
Axial optimization of biomimetic nanoenzyme catalysts applied to oxygen reduction reactions†
Jingjing
Liu‡
a,
Jingjing
Jia‡
b,
Huiying
Wen‡
a,
Siqi
Li
a,
Yingjie
Wu
a,
Qi
Wang
a,
Ziwang
Kan
a,
Yan
Li
a,
Xia
Wu
a,
Jingxiang
Zhao
*b,
Song
Liu
 *a and
Bin
Li
*a
*a and
Bin
Li
*a
aCollege of Chemistry, Chemical Engineering and Resource Utilization, Northeast Forestry University, Harbin, 150040, China. E-mail: carlosliusong@nefu.edu.cn; libinzh62@163.com
bCollege of Chemistry and Chemical Engineering, and Key Laboratory of Photonic and Electronic Bandgap Materials, Ministry of Education, Harbin Normal University, Harbin, 150025, China. E-mail: zhaojingxiang@hrbnu.edu.cn
First published on 21st February 2023
Abstract
Inspired by the bio-oxygen oxidation/reduction processes of hemoglobin, iron-based transition metal-like enzyme catalysts have been explored as oxygen reduction reaction (ORR) electrocatalysts. We synthesized a chlorine-coordinated monatomic iron material (FeN4Cl-SAzyme) via a high temperature pyrolysis method as a catalyst for the ORR. The half-wave potential (E1/2) was 0.885 V, which exceeded those of Pt/C and the other FeN4X-SAzyme (X = F, Br, I) catalysts. Furthermore, through density functional theory (DFT) calculations, we systematically explored the better performance reason of FeN4Cl-SAzyme. This work offers a promising approach toward high-performance single atom electrocatalysts.
Nature uses metalloenzymes to activate and reduce oxygen (O2) with excellent activity under mild conditions.1–7 In particular, O2 reduction to water can smoothly occur on the Fe site of cytochrome c oxidases.8–14 Bioinspired catalysts with metalloenzyme-like sites have been designed and investigated for this electrochemical process.15–24 For example, Hu found an efficient metalloenzyme-like electrocatalyst with single atomic Fe–N–C active sites. Despite their outstanding 4e− selectivity, the SAzymes generally showed moderate ORR activity with the half-wave potential < 0.851 V versus RHE.25
In the Fe active site structure of cytochrome c oxidases, four nitrogens coordinate with Fe in the molecular plane and a histidine imidazole group binding to the axial position.26 Precisely, the axial group can adjust the electron density of the Fe site. Rui Cao reported an enzyme-mimetic iron porphyrin complex. It was found that by introducing this enzyme-mimicking axial imidazole ligand, the oxygen reduction half-wave potential of the complex was increased by 160 mV.27 Furthermore, the influence of the axial heteroatom can assist the CO2 adsorption and the CO* desorption by tuning the electronic structure of the Fe active sites.28 Similarly, the O2 adsorption and the H2O desorption were also assisted by tuning the electronic structure of the Fe active sites. Recently, N,S-ligated single-atom Fe with an axial fifth hydroxyl (OH) ligand (Fe–N3S1OH) has been reported by Prof. Yi-Wang Chen. It is confirmed that the axial OH in Fe–N3S1OH can optimize the 3d orbital of the Fe center to enhance O2 adsorption.29 Therefore, the proper axial coordination atom of Fe SAzymes will enhance the ORR performance.30–32
Herein, we synthesized a series of enzyme-inspired single Fe atom electrocatalysts with different axial coordination atoms. Finally, FeN4Cl-SAzyme has good ORR catalytic performance.
The synthesis of FeN4Cl-SAzyme is based on high-temperature pyrolysis (Fig. 1a). The precursor was first prepared by mixing melamine, α-cellulose, iron nitrate nonahydrate and thiourea. In particular, the Cl species was introduced through employing hydrochloric acid. Then, a two-step carbonization (550 °C and 1000 °C) under an argon atmosphere was carried out. For comparison, the acid was omitted or the hydrochloric acid was replaced by hydrofluoric, hydrobromic or hydroiodic acid to obtain other electrocatalysts, which were denoted as FeN4-SAzyme and FeN4X-SAzyme (X = F, Br, I), respectively. As shown by scanning electron microscopy (SEM) and transmission electron microscopy (TEM), the as-prepared catalysts have a two-dimensional structure (Fig. 1b and c, Fig. S1, ESI†). Furthermore, the spectra of the catalysts were obtained by FTIR spectroscopy confirming the catalyst-like graphene structure33–35 (Fig. S2, ESI†). Atomic force microscopy (AFM) further confirmed the morphology with a thickness of about 2.8 nm, suggesting a few layers of graphene-like sheets (Fig. 1d). The uniformity was studied by EDS elemental mapping, which shows that C, N, O, Fe and Cl are uniformly distributed. Moreover, the crystallinity and disorder are shown in the X-ray diffraction (XRD) and Raman spectroscopy (Fig. S3 and S4, ESI†). There were only two peaks at 26° and 44° (Fig. S3, ESI†), belonging to the (002) and (101) planes of graphitic carbon, which indicates no iron related species particles. The intensity ratio of the D band (1350 cm−1) and G band (1580 cm−1) in the FeN4Cl-SAzyme's Raman spectroscopy is 0.9561, which has the smallest ratio among all catalysts (Table S1, ESI†), indicating the high degree of graphitization. Furthermore, the aberration-corrected high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) image (Fig. 1e) clearly confirmed the single Fe atom distribution (yellow dots, diameter about 0.2 nm) in the catalyst.
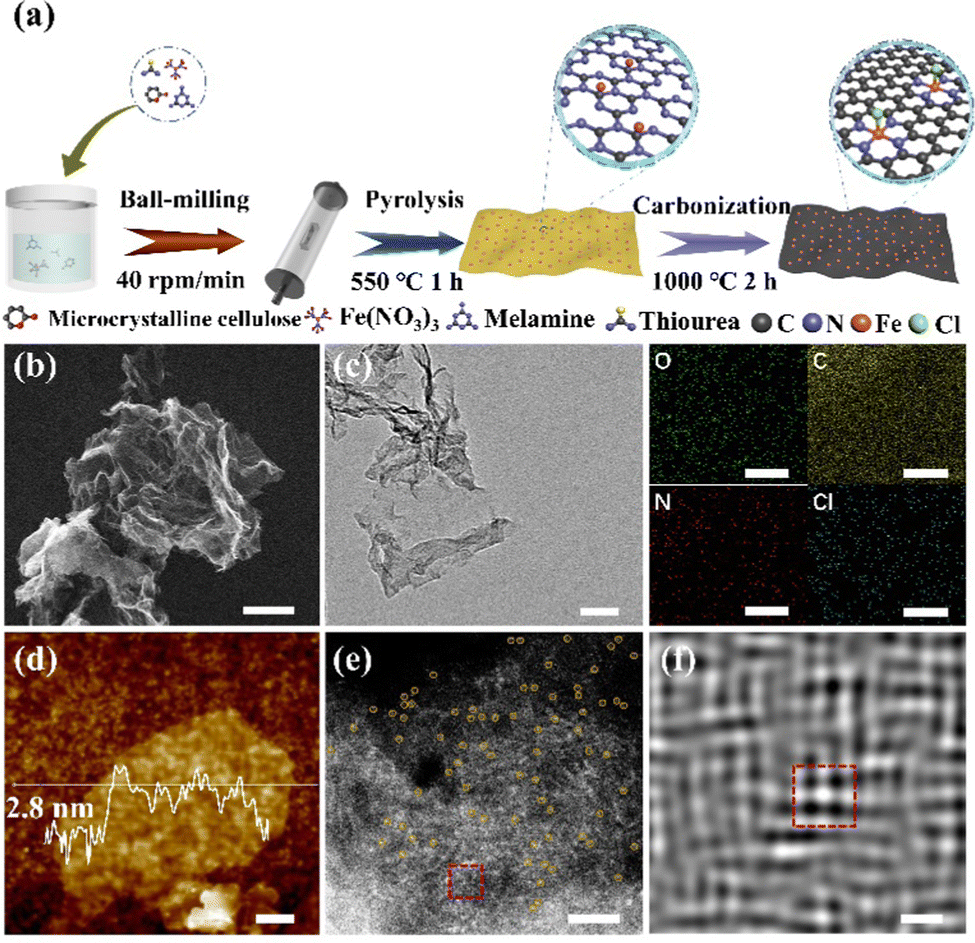 | ||
| Fig. 1 (a) The schematic synthesis illustration of FeN4Cl-SAzyme. (b) SEM image, scale bar: 2 μm. (c) TEM and corresponding EDS elemental mapping images, scale bar: 50 nm. (d) AFM image of FeN4Cl-SAzyme. Scale bar: 0.2 μm. (e) HAADF-STEM image of FeN4Cl-SAzyme. Scale bar: 2 nm. (f) The zoomed-in image of the single Fe atom area marked with the red dashed frames in the bottom of (e). Scale bar: 0.2 nm. | ||
In addition, the single Fe atom was coordinated with four atoms or five atoms (another atom in the back axial position) in Fig. 1f, which is obtained by Fourier transformation. Moreover, the Fe content determined by an inductively coupled optical emission spectrometer (ICP-OES) was approximately 2.0 wt%. Additionally, the nitrogen adsorption and desorption isotherm curves showed that the FeN4Cl-SAzyme has similar Brunauer–Emmett–Teller (BET) surface area and pore size (BJH) (Fig. S5 and Table S2, ESI†) to FeN4X-SAzyme (X = F, Br, I). This indicates that FeN4Cl-SAzyme has a porous structure that facilitates electron transport.
XPS was carried out to investigate the elemental information in the FeN4-SAzyme and FeN4X-SAzyme (Fig. 2a–c and Fig. S6–S12, ESI†). From the C 1s spectrum, the C 1s spectrum was deconvoluted into three peaks at 284.8 eV, 286.2 and 285.1 eV. These corresponded to the contribution from C–C, C–O and C–N bonds, respectively (Fig. S9b, S10b, S11b, and S12b, ESI†). The high-resolution XPS O 1s spectrum deconvoluted into three peaks, 532.9 eV, 532 eV, and 531.4 eV, which are assigned to C–O–H, C–O–C and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (Fig. 2a and Fig. S9c, S10c, S11c, S12c, ESI†). There was no Fe–O peak, which indicates that the Fe mainly coordinated with other elements and the N 1s spectrum can be deconvoluted into pyridinic-N (∼398.6 eV), pyrrolic-N (∼400.2 eV), graphitic N (∼401.1 eV), oxidized N (∼402.3 eV), and Fe–NX (∼399.5 eV) (Fig. 2b and Fig. S9d, S10d, S11d, S12d, ESI†). The solid coordination of the Fe atom with circumambient N atoms in the graphene-like substrate matrix stabilizes the atomically dispersed Fe sites. Moreover, the Fe 2p XPS spectra showed the three characteristic peaks of Fe2+ 2p3/2 (712 eV), Fe2+ 2p1/2 (724 eV) and Fe3+ 2p3/2 (716 eV) (Fig. 2c and Fig. S9e, S10e, S11e, S12e, ESI†). Simultaneously, the X-ray diffraction pattern of FeN4Cl-SAzyme displayed no peaks of Fe nanoparticles. Importantly, there was a remarkable peak of 198.3 eV, corresponding to Fe–Cl (Fig. S8, ESI†). Thus, the XPS results for FeN4Cl-SAzymes suggested that the Fe atoms mainly bind to the N and Cl.
O (Fig. 2a and Fig. S9c, S10c, S11c, S12c, ESI†). There was no Fe–O peak, which indicates that the Fe mainly coordinated with other elements and the N 1s spectrum can be deconvoluted into pyridinic-N (∼398.6 eV), pyrrolic-N (∼400.2 eV), graphitic N (∼401.1 eV), oxidized N (∼402.3 eV), and Fe–NX (∼399.5 eV) (Fig. 2b and Fig. S9d, S10d, S11d, S12d, ESI†). The solid coordination of the Fe atom with circumambient N atoms in the graphene-like substrate matrix stabilizes the atomically dispersed Fe sites. Moreover, the Fe 2p XPS spectra showed the three characteristic peaks of Fe2+ 2p3/2 (712 eV), Fe2+ 2p1/2 (724 eV) and Fe3+ 2p3/2 (716 eV) (Fig. 2c and Fig. S9e, S10e, S11e, S12e, ESI†). Simultaneously, the X-ray diffraction pattern of FeN4Cl-SAzyme displayed no peaks of Fe nanoparticles. Importantly, there was a remarkable peak of 198.3 eV, corresponding to Fe–Cl (Fig. S8, ESI†). Thus, the XPS results for FeN4Cl-SAzymes suggested that the Fe atoms mainly bind to the N and Cl.
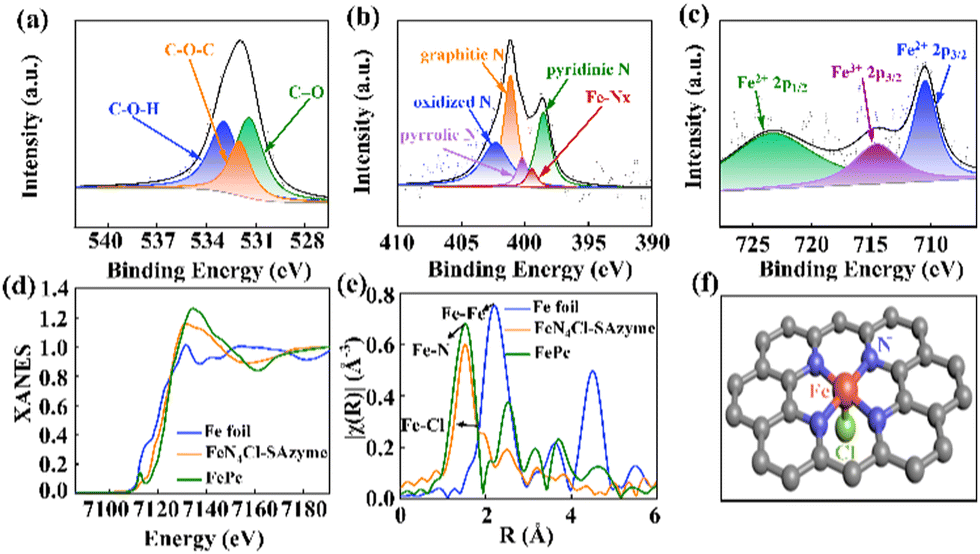 | ||
| Fig. 2 The XPS (a) O 1s, (b) N 1s, and (c) Fe 2p spectra of FeN4Cl-SAzyme. (d) Fe K-edge XANES spectra of Fe foil, FePc and FeN4Cl-SAzyme. (e) Fourier transformation of the EXAFS spectra of Fe foil, FePc and FeN4Cl-SAzyme. (f) The structure of the iron site in FeN4Cl-SAzyme. | ||
The coordination environment of the Fe atoms was further determined by synchrotron X-ray absorption spectroscopy measurements (XAS) (Fig. 2d and e, Fig. S13, ESI†). The normalized FeN4Cl-SAzyme K-edge absorption threshold was shifted to higher energies than Fe and close to FePc from the Fe K-edge X-ray absorption near-edge structure (XANES) spectra (Fig. 2d), which indicates that the Fe atoms are positively charged and attributed to the result of Fe bonding to N. Furthermore, the pre-edge peak at 7117 eV, the fingerprint of square-planar Fe–N4 moieties (D4h symmetry),36 had almost disappeared, suggesting that the Fe–N4 planar structure was distorted. Moreover, the Fourier transform of the extended X-ray absorption fine structure (FT-EXAFS) was also used to record the quantitative structural information of the Fe atom. Two major peaks, located around 1.59 Å and 1.93 Å, corresponded to the results of Fe–N and Fe–Cl coordination. No signal corresponding to Fe–Fe coordination was detected in FeN4Cl-SAzyme (around 2.14 Å) in Fig. 2e, suggesting that there is no Fe–Fe bond formed. The fitting of EXAFS is shown in Fig. S14 and S15 (ESI†). The simulation for EXAFS reveals that the Fe in the FeN4Cl-SAzyme is coordinated by a Cl atom and four N atoms. The fit parameters are given in Table S3 (ESI†). Besides, wavelet transform (WT) was used to analyze the Fe K edge EXAFS oscillations. In Fig. S16 (ESI†), the WT maximum at 4.3 Å−1 for FeN4Cl-SAzyme could be assigned to Fe–Cl bonding, and no intensity maximum corresponding to Fe–Fe is detected, in contrast to the WT plots of Fe foil, and FePc. This confirmed the HAADF-STEM results and indicated that Fe atoms are monodisperse. We speculated that its structure was as shown in Fig. 2f. Similarly, other catalysts also have FeN4X-SAzyme (X = F, Br, I) structures.
Rotating disc electrode (RDE) measurements were performed to evaluate the ORR performance of the catalysts. CV experiments were performed to investigate the electrocatalytic activity of the FeN4X-SAzyme in a voltage range from 0.2 V to 1.5 V vs. RHE in Fig. S17 (ESI†). A characteristic peak can be observed for all samples, FeN4Cl-SAzyme has the largest peak current density indicating that the ORR activity was higher than that for the FeN4F-SAzyme, FeN4Br-SAzyme and FeN4I-SAzyme. Otherwise, Fig. 3a exhibits the linear sweep voltammetry (LSV) curves of Pt/C and FeN4Cl-SAzyme, which demonstrates the E1/2 of 0.885 V for FeN4Cl-SAzyme and 0.873 V for Pt/C. Besides, the E1/2 for FeN4-SAzyme and FeN4X-SAzyme (X = F, Br, I) is 0.857 V, 0.854 V, 0.823 V and 0.869 V, respectively (Fig. S18a, ESI†). Tafel slope of FeN4Cl-SAzyme was estimated to be 97 mV dec−1, less than that of Pt/C (99 mV dec−1), FeN4-SAzyme (100 mV dec−1), FeN4F-SAzyme (118 mV dec−1), FeN4Br-SAzyme (116 mV dec−1) and FeN4I-SAzyme (103 mV dec−1) (Fig. 3b and Fig. S18b, ESI†). Thus, the ORR rate-determining step on FeN4Cl-SAzyme could be the first step (* + O2 + e− + H2O → *OOH + OH−). In order to determine the number of electrons transferred, LSV scans with different rotational speeds were performed, according to the Koutecky–Levich (K–L) equation, proving that this reaction is a 4-electron transfer ORR reaction (Fig. 3c and Fig. S19, ESI†). Furthermore, the FeN4Cl-SAzyme electrode exhibited a good methanol tolerance (Fig. 3d). The cyclic voltammetric (CV) curves were fitted to obtain the bilayer capacitance and the electrochemically active surface area (ECSA) (Fig. S20, ESI†) of the materials was analyzed. Ultimately, the results showed that the Cdl of FeN4Cl-SAzyme is 4.2 mF cm−2, which is a similar Cdl to the other catalysts (Fig. S21, ESI†).
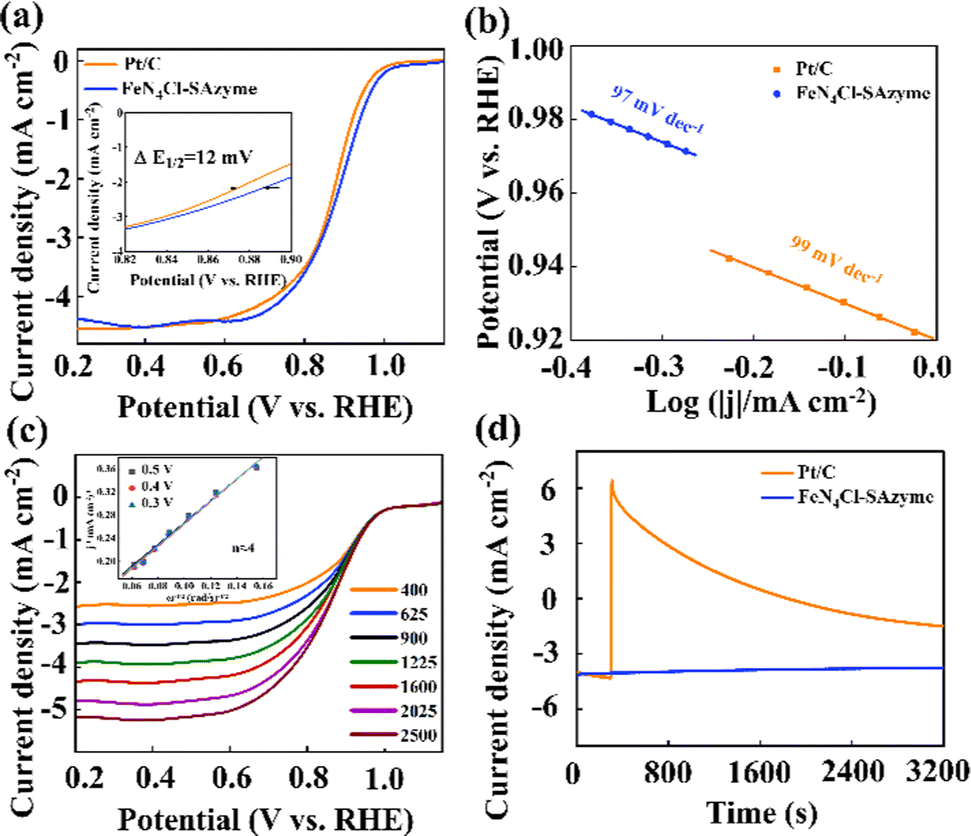 | ||
| Fig. 3 (a) The LSV curves of Pt/C and FeN4Cl-SAzyme. (b) Tafel slopes of Pt/C and FeN4Cl-SAzyme. (c) Electron transfer number of FeN4Cl-SAzyme. (d) Affect of methanol on the electrocatalytic activity of FeN4Cl-SAzyme and Pt/C. | ||
Finally, we tested the electrocatalytic stability of FeN4Cl-SAzyme by cycling the catalyst under an O2-saturated 0.1 M KOH atmosphere between 0.2 and 1.15 V vs. RHE. After 5000 cycles, the half-wave potential of FeN4Cl-SAzyme exhibited a slight negative shift of approximately 12 mV for 0.1 M KOH, which was almost negligible, suggesting the outstanding stability. However, the negative shift of Pt/C was about 22 mV after merely 5000 cycles in alkaline media (Fig. S22, ESI†). Subsequently, the SEM image and the XRD of FeN4Cl-SAzyme after the electrocatalytic stability test were recorded, and the results show that there were no significant changes compared with before (Fig. S23, ESI†).
To investigate mechanisms for theORR on the FeN4X-SAzyme, we performed the XPS measurements before and after the experiment. There was an extra peak at approximately 534.1 eV after the reaction (Fig. 4a), suggesting that the Fe–OOH spectrum appears. Similarly, the other electrocatalysts also showed the same change (Fig. S24–S26 and Table S4, ESI†). This indicated the efficient adsorption of O2. Furthermore, the ratio of Fe–OOH on FeN4Cl-SAzyme was higher than the other three electrocatalysts, which may result in the better ORR performance. Moreover, the appearance of the Fe–OOH was mainly due to the rate-determining step of * + O2 + e− + H2O → *OOH + OH−. Interestingly, this result was similar to the oxidation process of the cytochrome c oxidases. Subsequently, we performed DFT computations to explore the catalytic mechanism of the 4e pathway (Fig. 4b and c).
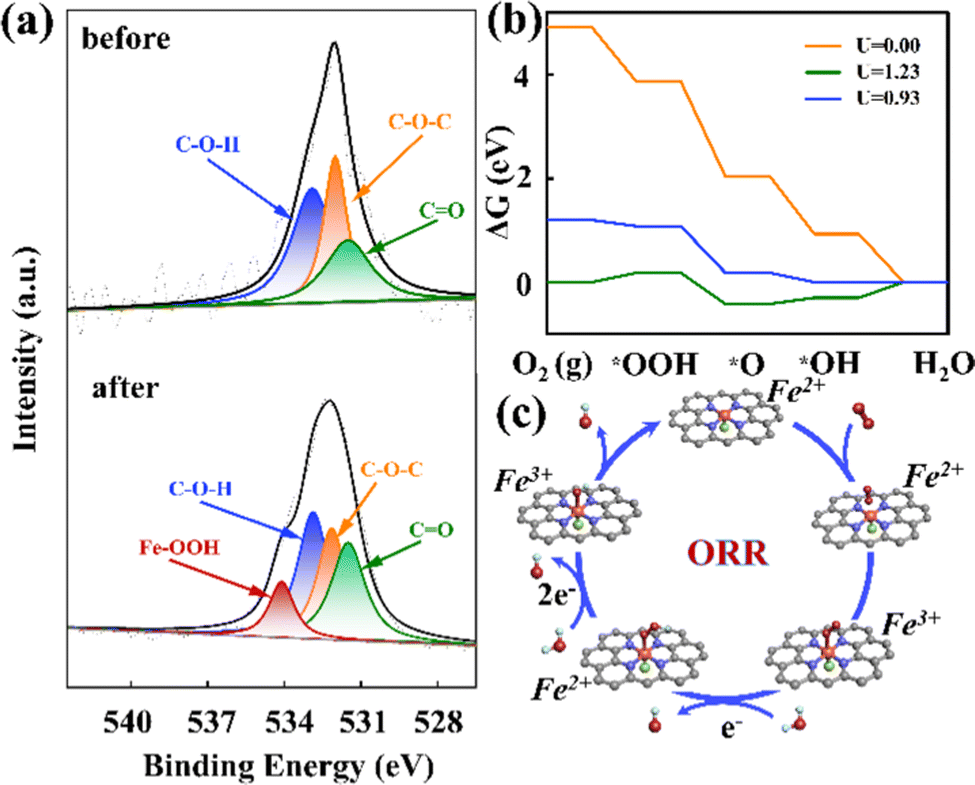 | ||
| Fig. 4 (a) O-spectrum test before and after the reaction of FeN4Cl-SAzyme. (b) Calculated Gibbs free energy diagrams of the 4e− ORR pathway on FeN4Cl-SAzyme. (c) Mechanistic pathway of the catalyst toward oxygen reduction reactions. | ||
According to the computed free energy profiles, we found that the ORR on F−, Br−, and I− coordinated Fe active sites is controlled by OOH* formation with a large overpotential of 1.01 V, 1.20 V and 1.18 V (Fig. S27–S29, ESI†), respectively. In contrast, on the FeN4Cl catalyst, the potential-determining step locates at the last electron transfer step (*OH + e− → OH− + *), and the limiting potential was computed to be 0.93 eV, corresponding to the overpotential of 0.30 V, which is much lower than those of the other three candidates and the state-of-the-art Pt catalysts (0.45 V), thus suggesting its superior ORR catalytic performance. For comparison, we also explored the ORR activity of the pristine FeN4. The results showed that the last step (i.e., OH* desorption) is uphill by 0.44 eV, indicating the strong binding strength of OH* on the FeN4 catalyst. On the contrary, the desorption of OH* only requires a low energy input of 0.30 eV on the FeN4Cl catalyst, suggesting its higher ORR activity, which is well consistent with the electron density differences for OH* adsorbed on the two catalysts. Clearly, the Cl coordination effectively modulates the charge of Fe in FeN4Cl (Fig. S30, ESI†), weakening the adsorption strength of the Fe site with OH* and thus achieving a better catalytic performance.
Furthermore, we took the Cl dopant as an example to examine the possibility of its doping into the carbon substrate, including C and N sites. The results showed that the interaction between Cl and C/N is not stable, as it will be optimized to the Fe site after full atomic relaxation. This is expected, because the Fe site exhibits larger positive charge than the C/N one, which is conducive to attracting the negatively charged Cl.
The ORR activity increased following the change of metal center from hydroperoxynitrite (Fe2+–O–O2−) to peroxynitrite (Fe3+–O–O−) after oxygen adsorption. In short, the valence state of the active center is changed. This corresponded to an activation effect, which makes it more favorable for the reaction to proceed next when more Fe3+ is generated. XPS analysis was performed on all synthesized samples to understand the bonding changes of the reactants (Fig. S31–S34, ESI†). Finally, it was found that the Cl-ligated Fe–N4 catalyst has a high Fe3+/Fe2+ ratio (Table S5, ESI†), which indirectly proves the reason for the excellent catalyst performance.
In summary, we prepared an atomically dispersed FeN4X-SAzyme (F, Cl, Br, I) catalyst by a pyrolysis method to achieve a significant enhancement of ORR activity. This strategy helped to obtain an efficient electrocatalyst. Subsequently, DFT showed that the Cl coordination effectively modulates the charge of Fe in FeN4Cl-SAzyme, weakening the adsorption strength of the Fe site with OH* and thus achieving a better catalytic performance in alkaline conditions. Finally, this study not only indicated that the activity of ORR can be improved by coordination with chlorine and nitrogen, but also provided inspiration for the future development of other substituted Pt catalysts.
This work was financially supported by the Young Elite Scientists Sponsorship Program by CAST (No. YESS20210262), the National Nature Science Foundation of China (No. 62001097), the China Postdoctoral Science Foundation-Funded Project (No. 2021M690571), the Heilongjiang Postdoctoral Fund (No. LBH-Z21096), the Provincial Natural Science Foundation Joint Guidance Project (No. LH2020F001), and the Fundamental Research Funds for the Central Universities (No. 2572020BU04).
Conflicts of interest
There are no conflicts to declare.References
- Y. Qin, J. Wen, X. Wang, L. Jiao, X. Wei, H. Wang, J. Li, M. Liu, L. Zheng, L. Hu, W. Gu and C. Zhu, ACS Nano, 2022, 16, 2997–3007 CrossRef CAS PubMed.
- S. Dey, B. Mondal, S. Chatterjee, A. Rana, S. Amanullah and A. Dey, Nat. Rev. Chem., 2017, 1, 1–20 CrossRef.
- X. Yang, H. Ben and A. J. Ragauskas, Asian J. Org. Chem., 2021, 10, 2473–2485 CrossRef CAS.
- P.-J. Wei, G.-Q. Yu, Y. Naruta and J.-G. Liu, Angew. Chem., Int. Ed., 2014, 53, 6659–6663 CrossRef CAS PubMed.
- W. Liu, P. Wang, Y. Ao, J. Chen, X. Gao, B. Jia and T. Ma, Adv. Mater., 2022, 34, 2202508 CrossRef CAS PubMed.
- W. Liu, P. Wang, J. Chen, X. Gao, H. Che, B. Liu and Y. Ao, Adv. Funct. Mater., 2022, 32, 2205119 CrossRef CAS.
- H. Che, P. Wang, J. Chen, X. Gao, B. Liu and Y. Ao, Appl. Catal., B, 2022, 316, 121611 CrossRef CAS.
- K. Liu, J. Fu, Y. Lin, T. Luo, G. Ni, H. Li, Z. Lin and M. Liu, Nat. Commun., 2022, 13, 1–8 CAS.
- X. Wan, W. Chen, J. Yang, M. Liu, X. Liu and J. Shui, ChemElectroChem, 2019, 6, 304–315 CrossRef CAS.
- Y. Han, Q. K. Li, K. Ye, Y. Luo, J. Jiang and G. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 15271–15278 CrossRef CAS PubMed.
- X. Xie, L. Shang, X. Xiong, R. Shi and T. Zhang, Adv. Energy Mater., 2021, 12, 2102688 CrossRef.
- Y.-B. Chen, J. Li, Y.-P. Zhu, J. Zou, H. Zhao, C. Chen, Q. Cheng, B. Yang, L.-L. Zou and Z.-Q. Zou, J. Mater. Chem. A, 2022, 10, 9886–9891 RSC.
- Q. Qu, S. Ji, Y. Chen, D. Wang and Y. Li, Trends Chem., 2021, 3, 954–968 CrossRef CAS.
- K. Holst-Olesen, L. Silvioli, J. Rossmeisl and M. Arenz, ACS Catal., 2019, 9, 3082–3089 CrossRef CAS.
- Y. Yao, Y. You, G. Zhang, J. Liu, H. Sun, Z. Zou and S. Sun, ACS Appl. Mater. Interfaces, 2016, 8, 6464–6471 CrossRef CAS PubMed.
- C. Sun, X. Chen, D. Zheng, W. Yao, H. Tan, Y. Zhang and S. Liu, Green Chem., 2021, 23, 9014–9023 RSC.
- S. Liu, H. B. Yang, S. F. Hung, J. Ding, W. Cai, L. Liu, J. Gao, X. Li, X. Ren, Z. Kuang, Y. Huang, T. Zhang and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 798–803 CrossRef CAS PubMed.
- G. Xiao, R. Lu, J. Liu, X. Liao, Z. Wang and Y. Zhao, Nano Res., 2021, 15, 3073–3081 CrossRef.
- Q. Sun, K. Zhu, X. Ji, D. Chen, C. Han, T.-T. Li, Y. Hu, S. Huang and J. Qian, Sci. China Mater., 2022, 65, 1453–1462 CrossRef CAS.
- H. Huang, D. Yu, F. Hu, S. C. Huang, J. Song, H. Y. Chen, L. L. Li and S. Peng, Angew. Chem., Int. Ed., 2022, 61, e202116068 CAS.
- F. Hu, D. Yu, M. Ye, H. Wang, Y. Hao, L. Wang, L. Li, X. Han and S. Peng, Adv. Energy Mater., 2022, 12, 2200067 CrossRef CAS.
- L. Li, D. Yu, P. Li, H. Huang, D. Xie, C.-C. Lin, F. Hu, H.-Y. Chen and S. Peng, Energy Environ. Sci., 2021, 14, 6419–6427 RSC.
- L. Wang, Y. Hao, L. Deng, F. Hu, S. Zhao, L. Li and S. Peng, Nat. Commun., 2022, 13, 5785 CrossRef CAS PubMed.
- C. Han, X. Zhang, Q. Sun, D. Chen, T. Miao, K. Su, Q. Li, S. Huang and J. Qian, Inorg. Chem. Front., 2022, 9, 2557–2567 RSC.
- W.-J. Jiang, W.-L. Hu, Q.-H. Zhang, T.-T. Zhao, H. Luo, X. Zhang, L. Gu, J.-S. Hu and L.-J. Wan, Chem. Commun., 2018, 54, 1307–1310 RSC.
- J. Dasgupta, U. Sen, D. Choudhury, P. Datta, A. Chakrabarti, S. B. Chakrabarty, A. Chakrabarty and J. K. Dattagupta, Biochem. Biophys. Res. Commun., 2003, 303, 619–623 CrossRef CAS PubMed.
- L. Xie, X. P. Zhang, B. Zhao, P. Li, J. Qi, X. Guo, B. Wang, H. Lei, W. Zhang, U. P. Apfel and R. Cao, Angew. Chem., Int. Ed., 2021, 60, 7576–7581 CrossRef CAS PubMed.
- Z. Li, R. Wu, S. Xiao, Y. Yang, L. Lai, J. S. Chen and Y. Chen, Chem. Eng. J., 2022, 430, 132882 CrossRef CAS.
- L. Li, S. Huang, R. Cao, K. Yuan, C. Lu, B. Huang, X. Tang, T. Hu, X. Zhuang and Y. Chen, Small, 2022, 18, 2105387 CrossRef CAS PubMed.
- Y. Han, Y. Wang, R. Xu, W. Chen, L. Zheng, A. Han, Y. Zhu, J. Zhang, H. Zhang, J. Luo, C. Chen, Q. Peng, D. Wang and Y. Li, Energy Environ. Sci., 2018, 11, 2348–2352 RSC.
- K. M. Zhao, S. Liu, Y. Y. Li, X. Wei, G. Ye, W. Zhu, Y. Su, J. Wang, H. Liu, Z. He, Z. Y. Zhou and S. G. Sun, Adv. Energy Mater., 2022, 12, 2103588 CrossRef CAS.
- L. Hu, C. Dai, L. Chen, Y. Zhu, Y. Hao, Q. Zhang, L. Gu, X. Feng, S. Yuan, L. Wang and B. Wang, Angew. Chem., Int. Ed., 2021, 60, 27324–27329 CrossRef CAS PubMed.
- B. Kartick, S. K. Srivastava and I. Srivastava, J. Nanosci. Nanotechnol., 2013, 13, 4320–4324 CrossRef CAS PubMed.
- M. Naebe, J. Wang, A. Amini, H. Khayyam, N. Hameed, L. H. Li, Y. Chen and B. Fox, Sci. Rep., 2014, 4, 4375 CrossRef PubMed.
- G. Bai, J. Wang, Z. Yang, H. Wang, Z. Wang and S. Yang, RSC Adv., 2014, 4, 47096–47105 RSC.
- W. Liu, L. Zhang, X. Liu, X. Liu, X. Yang, S. Miao, W. Wang, A. Wang and T. Zhang, J. Am. Chem. Soc., 2017, 139, 10790–10798 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc06197j |
| ‡ Jingjing Liu, Jingjing Jia and Huiying Wen contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |