
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering multifunctional metal/protein hybrid nanomaterials as tools for therapeutic intervention and high-sensitivity detection†
Antonio
Aires‡
a,
David
Maestro‡
b,
Jorge
Ruiz del Rio
b,
Ana R.
Palanca
 bc,
Elena
Lopez-Martinez
a,
Irantzu
Llarena
a,
Kalotina
Geraki
d,
Carlos
Sanchez-Cano
*a,
Ana V.
Villar
*be and
Aitziber L.
Cortajarena
*af
bc,
Elena
Lopez-Martinez
a,
Irantzu
Llarena
a,
Kalotina
Geraki
d,
Carlos
Sanchez-Cano
*a,
Ana V.
Villar
*be and
Aitziber L.
Cortajarena
*af
aCenter for Cooperative Research in Biomaterials (CIC biomaGUNE), Basque Research and Technology Alliance (BRTA), Paseo de Miramón 194, 20014, Donostia-San Sebastián, Spain. E-mail: alcortajarena@cicbiomagune.es; csanchez@cicbiomagune.es
bInstituto de Biomedicina y Biotecnología de Cantabria (IBBTEC), CSIC-Universidad de Cantabria, Albert Einstein 22, 39011, Santander, Spain. E-mail: anavictoria.villar@unican.es
cDepartamento de Anatomía y Biología Celular, Universidad de Cantabria, Avd. Herrera Oria s/n, 39011, Santander, Spain
dDiamond Light Source, Harwell Science and Innovation Campus, RG20 6RE, UK, England
eDepartamento de Fisiología y Farmacología, Universidad de Cantabria, Avd. Herrera Oria s/n, 39011, Santander, Spain
fIkerbasque, Basque Foundation for Science, Mª Díaz de Haro 3, 48013 Bilbao, Spain
First published on 14th December 2020
Abstract
Protein-based hybrid nanomaterials have recently emerged as promising platforms to fabricate tailored multifunctional biologics for biotechnological and biomedical applications. This work shows a simple, modular, and versatile strategy to design custom protein hybrid nanomaterials. This approach combines for the first time the engineering of a therapeutic protein module with the engineering of a nanomaterial-stabilizing module within the same molecule, resulting in a multifunctional hybrid nanocomposite unachievable through conventional material synthesis methodologies. As the first proof of concept, a multifunctional system was designed ad hoc for the therapeutic intervention and monitoring of myocardial fibrosis. This hybrid nanomaterial combines a designed Hsp90 inhibitory domain and a metal nanocluster stabilizing module resulting in a biologic drug labelled with a metal nanocluster. The engineered nanomaterial actively reduced myocardial fibrosis and heart hypertrophy in an animal model of cardiac remodeling. In addition to the therapeutic effect, the metal nanocluster allowed for in vitro, ex vivo, and in vivo detection and imaging of the fibrotic disease under study. This study evidences the potential of combining protein engineering and protein-directed nanomaterial engineering approaches to design custom nanomaterials as theranostic tools, opening up unexplored routes to date for the next generation of advanced nanomaterials in medicine.
Introduction
Despite tremendous advancement in diagnosis, treatment, and prevention procedures, highly prevalent diseases such as cancer and neurodegenerative and cardiovascular diseases are still a leading cause of death worldwide.3 Therefore, the development of new nanotechnological tools for relevant application in biomedicine is still an ongoing research goal addressed by different methodological approaches. In this context, the specific targeting of disease-related pathways and molecules, mostly proteins, is a promising strategy.Current techniques for tracking, imaging, and inhibiting proteins rely mostly on antibodies. These highly specific proteins are extremely useful tools for different applications, but present some limitations that hamper their use in some cases, including high toxicity, the immunogenicity observed in patients, and inefficient access to cytoplasmic targets.4 The aforementioned challenges could be overcome by the use of recombinantly produced alternatives, for example, molecules based on simpler protein recognition modules. Engineered proteins can go beyond what is possible with antibodies, as their sequence and structure can be easily modified to modulate their targeting capacities or intracellular localization. Furthermore, modular design strategies can be used to combine recognition units with other functionalities, thus generating powerful new tools for therapeutic intervention and diagnosis.
In this context, small engineered protein domains, inspired by natural binding domains, such as tetratricopeptide repeats, ankyrin repeats, and other repeat protein scaffolds, show great promise as the next generation of therapeutic biologics.5–8 For example, the engineered consensus tetratricopeptide repeat (CTPR) scaffold is a domain comprising the repetition of 34 amino acid structural modules, which are stabilized by predictable inter-repeat local interactions.9 These interactions determine the CTPR protein structure, with a clear distinction between residues that encode their stability or their functionality.10 CTPR proteins can be engineered to display different ligand recognition properties, as antibody mimics.11 In addition to their binding capabilities, CTPR domains can also be engineered to synthesize metal nanoclusters (NCs) by introducing metal binding sites within protein scaffolds.12–15 Of particular interest for biomedical applications are small noble metal nanoclusters, such as silver and gold, stabilized within biomolecules.16 The resulting CTPR-stabilized metal nanoclusters (Prot-NC) are highly photoluminescent, photo-stable, and biocompatible. Furthermore, these Prot-NCs are readily internalized by living cells, without compromising the NC integrity and their luminescence properties.14 Therefore, CTPR protein hybrid nanomaterials show great promise as the basis to construct tailored multifunctional nanomaterials for biotechnological and biomedical applications.
This work presents, as the first proof of concept, the fusion of a CTPR390 module [1] (as the protein recognition module)10,17 to target and inhibit Hsp90,18 and a fluorescent metal nanocluster stabilizing module, for in vitro, ex vivo, and in vivo detection and imaging (or tracking). The main goal is to explore the development and application of multifunctional hybrid nanomaterials based on the rational assembly of engineered protein modules with specified properties as building blocks. Remarkably, since the modules used are based on the same structural scaffold, the CTPR protein, they can be easily fused into single proteins to form new molecular tools for therapeutic intervention and high-sensitivity detection technologies. This multifunctional molecule is validated for therapeutic intervention and for monitoring replacement myocardial fibrosis secondary to heart damage. Replacement myocardial fibrosis has prognostic value usually associated with risk factors of poor cardiac recovery,19–21 that strongly affects heart function by overexpression and deposition of extracellular matrix pro-fibrotic components, mainly collagen I. However, therapies to directly target excessive fibrosis remain limited.20,22 The main target cytokine that triggers the overexpression of collagen fibers is transforming growth factor beta (TGFβ),23,24 and the blockade of its cascade through the inhibition of the chaperone Hsp90 reduces collagen synthesis and deposition in damaged hearts.25,26 In this sense, the correlation between the inhibition of Hsp90 and the reduction of the pro-fibrotic effects of TGFβ in cardiac fibrosis opened a new route for the development of anti-fibrotic therapies.8,25 As such, the designed Hsp90-binding module (CTPR390; [1]) inhibited Hsp90 preserving its chaperone activity and thus the cellular homeostasis,8,10,17 resulting in TGFβ signaling deregulation and collagen synthesis reduction.8 The added value of labelling the biologic drug 1 with gold nanoclusters (AuNCs) is demonstrated in this work by using Au as a unique tracing signal of the molecule when imaging the heart tissue obtained from treated animals by synchrotron X-ray fluorescence (SXRF). SXRF is a powerful technique that allows simultaneous mapping of the distribution of different elements in cells and tissues.27,28 The approach shown here enables the specific labelling of the protein-based biologic drug of interest, generating a theranostic tool that reduces the effects of the disease and can be unequivocally traced and quantified in tissue. This strategy permits assessment of the correlation between the tissue localization and the therapeutic effect of the biologic drug. Overall, this work reveals the first design of a hybrid protein-nanocluster biologic drug and the potential of this nanocomposite as a theranostic tool, opening up unexplored routes to date for the next generation of advanced biocompatible multimodal nanomaterials.
Results and discussion
To explore the development of multifunctional nanomaterials based on engineered proteins as new tools for therapeutic intervention and high-sensitivity monitoring of pathological myocardial fibrosis, fusion between the CTPR390 module [1], designed as a protein recognition moiety to target and inhibit the Hsp90 protein, and a fluorescent metal nanocluster stabilizing module [2], for ex vivo, in vitro, and in vivo detection and imaging, was performed (Fig. 1). The 1 module was previously developed and tested for fibrosis inhibition,8,17 and the nanocluster stabilizing module [2] was previously designed based on the crystal structure of CTPR protein (PDB ID: 2HYZ)2 for the synthesis and stabilization of iron–sulfur nanoclusters.12 Since thiols are known to bind strongly to metals, such as gold,29,30 a metal coordination site carrying four cysteine residues (Cys) was modelled on the CTPR concave surface.12 This site is expected to result in a stronger nanocluster coordination compared to previously reported sites comprising histidines.14 The initial design gave rise to the nanocluster stabilization module C2NC [2], which was the module intended to generate the final hybrid system. This module was first tested for its ability to stabilize gold nanoclusters, using a slightly modified green biocompatible chemistry method that had been previously described,14 in the context of a protein with two flanking wild type CTPR repeats [3] (Fig. S1†), since a CTPR with two repeats is marginally stable.31 The 3-AuNC complex was purified by size-exclusion chromatography, and the NC growth was followed by the appearance of a single peak fluorescence emission with a maximum at 440 nm (excitation at the maximum absorbance wavelength, 374 nm; Fig. 2A). Extensive control experiments confirmed the fluorescence emission of 3-AuNCs after reduction with sodium ascorbate, as the fluorescence was observed only when all the reagents were present (Fig. S1†). | ||
| Fig. 1 Multifunctional proteins designed for the treatment and monitoring of myocardial fibrosis. (A) General scheme of the chimeric multifunctional protein scaffold design and the synthesis and stabilization of fluorescent AuNCs. The crystal structure of the designed CTPR390 [1] recognition module (green) in complex with the C-terminal Hsp90 peptide (orange) (PDB ID: 3KD7) (center).1 A nanocluster stabilization site is introduced on a modified CTPR module in which four cysteines have been introduced [2] (cyan) on the structure of the CTPR (PDB ID: 2HYZ).2 Chimeric bifunctional protein [4] made by the fusion of the 1 and 2 modules. Synthesis and stabilization of fluorescent 4-AuNCs. (B) Schematic representation of the general approach for the treatment and monitoring of myocardial fibrosis. Representation of therapeutic 4-AuNC intraperitoneal injection into a fibrotic mouse. Representative examples of the therapeutic efficacy in the reduction of myocardial fibrosis. In vitro, ex vivo, and in vivo monitoring of the myocardial fibrosis. In vitro imaging of 4-AuNC treated primary myocardial fibroblasts by confocal microscopy (CM). Ex vivo4-AuNC fluorescence detection with an in vivo imaging system (IVIS) by excitation at 440 nm of 3 controls and one 4-AuNC treated heart. 4-AuNC detection by synchrotron X-ray fluorescence (SXRF) and inductively coupled plasma mass spectrometry ICP-MS techniques of in vivo4-AuNC treated mice. | ||
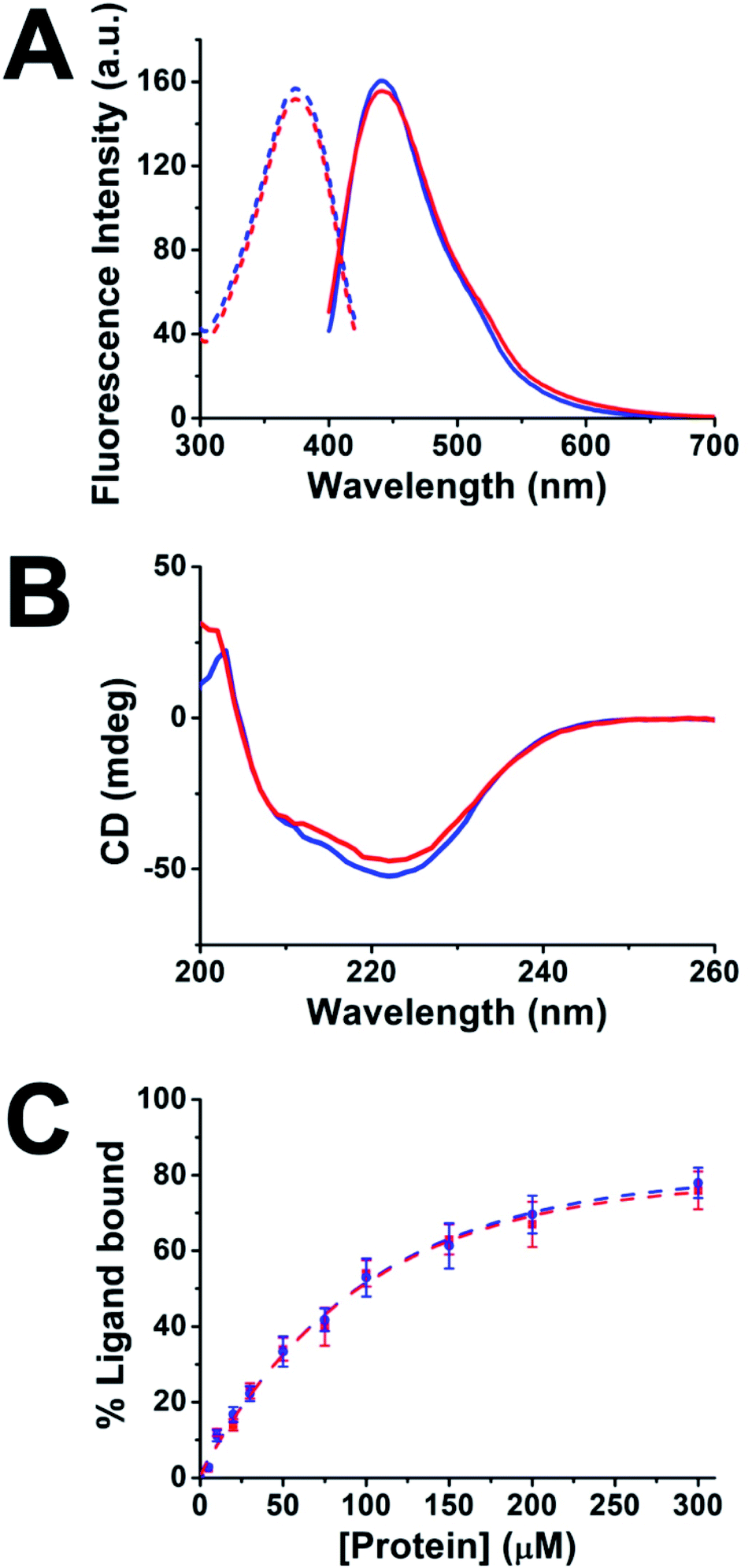 | ||
Fig. 2 (A) Fluorescence spectra of 3-AuNCs (blue) and 4-AuNCs (red). The excitation spectra are shown in dashed lines and the emission spectra in solid lines. (B) CD spectra of 4 (solid blue line) and 4-AuNCs (solid red line) recorded at 10 μM protein concentration at 25 °C. (C) Ligand binding activity of 4 (blue circles) and 4-AuNC (red squares). Fluorescence anisotropy of the fluorescein-labelled 24-mer C-terminal peptide of Hsp90. Dashed lines show the fit to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding model to calculate the binding affinity. The dissociation constant (KD) was KD = 155 ± 11 μM for 4, and KD = 160 = 15 μM for 4-AuNCs. :1 binding model to calculate the binding affinity. The dissociation constant (KD) was KD = 155 ± 11 μM for 4, and KD = 160 = 15 μM for 4-AuNCs. | ||
Upon demonstrating the capability of 2 to allow the synthesis/stabilization of blue-emitting AuNCs, 1 and 2 were fused using a modular cloning strategy (Fig. 1A).9 The resulting protein 4 was used for the synthesis/stabilization of AuNCs (Fig. 1A). After synthesizing the multifunctional 4-AuNC hybrid nanomaterial, the fluorescence emission and structural integrity of the protein scaffold were verified. Remarkably, excitation of 4-AuNCs with UV light caused a fluorescence emission with comparable efficiency to that of the NC obtained from 3 under the same experimental conditions (Fig. 2A). In addition, the circular dichroism (CD) spectra of 4-AuNCs revealed that the α-helical structure of the protein scaffold was not affected by the AuNC formation, which is critical for their application as a biologic drug (Fig. 2B). Then, the binding of the protein to Hsp90 (ref. 18) was verified by fluorescence anisotropy using a fluorescein labelled 24-mer Hsp90 peptide (Fig. 2C). The KD value obtained for the interaction between 4-AuNCs and the Hsp90 peptide was 160 ± 15 μM, similar to the value determined for the protein 4 alone (155 ± 11 μM) and to the value described for 1 (200 μM).17 These results confirmed that the binding domain module 1 present in 4 was not affected by the AuNC formation, which is key for using this novel methodology to develop new theranostic tools.
The multifunctional 4-AuNC hybrid nanomaterial was fully characterized, showing a fluorescence quantum yield (QY) of 10.05%, significantly higher than the QY reported for other protein-stabilized AuNCs (3.5–6.0%).13,32–34 The origin of this high QY is probably associated with the characteristics of the protein scaffold, since the QY of the AuNCs is strongly affected by the capping agents.35,36 The cysteine residues that compose the gold binding site were placed onto the protein concave surface and, thus the resulting AuNC should be shielded and rigidified by the protein scaffold. This decreases the non-radiative deactivation process and improves the stability of the AuNCs, leading to high QY values.37 In addition, the presence of electron-rich cysteine residues on the concave surface of the protein scaffold can largely promote the photoluminescence of the protein-stabilized AuNCs.36,38 The excited-state dynamics of the 4-AuNC was analyzed with time-resolved photoluminescence measurements (Fig. S2†). Multi-exponential fits of the photoluminescence decay curves, provided 2.53 ns amplitude-weighted average lifetimes that correspond to transitions between singlet states in AuNCs.39–41 XPS indicated that the Au cores of the 4-AuNC were mainly composed of Au1+ and Au0 species with a ratio 2:1 (Fig. S3†). In order to use the Au atoms as a specific tracer of the multifunctional molecule for SXRF a precise quantification of the NC size and the number of Au atoms per protein was performed. MALDI-TOF and ICP-MS were employed to define the number of Au atoms in the multifunctional 4-AuNC hybrid nanomaterial. The MALDI-TOF mass spectrum of 4-AuNC showed a broad peak at a larger molecular weight than that of the protein 4, which proved the stabilization of AuNCs by the protein and revealed certain polydispersity in size for the AuNC (Fig. S4 and Table S1†). An average of 8 ± 4 gold atoms per protein were estimated for the multifunctional 4-AuNC hybrid nanomaterial. Accordingly, ICP-MS determination of Au from 4-AuNCs indicated 8 ± 1 Au atoms per protein (Table S2†). This size is in concordance with that of previously described blue fluorescent AuNCs.33,42,43 Finally, the homogeneity in the morphology and size of the 4-AuNC was evaluated by scanning transmission electron microscopy (STEM) (Fig. S5†). The STEM images showed homogeneous NCs with diameters of 1.0 ± 0.2 nm (Fig. S5†).
The multifunctional 4-AuNC hybrid nanomaterial also showed good stability and biocompatibility, which are required for any biomedical application. 4-AuNCs were stable over a month in PBS at 4 °C (Fig. S6A†) and over a week under physiological conditions (PBS, and human plasma at 37 °C) (Fig. S6B†). In addition, the biocompatibility of the 4-AuNC was evaluated in cell cultures. NIH-3T3 fibroblast cells and primary cardiac fibroblasts extracted from adult mice were incubated with different concentrations of 4-AuNCs under standard cell culture conditions. Following 5 days of incubation, 4-AuNCs did not present any evident cytotoxicity even at the highest concentration tested (Fig. S7†). Furthermore, functional validation of 4-AuNCs as a biologic anti-fibrotic drug was also performed in vitro using primary cardiac fibroblasts extracted from adult mice. Fibroblasts treated with 4-AuNCs showed a significant reduction in gene expression of key pro-fibrotic markers, including collagen I and III, and fibronectin in Fig. S8.† Moreover, 4-AuNCs exhibited an efficient internalization (Fig. S9 and Movies S1 and S2†) while did not inhibit the Hsp90 chaperone activity (Fig. S10†) after 24 h of incubation in primary cardiac fibroblasts extracted from adult mice. These results are in agreement with previous observations for the 1 protein without AuNCs.8
Hereafter, the use of 4-AuNCs as a biologic anti-fibrotic drug to reduce myocardial fibrosis in vivo was evaluated in an angiotensin II (Ang II) mice model. It has been widely demonstrated that Ang II administration induces cardiovascular damage through pathological remodelling including hypertrophy and fibrosis.44 To induce fibrosis, mice were continuously infused with Ang II for 15 days through a subcutaneous osmotic mini-pump. The hypertension promoted by Ang II generates mechanical damage in the heart and in addition to mechanical stress, it activates pathological remodelling (hypertrophy and fibrosis) to end-organ damage in the short term. These reparative mechanisms, mainly fibrosis, in the long term lead to cardiac dysfunction and in the worst scenario cardiac failure.45 Consistent with previous reports, significant and widespread fibrosis was observed throughout the myocardium 15 days after Ang II administration.46 However, a reduction of the collagen area was observed in the hearts of mice treated with 4-AuNCs (Fig. 3A and B). With only one single dose of 4-AuNCs (200 μl at 1 mM), fibrosis remained limited 4 days after the treatment and it showed a more significant reduction 8 days after the 4-AuNC administration (Fig. 3A and B). At a shorter time after the administration (3 hours) reduction of the Ang II-enhanced fibrosis (Fig. 3A and B) and reversion of the hypertrophy (Fig. S11†) were not observed. These results were in accordance with previously described data for the 1 protein without AuNCs.8
 | ||
| Fig. 3 In vivo 4-AuNC effect on cardiac fibrosis. Histology, gene, and protein expression of cardiac markers related to fibrosis in the Ang II mouse model treated with 4-AuNCs. (A) Masson's trichrome staining of heart cross sections to evaluate fibrosis (mainly collagen I) and details of these sections. Muscle visualization in pink and fibrotic areas in purple. Groups of mice under study: Sham control mice with vehicle or with 4-AuNC administration at 3 hours, 4 and 8 days after the treatment (left panel); Ang II fibrotic mice with vehicle or with 4-AuNC administration at 3 hours, 4 and 8 days after the treatment (right panel). (B) Total collagen I area measured in the heart cross sections from the mice under study at 3 hours, 4 days or 8 days after the anti-fibrotic 4-AuNC administration. (C and D) Expression of the cardiac stress inducible genes encoding atrial natriuretic peptides Nppa and Nppb 8 days after the biologic 4-AuNC anti-fibrotic drug administration. (E and F) Gene-expression changes for COL III (D) and FN (E) in 4-AuNC treated mice (8 days). (G and H) Expression and quantification of COL III and FN proteins of 4-AuNC treated mice (8 days) in arbitrary units (A.U.). (*p < 0.05, **p < 0.005, ***p < 0.001; Mann–Whitney test). | ||
The greatest reduction of myocardial fibrotic areas was observed 8 days after the 4-AuNC treatment. Thus, the study at the molecular level was focused on this time point showing the reduction in gene expression of myocardial stress biomarkers (Nppa and Nppb) (Fig. 3C and D), and the reduction in gene and protein expression of the structural pro-fibrotic proteins (COL III, and FN) in 4-AuNC treated mice, compared to vehicle-treated mice (Fig. 3E–H). In addition to the reduced cardiac fibrosis by 4-AuNC treatment, a study of cardiac hypertrophy (heart weight to body weight ratio) was performed. Fig. S11† shows that the myocardial hypertrophy was notably reduced 8 days after 4-AuNC treatment.
Remarkably, the presence of Au made possible the detection of the multifunctional 4-AuNC hybrid nanomaterial, in organs, tissues, and fluids using a number of different techniques. As such, imaging of ex vivo hearts injected with 4-AuNCs showed a uniform distribution of the 4-AuNC within the organ (Fig. 1B). ICP-MS allowed the assessment of the biodistribution of 4-AuNC in mice (Fig. S12†), showing that 8 days after injection 4-AuNCs were accumulated mostly in the kidneys, liver, and spleen of both healthy and fibrotic individuals. These results suggest the presence of a regular activity of the hepatic metabolism and a desirable renal clearance in treated mice with or without fibrosis, as was expected considering the hydrodynamic size of the multifunctional 4-AuNC hybrid nanomaterial (8 ± 1 nm). In contrast, Au was found in low quantities in the stomach, lungs, intestines, urine, and blood, and was not detected in the brain and cerebellum, indicating that 4-AuNCs do not cross the brain blood barrier. Interestingly, significant differences were observed in the accumulation of Au in the cardiovascular areas of fibrotic individuals, such as atria, right and left ventricles, and the aorta, compared to healthy controls (Fig. S12†). These cardiovascular areas have been consistently reported as damaged areas in an Ang II murine model.47,48
SXRF using microfocused synchrotron radiation also permitted quantification of the accumulation of Au in tissue samples, while providing lateral resolution not available from ICP-MS. This technique allows the assessment of the localization of 4-AuNCs within the hearts of Ang II and healthy mice with a μm resolution (Fig. 4A–E and S13–21, and Table S3†). Three different time-points, 3 hours, 4 days and 8 days, were selected to evaluate the accumulation of 4-AuNC by SXRF in the hearts of the mice after the administration of 4-AuNCs (Fig. S15–S21†). Au elemental maps acquired using SXRF showed a higher accumulation of 4-AuNC in the hearts of fibrotic mice in comparison with the hearts of healthy mice. Furthermore, this accumulation was time dependent, and could be correlated with the antifibrotic response of 4-AuNCs (Fig. S23 and S24†). The two time-points that presented the maximal difference in the anti-fibrotic response (according to the collagen reduction and fibrotic biomarker quantification – Fig. 3) compared to that of the vehicle treated mice, 3 hours (lack of response) and 8 days (highest anti-fibrotic response) were analysed more in detail by SXRF. Initially, Au elemental maps acquired using SXRF confirmed the absence of 4-AuNCs in the hearts of vehicle treated mice, and the presence of low quantities of Au in those heart sections extracted from healthy mice treated with the 4-AuNCs, independent of the time after the treatment (1.66 ± 0.11 and 0.8 ± 0.02 ng mg−1, respectively 3 hours or 8 days after the treatment). In contrast, the hearts of Ang II mice treated with 4-AuNCs seemed to accumulate high quantities of Au (5.62 ± 0.22 and 27.22 ± 6.27 ng mg−1, respectively 3 hours or 8 days after the treatment) in a time dependent manner (4-fold increase between 3 hours and 8 days after the treatment; Fig. 4E). Remarkably, Au accumulation was independent of the area of the hearts studied (i.e. septum and posterior wall, top or bottom; Fig. 4A–C and S14–S20†). But, while the distribution of 4-AuNCs within these areas was homogeneous in healthy mice or Ang II mice 3 hours after the treatment with 4-AuNCs, Au maps acquired from the hearts of Ang II mice 8 days after the treatment showed a distinct punctuated pattern (with hot-spots containing over 50 times more gold than the average found in the whole area studied; Fig. 4D and S14–S21†). Such accumulation patterns are similar to the ones obtained from Ang II hearts when they are stained with the Masson's trichrome staining, which highlights fibrotic areas with a high concentration of collagen (Fig. 3A). Therefore, it might be possible that 4-AuNCs respond to distinct collagen deposit-domains within specific myocardial areas, thus tracking the damage. To confirm the accumulation of the multifunctional 4-AuNC hybrid nanomaterial in the fibrotic areas and explore its potential as a bio-marker of the myocardial fibrosis disease, partial Masson's trichrome staining was applied to specifically stain, with tungsten (W), the collagen-rich fibrotic regions. The experimental design was based on the first steps of the Masson's trichrome assay in which phosphotungstic acid (PTA) binds to collagen and fibres of connective tissues at low pH. Once the tungsten–collagen interaction was fixed on the fresh tissue, the co-localization of Au and W was assessed using SXRF maps obtained from PTA-stained heart sections. PTA is widely used as a contrast agent for X-ray based techniques, and is currently being tested as a possible contrast agent to study collagen distribution in cartilage using contrast-enhanced micro-computed tomography.49–51 High levels of tungsten were found in the hearts of Ang II mice when compared with those from the healthy mice (0.192 ± 0.003 and 0.079 ± 0.001 ng W per ng Zn, respectively; normalized to Zn levels as it indicates cellular areas due to high levels in nuclear regions; Fig. S22 and S24†). These results were expected, as increased collagen levels in fibrotic hearts were already observed (Fig. 3A). However, when fibrotic (Ang II) and healthy (Sham) mice were analyzed 8 days after 4-AuNC treatment, the hearts showed similar levels of W to the hearts of vehicle treated healthy (Sham) mice (0.096 ± 0.001 and 0.083 ± 0.001 ng W per ng Zn, respectively; Fig. S24†). Furthermore, only fibrotic (Ang II) mice accumulated high amounts of Au (as expected; Fig. S23†), and the correlation between the W and Au signals was much higher in the fibrotic heart samples treated with 4-AuNCs when compared with the healthy analogues (R-Pearson's coefficients 0.35 and −0.2 respectively; Fig. S24†). These results suggest that the reduced fibrosis levels observed upon treatment with 4-AuNCs might be related to the accumulation of the multifunctional 4-AuNC hybrid nanomaterial in fibrotic areas. Such accumulation could be due to the presence of high levels of Hsp90 (the target of 4-AuNCs) related to collagen production, as the expression of Hsp90α (inducible isoform under pathological conditions) was significantly augmented in the heart of AngII-fibrotic mice, while there was no variation in Hsp90β (constitutively expressed isoform) (Fig. S25†).8
 | ||
| Fig. 4 (A–C) Heart sections from mice showing the areas studied with SXRF. (A) Ang II fibrotic mouse treated with 4-AuNCs; (B) vehicle treated Ang II fibrotic mouse; (C) Sham healthy mouse treated with 4-AuNCs. (D) Au SXRF elemental maps showing localization of the Au element from 4-AuNCs in healthy (Sham) or fibrotic (Ang II) hearts of mice 3 hours and 8 days after 4-AuNC treatment; Raster scan: 5 × 5 μm2 step size, 1 s dwell time. Scale bar 100 μm. Concentration range in images: 0–2 ng mm−2 Au. (E) Quantity of Au accumulated in hearts of healthy (Sham) or fibrotic (Ang II) mice 3 hours and 8 days after 4-AuNC treatment. | ||
Conclusions
In summary, a simple protein engineering-based versatile approach to generate custom protein–metal hybrid nanomaterials for biomedical applications has been demonstrated. The main goal in this work was to explore the development and application of synthetic strategies to obtain multifunctional hybrid nanomaterials based on the rational assembly of engineered protein modules with specified properties as building blocks. As the sequence and structure of these engineered protein modules can be easily modified to modulate their targeting capacities and to introduce other functionalities, this modular design strategy can be used to combine recognition units and other functionalities, depending on the specific application, thus generating powerful tailored multifunctional nanomaterials for therapeutic intervention and diagnosis.Here, as the first proof of concept, an engineered multifunctional system was constructed, by combination of two designed modules, a recognition module, and a metal nanocluster stabilizing module, in a chimeric protein. In particular, a new tool for the therapeutic intervention and monitoring of myocardial fibrosis was generated, by the fusion of the CTPR390 module [1], a protein recognition moiety that targets and inhibits the TGFβ pro-fibrotic cascade protein, Hsp90, and a fluorescent metal nanocluster stabilizing module, for ex vivo, in vitro, and in vivo detection and imaging. The resulting protein-based hybrid nanomaterial (4-AuNC) was evaluated as a biologic anti-fibrotic drug to reduce myocardial fibrosis in vivo in an angiotensin II (Ang II) mouse model. A single dose administration of 4-AuNCs caused a significant reduction in the myocardial fibrosis and heart hypertrophy 8 days after treatment, due to the Hsp90 inhibition function. This inhibition preserved the Hsp90 chaperone function, thus minimizing the effects on cellular homeostasis, which opens a new route for the development of anti-fibrotic therapies. In addition, 4-AuNCs allowed ex vivo, in vitro, and in vivo detection and imaging of the fibrosis disease, due to the selective labelling of the biological molecule with a biocompatible metal nanocluster. Overall, the strategy presented here shows great promise of engineered protein-based hybrid nanomaterials and their potential use as theranostic tools, opening up unexplored routes to date for the synthesis of the next generation of advanced nanocomposites and their application in medicine.
Ethical statement
Live animal studies were approved by the University of Cantabria Institutional Laboratory Animal Care and Use Committee in compliance with the Guide for the Care and Use of Laboratory Animals (ILAR, 1985) and were conducted in accordance with the “European Directive for the Protection of Vertebrate Animals Used for Experimental and Other Scientific Purposes” (European Communities Council Directive 86/606/EEC).Author contributions
Conceptualization, A. L. C., and A. V. V.; methodology, A. A., D. M., JRdR., A. R. P., E. L-.M., I. L. L., K. G., and C. S.-C.; formal analysis, A. A., D. M., C. S.-C., A. V. V., and A. L. C.; investigation: protein hybrid synthesis and characterization and analytical studies A. A.; investigation: in vitro and in vivo studies, D. M., JRdR., A. R. P., and A. V. V.; investigation: XRF studies, C. S.-C., E. L.-M., K. G., A. V. V., and A. L. C.; writing-original draft preparation, A. A., D. M., C. S.-C., A. V. V., and A. L. C.; funding acquisition, A. V. V. and A. L. C. All the authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the European Research Council ERC-CoG-648071-ProNANO and ERC-PoC-841063-NIMM; Agencia Estatal de Investigación, Spain (PID2019-111649RB-I00; RTI2018-095214-B-I00; BIO2016-77367-C2-1-R; and BIO2015-72124-EXP); the Basque Government (Elkartek KK-2017/00008; RIS3-2019222005); and IDIVAL InnVal 17/22. C. S. C. thanks Gipuzkoa Foru Aldundia (Gipuzkoa Fellows program; grant number 2019-FELL-000018-01/62/2019) for financial support. E.L.-M. thanks the Spanish Ministry of Science and Innovation for the FPI grant (BES-2017-079646). This work was performed under the Maria de Maeztu Units of Excellence Program from the Spanish State Research Agency – Grant No. MDM-2017-0720 (CIC biomaGUNE). SXRF maps were acquired at the I18 beamline in the frame of the Diamond proposals SP20603. We thank Dr Marco Moller, Dr J. Calvo, Dr Otaegui, and Dr Desire Di Silvio at CIC biomaGUNE for support with the acquisition of electron microscopy images, mass spectrometry, and X-ray photoelectron spectroscopy measurements.Notes and references
- A. L. Cortajarena, J. Wang and L. Regan, FEBS J., 2010, 277, 1058–1066 CrossRef CAS.
- T. Kajander, A. L. Cortajarena, S. Mochrie and L. Regan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2007, 63, 800–811 CrossRef CAS.
- A. Becker, M. Leskau, B. L. Schlingmann-Molina, S. C. Hohmeier, S. Alnajjar, H. Murua Escobar and A. Ngezahayo, Sci. Rep., 2018, 8, 14963 CrossRef.
- A. Bradbury and A. Plückthun, Nature, 2015, 518, 27–29 CrossRef CAS.
- Y. L. Boersma and A. Plückthun, Curr. Opin. Biotechnol., 2011, 22, 849–857 CrossRef CAS.
- A. Plückthun, Annu. Rev. Pharmacol. Toxicol., 2015, 55, 489–511 CrossRef.
- L. Regan, M. R. Hinrichsen and C. Oi, Expert Rev. Proteomics, 2016, 13, 481–493 CrossRef CAS.
- R. A. Caceres, T. Chavez, D. Maestro, A. R. Palanca, P. Bolado, F. Madrazo, A. Aires, A. L. Cortajarena and A. V. Villar, J. Mol. Cell. Cardiol., 2018, 123, 75–87 CrossRef CAS.
- A. L. Cortajarena, S. G. J. Mochrie and L. Regan, Protein Sci., 2011, 20, 1042–1047 CrossRef CAS.
- A. L. Cortajarena, F. Yi and L. Regan, ACS Chem. Biol., 2008, 3, 161–166 CrossRef CAS.
- A. L. Cortajarena, T. Y. Liu, M. Hochstrasser and L. Regan, ACS Chem. Biol., 2010, 5, 545–552 CrossRef CAS.
- S. H. Mejias, Z. Bahrami-Dizicheh, M. Liutkus, D. J. Sommer, A. Astashkin, G. Kodis, G. Ghirlanda and A. L. Cortajarena, Chem. Commun., 2019, 55, 3319–3322 RSC.
- P. Couleaud, S. Adan-Bermudez, A. Aires, S. H. Mejías, B. Sot, A. Somoza and A. L. Cortajarena, Biomacromolecules, 2015, 16, 3836–3844 CrossRef CAS.
- A. Aires, I. Llarena, M. Moller, J. Castro-Smirnov, J. Cabanillas-Gonzalez and A. L. Cortajarena, Angew. Chem., Int. Ed., 2019, 58, 6214–6219 CrossRef CAS.
- A. Aires, V. Fernández-Luna, J. Fernández-Cestau, R. D. Costa and A. L. Cortajarena, Nano Lett., 2020, 20, 2710–2716 CrossRef CAS.
- Y. Tao, M. Li, J. Ren and X. Qu, Chem. Soc. Rev., 2015, 44, 8636–8663 RSC.
- A. L. Cortajarena, T. Kajander, W. Pan, M. J. Cocco and L. Regan, Protein Eng., Des. Sel., 2004, 17, 399–409 CrossRef CAS.
- C. Scheufler, A. Brinker, G. Bourenkov, S. Pegoraro, L. Moroder, H. Bartunik, F. U. Hartl and I. Moarefi, Cell, 2000, 101, 199–210 CrossRef CAS.
- G. Balčiūnaitė, D. Palionis, E. Žurauskas, V. Skorniakov, V. Janušauskas, A. Zorinas, T. Zaremba, N. Valevičienė, A. Aidietis, P. Šerpytis, K. Ručinskas, P. Sogaard and S. Glaveckaitė, BMC Cardiovasc. Disord., 2020, 20, 275 CrossRef.
- J. G. Travers, F. A. Kamal, J. Robbins, K. E. Yutzey and B. C. Blaxall, Circ. Res., 2016, 118, 1021–1040 CrossRef CAS.
- P. Kong, P. Christia and N. G. Frangogiannis, Cell. Mol. Life Sci., 2014, 71, 549–574 CrossRef CAS.
- Z. Fan and J. Guan, Biomater. Res., 2016, 20, 016–0060 CrossRef.
- M. Dobaczewski, W. Chen and N. G. Frangogiannis, J. Mol. Cell. Cardiol., 2011, 51, 600–606 CrossRef CAS.
- H. Khalil, O. Kanisicak, V. Prasad, R. N. Correll, X. Fu, T. Schips, R. J. Vagnozzi, R. Liu, T. Huynh, S. J. Lee, J. Karch and J. D. Molkentin, J. Clin. Invest., 2017, 127, 3770–3783 CrossRef.
- R. Garcia, D. Merino, J. M. Gomez, J. F. Nistal, M. A. Hurle, A. L. Cortajarena and A. V. Villar, Cell. Signalling, 2016, 28, 1563–1579 CrossRef CAS.
- R. Datta, T. Bansal, S. Rana, K. Datta, S. Chattopadhyay, M. Chawla-Sarkar and S. Sarkar, Cell. Signalling, 2015, 27, 2410–2424 CrossRef CAS.
- M. J. Pushie, I. J. Pickering, M. Korbas, M. J. Hackett and G. N. George, Chem. Rev., 2014, 114, 8499–8541 CrossRef CAS.
- R. Zhang, L. Li, Y. Sultanbawa and Z. P. Xu, Am. J. Nucl. Med. Mol. Imaging, 2018, 8, 169–188 CAS.
- Y. Ding, G. Gu, X.-H. Xia and Q. Huo, J. Mater. Chem., 2009, 19, 795–799 CAS.
- B. Maity, S. Abe and T. Ueno, Nat. Commun., 2017, 8, 14820 CrossRef CAS.
- T. Kajander, A. L. Cortajarena, E. R. G. Main, S. G. J. Mochrie and L. Regan, J. Am. Chem. Soc., 2005, 127, 10188–10190 CrossRef CAS.
- H. Wei, Z. Wang, L. Yang, S. Tian, C. Hou and Y. Lu, Analyst, 2010, 135, 1406–1410 RSC.
- H. Kawasaki, K. Hamaguchi, I. Osaka and R. Arakawa, Adv. Funct. Mater., 2011, 21, 3508–3515 CrossRef CAS.
- J. Xie, Y. Zheng and J. Y. Ying, J. Am. Chem. Soc., 2009, 131, 888–889 CrossRef CAS.
- S. E. Crawford, C. M. Andolina, A. M. Smith, L. E. Marbella, K. A. Johnston, P. J. Straney, M. J. Hartmann and J. E. Millstone, J. Am. Chem. Soc., 2015, 137, 14423–14429 CrossRef CAS.
- Z. Wu and R. Jin, Nano Lett., 2010, 10, 2568–2573 CrossRef CAS.
- A. Cantelli, G. Guidetti, J. Manzi, V. Caponetti and M. Montalti, Eur. J. Inorg. Chem., 2017, 2017, 5068–5084 CrossRef CAS.
- R. Jin, H. Qian, Z. Wu, Y. Zhu, M. Zhu, A. Mohanty and N. Garg, J. Phys. Chem. Lett., 2010, 1, 2903–2910 CrossRef CAS.
- C. C. Huang, Z. Yang, K. H. Lee and H. T. Chang, Angew. Chem., 2007, 46, 6824–6828 CrossRef CAS.
- M. A. Muhammed, P. K. Verma, S. K. Pal, R. C. Kumar, S. Paul, R. V. Omkumar and T. Pradeep, Chemistry, 2009, 15, 10110–10120 CrossRef CAS.
- A. M. Hussain, S. N. Sarangi, J. A. Kesarwani and S. N. Sahu, Biosens. Bioelectron., 2011, 29, 60–65 CrossRef CAS.
- P. Khandelwal, D. K. Singh, S. Sadhu and P. Poddar, Nanoscale, 2015, 7, 19985–20002 RSC.
- H. Ding, H. Li, X. Wang, Y. Zhou, Z. Li, J. K. Hiltunen, J. Shen and Z. Chen, Chem. Mater., 2017, 29, 8440–8448 CrossRef CAS.
- J. M. Schnee and W. A. Hsueh, Cardiovasc. Res., 2000, 46, 264–268 CrossRef CAS.
- N. Munoz-Durango, C. A. Fuentes, A. E. Castillo, L. M. Gonzalez-Gomez, A. Vecchiola, C. E. Fardella and A. M. Kalergis, Int. J. Mol. Sci., 2016, 17, 797 CrossRef.
- D. J. Glenn, M. C. Cardema, W. Ni, Y. Zhang, Y. Yeghiazarians, D. Grapov, O. Fiehn and D. G. Gardner, Am. J. Physiol.: Heart Circ. Physiol., 2015, 308, 5 CrossRef.
- W. J. Li, Y. Liu, J. J. Wang, Y. L. Zhang, S. Lai, Y. L. Xia, H. X. Wang and H. H. Li, Life Sci., 2016, 149, 18–24 CrossRef CAS.
- Q. Wang, Y. Yu, P. Zhang, Y. Chen, C. Li, J. Chen, Y. Wang and Y. Li, Basic Res. Cardiol., 2017, 112, 47 CrossRef.
- H. J. Nieminen, T. Ylitalo, S. Karhula, J. P. Suuronen, S. Kauppinen, R. Serimaa, E. Hæggström, K. P. H. Pritzker, M. Valkealahti, P. Lehenkari, M. Finnilä and S. Saarakkala, Osteoarthr. Cartil., 2015, 23, 1613–1621 CrossRef CAS.
- C. M. Disney, K. Madi, A. J. Bodey, P. D. Lee, J. A. Hoyland and M. J. Sherratt, Sci. Rep., 2017, 7, 16279 CrossRef CAS.
- L. Han, S. S. Karhula, M. A. Finnilä, M. J. Lammi, J. H. Ylärinne, S. Kauppinen, L. Rieppo, K. P. H. Pritzker, H. J. Nieminen and S. Saarakkala, PLoS One, 2017, 12, e0171075 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05215a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |