDL by ZnEt2-DBU and subsequent protonation using anhydrous HCl. | ||
These findings combined suggest that scenario (ii), the deprotonation of PDL, is correct and hence that DBU does not initiate polymerization of PDL to generate a zwitterionic propagating structure to yield cyclic polymers. 1H NMR spectroscopy of DBU added to a solution containing ZnEt2 also supports this conclusion. An upfield shift of the peaks originating from DBU and a downfield shift of the peaks originating form ZnEt2 is observed, suggesting that DBU coordinates to ZnEt2 forming a classical Lewis pair adduct (Fig. S8†). Such adducts are competitive with the ability of DBU as Lewis base to initiate the reaction by a nucleophilic attack.5 Thus, the polymerization is likely initiated by deprotonation, either directly by the ZnEt2 or by the DBU or through a combination of both. The pKa of the α-CH2 to the esters is generally too high (ethyl acetate; pKa = 25.6; water)44 to be deprotonated by DBU directly (pKaH = 11.5–11.9; water).45,46 However, when the ester of a lactone is coordinated to a Lewis acid, the electron density is withdrawn, decreasing the pKa of the α-CH2, which in turn could enable DBU to initiate the reaction by deprotonation (Fig. S9,† route 1). Alternatively and more probable, ZnEt2, exchanges one DBU ligand with a PDL molecule and initiates the reaction by deprotonation of the α-CH2 to the ester, by one of the anionic ethyl ligands (ethane, pKa = 50.6; water)47 (Fig. S9,† route 2). After initiation, the propagating mechanism is entirely anionic and not similar to a zwitterionic LP mechanism. In this context it should be noted that a bifunctional initiation by one ZnEt2 is unlikely, since the reaction of isopropanol or water with ZnEt2 only yields the substitution of one ethyl group, and not two, even at excess stoichiometry of isopropanol.48
Polymer topology and end groups
To further study the polymer structure and compare this to the suggested initiation mechanism, low number average molecular weight PPDL samples (Mn,SEC = 22.6 kg mol−1, Đ = 1.85) were synthesized by the ZnEt2-DBU LP in toluene at 120 °C and analyzed using 1H and 13C NMR spectroscopy (Fig. 2a, b, and S10†). The synthesized polymers were compared to PPDL synthesized by an anionic initiator (tBuOLi), which produces strictly linear polymers (Fig. 2b). The presence of end-groups, CH2OH group at 3.64 ppm in 1H NMR (Fig. 2b), and a ketone at 205.5 ppm in 13C NMR (Fig. 2a), supports that ZnEt2-DBU initiated the polymerization from the α-position on PDL to form linear polymers. Two ketones of different shifts were observed by 13C NMR, suggesting the formation of two stereoisomers during PDL initiation. Assignments of the proton and carbon groups are motivated by HSQC, HMBC and COSY NMR spectroscopic methods (Fig. S11–S13†). Moreover, ZnEt2 self-initiated polymerization of εCL displayed end groups in a similar fashion, indicating the deprotonation mechanisms in the absence of DBU holds true (Fig. S14†).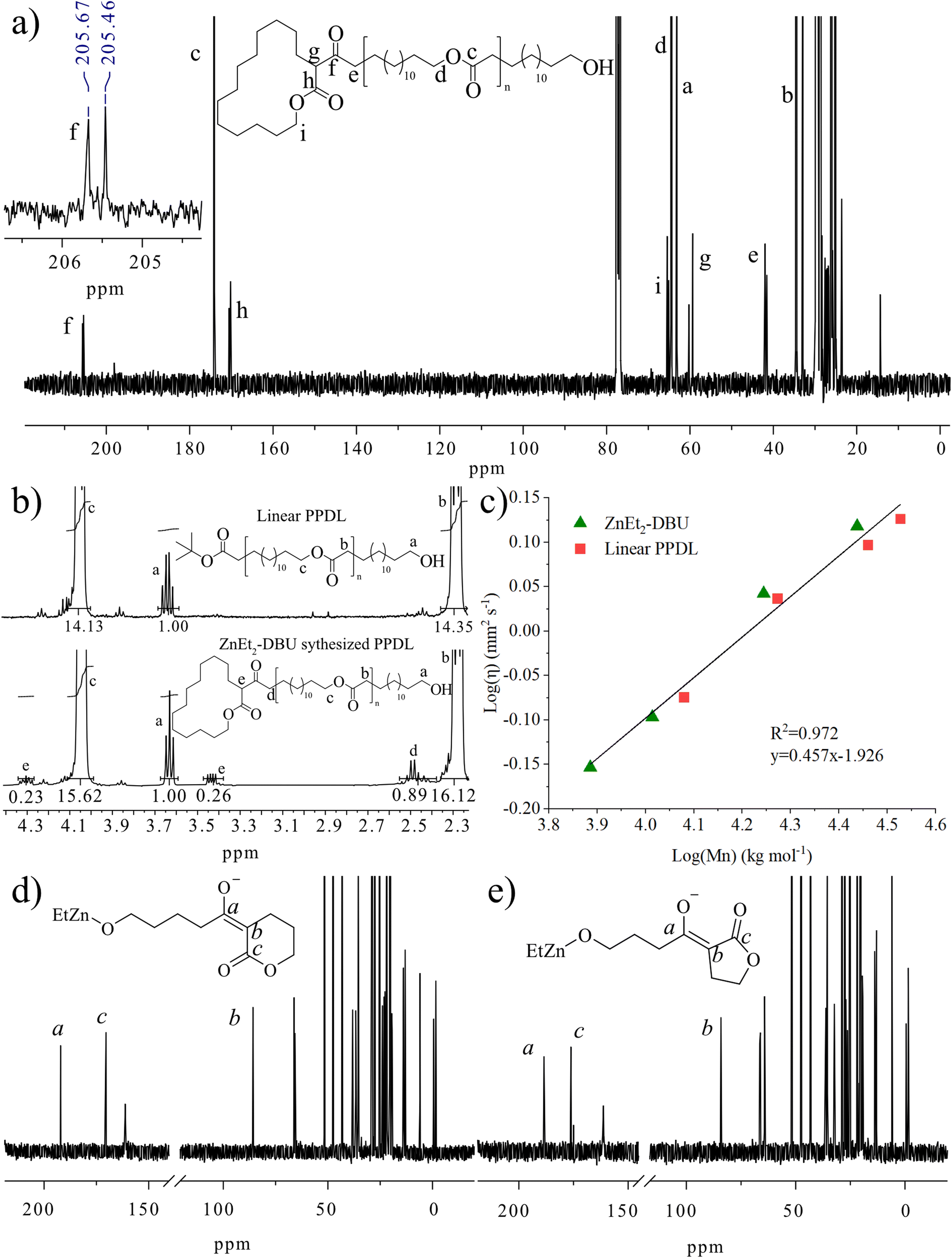 | ||
| Fig. 2 Analysis of low number average molecular weight PPDL (a) synthesized by ZnEt2-DBU LP at 120 °C (Mn,SEC = 22.6 kg mol−1, Đ = 1.85) and analyzed using 13C NMR (298 K, 101 MHz, CDCl3) and (b) synthesized by ZnEt2-DBU or tBuOLi at 120 °C analyzed and by 1H NMR (298 K, 400 MHz, CDCl3). (c) by viscometry (Ubbelohde, 0.009582 mm2 s−2, 0.1 g mL−1 CHCl3) analysis. Mn was derived from respective 1H NMR integration of CH2OH end group and repeating polymer units. (d) In situ13C NMR analysis of the reaction product of δVL with ZnEt2-DBU LP for [δVL]/[ZnEt2]/[DBU] = 1/1/2 in toluene at room temperature after 48 hours. (e) In situ13C NMR analysis of the reaction product of γBL with ZnEt2-DBU LP for [γBL]/[ZnEt2]/[DBU] = 1/1/2 in toluene at room temperature after 48 hours. | ||
A mechanistic detail to consider, is that the nucleophilic attack of the α-CH2, to the carbonyl, to ring-open another lactone, yields a β-keto ester structure. Similar structures commonly have pKa values below 10.65 (water).49 This would mean that the β-keto ester structure can be deprotonated much more prevalently by DBU (pKaH = 11.5–11.9; water)45,46 to form a new anionic specie (Scheme 3). Important to note, that it is the use of 2 equivalents of DBU to ZnEt2, that enables the presence of free DBU to deprotonate the β-keto ester. It is probable that deprotonation of the β-keto ester significantly affects the structure of the propagating end group through coordination of the zinc by both carbonyls in the β-keto ester. Moreover, it is hypothesized that the deprotonation reaction generates a resonance structure that inhibits the ring opening of the initial lactone at high conversions (Scheme 4).
 | ||
| Scheme 3 Deprotonation of the generated β-keto ester during the polymerization of PDL by the ZnEt2-DBU Lewis pair. | ||
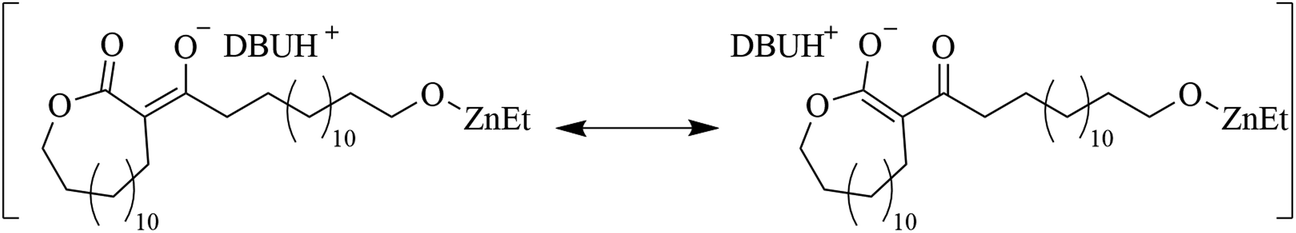 | ||
| Scheme 4 Resonance structures of deprotonated β-keto ester. | ||
As a further proof of the linear PPDL structure generated by the ZnEt2-DBU catalyzed polymerization of PDL, Ubbelhode viscometry was employed. Polymers of different topologies possess vastly different viscosities in solution. Cyclic polymers of polystyrene, for example, possess lower intrinsic viscosity to their linear counterparts, due to the difference in hydrodynamic volume.50–52 Therefore, a series of PPDL of different number average molecular weights synthesized by ZnEt2-DBU were dissolved in CHCl3 (0.1 g mL−1) and their viscosity analyzed and compared to a linear PPDL sample synthesized using an tBuOLi anionic initiator in toluene at 120 °C (Table S2†). A linear correlation between 1H NMR derived number average molecular weight and viscosity for all samples, strongly suggests that neither cyclic nor branched polymers are present (Fig. 2c). The same linear correlation persists with the 1H NMR and SEC derived number average molecular weight and viscosity (Fig. S15a and S15b†). Moreover, the 1H and SEC derived number average molecular weight are linear with each other, clearly testifying to presence of only linear polymers (Fig. S15c†).
In situ 13C NMR analysis of δVL and γBL treated with stoichiometric ZnEt2 and two equivalences of DBU at room temperature in toluene, revealed the formation of two shifts at ∼190 ppm and ∼85 ppm over time (Fig. S16 and S17†), to yield 13C NMR signals reminiscent of a β-keto ester (Fig. 2d and e), which are commonly seen in Claisen condensation products.53,54 Moreover, isolated P(δVL) synthesized by reaction with ZnEt2-DBU at room temperature in toluene (Fig. S18–S20†) displayed near identical 1H NMR end group signal as PPDL (Fig. S10†). This demonstrates the adaptability of ZnEt2-DBU and suggests that possibly many other lactones could be polymerized to form β-keto ester end groups via the deprotonation reaction.
Polymerization kinetics
One fascinating aspect of ZnEt2-DBU catalyzed polymerization of PDL are the associated polymerization kinetics and their non-linear behavior. To study the effects of temperature, [PDL] = 2 M in toluene was polymerized by ZnEt2-DBU at 60, 90, 105 and 120 °C (Fig. 3a). A long initiation period is most notable at all polymerization temperatures, followed by an exponential like rate increase to yield relatively fast propagation rates. At 60 °C, nearly no conversion of PDL was achieved within 5 hours. When the reaction temperature was increased from 90 °C to 105 °C the initiation period was shortened almost five-fold, and when the polymerization temperature was increased from 105 °C to 120 °C the initiation period was again halved, suggesting that initiation period decreases, near logarithmic, with temperature. At the same time, no drastic difference is observed in the propagation rate. Initiation periods are common in LP catalysis,4,5,7,18 where the polymerization temperature has been employed to shorten the initiation period.4,7 Our interpretation of the correlation with temperature, is the increased energy required to overcome the energy barrier of abstraction.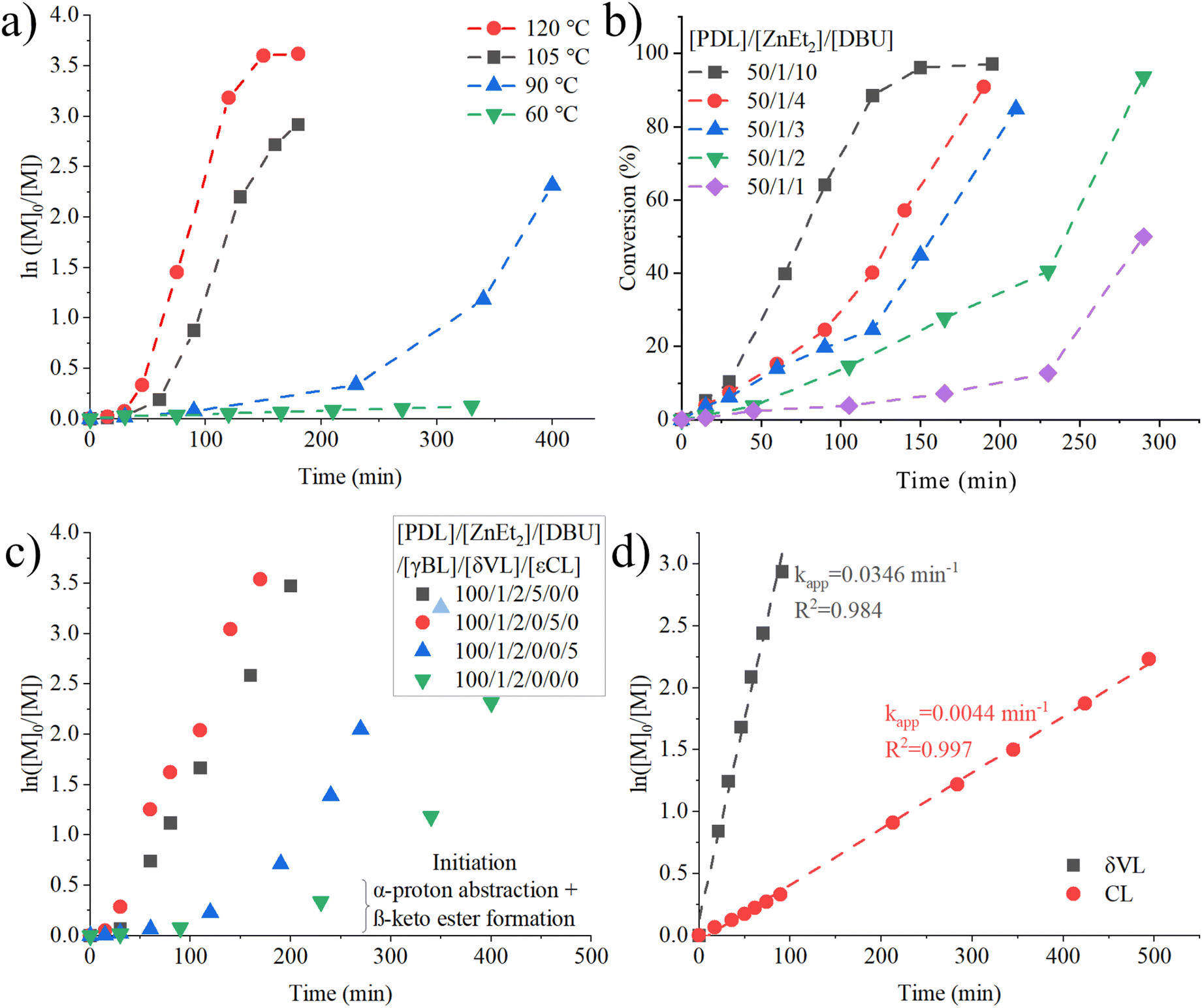 | ||
Fig. 3 Polymerization of [PDL]0 = 2 M in toluene for (a) [PDL]/[ZnEt2]/[DBU] = 100/1/2 at temperatures of 90, 105 and 120 °C, (b) [PDL]/[ZnEt2] = 50/1 at 90 °C with ZnEt2 equivalences to DBU ranging 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:10, and (c) ROP at 90 °C with or without five equivalences strained lactone to PDL. (d) polymerization of δVL and εCL by [monomer]/[ZnEt2]/[DBU] = 50/1/2 with [monomer]0 = 2 M in toluene at room temperature. :1 to 1:10, and (c) ROP at 90 °C with or without five equivalences strained lactone to PDL. (d) polymerization of δVL and εCL by [monomer]/[ZnEt2]/[DBU] = 50/1/2 with [monomer]0 = 2 M in toluene at room temperature. | ||
A significant impact on the initiation period of PDL polymerized at 90 °C by the number of equivalences of DBU to ZnEt2 was also observed (Fig. 3b). An increase in ratio of DBU to ZnEt2 shortens the initiation period, whilst the propagating rate is not affected. This holds true for all ratios, except when equimolar amounts of DBU to ZnEt2 are used, where the both initiation and propagating rates were much slower. It has previously been suggested that at stoichiometries of 1:1 for DMAP:ZnR2, 50% 2:1 DMAP:ZnR2 forms and while 50% of ZnR2 (R = Et, C6H5, or C6F5) is essentially free and inactive towards ring-opening polymerization of lactide, which could explain the low propagation rate even after the induction period for the equimolar LP run.5 Our study also evaluated an 2-fold excess of ZnEt2 to DBU, but this did not yield any conversion within 4 hours at 90 °C with [PDL] = 2 M in toluene, supporting the notion that ZnEt2 needs to be stabilized/assisted by DBU before initiation can occur. The equivalences of DBU to ZnEt2 do not affect the number average molecular weights achieved and show a linear behavior between conversion and number average molecular weight (Fig. S21†), further strengthening the notion of an anionic mechanism and initiator behavior of ZnEt2. These results strongly indicate that DBU plays a prominent role in the activation mechanism, but the propagation mechanism is independent of excess DBU. It should be noted that LP catalyzed polymerization rates can be favored both by an excess of Lewis base to Lewis acid,4,7,15 or an excess Lewis acid to Lewis base.6,16,17,25 It is likely that the ability of the LP to form adducts dictates whether excess Lewis acid is beneficial or not.
An unique observation, was the overall reduction in polymerization time needed to achieve full conversion of PDL when 5 equivalences of either δVL, εCL or γBL were added to a solution of PDL containing the ZnEt2-DBU at 90 °C (Fig. 3c). With the addition of even a small amount of strained lactone, the initiation period was halved for εCL and vanished almost completely for δVL and γBL, while the final propagation rate was not affected. Strained lactones possess a pKa value much lower than that of unstrained/linear esters (pKa = 13.5 γ-valerolactone; pKa = 25.6 EtOAc; gas-phase).55
εCL is a well-studied example of enthalpy-driven ROP, as εCL encompasses a large release of ring strain during polymerization, driving the reaction, whilst the entropy of polymerization is negative.20,21 With the exception of Novozyme-435 enzyme catalyzed reactions,56 studies on the copolymerization of PDL with εCL report that εCL is fully consumed within a fraction of the time that it takes for the PDL.25,57–60 The increased reactivity and acidity of δVL, εCL, and γBL likely promote the deprotonation and subsequent β-keto ester formation to accelerate the formation of the anionic initiating specie, yielding an overall shorter reaction time, with propagation rates remaining the same. To confirm that the strained lactone accelerated polymerization of PDL did occur via a Claisen condensation by deprotonation of and initiation from the strained lactone, low molecular weight PPDL was synthesized using a ZnEt2/DBU/γBL = 1/2/3 initiation system and purified by double precipitation in MeOH (Fig. S22†). The addition of γBL clearly impacts the end groups, but the overall character of β-keto ester end group remains. This indeed strongly advocates that the strained lactones are deprotonated preferably and transformed to initiators to be present as end groups on the isolated polymer.
The ability of the ZnEt2-DBU LP to polymerize strained lactones δVL, εCL and γBL at room temperature was also evaluated (Fig. 3d). γBL showed <1% conversion, which is not surprising considering its low thermodynamic ceiling temperature and stable monomer structure.42,61 δVL and εCL on the other hand, displayed fast linear kinetics at room temperature. It is likely that the relatively low pKa values of these lactones in addition to their ease of ring opening, severely accelerate β-keto ester formation, mitigating any initiation period as was observed for PDL. SEC analysis suggests that initiation efficiency for εCL was significantly lower (Mn = 26.7 kg mol−1, Đ = 2.64) (Fig. S23†) than for δVL (Mn = 16.8 kg mol−1, Đ = 1.72) (Fig. S19†), as δVL displayed a lower number average molecular weight. This indicates that δVL is deprotonated more efficiently than εCL, which may be supported by free energies of deprotonation observed by gas phase Fourier transform ion cyclotron resonance mass spectrometry, which measured δVL to be more acidic than εCL.62
Conclusions
Our results show that the Lewis-pair (LP) polymerization of δVL, εCL and PDL with ZnEt2-DBU proceeded by an anionic propagation mechanism, starting from the acidic α-protons on the lactone and not via initiation through acyl transfer to generate a zwitterionic structure. In situ1H NMR analysis revealed that the ethyl groups in ZnEt2 decompose during the polymerization, and FT-IR showed that the PDL carbonyl gains electron density by the shift to lower wavenumbers. In situ13C NMR analysis showed signals which resemble β-keto ester structures and the analysis of isolated PPDL by 13C NMR supported this notion by revealing the formation of a ketone. 1H NMR of isolated PPDL, P(δVL), and γBL-PPDL-OH showed signals that resemble a β-keto ester. Despite the variety in reaction conditions, all obtained polymers were linear. The reaction kinetics revealed an initiation period for PDL that could be shortened drastically by temperature, the equivalence of base, and the presence of lactones with more acidic α-protons to the carbonyl, such as γBL, δVL or εCL, evident of increased rate for the α-proton abstraction. The polymerization kinetics of δVL or εCL were linear at room temperature. Hence, ZnEt2-DBU catalyzed ring-opening polymerization of (macro)lactones proceeds rapidly and leads to the formation of unique end groups for post-functionalization. The more considerable consequence is that specific LP systems suggested bringing cyclic polymers might, in fact, instead, be linear with a Claisen type condensation as initiation from the monomer.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding from the Swedish Research Council (Grant Number 2020-03455) and the Knut and Alice Wallenberg Foundation (KAW) through the Wallenberg Wood Science Center for this work.References
- M. L. McGraw and E. Y.-X. Chen, Macromolecules, 2020, 53, 6102–6122 CrossRef CAS.
- M. Hong, J. Chen and E. Y.-X. Chen, Chem. Rev., 2018, 118, 10551–10616 CrossRef CAS PubMed.
- E. Piedra-Arroni, C. Ladavière, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2013, 135, 13306–13309 CrossRef CAS PubMed.
- B. Wang, L. Pan, Z. Ma and Y. Li, Macromolecules, 2018, 51, 836–845 CrossRef CAS.
- B. Wang, Y. Wei, Z.-J. Li, L. Pan and Y.-S. Li, ChemCatChem, 2018, 10, 5287–5296 CrossRef CAS.
- Q. Wang, W. Zhao, J. He, Y. Zhang and E. Y.-X. Chen, Macromolecules, 2017, 50, 123–136 CrossRef CAS.
- X.-Q. Li, B. Wang, H.-Y. Ji and Y.-S. Li, Catal. Sci. Technol., 2016, 6, 7763–7772 RSC.
- P. Walther, W. Frey and S. Naumann, Polym. Chem., 2018, 9, 3674–3683 RSC.
- A. Balint and S. Naumann, Polym. Chem., 2021, 12, 5320–5327 RSC.
- I. S. R. Karmel, M. Khononov, M. Tamm and M. S. Eisen, Catal. Sci. Technol., 2015, 5, 5110–5119 RSC.
- Y. A. Chang and R. M. Waymouth, Polym. Chem., 2015, 6, 5212–5218 RSC.
- Y. Zhang, G. M. Miyake and E. Y.-X. Chen, Angew. Chem., Int. Ed., 2010, 49, 10158–10162 CrossRef CAS PubMed.
- S. Naumann, P. B. V. Scholten, J. A. Wilson and A. P. Dove, J. Am. Chem. Soc., 2015, 137, 14439–14445 CrossRef CAS PubMed.
- S. Naumann and D. Wang, Macromolecules, 2016, 49, 8869–8878 CrossRef CAS.
- Y. Nakayama, S. Kosaka, K. Yamaguchi, G. Yamazaki, R. Tanaka and T. Shiono, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 297–303 CrossRef CAS.
- X. Wang, Y. Zhang and M. Hong, Mol. J. Synth. Chem. Nat. Prod. Chem., 2018, 23, 442 Search PubMed.
- W. Nzahou Ottou, E. Conde-Mendizabal, A. Pascual, A.-L. Wirotius, D. Bourichon, J. Vignolle, F. Robert, Y. Landais, J.-M. Sotiropoulos, K. Miqueu and D. Taton, Macromolecules, 2017, 50, 762–774 CrossRef CAS.
- Y. Zhang, G. M. Miyake, M. G. John, L. Falivene, L. Caporaso, L. Cavallo and E. Y.-X. Chen, Dalton Trans., 2012, 41, 9119–9134 RSC.
- P. Hodge, Chem. Rev., 2014, 114, 2278–2312 CrossRef CAS PubMed.
- A. Duda, A. Kowalski, S. Penczek, H. Uyama and S. Kobayashi, Macromolecules, 2002, 35, 4266–4270 CrossRef CAS.
- A. Duda and A. Kowalski, Handbook of Ring-Opening Polymerization, John Wiley & Sons, Ltd, 2009, pp. 1–51 Search PubMed.
- M. P. F. Pepels, R. G. Kleijnen, J. G. P. Goossens, A. B. Spoelstra, R. Tandler, H. Martens, M. Soliman and R. Duchateau, Polymer, 2016, 88, 63–70 CrossRef CAS.
- J. Cai, C. Liu, M. Cai, J. Zhu, F. Zuo, B. S. Hsiao and R. A. Gross, Polymer, 2010, 51, 1088–1099 CrossRef CAS.
- M. P. F. Pepels, R. A. C. Koeken, S. J. J. van der Linden, A. Heise and R. Duchateau, Macromolecules, 2015, 48, 4779–4792 CrossRef CAS.
- P. Walther and S. Naumann, Macromolecules, 2017, 50, 8406–8416 CrossRef CAS.
- A. Ramos, F. Carrillo-Hermosilla, R. Fernández-Galán, D. Elorriaga, J. Naranjo, A. Antiñolo and D. García-Vivó, Organometallics, 2022, 41, 2949–2957 CrossRef CAS PubMed.
- X. Xiao, H. Wang, Z. Huang, J. Yang, X. Bian and Y. Qin, Org. Lett., 2006, 8, 139–142 CrossRef CAS PubMed.
- Y.-J. Cherng, J.-M. Fang and T.-J. Lu, J. Org. Chem., 1999, 64, 3207–3212 CrossRef CAS PubMed.
- H. Huang, H. Zong, G. Bian and L. Song, Tetrahedron: Asymmetry, 2015, 26, 835–839 CrossRef CAS.
- K. Yearick and C. Wolf, Org. Lett., 2008, 10, 3915–3918 CrossRef CAS PubMed.
- E. Folkertsma, S. H. Benthem, J. T. B. H. Jastrzebski, M. Lutz, M. Moret and R. J. M. Klein Gebbink, Eur. J. Inorg. Chem., 2018, 2018, 1167–1175 CrossRef CAS PubMed.
- G. Gao, X.-F. Bai, F. Li, L.-S. Zheng, Z.-J. Zheng, G.-Q. Lai, K. Jiang, F. Li and L.-W. Xu, Tetrahedron Lett., 2012, 53, 2164–2166 CrossRef CAS.
- R. Hilgenkamp and C. K. Zercher, Org. Lett., 2001, 3, 3037–3040 CrossRef CAS PubMed.
- K. Żółtowska, M. Sobczak and E. Olędzka, Molecules, 2015, 20, 2816–2827 CrossRef PubMed.
- L. Sipos and M. Zsuga, J. Macromol. Sci., Part A: Pure Appl.Chem., 1997, 34, 1269–1284 CrossRef.
- Z. Grobelny, S. Golba and J. Jurek-Suliga, Polym. Bull., 2019, 76, 3501–3515 CrossRef CAS.
- I. Gitsov, I. B. Rashkov and I. M. Panayotov, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 2115–2126 CrossRef CAS.
- H. R. Kricheldorf and I. Kreiser-Saunders, Makromol. Chem., 1990, 191, 1057–1066 CrossRef CAS.
- A. K. Franz, Angew. Chem., Int. Ed., 2012, 51, 10701–10702 CrossRef CAS.
- B. V. Lebedev, Russ. Chem. Rev., 1996, 65, 1063 CrossRef.
- P. Olsén, K. Odelius and A.-C. Albertsson, Biomacromolecules, 2016, 17, 699–709 CrossRef PubMed.
- M. Hong and E. Y.-X. Chen, Nat. Chem., 2016, 8, 42–49 CrossRef CAS PubMed.
- A. H. M. Leung, S. D. Pike, A. J. Clancy, H. C. Yau, W. J. Lee, K. L. Orchard, M. S. P. Shaffer and C. K. Williams, Chem. Sci., 2018, 9, 2135–2146 RSC.
- T. L. Amyes and J. P. Richard, J. Am. Chem. Soc., 1996, 118, 3129–3141 CrossRef CAS.
- R. Srivastava, J. Mol. Catal. A: Chem., 2007, 264, 146–152 CrossRef CAS.
- F. Ravalico, S. L. James and J. S. Vyle, Green Chem., 2011, 13, 1778–1783 RSC.
- W. L. Jorgensen, J. M. Briggs and J. Gao, J. Am. Chem. Soc., 1987, 109, 6857–6858 CrossRef CAS.
- R. Herold, S. Aggarwal and V. Neff, Can. J. Chem., 1963, 41, 1968–1980 CrossRef.
- J. W. Bunting and J. P. Kanter, J. Am. Chem. Soc., 1993, 115, 11705–11715 CrossRef CAS.
- Y. Jeong, Y. Jin, T. Chang, F. Uhlik and J. Roovers, Macromolecules, 2017, 50, 7770–7776 CrossRef CAS.
- G. B. McKenna and D. J. Plazek, Polym. Commun., 1986, 27, 304–306 CAS.
- G. Hadziioannou, P. M. Cotts, G. ten Brinke, C. C. Han, P. Lutz, C. Strazielle, P. Rempp and A. J. Kovacs, Macromolecules, 1987, 20, 493–497 CrossRef CAS.
- Z. Chen, A. Y. Hong and X. Linghu, J. Org. Chem., 2018, 83, 6225–6234 CrossRef CAS PubMed.
- M. R. DeGraffenreid, S. Bennett, S. Caille, F. Gonzalez-Lopez de Turiso, R. W. Hungate, L. D. Julian, J. A. Kaizerman, D. L. McMinn, Y. Rew, D. Sun, X. Yan and J. P. Powers, J. Org. Chem., 2007, 72, 7455–7458 CrossRef CAS PubMed.
- R. Gómez-Bombarelli, M. González-Pérez, M. T. Pérez-Prior, E. Calle and J. Casado, J. Org. Chem., 2009, 74, 4943–4948 CrossRef PubMed.
- A. Kumar, B. Kalra, A. Dekhterman and R. A. Gross, Macromolecules, 2000, 33, 6303–6309 CrossRef CAS.
- J.-H. Bai, X.-Q. Wang, J.-H. Wang and L.-F. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1189–1196 CrossRef CAS.
- V. Ladelta, J. D. Kim, P. Bilalis, Y. Gnanou and N. Hadjichristidis, Macromolecules, 2018, 51, 2428–2436 CrossRef CAS.
- C. Li, W. Xu, Y. Lu and R. A. Gross, Macromol. Rapid Commun., 2020, 41, 2000417 CrossRef CAS PubMed.
- M. P. F. Pepels, M. Bouyahyi, A. Heise and R. Duchateau, Macromolecules, 2013, 46, 4324–4334 CrossRef CAS.
- K. N. Houk, A. Jabbari, H. K. Hall and C. Alemán, J. Org. Chem., 2008, 73, 2674–2678 CrossRef CAS PubMed.
- J. M. Karty, G. A. Janaway and J. I. Brauman, J. Am. Chem. Soc., 2002, 124, 5213–5221 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, reaction mechanisms, and characterization of samples by NMR, MALDI-ToF, and FT-IR analysis. See DOI: https://doi.org/10.1039/d3py00310h |
| This journal is © The Royal Society of Chemistry 2023 |