
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvances in single-cell multi-omics profiling
Dongsheng
Bai
a,
Jinying
Peng
*a and
Chengqi
Yi
 *abc
*abc
aState Key Laboratory of Protein and Plant Gene Research, School of Life Sciences, Peking University, Beijing, 100871, China. E-mail: chengqi.yi@pku.edu.cn; jypengpku@pku.edu.cn
bPeking-Tsinghua Center for Life Sciences, Peking University, Beijing, 100871, China
cSynthetic and Functional Biomolecules Center, College of Chemistry and Molecular Engineering, Peking University, Beijing, 100871, China
First published on 20th January 2021
Abstract
Single-cell profiling methods are developed to dissect heterogeneity of cell populations. Recently, multiple enzymatic or chemical treatments have been integrated into single-cell multi-omics profiling methods with high compatibility. These methods have been verified to identify rare or new cell types with high confidence. Single-cell multi-omics analysis can also provide tools to solve the complex regulatory network associated with genome coding, epigenome regulation, and transcripotome expression in a single cell. However, acquiring high-quality single-cell data still faces inherent technical challenges, and co-assays with some other layers of cell identify such as transcription factors binding, histone modifications etc., profiles need new technological breakthroughs to facilitate a more thorough understanding of a single cell. In this review, we summarize the recent advances of single-cell multi-omics methods and discuss the challenges and opportunities in this filed.
Introduction
The phenotypic diversity of organisms is mirrored by genomic heterogeneity at the single-cell level. Various dynamic heritable contexts drive the spatiotemporal expression of genes. Comprehensive genome or transcriptome mapping in different cell types and tissues using bulk approaches has established a brand new field for understanding cell biology and human diseases; however, undefined or rare cell types involved in complex combinations of tissues or biological processes require genetic and genomic perspectives at the single-cell level.Single-cell methods could reveal behaviors that have been hidden by bulk approaches. Dynamic changes in DNA, such as single nucleotide variations (SNV) or copy number variations (CNV), are the driving force of phenotypic diversity in evolution or disease development, and single-cell genome analysis could characterize the genomic heterogeneity of cell populations. Increasing evidence suggests that gene expression is also unique in each cell type and even in individual cells. Transcription is a stochastic biological process that requires more precise profiling to elucidate cell status and the underlying regulatory mechanisms of gene expression, which can be achieved by single-cell transcriptomics analysis.
Diverse cell phenotypes are translated from almost identical genome content via specific spatiotemporal gene expression; “the power of many” is derived from different physical and chemical states of genomic DNA sequences. Epigenetics aims to link modifications, associations, and conformations of the genome with transcriptomic status. Understanding trans-generational heritable memory and gene expression regulatory mechanisms largely relies on profiling the epigenetic status, including covalently modified DNA, RNA, histones, their regulators (writers, readers, erasers), transcription factor binding, and high-dimensional structures of assembled chromatin. Multiple layers perform in concert to ensure sophistication and exquisiteness. Similar to genomic or transcriptomic profiles, the average signals acquired from bulk approaches are limited in some applications. Single-cell epigenetics analysis opens a new avenue to illustrate the link between near-identical genetic codes and diverse expression outputs.
Over the past decades, tremendous progress including the innovation of sequencing platform has supported the rapid advancement of genomics and epigenomics, especially the next generation sequencing (NGS) technologies. There are several sequencing platforms with various throughput, library structure and cost including SOLiD, Ion Torrent, BGI, and Illumina. Illumina instruments have been the most popular platform to process and generate massive data sets that leading to a comprehensive understanding of genome coding, transcription correlated with phenotypic diversity.
Since the first performance of single-cell RNA-seq in 2009,1 many other single-cell sequencing methodologies have been developed for the individual profiling of multiple “omics.”2
Single-cell methods are employed to distinguish between different cell types in large cell populations such as mammalian brain tissues. Single-cell RNA sequencing and single-cell DNA methylome sequencing have been applied to identify cell subtypes in important biological processes or complex tissues. However, methods targeting the individual layers of a cell's identity cannot provide reliable and detailed classification within complex cell populations, since different layers have diverse dynamic or stable behaviors even under the same conditions.
Recently, multi-omics profiling of the genome, transcriptome, and epigenome has taken single-cell analysis to the next level, providing more robust approaches for the classification of different cell types. Parallel profiles of multiple layers help to disentangle the cell type-specific causal networks between or among genome coding, epigenome regulation, and transcriptome expression.
Main body
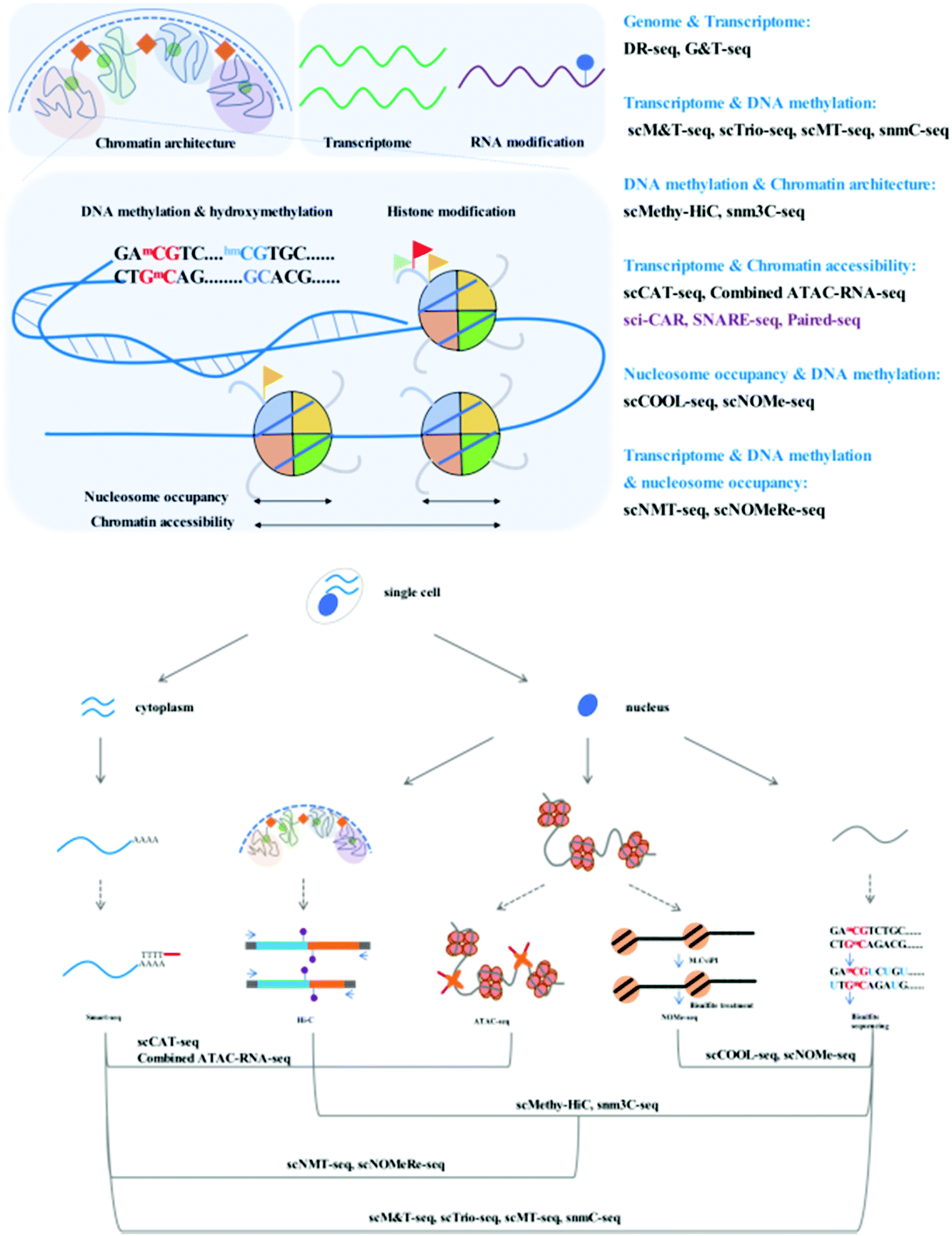
Through adaption or combination of current single-cell analysis assays, single-cell multi-omics integrates multiple treatments or reactions with high compatibility and efficiency (Fig. 1).Simultaneous analysis of the genome and transcriptome
A central question in the field of cell biology is how genotypes affect phenotypes. Direct measurement of the genome and the transcriptome in parallel is required to clarify the quantitative correlation between single-cell genetic variations and gene expression.Single-cell genomics and transcriptomics are developing rapidly, and the many technical challenges associated with these single-cell analyses, including biomolecule isolation, whole-genome amplification (WGA), and data analysis, have been well addressed. Whole-genome amplification is a critical technical challenge of single-cell genome sequencing methods. Pure PCR methods [such as degenerate oligonucleotide-primed (DOP)-PCR], isothermal methods [such as multiple displacement amplification (MDA)], and hybrid methods [such as multiple annealing and looping-based amplification cycles (MALBAC)] display different performances with respect to genome coverage, uniformity, and error rate.3 MALBAC, a widely accessible single-cell WGA method, boasts uniform and high genome coverage with a limited error rate due to the introduction of quasilinear preamplification, which reduces the bias from nonlinear amplification.4 The development of single-cell RNA-seq has enabled the detection of subtle differences in gene expression among single cells within the same population. Common steps involved in the single-cell RNA-seq workflow include cell lysis, reverse transcription, second-strand synthesis, and cDNA amplification. Smart-seq2 has been developed for single-cell RNA analysis with improved transcript capture efficiency.5 In comparison with the previous version, Smart-seq,6 this modified workflow includes a series of improvements associated with reverse transcription, template switching, and preamplification. Owing to its high versatility, Smart-seq2 has been widely integrated into many multi-omics techniques as an RNA library construction tool. Moreover, DR-seq (gDNA–mRNA sequencing) and G&T-seq (genome and transcriptome sequencing) have been developed to simultaneously profile the genome and transcriptome by integrating available single-cell WGA and single-cell RNA sequencing methods.7,8
To avoid unnecessary loss and contamination in DR-seq, genomic (g)DNA and messenger (m)RNA are released and amplified in one tube without physical separation. Following hand-picked single-cell lysis and reverse transcription using a specific adaptor, gDNA and complementary (c)DNA are amplified using the quasilinear amplification strategy. The DNA amplicons are randomly divided into two tubes and separated gDNA and mRNA libraries are constructed. As a result of the no-separation preamplification strategy, computational masking of coding regions must be performed during DR-seq to determine CNV. Conversely, during G&T-seq, gDNA and mRNA are physically separated after cell lysis, and amplification is carried out in different tubes. Modified Smart-seq2 with mRNA separation and first-strand synthesis on beads enables whole transcriptome full-length profiling, whereas the gDNA library from a single cell can be prepared using the whole genome amplification method of choice.
Both G&T-seq and DR-seq can be applied to studying how chromosomal aneuploidies and interchromosomal fusions influence gene expression at the single-cell level. In comparison with DR-seq, G&T-seq has the potential for loss of DNA or mRNA molecules but enables sequencing of full-length transcripts and allows the possibility of automation for high-throughput processing.
Simultaneous analysis of the epigenome and transcriptome
Epigenetics are recognized to play regulatory roles in the transcriptomic status via dynamic modification patterns within the genome. Simultaneous profiling of the single-cell transcriptome and epigenome has been developed, the technical challenges of which are similar to those mentioned above.DNA methylome and transcriptome
Gene expression in eukaryotes is controlled by several mechanisms, of which DNA methylation is a common and critical epigenetic modification in numerous cellular processes. Bisulfite sequencing is the tool of choice for profiling the DNA methylome since it has single-base resolution and quantitative ability. Recent advances have established single-cell methylome profiling methods based on reduced representative (RRBS)9 and post-bisulfite adaptor tagging (PBAT)10 bisulfite sequencing strategies. Using the PBAT strategy, single-cell bisulfite sequencing (scBS-seq)11 has been designed to cover as many whole-genome CpG sites as possible, while single-cell RRBS12 captures informative CpG sites in regulatory elements, which is more cost-effective.The relationship between DNA methylation and the transcriptome in the same cell requires direct measurement for clear demonstration. There exist many powerful tools that have been developed to simultaneously study DNA methylation and RNA profiles. Parallel profiling of the methylome and transcriptome in single cells has been used to investigate the correlation between mRNA transcription and DNA methylation in regulatory elements or gene bodies.
scM&T-seq (single-cell methylome and transcriptome sequencing) is the first reported method that combines DNA methylation and RNA profiling assays.13 Similar to G&T-seq, scM&T-seq separates gDNA and RNA from a single cell. Bisulfite treatment of gDNA can be performed without affecting RNA integrity, and scBS (single cell bisulfite sequencing) and single-cell RNA libraries are subsequently prepared as previously described in scBS-seq and Smart-seq2 with slight modifications.
Similar quality data to those from individual single-cell profiling assays can be attained with scMT-seq (single-cell methylome and transcriptome sequencing)14 and scTrio-seq (single-cell triple omics sequencing technique),15 which isolate an intact nucleus from the cytoplasmic fraction and use it to prepare a bisulfite library using a modified scRRBS workflow, while the cytoplasmic RNA library is prepared by Smart-seq2. Both scMT-seq and scTrio-seq also have similar limitations. Although scRRBS is a mature, cost-effective assay that reliably covers informative CGIs, it can only cover approximately 1% of CpG sites across the whole genome, while scM&T-seq has a much wider coverage owing to the use of the single-cell whole-genome bisulfite sequencing strategy. Wider coverage of DNA CpG methylation status results in a more comprehensive analysis in conjunction with RNA transcriptomics. The existing methods mentioned above perform physical separation of gDNA and RNA to allow joint profiling of the DNA methylome and transcriptome by preparing parallel sequencing libraries. One recently reported method, mCT-seq (methylcytosine and transcriptome sequencing)16 partitions RNA and DNA molecules by modified reverse transcription of RNA and amplification of cDNA with 5-methyl-dCTP incorporation, which requires no physical manipulation. Fully methylated cytosines in RNA-originating amplicons can be distinguished from gDNA that contain unmodified cytosines. snmCT-seq can be applied to single cells or single nuclei with a ruptured cell membrane.
 | ||
| Fig. 1 Assays for single-cell multi-omics profiling developed to profile DNA sequences, gene expression and epigenetic information layers simultaneously at the single-cell level. Single-cell multi-omics methods developed for the profiling of “one cell one tube” at a time are highlighted in black, and high throughput methods are highlighted in purple. | ||
Chromatin accessibility and the transcriptome
Chromatin is dynamically compacted or remodeled during different cellular programming events. Chromatin accessibility is measured by physical contact frequency and intensity of DNA chromatin with transcription factors, transcription machinery, and other DNA binding proteins. The interactive network of accessible chromatin across the whole genome regulates gene expression via defined regulatory elements such as promoters, enhancers, insulators, and other transcription factor binding sites. Mapping chromatin accessibility is a powerful tool for the dissection of cell heterogeneity and the delineation of candidate regulatory elements. Many epigenomic techniques, such as ATAC-seq,17 DNase-seq,18 and MNase-seq,19 have been developed to profile the chromatin accessibility landscape and physical contacts. These assays selectively digest and map exposed or nucleosome-bound DNA regions; however, it is important to keep in mind that only a merged snapshot of dynamic chromatin architecture averaged from thousands of cells can be obtained. DNase-seq and MNase-seq require a large number of cells and possess technical or principle challenges with respect to single-cell analysis. ATAC-seq is the easiest and fastest method since it contains no sonication, extraction, or enzymatic digestion steps; it uses hyperactive Tn5 transposase to perform simultaneous fragmentation and tagging with pre-loaded sequencing adaptors. The frequency of mapped regions correlates with chromatin accessibility. Most importantly, ATAC-seq is possible using modified protocols performed with single cells.20,21 The combined analysis of accessibility and expression can be used to identify distal regulatory elements that may be involved in driving specific gene expression.A number of methods for parallel profiling of chromatin accessibility and gene expression in a single cell have recently been published; for example, scCAT-seq (single-cell chromatin accessibility and transcriptome sequencing)22 made a combination of single-cell ATAC-seq and single-cell RNA-seq to jointly profile chromatin accessibility and gene expression. This method uses gentle lysis to release mRNA into the supernatant and physically separates cytoplasmic mRNA and nuclear chromatin into different tubes. The Smart-seq2 workflow is employed to construct an RNA sequencing library, and for chromatin accessibility, the standard ATAC-seq workflow is used to construct a sequencing library. Accordingly, a functionally relevant regulatory relationship between cis-regulatory elements and their putative target genes can be elucidated. scCAT-seq may be a promising tool for the characterization of distinct cell states or the identification of new cell types in complex cell populations. Combined ATAC–RNA-seq23 is another assay similar to scCAT-seq, which physically separates the nuclear chromatin and mRNA following Smart-seq2 and ATAC-seq library construction; however, it involves cell fixation and bulk cell tagmentation to reduce cost and simplify the protocol.
Simultaneous profiling of multi-omics in a single cell could provide comprehensive and quantitative results due to the high library preparation efficiency and sequencing depth. However, the current “one by one” strategy suffers from high cost and can profile only tens or hundreds of cells with the increased workload, thus imposing limitations on the feasibility of measuring complex tissue samples (such as brain neurons) consisting of highly heterogeneous cell populations. Improving the throughput of single-cell profiling is necessary to obtain a robust view of cell type composition in the context of complex samples.
To achieve high-throughput single-cell analysis, the first step is to uniquely label each cell. The single-cell combinatorial indexing (sci) method24 has been developed to index thousands to millions of cells using a combinatorial nucleic acid barcoding strategy. The sci method is compatible with current low-throughput single-cell profiling assays such as scATAC-seq20 and scRNA-seq.25 Recently, several high-throughput single-cell chromatin states and gene expression joint analysis methods have been developed which can provide deep insight into the diversity among tissues or heterogeneity of individual cells (Fig. 2).
sci-CAR (combinatorial indexing based coassay that jointly profiles chromatin accessibility and mRNA)26 is the first method to jointly profile chromatin accessibility and gene expression using a single-cell combinatorial indexing strategy, and is a successful combination of the previously published sci-ATAC-seq and sci-RNA-seq. In the sci-CAR workflow, the extracted nuclei are distributed in plate wells; the first RNA-seq index is introduced by reverse transcription and the first ATAC-seq index is introduced by Tn5 transposase-mediated tagmentation. After “pool and split” treatment, the first-round indexed nuclei are randomly sorted into plate wells. cDNA is synthesized and nuclei are lysed, after which the lysate is split for separate RNA-seq and ATAC-seq library construction. The second-round indexing is introduced by PCR using indexed primers. Each cell will have a unique combination of indexes, which allows for single-cell analysis. Following assessment of the proof of principle in a well-established tissue culture model, sci-CAR can be applied to the identification of different cell types within tissues using transcriptomics and chromatin accessibility profiles, establishing a correlation between cis-regulatory elements and their target genes in a single cell. Paired-seq27 is another recently developed method for high-throughput joint profiling of accessible chromatin and gene expression. In comparison with sci-CAR, paired-seq introduces one modified combinatorial indexing strategy based on multiple-round ligation and enables unique labeling of millions of cells during a single experiment, which is much higher throughput. Nuclei are distributed in separate wells with Tn5 preloaded with first-round barcodes, and tagmentation then introduces the first barcode into chromatin DNA, and reverse transcription introduces the first barcode into cDNA. Multiple rounds of barcoding ligation are performed using the “split and pool” strategy. To construct separate sequencing libraries, a clever “library dedication” strategy is employed to separately amplify the ATAC-seq and RNA-seq libraries. Paired-seq has been applied to study complex mouse forebrain tissues during different developmental stages, identifying major cell types in the context of these tissues and uncovering the dynamic cell composition during fetal development.
Another strategy for improving the throughput of single-cell profiling, such as SNARE-seq (droplet-based single-nucleus chromatin accessibility and mRNA expression sequencing), can be implemented on the micro-droplet platform, which uses a microfluidic device to compartmentalize droplets containing a single cell, lysis buffer, enzymes, and microbeads modified with barcoded primers on its surface. In SNARE-seq,28 compartmentalization is followed by Tn5 capture in permeabilized nuclei; nuclei in droplets are lysed, and tagmented chromatin DNA and mRNA are released for capture by barcoded beads. The RNA-seq and ATAC-seq libraries can be constructed, sequenced, and matched using their shared cell barcode. In comparison with sci-CAR and paired-seq, SNARE-seq has a similar performance to other single-nucleus RNA-seq methods and enables capture of exposed chromatin DNA of a much wider coverage.
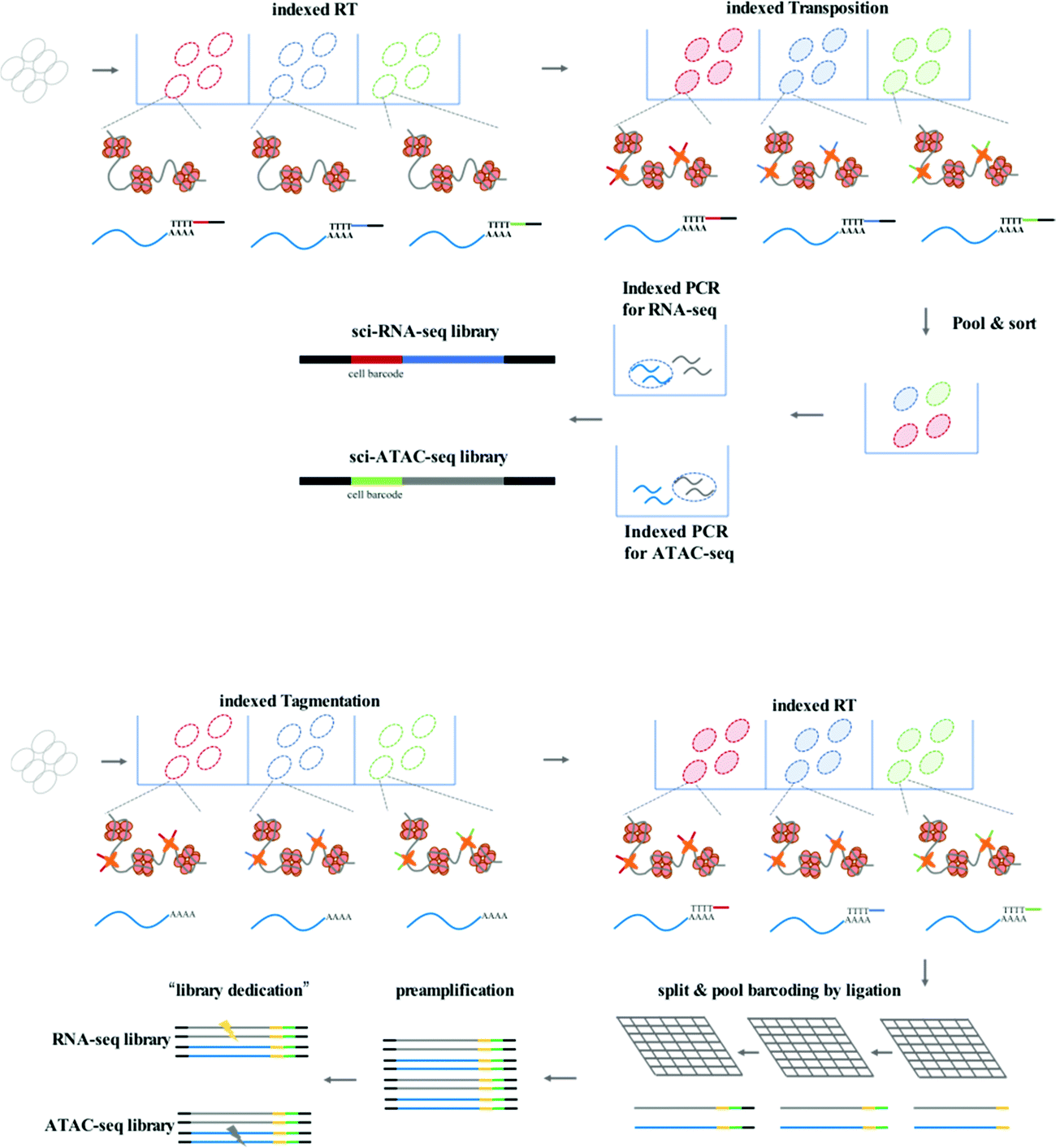 | ||
| Fig. 2 Workflow of sci-CAR and Paired-seq. | ||
Simultaneous analysis of different epigenetic layers
We have summarized single-cell profiling methods that can simultaneously profile the DNA methylome and transcriptome, in addition to chromatin accessibility. These methods aim to uncover the relationship between epigenetic regulatory signals and gene expression. To achieve simultaneous measurement of different epigenetic layers and dissect their crosstalk, many single-cell or single-nucleus multi-epigenomics sequencing methods have been published with a view to providing the opportunity to understand the relationship between DNA methylation and chromatin state.NOMe-seq (nucleosome occupancy and methylome sequencing)29 is a high-resolution nucleosome positioning and DNA methylation joint profiling assay. Single-cell COOL-seq (Chromatin Overall Omic-scale Landscape Sequencing)30 has been combined with NOMe-seq and PBAT-seq,10 and the necessary modifications have been made to improve the robustness at the single-cell level.
Following in vitro methylation in single nuclei with GpC methyltransferase, the released chromatin DNA is subjected to the single-cell PBAT workflow. Another method, scNOMe-seq,31 has a similar workflow to that of scCOOL-seq. Since GpC dinucleotides are not methylated in vivo, CpG methylation can be distinguished from GpC methylation in nucleosome-free regions, which is artificially introduced. Combining GpC and CpG sequencing results, the profiles of the CpG methylation pattern and nucleosome-free regions with GpC methylation can be identified. The sparse coverage of single-cell sequencing methods is a limitation of single-cell ATAC-seq;20,21 the true nucleosome-free regions cannot be distinguished from negative signals derived from read drop-out. Single-cell NOMe-seq can distinguish between different nucleosome-occupancy states independent of the recovered reads or allelic drop-out events.
These multimodal epigenetic analyses have been further adapted to achieve tri-omics with transcriptome sequencing. snNMT-seq (single-cell nucleosome, methylation and transcription sequencing)32 enables the joint analysis of chromatin accessibility, DNA methylome, and transcriptome. This method combines scM&T-seq13 and single-cell NOMe-seq31 mentioned above; the cytoplasmic RNA and nuclei are physically separated, the RNA is subjected to the Smart-seq2 workflow to construct the RNA library, and the nuclei are used to measure chromatin accessibility and DNA methylation using NOMe-seq at the single-nucleus level. Moreover, scNOMeRe-seq (single-cell nucleosome occupancy, methylome and RNA expression sequencing)33 provides yet another choice for the achievement of simultaneous analysis of chromatin accessibility, DNA methylome, and transcriptome. It combines scNOMe-seq with multiple annealing and dC-tailing-based quantitative single-cell RNA sequencing (MATQ-seq),34 which involves multiple annealing and looping-based amplification cycles (MALBAC) and operates with high sensitivity and accuracy at the single-cell level.
The function of chromatin or chromosomes is to efficiently fold and hierarchically package long DNA into a small nucleus. The chromosomal organization within the tiny nucleus has been linked to transcription, DNA replication, and repair. Now, chromosomal conformation capture techniques can be used to assess the three-dimensional chromosomal architecture with different range-scale contacts. Hi-C35 is a powerful method that measures pairwise contact frequencies from the whole genome via high-throughput sequencing. Hi-C data from bulk samples have enhanced our understanding of the general principles of organization and function of the 3D genome. However, it is important to keep in mind that these Hi-C data only provide average signals from large cell populations, and nuclear organization is not uniform even within the same cultured cell lines based on microscopy analysis. With the development of single-cell techniques, single-cell Hi-C36 has been designed to capture a snapshot of one cell at a time to reflect the dynamic behaviors among different cell types or different phases of the cell cycle.
Recent efforts regarding single-cell contact profiles have focused on reconstructing the 3D genomic architecture of different cell types; however, it is difficult to make a clear judgment whether single-cell contact profiles are suitable for the identification of cell types. Single-cell methylome analysis has been proven to possess such cell-type classification ability. snm3C-seq (single-nucleus methyl-3C sequencing)37 and scMethyl-HiC38 have been developed to verify the robustness of single-cell 3C or Hi-C data for cell type-dependent signatures via the simultaneous profiling of DNA methylation and capture of chromosome conformation maps. Fixed nuclei are treated as described for the in situ 3C39 or in situ Hi-C35 workflows, the ligated single nuclei are sorted and subjected to digestion and bisulfite treatment, and the recovered DNA is processed using an efficient single-cell DNA methylome sequencing method. The results provide an indication that single-cell Hi-C profiles display high confidence in cell-type classification but are highly dependent on genome coverage and bioinformatics processing.
Challenges and opportunities
“Omics” aims to simultaneously profile and analyze a wealth of biomolecules including genome, transcripotome, epigenome, proteome and metabolome, these different kinds of “omics” constitute a whole picture of cells, tissues and organs. There have been professional reviews40,41 on the proteome and metabolome which are mainly dependent on progress of analytical chemistry methods such as mass spectrum, while genome, transcripotome and epigenome within these genetic code materials largely benefited from advances in sequencing technologies. Recently, there have been many technologies developed for individual or multiple omics sequencing with fine-grained picture of single cells.Single-cell multi-omics analysis can solve the cell type-specific complex regulatory network associated with the genome, epigenome, and transcriptome; however, acquiring high-quality single-cell sequencing data faces inherent technical challenges. Currently, single-cell analysis methods suffer from a severe “drop-out” phenomenon, meaning allele drop-out for DNA-seq and negative mRNA detection for RNA-seq (Fig. 3a). This sparsity of genome or transcriptome coverage hinders downstream analysis. The major reasons are loss of nucleic acid material during treatments and amplification bias due to trace input from a single cell. As a result of the low genome coverage and high signal noise/error, some single-cell analysis methods have limited compatibility with other single-cell workflows. Thus, there is no reliable multi-omics method for joint analysis involving histone modifications and other DNA modifications except 5mC, since no single-cell Chip-seq methods were yet available. Single-cell analysis of histone modifications and other transcription factor (TF) binding is challenging; however, recently published single-cell ItChip-seq has been developed to profile histone modifications and TF binding regions, providing a tool to elucidate another important epigenetic layer of information associated with transcriptional regulatory networks.42 The feasibility of multi-omics analysis, including histone modification or other TF binding sites analysis, can be studied (Fig. 3b).
DNA methylation and demethylation play important roles in mammalian development and disease. The cycle of DNA methylation and demethylation involves several intermediates: 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-caboxylcytosine (5caC). Recently, several bisulfite-free methods have been developed for profiling DNA epigenetic modifications.43–47 In comparison with traditional bisulfite treatment-based methods, bisulfite-free methods use mild enzymatic or chemical reactions and may show high compatibility with other omics sequencing methods.48 Parallel analysis of 5mC and its oxidized derivatives can facilitate understanding of the mechanism of active demethylation and its regulatory role in development or disease. Chemical tags on gDNA can regulate gene expression without changing the DNA sequences and recently, researchers have found that epigenetic modifications on mRNA are linked to RNA stability and translation.49 The joint analysis of RNA modifications and other omics (such as transcriptomics) may facilitate a more thorough understanding of their biological functions. m6A is the most-studied RNA modification, although the profiling methods used suffer from limited resolution and poor reproducibility. There still exists no single-cell RNA m6A profiling method due to the lack of an efficient and mild enzymatic or chemical transition reaction for m6A.
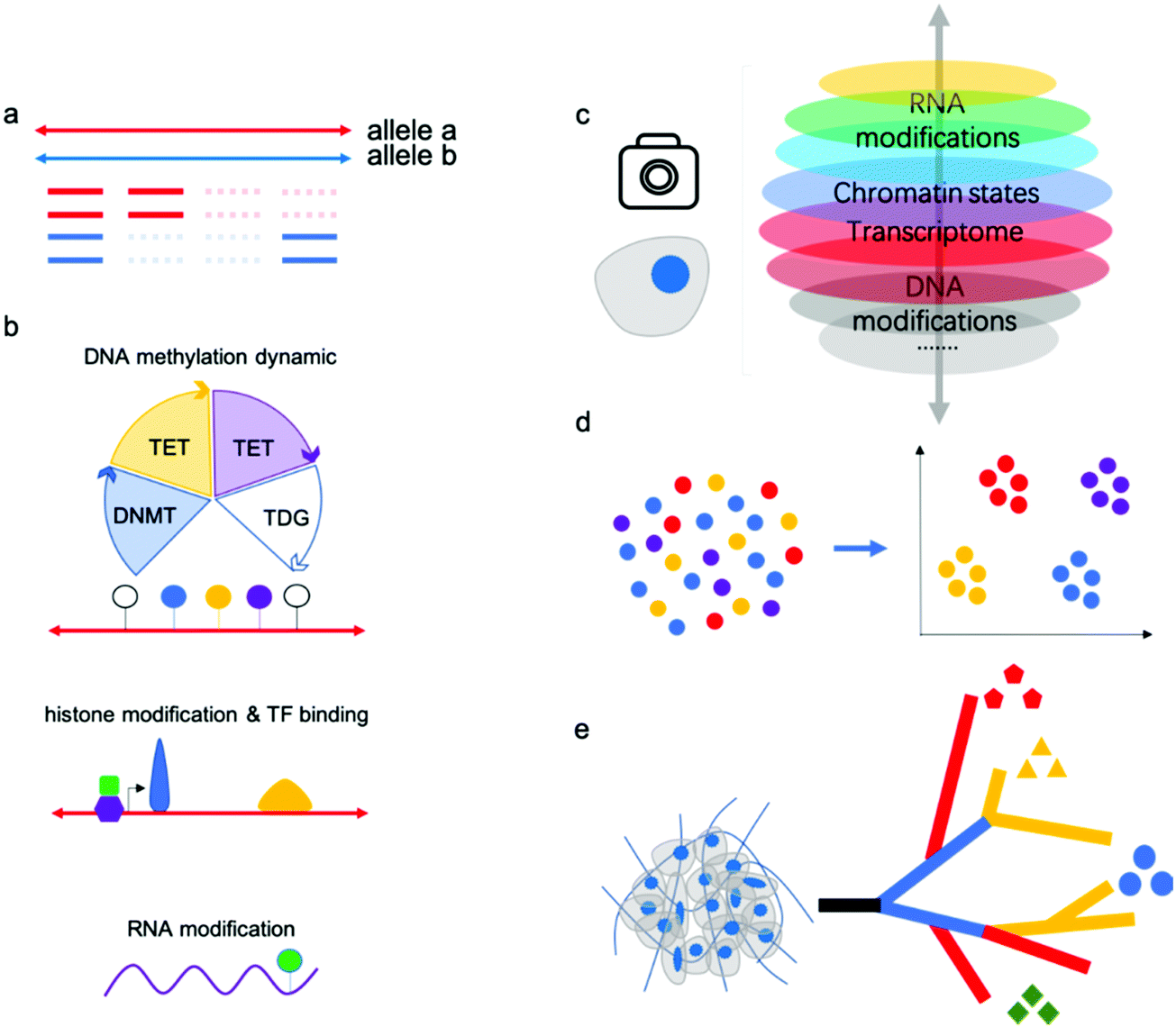 | ||
| Fig. 3 (a) Single-cell analysis methods suffer from a severe “drop-out.” (b) Co-assays of other layers of cell identity, such as DNA demethylation dynamics, histone modifications or transcription factor binding sites, and RNA modifications, with mRNA sequencing data will enhance the comprehensive understanding of gene expression regulatory mechanism and networks. (c) Single-cell multi-omics analysis can solve the complex regulatory network associated with the genome, epigenome, and transcriptome in a single cell. (d) Single-cell multi-omics analysis has been verified to classify cell subtypes with high confidence. (e) Single-cell multi-omics will assist in elucidating intratumor heterogeneity and tumor evolution by reconstructing cell lineage trajectories. | ||
Many single-cell multi-omics methods have been applied to the study of important biological processes or tissues, such as early embryo development and the mammalian brain atlas. Previous methods such as single-cell RNA sequencing have identified many new cell types in these samples; however, RNA signatures are easily affected by the environment. Epigenetic modifications or chromatin states have been verified to classify cell subtypes with high confidence, and joint analysis will provide even more robust results (Fig. 3c,d).
Further, single-cell methods have also begun to explore key questions related to tumor progression, including cancer cell diversity and evolution, which are difficult to address using bulk approaches. As is well known, cancers are extremely complex with high heterogeneity; single-cell multi-omics will assist in dissecting intratumor heterogeneity and tumor evolution by reconstructing cell lineage trajectories (Fig. 3e).
Conclusion
The rapid development of sequencing technologies has enhanced the study of cell processes, with single-cell multi-omics displaying unprecedented resolution and precision. Recent progress has provided powerful tools to explore the regulation of genome expression associated with genome instability and epigenetic modifications in the same cell, enhancing our understanding of heterogeneity among cell population. To improve the performance of the workflow and maximize the quality of single-cell sequencing results, it is imperative to create new technologies that decrease drop-out, integrate additional layers of cell identity, and have the potential for high-throughput processing. Single-cell co-assays based on next-generation sequencing will further expand our increasingly comprehensive understanding of cells’ biological networks.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Ministry of Science and Technology of China (2019YFA0110900, 2019YFA0802200), the National Natural Science Foundation of China (no. 91953201, 21825701 to C. Y.), and the Fok Ying Tung Education Foundation (no. 161018 to C. Y.).References
- F. Tang, C. Barbacioru, Y. Wang, E. Nordman, C. Lee, N. Xu, X. Wang, J. Bodeau, B. B. Tuch, A. Siddiqui, K. Lao and M. A. Surani, Nat. Methods, 2009, 6, 377–382 CrossRef CAS.
- D. Bai and C. Yi, Small Methods, 2019, 3, 1900137 CrossRef CAS.
- C. Gawad, W. Koh and S. R. Quake, Nat. Rev. Genet., 2016, 17, 175–188 CrossRef CAS.
- C. Zong, S. Lu, A. R. Chapman and X. S. Xie, Science, 2012, 338, 1622–1626 CrossRef CAS.
- S. Picelli, O. R. Faridani, A. K. Bjorklund, G. Winberg, S. Sagasser and R. Sandberg, Nat. Protoc., 2014, 9, 171–181 CrossRef CAS.
- D. Ramskold, S. Luo, Y. C. Wang, R. Li, Q. Deng, O. R. Faridani, G. A. Daniels, I. Khrebtukova, J. F. Loring, L. C. Laurent, G. P. Schroth and R. Sandberg, Nat. Biotechnol., 2012, 30, 777–782 CrossRef.
- S. S. Dey, L. Kester, B. Spanjaard, M. Bienko and A. van Oudenaarden, Nat. Biotechnol., 2015, 33, 285–289 CrossRef CAS.
- I. C. Macaulay, W. Haerty, P. Kumar, Y. I. Li, T. X. Hu, M. J. Teng, M. Goolam, N. Saurat, P. Coupland, L. M. Shirley, M. Smith, N. Van der Aa, R. Banerjee, P. D. Ellis, M. A. Quail, H. P. Swerdlow, M. Zernicka-Goetz, F. J. Livesey, C. P. Ponting and T. Voet, Nat. Methods, 2015, 12, 519–522 CrossRef CAS.
- A. Meissner, A. Gnirke, G. W. Bell, B. Ramsahoye, E. S. Lander and R. Jaenisch, Nucleic Acids Res., 2005, 33, 5868–5877 CrossRef CAS.
- F. Miura, Y. Enomoto, R. Dairiki and T. Ito, Nucleic Acids Res., 2012, 40, e136 CrossRef CAS.
- S. A. Smallwood, H. J. Lee, C. Angermueller, F. Krueger, H. Saadeh, J. Peat, S. R. Andrews, O. Stegle, W. Reik and G. Kelsey, Nat. Methods, 2014, 11, 817–820 CrossRef CAS.
- H. Guo, P. Zhu, X. Wu, X. Li, L. Wen and F. Tang, Genome Res., 2013, 23, 2126–2135 CrossRef CAS.
- C. Angermueller, S. J. Clark, H. J. Lee, I. C. Macaulay, M. J. Teng, T. X. Hu, F. Krueger, S. Smallwood, C. P. Ponting, T. Voet, G. Kelsey, O. Stegle and W. Reik, Nat. Methods, 2016, 13, 229–232 CrossRef CAS.
- Y. Hu, K. Huang, Q. An, G. Du, G. Hu, J. Xue, X. Zhu, C. Y. Wang, Z. Xue and G. Fan, Genome Biol., 2016, 17, 88 CrossRef.
- Y. Hou, H. Guo, C. Cao, X. Li, B. Hu, P. Zhu, X. Wu, L. Wen, F. Tang, Y. Huang and J. Peng, Cell Res., 2016, 26, 304–319 CrossRef CAS.
- C. Luo, H. Liu, B.-A. Wang, A. Bartlett, A. Rivkin, J. R. Nery and J. R. Ecker, bioRxiv, 2018, 434845 Search PubMed.
- J. D. Buenrostro, P. G. Giresi, L. C. Zaba, H. Y. Chang and W. J. Greenleaf, Nat. Methods, 2013, 10, 1213–1218 CrossRef CAS.
- A. P. Boyle, S. Davis, H. P. Shulha, P. Meltzer, E. H. Margulies, Z. Weng, T. S. Furey and G. E. Crawford, Cell, 2008, 132, 311–322 CrossRef CAS.
- F. Schlesinger, A. D. Smith, T. R. Gingeras, G. J. Hannon and E. Hodges, Genome Res., 2013, 23, 1601–1614 CrossRef CAS.
- D. A. Cusanovich, R. Daza, A. Adey, H. A. Pliner, L. Christiansen, K. L. Gunderson, F. J. Steemers, C. Trapnell and J. Shendure, Science, 2015, 348, 910–914 CrossRef CAS.
- J. D. Buenrostro, B. Wu, U. M. Litzenburger, D. Ruff, M. L. Gonzales, M. P. Snyder, H. Y. Chang and W. J. Greenleaf, Nature, 2015, 523, 486–490 CrossRef CAS.
- L. Q. Liu, C. Y. Liu, A. Quintero, L. Wu, Y. Yuan, M. Y. Wang, M. N. Cheng, L. Z. Leng, L. Q. Xu, G. Y. Dong, R. Li, Y. Liu, X. Y. Wei, J. S. Xu, X. W. Chen, H. R. Lu, D. S. Chen, Q. L. Wang, Q. Zhou, X. X. Lin, G. B. Li, S. P. Liu, Q. Wang, H. R. Wang, J. L. Fink, Z. L. Gao, X. Liu, Y. Hou, S. D. Zhu, H. M. Yang, Y. M. Ye, G. Lin, F. Chen, C. Herrmann, R. Eils, Z. C. Shang and X. Xu, Nat. Commun., 2019, 10(1), 1–10 CrossRef.
- M. Reyes, K. Billman, N. Hacohen and P. C. Blainey, Adv. Biosyst., 2019, 3(11), 1900065(1 of 6)–1900065(6 of 6) Search PubMed.
- S. A. Vitak, K. A. Torkenczy, J. L. Rosenkrantz, A. J. Fields, L. Christiansen, M. H. Wong, L. Carbone, F. J. Steemers and A. Adey, Nat. Methods, 2017, 14, 302–308 CrossRef CAS.
- J. Cao, J. S. Packer, V. Ramani, D. A. Cusanovich, C. Huynh, R. Daza, X. Qiu, C. Lee, S. N. Furlan, F. J. Steemers, A. Adey, R. H. Waterston, C. Trapnell and J. Shendure, Science, 2017, 357, 661–667 CrossRef CAS.
- J. Cao, D. A. Cusanovich, V. Ramani, D. Aghamirzaie, H. A. Pliner, A. J. Hill, R. M. Daza, J. L. McFaline-Figueroa, J. S. Packer, L. Christiansen, F. J. Steemers, A. C. Adey, C. Trapnell and J. Shendure, Science, 2018, 361, 1380–1385 CrossRef CAS.
- C. Zhu, M. Yu, H. Huang, I. Juric, A. Abnousi, R. Hu, J. Lucero, M. M. Behrens, M. Hu and B. Ren, Nat. Struct. Mol. Biol., 2019, 26, 1063–1070 CrossRef CAS.
- S. Chen, B. B. Lake and K. Zhang, Nat. Biotechnol., 2019, 37, 1452–1457 CrossRef CAS.
- T. K. Kelly, Y. Liu, F. D. Lay, G. Liang, B. P. Berman and P. A. Jones, Genome Res., 2012, 22, 2497–2506 CrossRef CAS.
- F. Guo, L. Li, J. Li, X. Wu, B. Hu, P. Zhu, L. Wen and F. Tang, Cell Res., 2017, 27, 967–988 CrossRef CAS.
- S. Pott, eLife, 2017, 6, e23203 CrossRef.
- S. J. Clark, R. Argelaguet, C. A. Kapourani, T. M. Stubbs, H. J. Lee, C. Alda-Catalinas, F. Krueger, G. Sanguinetti, G. Kelsey, J. C. Marioni, O. Stegle and W. Reik, Nat. Commun., 2018, 9, 781 CrossRef.
- Y. Wang, P. Yuan, Z. Yan, M. Yang, Y. Huo, Y. Nie, X. Zhu, L. Yan and J. Qiao, bioRxiv, 2019, 803890 Search PubMed.
- K. W. Sheng, W. J. Cao, Y. C. Niu, Q. Deng and C. H. Zong, Nat. Methods, 2017, 14(3), 267–270 CrossRef CAS.
- E. Lieberman-Aiden, N. L. van Berkum, L. Williams, M. Imakaev, T. Ragoczy, A. Telling, I. Amit, B. R. Lajoie, P. J. Sabo, M. O. Dorschner, R. Sandstrom, B. Bernstein, M. A. Bender, M. Groudine, A. Gnirke, J. Stamatoyannopoulos, L. A. Mirny, E. S. Lander and J. Dekker, Science, 2009, 326, 289–293 CrossRef CAS.
- T. Nagano, Y. Lubling, T. J. Stevens, S. Schoenfelder, E. Yaffe, W. Dean, E. D. Laue, A. Tanay and P. Fraser, Nature, 2013, 502, 59–64 CrossRef CAS.
- D. S. Lee, C. Luo, J. Zhou, S. Chandran, A. Rivkin, A. Bartlett, J. R. Nery, C. Fitzpatrick, C. O'Connor, J. R. Dixon and J. R. Ecker, Nat. Methods, 2019, 16, 999–1006 CrossRef CAS.
- G. Li, Y. Liu, Y. Zhang, N. Kubo, M. Yu, R. Fang, M. Kellis and B. Ren, Nat. Methods, 2019, 16, 991–993 CrossRef CAS.
- Z. Duan, M. Andronescu, K. Schutz, C. Lee, J. Shendure, S. Fields, W. S. Noble and C. Anthony Blau, Methods, 2012, 58, 277–288 CrossRef CAS.
- A. F. Altelaar, J. Munoz and A. J. Heck, Nat. Rev. Genet., 2013, 14, 35–48 CrossRef CAS.
- G. J. Patti, O. Yanes and G. Siuzdak, Nat. Rev. Mol. Cell Biol., 2012, 13, 263–269 CrossRef CAS.
- S. S. Ai, H. Q. Xiong, C. Li, Y. J. Luo, Q. Shi, Y. X. Liu, X. H. Yu, C. Li and A. B. He, Nat. Cell Biol., 2019, 21(9), 1164–1172 CrossRef CAS.
- B. Xia, D. L. Han, X. Y. Lu, Z. Z. Sun, A. K. Zhou, Q. Z. Yin, H. Zeng, M. H. Liu, X. Jiang, W. Xie, C. He and C. Q. Yi, Nat. Methods, 2015, 12, 1047–1050 CrossRef CAS.
- C. X. Zhu, Y. Gao, H. S. Guo, B. Xia, J. H. Song, X. L. Wu, H. Zeng, K. Kee, F. C. Tang and C. Q. Yi, Cell Stem Cell, 2017, 20(5), 720–731 CrossRef CAS.
- H. Zeng, B. He, B. Xia, D. S. Bai, X. Y. Lu, J. B. Cai, L. Chen, A. K. Zhou, C. X. Zhu, H. W. Meng, Y. Gao, H. S. Guo, C. He, Q. Dai and C. Q. Yi, J. Am. Chem. Soc., 2018, 140, 13190–13194 CrossRef CAS.
- Y. B. Liu, P. Siejka-Zielinska, G. Velikova, Y. Bi, F. Yuan, M. Tomkova, C. S. Bai, L. Chen, B. Schuster-Bockler and C. X. Song, Nat. Biotechnol., 2019, 37(4), 424–429 CrossRef CAS.
- E. K. Schutsky, J. E. DeNizio, P. Hu, M. Y. Liu, C. S. Nabel, E. B. Fabyanic, Y. Hwang, F. D. Bushman, H. Wu and R. M. Kohli, Nat. Biotechnol., 2018, 36(11), 1083–1090 CrossRef CAS.
- H. Zeng, B. He and C. Q. Yi, ChemBioChem, 2019, 20, 1898–1905 CrossRef CAS.
- I. A. Roundtree, M. E. Evans, T. Pan and C. He, Cell, 2017, 169, 1187–1200 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |