
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe pursuit of mechanism of action: uncovering drug complexity in TB drug discovery
Tianao
Yuan†
 ,
Joshua M.
Werman†
and
Nicole S.
Sampson
*
,
Joshua M.
Werman†
and
Nicole S.
Sampson
*
Department of Chemistry, Stony Brook University, Stony Brook, NY 11794-3400, USA. E-mail: nicole.sampson@stonybrook.edu; Fax: +1-631-632-5738; Tel: +1-631-632-7952
First published on 13th January 2021
Abstract
Whole cell-based phenotypic screens have become the primary mode of hit generation in tuberculosis (TB) drug discovery during the last two decades. Different drug screening models have been developed to mirror the complexity of TB disease in the laboratory. As these culture conditions are becoming more and more sophisticated, unraveling the drug target and the identification of the mechanism of action (MOA) of compounds of interest have additionally become more challenging. A good understanding of MOA is essential for the successful delivery of drug candidates for TB treatment due to the high level of complexity in the interactions between Mycobacterium tuberculosis (Mtb) and the TB drug used to treat the disease. There is no single “standard” protocol to follow and no single approach that is sufficient to fully investigate how a drug restrains Mtb. However, with the recent advancements in -omics technologies, there are multiple strategies that have been developed generally in the field of drug discovery that have been adapted to comprehensively characterize the MOAs of TB drugs in the laboratory. These approaches have led to the successful development of preclinical TB drug candidates, and to a better understanding of the pathogenesis of Mtb infection. In this review, we describe a plethora of efforts based upon genetic, metabolomic, biochemical, and computational approaches to investigate TB drug MOAs. We assess these different platforms for their strengths and limitations in TB drug MOA elucidation in the context of Mtb pathogenesis. With an emphasis on the essentiality of MOA identification, we outline the unmet needs in delivering TB drug candidates and provide direction for further TB drug discovery.
Introduction
The unmet need for TB drugs and current screening strategies for drug discovery
After almost 70 years since its discovery,1,2 isoniazid (INH) has remained the most clinically successful drug for Tuberculosis (TB) treatment. In 1882 Robert Koch discovered the pathogen responsible for TB, yet in 2020, TB still represents the leading cause of death from a single infectious agent around the world.3 There are an estimated 10 million people globally that developed active TB disease in 2018 and over half a million of those cases were diagnosed with drug resistant TB.3 The increasing emergence of drug resistant Mycobacterium tuberculosis (Mtb) infection and the prolonged drug regimen required to treat drug resistant TB underscore the unmet need in TB drug discovery and development that currently exists.Over the past 50 years, only two new drugs have been approved by the U.S. Food and Drug Administration (FDA) for the treatment of TB. Bedaquiline (BDQ) was approved as a second-line drug for multidrug-resistant TB treatment in 20124 and pretomanid was approved in 2019 as part of a three-drug regimen for the treatment of highly drug resistant TB.5
At the beginning of the genomic era, the focus of antibacterial drug development shifted from traditional cell-based phenotypic screens to enzyme-based biochemical screens. Through the use of large compound libraries, compounds were screened in a high throughput manner for activity against the protein products of essential genes required for growth or survival of the bacterial organism. However, these screening campaigns have not proved very successful at delivering desirable drugs in areas of need.6,7 Thereafter, the focus in the antibiotic drug discovery field has shifted back to whole cell-based phenotypic screens to identify bacterial growth inhibitors directly.8 Notably, both newly approved TB drugs were discovered via whole cell-based phenotypic screens.9,10
The advantages that are provided by whole cell-based phenotypic screening include exclusion of compounds that are unable to penetrate through the hydrophobic mycobacterial cell wall, selection of compounds that inhibit vulnerable cellular targets, selecting hit compounds that target multiple cellular functions, and selecting prodrugs that require intracellular bioactivation.8 In fact, there have been analyses arguing that antibiotics that target a single protein are less successful than those directed against multiple molecular targets.11,12
However, whole cell-based phenotypic screening is considered to be largely empirical. While phenotypic screening is on the whole more efficient in translating compound activity into potential therapeutic impact, the mechanisms of action of these compounds are not immediately revealed. Compound libraries used in phenotypic screens have evolved to include compounds of greater structural diversity, for example, there has been renewed interest in natural products for their antibacterial properties. As new technologies have become available, screening strategies have become more comprehensive, leading to the identification of a greater number of hit compounds with novel structures and mechanisms. However, without consideration of an optimal mechanism of action (MOA), drug discovery programs generally carry high attrition rates downstream.13 Attrition occurs because not all essential targets in laboratory-grown bacteria have equal potential to be exploited as antibacterial drug targets in vivo.
In vitro broth media conditions used in whole cell-based phenotypic screens greatly differ from the pulmonary micro-environment where Mtb resides in the host. This difference results in a high risk of selecting compounds that inhibit an essential target in vitro which is dispensable in vivo.14 As a result, different culture models for whole cell-based phenotypic screens have been developed to more closely mimic the environment in which Mtb resides in the host, in the hope of improving drug discovery outcomes.6,8 However, as these culture models increase in complexity, the identification and assignment of MOAs becomes more complex, and requires a longer time to unravel.15
The essential role of MOA identification in successfully delivering TB drug candidates
In order to elucidate the MOA of an inhibitor, a vast amount of work is often required.16 There is no generic method to elucidate the MOA of all inhibitors and usually multiple approaches are required to uncover the mechanisms of a new compound's activity.17Understanding MOA plays an essential role in antibiotic development. Although regulatory approval for antibiotics does not require a clearly defined MOA, there is strong motivation to understand the MOA or to identify the molecular target(s) of an antibiotic for advancing hit compounds and lead optimization.18 Understanding enables exclusion of compounds that elicit bactericidal effects in a non-specific manner. Knowledge of the molecular target(s) also provides a basis for a comprehensive structure–activity relationship (SAR) study to improve activity and pharmacological properties, which can facilitate the rational design and development of next generation leads. This format of drug development simultaneously enables minimization of off-target effects in the host. Understanding MOA helps to lower the risk of compounds failing in the later stages of development due to host interference, general toxicity, and target drugability. As TB is generally treated with a combination of therapeutic agents, it is crucial to understand the molecular targets of each drug used so that an optimized combination may be designed to address the issues of bacterial persistence and drug resistance.19 All of these factors highlight the need and importance of target identification and MOA assignment in antibiotic drug discovery (Fig. 1).
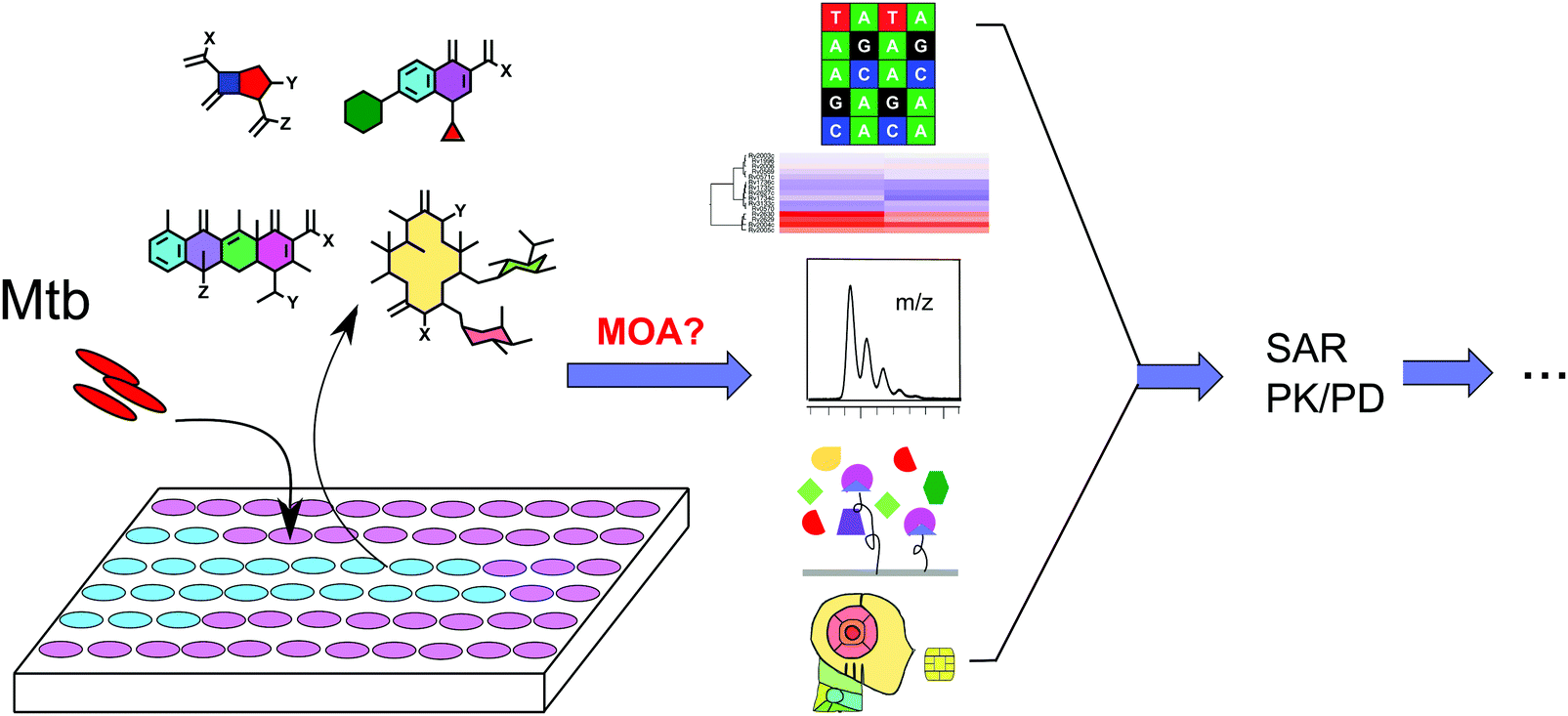 | ||
| Fig. 1 Schematic representation of phenotypic screening-based TB drug discovery. Hit compounds are identified from whole cell-based phenotypic screens. Their MOAs can be assessed through genetic, metabolomic, biochemical, and computational approaches. A clearly defined MOA will lay a good foundation for the following SAR and pharmacokinetics (PK)/pharmacodynamics (PD) studies. | ||
Prior to the genomic era, MOA studies for TB drugs were predominantly conducted through biochemical approaches. As detailed in several reviews outlining the history of TB drug development,20,21 using INH as an example, the majority of MOA discovery focused on examining the intracellular levels of extractable cell components or the production of key metabolites, such as cell wall associated lipids and sugars.2 Serendipity aside, the real breakthrough in TB drug MOA research occurred with the advent of the genomic era when DNA sequencing technology enabled entire genomes to be characterized and the introduction of foreign DNA through plasmid transformation became possible.22–24 These approaches have provided researchers a powerful tool in defining the primary molecular targets and the MOAs of TB drugs.
Benefiting from advancement in -omic technologies, the research strategies involved in the MOA identification of TB drugs have evolved from direct biochemical methods, to genetic interaction, chemical-genetics interaction profiling, metabolomic profiling, and computational machine learning. The integration of all these approaches from compound screening to MOA assessment has yielded over 10 new TB drug candidates that are currently undergoing clinical testing.25
While many such technologies are applied generally in drug MOA elucidation in the entire drug discovery field, in this review, we specifically summarize the methodologies applied in the assessment of MOA in TB drug discovery with an emphasis on Mtb physiology and Mtb infection pathogenicity. We highlight the latest technologies and advances for target identification in the field and discuss comparative advantages and disadvantages of each approach. In doing so, we hope to stimulate more innovative strategies to deliver drug candidates in TB drug discovery and expand our understanding of Mtb pathogenicity.
Spontaneous resistant mutants and WGS
Acquired drug resistance in Mtb
When streptomycin was introduced into the TB treatment regimen in the 1940s, the emergence of Mtb drug resistance in patients was quickly observed.26 While intrinsic drug resistance plays a role in mediating antibiotic-induced stress in Mtb, acquired drug resistance accounts for the majority of the molecular mechanisms of drug resistance.27 Acquired drug resistance in Mtb arises predominantly through spontaneous mutations in chromosomal genes, whereas mobile horizontal gene transfer (HGT), a common source of acquired drug resistance in many bacteria, is rare in Mtb.28Spontaneous mutations arise in all prokaryotes at a constant rate of 0.0033 times per replication.29 While most of the individual cells carrying mutations do not survive, the ones that persist provide advantages, which allow the organism to more effectively adapt to the environment, and therefore accumulate in the population. In Mtb specifically, antibiotic resistance occurs at a rate of 10−7–10−9 mutations per cell division.30 Even though the causes and mechanisms for acquired drug resistance are different, isolating drug resistant colonies and following with genetic analysis provides a starting point for drug MOA hypotheses.
Unraveling drug antibacterial mechanism through genome analysis
Mutations usually occur in the form of single nucleotide missense changes, premature termination, nonsense mutations, or frameshift mutations which disrupt the bactericidal effect of antibiotics. The mechanisms for antibiotic resistance can be summarized into seven categories.31,32 They are a gene mutation that results in:• reduced binding of the drug but preserves function of the target
• increased expression of the target
• modified or increased catabolism of the drug
• decreased activation of the prodrug
• decreased drug uptake or increased drug export
• expression of a compensatory pathway
• loss of function in an enzyme that undoes the action targeted by the drug.
With advancements in whole genome sequencing (WGS) technology, raising spontaneous resistant mutants in vitro on solid media has become the most common and straightforward approach to study the molecular mechanisms of TB drugs (Fig. 2). This approach, which can be widely applied, provides a high throughput platform to study the MOAs of the hits that arise from such screens.33 Mutations that confer resistance then need to be introduced into the mycobacteria through a variety of genetic approaches34 and the drug resistance of the engineered strain must be confirmed to have a similar drug phenotype to the spontaneously resistant strain. Other genetic techniques, including conditional expression of a target gene can be applied to further validate the interaction between the drug and its putative target.35
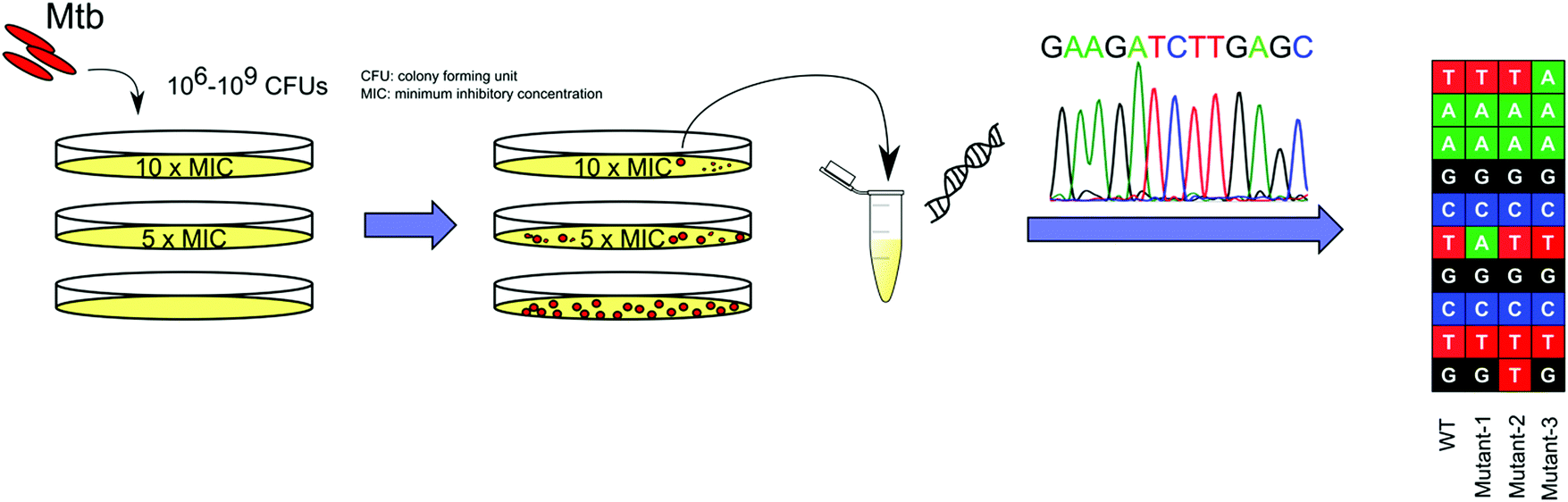 | ||
| Fig. 2 Schematic representation of WGS of spontaneous drug resistant mutants for TB drug MOA identification. 106–109 CFUs of Mtb are plated onto agar plates containing a range of concentrations of the drug, from 2×–10× MIC, for 8–10 weeks at 37 °C. Drug resistant colonies are picked and cultured in broth media, followed by the extraction of genomic DNA for WGS. CFU: colony forming unit. MIC: minimum inhibitory concentration. | ||
INH was identified in the 1950s.36 However, even with many meticulously planned biochemical experiments examining the mode-of-action of INH, it would not be until the late 1990s to early 2000, through the use of INH resistant Mtb strains, that researchers were able to reveal the MOA of INH. The Mtb catalase peroxidase KatG activates INH to an isonicotinoyl radical which forms an adduct with NAD, and the INH–NAD adduct inhibits InhA, the enoyl-ACP reductase of the fatty acid synthase type II system.2 BDQ was the first TB drug approved by the FDA in nearly 50 years. The primary target of BDQ, the ATP synthase encoded by atpE, was identified from WGS of in vitro generated resistant strains.9
Challenges in TB drug MOA assignment through resistant mutant generation
Due to the different mechanisms of drugs, and the essentiality and vulnerability of molecular targets or cellular machinery, spontaneous mutants are often difficult to generate. Mutants also may be only partially resistant to the drug thus the mutations may not indicate the direct molecular targets of the drugs. Moreover, non-specific mutations in the genome are commonly observed, further complicating identification of the molecular target. In many scenarios, a TB drug can work through a multifaceted mechanism, disrupting multiple biological pathways. As a result, the mutations may not point to a single target. Resistance mutations of prodrugs often only occur in the gene encoding the bioactivating enzyme, leaving the real target unidentified, such as the case for the newly approved TB drug pretomanid.37For compounds with MOAs targeting multiple proteins, depending on the resistance rate of each target, detecting spontaneously resistant strains can be more difficult. This difficulty is most likely due to the low probability of multiple protective mutations occurring in a single cell within a manageable population size. First described in the 1950's, a method to induce mutations chemically by addition of a mutagen, such as 5-bromouracil, enables elevation of the mutation rate within the population, thereby increasing the likelihood of isolating drug resistant strains.41 This approach was applied in a recent study to unravel the molecular target of a class of quinoline compounds in Mtb, where with elevated mutation rates, single nucleotide polymorphisims (SNPs) were identified in Rv2439c which encodes a glutamate kinase in proline biosynthesis.42 The quinoline compounds function by activating the glutamate kinase, which leads to an overproduction of proline, and the associated redox imbalance causes bacterial death.
It is worth noting that not all mutations that rescue mycobacteria from antibiotic treatment occur in an essential gene. A recent phenotypic screen identified a class of 4-hydroxyquinolines for their bactericidal activities against both replicating and non-replicating Mtb. Mechanistic investigations under replicating conditions revealed that SNPs in the non-essential gene Rv1239c encoding a putative Mg2+/Co2+ transporter CorA in Mtb could rescue the bacteria from compound treatment in replicating conditions.43 Transposon disruption of Rv1239c or direct chemical inhibition of CorA did not affect Mtb viability indicating CorA was not the molecular target of the 4-hydroxyquinolines. Instead, subsequent experiments showed that the compounds functioned by depleting the intracellular Mg2+ and the altered function of CorA might favor the Mg2+ influx into the bacteria to mitigate the bactericidal activity. However, SNPs in Rv1239 were not able to protect Mtb from 4-hydroxyquinoline treatment under non-replicating conditions suggesting a different MOA. WGS of drug resistant strains raised under non-replicating conditions did not yield informative results reflecting the limitation of using resistance mutants to rationalize bacterial chemical phenotypes.
Moreover, our understanding of Mtb's genome is still limited; of 4000 genes in the Mtb genome, 25% encode hypothetical proteins with unknown biochemical functions.44 Thus, it is challenging to pinpoint the MOA of an individual compound solely relying on resistant mutant sequencing. As the puzzles of many drug mechanisms remain unsolved, researchers need to employ more comprehensive and multifaceted approaches to answer the complex questions posited in/by target validation.
Transcriptional profiling
Bacterial transcriptional adaptation to antibiotic perturbations
When generating resistant mutants is not possible or useful, what other genetic approaches can be applied to uncover mechanistic information? Bacteria often respond to chemical or physical perturbations by altering the level or activity of transcription factors and initiate rewiring of cellular regulatory networks.45,46 The alteration of the transcriptome often relies on DNA binding proteins, activators or repressors, which interact with a specific DNA sequence in promoter regions. Genome wide transcriptional responses of Mtb to an antibiotic can be used as a supplementary tool to understand the expression level of key genes or transcription of critical metabolic pathways, which are critical for adaption to the disruption caused by antibiotic treatment. This information helps to reveal the primary target and the events downstream of primary target inhibition that actively contribute to drug action. As such, transcriptional profiling has been widely utilized to predict and validate the MOA of TB drugs during lead optimization47,48 and has been utilized to understand and predict drug synergy in order to yield better therapeutic outcomes.49Predicting drug MOAs by transcriptional profiling in Mtb
Transcriptional profiling has become a mainstay in TB drug discovery since its introduction to Mtb research in early 2000. Through microarray-based genome wide transcriptional profiling, Barry and colleagues provided an encyclopedic summary of Mtb's transcriptional response to 430 antibiotics and to inhibitory growth conditions.47 This work revealed 150 gene clusters in Mtb's transcriptome that are jointly regulated to adapt to metabolic perturbations. These gene networks serve as signatures that are specific to certain types of metabolic inhibition, such as cell wall synthesis, cell respiration, DNA transcription, and translation. Drawing on the work of Barry et al., with the broad application of RNA-seq technology, transcriptional profiling has been applied to study Mtb's pathogenesis in terms of how Mtb responds and adapts to different growth and stress conditions,50–52 and more importantly, has laid an essential foundation for the use of transcriptional profiling to understand and predict the MOAs of TB drugs (Fig. 3).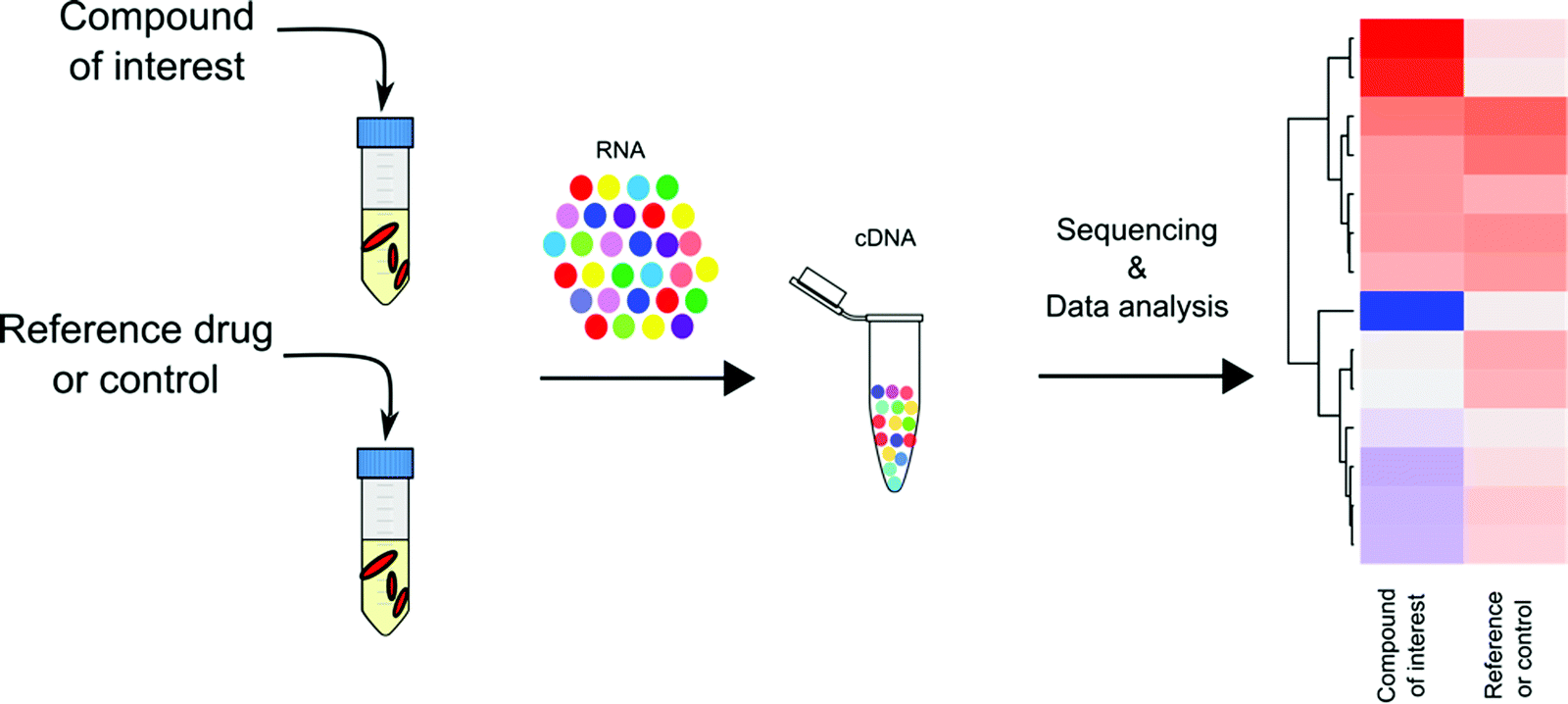 | ||
| Fig. 3 General workflow of transcriptional profiling for TB drug MOA assessment. Mycobacterial total RNA is isolated from Mtb cultures with or without drug treatment and purified RNA is converted to cDNA. The resulting cDNA libraries are subjected to quantitative sequencing analysis for transcriptional profiling. | ||
Advantages of transcriptional profiling in TB drug MOA prediction
When compared to WGS of resistant mutants, transcriptional profiling can reveal additional information on the MOA of an antibiotic and its impact on the global mycobacterial transcriptome. Pretomanid works against both replicating and non-replicating Mtb and its activity requires bio-reductive activation of the aromatic nitro group and concomitant nitric oxide release.10,37,53 The bactericidal activity under aerobic conditions is associated with pretomanid's inhibition of mycolic acid biosynthesis.10 To elucidate its killing effect under hypoxia, transcriptional profiles of Mtb under pretomanid treatment were examined.54 Through this approach, cell wall synthesis inhibition was validated and a new aspect of the MOA: inhibition of respiration causing an energy production constraint, was discovered. Upon drug treatment, genes involved in respiration, such as the cyd operon encoding the cytochrome bd complex, were upregulated, and consequently the cellular redox and energy homeostasis were disrupted. These observations led to a proposed mechanism of pretomanid under hypoxia that the nitric oxide released from the nitro bio-reduction of pretomanid poisoned the respiratory electron transport chain and disrupted energy homeostasis.Transcriptional profiling can help decipher how bacteria respond to mediate the bactericidal or bacteriostatic effects caused by antibiotic treatment. As demonstrated in a transcriptional network analysis study of BDQ on Mtb, after 96 h of BDQ treatment, in addition to genes in the ATP synthase operon which is the primary target of BDQ, upregulation of two gene regulators Rv0324 and Rv0880 aid in tolerance of Mtb to BDQ.55 Upon their upregulation, Mtb enters into a BDQ-tolerant state, which manifests as delayed killing during the early stage of drug treatment.56 Genetic disruption of these two regulators accelerated the killing effect of BDQ. Therefore, transcriptional profile analysis revealed two critical genes as nontraditional drug targets that have the potential to reduce the treatment duration of BDQ TB therapy. These two genes would likely be dismissed as drug targets due to their non-essentiality to Mtb growth, yet here their importance for BDQ MOA suggests a potential therapeutic strategy to sensitize Mtb to BDQ.
Small molecules like vitamin C and cysteine have been shown in recent studies to induce enhanced production of reactive oxygen species (ROS) in Mtb, such that vitamin C treatment can sterilize Mtb culture57 and cysteine can sensitize Mtb to INH or rifampicin treatment.58 As demonstrated by Vilcheze et al., these small molecules may not have a primary target in Mtb, instead they rather act in a more general way by disrupting cellular machineries. Transcriptional analysis was used as a primary approach to investigate the mechanisms involved in the bacterial killing by these small molecules. Critical pathways and essential genes in the intermediary metabolism, respiration, and lipid biosynthesis that play significant roles in Mtb's response were identified. Subsequent biochemical experiments that were designed based on the information revealed by transcriptional profiling eventually led to the elucidation of the MOAs of these small molecules. Vitamin C causes bursts in oxidative stress levels in Mtb by generating Fe2+ from Fe3+ and subsequent production of ROS in the presence of oxygen via Fenton reactions, while cysteine enhances respiration and oxygen consumption in Mtb initiating elevated production of ROS. A similar mechanistic study was undertaken for a bicyclic 2-pyridone compound that was shown to reverse INH resistance in Mtb by disrupting respiration homeostasis.59
As bacterial strains can be engineered with controlled transcription levels of certain pathways, transcriptional profiling provides utility for target confirmation in drug mechanistic investigation. In a target-based whole cell screen, Zheng et al. selectively targeted the DosRST signaling pathway by using a DosRST-dependent fluorescent Mtb reporter strain.60 After the hit compounds had been identified, transcriptional profiling studies were undertaken to investigate the inhibitory mechanism of the hit compounds by comparing the transcriptional response of wild-type Mtb upon compound treatment with the response of the ΔdosR mutant. Among the 55 genes that were downregulated by hit compound HC102A, 48 genes were also down regulated by ΔdosR mutation compared to wild-type Mtb. When the ΔdosR mutant was treated with HC102A, no additional genes were repressed when compared to ΔdosR mutant without compound treatment, validating that HC102A phenocopies ΔdosR mutation and thus has a highly specific on target effect. Following the comprehensive transcriptional profiling studies, well designed biochemical based approaches were undertaken that elucidated the specific inhibitory mechanisms, which included inhibiting DosST sensor kinase heme and DosST autophosphorylation.
Transcriptional analysis and the related “real-time quantitative reverse transcription-PCR” (RT-qRT-PCR) method provide the versatility needed for drug MOA study in Mtb under a wide array of conditions. In addition to the heterogeneity of Mtb infection in the host,61 the metabolic state and transcriptome of Mtb in vivo are different than in broth culture and dynamically change.51,62,63 Thus, transcriptional and metabolic responses to TB drugs can be very condition dependent.64–66 Therefore, evaluations of drug MOA under in vitro growth conditions may not fully capture the entire spectrum of how a drug works or how Mtb responds to drug treatment during the in vivo infection. Transcriptional profiling of different in vitro models provides the possibility to evaluate Mtb's gene expression changes induced by antibiotics during different metabolic states, thus facilitating more comprehensive in vivo drug MOA studies.
Limitations of transcriptional profiling in TB drug MOA prediction
Transcriptional profiling is a powerful tool in drug MOA evaluations to comprehend the effects of drug treatment on bacteria on a transcriptional level but is considered to be an indirect approach for drug MOA assessment. MOA predictions based on transcriptional profiling require further experimental validation or supporting evidence.The transcriptional response of Mtb to antibiotics is dynamic during the course of drug treatment, and thus many factors affect the outcome. Different durations of drug treatment, different drug concentrations, and different growth conditions of Mtb may all yield different transcriptional responses.
Genome wide transcriptional profiling data analysis is based on the comparison of ∼4000 measurements across the Mtb genome. Since it is a comparative analysis, the reference transcriptome or strain used in the experiment is essential for obtaining informative results. The reproducibility of transcriptome analyses pose a major challenge, as technical and procedural errors will dramatically affect experimental outcomes and complicate data analyses.67 Furthermore, bacterial responses to antibiotic treatment are a combination of drug on-target inhibition and global stress responses; in order to understand the MOA of a drug from transcriptional profiling it is essential to distinguish the nonspecific stress responses from the real drug inhibitory response.
Bacterial reporter strains
Genetically engineered Mtb reporter strains for drug MOA prediction
Another aspect of transcriptional response-based drug MOA prediction and assessment is the utilization of genetically engineered bacterial reporter strains. As detailed in recent reviews,68,69 specifically designed reporter strains of Mtb have been widely utilized in target-based whole cell screens for TB drug discovery and studies of pathogen–host interactions. A typical reporter strain contains a gene of interest or a promoter and a signal generating gene. The inhibition or induction of the gene of interest can be studied by monitoring the signal generated. The identification of inhibition-specific promoter genes in Mtb, such as iniBAC for cell wall synthesis inhibition70 and recA for DNA damage,71 have facilitated the exploitation of these promoters in constructing reporter strains. When a certain cellular metabolism is inhibited, the promoter is induced, the reporter is then expressed, and a readout signal such as luminescence or fluorescence is generated.Three reporter strains generated by Naran et al. via the coupling of the bioluminescent luxCDABE operon to the Mtb iniBAC, recA, and radA promoters, which control expression of genes responsible for mycobacterial cell wall biosynthesis or DNA metabolism, provided a quantitative measurement in real time of the rate of induction of these promoters over a 10–12 day incubation period.72 Based upon the time point at which luminescence occurs during the course of drug treatment, and the fold change of luminescence readout compared to control groups, this approach may indicate whether a compound is directly targeting a cellular process or if it is a secondary or downstream effect of the drug MOA. TB drugs directly targeting cell wall biosynthesis or DNA metabolism showed early induction of promoters during the first 4 days of treatment, whereas compounds that might have associated downstream effects on cell wall biosynthesis or DNA metabolism showed delayed responses. Moreover, the reporter strains based on recA and radA promoters distinguish between compounds that inhibit cell growth by inhibiting DNA metabolism from the general genotoxins, thereby eliminating compounds that possess cytotoxicity and act in a nonspecific manner.
Strengths and limitations of Mtb reporter strains for drug MOA prediction
The readout of these inducible reporter strains is continuous and in real time, and unlike quantitative RT-PCR or transcriptome analysis, does not require cell disruption and can deliver results continuously over an extended duration. It is also possible to use reporter strains to study the dynamic changes of drug treatment during Mtb infection in a host system, by incorporating genes encoding fluorescent proteins and imaging tools, such that a better understanding of how an antibiotic acts against Mtb in vivo can be achieved.Despite the advantages that these reporter strains provide in antibiotic MOA assessment such as rapid, quantitative and in real time detection, a positive signal from the reporter does not provide definitive proof of the MOA. Similar to transcriptional profiling, these reporter strains do not pinpoint the molecular target of the antibiotic and can only be used with a bias to examine suspected metabolic pathways that the compound is potentially affecting.
Moreover, the scope of the pathways that can be examined are limited and the construction of these reporter strains is highly dependent on the promoter genes chosen. As our understanding of Mtb's bacterial machinery and their regulatory mechanisms is still limited, available and suitable inhibition-specific inducible promoters that can be applied to this system are limited.73
Chemical genetics interaction
Reverse chemical genetics for antibiotic MOA assessment
Selecting spontaneous resistant mutants against compounds of interest and then linking the drug resistance phenotype to genotype is considered a forward chemical genetics approach. Conversely, if a genome wide collection of mutants is available, the phenotype of drug susceptibility can be mapped to specific mutants by screening the compound of interest against the genome wide library of mutants. This strategy is considered a reverse chemical genetic approach74 and is referred to as the approach of chemical genetics interaction in this article.Mapping drug phenotype to gene deletion mutants was first established in yeast as a method to identify drug targets.75 The same principles of chemical genetic interactions were soon adapted in the MOA assessment of antibiotics in bacterial organisms like E. coli76,77 and S. aureus.74 Genetic methods, such as antisense-based genetic overexpression or genetic knock-down were used to generate mutants with altered gene expression levels in order to test their fitness in the presence of compounds of interest at lethal concentrations. The central hypothesis of these approaches is that the chemical genetics interaction between a compound of interest and its molecular target will cause a change in drug susceptibility between wild-type and a genetic mutant related to the target. The hypo- or hypersensitivity to antibiotics will then lead to a fitness change in terms of bacterial growth and abundance, which can be quantified by sequencing of barcodes that are engineered with the genetic modification and unique to each specific mutant. Consequently, a chemical genetics interaction profile can be generated and the potential molecular target(s) of the compound of interest can be proposed.
Mtb mutants with altered gene expression levels for biased drug MOA prediction
In biased drug screens targeting particular metabolic pathways, a small library of mutant strains from that targeting pathway can be used to assess with which potential proteins the hit compounds are interacting. For example, using a library of cholesterol analogs to target cholesterol metabolism in Mtb, Yang and coworkers discovered a number of azasteroids that could sensitize Mtb to existing TB drugs.78 In order to elucidate the MOA of these compounds, a small library of knock out or transposon mutants of genes regulated by cholesterol were utilized to characterize bacterial fitness changes after drug treatment.78 They discovered that azasteroid compounds lost their drug potentiation activities against fadE18, melF, and melH transposon mutants. These three genes are all transcriptionally repressed by the Mce3R regulator, indicating the involvement of Mce3R regulon in the MOA of azasteroid. From a growth inhibitor high-throughput screen in Mtb infected macrophages, VanderVen and coworkers identified a group of compounds that are conditionally active against Mtb in cholesterol-containing media indicating a cholesterol metabolism associated MOA. An Mtb Δicl1 mutant was utilized to examine the compounds’ ability to restore the growth of the Δicl1 mutant in cholesterol-containing media. When grown on cholesterol, the Δicl1 mutant is not able to detoxify propionyl-CoA accumulated from cholesterol catabolism. As a result, the Δicl1 mutant is not able to grow in cholesterol-containing media by itself. Restored growth of the Δicl1 mutant in cholesterol media thereby indicated that the MOA is associated with inhibition of propionyl-CoA producing enzymes from either the cholesterol catabolism pathway or the downstream methylcitrate cycle.79Genome wide chemical genetics interaction profiles (CGIPs) in Mtb for drug MOA identification
The use of genetic methods to screen for inhibitors of essential genes in bacteria can be challenging to achieve. Prior to 2019, several groups undertook using saturated transposon mutant libraries for differential drug sensitivity screening to generate a CGIP in either Mycobacterium smegmatis (M. smeg)80,81 or Mtb.82 These CGIPs were used to identify the primary and secondary targets of TB drugs and to identify the genetic determinants of intrinsic antibiotic susceptibility. However, as transposon insertion leads to loss of function of gene products, some mutants carrying insertions in essential genes under certain culture conditions will not be represented in these libraries. As a result, the mutant pool size is limited and may not include essential targets that are most desirable.An alternative approach is depletion of an essential gene rather than elimination of the target. Subsequent screening of compound libraries to find hits that target these hypersensitive strains provides a chemical-genetic interaction profile. In 2019, a library of 2014 hypomorphic strains representing 474 of approximately 625 essential genes in Mtb was constructed by Johnson and coworkers using conditional proteolysis or transcriptional control.83 Using a pool of 152 of these hypomorphic alleles, they screened against a library of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 bioactive small molecules and generated more than 7 million CGIPs in Mtb. This robust library generation strategy identified both antimycobacterial compounds and their candidate targets simultaneously (Fig. 4). The CGIPs generated from the screening uncovered compounds with novel targets, but also identified new chemical classes that act on the same targets as existing Mtb drugs.
000 bioactive small molecules and generated more than 7 million CGIPs in Mtb. This robust library generation strategy identified both antimycobacterial compounds and their candidate targets simultaneously (Fig. 4). The CGIPs generated from the screening uncovered compounds with novel targets, but also identified new chemical classes that act on the same targets as existing Mtb drugs.
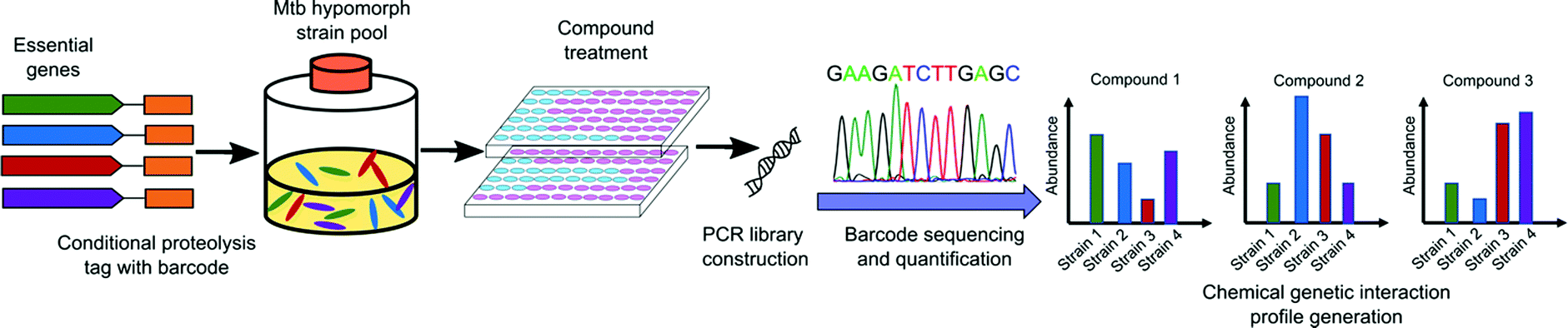 | ||
| Fig. 4 Schematic representation of large-scale chemical-genetic interaction screening strategy for the discovery of new Mtb inhibitors. Mtb hypomorph strains are generated by integrating the conditional proteolysis tag, caseinolytic protease (Clp) DAS tag, with barcode at the C terminus of target genes of interest into the chromosome. Compounds are screened against pools of hypomorph strains. After compound treatment, chromosomal barcodes are quantified for changes in abundance relative to vehicle controls and sequenced by PCR amplification. The CGIP for each compound is represented as the change in strain abundance relative to control.84 | ||
In this large-scale chemical genetics screening campaign, Johnson et al. prioritized compounds that did not strongly inhibit wild-type Mtb, but were strongly active against at least one hypomorph. Due to the static equilibrium conditions of MIC determination assays, solely MIC based compound prioritization strategy may bias toward selection of extracellular targets. Less stringent MIC cutoffs in initial whole-cell screens may identify hits that can be developed into lead compounds with improved potency against vulnerable intracellular targets.8 This approach led to the discovery of compound BRD-8000.3 which possesses a nanomolar MIC against wild-type Mtb by targeting an essential uncharacterized efflux pump in Mtb, EfpA.83 Then, using the CGIP of BRD-8000.3 as a reference, Johnson and coworkers later reported a new compound, BRD-9327, which inhibited the same target in Mtb but displayed a different mode of action and resistance mechanism than compound BRD-8000.3. These two compounds are synergistic and in Mtb, resistance to one increases the sensitivity to the other, thereby presenting a potential drug combination to restrain acquired drug resistance in Mtb.84
More recently, CRISPR technology has been employed in chemical genetics to advance drug discovery and target validation. This technology can be utilized to knockout genes and to either inhibit or activate the expression of a gene without changing the genome. In drug–target validation, CRISPR interference editing provides an efficient and scalable platform for controllable gene expression silencing which is advantageous compared to other methods of transcriptional control due to lack of disruption of DNA sequence in the genome. It has been demonstrated in Mtb that CRISPR interference can achieve a 20 to 100-fold knockdown of target gene expression with minimum proteotoxicity and has been shown to be more effective than conditional proteolysis, thereby offering a great tool for generation of hypomorphic mutants of essential genes at genome scale.85
Advantages and limitations for CGIP in identifying drug MOAs
Chemical genetics interaction profiling provides a systematic chemical biologic method to identify a molecular target from a genetic library. CGIP also affords insights into the secondary and tertiary molecular targets of a drug, which is integral to understanding a drug's MOA. As an approach to compound screening, CGIP facilitates rapid target identification and verification. CGIP allows hit compounds to be selected and prioritized on the basis of MOA, instead of potency alone. Additionally, CGIP enables a rapid expansion of the chemical entities inhibiting the same target, which may work through different modes of interaction, to tackle drug resistance.However, some gene depletion strains can possess hypersensitivity to many bioactive agents, as manifested by the disruption of FecB, a putative iron di-citrate-binding protein, which confers resistance of Mtb to many antibiotic agents.82 This may reflect the intrinsic synthetic lethality between the depletion of genes which interact with the drug target and the chemical inhibition of the target protein. Another limitation of this approach is that efflux pumps and detoxification mechanisms in the cells may prevent the primary molecular target from being exposed by reducing the effective cellular drug concentration.74
Metabolomic profiling
Metabolic adaptations to antibiotic perturbations can be studied
Metabolomics refers to the strategic combination of analytical methods in order to study the global change of small molecules in an organism as they are broken down or synthesized, otherwise known as metabolites.86 The dynamic changes in these endogenous molecules are generally in response to standard biological perturbations, such as stress or nutrient shifts, but unique metabolic responses are observed after the introduction of antibiotics to the bacterial organism.87 The total pool of small molecules in biological systems can be characterized and identified by analytical chemistry methodologies that generate signature liquid chromatography (LC) and mass spectrometry (MS) profiles.Traditionally, metabolomic profiling has been applied in the biochemical characterization of novel metabolic enzymes and pathways. This approach has led to the discovery of the biochemical functions of many essential enzymes in Mtb, including enzymes in central carbon metabolism and lipid and amino acid biosynthesis.88
Conversely, the dynamic change and intracellular levels of small molecules can be mapped to a genotype as well. Understanding the changes in the levels of these biological molecules has been exploited as a useful tool in the identification of antibiotic function and more specifically in validating the function of an metabolic enzyme target.89 In this vein, accumulation of substrates and decreases in metabolic products can generally signify that, either directly or indirectly, bacterial metabolic processes are being disrupted.
Metabolic enzyme inhibitors are optimal candidates for MS-based MOA studies due to the direct correlation between substrate or product concentration in the inhibited pathway and enzyme-target-identity. Popular drug targets in Mtb include folate and cell wall biosynthesis, transcription, translation, and DNA supercoiling.90 These targets, among numerous other pathways, have been identified or verified by global metabolomic studies.
Unraveling drug MOAs by metabolomic profiling in Mtb
Through early trials examining pretomanid efficacy in clinical isolates of Mtb drug resistant strains, it was apparent that pretomanid did not have the same resistance profile as other commonly used antibiotics, insinuating a novel mode of action. Subsequent sequencing of pretomanid resistant mutants implicated F420-dependent glucose-6-phosphate dehydrogenase (G6PD) in susceptibility,37 but the primary mode of action of pretomanid was not fully characterized. G6PD is not the direct target of pretomanid, rather G6PD is required to replenish the reduced F420 cofactor required for deazaflavin dependent nitroreductase (Ddn) activation of pretomanid.91More recently, the Mtb metabolome under treatment with pretomanid and nine other antibiotics was analyzed by gas chromatography (GC)-MS.92 The analysis confirmed previous findings of a unique mode of action and provided further insights into the MOA of pretomanid. An entirely distinct set of responses was observed under pretomanid treatment in comparison to other classes of antibiotics, and the metabolomic data acquired illuminated the cause of the unique set of responses. The increased turnover of G6PD to recycle the F420 cofactor leads to increased production of downstream metabolites of glucose-6-phosphate, phosphorylated aldehyde sugars, which through glycolytic mechanisms leads to the accumulation of methylglyoxal – a cytotoxic molecule detrimental to the overall fitness of the bacteria.92 Whereas resistant mutant sequencing revealed the susceptible enzyme, G6PD, metabolomic profiling was required to identify the mode of bactericidal action.
Through WGS of resistant mutants, BDQ was thought to enact its antimycobacterial effects predominantly through the inhibition of mycobacterial ATP synthase. Drug–target inhibition is often oversimplified as the “sole basis of antibiotic induced cell death,” which is readily apparent in target-based drug design.93 Wang et al. sought to elucidate secondary downstream effects of BDQ treatment via metabolomic analyses. The dose and time dependent profiles of 130 metabolites, most of which are involved in adenosine phosphate-dependent reactions, were examined in response to antibiotic treatment. A near quantitative correlation between ATP and glutamine levels revealed glutamine synthetase as a collateral target to ATP synthase inhibition, enhancing BDQ's anti-mycobacterial activity.94 Whereas genomic based techniques were only able to identify mutations in the atpE gene, metabolite analysis exposed both ATP synthase inhibition as well glutamine synthesis vulnerability.
An amidino-urea scaffold was identified from a high-throughput screening for its antimycobacterial activity both in vitro and in TB animals by Ballinger and coworkers. To explore the MOA of compound 8918, its impact on the Mtb metabolome was examined in parallel with four TB drugs with known mechanisms.31 Several molecules from coenzyme A (CoA) biosynthesis and CoA utilizing pathways were discovered to be affected specifically by the amidino-urea compound in a concentration dependent manner. Subsequently, WGS of spontaneous resistant mutants confirmed the target of 8918 to be the phosphopantetheinyl transferase (PptT) in lipid biosynthesis, that is responsible for pantothenate transfer from CoA to acyl carrier proteins (ACPs) and enables ACPs to synthesize structural lipids. Compound 8918 was the first reported bactericidal compound against Mtb which functioned by inhibiting the CoA metabolism pathway.
Some drugs in bacteria activate compensatory mechanisms to endure antibiotic stresses, this response type can be utilized to elucidate the genuine drug target and to understand drug resistance. In the case of fluoroquinolones (FQ) which inhibit bacterial DNA gyrase, metabolomic studies identified an increase in cellular dTDP-rhamnose levels as a survival mechanism to mediate resistance to FQs. Increased concentration of dTDP-rhamnose, an activated sugar metabolite used for modifying polysaccharide capsule, results in increased DNA gyrase A transcription, thereby increasing the amount of FQ target in the cell for sequestration of antibiotic. gyrA expression is induced by a respiratory metabolic transcription factor AroA. In turn, gyrA expression inhibits dTDP-rhamnose production.95 This gene circuit is an example of the interplay between DNA replication and metabolism.
Strengths and challenges of metabolomic profiling for unraveling TB drug MOAs
The overview of global biochemical events that is revealed through metabolomics enables researchers to obtain a clearer picture of the molecular events occurring during antibiotic treatment. Similar to transcriptome analysis, control parameters and experimental design are key to achieve comprehensive and informative global metabolomic studies due to the dynamic nature of metabolism during bacterial growth.96 Coupled with extensive knowledge of metabolic pathways, “blockages” caused by exogenously inhibited gene function can be traced back to their source. It is through these efforts that MS metabolomics has been effectively utilized to uncover the mechanisms of action of current TB therapies.Metabolomic analysis has the capability to be implemented in a high-throughput manner. Experiments can be automated on a μ-volume scale in 96–384 well plates when appropriate methods are utilized, enabling drugs of interest to be examined in a robust manner.97 The ability to customize metabolomics, specifically data set parameters, by screening for certain metabolites through the use of specialized mass spectrometry instruments adds to its utility.98
The ability to choose between targeted and untargeted studies facilitates the dual modality of metabolomics for use in both large- and small-sample size experiments. The global analyses of eight small molecule inhibitors with known MOAs by Vincent et al.96 and the pseudo-targeted screening of nearly 1300 chemically diverse compounds by Campos et al.99 are excellent examples of the targeted/untargeted dichotomy that is afforded by MS based metabolomics.
However, challenges still remain. In untargeted metabolomics, which is useful as a frontline strategy to uncover global information, the sheer size of data sets can be difficult to analyze. The use of pseudo- and targeted experiments, in which ions of particular molecules are selected for, assuages many of the complications associated with big data. Although metabolomic profiling is a useful tool in bridging the gap between intracellular biomolecular changes and drug–target inhibition, the accumulation of metabolic molecules is often a reflection of the disruption of a certain cellular machinery. Tracing these downstream effects to their primary drug target is challenging and requires a combination of genetic and biochemical approaches.
Biochemical affinity purification: target pull-down
Direct capture of drug-interacting proteins through chemical strategies
The biochemical affinity purification or target pull-down assay is a more tangible and direct method of drug–target identification in comparison to the other techniques described in this review. This technique has been applied in drug target identification and drug off-target evaluation since the 1950s, and this approach has been extensively reviewed in the field of chemical biology.100–102 Like Paul Ehrlich's magic bullet theory, the target pull-down approach is established on the concept that most drugs inhibit a target protein by direct binding in an exclusive and highly specific manner. The goal of a pull-down assay is to capture and characterize the protein(s) that an inhibitor strongly interacts with in a cellular context.The general workflow of a traditional target pull-down assay consists of immobilizing the small molecule of interest onto a solid matrix through a linker. After cell lysates have been passed through the solid matrix, and the matrix washed, the immobilized protein can be eluted, isolated, and characterized. With the development of analytical technologies and chemical synthetic approaches, several alternative methodologies have been developed, where in place of immobilization, the small molecule can be modified with an analytical handle or tag. In this scheme, the intact cell is treated with the small molecule which binds the target protein. The target is then isolated from cell lysates via the handle or tag (Fig. 5).103 Once the inhibitor–enzyme complex is separated from cell lysates, the sequestered protein can be identified through a variety of methods. Briefly, these include examination of whole proteins via LC-MS, SDS-PAGE, or native-PAGE, as well as trypsin digestion followed by identification of peptide fragments. Peptide fragments may be analyzed via MS methods such as Matrix Assisted Laser Desorption Ionization (MALDI) and with the aid of software, protein identity can be ascertained.104 This approach is also often referred as chemical proteomics.
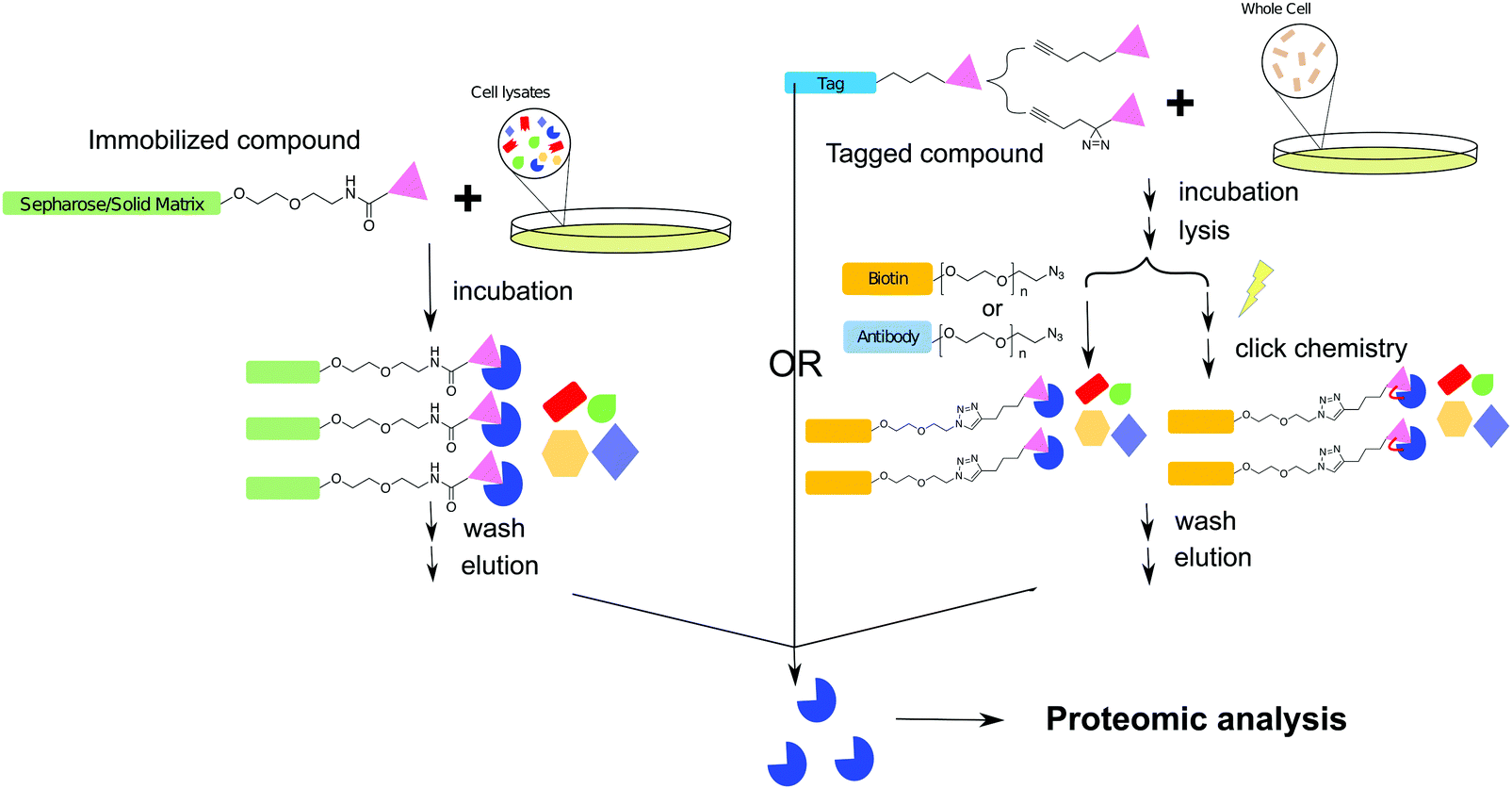 | ||
| Fig. 5 Workflow schemes of affinity purification for target protein isolation. The direct pull-down is achieved by immobilizing the compound of interest on a solid support. After incubating with cell lysates and washing out non-specifically bound entities, the target protein is isolated for proteomic analysis. In click chemistry or photo crosslinking assisted target pull-down, the functionalized compound of interest can be used in the context of whole cells. After incubation and cell lysis, the compound bound proteins are captured through covalent interactions with the capture ligand. After subsequent washing, capture and elution steps, the identities of isolated proteins are characterized by proteomic analysis. | ||
Direct target pull-down in the identification of TB drug targets
As demonstrated by Schmitt and coworkers, a target pull-down approach was undertaken to identify the protein target of the natural product cyclomarin in Mtb.105 Initial generation of spontaneous resistant mutants was not successful, indicating a low mutation frequency, and thus, the need for an alternative approach to target identification. From cell lysates the target protein was captured via sepharose immobilized cyclomarin and identified to be ClpC1 (Rv3596) by proteomic analysis. ClpC1 is a part of the ATPase superfamily and functions as a regulatory subunit with chaperone activity that delivers proteins to the protease subunits of a protease complex. Interestingly, cyclomarin is also an effective antibiotic for treatment of P. falciparium, the causative parasite of malaria. The MOA was discovered through an analogous pull-down experiment [compared to the Mtb MOA assessment] and implicated a diadenosine triphosphate hydrolase, unrelated to the Clp protease complex previously confirmed as the target in Mtb.106 Thus, the cyclomarin MOA is a curious twist on the concept of polypharmacology because it targets two unrelated proteins that are present in two taxonomically unrelated pathogens.Click chemistry and photo crosslinking enabled target pull-down in identifying TB drug targets
With the discovery and development of click chemistry,107 azido–alkyne functionalization often is applied to small molecules for affinity purification of their drug targets. The compound of interest is modified to contain either an azide or alkyne functional group with the aim of not reducing or changing the original bioactivity. The fundamentals of copper catalyzed azide–alkyne click chemistry have been extensively reviewed.108,109 Copper free click cycloaddition is an alternative biorthogonal method often utilized under biological conditions which has also been adapted for target pull-down assays.110 The complimentary functional group can be coupled to a secondary molecule such as an antibody or biotin, which is then, respectively, captured via a primary antibody or streptavidin bound immobilized surface (Fig. 4).111Zhao et al. used a click chemistry based pull-down experiment to verify the target of a benzoxazinone inhibitor discovered via a target-based biochemical inhibitor screen against MarP, a serine protease that regulates intra-bacterial pH homeostasis in Mtb.112 After treating Mycobacterium bovis BCG and performing the target pull-down protocol, the alkyne modified antibiotic bound to target was “clicked” to an affinity handle and the complex captured with streptavidin beads. MarP was identified by immunoblotting with antibody raised against the purified protein. Analysis of the SDS-PAGE gel with fluorescently tagged streptavidin revealed several different bands. Peptide mass fingerprinting of these bands identified high temperature requirement A1 (HtrA1) as an additional protein that interacts with the benzoxazinone inhibitor. This alternative target, which was undetected in earlier genetic screens against benzoxazinone inhibitors, is an essential enzyme in Mtb and thus warrants further study as an antimycobacterial target.
Suicidal inhibitors modify the target protein through covalent interactions, thereby presenting a class of suitable candidates for affinity pull-down without cross-linker modifications. Suicidal inhibitors can be designed from a target-based approach. Thus, the pull-down may bias towards the designated protein target, but they can still be explored in the verification of MOA intracellularly. There are many examples of suicidal inhibitors against Mtb including benzothiazinones (BTZs) inhibiting DprE1113 and oxathiazol-2-ones inhibiting Mtb proteasomes.114 In contrast, noncovalent interactions between the small molecule and target protein in the cell are affected by many factors, such as drug residence time and thermodynamic binding affinity. A pitfall of noncovalent inhibitors is that interactions are not permanent and so compounds that do not covalently bind to their target may not function as optimal candidates for pull-down experiments as target dissociation will prevent the identification of the target. To circumvent this issue, photo crosslinking chemical moieties have also been included in the construction of small molecule probes for target pull-down assays. Photoreactive covalent linkers, such as benzophenone or diazirine moieties, which upon irradiation with ultraviolet light insert into C–H, N–H, or O–H bonds,115 are highly practical reagents for resolving the matter of covalent attachment of inhibitors with some caveats.
Although not in Mtb, a combination of click chemistry and photo crosslinking based affinity pull-down was used to reveal the antibiotic mechanism for a class of kinase inhibitors against methicillin resistant Staphylococcus aureus.116 To determine target identity and MOA, the lead compound PK150 was modified to contain a diazirine photo-linker and alkyne tag. After incubation with intact bacteria, photoreaction and conjugation to a rhodamine–biotin–azide tag through click chemistry facilitated protein capture. Enrichment via avidin beads and subsequent analysis by tandem LC-MS implicated SbsB, an essential membrane serine endopeptidase from the protein secretion pathway and MenG, which catalyzes the final step in menaquinone synthesis, a vitamin crucial for bacterial respiration and energy metabolism as the targets of PK150.116 This dual functionalization by combination of diazirine photo-linker and alkyne tag ensured a successful capture of the potential target proteins and can be applied to pull-down experiments in Mtb for non-covalent inhibitors.
Advantages of target pull-down in drug MOA unraveling and considerations in experimental design
While the pull-down assay is often considered the proverbial “fishing expedition” in the drug–target validation space, it has the ability to discern novel mechanistic information that may be overlooked by other target identification techniques. Tetrahydropyrazolo[1,5-α]pyrimidine-3-carboxamide (THPP) was originally identified for its antimycobacterial activity through a large phenotypic screening campaign.117 Original MOA assignment through WGS of drug resistant mutants revealed that the compound functioned via targeting MmpL3, an essential trehalose monomycolate lipid transporter in Mtb.118 However, through a chemical proteomics strategy where a THPP analogue was covalently immobilized on sepharose beads, EchA6 was identified as the only protein that bound selectively to the compound compared to negative controls.119 A series of biochemical evaluations validated the conclusion that EchA6 is the molecular target of THPP. In contrast, MmpL3 may act as a THPP transporter which accounts for the drug resistance obtained from spontaneous mutagenesis.The design of target pull-down probes must be meticulous. Key to a successful target pull-down, the modification of the small molecule not only has to retain the antibacterial activity of the parental compound, but also must minimize nonspecific interactions with other proteins. For photo crosslinking type probes, the covalent linker must be small enough to not interfere with binding, and the irradiation resulting in photoreaction should be rapid and not interfere with biological functions. Benzophenones undergo photoreaction at long wavelengths, avoiding interfering absorbance by abundant nucleotides and their subsequent damage, but due to their bulkiness may interfere with inhibitor binding and require longer irradiation times. Aryl-azides are small but undergo photoreaction at short wavelengths that are harmful to biological molecules, and the reactive nitrene species formed by photoreaction often decreases photolabeling yields. Diazirines represent the optimal balance between wavelength and size of the covalent linkers, their predominant drawback being the intricate synthetic route to make them.120
The tag used to capture the enzyme-inhibitor complex may also complicate the pull-down assay. When directly linked to the inhibitor, the tag can cause decreased target affinity due to its size. The concern of reduced affinity can be resolved by separating the tag from the inhibitor and instead, including a small reactive functionality to generate the tag after the enzyme-inhibitor complex has been formed. The azide–alkyne click reaction, as previously described, is an exemplary reactive pair for this type of tag attachment.
As nonspecific binding is difficult to avoid in affinity purification, control experiments and comparison groups are critical for identifying the real protein target in the cell. Enrichment and statistical analysis are often used to eliminate the interfering components. After the potential molecular target has been identified, the chemotype needs to be confirmed by the genotype, i.e. manipulation of the tentative target's gene expression level should yield a change in drug susceptibility. We refer to several reviews detailing the efforts that can be taken to ensure the success of the experiment.121,122 Overall, the target pull-down coupled with MS proteomic analysis offers the most comprehensive and direct method of determining the interactions of an inhibitor and its corresponding targets and is a valuable tool in MOA discovery.
Machine learning and computational inference
Computational machine learning in predicting antibiotics’ MOAs
Given recent advancements in artificial intelligence technologies, machine learning has become more and more popular in the field of antibiotic drug discovery,123 to virtually screen antimicrobial agents and predict the MOAs and efficacies of hits. Machine learning (ML) involves the feeding of data sets, called training datasets, to programmer generated computer algorithms. These data sets are utilized to train the program in order to make conclusions based on experimentally obtained data and generally are derived from existing bioinformatic databases. Adaptive improvements are more effectively and successfully accomplished with large data sets of high-quality information. Deep learning (DL) is a subset of ML that has been gaining traction due to increased computing power and the increasing scale and complexity that is afforded by such advancements. The principles of ML algorithms are summarized in depth in reviews by Chen et al.124 and Jung.125This section will be centered on the utilization of ML techniques to determine or predict the target of antibacterial compounds from phenotypic screening assays, although not directly in Mtb. Prediction of targets can be made using a multitude of different types of data, from metabolomic, proteomic, and transcriptional, to other forms of genetic data.126 A wide range of suitable data enables the application of ML/DL to a variety of biochemical experiments and makes this data analysis technique versatile and in reach of many laboratories through collaborative efforts.
The general approach of ML is to monitor the global changes in biochemical state caused by the introduction of the antibiotic and to relate these results to the causative molecular target(s). These computational results can later be verified by experimentally by altering the gene expression level of the proposed target(s). Protein function annotation can guide the prediction of causal events. The genomic sequencing of the bacterial genome and the accurate annotation of the proteins encoded contributes greatly to the success of ML. Application of this combination has been successful in the mechanistic verification of antibiotics in E. coli. Yang et al. utilized machine learning with carefully curated biological network models in order to elucidate direct mechanistic insights into how antibiotics perturb the biological environment of an organism.127 Their approach differs from traditional modern ML predictive techniques which generally lack a biological network model to enrich predictive models. The lethality of three antibiotics with distinct mechanisms of action were screened in E. coli using MS to analyze metabolomic perturbations. Benchmarking studies were used to compare lethality contributions with and without biological network modeling. Inclusion of networks allowed depiction of the contributions of specific metabolic pathways to the lethality of the antibiotics.
Gardner et al. utilized machine learning to identify drug MOA via multiple linear regression modeling of a nine-transcript subnetwork of the SOS pathway in E. coli. Quantitative RT-PCR was used to monitor transcript levels of the set of nine genes under a variety of perturbation combinations and to train the algorithm. The small molecule chemotherapeutic mitomycin C (MMC) was then used to treat E. coli culture, and the same nine gene transcripts were monitored in order to understand the transcriptional effects of MMC on the SOS pathway. All genes in the network that were examined underwent large transcriptional changes, but the recA gene was correctly identified as the MMC transcriptional target by the ML method.128
ML has yet to be widely applied in drug mechanistic investigations in Mtb. However, with more and more data available from the systematic studies of the Mtb metabolome, transcriptome, genome, and their corresponding phenotypes, ML will be on the forefront of TB drug discovery. By utilizing a “systems” approaches to reveal underlying drug mechanisms, ML, unlike other methods, can provide information on how events downstream of primary target inhibition actively participate in antibiotic lethality.129 This tool is most powerful with comprehensive experimental data sets of systems under study, thus highlighting the potential of ML for deepening understanding of Mtb pathogenesis and drug MOA.
Computational inference in predicting drug MOAs in Mtb
Computationally aided target inference explores chemical and structural databases, to predict drug MOA based on existing similar experimental results. At GlaxoSmithKline researchers exploited an integrative three-dimensional computational analysis, based on chemical structure, protein ligand complex structure, and historical in-house registrations from biochemical assays, to predict drug targets from two large phenotypic screening campaigns in Mtb.130,131 The algorithm compares the chemical structural similarity of input compounds to compounds with experimentally validated targets from the ChEMBL database. It searches the structural similarity of input compounds to ligand structures from protein complexes in the Protein Data Bank, and it also predicts target information based on existing in-house biochemical assays. This integrative approach allows the target prediction of hundreds of hit compounds from large screening campaigns.Concluding remarks and future perspectives
Over the past five decades, only two new small molecule therapeutics have been approved for the treatment of TB disease. With the hundreds to thousands of compounds in the development pipeline, it is difficult to perceive this reality. Why is it so difficult to deliver new TB drugs to market, especially considering the largely unmet needs that exist in TB treatment?There are multiple contributing factors that complicate TB drug development. When we consider the history of the tuberculosis pathogen, which has historical mentions dating back nearly 3300 years to the first written records in India and China,132 it is considered to be one of the most successful pathogens in human history. Mtb manages to escape eradication by human immune defenses to reside in a niche within the hosts lungs.133 This niche, characterized as a granulomatous cellular environment, is difficult for antibiotics to penetrate.134 Coupled with a slow growth rate, metabolic heterogeneity, and the formation of persistent mycobacteria, it is exceedingly challenging for antibiotics to sterilize infected tissue.135 In addition, there is a high attrition rate of compounds in the pipeline due to unfavorable PK/PD and safety properties. Comprehension of the MOA of hit/lead compounds in the pipeline and how the MOA correlates to physiological metabolic state of Mtb is crucial for development of improved compounds.
Novel targets and thus MOAs are essential to develop improved compounds and to combat existing drug resistance, and target validation for clinical use is equally crucial. MOA assignments for compounds from phenotypic screens can unveil novel drug targets, such as MmpL3 and DprE1, that are vulnerable to small molecule inhibition and essential for bacterial growth.8 These molecular targets can potentially facilitate a new series of target-based drug discovery programs to deliver new chemical entities with improved drug profiles.
Each strategy described in this review for MOA identification of TB drugs has associated strengths and limitations (Table 1). Some techniques may be more accessible, while others may require established platforms and prove to be technically challenging. A single appropriate approach most likely only serves to provide a starting point for hypothesis and validation. A comprehensive and indisputable assessment requires several lines of evidence including biochemical, genetic, metabolomic and structural-biological confirmations. The mechanisms with which TB drugs impart their activity are often complex. Even after years of clinical use, new aspects of how TB drugs function are still being uncovered. A single approach to uncover MOA, such as those described in this review, will not be sufficient, and a combined and comprehensive approach is required to effectively identify the mechanisms of a single agent.
| Approaches | Advantage | Limitations |
|---|---|---|
| WGS of spontaneous resistant mutant | • Potential to reveal the direct drug target | • Direct drug target mutations are not guaranteed |
| • Commonly used and relatively straightforward | • Non-specific mutations may arise | |
| • With next generation sequencing, the approach has the potential for high-throughput screening | • Mutations in drug modifying genes i.e. pro-drugs | |
| • Mutations in related but non-target genes i.e. transporters | ||
| • Resistant mutants can be hard to raise | ||
| • Further confirmation necessary | ||
| Transcriptional profiling | • Reveals global response to the drug | • May not reveal the direct drug target |
| • Reveals the downstream effect of primary drug target inhibition | • Nonspecific drug-stress response interference | |
| • Experimental design can dramatically affect the outcome | ||
| Reporter strain | • Fast and continuous readout | • The inhibition-specific promoter genes in Mtb are limited |
| • Can be used to dynamically study the drug MOA in real time | • Does not pinpoint the molecular target of a drug | |
| Chemical genetics interaction profiling | • High throughput | • Efflux pump and detoxification mechanism interference |
| • Can be used to access genome wide drug–target interactions | • Nonspecific hyper- or hyposensitivity | |
| • Reveals downstream molecular targets of a drug | ||
| Metabolomic profiling | • Insight into target inhibition at molecular level | • Tracing upstream metabolites to primary drug target can be challenging |
| • High throughput | • Based on comparison and requires an annotated database that includes identified metabolites | |
| • Global metabolome analysis reveals additional information secondary to primary target inhibition | • Data analysis can be challenging | |
| • Requires extensive mass spec. setup for analysis conditions | ||
| Target pull-down | • Direct and straightforward – capture and analysis of target | • Chemical modification of the drug is needed |
| • Non-specific binding interference | ||
| • Requires secondary analysis i.e. proteomics | ||
| Machine learning and computational inference | • Fast | • Established on the basis of large data sets |
| • Wet lab experiments not required | • Further confirmation is needed | |
| • High throughput | • Prior knowledge/expertise in computer languages required |
Looking back, an antibiotics’ PK/PD properties and safety profiles are directly linked to its MOA. The on target and off target effects of an antibiotic are reflective of the cellular machineries that are perturbed. Looking ahead, it is essential to continue current work to further understand the physiology and pathogenicity of Mtb to develop more comprehensive databases for systems-based assessment of drug MOA. Completing the cycle, drug MOA identification in Mtb will improve our understanding of the pathogenic bacterial machineries of Mtb and motivate more advanced screening strategies for new inhibitory chemical entities.136 Novel approaches, such as exploiting the synthetic lethality gene network in bacteria,137 utilizing machine-learning and artificial intelligence for revealing lethality mechanisms,127,138 and computationally aided compound analysis139,140 should all be used to elucidate MOA of Mtb drug candidates. Wider application of these approaches will advance translation of compounds into the clinic for treatment of TB, a disease which remains persistent and deadly.
Glossary of abbreviations
| ACPs | Acyl carrier proteins |
| BDQ | Bedaquiline |
| CFU | Colony forming unit |
| CGIP | Chemical genetics interaction profile |
| CoA | Coenzyme A |
| DL | Deep learning |
| FDA | Food and Drug Administration |
| FQ | Fluroquinolone |
| G6PD | Glucose-6-phosphate dehydrogenase |
| GC | Gas chromatography |
| INH | Isoniazid |
| LC | Liquid chromatography |
| M. smeg | Mycobacterium smegmatis |
| MIC | Minimum inhibitory concentration |
| ML | Machine learning |
| MMC | Mitomycin C |
| MOA | Mechanism of action |
| MS | Mass spectrometry |
| Mtb | Mycobacterium tuberculosis |
| PD | Pharmacodynamics |
| PK | Pharmacokinetics |
| PZA | Pyrazinamide |
| ROS | Reactive oxygen species |
| RT-qRT-PCR | Real-time quantitative reverse transcription-polymerase chain reaction |
| SAR | Structure–activity relationship |
| SNPs | Single nucleotide polymorphisims |
| SOS | Cellular global response to DNA damage |
| TB | Tuberculosis |
| THPP | Tetrahydropyrazolo[1,5-α]pyrimidine-3-carboxamide |
| WGS | Whole genome sequencing |
Conflicts of interest
The authors have patent interest in tuberculosis therapeutic treatments.Acknowledgements
The author's research described in this review is funded by NIH grant RO1AI134054 to N. S. S.References
- H. H. Fox, Science, 1952, 116, 129–134 CrossRef CAS.
- C. Vilcheze and W. R. Jacobs Jr, Annu. Rev. Microbiol., 2007, 61, 35–50 CrossRef CAS.
- WHO, Global Tuberculosis Report, https://www.who.int/tb/publications/global_report/en/, (accessed 30thDecember, 2019).
- FDA, SIRTURO (bedaquiline) label, https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/204384s000lbl.pdf, (accessed 30th December, 2019).
- FDA, Drug Approval Package: Pretomanid, https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/212862Orig1s000TOC.cfm, (accessed 30th December, 2019).
- M. N. Gwynn, A. Portnoy, S. F. Rittenhouse and D. J. Payne, Ann. N. Y. Acad. Sci., 2010, 1213, 5–19 CrossRef.
- H. Brotz-Oesterhelt and P. Sass, Future Microbiol., 2010, 5, 1553–1579 CrossRef.
- T. Yuan and N. S. Sampson, Chem. Rev., 2018, 118, 1887–1916 CrossRef CAS.
- K. Andries, P. Verhasselt, J. Guillemont, H. W. Gohlmann, J. M. Neefs, H. Winkler, J. Van Gestel, P. Timmerman, M. Zhu, E. Lee, P. Williams, D. de Chaffoy, E. Huitric, S. Hoffner, E. Cambau, C. Truffot-Pernot, N. Lounis and V. Jarlier, Science, 2005, 307, 223–227 CrossRef CAS.
- C. K. Stover, P. Warrener, D. R. VanDevanter, D. R. Sherman, T. M. Arain, M. H. Langhorne, S. W. Anderson, J. A. Towell, Y. Yuan, D. N. McMurray, B. N. Kreiswirth, C. E. Barry and W. R. Baker, Nature, 2000, 405, 962–966 CrossRef CAS.
- L. L. Silver, Nat. Rev. Drug Discovery, 2007, 6, 41–55 CrossRef CAS.
- A. L. Hopkins, Nat. Chem. Biol., 2008, 4, 682–690 CrossRef CAS.
- D. C. Swinney and J. Anthony, Nat. Rev. Drug Discovery, 2011, 10, 507–519 CrossRef CAS.
- K. Pethe, P. C. Sequeira, S. Agarwalla, K. Rhee, K. Kuhen, W. Y. Phong, V. Patel, D. Beer, J. R. Walker, J. Duraiswamy, J. Jiricek, T. H. Keller, A. Chatterjee, M. P. Tan, M. Ujjini, S. P. Rao, L. Camacho, P. Bifani, P. A. Mak, I. Ma, S. W. Barnes, Z. Chen, D. Plouffe, P. Thayalan, S. H. Ng, M. Au, B. H. Lee, B. H. Tan, S. Ravindran, M. Nanjundappa, X. Lin, A. Goh, S. B. Lakshminarayana, C. Shoen, M. Cynamon, B. Kreiswirth, V. Dartois, E. C. Peters, R. Glynne, S. Brenner and T. Dick, Nat. Commun., 2010, 1, 57 CrossRef.
- L. L. Silver, Clin. Microbiol. Rev., 2011, 24, 71–109 CrossRef CAS.
- M. Schenone, V. Dancik, B. K. Wagner and P. A. Clemons, Nat. Chem. Biol., 2013, 9, 232–240 CrossRef CAS.
- M. A. Farha and E. D. Brown, Nat. Prod. Rep., 2016, 33, 668–680 RSC.
- K. Katsuno, J. N. Burrows, K. Duncan, R. Hooft van Huijsduijnen, T. Kaneko, K. Kita, C. E. Mowbray, D. Schmatz, P. Warner and B. T. Slingsby, Nat. Rev. Drug Discovery, 2015, 14, 751–758 CrossRef CAS.
- M. Tyers and G. D. Wright, Nat. Rev. Microbiol., 2019, 17, 141–155 CrossRef CAS.
- S. Chakraborty and K. Y. Rhee, Cold Spring Harbor Perspect. Med., 2015, 5, a021147 CrossRef.
- Global Alliance for TB Drug Development, Tuberculosis, 2008, 88, 85–86 CrossRef.
- J. M. Heather and B. Chain, Genomics, 2016, 107, 1–8 CrossRef CAS.
- S. B. Snapper, L. Lugosi, A. Jekkel, R. E. Melton, T. Kieser, B. R. Bloom and W. R. Jacobs Jr, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 6987–6991 CrossRef CAS.
- S. T. Cole, R. Brosch, J. Parkhill, T. Garnier, C. Churcher, D. Harris, S. V. Gordon, K. Eiglmeier, S. Gas, C. E. Barry 3rd, F. Tekaia, K. Badcock, D. Basham, D. Brown, T. Chillingworth, R. Connor, R. Davies, K. Devlin, T. Feltwell, S. Gentles, N. Hamlin, S. Holroyd, T. Hornsby, K. Jagels, A. Krogh, J. McLean, S. Moule, L. Murphy, K. Oliver, J. Osborne, M. A. Quail, M. A. Rajandream, J. Rogers, S. Rutter, K. Seeger, J. Skelton, R. Squares, S. Squares, J. E. Sulston, K. Taylor, S. Whitehead and B. G. Barrell, Nature, 1998, 393, 537–544 CrossRef CAS.
- Clinical Pipeline, https://www.newtbdrugs.org/pipeline/clinical, (accessed 6Apr2020, 2020).
- J. Crofton and D. A. Mitchison, Br. Med. J., 1948, 2, 1009–1015 CrossRef CAS.
- P. E. Almeida Da Silva and J. C. Palomino, J. Antimicrob. Chemother., 2011, 66, 1417–1430 CrossRef CAS.
- S. H. Gillespie, Antimicrob. Agents Chemother., 2002, 46, 267–274 CrossRef CAS.
- J. W. Drake, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 7160–7164 CrossRef CAS.
- H. L. David, Appl. Microbiol., 1970, 20, 810–814 CrossRef CAS.
- E. Ballinger, J. Mosior, T. Hartman, K. Burns-Huang, B. Gold, R. Morris, L. Goullieux, I. Blanc, J. Vaubourgeix, S. Lagrange, L. Fraisse, S. Sans, C. Couturier, E. Bacque, K. Rhee, S. M. Scarry, J. Aube, G. Yang, O. Ouerfelli, D. Schnappinger, T. R. Ioerger, C. A. Engelhart, J. A. McConnell, K. McAulay, A. Fay, C. Roubert, J. Sacchettini and C. Nathan, Science, 2019, 363, eaau8959 CrossRef CAS.
- S. M. Gygli, S. Borrell, A. Trauner and S. Gagneux, FEMS Microbiol. Rev., 2017, 41, 354–373 CrossRef CAS.
- T. R. Ioerger, T. O'Malley, R. Liao, K. M. Guinn, M. J. Hickey, N. Mohaideen, K. C. Murphy, H. I. Boshoff, V. Mizrahi, E. J. Rubin, C. M. Sassetti, C. E. Barry 3rd, D. R. Sherman, T. Parish and J. C. Sacchettini, PLoS One, 2013, 8, e75245 CrossRef CAS.
- W. R. Jacobs Jr, Microbiol. Spectrum, 2014, 2, MGM2–0037-2013 CrossRef.
- J. C. Evans and V. Mizrahi, Front. Microbiol., 2015, 6, 812 CrossRef.
- J. Bernstein, W. A. Lott, B. A. Steinberg and H. L. Yale, Am. Rev. Tuberc., 1952, 65, 357–364 CAS.
- U. H. Manjunatha, H. Boshoff, C. S. Dowd, L. Zhang, T. J. Albert, J. E. Norton, L. Daniels, T. Dick, S. S. Pang and C. E. Barry 3rd, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 431–436 CrossRef CAS.
- A. Scorpio and Y. Zhang, Nat. Med., 1996, 2, 662–667 CrossRef CAS.
- P. Gopal, G. Gruber, V. Dartois and T. Dick, Trends Pharmacol. Sci., 2019, 40, 930–940 CrossRef CAS.
- R. M. Anthony, A. L. den Hertog and D. van Soolingen, J. Antimicrob. Chemother., 2018, 73, 1750–1754 CrossRef CAS.
- S. Benzer and E. Freese, Proc. Natl. Acad. Sci. U. S. A., 1958, 44, 112–119 CrossRef CAS.
- G. G. Makafe, M. Hussain, G. Surineni, Y. Tan, N. K. Wong, M. Julius, L. Liu, C. Gift, H. Jiang, Y. Tang, J. Liu, S. Tan, Z. Yu, Z. Liu, Z. Lu, C. Fang, Y. Zhou, J. Zhang, Q. Zhu, J. Liu and T. Zhang, Cell Chem. Biol., 2019, 26, 1187–1194.e1185 CrossRef CAS.
- L. Lopez Quezada, S. Silve, M. Kelinske, A. Liba, C. Diaz Gonzalez, M. Kotev, L. Goullieux, S. Sans, C. Roubert, S. Lagrange, E. Bacque, C. Couturier, A. Pellet, I. Blanc, M. Ferron, F. Debu, K. Li, J. Aube, J. Roberts, D. Little, Y. Ling, J. Zhang, B. Gold and C. Nathan, mBio, 2019, 10, e01405 CrossRef.
- Z. Yang, X. Zeng and S. K. Tsui, BMC Genomics, 2019, 20, 394 CrossRef.
- A. N. Brooks, S. Turkarslan, K. D. Beer, F. Y. Lo and N. S. Baliga, Wiley Interdiscip. Rev.: Syst. Biol. Med., 2011, 3, 544–561 CAS.
- J. C. Perez and E. A. Groisman, Cell, 2009, 138, 233–244 CrossRef CAS.
- H. I. Boshoff, T. G. Myers, B. R. Copp, M. R. McNeil, M. A. Wilson and C. E. Barry 3rd, J. Biol. Chem., 2004, 279, 40174–40184 CrossRef CAS.
- P. Murima, P. F. de Sessions, V. Lim, A. N. Naim, P. Bifani, H. I. Boshoff, V. K. Sambandamurthy, T. Dick, M. L. Hibberd, M. Schreiber and S. P. Rao, PLoS One, 2013, 8, e69191 CrossRef CAS.
- S. Ma, S. Jaipalli, J. Larkins-Ford, J. Lohmiller, B. B. Aldridge, D. R. Sherman and S. Chandrasekaran, mBio, 2019, 10, e02627 CAS.
- M. I. Voskuil, D. Schnappinger, K. C. Visconti, M. I. Harrell, G. M. Dolganov, D. R. Sherman and G. K. Schoolnik, J. Exp. Med., 2003, 198, 705–713 CrossRef CAS.
- I. Keren, S. Minami, E. Rubin and K. Lewis, mBio, 2011, 2, e00100 CrossRef.
- W. Lin, P. F. de Sessions, G. H. Teoh, A. N. Mohamed, Y. O. Zhu, V. H. Koh, M. L. Ang, P. C. Dedon, M. L. Hibberd and S. Alonso, Infect. Immun., 2016, 84, 2505–2523 CrossRef CAS.
- R. Singh, U. Manjunatha, H. I. Boshoff, Y. H. Ha, P. Niyomrattanakit, R. Ledwidge, C. S. Dowd, I. Y. Lee, P. Kim, L. Zhang, S. Kang, T. H. Keller, J. Jiricek and C. E. Barry 3rd, Science, 2008, 322, 1392–1395 CrossRef CAS.
- U. Manjunatha, H. I. Boshoff and C. E. Barry, Commun. Integr. Biol., 2009, 2, 215–218 CrossRef CAS.
- E. J. R. Peterson, S. Ma, D. R. Sherman and N. S. Baliga, Nat. Microbiol., 2016, 1, 16078 CrossRef CAS.
- A. Koul, L. Vranckx, N. Dhar, H. W. Gohlmann, E. Ozdemir, J. M. Neefs, M. Schulz, P. Lu, E. Mortz, J. D. McKinney, K. Andries and D. Bald, Nat. Commun., 2014, 5, 3369 CrossRef.
- C. Vilcheze, T. Hartman, B. Weinrick and W. R. Jacobs Jr, Nat. Commun., 2013, 4, 1881 CrossRef.
- C. Vilcheze, T. Hartman, B. Weinrick, P. Jain, T. R. Weisbrod, L. W. Leung, J. S. Freundlich and W. R. Jacobs Jr, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 4495–4500 CrossRef CAS.
- K. Flentie, G. A. Harrison, H. Tukenmez, J. Livny, J. A. D. Good, S. Sarkar, D. X. Zhu, R. L. Kinsella, L. A. Weiss, S. D. Solomon, M. E. Schene, M. R. Hansen, A. G. Cairns, M. Kulen, T. Wixe, A. E. G. Lindgren, E. Chorell, C. Bengtsson, K. S. Krishnan, S. J. Hultgren, C. Larsson, F. Almqvist and C. L. Stallings, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 10510–10517 CrossRef CAS.
- H. Zheng, C. J. Colvin, B. K. Johnson, P. D. Kirchhoff, M. Wilson, K. Jorgensen-Muga, S. D. Larsen and R. B. Abramovitch, Nat. Chem. Biol., 2017, 13, 218–225 CrossRef CAS.
- A. M. Cadena, S. M. Fortune and J. L. Flynn, Nat. Rev. Immunol., 2017, 17, 691–702 CrossRef CAS.
- K. H. Rohde, D. F. Veiga, S. Caldwell, G. Balazsi and D. G. Russell, PLoS Pathog., 2012, 8, e1002769 CrossRef CAS.
- D. Schnappinger, S. Ehrt, M. I. Voskuil, Y. Liu, J. A. Mangan, I. M. Monahan, G. Dolganov, B. Efron, P. D. Butcher, C. Nathan and G. K. Schoolnik, J. Exp. Med., 2003, 198, 693–704 CrossRef CAS.
- K. N. Adams, K. Takaki, L. E. Connolly, H. Wiedenhoft, K. Winglee, O. Humbert, P. H. Edelstein, C. L. Cosma and L. Ramakrishnan, Cell, 2011, 145, 39–53 CrossRef CAS.
- Y. Liu, S. Tan, L. Huang, R. B. Abramovitch, K. H. Rohde, M. D. Zimmerman, C. Chen, V. Dartois, B. C. VanderVen and D. G. Russell, J. Exp. Med., 2016, 213, 809–825 CrossRef CAS.
- J. P. Sarathy, L. E. Via, D. Weiner, L. Blanc, H. Boshoff, E. A. Eugenin, C. E. Barry 3rd and V. A. Dartois, Antimicrob. Agents Chemother., 2018, 62, e02266 Search PubMed.
- D. Schnappinger, Cold Spring Harbor Perspect. Med., 2015, 5, a021139 CrossRef.
- R. B. Abramovitch, IUBMB Life, 2018, 70, 818–825 CrossRef CAS.
- N. J. MacGilvary and S. Tan, Pathog. Dis., 2018, 76, fty017 Search PubMed.
- D. Alland, A. J. Steyn, T. Weisbrod, K. Aldrich and W. R. Jacobs Jr, J. Bacteriol., 2000, 182, 1802–1811 CrossRef CAS.
- K. L. Smollett, K. M. Smith, C. Kahramanoglou, K. B. Arnvig, R. S. Buxton and E. O. Davis, J. Biol. Chem., 2012, 287, 22004–22014 CrossRef CAS.
- K. Naran, A. Moosa, C. E. Barry 3rd, H. I. Boshoff, V. Mizrahi and D. F. Warner, Antimicrob. Agents Chemother., 2016, 60, 6748–6757 CrossRef CAS.
- H. P. Fischer, N. A. Brunner, B. Wieland, J. Paquette, L. Macko, K. Ziegelbauer and C. Freiberg, Genome Res., 2004, 14, 90–98 CrossRef CAS.
- T. Roemer, J. Davies, G. Giaever and C. Nislow, Nat. Chem. Biol., 2011, 8, 46–56 CrossRef.
- G. Giaever, P. Flaherty, J. Kumm, M. Proctor, C. Nislow, D. F. Jaramillo, A. M. Chu, M. I. Jordan, A. P. Arkin and R. W. Davis, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 793–798 CrossRef CAS.
- R. Pathania, S. Zlitni, C. Barker, R. Das, D. A. Gerritsma, J. Lebert, E. Awuah, G. Melacini, F. A. Capretta and E. D. Brown, Nat. Chem. Biol., 2009, 5, 849–856 CrossRef CAS.
- M. A. Farha and E. D. Brown, Chem. Biol., 2010, 17, 852–862 CrossRef CAS.
- X. Yang, T. Yuan, R. Ma, K. I. Chacko, M. Smith, G. Deikus, R. Sebra, A. Kasarskis, H. van Bakel, S. G. Franzblau and N. S. Sampson, ACS Infect. Dis., 2019, 5, 1239–1251 CrossRef CAS.
- B. C. VanderVen, R. J. Fahey, W. Lee, Y. Liu, R. B. Abramovitch, C. Memmott, A. M. Crowe, L. D. Eltis, E. Perola, D. D. Deininger, T. Wang, C. P. Locher and D. G. Russell, PLoS Pathog., 2015, 11, e1004679 CrossRef.
- D. C. Alexander, J. R. Jones and J. Liu, Antimicrob. Agents Chemother., 2003, 47, 3208–3213 CrossRef CAS.
- R. L. Campen, D. F. Ackerley, G. M. Cook and R. F. O'Toole, Tuberculosis, 2015, 95, 432–439 CrossRef CAS.
- W. Xu, M. A. DeJesus, N. Rucker, C. A. Engelhart, M. G. Wright, C. Healy, K. Lin, R. Wang, S. W. Park, T. R. Ioerger, D. Schnappinger and S. Ehrt, Antimicrob. Agents Chemother., 2017, 61, e01334 Search PubMed.
- E. O. Johnson, E. LaVerriere, E. Office, M. Stanley, E. Meyer, T. Kawate, J. E. Gomez, R. E. Audette, N. Bandyopadhyay, N. Betancourt, K. Delano, I. Da Silva, J. Davis, C. Gallo, M. Gardner, A. J. Golas, K. M. Guinn, S. Kennedy, R. Korn, J. A. McConnell, C. E. Moss, K. C. Murphy, R. M. Nietupski, K. G. Papavinasasundaram, J. T. Pinkham, P. A. Pino, M. K. Proulx, N. Ruecker, N. Song, M. Thompson, C. Trujillo, S. Wakabayashi, J. B. Wallach, C. Watson, T. R. Ioerger, E. S. Lander, B. K. Hubbard, M. H. Serrano-Wu, S. Ehrt, M. Fitzgerald, E. J. Rubin, C. M. Sassetti, D. Schnappinger and D. T. Hung, Nature, 2019, 571, 72–78 CrossRef CAS.
- E. O. Johnson, E. Office, T. Kawate, M. Orzechowski and D. T. Hung, ACS Infect. Dis., 2020, 6, 56–63 CrossRef CAS.
- J. M. Rock, F. F. Hopkins, A. Chavez, M. Diallo, M. R. Chase, E. R. Gerrick, J. R. Pritchard, G. M. Church, E. J. Rubin, C. M. Sassetti, D. Schnappinger and S. M. Fortune, Nat. Microbiol., 2017, 2, 16274 CrossRef CAS.
- I. du Preez, L. Luies and D. T. Loots, Tuberculosis, 2019, 115, 126–139 CrossRef CAS.
- D. G. Robertson and U. Frevert, Clin. Pharmacol. Ther., 2013, 94, 559–561 CrossRef CAS.
- M. Drapal and P. D. Fraser, Microorganisms, 2019, 7, 148 CrossRef CAS.
- R. S. Jansen and K. Y. Rhee, Trends Pharmacol. Sci., 2017, 38, 393–405 CrossRef CAS.
- Z. S. Bhat, M. A. Rather, M. Maqbool and Z. Ahmad, Biomed. Pharmacother., 2018, 103, 1733–1747 CrossRef CAS.
- A. E. Mohamed, F. H. Ahmed, S. Arulmozhiraja, C. Y. Lin, M. C. Taylor, E. R. Krausz, C. J. Jackson and M. L. Coote, Mol. BioSyst., 2016, 12, 1110–1113 RSC.
- R. Baptista, D. M. Fazakerley, M. Beckmann, L. Baillie and L. A. J. Mur, Sci. Rep., 2018, 8, 5084 CrossRef.
- P. Belenky, J. D. Ye, C. B. Porter, N. R. Cohen, M. A. Lobritz, T. Ferrante, S. Jain, B. J. Korry, E. G. Schwarz, G. C. Walker and J. J. Collins, Cell Rep., 2015, 13, 968–980 CrossRef CAS.
- Z. Wang, V. Soni, G. Marriner, T. Kaneko, H. I. M. Boshoff, C. E. Barry 3rd and K. Y. Rhee, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 19646–19651 CrossRef CAS.
- M. Zampieri, M. Zimmermann, M. Claassen and U. Sauer, Cell Rep., 2017, 19, 1214–1228 CrossRef CAS.
- I. M. Vincent, D. E. Ehmann, S. D. Mills, M. Perros and M. P. Barrett, Antimicrob. Agents Chemother., 2016, 60, 2281–2291 CrossRef CAS.
- M. Rohman and J. Wingfield, Methods Mol. Biol., 2016, 1439, 47–63 CrossRef CAS.
- K. Dettmer, P. A. Aronov and B. D. Hammock, Mass Spectrom. Rev., 2007, 26, 51–78 CrossRef CAS.
- A. I. Campos and M. Zampieri, Mol. Cell, 2019, 74, 1291–1303.e1296 CrossRef CAS.
- B. Lomenick, R. W. Olsen and J. Huang, ACS Chem. Biol., 2011, 6, 34–46 CrossRef CAS.
- W. Zheng, G. Li and X. Li, Arch. Pharmacal Res., 2015, 38, 1661–1685 CrossRef CAS.
- F. Cong, A. K. Cheung and S. M. Huang, Annu. Rev. Pharmacol. Toxicol., 2012, 52, 57–78 CrossRef CAS.
- A. McFedries, A. Schwaid and A. Saghatelian, Chem. Biol., 2013, 20, 667–673 CrossRef CAS.
- R. Aebersold and D. R. Goodlett, Chem. Rev., 2001, 101, 269–295 CrossRef CAS.
- E. K. Schmitt, M. Riwanto, V. Sambandamurthy, S. Roggo, C. Miault, C. Zwingelstein, P. Krastel, C. Noble, D. Beer, S. P. Rao, M. Au, P. Niyomrattanakit, V. Lim, J. Zheng, D. Jeffery, K. Pethe and L. R. Camacho, Angew. Chem., Int. Ed., 2011, 50, 5889–5891 CrossRef CAS.
- N. Burstner, S. Roggo, N. Ostermann, J. Blank, C. Delmas, F. Freuler, B. Gerhartz, A. Hinniger, D. Hoepfner, B. Liechty, M. Mihalic, J. Murphy, D. Pistorius, M. Rottmann, J. R. Thomas, M. Schirle and E. K. Schmitt, ChemBioChem, 2015, 16, 2433–2436 CrossRef.
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef CAS.
- L. Liang and D. Astruc, Coord. Chem. Rev., 2011, 255, 2933–2945 CrossRef CAS.
- S. I. Presolski, V. P. Hong and M. G. Finn, Curr. Protoc. Chem. Biol., 2011, 3, 153–162 CrossRef.
- J. C. Jewett and C. R. Bertozzi, Chem. Soc. Rev., 2010, 39, 1272–1279 RSC.
- L. I. Willems, W. A. van der Linden, N. Li, K. Y. Li, N. Liu, S. Hoogendoorn, G. A. van der Marel, B. I. Florea and H. S. Overkleeft, Acc. Chem. Res., 2011, 44, 718–729 CrossRef CAS.
- N. Zhao, C. M. Darby, J. Small, D. A. Bachovchin, X. Jiang, K. E. Burns-Huang, H. Botella, S. Ehrt, D. L. Boger, E. D. Anderson, B. F. Cravatt, A. E. Speers, V. Fernandez-Vega, P. S. Hodder, C. Eberhart, H. Rosen, T. P. Spicer and C. F. Nathan, ACS Chem. Biol., 2015, 10, 364–371 CrossRef CAS.
- C. Trefzer, H. Skovierova, S. Buroni, A. Bobovska, S. Nenci, E. Molteni, F. Pojer, M. R. Pasca, V. Makarov, S. T. Cole, G. Riccardi, K. Mikusova and K. Johnsson, J. Am. Chem. Soc., 2012, 134, 912–915 CrossRef CAS.
- G. Lin, D. Li, L. P. de Carvalho, H. Deng, H. Tao, G. Vogt, K. Wu, J. Schneider, T. Chidawanyika, J. D. Warren, H. Li and C. Nathan, Nature, 2009, 461, 621–626 CrossRef CAS.
- L. Dubinsky, B. P. Krom and M. M. Meijler, Bioorg. Med. Chem., 2012, 20, 554–570 CrossRef CAS.
- P. Le, E. Kunold, R. Macsics, K. Rox, M. C. Jennings, I. Ugur, M. Reinecke, D. Chaves-Moreno, M. W. Hackl, C. Fetzer, F. A. M. Mandl, J. Lehmann, V. S. Korotkov, S. M. Hacker, B. Kuster, I. Antes, D. H. Pieper, M. Rohde, W. M. Wuest, E. Medina and S. A. Sieber, Nat. Chem., 2019, 12, 145–158 CrossRef.
- L. Ballell, R. H. Bates, R. J. Young, D. Alvarez-Gomez, E. Alvarez-Ruiz, V. Barroso, D. Blanco, B. Crespo, J. Escribano, R. Gonzalez, S. Lozano, S. Huss, A. Santos-Villarejo, J. J. Martin-Plaza, A. Mendoza, M. J. Rebollo-Lopez, M. Remuinan-Blanco, J. L. Lavandera, E. Perez-Herran, F. J. Gamo-Benito, J. F. Garcia-Bustos, D. Barros, J. P. Castro and N. Cammack, ChemMedChem, 2013, 8, 313–321 CrossRef CAS.
- M. J. Remuinan, E. Perez-Herran, J. Rullas, C. Alemparte, M. Martinez-Hoyos, D. J. Dow, J. Afari, N. Mehta, J. Esquivias, E. Jimenez, F. Ortega-Muro, M. T. Fraile-Gabaldon, V. L. Spivey, N. J. Loman, M. J. Pallen, C. Constantinidou, D. J. Minick, M. Cacho, M. J. Rebollo-Lopez, C. Gonzalez, V. Sousa, I. Angulo-Barturen, A. Mendoza-Losana, D. Barros, G. S. Besra, L. Ballell and N. Cammack, PLoS One, 2013, 8, e60933 CrossRef CAS.
- J. A. Cox, K. A. Abrahams, C. Alemparte, S. Ghidelli-Disse, J. Rullas, I. Angulo-Barturen, A. Singh, S. S. Gurcha, V. Nataraj, S. Bethell, M. J. Remuinan, L. Encinas, P. J. Jervis, N. C. Cammack, A. Bhatt, U. Kruse, M. Bantscheff, K. Futterer, D. Barros, L. Ballell, G. Drewes and G. S. Besra, Nat. Microbiol., 2016, 1, 15006 CrossRef CAS.
- M. W. Halloran and J. P. Lumb, Chemistry, 2019, 25, 4885–4898 CrossRef CAS.
- M. Fonovic and M. Bogyo, Expert Rev. Proteomics, 2008, 5, 721–730 CrossRef CAS.
- M. H. Wright and S. A. Sieber, Nat. Prod. Rep., 2016, 33, 681–708 RSC.
- H. Chen, O. Engkvist, Y. Wang, M. Olivecrona and T. Blaschke, Drug Discovery Today, 2018, 23, 1241–1250 CrossRef.
- R. Chen, X. Liu, S. Jin, J. Lin and J. Liu, Molecules, 2018, 23, 2208 CrossRef.
- A. Jung, arXiv e-prints, 2018, arXiv:1805.05052.
- T. Ching, D. S. Himmelstein, B. K. Beaulieu-Jones, A. A. Kalinin, B. T. Do, G. P. Way, E. Ferrero, P. M. Agapow, M. Zietz, M. M. Hoffman, W. Xie, G. L. Rosen, B. J. Lengerich, J. Israeli, J. Lanchantin, S. Woloszynek, A. E. Carpenter, A. Shrikumar, J. Xu, E. M. Cofer, C. A. Lavender, S. C. Turaga, A. M. Alexandari, Z. Lu, D. J. Harris, D. DeCaprio, Y. Qi, A. Kundaje, Y. Peng, L. K. Wiley, M. H. S. Segler, S. M. Boca, S. J. Swamidass, A. Huang, A. Gitter and C. S. Greene, J. R. Soc., Interface, 2018, 15, 20170387 CrossRef.
- J. H. Yang, S. N. Wright, M. Hamblin, D. McCloskey, M. A. Alcantar, L. Schrubbers, A. J. Lopatkin, S. Satish, A. Nili, B. O. Palsson, G. C. Walker and J. J. Collins, Cell, 2019, 177, 1649–1661.e1649 CrossRef CAS.
- T. S. Gardner, D. di Bernardo, D. Lorenz and J. J. Collins, Science, 2003, 301, 102–105 CrossRef CAS.
- J. H. Yang, S. C. Bening and J. J. Collins, Curr. Opin. Microbiol., 2017, 39, 73–80 CrossRef CAS.
- F. Martinez-Jimenez, G. Papadatos, L. Yang, I. M. Wallace, V. Kumar, U. Pieper, A. Sali, J. R. Brown, J. P. Overington and M. A. Marti-Renom, PLoS Comput. Biol., 2013, 9, e1003253 CrossRef.
- M. J. Rebollo-Lopez, J. Lelievre, D. Alvarez-Gomez, J. Castro-Pichel, F. Martinez-Jimenez, G. Papadatos, V. Kumar, G. Colmenarejo, G. Mugumbate, M. Hurle, V. Barroso, R. J. Young, M. Martinez-Hoyos, R. Gonzalez del Rio, R. H. Bates, E. M. Lopez-Roman, A. Mendoza-Losana, J. R. Brown, E. Alvarez-Ruiz, M. A. Marti-Renom, J. P. Overington, N. Cammack, L. Ballell and D. Barros-Aguire, PLoS One, 2015, 10, e0142293 CrossRef.
- I. Barberis, N. L. Bragazzi, L. Galluzzo and M. Martini, J. Prev. Med. Hyg., 2017, 58, E9–E12 CAS.
- C. J. Martin, A. F. Carey and S. M. Fortune, Semin. Immunopathol., 2016, 38, 213–220 CrossRef CAS.
- V. Dartois, Nat. Rev. Microbiol., 2014, 12, 159–167 CrossRef CAS.
- B. Gold and C. Nathan, Microbiol. Spectrum, 2017, 5, TBTB2–0031-2016 CrossRef CAS.
- Q. Wang, H. I. M. Boshoff, J. R. Harrison, P. C. Ray, S. R. Green, P. G. Wyatt and C. E. Barry 3rd, Science, 2020, 367, 1147–1151 CrossRef CAS.
- L. Pasquina, J. P. Santa Maria Jr, B. McKay Wood, S. H. Moussa, L. M. Matano, M. Santiago, S. E. Martin, W. Lee, T. C. Meredith and S. Walker, Nat. Chem. Biol., 2016, 12, 40–45 CrossRef CAS.
- J. M. Stokes, K. Yang, K. Swanson, W. Jin, A. Cubillos-Ruiz, N. M. Donghia, C. R. MacNair, S. French, L. A. Carfrae, Z. Bloom-Ackerman, V. M. Tran, A. Chiappino-Pepe, A. H. Badran, I. W. Andrews, E. J. Chory, G. M. Church, E. D. Brown, T. S. Jaakkola, R. Barzilay and J. J. Collins, Cell, 2020, 180, 688–702.e613 CrossRef CAS.
- Y. Yamanishi, M. Araki, A. Gutteridge, W. Honda and M. Kanehisa, Bioinformatics, 2008, 24, i232–240 CrossRef CAS.
- Z. He, J. Zhang, X. H. Shi, L. L. Hu, X. Kong, Y. D. Cai and K. C. Chou, PLoS One, 2010, 5, e9603 CrossRef.
Footnote |
| † Contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |