
Breaking bonds and breaking rules: inert-bond activation by [(iPr3P)Ni]5H4 and catalytic stereospecific norbornene dimerization†
Junyang
Liu
 ,
Manar M.
Shoshani‡
,
Kethya
Sum
and
Samuel A.
Johnson
*
,
Manar M.
Shoshani‡
,
Kethya
Sum
and
Samuel A.
Johnson
*
Department of Chemistry and Biochemistry, University of Windsor, Sunset Avenue 401, Windsor, ON N9B 3P4, Canada. E-mail: sjohnson@uwindsor.ca; Fax: (+1) 519-973-7098
First published on 23rd January 2023
Abstract
The facile carbon atom abstraction reaction by [(iPr3P)Ni]5H6 (1) with various terminal alkenes to give [(iPr3P)Ni]5H4(μ5-C) (2) occurs via a common highly reactive intermediate [(iPr3P)Ni]5H4 (3), which was isolated by the reaction of 1 with norbornene. Temperature dependent 1H and 31P{1H} NMR chemical shifts of 3 are consistent with a thermally populated triplet excited state only 2 kcal mol−1 higher energy than the diamagnetic ground state. Complex 3 catalyzes the dimerization of norbornene to stereoselectively provide exclusively (Z) anti-(bis-2,2'-norbornylidene).
The cost and environmental impact of the relatively low-abundance late transition metals, such a Pd, Rh and Ir, provide an impetus for their replacement in catalysis by the abundant 1st row congeners.1–3 This is particularly difficult for modern green chemical conversions that target the catalytic transformations of traditionally inert bonds such as C–H,1,4–14 C–C,1,15–23 and C–O.1,24–28 One of the oft-touted strategies to make this possible is cooperative reactivity, where multiple metal atoms in a cluster work simultaneously to effect transformations that would be difficult or impossible for monometallic complexes. Although this strategy is commonly utilized by Nature,29–34 clusters capable of such reactivity are underdeveloped.35–41
Our group has previously reported the synthesis and characterization of [(iPr3P)Ni]5H6 (1),38 a pentanuclear nickel hydride cluster, which activates C–O, C–S, C–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds under ambient conditions.42–44 Late transition metal clusters supported solely by phosphines and hydrides have few examples,45–48 outside of group 11,49,50 and a fundamental understanding of the potential of complexes like 1 in modern catalysis is lacking. A remarkable reactivity demonstrated by 1 is the facile cleavage of CC and C–H bonds in terminal alkenes to abstract a C atom to form an interstitial carbide cluster at temperatures as low as 243 K using ethylene, as shown in Scheme 1.37 This reaction even occurs in the presence of typically more reactive functional groups and provides the thermally robust carbide cluster [(iPr3P)Ni]5H4(μ5-C) (2). This is not catalytic, but does demonstrate the potential of these nickel clusters to effect dramatic low-barrier C–H and skeletal bond changes with hydrocarbons that are not accessible with mononuclear complexes. An intermediate observed with isobutylene, shown on the bottom right of Scheme 1, reveals how all five Ni centres work cooperatively to bind the multiply C–H activated substrate prior to CC bond cleavage.
C bonds under ambient conditions.42–44 Late transition metal clusters supported solely by phosphines and hydrides have few examples,45–48 outside of group 11,49,50 and a fundamental understanding of the potential of complexes like 1 in modern catalysis is lacking. A remarkable reactivity demonstrated by 1 is the facile cleavage of CC and C–H bonds in terminal alkenes to abstract a C atom to form an interstitial carbide cluster at temperatures as low as 243 K using ethylene, as shown in Scheme 1.37 This reaction even occurs in the presence of typically more reactive functional groups and provides the thermally robust carbide cluster [(iPr3P)Ni]5H4(μ5-C) (2). This is not catalytic, but does demonstrate the potential of these nickel clusters to effect dramatic low-barrier C–H and skeletal bond changes with hydrocarbons that are not accessible with mononuclear complexes. An intermediate observed with isobutylene, shown on the bottom right of Scheme 1, reveals how all five Ni centres work cooperatively to bind the multiply C–H activated substrate prior to CC bond cleavage.
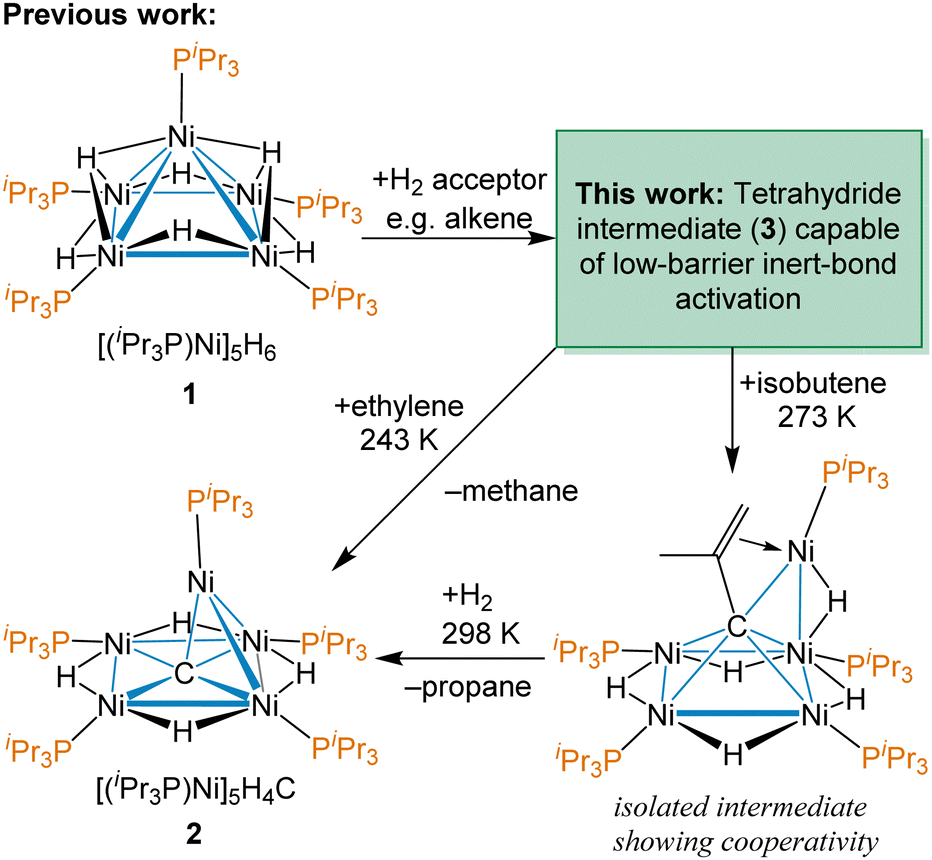 | ||
| Scheme 1 Previous work on facile C–H bond rearrangement and carbon atoms abstraction from terminal alkenes by the pentanuclear cluster [Ni(PiPr3)]5H6 (1). | ||
Although no common intermediate had been observed in the formation of carbide 2 from different alkenes, it seemed likely that the initial reaction of alkenes with [(iPr3P)Ni]5H6 (1) would occur with hydrogenation of an equivalent of alkene to give the electron deficient tetrahydride [(iPr3P)Ni]5H4 (3). This work details synthetic efforts to isolate and characterize this highly reactive species, capable of facile C–H and CC bond activation.
A shared unusual minor 1H NMR signal of a trace intermediate was observed in the reactions of 1 with a variety of alkenes. For example, when the reaction of [(iPr3P)Ni]5H6 (1) with isobutene in THF-d8 was monitored by 1H NMR, in addition to the previously reported and isolated intermediate shown in Scheme 1, an unusual septet at δ 4.30 with J = 7.3 Hz was observed early in the course of the reaction. The signal disappeared upon complete conversion of 1 to 2. The coupling was suggestive of the PCH environment of a Ni bound iPr3P, which would normally occur between 1.9–2.4 ppm in diamagnetic pentanuclear Ni clusters. The shift could be explained by an unusual bonding environment (e.g. an agostic interaction) or a thermally accessible paramagnetic triplet state influencing the chemical shift; previous work has shown that 1 has a low-lying triplet state 8.3 kcal mol−1 higher energy than its ground state.38 To test if replacement of the hydrides by deuterides affected the chemical shift of the septet observed at δ 4.30 and gain insight into how many hydrides are in this intermediate, the reaction of 1 with isobutylene was done in benzene-d6. Over the course of 30 minutes the minor species 3 is seen to convert to isotopologues putatively assigned at this point as [(iPr3P)Ni]5H3D (3-d1), [(iPr3P)Ni]5H2D2 (3-d2), [(iPr3P)Ni]5HD3 (3-d3) and [(iPr3P)Ni]5D4 (3-d4), as shown in Scheme 2.
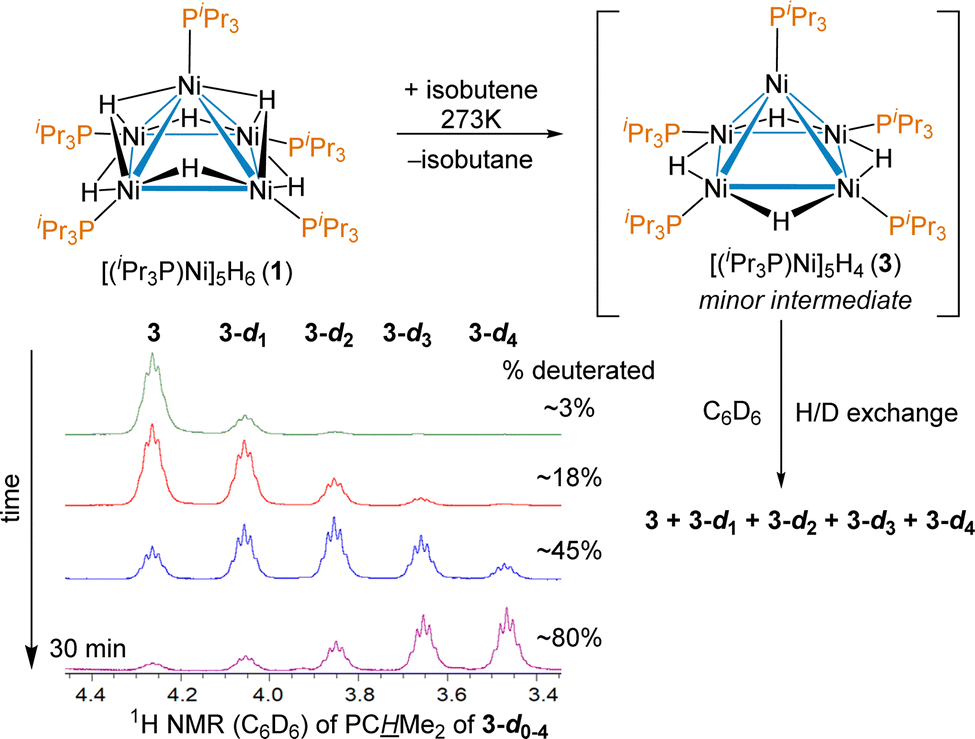 | ||
| Scheme 2 Reaction of 1 with isobutene in C6D6, with the distinct 1H NMR signals of the deuterated isotopologues of intermediate [(iPr3P)Ni]5H4 (3) with a large isotope shift. | ||
The large ∼0.2 ppm isotope shift for each deuteride on the chemical shift of the PCH environment supports a low-lying triplet state,51 and the number of isotopologues confirm that 3 is indeed a tetrahydride. The δ 4.30 resonance of 3 was also observed as a minor intermediate in reaction between 1 and other olefins such as 1-hexene and styrene, which suggests no hydrocarbyl or alkene is bound, providing additional support for the putative assignment. The synthesis, isolation and complete characterization of 3 with many alkenes and alkynes proved impossible, as it was only ever observed as a minor component of the reaction mixture and consumed nearly as quickly as it is formed (see ESI† for details); however, the persistence of observable concentrations of 3 in solution even at room temperature suggested it should be isolable.
Norbornene was tested due to its −33 kcal mol−1 heat of hydrogenation,52 its modest steric hindrance and the volatility of its hydrogenation product, norbornane. Reaction of 1 with 10 equiv. of norbornene provided complete consumption of norbornene immediately with the formation of 3 as observed by 1H NMR; the use of an excess of norbornene was required due to a catalytic dimerization of norbornene by 3 (vide infra). Monitored using low-temperature 1H NMR, the hydrogenation of norbornene occurs rapidly at temperatures as low as 253 K to give quantitative conversion to 3. The thermal decomposition of 3 that gives back sub-stoichiometric 1 was minimal at this temperature. The reaction of 1 with a dropwise addition of norbornene at 253 K and further cooling the reaction mixture at 233 K provided the successful isolation of crystals of 3 suitable for X-ray diffraction (Scheme 3). Despite near quantitative yields at 253 K by 1H NMR, isolated yields were typically ∼40%. The product was not stable at room temperature and rapidly decomposes in all but very dilute solutions.
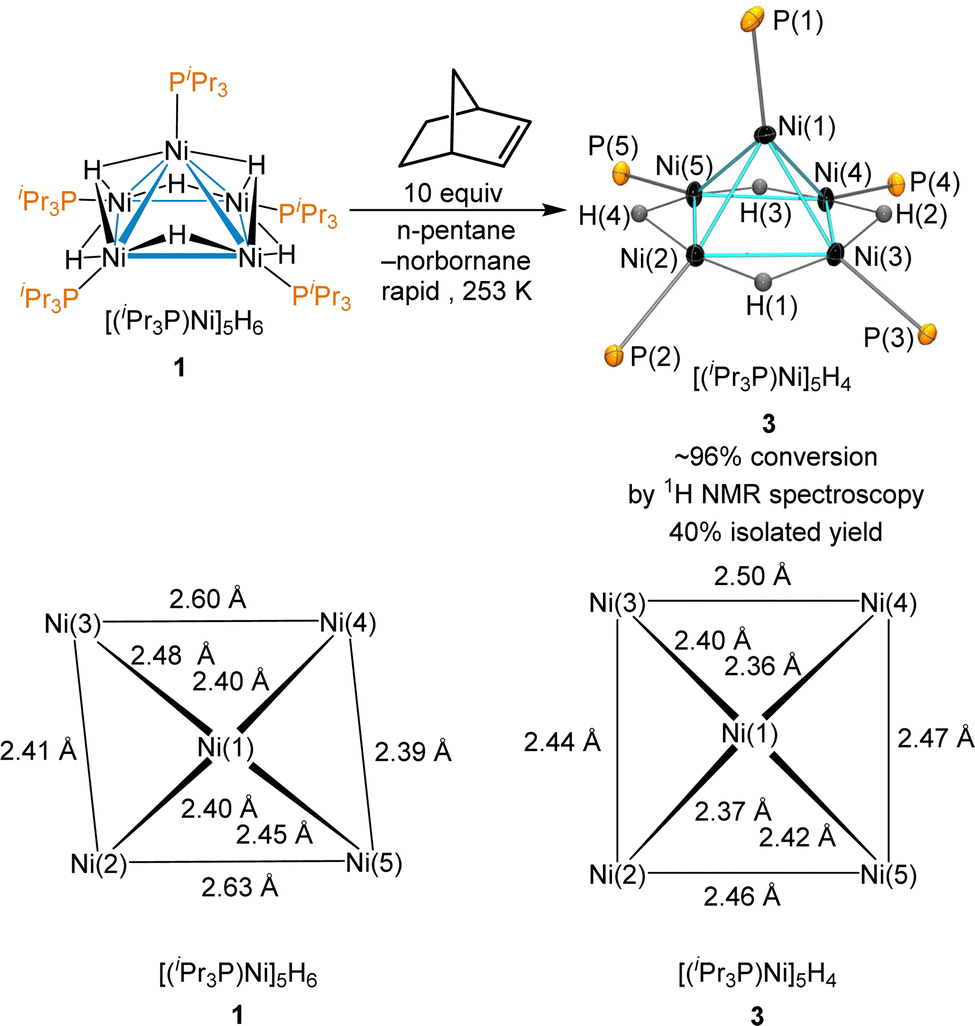 | ||
| Scheme 3 Formation of 3 from reaction between 1 and excess norbornene and ORTEP depiction of [(iPr3P)Ni]5H4 (3) as determined by X-ray crystallography, shown with 50% probability ellipsoids. Isopropyl groups attached to phosphorus and hydrogens (except hydrides) are omitted for clarity. Selected bond distances (Å) and angles (°): Ni(1)–Ni(2) 2.3726(5), Ni(1)–Ni(3) 2.3996(5), Ni(1)–Ni(4) 2.3626(5), Ni(1)–Ni(5) 2.4230(4), Ni(2)–Ni(3)2.442(4), Ni(2)–(5) 2.4647(6), Ni(3)–Ni(4) 2.4969(6), N(4)–Ni(5) 2.4727(5), Ni(5)–Ni(2)–Ni(3) 94.73, Ni(2)–Ni(3)–Ni(4) 85.95, Ni(3)–Ni(4)–Ni(5) 93.16, Ni(4)–Ni(5)–Ni(2) 85.99, Ni(2)–Ni(1)–P(1) 121.35, Ni(1)–Ni(2)–P(2) 170.7, Ni(1)–Ni(3)–P(3) 158.55, Ni(1)–Ni(4)–P(4) 144.59, Ni(1)–Ni(5)–P(5) 144.71. | ||
The unit cell of 3 is nearly identical to that of 1, but the Ni–Ni bond distances are significantly different, as shown on the bottom of Scheme 3. Cluster 3 has an electron count of 64 electrons and adopts a square pyramidal geometry, with each edge of the basal Ni4 moiety spanned by a μ2-hydride. The largest deviation C4 symmetry is from the Ni(1)–P(1) bond leaning towards the edge of Ni(1)–Ni(2), and the P(2) lies significantly below the plane of the square Ni4 base, with a Ni(1)–Ni(2)–P(2) angle of 170.7°.
Multinuclear NMR of 3 were obtained in THF-d8 and fully assigned. All resonances associated with 3 are observed to be temperature dependent and move toward their diamagnetic values upon cooling, with resonances of nuclei directly bonded to Ni spanning a larger range, particularly phosphorus and the hydrides. At 293 K, the 31P{1H} NMR of 3 features a single broad singlet at δ 569. The 298 K 1H NMR features a broad hydride resonance at δ 50.1, the PCH septet at δ 4.31 and the isopropyl methyl doublet at δ 1.41. At 193 K, the 31P{1H} NMR of 3 features a broad singlet at δ 152, while the 1H NMR hydride resonance is at δ −9.5, the PCH septet at δ 2.32 and the isopropyl methyl doublet at δ 1.33. At no temperature could the basal phosphines be distinguished from the capping phosphine; similar rapid fluxionality was also observed previously in 1. In toluene-d8 a mixture of the 3-dn (n = 0–4) isotopologues were obtained from H/D exchange. The temperature dependence of the 31P{1H} chemical shifts for of 3 in THF-d8 and the 3-dn isotopologues in toluene-d8 are shown in Fig. 1.
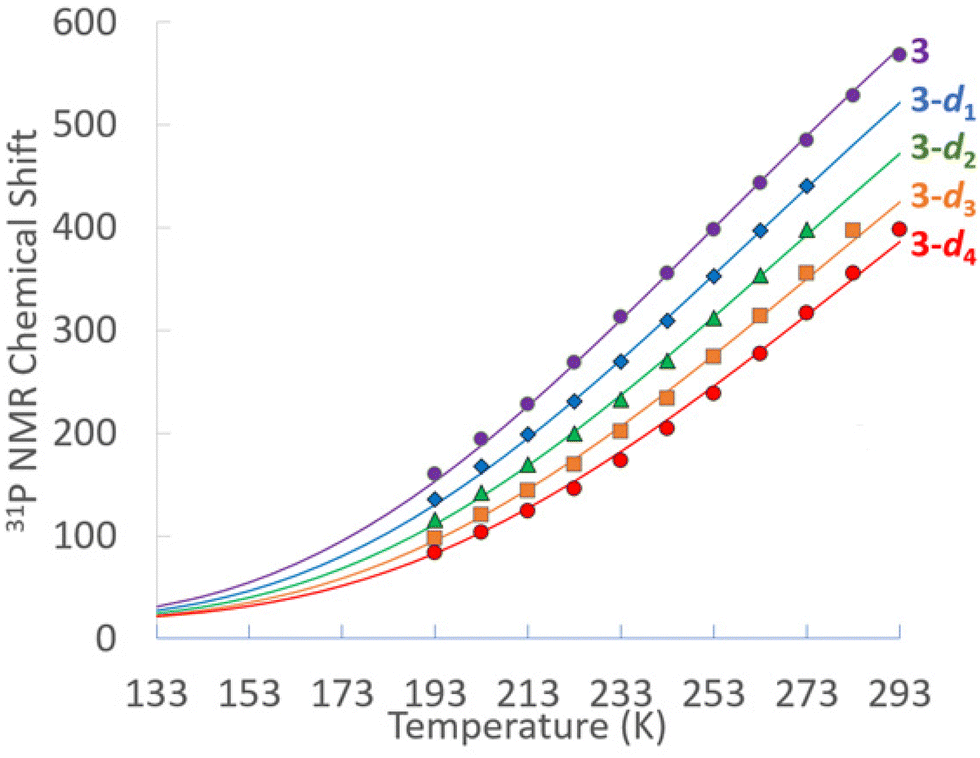 | ||
| Fig. 1 The temperature dependence of the 31P{1H} chemical shifts of 3 in THF-d8 (purple circles) and the 3-d1 (blue diamonds), 3-d2 (green triangles), 3-d3 (orange squares) and 3-d4 (red circles) isotopologues in toluene-d8. The solid lines show a fit based on the Boltzmann population of a low-lying triplet state. | ||
The solid lines fits in Fig. 1 are plot based of the occupation of a paramagnetic triplet state that features both 1/T dependent Fermi contact and 1/T2 dependent pseudocontact shifts. The ΔH associated with occupying the triplet state is modeled to be only 2.1 kcal mol−1. With the ΔS set to R![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) ln(3) to accommodate the entropic favourability of the threefold degenerate triplet state,533 has an estimated ΔG298
ln(3) to accommodate the entropic favourability of the threefold degenerate triplet state,533 has an estimated ΔG298![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K for population its triplets state of only 1.5 kcal mol−1. As would be expected, the 1H NMR hydride signals for 3 in toluene-d8 also have shifts for the isotopologues 3-d1, 3-d2 and 3-d3.
K for population its triplets state of only 1.5 kcal mol−1. As would be expected, the 1H NMR hydride signals for 3 in toluene-d8 also have shifts for the isotopologues 3-d1, 3-d2 and 3-d3.
The excess of norbornene used in the synthesis of 3 is required due to the catalytic dimerization of norbornene by 3. As shown in Scheme 4, this reaction can be carried out using 5 mol% 3 at −20 °C. This organic product was obtained as an oil by filtering the sample through a silica plug and identified as a single isomer of bis-2,2′-norbornylidene by both 1H and 13C{1H} NMR. This appears to be the first highly-selective norbornene dimerization catalyst, with previous Ni examples all requiring elevated temperatures and yielding mixtures of isomers54–56 The mechanism likely features oxidative coupling of two norbornene molecules to 3, followed by C–H bond activation and reductive elimination to give the C2 product. In comparison, a solution of the mononuclear Ni(0) source [Ni(iPr3P)2]2N2 and norbornene provided no catalysis. The isomer of the norbornene dimer produced was identified through its complexation with a [SIPr]Au(I) cation as its SbF6− salt. Crystals suitable for X-ray diffraction were grown from slow diffusion of n-hexane into a CH2Cl2 solution.
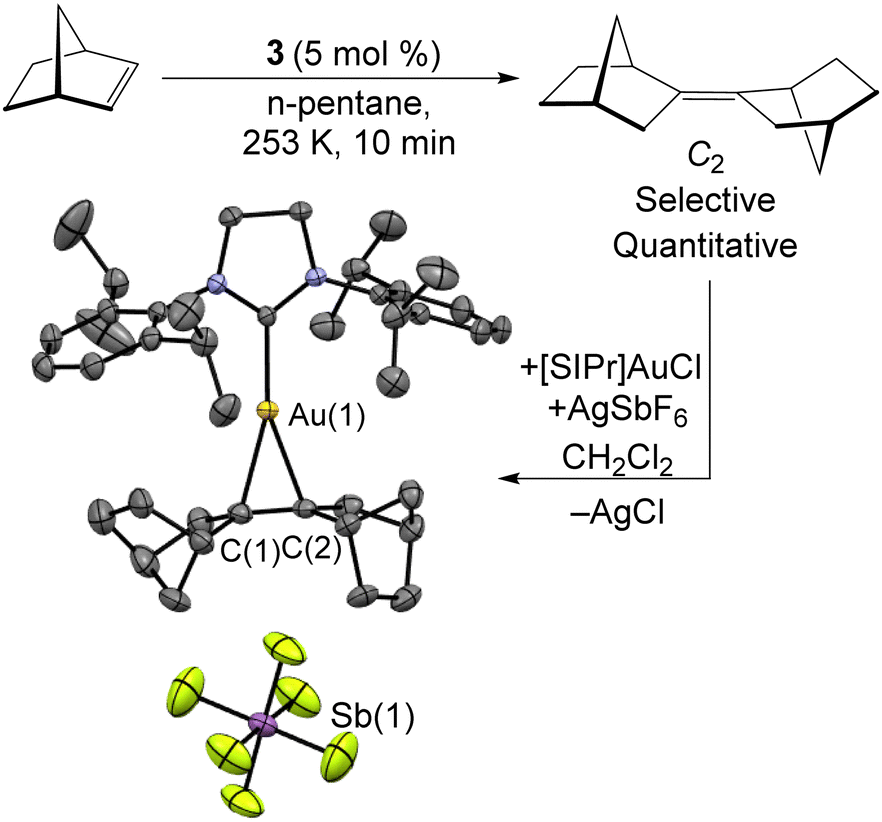 | ||
| Scheme 4 Selective dimerization of norbornene catalyzed by 3. The isomer formed was determined by complexation to [SIPr]Au(I), where [SIPr] = 1,3-bis(2,6-diisopropylphenyl)-2-imidazolidinylidene. | ||
In summary, the isolation of complex 3 confirms it as the most likely intermediate in previously reported carbon atom abstraction from alkenes, C–O cleavage, and C–H/D exchange using 1 as a precursor. This new work suggests that these atypical reactive clusters not prone to following classic rules for cluster electron counting, with evidence for a 4e-span of electron counts and oxidation states from [(iPr3P)Ni]5H4 (3), [(iPr3P)Ni]5H6 (1) and [(iPr3P)Ni]5H8.43 The very low-lying triplet states in the presence of strong field ligands is atypical of organometallic chemistry. The selective dimerization of norbornene demonstrates the potential of the Ni5 core to form C–C bonds and rearrange C–H bonds with minimal activation barriers. Further work needs to be done to fully understand the potential of this and related clusters in catalytic reactions.
SAJ acknowledges support from the Natural Sciences and Engineering Research Council (NSERC) of Canada in the form of a Discovery Grant.
Conflicts of interest
There are no conflicts to declare.References
- B. Su, Z.-C. Cao and Z.-J. Shi, Acc. Chem. Res., 2015, 48, 886–896 CrossRef CAS PubMed.
- P. Chirik and R. Morris, Acc. Chem. Res., 2015, 48, 2495 CrossRef CAS PubMed.
- R. M. Bullock, J. G. Chen, L. Gagliardi, P. J. Chirik, O. K. Farha, C. H. Hendon, C. W. Jones, J. A. Keith, J. Klosin, S. D. Minteer, R. H. Morris, A. T. Radosevich, T. B. Rauchfuss, N. A. Strotman, A. Vojvodic, T. R. Ward, J. Y. Yang and Y. Surendranath, Science, 2020, 369, eabc3183 CrossRef CAS PubMed.
- J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS PubMed.
- J. F. Hartwig, J. Am. Chem. Soc., 2015, 138, 2–24 CrossRef PubMed.
- R. G. Bergman, Nature, 2007, 446, 391–393 CrossRef CAS PubMed.
- H. Azizollahi and J.-A. García-López, Molecules, 2020, 25, 5900 CrossRef CAS PubMed.
- J. Loup, U. Dhawa, F. Pesciaioli, J. Wencel-Delord and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 12803–12818 CrossRef CAS PubMed.
- G. Pototschnig, N. Maulide and M. Schnürch, Chem. – Eur. J., 2017, 23, 9206–9232 CrossRef CAS PubMed.
- T. Rogge, N. Kaplaneris, N. Chatani, J. Kim, S. Chang, B. Punji, L. L. Schafer, D. G. Musaev, J. Wencel-Delord, C. A. Roberts, R. Sarpong, Z. E. Wilson, M. A. Brimble, M. J. Johansson and L. Ackermann, Nat. Rev. Methods Primers, 2021, 1, 43 CrossRef CAS.
- S. K. Sinha, S. Guin, S. Maiti, J. P. Biswas, S. Porey and D. Maiti, Chem. Rev., 2022, 122, 5682–5841 CrossRef CAS PubMed.
- R. Shang, L. Ilies and E. Nakamura, Chem. Rev., 2017, 117, 9086–9139 CrossRef CAS PubMed.
- N. V. Tzouras, I. K. Stamatopoulos, A. T. Papastavrou, A. A. Liori and G. C. Vougioukalakis, Coord. Chem. Rev., 2017, 343, 25–138 CrossRef CAS.
- B. Liu, A. M. Romine, C. Z. Rubel, K. M. Engle and B.-F. Shi, Chem. Rev., 2021, 121, 14957–15074 CrossRef CAS PubMed.
- F. Song, T. Gou, B.-Q. Wang and Z.-J. Shi, Chem. Soc. Rev., 2018, 47, 7078–7115 RSC.
- A. P. Y. Chan and A. G. Sergeev, Coord. Chem. Rev., 2020, 413, 213213–213239 CrossRef CAS.
- H. Lu, T.-Y. Yu, P.-F. Xu and H. Wei, Chem. Rev., 2020, 121, 365–411 CrossRef PubMed.
- J. Wang, S. A. Blaszczyk, X. Li and W. Tang, Chem. Rev., 2020, 121, 110–139 CrossRef PubMed.
- M. Murakami and Y. Ito, in Activation of Unreactive Bonds and Organic Synthesis, ed. S. Murai, H. Alper, R. A. Gossage, V. V. Grushin, M. Hidai, Y. Ito, W. D. Jones, F. Kakiuchi, G. Koten, Y.-S. Lin, Y. Mizobe, S. Murai, M. Murakami, T. G. Richmond, A. Sen, M. Suginome and A. Yamamoto, Springer Berlin, Heidelberg, 1999, ch. 5, pp. 97–129 Search PubMed.
- M. D. R. Lutz and B. Morandi, Chem. Rev., 2020, 121, 300–326 CrossRef PubMed.
- F. Chen, T. Wang and N. Jiao, Chem. Rev., 2014, 114, 8613–8661 CrossRef CAS PubMed.
- G. Fumagalli, S. Stanton and J. F. Bower, Chem. Rev., 2017, 117, 9404–9432 CrossRef CAS PubMed.
- L. Souillart and N. Cramer, Chem. Rev., 2015, 115, 9410–9464 CrossRef CAS PubMed.
- T. B. Boit, A. S. Bulger, J. E. Dander and N. K. Garg, ACS Catal., 2020, 10, 12109–12126 CrossRef CAS PubMed.
- J. Cornella, C. Zarate and R. Martin, Chem. Soc. Rev., 2014, 43, 8081–8097 RSC.
- A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439–443 CrossRef CAS PubMed.
- M. Tobisu and N. Chatani, Acc. Chem. Res., 2015, 48, 1717–1726 CrossRef CAS PubMed.
- E. Bisz and M. Szostak, ChemSusChem, 2017, 10, 3964–3981 CrossRef CAS PubMed.
- M. Can, F. A. Armstrong and S. W. Ragsdale, Chem. Rev., 2014, 114, 4149–4174 CrossRef CAS PubMed.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- D. C. Rees, F. Akif Tezcan, C. A. Haynes, M. Y. Walton, S. Andrade, O. Einsle and J. B. Howard, Philos. Trans. R. Soc. A, 2005, 363, 971–984 CrossRef CAS PubMed.
- O. Einsle, F. A. Tezcan, S. L. A. Andrade, B. Schmid, M. Yoshida, J. B. Howard and D. C. Rees, Science, 2002, 297, 1696–1700 CrossRef CAS PubMed.
- B. M. Hoffman, D. Lukoyanov, Z.-Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041–4062 CrossRef CAS PubMed.
- H. Dobbek, V. Svetlitchnyi, L. Gremer, R. Huber and O. Meyer, Science, 2001, 293, 1281–1285 CrossRef CAS PubMed.
- S. Hu, T. Shima and Z. Hou, Nature, 2014, 512, 413–415 CrossRef CAS PubMed.
- T. Shima, S. W. Hu, G. Luo, X. H. Kang, Y. Luo and Z. M. Hou, Science, 2013, 340, 1549–1552 CrossRef CAS PubMed.
- M. M. Shoshani and S. A. Johnson, Nat. Chem., 2017, 9, 1282–1285 CrossRef CAS.
- R. Beck, M. Shoshani and S. A. Johnson, Angew. Chem., Int. Ed., 2012, 51, 11753–11756 CrossRef CAS PubMed.
- T. Shima, J. Yang, G. Luo, Y. Luo and Z. Hou, J. Am. Chem. Soc., 2020, 142, 9007–9016 CrossRef CAS PubMed.
- S. Hu, T. Shima and Z. Hou, J. Am. Chem. Soc., 2020, 142, 19889–19894 CrossRef CAS PubMed.
- M. Kondo and S. Masaoka, Acc. Chem. Res., 2020, 53, 2140–2151 CrossRef CAS PubMed.
- M. M. Shoshani and S. A. Johnson, Inorg. Chem., 2015, 54, 11977–11985 CrossRef CAS PubMed.
- M. M. Shoshani, J. Y. Liu and S. A. Johnson, Organometallics, 2018, 37, 116–126 CrossRef CAS.
- M. M. Shoshani, V. Semeniuchenko and S. A. Johnson, Chem. – Eur. J., 2018, 24, 14282–14289 CrossRef CAS PubMed.
- D. Gregson, J. A. K. Howard, M. Murray and J. L. Spencer, J. Chem. Soc. Chem. Comm., 1981, 716–717 RSC.
- M. J. Ingleson, M. F. Mahon, P. R. Raithby and A. S. Weller, J. Am. Chem. Soc., 2004, 126, 4784–4785 CrossRef CAS PubMed.
- Y. Ohki, Y. Shimizu, R. Araake, M. Tada, W. M. C. Sameera, J.-I. Ito and H. Nishiyama, Angew. Chem., Int. Ed., 2016, 55, 15821–15825 CrossRef CAS PubMed.
- R. Araake, K. Sakadani, M. Tada, Y. Sakai and Y. Ohki, J. Am. Chem. Soc., 2017, 139, 5596–5606 CrossRef CAS PubMed.
- M. R. Churchill, S. A. Bezman, J. A. Osborn and J. Wormald, Inorg. Chem., 1972, 11, 1818–1825 CrossRef CAS.
- T. Nakajima, K. Nakamae, Y. Ura and T. Tanase, Eur. J. Inorg. Chem., 2020, 2211–2226 CrossRef CAS.
- A brief explanation of the cause of the hydride/deuteride isotope-shifts are provided in the ESI†.
- R. B. Turner, W. R. Meador and R. E. Winkler, J. Am. Chem. Soc., 1957, 79, 4116–4121 CrossRef CAS.
- W. Nicolazzi and A. Bousseksou, C. R. Chim, 2018, 21, 1060–1074 CrossRef CAS.
- C. Zhang, X. Zhang, J.-J. Zou and G. Li, Coord. Chem. Rev., 2021, 436, 213779 CrossRef CAS.
- D.-J. Huang and C.-H. Cheng, J. Organomet. Chem., 1995, 490, C1–C7 CrossRef CAS.
- A. Tenaglia, E. Terranova and B. Waegell, J. Mol. Catal., 1987, 40, 281–287 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and crystallographic data (PDF and CIF). CCDC 2222969 and 2222970. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc06681e |
| ‡ Current address: Department of Chemistry, University of Texas Rio Grande Valley, 1 W University Blvd, Brownsville, TX, 78520, USA. |
| This journal is © The Royal Society of Chemistry 2023 |