
Triphenylphosphine oxide promoting visible-light-driven C–C coupling via desulfurization†
Shea
Stewart
 ,
Robert
Maloney
and
Yugang
Sun
*
,
Robert
Maloney
and
Yugang
Sun
*
Department of Chemistry, Temple University, 1901 North 13th Street, Philadelphia, Pennsylvania 19122, USA. E-mail: ygsun@temple.edu
First published on 6th March 2023
Abstract
Triphenylphosphine oxide (TPPO) and triphenylphosphine (TPP) can form a complex in solution, promoting visible light absorption to trigger electron transfer within the complex and generate radicals. Subsequent radical reactions with thiols enable desulfurization to produce carbon radicals that react with aryl alkenes to yield new C–C bonds. Since ambient oxygen can easily oxidize TPP to TPPO, the reported method requires no explicit addition of a photocatalyst. This work highlights the promise of using TPPO as a catalytic photo-redox mediator in organic synthesis.
Photons can serve as “reactants” to lower the environmental impact of chemical synthesis by enabling transformations without harsh conditions such as elevated temperature, high pressures, and toxic reagents. For example, excitation of a photocatalyst enhances its redox reactivity towards species in reaction systems, promoting single electron transfer (SET) to form active ionic radicals. Tremendous efforts have been made to develop photocatalysis in recent decades, with a wide array of reports each year outlining increasingly complex chemical transformations enabled by simply illuminating reactions with visible light1–4 Among these efforts, the generation of phosphorous-centred radical cations via SET from phosphine to a photocatalyst has sprung up as an effective strategy to access reactive carbon-centred radicals.5–8 This strategy hinges on the formation of phosphoranyl radicals via ionic attack by deprotonated hydroxyl/mercapto groups onto phosphine radical cations. The resulting phosphoranyl radicals quickly undergo β-scission yielding phosphine oxide/sulfide and carbon radicals in a process known as deoxygenation/desulfurization. Carbon radicals from this process can be used to selectively form new C–C bonds. Visible-light-driven photocatalytic generation of phosphoranyl radicals has been commonly carried out using an Ir-centred metal-ligand complex5,6,9 as a light absorber, although other photocatalysts such as organic dye molecules have been evaluated as well.10 The high cost and recycling difficulty of the (homogeneous) molecular photocatalysts remains challenging to resolve. In addition, the resulting phosphine oxide/sulfide derived from the dissociation of phosphoranyl radicals are often considered as “useless” waste products.
Here, we report that C–C coupling via desulfurization (de-S) by TPP can proceed under short-wavelength visible light in the absence of photocatalysts. Systematic studies reveal that TPPO serves as a quasi-photocatalyst to enable absorption of short-wavelength visible light through the formation of a TPPO-TPP complex (Fig. S1, ESI†). Photoexcitation of the TPPO-TPP enables SET from TPP to TPPO, forming radical ions to trigger the sequential radical steps of reductive C–C coupling via de-S. In the overall reaction, TPPO behaves like a photocatalyst although it does not absorb visible light itself.
For instance, reductive C–C coupling between styrene (1) and methyl thioglycolate (2) can occur through de-S using TPP as the sulfur scavenger. A typical reaction involving 1.5 equivalents of TPP and 2 equivalents of 2 relative to 1 gives the desirable outcome of the C–C coupling product, methyl 4-phenylbutyrate (3) (Table 1, entry 1). The reaction proceeds in varying solvents and with no strict atmospheric control, indicating the robustness of the visible-light-driven de-S process (Table 1, entries 2-5). In contrast, no formation of 3 occurs in the dark while heating to 60 °C, confirming the crucial role of photoillumination in initiating the radical reactions (Table 1, entry 6). Because of the weak overlap between the absorption spectrum of the TPPO-TPP complex and the emission spectrum of the white LED (Fig. 1), the reaction kinetics are slow at room temperature, yielding only 30% of 3 after 24 hours (Table 1, entry 1). The yield reaches 92% after 5 days (Table 1, entry 7), or 99% after 18 hours while heating to 60 °C (Table 1, entry 8). Overall, the results in Table 1 indicate that photoillumination triggers a series of sequential radical reaction steps, which are favoured at elevated temperatures, for C–C coupling to occur. This conclusion is corroborated by lack of 3 upon addition of TEMPO and observation of a TEMPO-methyl acetate radical adduct (Fig. S2, ESI†).
|
|
||
|---|---|---|
| Entry | Conditions | Yield (%) |
a Standard reaction conditions referring to entry 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :46 μL 1, 157 mg b-TPP, 72 μL b-2, 2 mL acetonitrile (solvent), 0.8 W cm−2 white LED (Dolan-Jenner Fiber-Lite MI-LED B1), photoillumination for 24 hours, no atmospheric control, and room temperature. Changes are specified for each entry. All reactions were run without a stir bar to avoid contamination. The yield of 3 was determined using 1H-NMR with t-cinnamaldehyde as internal standard. :46 μL 1, 157 mg b-TPP, 72 μL b-2, 2 mL acetonitrile (solvent), 0.8 W cm−2 white LED (Dolan-Jenner Fiber-Lite MI-LED B1), photoillumination for 24 hours, no atmospheric control, and room temperature. Changes are specified for each entry. All reactions were run without a stir bar to avoid contamination. The yield of 3 was determined using 1H-NMR with t-cinnamaldehyde as internal standard.
|
||
| 1 | None | 30 |
| 2 | In trifluorobenzene, 60 °C, 18 h | 95 |
| 3 | In toluene, 60 °C, 18 h | 90 |
| 4 | In isopropyl alcohol, 60 °C, 18 h | 82 |
| 5 | Under argon (balloon) | 32 |
| 6 | Dark, 60 °C | 0 |
| 7 | 5 days | 92 |
| 8 | 60 °C, 18 h | 99 |
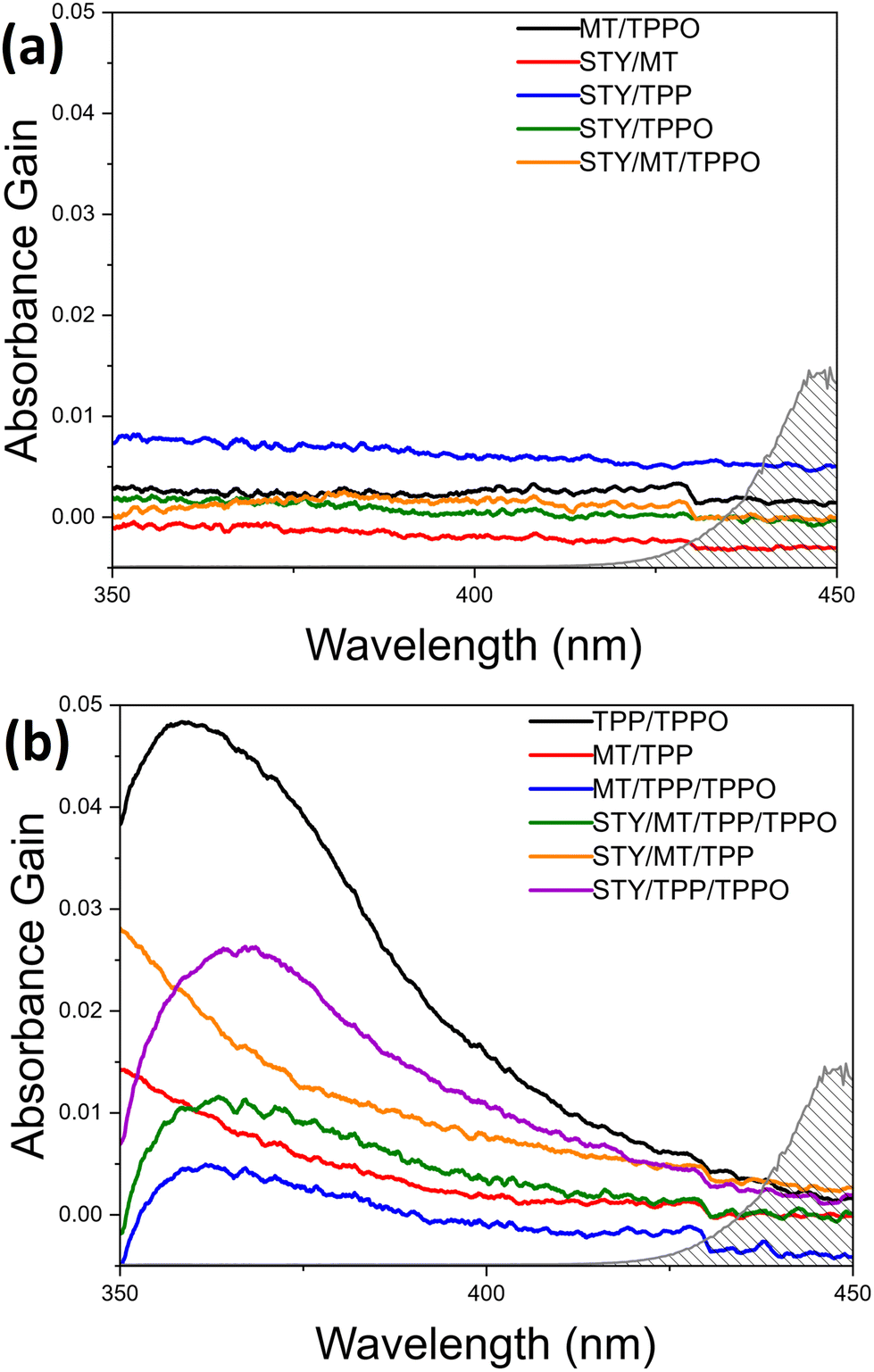 | ||
| Fig. 1 UV-Visible absorption spectra of (a) reagent combinations lacking interactions to yield visible light absorption and (b) reagent combinations exhibiting strong interactions to impart visible light absorption. The spectra in (b) and (c) were plotted by subtracting the absorption of each individual reagent from the measured absorption of the combinations. For example, the absorption spectrum of the TPP/TPPO mixture was measured followed by subtraction of both the TPP and TPPO absorptions in (a), resulting in the net absorption spectrum of TPP/TPPO shown in (c). The gray line with shading underneath represents the emission spectrum of the white LED lamp. All absorption spectra were obtained from solutions containing 0.27 mmol of each reagent in 2 mL of acetonitrile. The minor offsets of baselines are ascribed to the solvent-evaporation-induced heterogeneity in the solutions. | ||
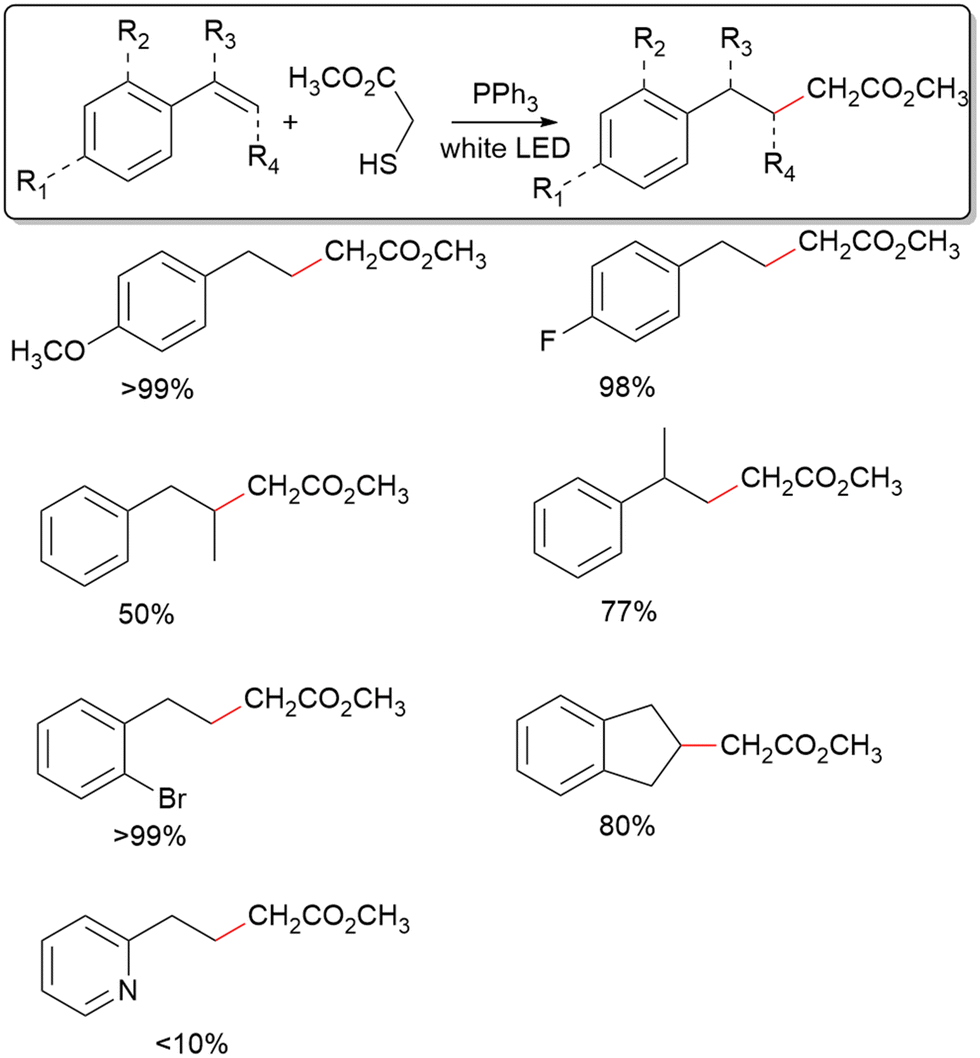
Several representative styrene derivatives have been tested to display the utility of this system for C–C coupling with aryl alkenes (Table 2). Both 4-methoxy and 4-fluoro styrene giving excellent yields shows little sensitivity of the method towards the electronic structure of the aromatic ring. Furthermore, the use of 2-bromo styrene and 1H-indene displays a moderate tolerance for steric hinderance about the phenyl ring. Using 2-vinylpyridine gives a very depressed yield, indicating low tolerance for radical intermediates possessing resonance forms where a heteroatom bears the radical. For α-methyl styrene and cis-β-styrene, fair yields have been achieved, showing modest tolerance for steric crowding at the alkene reaction site. In contrast, C–C coupling only occurs for mercaptans with an electron-withdrawing group adjacent to the radical formed after de-S (Fig. S3, ESI†). As such, of the mercaptans tested, only 2 and thioacetic acid show positive results towards C–C coupling. However, the success of thioacetic acid is notable as is it unveils the utility of desulfurization in forming both sp3–sp3 and sp2–sp3 bonds. The inefficient C–C coupling for mercaptans without electron withdrawing groups is likely because the radicals formed after C–C coupling should be significantly more stable than the radical resulting from de-S, which is not the case for the likes of benzyl mercaptan.
| Reaction conditions: 0.4 mmol of styrene derivatives, 157 mg b-TPP, 72 μL b-2, 2 mL acetonitrile (solvent), 0.8 W cm−2 (Dolan–Jenner Fiber-Lite MI-LED B1), photoillumination for 18 hours, no atmospheric control, and 60 °C. All reactions were run without a stir bar to avoid contamination. Reaction yields were determined using 1H-NMR with t-cinnamaldehyde as internal standard. |
|---|
|
To our knowledge, there are no reports of visible-light-driven phosphoranyl radical fragmentation in the absence of a photocatalyst.5–8 However, a rich literature outlines thiol-olefin and disulfide-olefin electron donor–acceptor (EDA) complexes that form within reaction solutions to absorb short-wavelength visible light and enable free-radical thiol-ene co-oxidation reactions in the presence of O2.11–14 These studies motivated us to study the possible formation of an EDA complex in the de-S C–C coupling reaction solution. Although 2 and 1 might form an EDA complex to absorb visible light, the corresponding oxidation by the soluble O2 cannot generate C–C coupling product. The formation C–C coupling after LED illumination of solutions with 2, 1, and TPP suggests the formation of different EDA complexes which absorb visible light. Survey spectral scans of 2, 1, TPP, and TPPO individually in acetonitrile in the range of 200–1100 nm show their optical transparency outside of the ultraviolet region (Fig. S4, ESI†). The spectra of different de-S reagent mixtures in acetonitrile were measured with 30 minute scans in the narrow range of 350–450 nm to improve the signal-to-noise ratios (Fig. 1).
The spectra of individual reagents are, expectedly, rather featureless with the onset absorbance being well into the ultraviolet (UV) region (Fig. S4, ESI†). Analysing the absorption spectra of the reagent combinations after subtracting out the spectra obtained for the corresponding individual reagents unveils the interactions between the reagents in solution. If the net signals are flat (or wavelength-independent), very weak interaction or no interaction exists between the different reagent molecules. The apparent wavelength-dependent net signals indicate the existence of strong intermolecular interactions including the formation of EDA complexes. Fig. 1a shows the net spectra of reagent combinations in which the signals are uniform, and no strong intermolecular interactions occur. Notably, these spectra include all combinations in which neither TPP/TPPO nor 2/TPP combinations exist. In Fig. 1b, however, a very obvious peak is observed for all mixtures containing both TPP/TPPO, ascribed to the formation of a TPPO-TPP EDA complex (black curve). This peak changes upon addition of 1 and 2 (purple, green, and blue curves), which is ascribed to the fact that both TPP15,16 and TPPO17,18 are Lewis bases that can coordinate with the acidic hydrogen of 2. Also, TPP is known to act as a nucleophile towards carbon double bonds.19 Therefore, the presence of 2 and 1 weakens the interactions between TPP and TPPO to shift the position of the net absorption peak and reduces the concentration of the TPP-TPPO complex to lower the peak intensity. Fig. 1b also shows the tailing profiles for solutions containing both TPP and 2, which may arise from S–H/π-interactions.20 The increased intensity of this absorption tail for the 1/2/TPP solution (orange vs. red curves) suggests additional interaction of 1 with 2/TPP. The appearance of strong net absorption in Fig. 1b highlights the importance of TPP in gaining visible-light absorption through strong interactions with other molecules. While the net absorption peak maximum for the mixtures with TPPO/TPP, 2/TPP, and 1/2/TPP is in the UV region, the peak tails dip into the visible region. The TPPO-TPP peak's proximity to the visible region is evidence that TPPO-TPP EDA complex is the key species responding to visible light for the visible-light-driven de-S processes. The importance of the TPPO-TPP complex for visible-light-driven de-S processes has been explored systematically by controlling the initial amounts of TPPO in the de-S reactions. The purchased TPP in the original bottle (b-TPP) contains significant amounts of TPPO impurity (Fig. S5, ESI†). Additionally, TPPO can be formed during reactions under ambient conditions (Fig. S5, ESI†). The purchased 2 in the original bottle (b-2) is labelled as 95% purity, containing dimethyl 2,2′-disulfanediyldiacetate (2-disulfide). The role of both impurities, TPPO and 2-disulfide, are evaluated for de-S by using reagents purified immediately prior to use (p-TPP and p-2) as references (Table 3). Comparing entries 1 vs. 3 and 2 vs. 4, it is evident that TPPO offers a substantial enhancement to the rate of de-S, roughly quadrupling the rate of reaction when comparing 1 to 3. In regards to 2-disulfide (entries 1 vs. 2 and 3 vs. 4), the b-2 exhibits higher yield of methyl acetate than the p-2. However, using dibenzyl disulfide and TPP with water as proton source yields less toluene (the de-S product) than using benzyl mercaptan (Fig. S6, ESI†). Such a contrast in yielding de-S products for different mercaptans and di-mercaptans indicates that neither mercaptans nor di-mercaptans are the key species for triggering the generation of reactive radicals. This is consistent with other reports in which visible light cannot drive disulfide homolysis to generate radicals.11 Therefore, the observations of de-S reactions in Table 3 indicate that the de-S process is initiated by reactive radicals from elsewhere, i.e., photoexcitation of the TPPO-TPP EDA complex. Because de-S must occur to provide the methyl acetate radical required for the C–C coupling, the TPPO-TPP complex should also accelerate formation of 3. The yield of 3 indeed increases in accordance with conditions which optimize the amount of TPPO (Table S1, ESI†). This further confirms the importance of the TPPO-TPP EDA complex in triggering the generation of reactive radicals through visible-light-driven SET between TPPO and TPP.
|
|
||
|---|---|---|
| Entry | Reagents | Methyl Acetate Yield (%) |
| Reaction conditions: 157 mg TPP, 72 μL 2, 2 mL acetonitrile (solvent), 0.8 W cm−2 white LED (Dolan-Jenner Fiber-Lite MI-LED B1), photoillumination for 6 hours, no atmospheric control, and temperature at 60 °C All reactions were run without a stir bar to avoid contamination. The yield of product, methyl acetate, was determined using 1H-NMR with t-cinnamaldehyde as an internal standard. b-TPP contained ∼1% TPPO as determined by 31P-NMR. b-2 contained ∼5% 2-disulfide according to the manufacturer. | ||
| 1 | p-TPP, p-2 | 6 |
| 2 | p-TPP, b-2 | 15 |
| 3 | b-TPP, p-2 | 26 |
| 4 | b-TPP, b-2 | 33 |
Based upon these findings, Fig. 2 highlights the possible major intermediate radicals and reaction pathway for the “photocatalyst-free” visible-light-driven de-S and subsequent C–C coupling. TPPO-TPP EDA complex is formed to enhance absorption of short-wavelength visible light. SET from TPP to TPPO occurs in the photoexcited complex, transiently forming TPP˙+ and TPPO˙− in a possibly bound state. 2 intercepts the excited state TPPO-TPP complex, yielding both TPP˙-OH and the 2-TPP˙ phosphoranyl radical. The phosphoranyl radical rapidly dissociates via β-scission to give triphenylphosphine sulfide and a methyl acetate radical. Styrene intercepts the methyl acetate radical, creating the new C–C bond. The resulting carbon radical abstracts H˙ from TPP˙-OH. This final step forms the final C–C coupling product and recovers TPPO. TPPO acts as a catalyst by coordinating with TPP to impart visible light absorption and allow charge separation to tigger a radical reaction cascade.
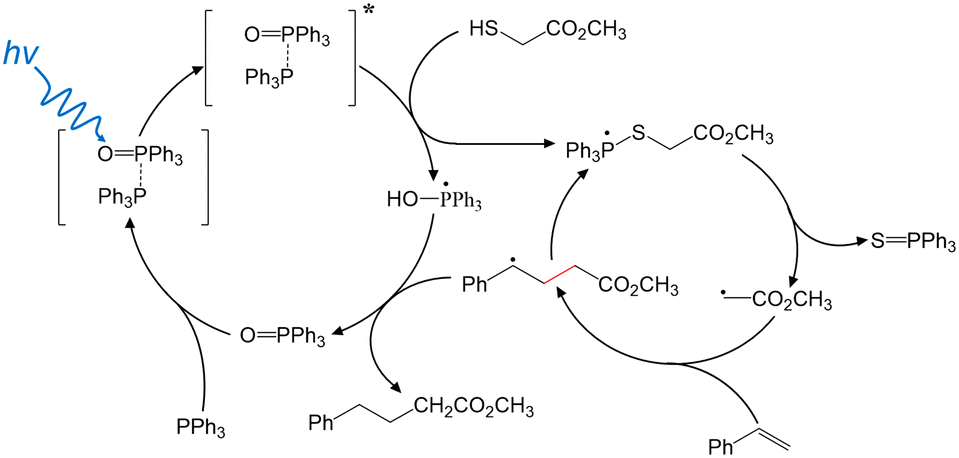 | ||
| Fig. 2 Plausible mechanism for TPPO-TPP enabled C–C coupling via de-S. | ||
In conclusion, TPPO behaves as “photocatalyst” for visible-light-driven reductive C–C coupling via de-S between mercaptans with electron-deficient groups and aryl alkenes through the formation of a TPPO-TPP EDA complex. The TPPO-TPP EDA complex is responsible for enhancing visible light absorption, driving SET from TPP to TPPO to form reactive radical ions that initiate a series of radical reactions towards the desired C–C coupling products. No delicately designed photocatalysts including metal–ligand complexes and dyes are needed in the present visible-light-driven C–C coupling reactions. Consequently, the reactions are not sensitive to atmospheric oxygen and can be carried out in air at room temperature, which is contrast to the photocatalytic C–C coupling reactions using conventional photocatalysts requiring anaerobic atmospheres.3,5 This method appreciably applies to a variety of styrene derivatives for C–C coupling reactions with 2. However, mercaptans with electron-rich groups such as benzyl mercaptan are not favourable for C–C coupling, although they do undergo the de-S process. Therefore, the strategy relying on the formation of TPPO-TPP EDA complex represents a general approach to achieve de-S of mercaptans under illumination of visible light. The discoveries of the present work open an alternative avenue to enable the photocatalytic C–C coupling reactions using conventional photocatalysts requiring anaerobic atmospheres.3,5 Interestingly, TPPO, an inevitable impurity formed during storage and reactions in aerobic environments, interacts with TPP, one important chemical for phosphoranyl radical chemistry, to form TPPO-TPP EDA complex, which absorbs short-wavelength visible light to enable forming phosphoranyl radicals. Because phosphoranyl radical fragmentation can produce a wide range of carbon radicals, the protocol reported in this work is promising for chemical synthesis under illumination of LED lamp at room temperature. The discoveries of the present work shed light on enabling visible-light-driven radical chemistry without using expensive molecular photocatalysts.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Melchionna and P. Fornasiero, ACS Catal., 2020, 10, 5493–5501 CrossRef CAS.
- K. P. S. Cheung, S. Sarkar and V. Gevorgyan, Chem. Rev., 2022, 122, 1543–1625 CrossRef.
- K. Teegardin, J. I. Day, J. Chan and J. Weaver, Org. Process Res. Dev., 2016, 20, 1156–1163 CrossRef CAS.
- M.-Y. Qi, M. Conte, M. Anpo, Z.-R. Tang and Y.-J. Xu, Chem. Rev., 2021, 121, 13051–13085 CrossRef CAS.
- J. A. Rossi-Ashton, A. K. Clarke, W. P. Unsworth and R. J. K. Taylor, ACS Catal., 2020, 10, 7250–7261 CrossRef CAS.
- X.-Q. Hu, Y.-X. Hou, Z.-K. Liu and Y. Gao, Org. Chem. Front., 2020, 7, 2319–2324 RSC.
- E. E. Stache, A. B. Ertel, T. Rovis and A. G. Doyle, ACS Catal., 2018, 8, 11134–11139 CrossRef CAS PubMed.
- M. Zhang, J. Xie and C. Zhu, Nat. Commun., 2018, 9, 3517 CrossRef.
- G.-N. Li, H.-C. Li, M.-R. Wang, Z. Lu and B. Yu, Adv. Synth. Catal., 2022, 364, 3927–3931 CrossRef CAS.
- H. Jiang, G. Mao, H. Wu, Q. An, M. Zuo, W. Guo, C. Xu, Z. Sun and W. Chu, Green Chem., 2019, 21, 5368–5373 RSC.
- Y. Deng, X.-J. Wei, H. Wang, Y. Sun, T. Noël and X. Wang, Angew. Chem., Int. Ed., 2017, 56, 832–836 CrossRef CAS.
- M. S. Kharasch, W. Nudenberg and G. J. Mantell, J. Org. Chem., 1951, 16, 524–532 CrossRef CAS.
- A. Fava, G. Reichenbach and U. Peron, J. Am. Chem. Soc., 1967, 89, 6696–6700 CrossRef CAS.
- H. H. Szmant, A. J. Mata, A. J. Namis and A. M. Panthananickal, Tetrahedron, 1976, 32, 2665–2680 CrossRef CAS.
- M. Shabbir, Z. Akhter, A. R. Ashraf, H. Ismail, A. Habib and B. Mirza, J. Mol. Struct., 2017, 1149, 720–726 CrossRef CAS.
- C. F. Albert, P. C. Healy, J. D. Kildea, C. L. Raston, B. W. Skelton and A. H. White, Inorg. Chem., 1989, 28, 1300–1306 CrossRef CAS.
- M. C. Etter and P. W. Baures, J. Am. Chem. Soc., 1988, 110, 639–640 CrossRef CAS.
- D. C. Batesky, M. J. Goldfogel and D. J. Weix, J. Org. Chem., 2017, 82, 9931–9936 CrossRef CAS PubMed.
- Y. C. Fan and O. Kwon, Chem. Commun., 2013, 49, 11588 RSC.
- C. R. Forbes, S. K. Sinha, H. K. Ganguly, S. Bai, G. P. A. Yap, S. Patel and N. J. Zondlo, J. Am. Chem. Soc., 2017, 139, 1842–1855 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, control syntheses, synthesis and characterization of substrate scope compounds. See DOI: https://doi.org/10.1039/d3cc00001j |
| This journal is © The Royal Society of Chemistry 2023 |