
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and characterization of carbene derivatives of Th@C3v(8)-C82 and U@C2v(9)-C82: exceptional chemical properties induced by strong actinide–carbon cage interaction†
Xinye
Liu‡
a,
Bo
Li‡
b,
Wei
Yang‡
a,
Yang-Rong
Yao
c,
Le
Yang
b,
Jiaxin
Zhuang
a,
Xiaomeng
Li
a,
Peng
Jin
 *b and
Ning
Chen
*a
*b and
Ning
Chen
*a
aCollege of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou, Jiangsu 215123, P. R. China. E-mail: chenning@suda.edu.cn
bSchool of Materials Science and Engineering, Hebei University of Technology, Tianjin 300130, P. R. China. E-mail: china.peng.jin@gmail.com
cDepartment of Chemistry, University of Texas at El Paso, 500 West University Avenue, El Paso, Texas 79968, USA
First published on 21st December 2020
Abstract
Chemical functionalization of endohedral metallofullerenes (EMFs) is essential for the application of these novel carbon materials. Actinide EMFs, a new EMF family member, have presented unique molecular and electronic structures but their chemical properties remain unexplored. Here, for the first time, we report the chemical functionalization of actinide EMFs, in which the photochemical reaction of Th@C3v(8)-C82 and U@C2v(9)-C82 with 2-adamantane-2,3′-[3H]-diazirine (AdN2, 1) was systematically investigated. The combined HPLC and MALDI-TOF analyses show that carbene addition by photochemical reaction afforded three isomers of Th@C3v(8)-C82Ad and four isomers of U@C2v(9)-C82Ad (Ad = adamantylidene), presenting notably higher reactivity than their lanthanide analogs. Among these novel EMF derivatives, Th@C3v(8)-C82Ad(I, II, III) and U@C2v(9)-C82Ad(I, II, III) were successfully isolated and were characterized by UV-vis-NIR spectroscopy. In particular, the molecular structures of first actinide fullerene derivatives, Th@C3v(8)-C82Ad(I) and U@C2v(9)-C82Ad(I), were unambiguously determined by single crystal X-ray crystallography, both of which show a [6,6]-open cage structure. In addition, isomerization of Th@C3v(8)-C82Ad(II), Th@C3v(8)-C82Ad(III), U@C2v(9)-C82Ad(II) and U@C2v(9)-C82Ad(III) was observed at room temperature. Computational studies suggest that the attached carbon atoms on the cages of both Th@C3v(8)-C82Ad(I) and U@C2v(9)-C82Ad(I) have the largest negative charges, thus facilitating the electrophilic attack. Furthermore, it reveals that, compared to their lanthanide analogs, Th@C3v(8)-C82 and U@C2v(9)-C82 have much closer metal–cage distance, increased metal-to-cage charge transfer, and strong metal–cage interactions stemming from the significant contribution of extended Th-5f and U-5f orbitals to the occupied molecular orbitals, all of which give rise to their unusual high reactivity. This study provides first insights into the exceptional chemical properties of actinide endohedral fullerenes, which pave ways for the future functionalization and application of these novel EMF compounds.
Introduction
Encapsulation of metal atoms or metal containing clusters inside hollow fullerene cages offers hybrid molecules called endohedral metallofullerenes (EMFs).1 These novel hybrid species have fascinating properties that are expected to have potential applications in the fields of magnetism,2 photovoltaic cells,3 and biomedicine.4 Application of EMFs often requires chemical modification of the pristine compounds, and the exohedral functionalization of EMFs has been extensively studied in recent years.5 For example, water-soluble derivatives of EMFs such as Gd@C60[C(COOH)2] and Gd@C82(OH)x have been proved to be effective as magnetic resonance imaging contrast agents and nanomedicine.4,6,7 Besides, the EMF derivatives form donor–acceptor conjugates for photovoltaics, and a series of Lu3N@C80-PCBX (X = M: methyl, B: butyl, H: hexyl, O: octyl) compounds were found to be useful for constructing high-performance organic photovoltaic devices.8,9To date, different types of chemical reactions for functionalization of EMFs have been reported, such as Diels–Alder reactions,10 Bingel–Hirsch reactions,11 1,3-dipolar cycloadditions,12,13 free-radical reactions,14 and photochemical reactions.15 These studies revealed that the encapsulated species exert a major impact on the chemical reactivity and selectivity of EMFs, which are substantially different from those of the empty fullerenes. For instance, Akasaka et al. reported that the photochemical reactivity of D2d(23)-C84 toward 2-adamantane-2,3′-[3H]-diazirine differs drastically from that of Sc2C2@D2d(23)-C84, although they share the same carbon cage geometry.16 Moreover, even the change of the encapsulated clusters was found to have a major impact on the reactivity and regioselectivity of the corresponding endohedral nitride cluster fullerenes.17,18 On the other hand, the metal positions and movements within fullerene cages could be altered by exohedral functionalization. Nagase et al. reported that the encaged metal atoms circulate three-dimensionally in pristine Ih-C80, but the two Ce atoms are fixed inside the Ih-C80 cage after the bis-silylation of Ce2@Ih-C80 with disilirane.19 Thus, the interaction between the cage and the encapsulated species plays an essential role in both the reactivity of the cage and the kinetics of the endohedral metal ions or clusters.
Mono-EMFs are the most commonly studied EMFs which feature relatively high product yield. For these EMFs, charge transfer between the metal ion and the fullerene cages plays an important role in their chemical reactivity. Akasaka, Xing Lu and coworkers investigated the adamantylidene addition reactions of a series of lanthanide-based mono-EMFs including M@C2v-C82 (M = Y, La, Ce, Gd and Sc) and La@Cs(6)-C82.20–25 Though the reactivity of M@C82 (M = Y, La, Ce, and Gd) toward adamantylidene was very similar to each other, affording only two M@C82Ad isomers, the same reaction of Sc@C2v-C82 afforded four monoadducts. The unique chemical property of Sc@C2v-C82 was rationalized by the close contact of Sc3+ to the cage and efficient charge back-donation from the cage to the metal, which results from the small radius of Sc3+.24 These studies indicate that the properties of the entrapped single metal ion in the cage may affect the chemical properties of the whole molecule.
Our recent studies of a series of uranium and thorium-based actinide EMFs have shown that they differ remarkably from the conventional lanthanide-based EMFs with respect to their molecular structure and electronic properties.26–30 In particular, a strong interaction involving both unique 4 electron metal-to-cage transfer and the covalent interactions was found between the encapsulated actinide metal ions and the host fullerene cages.30–35 Thus, we expect that the chemical reactivity of actinide EMFs could be different from those of lanthanide based EMFs and such studies will be of great importance for the future application of actinide EMFs. Herein, photochemical reactions of Th@C3v(8)-C82 and U@C2v(9)-C82 were performed in the presence of 2-adamantane-2,3′-[3H]-diazirine (AdN2, 1) to probe the chemical properties of actinide EMFs for the first time. The carbene addition reaction of Th@C3v(8)-C82 and U@C2v(9)-C82 afforded three isomers of Th@C3v(8)-C82Ad and four isomers of U@C2v(9)-C82Ad (Ad = adamantylidene), showing higher reactivity than their lanthanide analogs, i.e. M@C82 (M = Y, La, Ce, and Gd). Th@C3v(8)-C82Ad(I, II, III) and U@C2v(9)-C82Ad(I, II, III) were successfully isolated and characterized using UV-vis-NIR spectroscopy. In particular, the structures of Th@C3v(8)-C82Ad(I) and U@C2v(9)-C82Ad(I) were unambiguously elucidated by single crystal X-ray crystallography. Theoretical calculations were performed to further rationalize the exceptional chemical properties of these actinide EMFs.
Results and discussion
Formation and isolation of adamantylidene adducts
The toluene solution of Th@C3v(8)-C82 with excessive addition of 1 was irradiated with an ultrahigh-pressure mercury-arc lamp (cutoff < 350 nm) at room temperature. The reaction was monitored by analytical HPLC (Fig. 1a). Before irradiation, only one HPLC peak of Th@C3v(8)-C82 appeared at retention time around 63 min. After irradiation for 15 minutes, about 58% of Th@C3v(8)-C82 was consumed and three new peaks appeared at 22 min, 28 min and 35 min. The mass spectrometric results reveal that all of the three HPLC fractions show a strong signal at m/z = 1350, which could be ascribed to the signal of the monoadducts of Th@C3v(8)-C82Ad and agrees well with the theoretical isotopic distribution (Fig. S1†). Thus, the resulting reaction products can be assigned to monoadduct isomers of Th@C3v(8)-C82Ad(I), Th@C3v(8)-C82Ad(II), and Th@C3v(8)-C82Ad(III), respectively. These monoadducts were conveniently isolated and purified using the Buckyprep column. The conversion yields of Th@C3v(8)-C82Ad(I), Th@C3v(8)-C82Ad(II), and Th@C3v(8)-C82Ad(III) were estimated to be 63%, 22% and 15% judging from the HPLC peak area.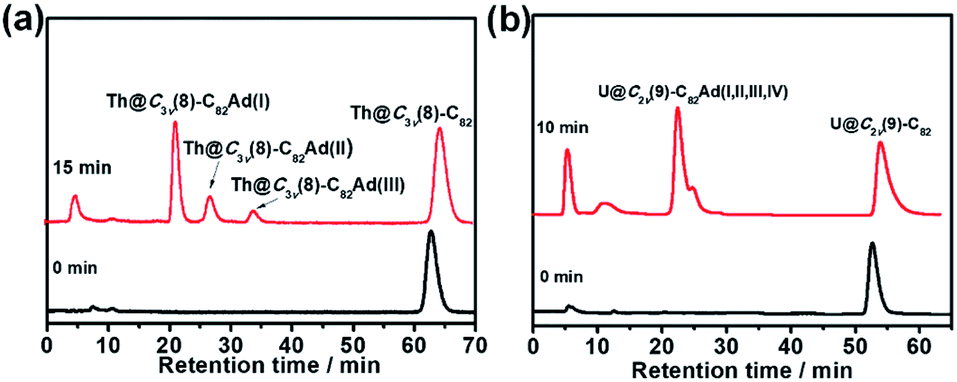 | ||
| Fig. 1 (a) HPLC tracing of the reaction between Th@C3v(8)-C82 with 1 at different times (black line: the signal of the pristine compound Th@C3v(8)-C82 before irradiation, red line: the signal of the resulting products after irradiation for 15 minutes). (b) HPLC tracing of the reaction between U@C2v(9)-C82 with 1 at different times (black line: the signal of the pristine compound U@C2v(9)-C82 before irradiation, red line: the signal of the resulting products after irradiation for 10 minutes). HPLC conditions: Buckyprep column (10 mm × 250 mm): flow rate, 4 mL min−1; toluene as the mobile phase; wavelength 310 nm. | ||
Similarly, photoirradiation on a toluene solution of U@C2v(9)-C82 in the presence of an excess of 1 results in the formation of the EMF adducts. The reaction was monitored by analytical HPLC (Fig. 1b). The single peak at retention time of 50 min should be assigned to U@C2v(9)-C82 before irradiation. After irradiation for 10 minutes, new peaks were observed between 20 and 26 min, which represent the monoadduct isomers, as confirmed by mass spectrometry results that show a strong signal at m/z = 1356 (Fig. S2†). A recycling HPLC separation process was further employed to isolate the mixture of four monoadduct isomers. As a result, U@C2v(9)-C82Ad(I), U@C2v(9)-C82Ad(II), U@C2v(9)-C82Ad(III) and U@C2v(9)-C82Ad(IV) were successfully obtained (Fig. 2). The conversions of U@C2v(9)-C82Ad(I), U@C2v(9)-C82Ad(II), U@C2v(9)-C82Ad(III) and U@C2v(9)-C82Ad(IV) were estimated from the HPLC peak area to be 60%, 20%, 15% and 5%, respectively.
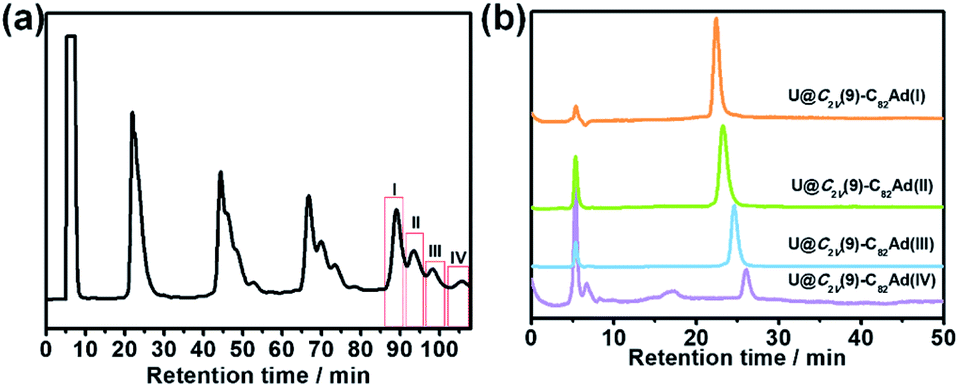 | ||
| Fig. 2 (a) Recycling HPLC profile for the separation of U@C2v(9)-C82Ad(I), U@C2v(9)-C82Ad(II), U@C2v(9)-C82Ad(III) and U@C2v(9)-C82Ad(IV). (b) HPLC chromatogram of isolated U@C2v(9)-C82Ad(I), U@C2v(9)-C82Ad(II), U@C2v(9)-C82Ad(III) and U@C2v(9)-C82Ad(IV). HPLC conditions: Buckyprep column (10 mm × 250 mm); flow rate, 4 mL min−1; toluene as the mobile phase; wavelength 310 nm. | ||
UV-vis-NIR spectroscopic characterization
UV-vis-NIR absorption spectroscopy has been proved to be an effective tool for the identification of the electronic structures of EMFs and their derivatives. Fig. 3a shows the UV-vis-NIR absorption spectra of Th@C3v(8)-C82 and its monoadduct isomers in CS2. All the monoadducts and Th@C3v(8)-C82 have similar broad absorption spectra, featured by absorption peaks at 650 and 750 nm. However, the intensity of the absorption peak at 650 nm gradually reduces in the order of Th@C3v(8)-C82Ad(I), Th@C3v(8)-C82Ad(II), Th@C3v(8)-C82Ad(III) and Th@C3v(8)-C82 (Fig. 3a), suggesting a minor impact of the addition of the Ad group to different positions of the carbon cage on the electronic structures of EMFs. Similarly, compared with Th@C3v(8)-C82, the three monoadducts show slightly different absorption patterns from 900 to 1100 nm. These results suggest that the electronic structure of Th@C3v(8)-C82 has been altered slightly by the addition of the Ad addend. In Fig. 3b, U@C2v(9)-C82 and its monoadducts, on the other hand, show sharp absorption peaks at 620 nm and a broad absorption range from 850 to 1100 nm. The absorptions of U@C2v(9)-C82 and its monoadducts are almost identical to each other, indicating a weaker impact of the addition of Ad group on the electronic structure of U@C2v(9)-C82. For comparison, the UV-vis-NIR absorption features of M@C82Ad (M = Ce, La, Y, Gd and Sc) were very similar to those of the pristine M@C82 (M = Ce, La, Y, Gd and Sc), indicating that those carbene adducts retain the essential electronic and structural characteristics of M@C82. Previous studies suggest that the formation of an open-cage structure has the sp2-character of the carbon atoms at the addition sites retained, thus maintaining the original π-system of the fullerene cage of the M@C82Ad isomers.16,21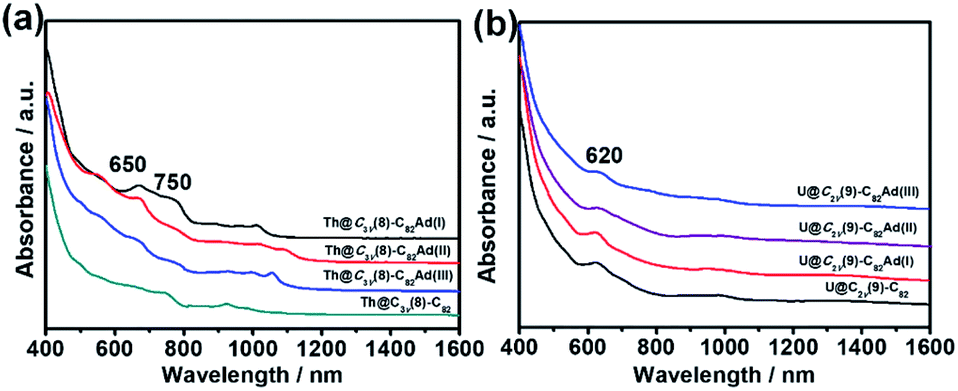 | ||
| Fig. 3 UV-vis-NIR absorption spectra of (a) Th@C3v(8)-C82 and Th@C3v(8)-C82Ad(I, II, III) and (b) U@C2v(9)-C82 and U@C2v(9)-C82Ad(I, II, III) in CS2. | ||
Electrochemical studies
Fig. 4 shows the cyclic voltammograms of Th@C3v(8)-C82Ad(I) and U@C2v(9)-C82Ad(I) measured in o-dichlorobenzene (o-DCB) solution using tetrabutylammonium hexafluorophosphate as a supporting electrolyte. Table 1 lists the electrochemical potentials of Th@C3v(8)-C82Ad(I), Th@C3v(8)-C82, U@C2v(9)-C82Ad(I) and U@C2v(9)-C82. The cyclic voltammogram of Th@C3v(8)-C82Ad(I) presents one oxidation peak and two reduction peaks. The first oxidation step with potential at 0.40 V is reversible. For reduction steps, the first reductive process at −1.18 V is irreversible, and the second at −1.72 V is reversible. In general, the oxidation process of Th@C3v(8)-C82Ad(I) is somewhat similar to that of Th@C3v(8)-C82 while the reduction processes show differences as Th@C3v(8)-C82 presents three irreversible reduction processes. As compared to Th@C3v(8)-C82, the first reduction and first oxidation potentials of Th@C3v(8)-C82Ad(I) were negatively shifted by 0.06 V and 0.13 V, respectively. Accordingly, the electrochemical gap of Th@C3v(8)-C82Ad(I) (1.58 V) is slightly larger than that of Th@C3v(8)-C82 (1.51 V). On the other hand, the cyclic voltammogram of U@C2v(9)-C82Ad(I) shows two oxidation peaks and four reduction peaks. The first oxidation process at 0.01 V is reversible, and the second at 0.80 V is irreversible. The reductive processes exhibit three reversible steps at −0.55 V, −1.47 V, −1.84 V, respectively and one irreversible reductive process at −2.39 V (Fig. 4b). The first oxidation and first reduction potentials of U@C2v(9)-C82Ad(I) were negatively shifted by 0.09 V and 0.12 V, respectively, compared to those of the pristine U@C2v(9)-C82. Therefore, the electrochemical gap of U@C2v(9)-C82Ad(I) (0.56 V) was close to that of U@C2v(9)-C82 (0.53 V). The small shifts of oxidation and reduction potentials indicate that the HOMO and LUMO of Th@C3v(8)-C82Ad(I) and U@C2v(9)-C82Ad(I) resemble those of pristine Th@C3v(8)-C82 and U@C2v(9)-C82, respectively. Small negative shifts of the redox potentials were also found in M@C82 (M = Ce, La, Y, Gd and Sc) carbene adducts, and the shift of the redox potentials could be ascribed to the weak electron-donating nature of the carbene adducts.20–24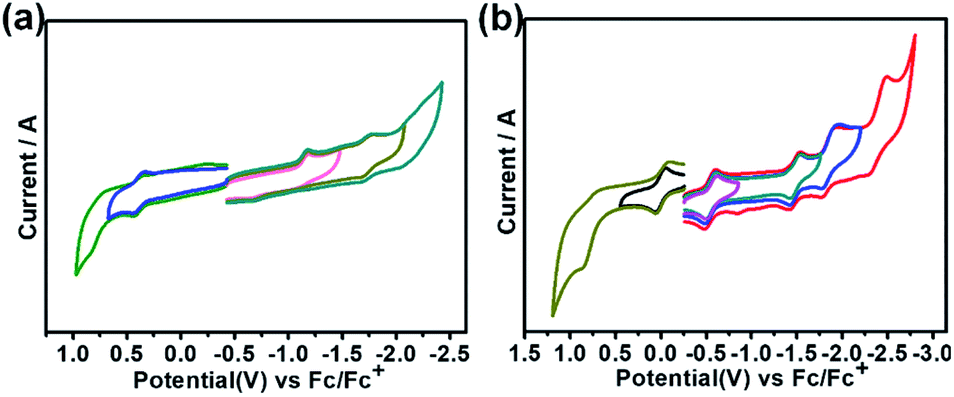 | ||
| Fig. 4 Cyclic voltammogram of (a) Th@C3v(8)-C82Ad(I) and (b) U@C2v(9)-C82(I) in o-dichlorobenzene (0.05 M (n-Bu)4NPF6; scan rate 100 mV s−1 for CV). | ||
| Compound | E 2+/+ | E +/0 | E 0/− | E −/2− | E 2−/3− | E 3−/4− | E 4−/5− | ECgap (V) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Half-wave potential in volts (reversible redox process). b Peak potential in volts (irreversible redox process). | |||||||||
| Th@C3v(8)-C82Ad(I) | +0.40a | −1.18b | −1.72a | 1.58 | This work | ||||
| Th@C3v(8)-C82 | +0.46a | −1.05b | −1.54b | −1.69b | 1.51 | 26 | |||
| U@C2v(9)-C82Ad(I) | +0.80b | +0.01a | −0.55a | −1.47a | −1.84a | −2.39b | 0.56 | This work | |
| U@C2v(9)-C82 | +0.10a | −0.43a | −1.42a | −1.76b | −1.77b | −2.21b | 0.53 | 28 | |
Isomerization of Th@C3v(8)-C82Ad(II), Th@C3v(8)-C82Ad(III), U@C2v(9)-C82Ad(II) and U@C2v(9)-C82Ad(III)
The stabilities of Ad adducts of Th@C3v(8)-C82 and U@C2v(9)-C82 were assessed at room temperature. As shown in Fig. 5a, the HPLC result of Th@C3v(8)-C82Ad(I) exhibited only one peak at 20 min after 30 days, indicating that the isomer Th@C3v(8)-C82Ad(I) was stable at room temperature. The isomerization process of Th@C3v(8)-C82Ad(II) was observed by HPLC (Fig. 5a). After 30 days, Th@C3v(8)-C82Ad(II) gradually isomerized to Th@C3v(8)-C82Ad(I) with a conversion rate of ca. 17%. Moreover, after it was stored at room temperature for 30 days, the chromatogram of Th@C3v(8)-C82Ad(III) shows three characteristic peaks with retention times at 20 min, 25 min and 35 min, corresponding to Th@C3v(8)-C82Ad(I), Th@C3v(8)-C82Ad(II) and Th@C3v(8)-C82Ad(III), respectively. This result demonstrated that Th@C3v(8)-C82Ad(III) was unstable and isomerized to Th@C3v(8)-C82Ad(II). But there was no direct evidence for the isomerization of Th@C3v(8)-C82Ad(III) to Th@C3v(8)-C82Ad(I), which could originate either from the direct transformation of isomer(III) or from a stepwise isomerization of III → II → I. Similarly, we investigated the stabilities of the three isomers of U@C2v(9)-C82Ad after keeping the samples at room temperature for 30 days. As shown in Fig. 5b, quite similar to what is observed for Th@C3v(8)-C82Ad, HPLC demonstrated the high stability of U@C2v(9)-C82Ad(I) by a sole peak after recycling. For U@C2v(9)-C82Ad(II), its isomerization to U@C2v(9)-C82Ad(I) was evidently observed with the splitting of the HPLC peak into two after a few cycles. Expectedly, for U@C2v(9)-C82Ad(III), we observed three peaks after the several cycles indicating the formation of isomer(I) and isomer(II). Note that for the isomerization of U@C2v(9)-C82Ad(III), HPLC profiles show that the percentages of the newly formed U@C2v(9)-C82Ad(I) and U@C2v(9)-C82Ad(II) were almost equivalent, indicating that the isomerization of U@C2v(9)-C82Ad(III) into U@C2v(9)-C82Ad(I) and U@C2v(9)-C82Ad(II) should coexist at the same time. For comparison, the stabilities of lanthanide EMFs were tested under thermal conditions in the previously reported work. In those cases, the interconversion was also observed in Ad carbene adducts of La@C2v(9)-C82, Sc2C2@C2v(5)-C80, D2d(23)-C84 and Sc2C2@D2d(23)-C84 under thermal conditions.16,21,36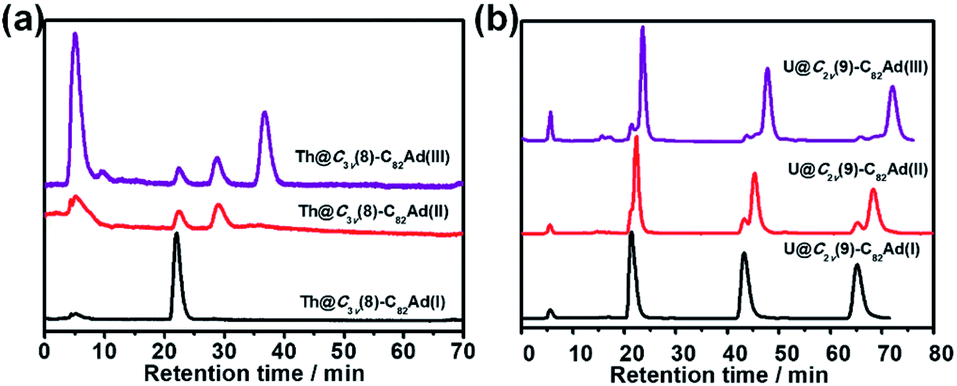 | ||
| Fig. 5 (a) HPLC traces of isomerization of Th@C3v(8)-C82Ad(I, II, III) and (b) recycling HPLC traces of isomerization of U@C2v(9)-C82Ad(I, II, III) at room temperature. HPLC conditions: Buckyprep column (10 mm × 250 mm); flow rate, 4 mL min−1; toluene as the mobile phase. | ||
X-ray crystallographic analysis of Th@C3v(8)-C82Ad(I) and U@C2v(9)-C82Ad(I)
The molecular structures of the two stable compounds, Th@C3v(8)-C82Ad(I) and U@C2v(9)-C82Ad(I), were unambiguously determined by single crystal X-ray diffraction analyses (Fig. 6). It is worth noting that they are the first examples of crystal structures of actinide fullerene derivatives. The X-ray structure of Th@C3v(8)-C82Ad(I) clearly shows that the addition occurred at the [6,6]-bond [C(1)–C(2)] which was connected to a pentagon and a hexagon at the terminal (Fig. 6a). The C1–C2 distance was determined to be 2.136 Å. This distance is notably longer than that of typical C–C single bond (1.5 Å) and double bond (1.4 Å), unambiguously confirming the [6,6]-open fullerene cage structure. The Th⋯C(1) and Th⋯C(2) distances are 2.565 Å and 2.568 Å, respectively, longer than the corresponding metal–carbon distances in other M@C82Ad (M = Sc, Y, Gd) and shorter than that in La@C82Ad,20,21,23,24 likely due to the intermediate ionic radius of Th4+. The Th ion in pristine Th@C3v(8)-C82 is located at the intersection of three hexagons, the distances between Th and close cage carbons range from 2.340 to 2.494 Å. Ad breaks one of the three [6,6]-bonds on the cage near the Th ion. Moreover, C1 and C2 were pulled out from the cage toward the Ad group, causing the deformation of the carbon cage. The distance between Th ion and the near cage carbon in Th@C3v(8)-C82Ad(I) ranges from 2.487 to 2.611 Å (Fig. 6c), much longer than the short Th–C distances in pristine Th@C3v(8)-C82. This indicates that the addition of the Ad group might reduce the metal–cage interactions between the Th and C82 cage. Interestingly, compared to the disorder of Th position in pristine Th@C3v(8)-C82, the Th ion is fully ordered in Th@C3v(8)-C82Ad(I).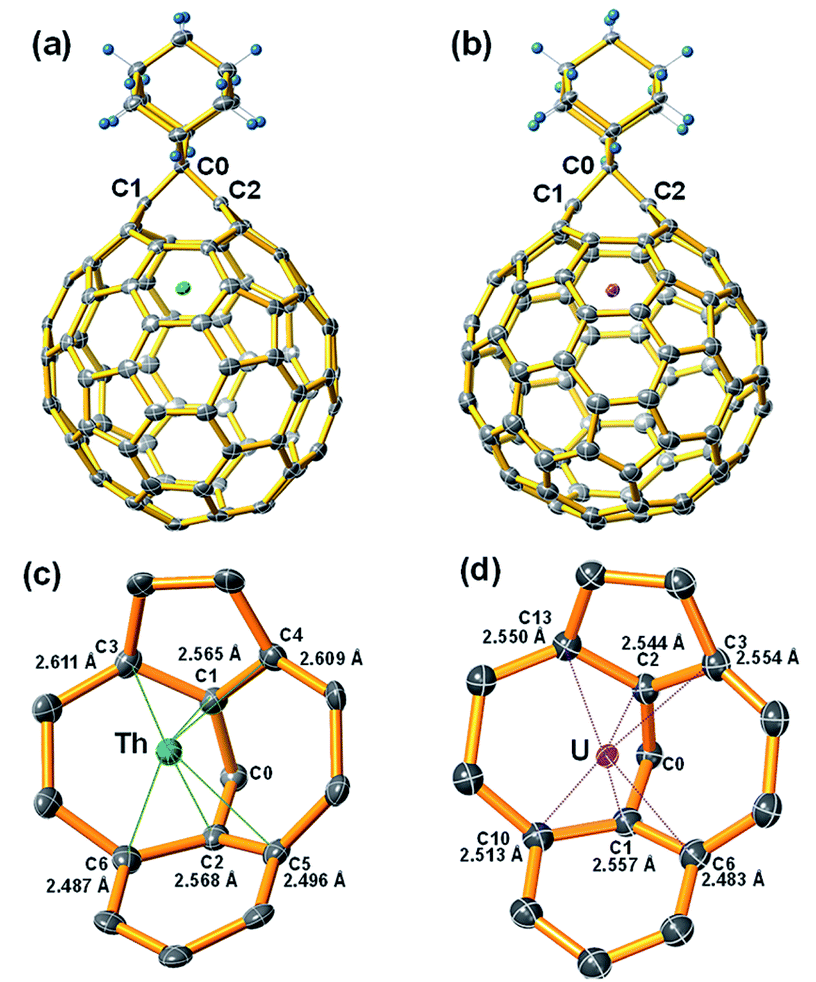 | ||
| Fig. 6 ORTEP drawings of (a) Th@C3v(8)-C82Ad(I) and (b) U@C2v(9)-C82Ad(I) showing thermal ellipsoids at the 50% probability level. (c and d) The view showing the interaction of the metal ion with the closest cage portion. | ||
Fig. 6b shows the single crystal X-ray structure of U@C2v(9)-C82Ad(I). Similar to Th@C3v(8)-C82Ad(I), the C82 carbon cage and the encaged U ion are fully ordered. The U ion in U@C2v(9)-C82 is located under the center of the hexagonal ring along the C2 axis. In U@C2v(9)-C82Ad(I), the Ad group adds to a [6,6]-bond of the hexagonal ring close to the U ion, which resembles the cases in M@C2v(9)-C82Ad (M = La, Gd, Ce, Y, Sc).20–24 The distance of C1–C2 is 2.071 Å, smaller than that of Th@C3v(8)-C82Ad(I) but close to M@C82Ad (M = Sc, Y, La, Gd), also suggesting an open-cage structure. The U⋯C(1) and U⋯C(2) distances are 2.557 Å and 2.544 Å respectively, and the distance between U atom and the nearest cage carbon in U@C2v(9)-C82Ad(I) is 2.483 Å. These distances of U@C2v(9)-C82Ad(I) are almost identical to those of Th@C3v(8)-C82Ad(I), due to the close ionic radius of U3+ and Th4+. These results are consistent with previous reports which demonstrated that the distances of M–C1 and M–C2 increase uniformly with increase in ionic radius, but the distance between C1 and C2 remains almost constant.24
Computational studies
DFT calculations were then carried out to understand the unusual chemical reactivities of Th@C3v(8)-C82 and U@C2v(9)-C82 toward the Ad addition. There are 17 different types of nonequivalent carbon types in Th@C3v(8)-C82 (Fig. 7). Table S1† summarizes their charge densities and POAV (p-orbital axis vector) values. Clearly, C1 and C2 have the largest negative charge densities among them. Besides, these two carbon atoms also have rather large POAV values which mean their large local strain on the cage surface, suggesting that they are the best sites toward electrophilic attack. We then optimized all the possible Ad adducts corresponding to 25 types of nonequivalent cage bonds. Consistent with our above analysis and the single crystal structure of Th@C3v(8)-C82Ad(I), the lowest-energy Th@C3v(8)-C82Ad isomer has the Ad group added at the C1–C2 bond, followed by the additions at the C3–C4 and C2–C3 sites (Table S2†). Their corresponding carbon atoms (C1 to C4) all have the largest negative charges, indicating that the observed reactivity and regioselectivity could be mainly understood by analyzing the charge density of cage carbons. According to the observed isomerization (II to I, III to II) and the cage connectivity (Fig. 7), we predict that Th@C3v(8)-C82Ad(II) and Th@C3v(8)-C82Ad(III) correspond to the addition at the C3–C4 and C2–C3 bonds, respectively. Fig. 8 depicts the optimized geometries of these three isomers, all of which have open-cage structures. Note that the optimized structure of Th@C3v(8)-C82Ad(I) isomer agrees very well with our single crystal data (selected distance parameters are compared in Table S3†). Fig. 9 shows the frontier molecular orbitals of Th@C3v(8)-C82 and Th@C3v(8)-C82Ad(I). The similar orbital distributions and HOMO–LUMO gap energies indicate that they have similar redox potentials, in line with our experimental results.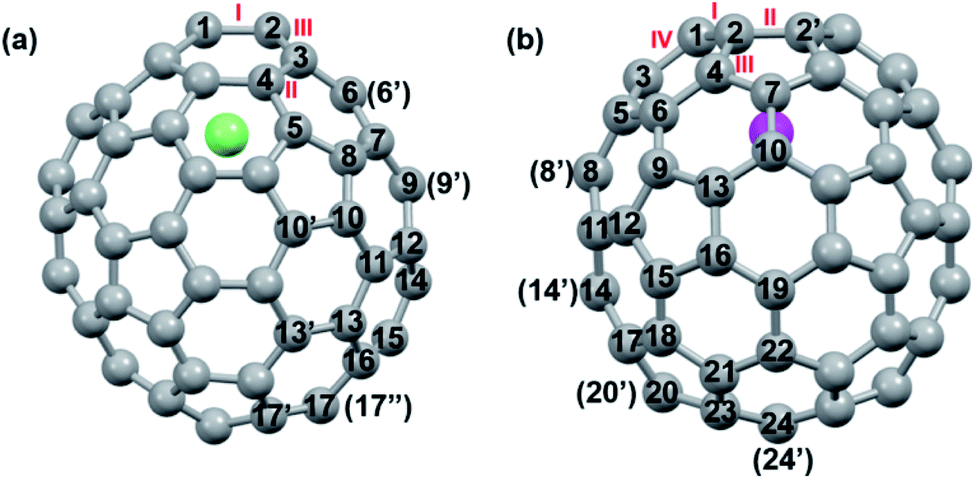 | ||
| Fig. 7 Different types of carbon atoms in (a) Th@C3v(8)-C82 and (b) U@C2v(9)-C82. | ||
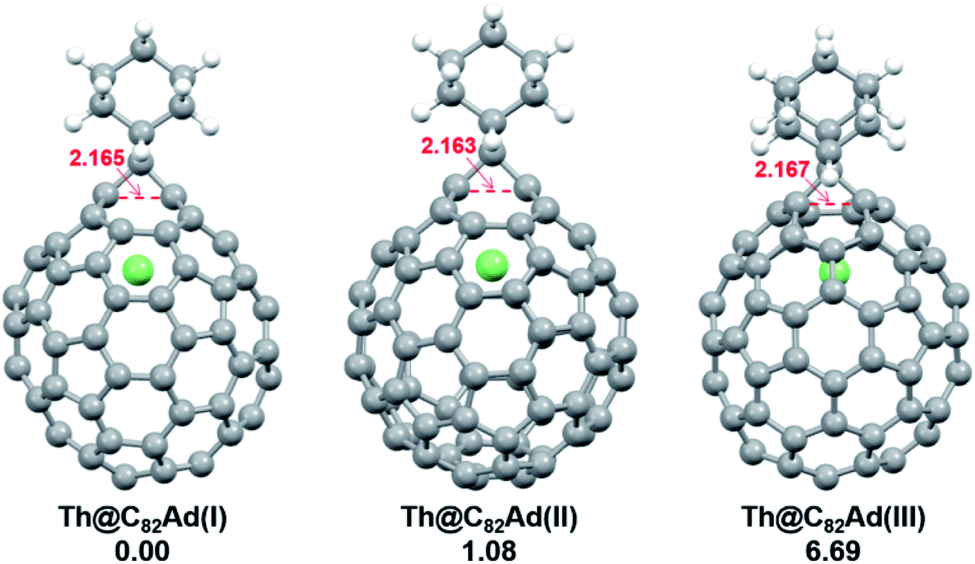 | ||
| Fig. 8 Optimized structures of the Th@C3v(8)-C82Ad(I–III) isomers with relative energies (kcal mol−1). The distances between the two addition site C atoms are given in Å. | ||
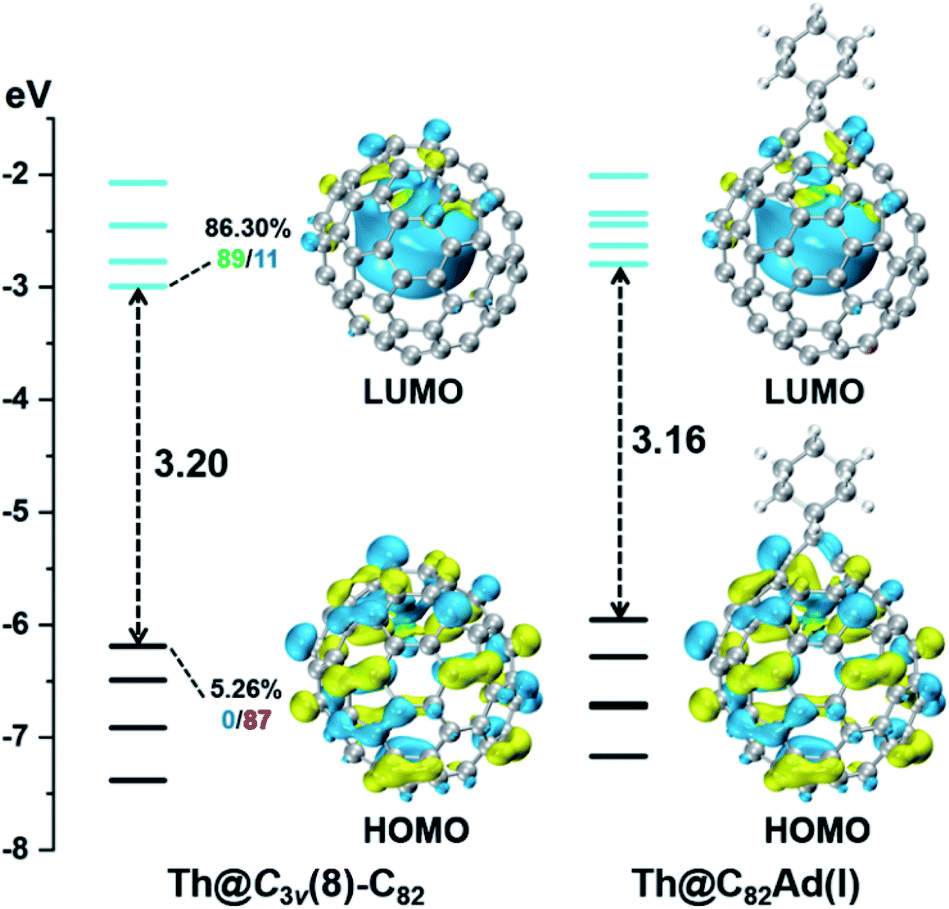 | ||
| Fig. 9 Frontier molecular orbitals of Th@C3v(8)-C82 and Th@C3v(8)-C82Ad(I) (occupied: black; unoccupied: cyan). For selected orbitals, the participation (%) of the Th AOs is first given, followed by two numbers showing the % contribution of the Th-5f (red), -6d (blue), or -7s (green) AOs to this metal hybrid orbital. | ||
We then turned to U@C2v(9)-C82, which has 24 different types of carbon atoms according to the C2v cage symmetry (Fig. 7). As shown in Table S4,† C1 and C2 have the largest negative charge densities, followed by C3 and C4, implying their high reactivity. We then optimized all possible Ad adducts corresponding to 35 types of nonequivalent cage bonds of U@C2v(9)-C82. Indeed, the addition at the C1–C2 bond led to the lowest-energy U@C2v(9)-C82Ad isomer, followed by the additions at the C2–C2′, C2–C4 and C1–C3 bonds (Fig. 7, 10 and Table S5†). As indicated by the long C–C bond distances, all of the four isomers have open-cage structures. The optimized structure of the C1–C2 adduct is consistent with our single crystal structure of U@C2v(9)-C82Ad(I) (Table S3†). According to the observed isomerization (II to I, III to I, III to II) and the cage connectivity (Fig. 7), we speculate that the obtained U@C2v(9)-C82Ad(II), U@C2v(9)-C82Ad(III) and U@C2v(9)-C82Ad(IV) correspond to the addition at the C2–C2′, C2–C4 and C1–C3 bonds, respectively. Note that the relative locations of the Ad group in isomers I, II and III suggest that the isomerization from III to I and II should coexist, which agrees perfectly with the fact that the newly formed U@C2v(9)-C82Ad(I) and U@C2v(9)-C82Ad(II) in isomerization have similar yield. Fig. 11 shows the frontier molecular orbitals of U@C2v(9)-C82 and U@C2v(9)-C82Ad(I). The similar orbital distributions and HOMO–LUMO gap energies indicate that they have similar redox potentials, thus supporting our experiments.
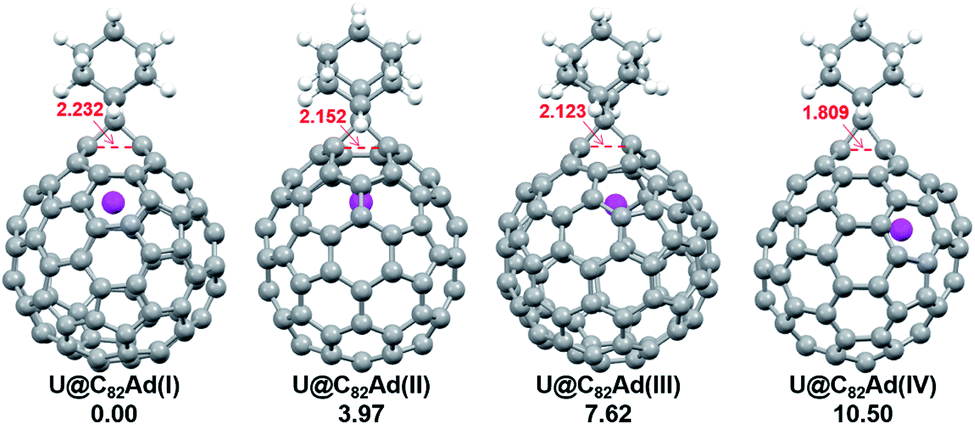 | ||
| Fig. 10 Optimized structures of the U@C2v(9)-C82Ad(I–IV) isomers with relative energies (kcal mol−1). The distances between the two addition site C atoms are given in Å. | ||
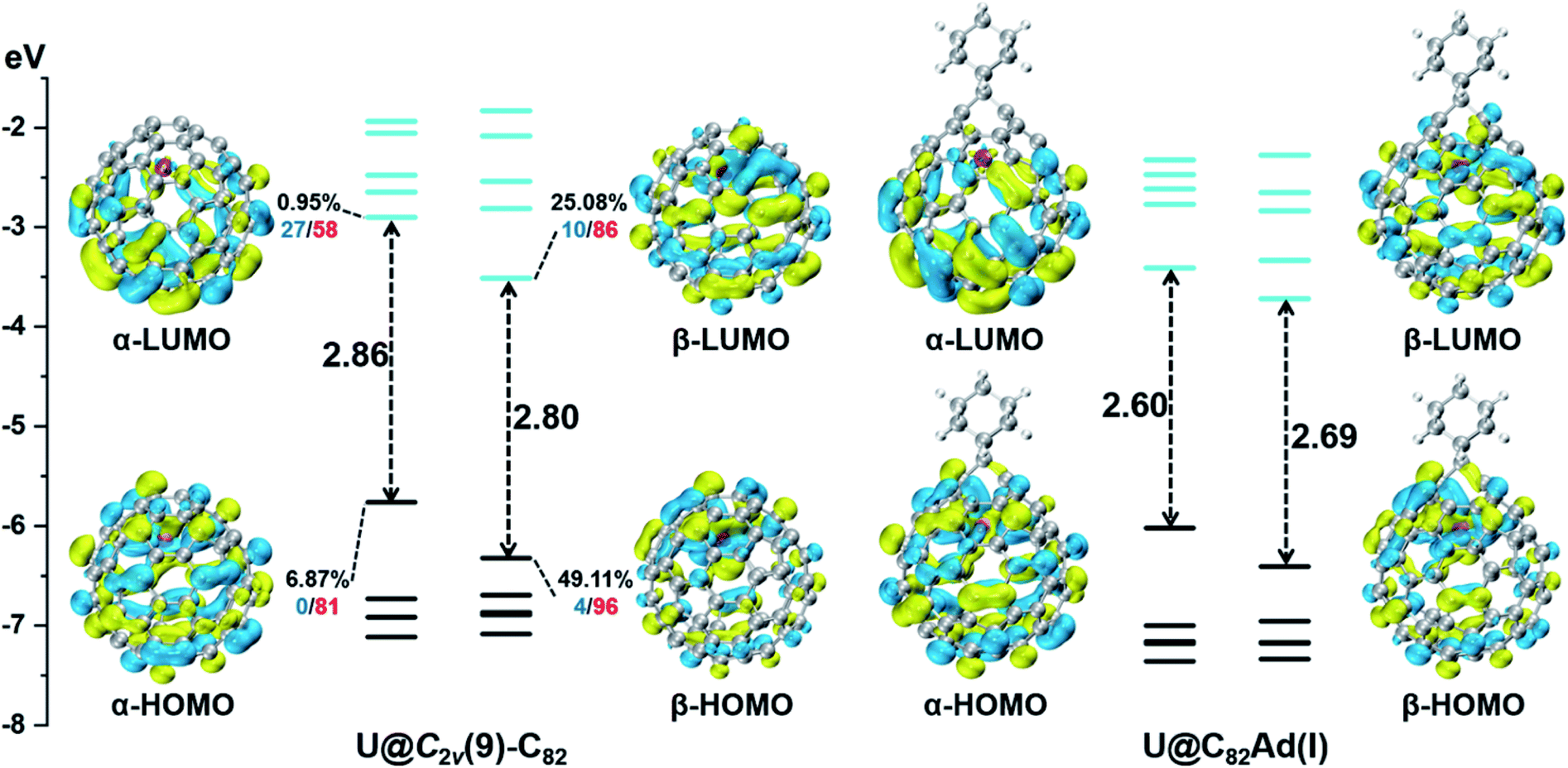 | ||
| Fig. 11 Frontier molecular orbitals of U@C2v(9)-C82 and U@C2v(9)-C82Ad(I) (occupied: black; unoccupied: cyan). For selected orbitals, the participation (%) of the U AOs is first given, followed by two numbers showing the % contribution of the U-5f (red), or -6d (blue) AOs to this metal hybrid orbital. | ||
In experiments, La@C2v(9)-C82 can only give rise to two Ad adducts, whereas both U@C2v(9)-C82 and Sc@C2v(9)-C82 yield four adducts, and three were obtained for Th@C3v(8)-C82. Their reactivity differences could be rationalized by comparing the endohedral structures, charge transfer and metal–cage interactions of corresponding pristine EMFs.
First, Table S6† compares their calculated nearest metal–cage distances, which show that Th@C3v(8)-C82, U@C2v(9)-C82 and Sc@C2v(9)-C82 all have much closer metal–cage contact than La@C2v(9)-C82. Second, different from the formal charges (Th: 4+ and U/Sc/La: 3+), the calculated Mulliken charges of encapsulated Th, U and Sc are +3.46, +2.65 and +2.69, respectively, more positive than that of La (+2.43) in La@C2v(9)-C82, indicating that more electrons were transferred to the outer cage from the former three metals. Third, for La@C2v(9)-C82, our calculations show that its metal contribution to the singly occupied MO (SOMO) is as small as 2.21% (La-5d: 22%, La-4f: 49%), whereas Sc@C2v(9)-C82 and U@C2v(9)-C82 have much larger metal contributions of 9.96% (Sc-3d: 77%) and 6.87% (U-5f: 81%) to their SOMOs, respectively. Similarly, Th@C3v(8)-C82 has substantial metal contribution of 5.26% (Th-5f: 87%) to its HOMO. As shown in Fig. 9 and 11, the significant contribution of Th-5f and U-5f orbitals to the occupied molecular orbitals should account for these strong interactions, because the 5f orbitals of early actinide elements are spatially more extended than the core-like 4f orbitals of lanthanide elements. Clearly, the latter three EMFs have much closer metal–cage distances, more metal-to-cage charge transfer, and stronger metal–cage orbital interactions, all of which greatly affect the distribution of charge density on the cage with more carbon atoms holding large negative charges (more negative than −0.08, Fig. 12) and become reactive toward Ad attack. As a result, they are rather reactive and afford more adducts than La@C2v(9)-C82 under the same reaction conditions.
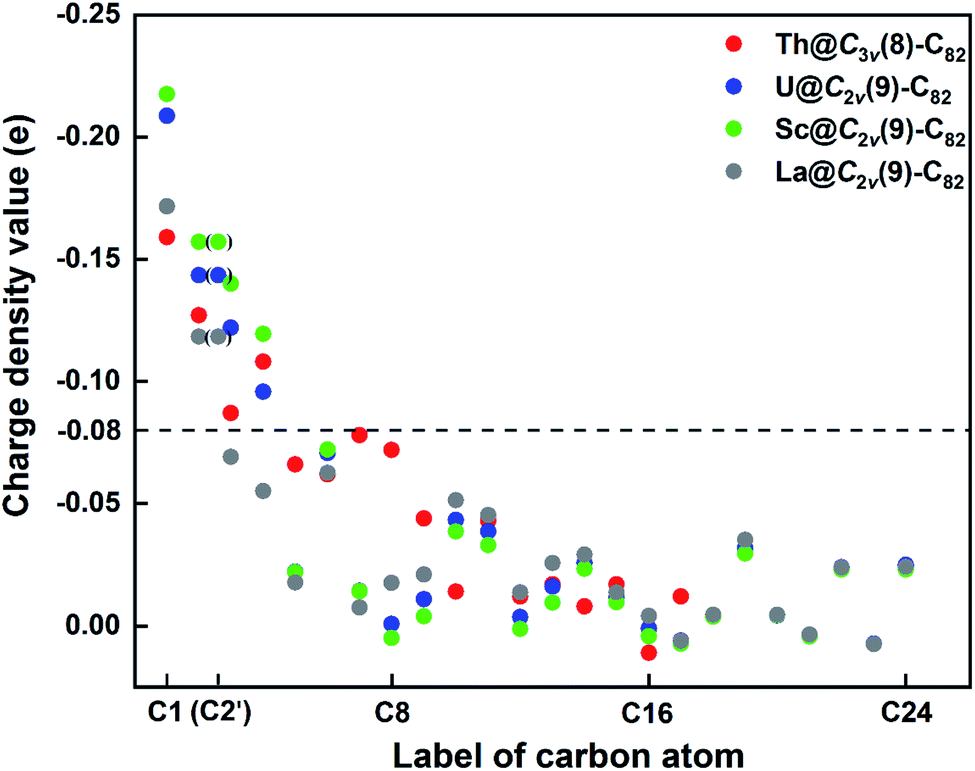 | ||
| Fig. 12 Charge density values of different carbon atoms in Th@C3v(8)-C82 and M@C2v(9)-C82 (M = U, Sc, La). Please refer to Fig. 7 for the atom numbering. | ||
Conclusions
In summary, we report the chemical reaction of two actinide endohedral fullerenes, i.e. Th@C3v(8)-C82 and U@C2v(9)-C82, for the first time. The combined HPLC and MALDI-TOF analyses suggest that, compared to their lanthanide analogs, Th@C3v(8)-C82 and U@C2v(9)-C82 show higher reactivity towards carbene addition via photochemical reaction and afforded three isomers of Th@C3v(8)-C82Ad and four isomers of U@C2v(9)-C82Ad (Ad = adamantylidene) respectively. Among them, Th@C3v(8)-C82Ad(I, II, III) and U@C2v(9)-C82Ad(I, II, III) were successfully isolated and were characterized by UV-vis-NIR spectroscopy. The electrochemical properties of Th@C3v(8)-C82Ad(I) and U@C2v(9)-C82Ad(I) were investigated by cyclic voltammetry. In particular, the molecular structures of Th@C3v(8)-C82Ad(I) and U@C2v(9)-C82Ad(I) were elucidated by single crystal X-ray crystallography, which shows that both compounds have a [6,6]-open cage structure. In addition, isomerization reaction of Th@C3v(8)-C82Ad(II), Th@C3v(8)-C82Ad(III), U@C2v(9)-C82Ad(II) and U@C2v(9)-C82Ad(III) were observed at room temperature. Computational studies suggest that C1 and C2 cage carbon atoms of both Th@C3v(8)-C82 and U@C2v(9)-C82 have the largest negative charge densities and are the best sites toward electrophilic attack, which agrees with the experimental observation. Furthermore, it reveals that Th@C3v(8)-C82 and U@C2v(9)-C82 have much closer metal–cage contact with the metal atom transferring more electrons to the outer cage. The significant contribution of Th-5f and U-5f orbitals to the occupied molecular orbitals should account for the stronger interactions between the actinide metal and cage carbon atom, and all these differences give rise to the unusual high reactivity compared to their lanthanide analogs.Experimental section
Materials and general instruments
Th@C3v(8)-C82 and U@C2v(9)-C82 were synthesized using an improved arc-discharge method and separated by multi-stage HPLC procedures.A Buckyprep column (10 × 250 mm, Cosmosil, Nacalai Tesque, Japan) was employed with toluene as the eluent for high performance liquid chromatography (HPLC) separation. UV-vis-NIR spectra of the purified Th@C3v(8)-C82Ad and U@C2v(9)-C82Ad were obtained with a Cary 5000 UV-vis-NIR spectrophotometer (Agilent, USA) in CS2 solution. The positive-ion mode matrix assisted laser desorption/ionization time-of-flight (MALDI-TOF) spectrophotometer (Bruker, Germany) was employed for the mass characterization, with trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile as the matrix. Cyclic voltammetry (CV) was recorded in 1,2-dichlorobenzene (o-DCB) using a CHI-660E instrument. A conventional three-electrode cell consisting of a platinum counter electrode, a glassy carbon working electrode, and a silver reference electrode was used for the measurement. (n-Bu)4NPF6 (0.05 M) was used as a supporting electrolyte. The CV was measured at a scan rate of 100 mV s−1.
Synthesis of adamantyl derivatives of Th@C3v(8)-C82 and U@C2v(9)-C82
AdN2 (1) was synthesized and purified as previously reported.37,38 In a typical procedure, 3 mg of Th@C3v(8)-C82 and an excess of 1 were dissolved in 15 mL of anhydrous toluene, and the mixture was degassed by freeze–pump–thaw cycles under reduced pressures and then irradiated with an ultrahigh-pressure mercury-arc lamp (cutoff < 350 nm) at room temperature (Scheme 1). The reaction was monitored by analytical HPLC. The three isomers Th@C3v(8)-C82Ad(I), Th@C3v(8)-C82Ad(II), and Th@C3v(8)-C82Ad(III) were isolated from the unreacted starting materials by one-step HPLC. A toluene solution of U@C2v(9)-C82 (5 mg) and an excess of 1 were degassed by freeze–pump–thaw cycles under reduced pressures. The mixture was photoirradiated using an ultrahigh-pressure mercury-arc lamp (cutoff < 350 nm) at room temperature (Scheme 1). The reaction was monitored by analytical HPLC. The resulting monoadducts were isolated by recycling HPLC using a Buckyprep column to obtain pure U@C2v(9)-C82Ad(I), U@C2v(9)-C82Ad(II), U@C2v(9)-C82Ad(III) and U@C2v(9)-C82Ad(IV).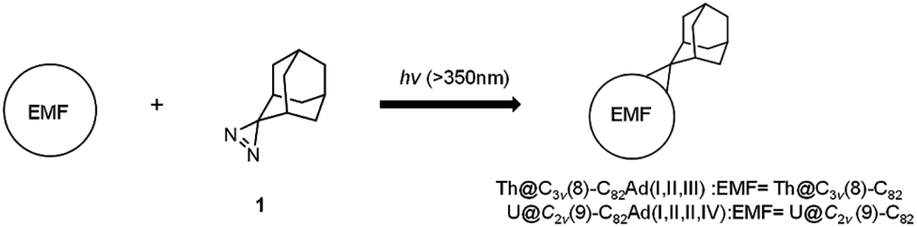 | ||
| Scheme 1 Reaction of 1 with Th@C3v(8)-C82 or U@C2v(9)-C82. | ||
X-ray crystallographic study
The black block crystals of 2(Th@C3v(8)-C82Ad)·5(CS2) were obtained by liquid–liquid bilayer diffusion methods of a solution of Th@C3v(8)-C82Ad(I) in CS2 and hexane as the poor solvent. X-ray data were collected at 173 K using a diffractometer (Bruker D8 Venture) equipped with a CCD collector. The multi-scan method was used for absorption correction. The structures were solved using direct methods39 and refined on F2 using full-matrix least-squares using the SHELXL2014 crystallographic software package.40 Hydrogen atoms were inserted at calculated positions and constrained with isotropic thermal parameters.Crystal data for 2(Th@C3v(8)-C82Ad)·5(CS2): C189H28S10Th2, Mr = 3082.79, 0.2 × 0.15 × 0.2 mm3, orthorhombic, space group Pbca (No. 61), a = 21.6511(4) Å, b = 21.3564(4) Å, c = 22.0916(4) Å, α = 90°, β = 90°, γ = 90°, V = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 214.9(3) Å3, Z = 4, ρcalcd = 2.005 g cm−3, μ(Cu Kα) = 11.854 mm−1, θ = 3.53–72.12°, T = 173(2) K, R1 = 0.0305, wR2 = 0.0761 for all data; R1 = 0.0289, wR1 = 0.0748 for 984 reflections (I > 2.0σ(I)) with 992 parameters. Goodness of fit indicator 1.060. Maximum residual electron density 3.852 e Å−3.
214.9(3) Å3, Z = 4, ρcalcd = 2.005 g cm−3, μ(Cu Kα) = 11.854 mm−1, θ = 3.53–72.12°, T = 173(2) K, R1 = 0.0305, wR2 = 0.0761 for all data; R1 = 0.0289, wR1 = 0.0748 for 984 reflections (I > 2.0σ(I)) with 992 parameters. Goodness of fit indicator 1.060. Maximum residual electron density 3.852 e Å−3.
The crystallographic data for this structure have been deposited at the Cambridge Crystallographic Data Centre (CCDC) with the deposition number 2031751.†
Black needle crystals of U@C2v(9)-C82Ad(I) were obtained by evaporating slowly a solution of U@C2v(9)-C82Ad(I) in CS2. X-ray data were collected at 120 K using a diffractometer (Bruker D8 Venture) equipped with a CCD collector. The multi-scan method was used for absorption correction. The structures were solved using direct methods39 and refined on F2 using full-matrix least-squares using the SHELXL2014 crystallographic software package.40 Hydrogen atoms were inserted at calculated positions and constrained with isotropic thermal parameters.
Crystal data for U@C2v(9)-C82Ad·1(CS2): C93H14S2U, Mr = 1433.19, 0.15 × 0.12 × 0.08 mm3, monoclinic, space group P21/c (No. 14), a = 11.3037(7) Å, b = 21.8879(12) Å, c = 20.9098(13) Å, α = 90°, β = 101.257(3)°, γ = 90°, V = 5073.9(5) Å3, Z = 4, ρcalcd = 1.876 g cm−3, μ(Cu Kα) = 10.273−1, θ = 2.953–72.533°, T = 120(2) K, R1 = 0.1287, wR2 = 0.2438 for all data; R1 = 0.0957, wR1 = 0.2823 for 7199 reflections (I > 2.0σ(I)) with 865 parameters. Goodness of fit indicator 1.050. Maximum residual electron density 4.689 e Å−3.
The crystallographic data for this structure have been deposited at the Cambridge Crystallographic Data Centre (CCDC) with the deposition number 2031857.†
Isomerization of Th@C3v(8)-C82Ad and U@C2v(9)-C82Ad at room temperature
Powders of Th@C3v(8)-C82Ad(I), Th@C3v(8)-C82Ad(II) and Th@C3v(8)-C82Ad(III) were placed in clear glass storage vials. After 30 days, the three samples in the storage vials were dissolved in toluene and analyzed by analytical HPLC respectively. The chromatogram of Th@C3v(8)-C82Ad(I) exhibited only one peak at 20 min. The chromatogram of Th@C3v(8)-C82Ad(II) exhibits two peaks at 20 min and 25 min, which can be ascribed to Th@C3v(8)-C82Ad(I) and Th@C3v(8)-C82Ad(II) with a relative ratio of 35% and 65%, respectively (estimated from the HPLC area). The chromatogram of Th@C3v(8)-C82Ad(III) shows three peaks with retention times at 20 min, 25 min and 35 min, which correspond to Th@C3v(8)-C82Ad(I), Th@C3v(8)-C82Ad(II) and Th@C3v(8)-C82Ad(III) with percentages of 15%, 30% and 55%, respectively (estimated from the HPLC area).Powders of U@C2v(9)-C82Ad(I), U@C2v(9)-C82Ad(II) and U@C2v(9)-C82Ad(III) were kept in glass bottles at room temperature. After 30 days, the three samples in the storage vials were dissolved in toluene and analyzed by analytical HPLC respectively. Recycling HPLC of U@C2v(9)-C82Ad(I) exhibited only one sample peak. Recycling HPLC of U@C2v(9)-C82Ad(II) exhibits two sample peaks, which correspond to U@C2v(9)-C82Ad(I) and U@C2v(9)-C82Ad(II) with percentages of 17% and 83%, respectively. Recycling HPLC of U@C2v(9)-C82Ad(III) shows three sample peaks corresponding to U@C2v(9)-C82Ad(I), U@C2v(9)-C82Ad(II) and U@C2v(9)-C82Ad(III) with relative percentages of 6%, 7% and 87%, respectively.
Computational methods
Based on the density functional theory (DFT), full geometry optimizations with symmetry constraints were carried out using the BLYP functional41,42 under the generalized-gradient approximation (GGA), as implemented in the DMol3 program.43,44 The double numerical plus d-functions (DND) basis set was used for C and the effective core potentials (ECP) basis set for Th and U.45,46 The convergence criteria adopted in geometry optimizations are that the energy is 2.0 × 10−5 Hartree (Ha), the gradient is 4.0 × 10−3 Ha Å−1 and the displacement is 5.0 × 10−3 Å. All the results were visualized with the aid of the Mercury program.47Conflicts of interest
There are no conflicts to declare.Acknowledgements
N. C. thanks the National Science Foundation China (NSFC 51302178, 91961109) and the NSF of Jiangsu Province (BK20200041), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD). P. J. thanks the Natural Science Foundation of Hebei Province of China (B2019202210).Notes and references
- A. A. Popov, S. Yang and L. Dunsch, Chem. Rev., 2013, 113, 5989–6113 CrossRef CAS.
- W. Yang, G. Velkos, F. Liu, S. M. Sudarkova, Y. Wang, J. Zhuang, H. Zhang, X. Li, X. Zhang, B. Buchner, S. M. Avdoshenko, A. A. Popov and N. Chen, Adv. Sci., 2019, 6, 1901352 CrossRef CAS.
- K. Wang, X. Liu, R. Huang, C. Wu, D. Yang, X. Hu, X. Jiang, J. C. Duchamp, H. Dorn and S. Priya, ACS Energy Lett., 2019, 4, 1852–1861 CrossRef CAS.
- J.-P. Zheng, M.-M. Zhen, C.-R. Wang and C.-Y. Shu, Chin. J. Anal. Chem., 2012, 40, 1607–1615 CrossRef CAS.
- P. Jin, Y. Li, S. Magagula and Z. Chen, Coord. Chem. Rev., 2019, 388, 406–439 CrossRef CAS.
- R. D. Bolskar, A. F. Benedetto, L. O. Husebo, R. E. Price, E. F. Jackson, S. Wallace, L. J. Wilson and J. M. Alford, J. Am. Chem. Soc., 2003, 125, 5471–5478 CrossRef CAS.
- H. Kato, Y. Kanazawa, M. Okumura, A. Taninaka, T. Yokawa and H. Shinohara, J. Am. Chem. Soc., 2003, 125, 4391–4397 CrossRef CAS.
- R. B. Ross, C. M. Cardona, D. M. Guldi, S. G. Sankaranarayanan, M. O. Reese, N. Kopidakis, J. Peet, B. Walker, G. C. Bazan, E. Van Keuren, B. C. Holloway and M. Drees, Nat. Mater., 2009, 8, 208–212 CrossRef CAS.
- R. B. Ross, C. M. Cardona, F. B. Swain, D. M. Guldi, S. G. Sankaranarayanan, E. Van Keuren, B. C. Holloway and M. Drees, Adv. Funct. Mater., 2009, 19, 2332–2337 CrossRef CAS.
- E. B. Iezzi, J. C. Duchamp, K. Harich, T. E. Glass, H. M. Lee, M. M. Olmstead, A. L. Balch and H. C. Dorn, J. Am. Chem. Soc., 2002, 124, 524–525 CrossRef CAS.
- O. Lukoyanova, C. M. Cardona, J. Rivera, L. Z. Lugo-Morales, C. J. Chancellor, M. M. Olmstead, A. Rodríguez-Fortea, J. M. Poblet, A. L. Balch and L. Echegoyen, J. Am. Chem. Soc., 2007, 129, 10423–10430 CrossRef CAS.
- C. M. Cardona, A. Kitaygorodskiy, A. Ortiz, M. Á. Herranz and L. Echegoyen, J. Org. Chem., 2005, 70, 5092–5097 CrossRef CAS.
- T. Cai, Z. Ge, E. B. Iezzi, T. E. Glass, K. Harich, H. W. Gibson and H. C. Dorn, Chem. Commun., 2005, 3594–3596 RSC.
- C. Shu, T. Cai, L. Xu, T. Zuo, J. Reid, K. Harich, H. C. Dorn and H. W. Gibson, J. Am. Chem. Soc., 2007, 129, 15710–15717 CrossRef CAS.
- Y. Iiduka, O. Ikenaga, A. Sakuraba, T. Wakahara, T. Tsuchiya, Y. Maeda, T. Nakahodo, T. Akasaka, M. Kako, N. Mizorogi and S. Nagase, J. Am. Chem. Soc., 2005, 127, 9956–9957 CrossRef CAS.
- M. Yamada, Y. Tanabe, J. S. Dang, S. Sato, N. Mizorogi, M. Hachiya, M. Suzuki, T. Abe, H. Kurihara, Y. Maeda, X. Zhao, Y. Lian, S. Nagase and T. Akasaka, J. Am. Chem. Soc., 2016, 138, 16523–16532 CrossRef CAS.
- A. Rodríguez-Fortea, J. M. Campanera, C. M. Cardona, L. Echegoyen and J. M. Poblet, Angew. Chem., Int. Ed., 2006, 45, 8176–8180 CrossRef.
- N. Chen, E.-Y. Zhang, K. Tan, C.-R. Wang and X. Lu, Org. Lett., 2007, 9, 2011–2013 CrossRef CAS.
- M. Yamada, T. Nakahodo, T. Wakahara, T. Tsuchiya, Y. Maeda, T. Akasaka, M. Kako, K. Yoza, E. Horn, N. Mizorogi, K. Kobayashi and S. Nagase, J. Am. Chem. Soc., 2005, 127, 14570–14571 CrossRef CAS.
- X. Lu, H. Nikawa, L. Feng, T. Tsuchiya, Y. Maeda, T. Akasaka, N. Mizorogi, Z. Slanina and S. Nagase, J. Am. Chem. Soc., 2009, 131, 12066–12067 CrossRef CAS.
- Y. Maeda, Y. Matsunaga, T. Wakahara, S. Takahashi, T. Tsuchiya, M. O. Ishitsuka, T. Hasegawa, T. Akasaka, M. T. H. Liu, K. Kokura, E. Horn, K. Yoza, T. Kato, S. Okubo, K. Kobayashi, S. Nagase and K. Yamamoto, J. Am. Chem. Soc., 2004, 126, 6858–6859 CrossRef CAS.
- Y. Takano, M. Aoyagi, M. Yamada, H. Nikawa, Z. Slanina, N. Mizorogi, M. O. Ishitsuka, T. Tsuchiya, Y. Maeda, T. Akasaka, T. Kato and S. Nagase, J. Am. Chem. Soc., 2009, 131, 9340–9346 CrossRef CAS.
- T. Akasaka, T. Kono, Y. Takematsu, H. Nikawa, T. Nakahodo, T. Wakahara, M. O. Ishitsuka, T. Tsuchiya, Y. Maeda, M. T. H. Liu, K. Yoza, T. Kato, K. Yamamoto, N. Mizorogi, Z. Slanina and S. Nagase, J. Am. Chem. Soc., 2008, 130, 12840–12841 CrossRef CAS.
- M. Hachiya, H. Nikawa, N. Mizorogi, T. Tsuchiya, X. Lu and T. Akasaka, J. Am. Chem. Soc., 2012, 134, 15550–15555 CrossRef CAS.
- T. Akasaka, T. Kono, Y. Matsunaga, T. Wakahara, T. Nakahodo, M. O. Ishitsuka, Y. Maeda, T. Tsuchiya, T. Kato, M. T. H. Liu, N. Mizorogi, Z. Slanina and S. Nagase, J. Phys. Chem. A, 2008, 112, 1294–1297 CrossRef CAS.
- Y. Wang, R. Morales-Martinez, X. Zhang, W. Yang, Y. Wang, A. Rodriguez-Fortea, J. M. Poblet, L. Feng, S. Wang and N. Chen, J. Am. Chem. Soc., 2017, 139, 5110–5116 CrossRef CAS.
- X. Zhang, Y. Wang, R. Morales-Martínez, J. Zhong, C. de Graaf, A. Rodríguez-Fortea, J. M. Poblet, L. Echegoyen, L. Feng and N. Chen, J. Am. Chem. Soc., 2018, 140, 3907–3915 CrossRef CAS.
- W. Cai, R. Morales-Martínez, X. Zhang, D. Najera, E. L. Romero, A. Metta-Magaña, A. Rodríguez-Fortea, S. Fortier, N. Chen, J. M. Poblet and L. Echegoyen, Chem. Sci., 2017, 8, 5282–5290 RSC.
- X. Zhang, W. Li, L. Feng, X. Chen, A. Hansen, S. Grimme, S. Fortier, D. C. Sergentu, T. J. Duignan, J. Autschbach, S. Wang, Y. Wang, G. Velkos, A. A. Popov, N. Aghdassi, S. Duhm, X. Li, J. Li, L. Echegoyen, W. H. E. Schwarz and N. Chen, Nat. Commun., 2018, 9, 2753 CrossRef.
- W. Cai, L. Abella, J. Zhuang, X. Zhang, L. Feng, Y. Wang, R. Morales-Martínez, R. Esper, M. Boero, A. Metta-Magaña, A. Rodríguez-Fortea, J. M. Poblet, L. Echegoyen and N. Chen, J. Am. Chem. Soc., 2018, 140, 18039–18050 CrossRef CAS.
- Y. Li, L. Yang, C. Liu, Q. Hou, P. Jin and X. Lu, Inorg. Chem., 2018, 57, 7142–7150 CrossRef CAS.
- P. Jin, C. Liu, Y. Li, L. Li and Y. Zhao, Int. J. Quantum Chem., 2018, 118, e25501 CrossRef.
- Y. Li, L. Yang, Z. Li, Q. Hou, L. Li and P. Jin, Inorg. Chem., 2019, 58, 10648–10655 CrossRef CAS.
- Y. Li, L. Yang, Z. Wei, Q. Hou, L. Li and P. Jin, Int. J. Quantum Chem., 2019, 119, e25826 CrossRef.
- D. Hao, L. Yang, Z. Wei, Q. Hou, L. Li and P. Jin, Inorg. Chem., 2020, 59, 7039–7048 CrossRef CAS.
- H. Kurihara, X. Lu, Y. Iiduka, H. Nikawa, N. Mizorogi, Z. Slanina, T. Tsuchiya, S. Nagase and T. Akasaka, J. Am. Chem. Soc., 2012, 134, 3139–3144 CrossRef CAS.
- Q. Ye, I. V. Komarov, A. J. Kirby and M. Jones, J. Org. Chem., 2002, 67, 9288–9294 CrossRef CAS.
- Y. Schneider, J. Prévost, M. Gobin and C. Y. Legault, Org. Lett., 2014, 16, 596–599 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866–872 CrossRef CAS.
- A. Bergner, M. Dolg, W. Küchle, H. Stoll and H. Preuß, Mol. Phys., 1993, 80, 1431–1441 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: MALDI-TOF mass spectra and theoretical calculation results. CCDC 2031751 and 2031857. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc06111e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |