
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical methods for protein site-specific ubiquitination
Weijun
Gui
,
Gregory A.
Davidson
and
Zhihao
Zhuang
 *
*
Department of Chemistry and Biochemistry, University of Delaware, 214A Drake Hall, Newark, DE 19716, USA. E-mail: zzhuang@udel.edu
First published on 25th February 2021
Abstract
Ubiquitination is an important protein post-translational modification regulating many cellular processes in eukaryotes. Ubiquitination is catalyzed by a three-enzyme cascade resulting in the conjugation of the C-terminal carboxylate of ubiquitin (Ub) to the ε-amino group of a lysine residue in the acceptor protein via an isopeptide bond. In vitro enzymatic ubiquitination utilizing Ub ligases has been successfully employed to generate Ub dimers and polymers. However, limitations of the enzymatic approach exist, particularly due to the requirement of specific Ub ligase for any given target protein and the low catalytic efficiency of the Ub ligase. To achieve an in-depth understanding of the molecular mechanism of Ub signaling, new methods are needed to generate mono- and poly-ubiquitinated proteins at a specific site with defined polyubiquitin chain linkage and length. Chemical methods offer an attractive solution to the above-described challenges. In this review, we summarize the recently developed chemical methods for generating ubiquitinated proteins using synthetic and semisynthetic approaches. These new tools and approaches, as an important part of the Ub toolbox, are crucial to our understanding and exploitation of the Ub system for novel therapeutics.
1. Introduction
Ubiquitination, the attachment of ubiquitin (Ub) to the ε-amino group of a lysine residue in the acceptor protein, is a reversible protein post-translational modification (PTM) in eukaryotes, regulating almost every aspect of eukaryotic biological processes including protein degradation, protein trafficking, and DNA damage response.1–4 Arguably, the best-characterized role of the ubiquitination pathway is the degradation of cellular proteins by the 26S proteasome, for which the 2004 Noble Prize in Chemistry was awarded to Aaron Ciechanover, Avram Hershko, and Irwine Rose.5,6Ubiquitination is orchestrated by a series of three enzymes: Ub-activating enzyme (E1), Ub-conjugating enzyme (E2), and ubiquitin ligase (E3).7,8 The first step of ubiquitination is an ATP-dependent reaction catalyzed by E1 generating a thioester linkage between Ub's C-terminus and E1's active site cysteine.9,10 The Ub moiety is then transferred to an E2 through a transthiolation reaction from the charged E1.11 A E3 Ub ligase catalyzes the final step of the ubiquitination by transferring Ub from Ub-E2 to a lysine residue of the substrate protein, forming an isopeptide linkage between the C-terminus of Ub and substrate protein.11–13 The RING E3 ligases, around 600 members in humans, do not form a Ub covalent adduct, while the HECT E3 ligases (around 30 in humans) and RBR E3 ligases (around 12 in humans) form a Ub-thioester intermediate at the E3 active site. The human genome contains 2 E1s, 40 E2s, and more than 600 E3s.14,15
Ubiquitination can occur on many eukaryotic proteins including Ub itself. For Ub, any of the seven lysine residues (K6, K11, K27, K29, K33, K48, and K63) or the N-terminal methionine on Ub can be used to form the amide bond between Ub moieties.4 This results in polyUb chains of specific linkages. K11- and K48-linked polyUb chains are mainly involved in proteasomal degradation of targeted proteins.16,17 K63-linked polyUb chain regulates cell membrane trafficking, protein kinase activation, DNA repair and chromatin dynamics.18 PolyUb chain of M1 linkage regulates NF-κB activation and apoptosis.19 Our knowledge of other atypical polyUb chain linkages (K6, K27, K33) is still limited.20 Notably, we now know that both linear and branched polyUb chains exist in cells and are implicated in different cellular pathways.3 Deubiquitinating enzymes, referred to as DUBs, reverse the ubiquitination process by removing a Ub moiety or a polyUb chain from a receptor protein.21,22 DUBs can also recognize distinct Ub chain linkages and cleave Ub chains with either endo- or exo-cleavage modes, suggesting that Ub chain topology plays a crucial role in Ub-mediated cellular signaling.23
To replicate the ubiquitination process in vitro, enzymatic generation of K48- and K63-linked Ub dimers and polymers using Ub ligases was first reported by Cecile M. Pickart and coworkers in the 1990s,24,25 followed by the report of enzymatic generation of K11-linked dimer by Bremm et al. in the 2000s.26 Later, in vitro assembly of K6, K29, and K33 polyUb using specific E3 ligases were also reported.27–29 More recently, an E2 (Ubc9) mediated ubiquitination and ISGylation was reported.30 While in vitro enzymatic generation of Ub dimers and polymers remains a very useful tool to study the different facets of ubiquitination and deubiquitination, the major limitation of the enzymatic approaches is the requirement of specific Ub ligases for a given chain linkage and a particular target protein.
Understanding the recognition of polyUb by a large number of decoder proteins and the processing of polyUb chains by DUBs requires the generation of different types of polyUb chains of defined linkage in sufficient quantities for biochemical and biophysical studies. Chemical synthesis and semi-synthesis of ubiquitinated protein offer an attractive solution to this challenge.31 Several chemical methods including solid-phase peptide synthesis (SPPS),32–34 native chemical ligation (NCL),35–50 silver-mediated chemical ubiquitination51,52 have been developed to generate a native isopeptide linkage between Ub moieties in a polyUb chain or between Ub and a receptor protein (Fig. 1). Furthermore, chemical ubiquitination using α-halogen ketone ligation,53–56 disulfide exchange,57–61 Cu(I)-catalyzed azide–alkyne cycloaddition,62–64 thiol–ene coupling,65,66 oxime and hydrazone formation,66–70 conjugate addition,60,71–74 sortase-mediated,75 and cysteine-aminoethylation assisted chemical ubiquitination76,77 were also reported to generate ubiquitinated proteins with a non-native linkage. In this review, we summarize the chemical methods that have been developed for Ub chain formation and site-specific protein ubiquitination as well as the utility of the chemically synthesized ubiquitinated proteins in understanding their roles in the relevant cellular processes.
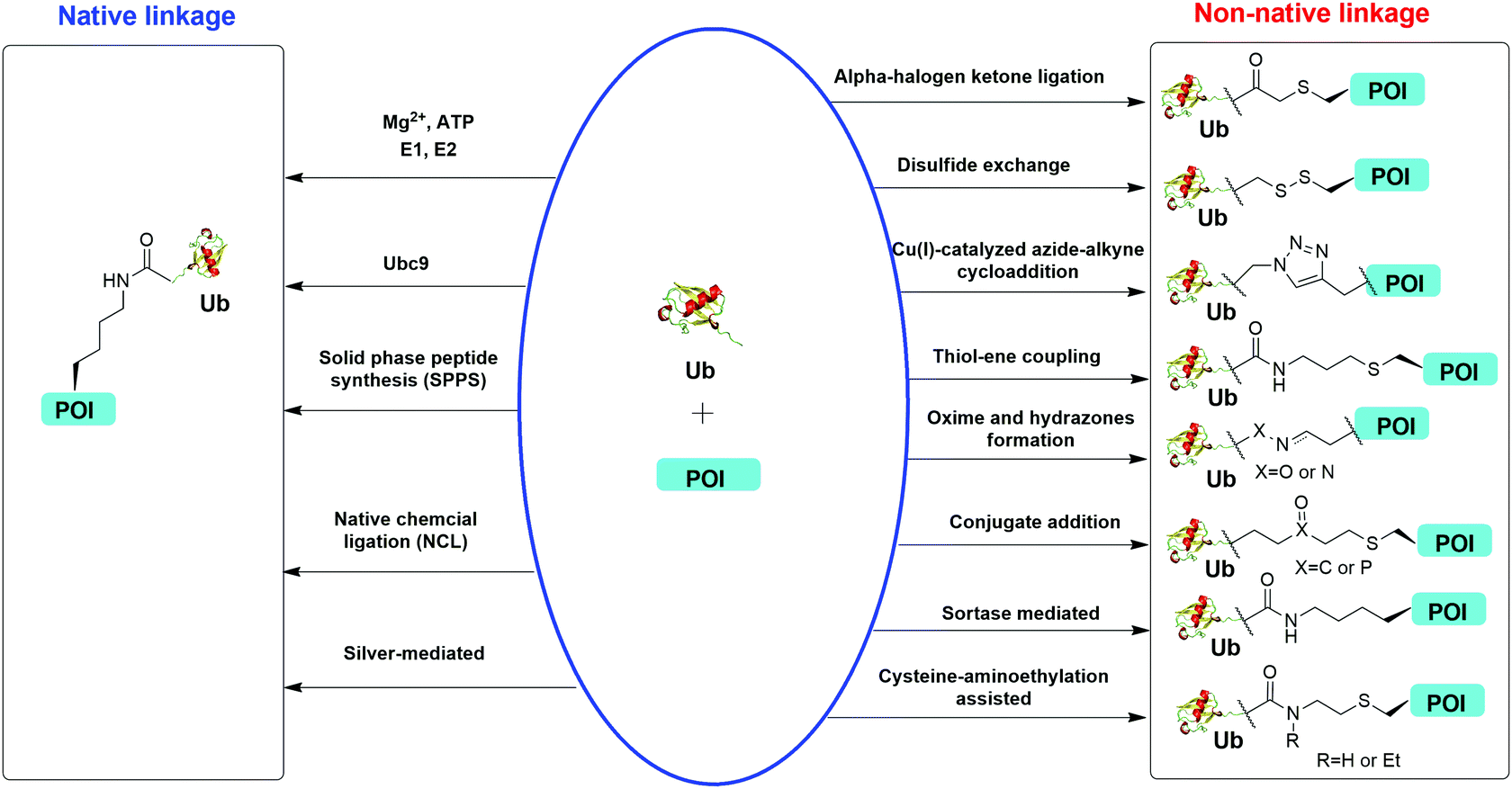 | ||
| Fig. 1 Overview of in vitro chemical methods for protein site-specific ubiquitination. POI stands for protein of interest. | ||
2. Chemical methods for protein site-specific ubiquitination through a native isopeptide linkage
2.1 Enzymatic ubiquitination using ubiquitin ligases
An initial breakthrough in enzymatic ubiquitination was the successful in vitro synthesis of Ub dimers and polymers using Ub-activating enzyme E1, Ub-conjugating enzyme E2 and ATP in the absence of an E3 Ub ligase by Pickart and coworkers. In 1990, Chen et al. reported that the E2 enzyme (E225K) is capable of catalyzing the formation of K48-linked polyUb chain in vitro in the absence of an E3 ligase (Fig. 2A).78 In this report, the authors showed that incubation of E1, E225K and radiolabeled Ub in the presence of Mg2+ and ATP led to the formation of radiolabeled Ub dimers and polymer products. Tryptic digestion of the Ub dimer and polymer confirmed the isopeptide linkage between the Ub moieties. Later in 2001, Hofmann et al. reported that a heterodimeric complex of Ubc13 (an E2 enzyme) and Mms2 (an E2 variant) can assemble Lys63-linked Ub chains in vitro without an E3 ligase (Fig. 2B).79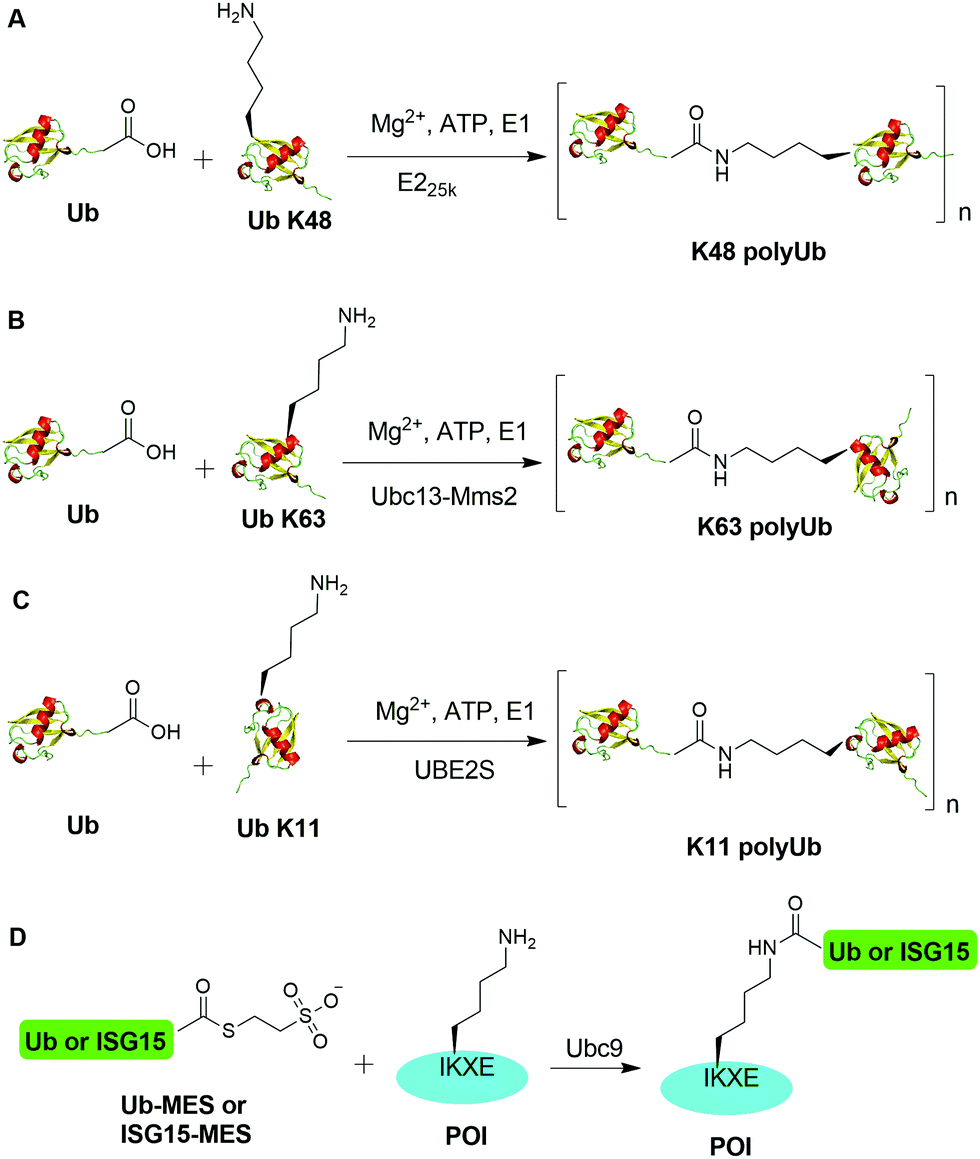 | ||
| Fig. 2 Enzymatic generation of polyubiquitin using Ub pathway enzymes. Enzymatic generation of K48-linked (A), K63-linked (B), and K11-linked (C) Ub polymers. (D) Lysine acylation using Ub conjugating enzyme Ubc9 for site-specific ubiquitination and ISGylation of recombinant proteins. | ||
In order to generate Ub dimers and polymers with distinguishable distal and proximal Ub for biochemical studies, mixed populations of lysine-blocked and C-terminally blocked Ub mutants were used to synthesize the K48- or K63-linked Ub chains of defined length with specific Ub chain orientation.80–82 For example, in order to generate K48-linked Ub dimer, a specific E2 (E225K) was incubated with a distal Ub with Lys48 mutated to Arg (UbK48R) and a proximal Ub mutant in which an additional Asp was added to wide-type Ub (UbD77) to cap the C terminus of proximal Ub. Using this method, milligram of Ub dimers of discrete lengths can be obtained in an one-step synthesis reaction.80In vitro synthesized Ub dimer and tetramer using this method have facilitated the structural understanding of different Ub chains, Ub chains recognition by Ub-binding proteins, and activity and specificity of DUBs that disassemble the polyUb chains.83–85 Besides the well-studied K48 and K63 Ub linkages, atypical Ub linkage (M1, K6, K11, K27, K29, K33) are also crucial for eukaryotic cells.86In vitro enzymatic assembly of K6, K11, K29, K33 polyUb chains using specific Ub ligases in conjunction with DUBs were described recently (see Fig. 2C for K11 Ub chain synthesis as an example).87
Recently, another breakthrough of enzymatic ubiquitination by the E2 SUMO-conjugating enzyme Ubc9 was reported by Hofmann et al. (Fig. 2D).30 The authors named this method as lysine acylation using conjugating enzymes (LACE), which enables site-specific modifications including ubiquitination and ISGylation of recombinant proteins with just Ubc9 in the absence of E1 and E3 enzymes. According to this report, the authors found that Ubc9, a SUMO-conjugating enzyme, recognizes and modifies the lysine embedded in a consensus SUMOylation motif ΨKXD/E (Ψ is a hydrophobic residue) in a recombinant protein, yielding an isopeptide linkage. By exploiting the unique activity of Ubc9, the authors achieved site-specific ubiquitination and ISGylation of targeted lysine residue in recombinant proteins including SUMO2 and α-synuclein with minimal modification of residues flanking the target lysine.
2.2 Chemical ubiquitination using solid-phase peptide synthesis
Solid-phase peptide synthesis (SPPS) has proven to be a powerful tool in generating peptides and proteins since its development in 1963.88 Using SPPS, total synthesis of Ub and ubiquitinated substrate protein were first achieved in the 1990s. In 1994, Ramage et al. reported the total chemical synthesis of Ub on an useful scale (100–200 mg) by stepwise SPPS (Fig. 3A).89 Total chemical synthesis of Ub enabled the preparation of Ub analogs by insertion of unnatural amino acids or isotopically labeled amino acids, allowing for a better understanding of cellular Ub signaling process. In 1999, Layfield et al. reported the synthesis of a linear fusion of Ub and 52-amino-acid ribosomal protein (UBICEP52) through a native peptide linkage between Ub C-terminal carboxylate and the N-terminal of ICEP52 using SPPS (Fig. 3B).32 Moreover, the authors generated Ub-Lys and Ub-Val by conjugating the ε-amino group of the lysine residue and α-amine of Val to the C-terminus of Ub using SPPS respectively. The study showed that the Ub moiety in the synthesized UBICEP52, Ub-Lys and Ub-Val can be cleaved by DUBs.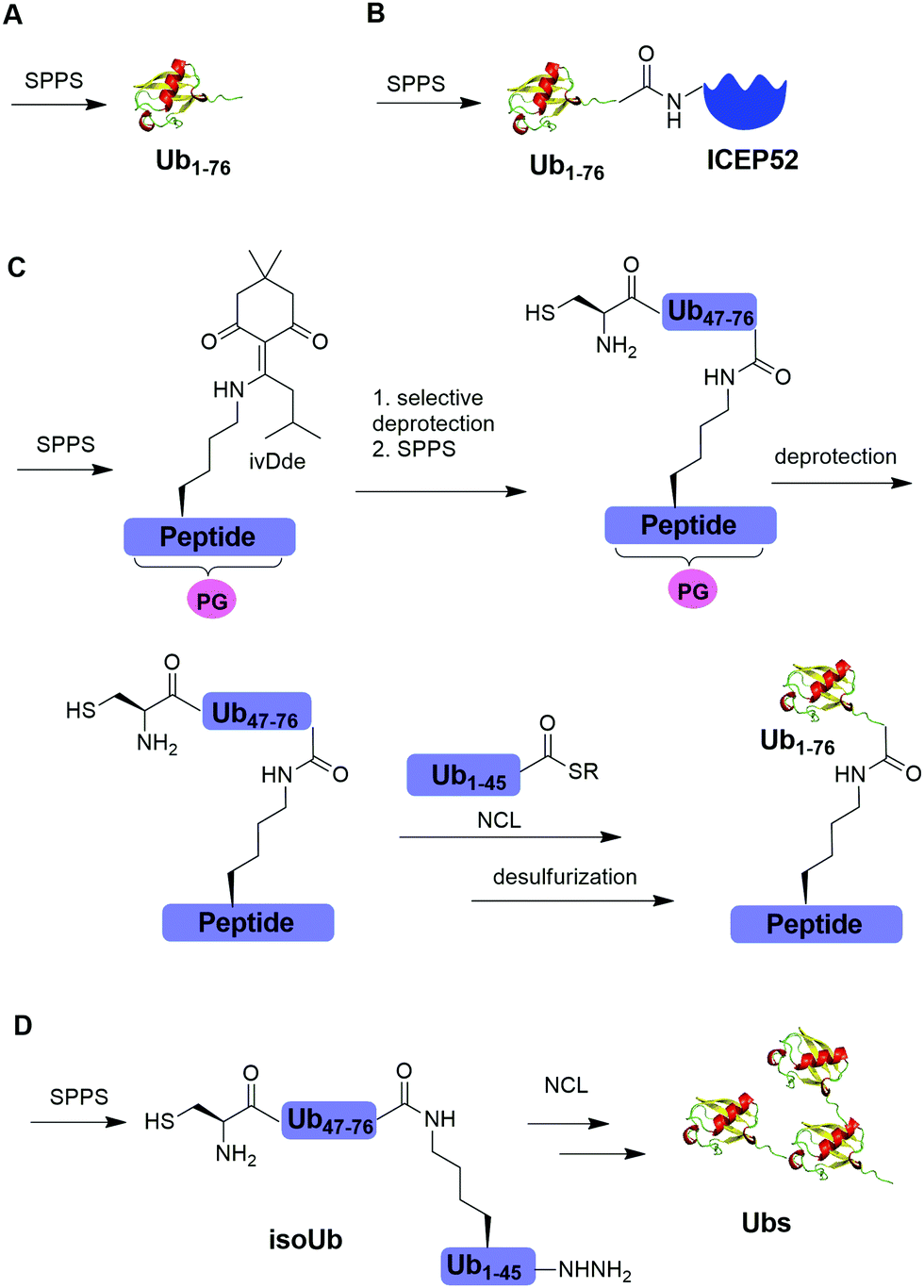 | ||
| Fig. 3 Chemical ubiquitination using SPPS. (A) Total synthesis of Ub using SPPS. (B) Generation of Ub-ICEP52 using SPPS. (C) Synthesis of the ubiquitinated peptide using SPPS. (D) Synthesis of atypical Ub chains using isoUb strategy. | ||
In 2011, Kumar et al. reported the generation of native isopeptide linked Ub-peptide using SPPS (Fig. 3C).33 In this report, the authors used SPPS to incorporate a 1-[4,4-dimethyl-2,6-dioxo-cyclohexylidene]-3-methylbutyl (ivDde) orthogonally protected Lys into the designated position of Ub chain. The other Lys residues in the Ub chain were protected by tert-butoxycarbonyl (Boc). Subsequently, selective deprotection of the ivDde allowed the SPPS elongation of C-terminal Ub. Followed by extension of N-terminus of Ub through the ligation of Cys with Ub1–45 thioester enabled the generation of the full-length native linked ubiquitinated peptide after desulfurization.
In 2017, Tang et al. reported an isoUb strategy for efficient synthesis of atypical Ub chains, especially for K11,K48-branched Ub chains (Fig. 3D).34 In this report, the authors used a premade isopeptide-linked 76-mer synthon (isoUb) that contains an N-terminal Cys and a C-terminal hydrazide as a key building block to assemble atypical Ub chains. Following ligation of the isoUb building block by NCL, several atypical Ub chains of defined linkage and length including K27-linked tetra-Ub and K11/K48-branched tri-, tetra-, penta-, and hexa-Ubs were prepared successfully.
2.3 Chemical ubiquitination using native chemical ligation
Native chemical ligation (NCL), characterized by the ligation of a N-terminal cysteine-containing peptide and a second peptide possessing a C-terminal thioester and the formation of a native peptide bond at the ligation junction, is a powerful chemical method of synthesizing native and modified proteins of moderate size.90 The total synthesis of Ub and diUb have been achieved using NCL coupled with SPPS. In 2005, the Kent group reported the successful generation of Ub using NCL and SPPS.91 However, NCL usually leaves a Cys residue at the ligation junction, limiting its application in generating native isopeptide linkage between Ub and protein of interest (POI). Auxiliary groups, which can be removed under mild conditions such as weak acid or UV light, provide a good solution to this problem.One example of using auxiliary group-mediated chemical ubiquitination is histone ubiquitination. Previous studies have shown that ubiquitination of histone H2A and H2B is of great importance in the DNA damage response,92,93 gene silencing,94 transcription elongation.95 To fully understand the cellular process that involves Ub modification of histones requires homogeneous ubiquitinated histones with good quantity and purity for biochemical and structural characterization. However, in vitro reconstitution of ubiquitinated histone using Ub ligases and associated factors only allows the production of ubiquitinated histones in a very limited quantity.96 Photocleavable auxiliary-mediated NCL coupled with expressed protein ligation (EPL) provides a wonderful example to generate ubiquitinated histones in vitro. In 2007, Chatterjee et al. reported a photocleavable auxiliary-mediated site-specific ubiquitination of H2B peptide at Lys120 position (Fig. 4A).35 In this report, the authors first used SPPS to incorporate ivDde-protected Lys at the designated position in the H2B peptide and Boc-protected Lys at other Lys positions. Then, the photocleavable auxiliary can be incorporated into Lys120 of H2B following a reaction sequence: selective deprotection of the designated Lys residue in the H2B peptide, amide coupling of lysine sidechain amine with 2-bromoacetic acid, and a nucleophilic substitution of the α-bromide by the amine group in the photocleavable auxiliary. Then, the exposed free thiol in the auxiliary facilitates the formation of a native amide bond between C-terminus of Ub1–75 and the modified Lys side chain in H2B (Fig. 4A). The photocleavable auxiliary group can be readily removed by 325 nm UV irradiation generating a native isopeptide linkage between Ub and H2B peptide. Later in 2008, using a similar photocleavable auxiliary strategy coupled with NCL and desulfurization, the Muir lab reported the successful generation of the full-length ubiquitinated H2B at Lys120 position.36 Importantly, the synthesized Ub-H2B can be reconstituted into nucleosomes and revealed that Ub-H2B at Lys120 activates methylation of histone H3 at Lys79 by the histone lysine methyltransferase hDot1L.
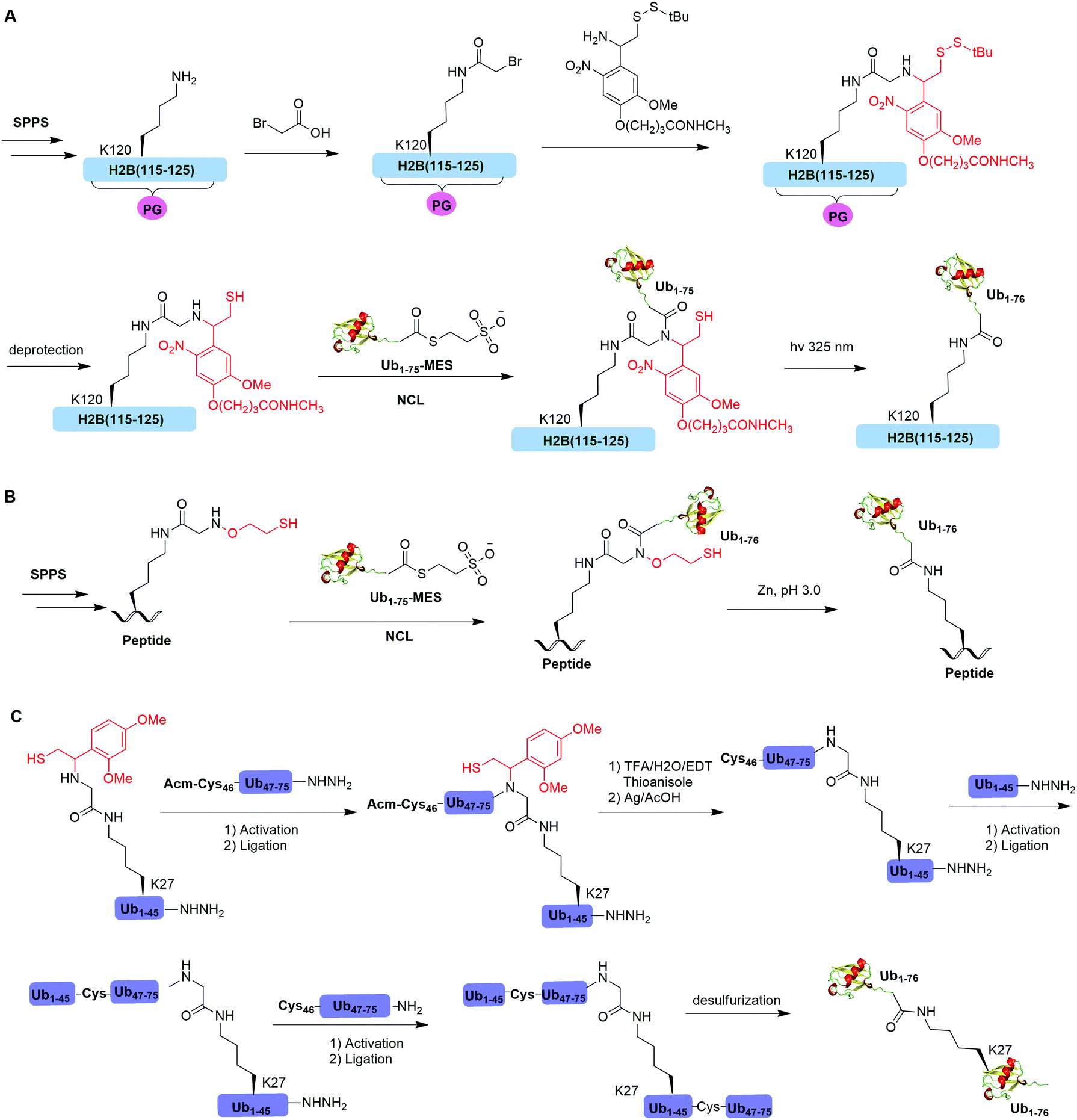 | ||
| Fig. 4 Auxiliary mediated NCL to generate natively-linked ubiquitinated peptides and proteins. (A) Photocleavable auxiliary- mediated NCL to generate ubiquitinated H2B peptide. (B) 2-(aminooxy)ethanethiol-mediated chemical ubiquitination. (C) 1-(2,4-Dimethoxyphenyl)-2-mercaptoethyl-mediated chemical ubiquitination. | ||
Two other auxiliary-mediated chemical ubiquitination methods were also reported. In 2014, Weller et al. reported a 2-(aminooxy)ethanethiol auxiliary-mediated chemical ubiquitination method (Fig. 4B).47 In this report, SPPS was used to generate a Ub peptide with a specific lysine modified by 2-(aminooxy)ethanethiol auxiliary group. Then, the Ub peptide undergoes NCL with Ub(1–75)-α-thioester. Finally, the auxiliary group can be removed by zinc in an acidic condition and with 6 M Gn·HCl generating a native isopeptide linkage at the ligation junction. Importantly, this auxiliary-mediated chemical ubiquitination method has several advantages in terms of ligation rate and compatibility with Cys residues in the POI. Notably, the post-ligation product with an N-substituted Gly76 in Ub resists the hydrolysis by DUBs. However, the removal of the auxiliary group using Zn under pH 3 limits its application to unstructured peptide or protein. Later, in 2016, Weller et al. optimized the removal strategy of the auxiliary by 4-mercaptophenylacetic acid-mediated cleavage of N–O bonds for chemical ubiquitination of folded proteins allowing for the generation of full-length SUMO-H2B.48
In 2016, Pan et al. reported that the trifluoroacetic acid (TFA)-labile 1-(2,4-dimethoxyphenyl)-2-mercaptoethyl can be used as an auxiliary to synthesize K27-linked Ub dimers and trimers (Fig. 4C).49 In this study, SPPS was used to generate Ub1–45 with K27 modified by the auxiliary group. After NCL with Acm-Cys46-Ub47–75-NHNH2, the auxiliary group can be removed by TFA/H2O/EDT/thioanisole and Ag/AcOH. Then full-length K27-linked Ub dimer can be successfully generated by NCL and desulfurization. Notably, using quasi-racemic crystallography the authors revealed that the crystal structure of K27-linked diUb and triUb are symmetric and triangular respectively.
Auxiliary-mediated chemical ubiquitination is a useful method to generate ubiquitinated proteins with a native isopeptide linkage. However, the ligation reaction exhibits slow kinetics due to steric hindrance imposed by the auxiliary-bearing secondary amine. To overcome this problem, another breakthrough in the chemical ubiquitination was made by the installation of a thiol handle at the γ- or δ-carbon of a lysine residue, which can undergo NCL with Ub-MES to generate a natively linked ubiquitinated protein after desulfurization. In 2009, Yang et al. reported a γ-thiol lysine mediated chemical ubiquitination through dual NCL (Fig. 5A).37 In this study, the authors used SPPS to incorporate a carboxybenzyl (Cbz) protected γ-thiol lysine residue into the peptide, which can undergo NCL two times with N-terminal peptide and Ub-MES respectively, yielding a natively linked Ub-peptide as the final product after desulfurization. Later in 2010, Yang et al. reported an optimized dual NCL strategy by using a photolabile NVOC protection group instead of the acid-labile Cbz to protect the ε-amino group of Lys, and successfully generated a native K48-linked diUb as the final product.39
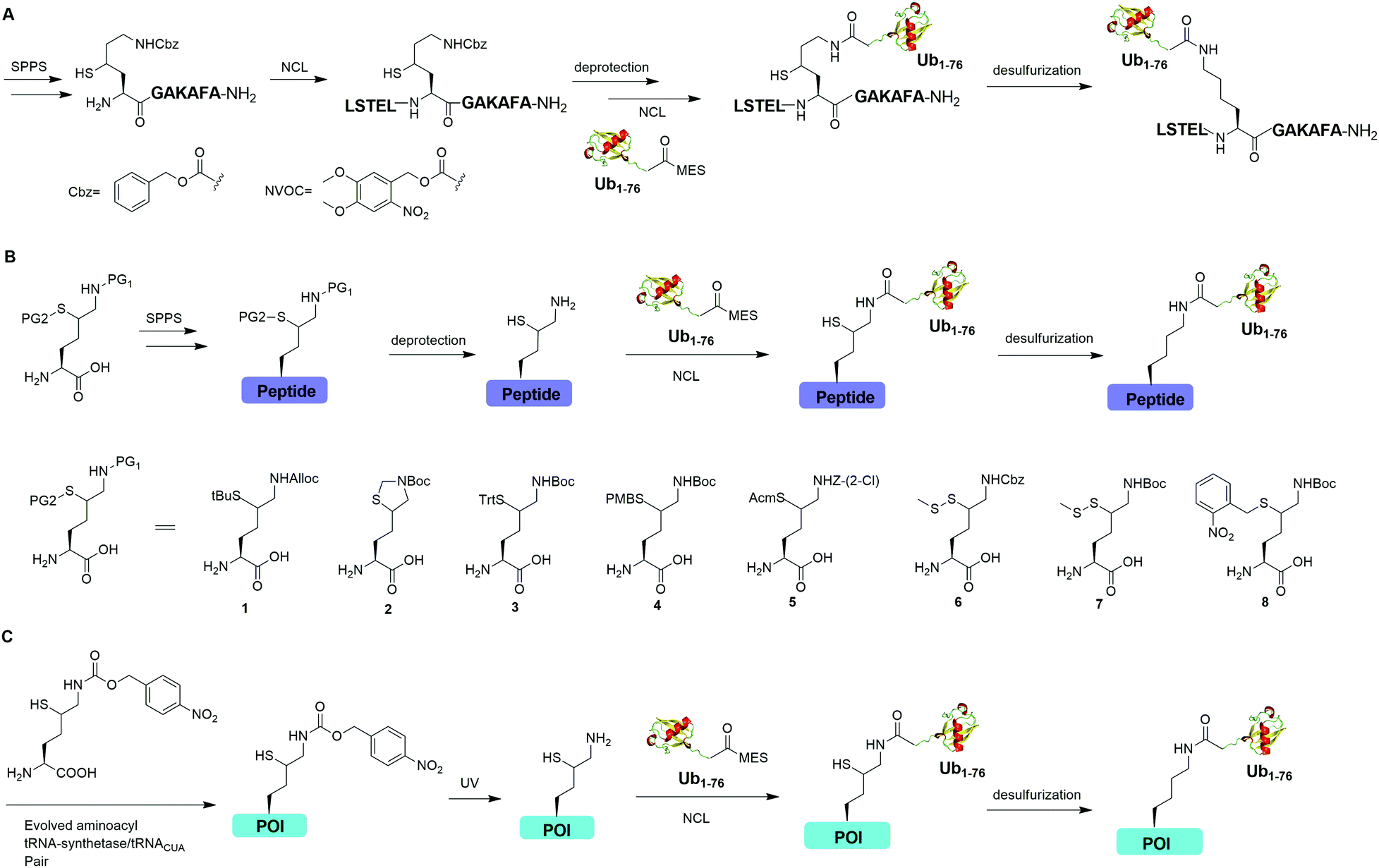 | ||
| Fig. 5 γ- or δ-Thiol lysine-mediated NCL to generate native ubiquitinated peptide and protein. (A) γ-Thiol lysine-mediated dual NCL to generate native Ub-peptide. (B) δ-Thiol lysine-mediated NCL to generate native Ub-peptide. (C) Genetic incorporation of δ-thiol-Nε-(p-nitrocarbobenzyloxy)lysine for NCL to generate ubiquitinated protein (POI) with a native isopeptide linkage. | ||
In 2009, Kumar et al. used SPPS to incorporate a protected δ-mercaptolysine residue into a target peptide. After selective deprotection, a δ-mercaptolysine-mediated NCL leads to a native linkage between Ub and the peptide after desulfurization (Fig. 5B).38 Later, Brik and coworkers reported several protection strategies for δ-mercaptolysine analogues (structures showed in Fig. 5B), in particular the thiazolidine protected δ-mercaptolysine, employed in the synthesis of various Ub chains and ubiquitinated substrates.31,40 δ-Mercaptolysine mediated chemical ubiquitination represents a very successful strategy to generate homogeneous site-specific ubiquitinated POI samples for decoding the function of ubiquitinated POI.31,42,97 Moreover, this strategy was also used in the generation of Ub-based chemical probes to study the activity of DUBs.98
In 2011, Virdee et al. reported a genetic approach using a pyrrolysyl-tRNA synthetase/tRNACUA pair to incorporate a photocaged unnatural amino acid δ-thiol-Nε-(p-nitrocarbobenzyloxy)lysine into specific lysine position of targeted protein, which enabled the traceless chemical ubiquitination of POI.43 In this report, a native isopeptide bond between the target protein and Ub was successfully generated through photo-deprotection to free the thiol–lysine residue followed by NCL with Ub-MES and desulfurization (Fig. 5C).
A notable example of δ-mercaptolysine-mediated chemical ubiquitination is that of α-synuclein (α-syn). α-Syn is a 140-residue intrinsically disordered presynaptic protein, involved in the pathogenesis of Parkinson's disease (PD) and several other neurodegenerative disorders.99–101 Interestingly, mono-, and poly-ubiquitination of α-syn have been associated with Lewy bodies of PD patients.102 While previous studies showed that monoubiquitination of α-syn inhibits rather than promotes α-syn fibrillization, the function of polyubiquitination of α-syn is not clear. In vitro enzymatic site-specific ubiquitination of α-syn at a single or multiple lysine residues remains difficult to achieve. Coexpression of α-syn with Ub ligases results in mainly mono- and di-ubiquitination at multiple sites.103,104 To overcome these challenges, Hejjaoui et al. in 2010 reported a δ-mercaptolysine-mediated monoubiquitination of α-syn at Lys6 (Fig. 6A).44 The authors used recombinantly expressed α-syn fragment comprising residues 19–140 with a Cys residue at the N-terminus and SPPS-generated peptide containing residues 1–18 of α-syn with a protected δ-mercaptolysine replacing the Lys6 in α-syn. Then NCL was used to connect the two pieces of peptide and generate a full-length α-syn mutant with the original Ala19 replaced by Cys. Followed by the removal of the protection group of δ-mercaptolysine in the α-syn intermediate, Ub can be attached to the α-syn mutant through NCL. Finally, global desulfurization to remove the thiol yielded ubiquitinated α-syn with a native isopeptide linkage between the C-terminus of Ub and the Lys6 of α-syn. Importantly, biochemical studies using the chemical synthesized Ub-α-syn supported the idea that ubiquitination of α-syn at its N-terminal region stabilizes the monomeric form of α-syn and prevents its oligomerization and fibrillogenesis.
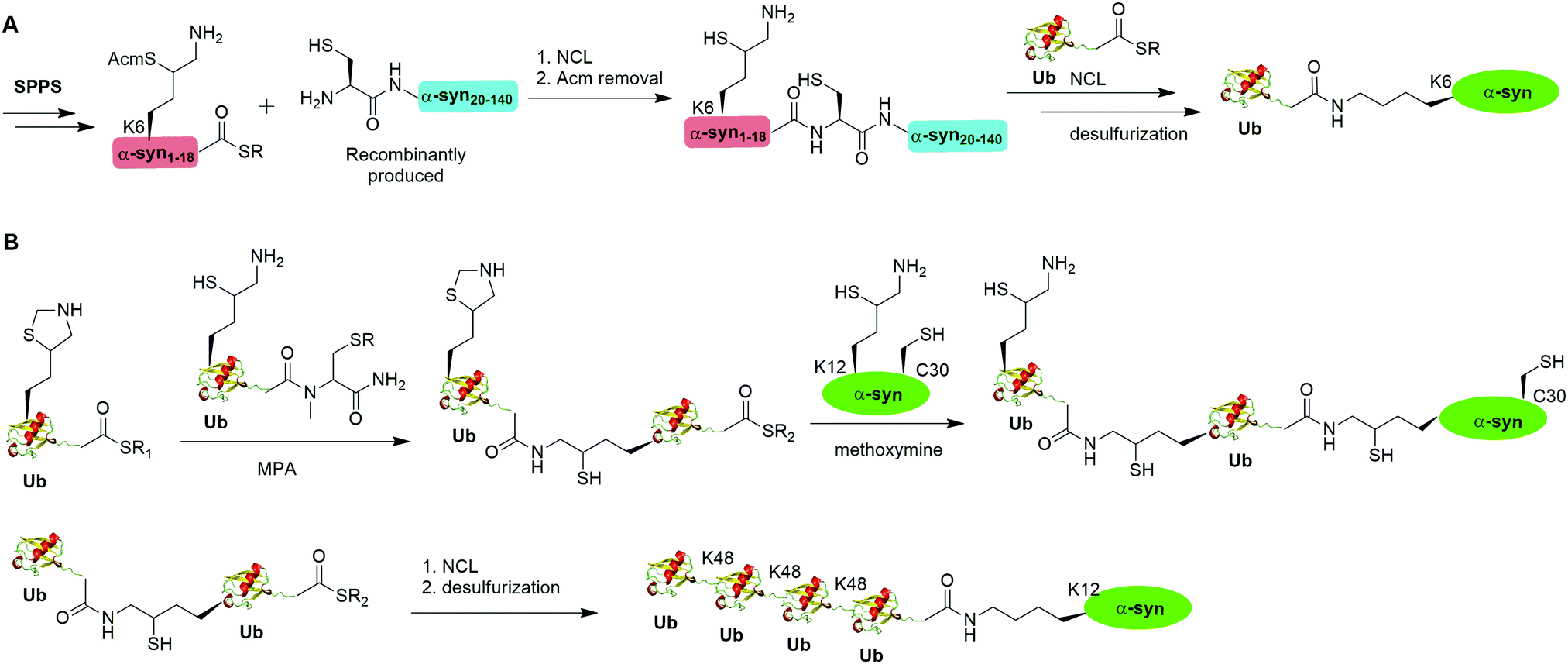 | ||
| Fig. 6 δ-Mercaptolysine-mediated NCL to generate mono- and ploy-ubiquitinated α-syn. (A) δ-Mercaptolysine-mediated NCL to generate monoubiquitinated α-syn. (B) δ-Mercaptolysine-mediated NCL to generate ployubiquitinated α-syn. | ||
By exploiting the δ-mercaptolysine-mediated chemical ubiquitination approach, the total synthesis of diUb of all seven linkages41 as well as K48-linked tetraUb45 were achieved by the Brik group. Further, Haj-Yahya et al. reported a semisynthetic strategy to generate polyubiquitinated α-syn (Fig. 6B).46 The authors used SPPS to incorporate a δ-mercaptolysine at the desired lysine position of Ub. After NCL with a Ub mutant containing a δ-mercaptolysine, a diUb intermediate was generated. Then, 4-mercaptophenylacetic acid was used to activate the C-terminus of the diUb intermediate followed by an NCL with the δ-mercaptolysine-containing α-syn generating a diUb-α-syn. Then the protected δ-mercaptolysine can be unmasked by methoxylamine and react with diUb mutant through a NCL reaction yielding a tetraUb-α-syn. After global desulfurization, a native isopeptide linked tetraUb-α-syn was generated. Biochemical studies using the generated Ub-α-syn species indicated that di- and tetra-ubiquitinated α-syn, but not mono-ubiquitinated α-syn, are prone to proteasome degradation and protected from cleavage by proteasome-associated deubiquitinases.
Another example of δ-mercaptolysine mediated chemical ubiquitination is α-globin. Previous studies showed ubiquitination of α-globin can lead to its degradation by the proteasomal system in cells.105–108 Studying the fate of mono- and poly-ubiquitination of α-globin at specific sites requires homogeneous samples with good quantity and purity. In 2019, Sun et al. reported the generation of K48-linked tetraUb α-globin at Lys105 position of the protein using the δ-mercaptolysine-mediated chemical ubiquitination strategy similar to the generation of polyUb α-syn.50 In this report, the authors revealed the diverse fate of Ub moieties in polyubiquitinated α-globin. The distal Ub in the tetra-Ub adduct was efficiently removed by DUBs while the proximal Ub was predominantly degraded with the substrate. The degradation efficiency was proportional to the Ub chain length with tetra-Ub globin being an efficient substrate for proteasome and little degradation of mono-Ub globin.
2.4 Silver-mediated chemical ubiquitination
In 2010, Virdee et al. reported a powerful combination of genetic code expansion and chemoselective chemical reactions to generate native K6- and K29-linked Ub dimer, which allowed structural characterization of K6-linked diUb and profiling of DUB specificity toward K6- and K29-linked Ub chains (Fig. 7A).51 In this report, the Boc-protected Lys was recombinantly encoded at the specific position in Ub using the PylRS/tRNACUA pair, while all other Lys residues were orthogonally protected with N-(benzyloxycarbonyloxy)succinimide (Cbz-OSu). After selective removal of the Boc-protecting group from the designated Lys, silver-mediated direct coupling reaction between the Lys and a Ub-thioester enables the generation of a native isopeptide bond between two Ubs. Finally, global deprotection of other Lys residues yields the native K6 and K29-linked Ub dimers.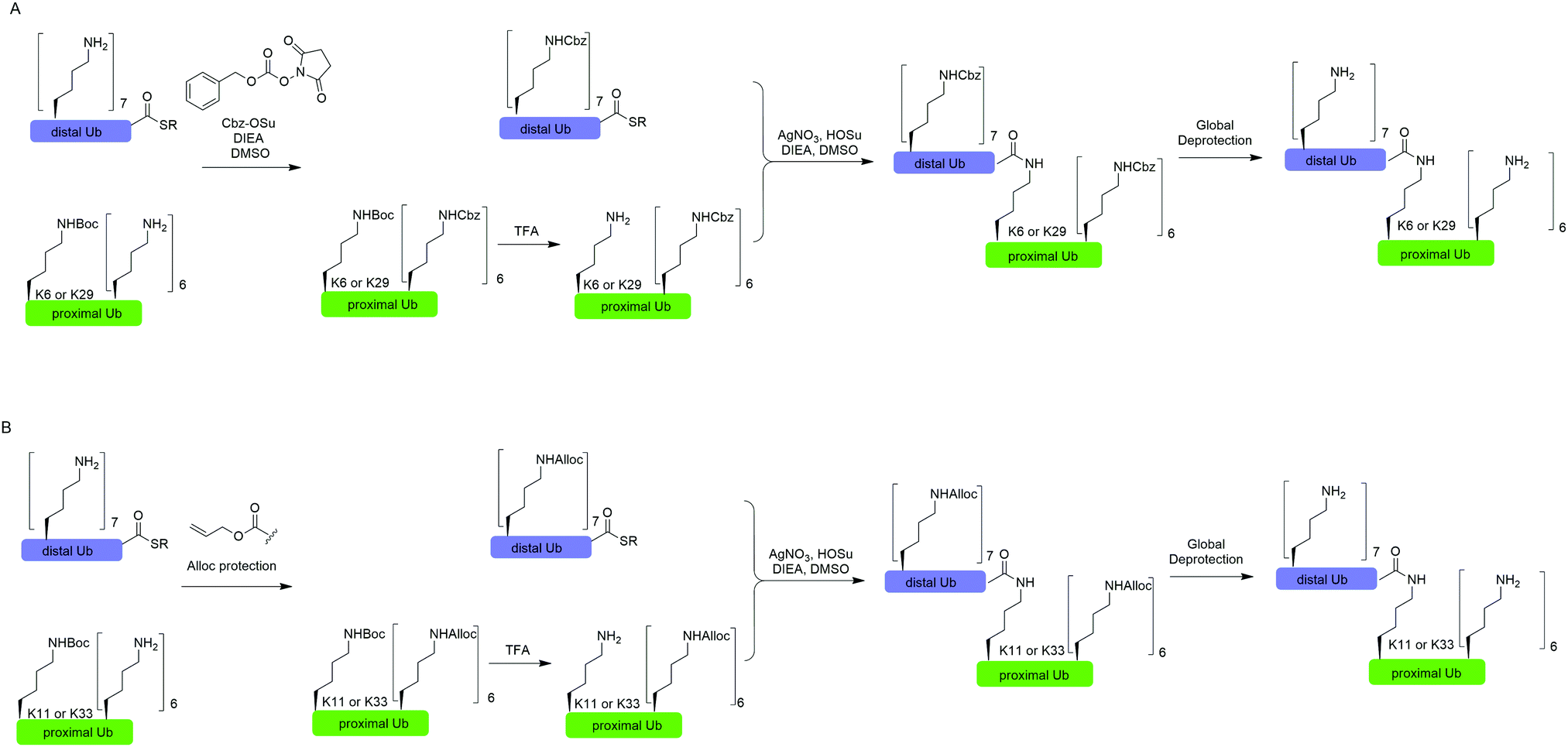 | ||
| Fig. 7 Silver-mediated chemical ubiquitination to generate K6- and K29-linked diUb (A) and K11- and K33-linked diUb (B). | ||
In 2011, Castañeda et al. reported the generation of native K11-linked di-, tri-, and tetra-Ubs, K33-linked diUb, and K11, K33-linked triUb using a similar method (Fig. 7B).52 In this report, instead of using Cbz, the authors used allyloxycarbonyl (Alloc) group to protect Lys residues in the Ub chain for the reason that Alloc can be removed under a milder condition.
3. Chemical ubiquitination through a non-native isopeptide linkage
Chemical methods allowed the preparation of ubiquitinated proteins with a native isopeptide linkage are often difficult to obtain. These methods work best for proteins of small size and can be readily refolded following reactions under harsh denaturing conditions. To address these limitations, several chemical ubiquitination methods have been developed to generate proteins with site-specific ubiquitination through a linkage mimicking the native one. These methods have expanded the repertoire of ubiquitinated proteins and provided facile methods for generating homogeneous ubiquitinated proteins with a good quantity.3.1 Alpha-halogen ketone-mediated chemical ubiquitination
Alpha-halogen ketone-mediated chemical ligation with thiol provides a facile method to generate ubiquitinated POI. Instead of incorporating an unnatural amino acid into the protein, the nucleophilicity of the thiol group in a cysteine residue was exploited for chemical ligation with Ub that contains a C-terminal reactive electrophile. In 2000, Yin et al. reported the generation of K11, K29, K48 and K63-linked nonhydrolyzable diUb analogues using 1,3-dichloroacetone as the cross-linking reagent (Fig. 8A).53 For the design of the Ub mutants, the authors used site-directed mutagenesis to replace the targeted Lys with Cys on the proximal Ub and the C-terminal Gly76 with Cys on the distal Ub respectively. 1,3-Dichloroacetone was used to react with the Cys in proximal Ub yielding a Ub-chloromethyl ketone adduct. Then an equimolar amount of distal UbG76C mutant was added to the mixture generating the desired diUb as the final product. Importantly, the study revealed that these final diUb products are resistant to DUB cleavage and inhibit the activity of several DUBs tested. The introduction of a Cys at the C-terminus of Ub for chemical ligation led to a relatively large deviation from the native diUb isopeptide linkage.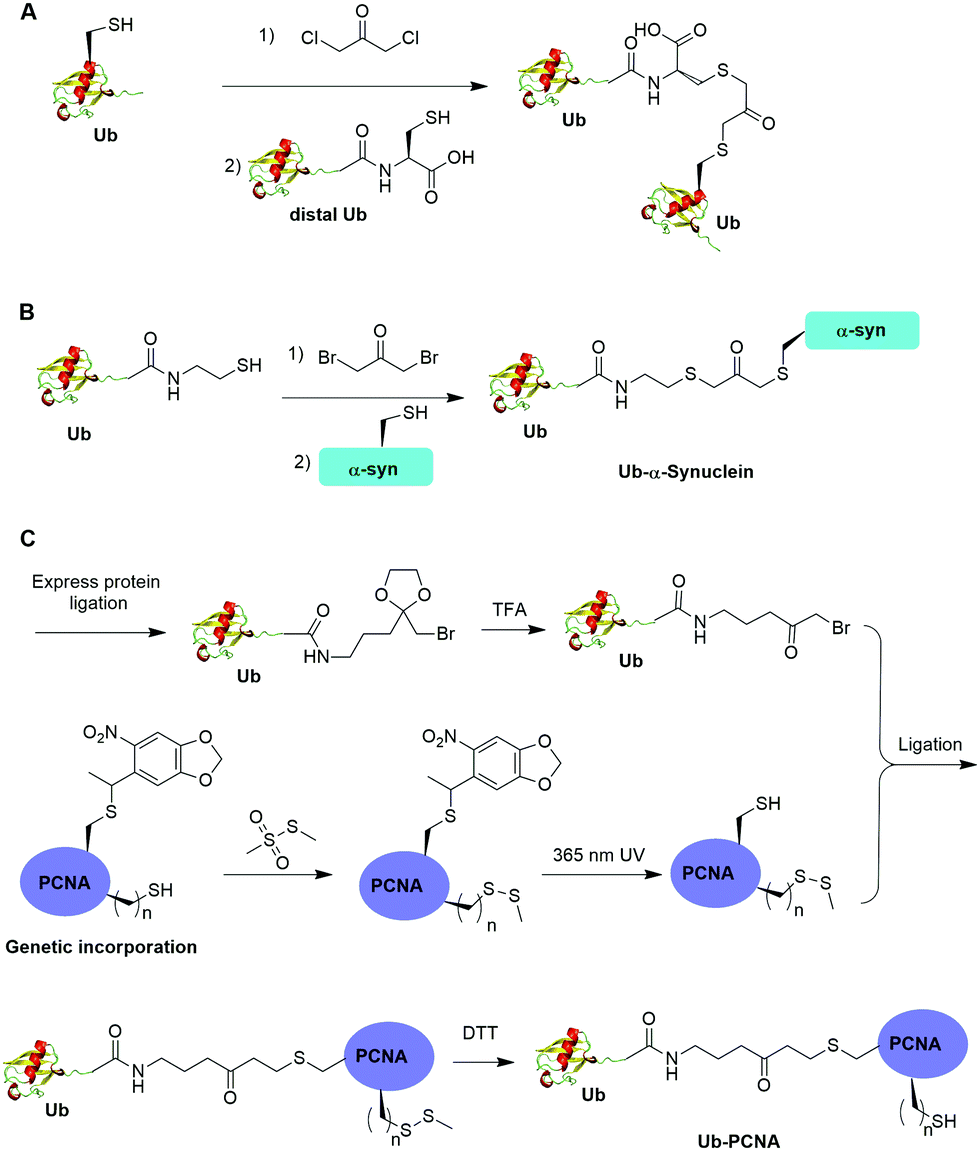 | ||
| Fig. 8 Chemical ubiquitination using α-halogen ketone ligation strategy. (A) Chemical ubiquitination using 1,3-dichloroacetone to generate diUb. (B) Chemical ubiquitination using 1,3-dibromoacetone to generate Ub-α-synuclein. (C) Chemical ubiquitination using an amine-containing α-bromo ketone linker to generate Ub-PCNA. | ||
In 2016, Lewis et al. reported the synthesis of nonhydrolyzable Ub-α-synuclein and SUMO-α-synuclein using 1,3-dibromoacetone as the cross-linking reagent (Fig. 8B).54 This method has advantages over the above mentioned 1,3-dichloroacetone method in term of reaction yield. Instead of using G76C Ub mutant, the authors generated Ub-aminoethanethiol using EPL. Then, the Ub-aminoethanethiol was incubated with 1,3-dibromoacetone, generating a Ub intermediate. After HPLC separation, the Ub intermediate was ligated to α-synuclein with a specific Lys to Cys mutant (K6C, K23C, K43C, or K96C) to generate site-specifically modified Ub-α-synuclein. Notably, the study suggested that ubiquitination at 23, 43, and 96 position of α-synuclein can inhibit its aggregation and toxicity. In contrast, ubiquitination at position 6 on α-synuclein led to the formation of irregular short fibers. Using this method, ubiquitination of H2B at position 120 was also achieved, which facilitated the structural determination of ubiquitinated nucleosome bound with the SAGA DUB module.56
One limitation of the above described α-halogen ketone-mediated chemical ubiquitination method is that the protein to be ubiquitinated either contains no native cysteines or all native cysteines needs to be mutated to serines in order for site-specific ubiquitination. In 2016, Yang et al. reported the generation of a nonhydrolyzable Ub-PCNA with the preservation of native cysteines on PCNA through α-bromoketone-mediated ligation (Fig. 8C).55 In this report, the authors first synthesized a linker molecule (3-(2-(bromomethyl)-1,3-dioxolan-2-yl)propan-1-amine). The linker molecule reacts with Ub-MESNA, generating a Ub intermediate that contains a protected α-bromoketone. Following the deprotection of the carbonyl group, the Ub intermediate can be ligated with an introduced Cys in PCNA. In order to achieve site-specific ubiquitination of PCNA with the preservation of native cysteines, a photocaged Cys was incorporated at the designated Lys164 position of PCNA using an amber codon strategy while the native cysteines were protected by methyl methanethiosulfonate (MMTS). After selective removal of the photocage protection group on Cys164 and ligation with the α-bromoketone in the Ub intermediate, DTT was added to remove the protection group on the native cysteines, generating Ub-PCNA as the final product with the preservation of the native cysteine residues in PCNA. PCNA acts as a scaffold for DNA polymerases including the Y-family polymerases that function in translesion DNA synthesis. Using an in vitro assay it was shown that the above chemically ubiquitinated PCNA stimulates the translesion synthesis activity of Polη similarly to that of the native monoubiquitinated PCNA. Notably by modifying the linker structure such as including a carbon–carbon double bond next to the carbonyl group, a Michael acceptor warhead can be conveniently introduced to the ubiquitinated protein species. Using this ligation strategy, the Zhuang group has successfully generated diUb activity-based probes, Ub-PCNA probes, and cell-permeable Ub probes.109–111
3.2 Chemical ubiquitination through a disulfide linkage
Disulfide-containing molecules as chemical linkers have been explored in protein bioconjugation.112 Ellman's reagent (5,5′-dithiobis-(2-nitrobenzoic acid) or DTNB) developed by George L. Ellman was known for its ability to facilitate rapid disulfide exchange reaction. In 2010, the Zhuang and Muir labs independently reported the respective chemical ubiquitination of PCNA and H2B through disulfide linkage. Chen et al. reported the chemical ubiquitination and SUMOylation of PCNA through a disulfide linkage at different sites of PCNA (Fig. 9A).57 In this report, the authors first used the expressed protein ligation method to generate Ub-cysteamine. Then DTNB was used to activate the thiol group in this Ub intermediate, yielding a DTNB-activated Ub-cysteamine. Then, the disulfide exchange reaction was carried out between the activated Ub species with a cysteine-light PCNA with a unique cysteine introduced at selected position (164, 127, 107 or 44), generating Ub-PCNA as the final product. Using a similar strategy, SUMOylated PCNA was also generated. Importantly, this report revealed that the monoubiquitination of PCNA at the selected positions on PCNA did not show significant difference in promoting polymerase exchange in translesion DNA synthesis.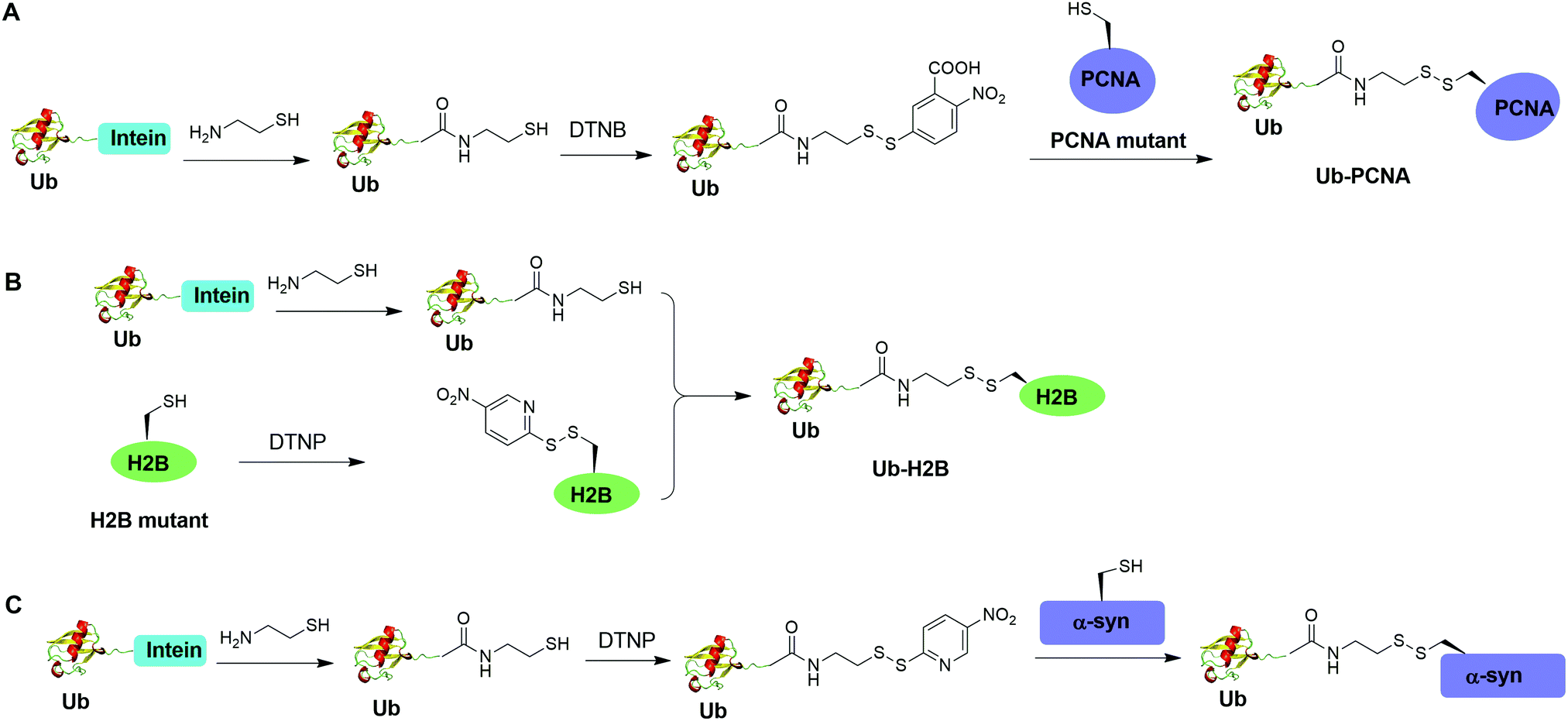 | ||
| Fig. 9 Chemical ubiquitination using a disulfide exchange ligation strategy. Generation of disulfide-linked Ub-PCNA (A), disulfide-linked Ub-H2B (B) and disulfide-linked Ub-α-syn (C). | ||
Chatterjee et al. reported a disulfide-directed histone ubiquitination method (Fig. 9B).58 In this report, instead of using DTNB to activate the thiol in Ub-cysteamine, the authors used 2,2′-dithiobis(5-nitropyridine) (DTNP) to activate the designated Cys in H2B. Then the rapid disulfide bond exchange reaction was carried out between Ub-cysteamine and DTNP-activated Cys in H2B, yielding Ub-H2B as the final product. Using this method, the author successfully generated Ub-H2B at specific positions on H2B (K108, K116, K120, and K125) and revealed that H2BK125 monoubiquitination stimulates hDot1L activity.
Meier et al. also reported site-specific chemical ubiquitination of α-synuclein through disulfide linkage (Fig. 9C).59 Using a similar method, the authors first generated Ub-cysteamine, followed by the activation of the thiol group with DTNP. Finally, ubiquitinated α-synuclein can be generated through a transthiolation reaction with the thiol group in the α-synuclein. The authors generated nine site-specifically ubiquitinated α-synuclein derivatives and demonstrated that different ubiquitination sites have varied effects on α-synuclein aggregation.
Moreover, disulfide-mediated chemical ubiquitination has also been used to generate polyUb-α-globin and polyUb-PCNA. Hemantha et al. generated polyUb-α-globin using a disulfide-directed chemical ubiquitination strategy (Fig. 10A).60 In this study, the authors utilized SPPS and NCL to generate polyUb (n = 2, 3, 4) building blocks containing a C-terminal cysteamine at proximal Ub. The designated Cys in α-globin was activated by DTNP, followed by the final attachment of a Ub chain to α-globin through a transthiolation reaction. Moreover, in this report the authors tested two other reactive groups introduced at the C-terminus of the proximal Ub as well. Both bromoacetamide and maleimide groups allowed efficient generation of polyUb α-globin. A cell-free reconstituted system was used to assess the proteasomal degradation of mono- and di-ubiquitinated α-globin. Interestingly, although monoubiquitination of α-globin was not sufficient to trigger α-globin degradation by proteasome, the di-Ub-α-globin can be rapidly degraded.
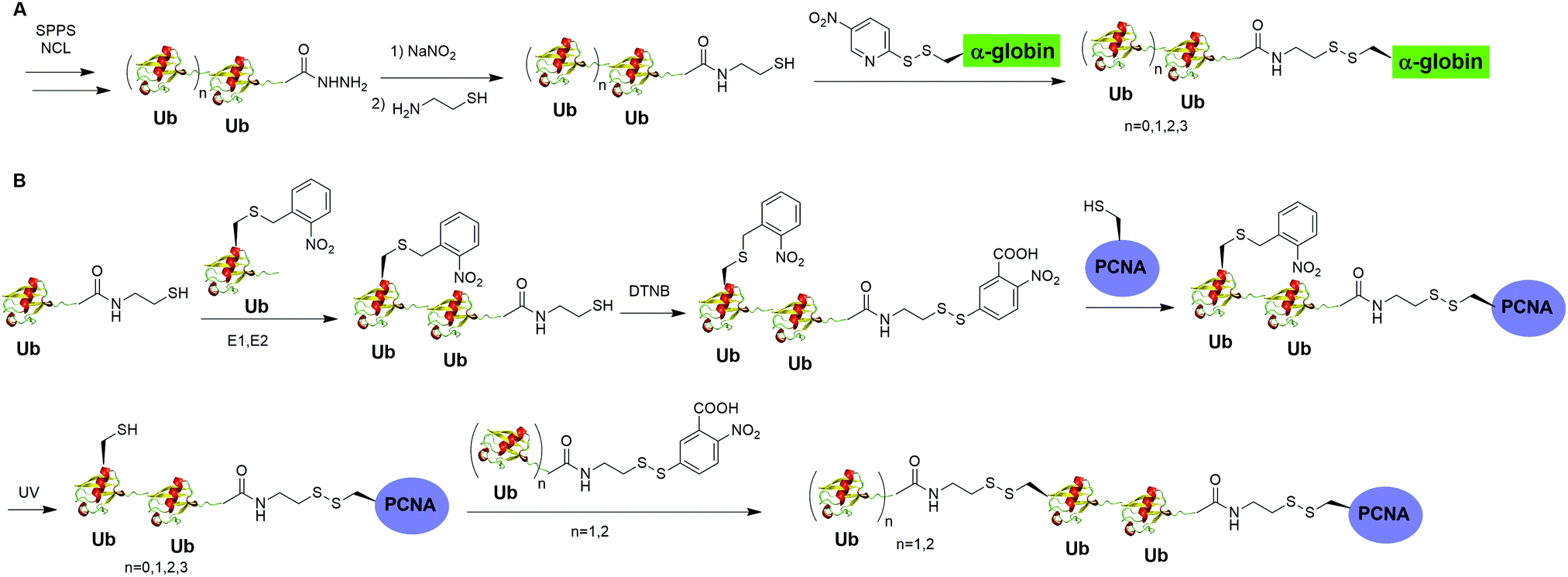 | ||
| Fig. 10 Chemical polyubiquitination using a disulfide exchange reaction. (A) Generation of disulfide-linked polyUb-α-globin. (B) Generation of disulfide-linked polyUb-PCNA. | ||
In DNA damage tolerance, K63-linked polyubiquitinated PCNA plays an important role in the selection of the error-free lesion bypass pathway in eukaryotes. In 2014, Yang et al. reported the generation of tetraUb-PCNA through a disulfide-mediated ligation strategy (Fig. 10B).61 In this report, the authors first utilized an intein-based strategy to generate Ub-cysteamine. For the distal Ub, the designated Lys in Ub was mutated to Cys and protected by a 2-nitrobenzyl photocage group to prevent uncontrolled disulfide formation. Then, mouse E1, human E2 (Ubc13-Mms2) were used to ligate these two Ub species, yielding a diUb building blocks. Then, the C-terminal cysteamine of diUb was activated via DTNB treatment and subsequently ligated to K164C PCNA through a transthiolation reaction. Next, the photocage 2-nitrobenzyl group in Ub was removed by UV irradiation and reacted with another activated diUb building block forming tetra-Ub-PCNA as the final product. Using this construct the authors tested in vitro DNA synthesis by DNA polymerases and polymerase switching mediated by ubiquitination of PCNA. It was found that K63-linked tetraUb-PCNA suppressed the DNA synthesis activity of Polη, likely by trapping Polη in a nonproductive mode. The ability of generating well-defined polyubiquitinated PCNA opens the door to identify the elusive reader proteins of polyubiquitinated PCNA in the error-free lesion bypass process.
3.3 Cu(I)-catalyzed azide–alkyne cycloaddition
The Cu(I)-catalyzed azide–alkyne cycloaddition reaction, also known as click reaction, is a rapid and biocompatible reaction. In 2010, Weikart et al. reported the generation of SUMO-PML11 using Cu(I)-catalyzed azide–alkyne cycloaddition (Fig. 11A).62 In this report, the authors first used site-directed mutagenesis to mutate the target Lys to Cys in PML11 for installation of an azide group through the nucleophilic reaction of thiol with N-(2-azidoethyl)-2-iodoacetamide. The authors used the expressed protein ligation to conjugate propargylamine to the C-terminus of SUMO. Finally, Cu(I)-catalyzed azide–alkyne cycloaddition between the azide-modified PML11 and SUMO-propargylamine yielding SUMO-PML11 as the final product. While the triazole linkage deviates from the native linkage in size and length, it resists DUB cleavage.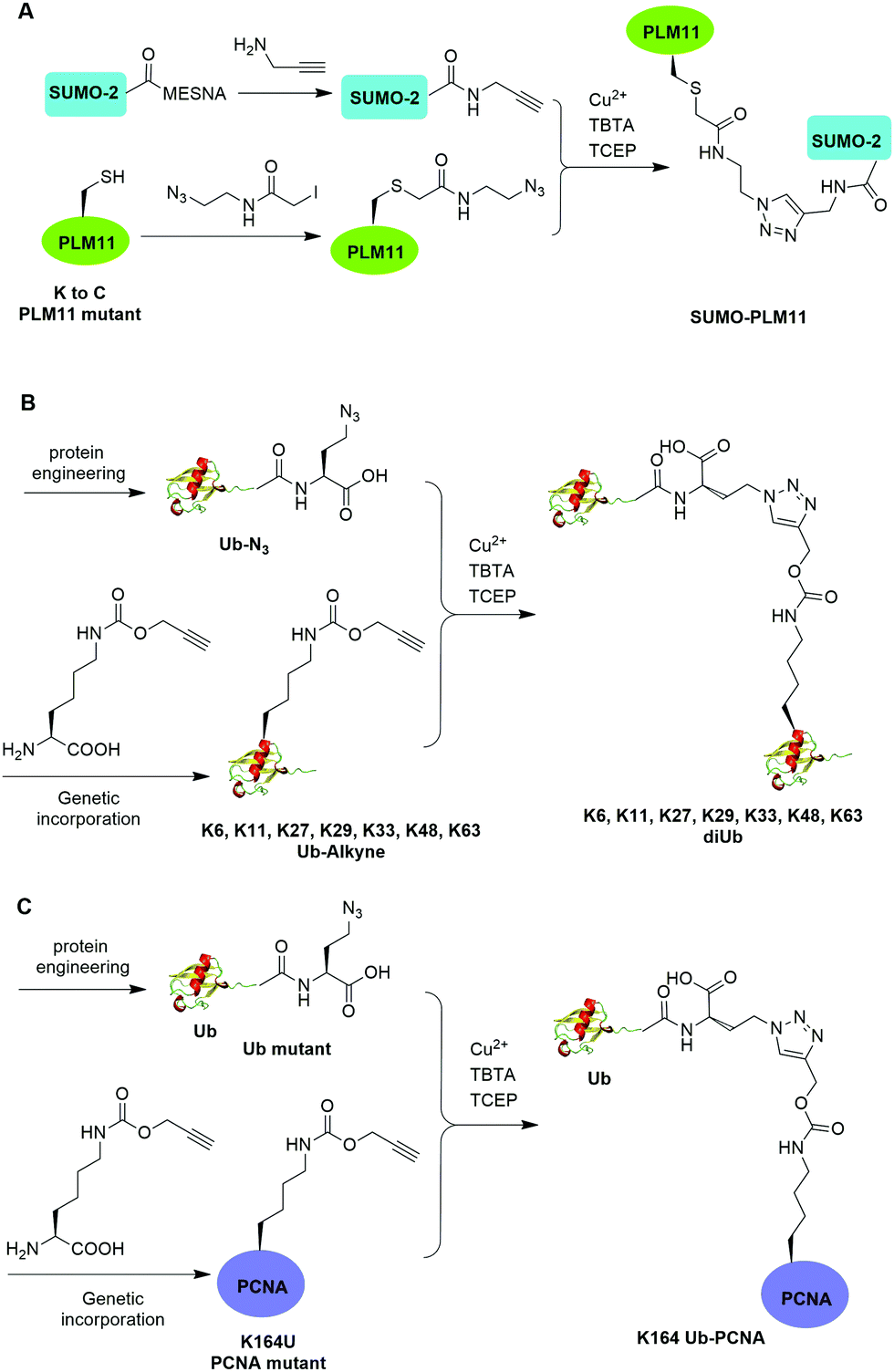 | ||
| Fig. 11 Chemical ubiquitination using Cu(I)-catalyzed azide–alkyne cycloaddition reaction. (A) Generation of SUMO-PLM11. (B) Generation of ubiquitin dimers. (C) Generation of Ub-PCNA. | ||
In the same year, Eger et al. reported the generation of Lys6, Lys11, Lys27, Lys29, Lys33, Lys48, and Lys63-linked diUb using Cu(I)-catalyzed azide–alkyne cycloaddition (Fig. 11B).63 For the design of distal Ub, the authors generated Ub mutant with the insertion of Ala and Ser between the initial Met and second amino acid Gln and mutated the last Gly to Met (MASUb76M). Then, MASUb76M was expressed in the Met auxotrophic E. coli stain B834(DE3). Using Met suppression and azidohomoalanine (Aha) feeding, Ub mutant with the C-terminal Met completely replaced by Aha and the first Met cleaved off by methionine aminopeptidase was obtained. For proximal Ub, the authors used amber codon suppression to incorporate the propargyl-protected Lys derivative (Plk) at the specific Lys positions in Ub. With distal and proximal Ub building blocks in hand, Cu(I)-catalyzed azide–alkyne cycloaddition yielded Ub dimers with a triazole linkage that is resistant to DUB cleavage. Moreover, Cu(I)-catalyzed azide–alkyne cycloaddition has been used to generate internal and terminal diUb activity-based probes. In 2013, McGouran et al. reported the generation of internal diUb probe of all eight linkages (M1, K6, K11, K27, K29, K33, K48, K63) through Cu(I)-catalyzed azide–alkyne cycloaddition, allowing for the profiling of DUB activities.113 More recently, terminal activity-based diUb probes and interactor affinity probes were also generated by Cu(I)-catalyzed azide–alkyne cycloaddition.114–116
Using a similar strategy, in 2011, Eger et al. reported the generation of a mono-ubiquitinated PCNA using Cu(I)-catalyzed azide–alkyne cycloaddition (Fig. 11C).64 In this report, the authors generated Ub and PCNA mutant containing unnatural amino acids that carry an azide (Aha) and an alkyne (Plk) in their side chains respectively. Followed by Cu(I)-catalyzed azide–alkyne cycloaddition, Ub-PCNA was successfully prepared with good purity and yield.
3.4 Chemical ubiquitination using thiol–ene coupling
The addition of thiyl radicals to alkenes also known as thiol–ene coupling (TEC) has been widely applied in bioconjugation due to its high reaction rate. With a reaction constant of 106 M−1 s−1, this reaction has the potential to serve as a powerful method for chemically modifying proteins. In 2012, Valkevich et al. reported the generation of Ub dimer and branched Ub trimers using TEC reaction (Fig. 12).66 In this study, the authors used UbD77 to react with an excess amount of allylamine catalyzed by a DUB YUH1 to generate distal Ub mutant (Ub-AA). For proximal Ub, site-directed mutagenesis was used to mutate the target Lys to Cys. With 365 nm UV irradiation, the water-soluble free radical initiator lithium acyl phosphinate (LAP) initiates the TEC reaction between the two Ub species generating a Ub dimer as the final product. Using this method, K6-, K11-, K29-, K33-, K48-, K63-linked diUbs were generated successfully except K27-linked diUb due to potential steric hindrance. Also, the authors successfully generated K6/K48, K11/K48, K48/K63 branched triUb. One advantage of the thioether linkage generated by thiol–ene coupling is that it mimics the native isopeptide and can be cleaved by DUBs in a similar efficiency as the native Ub dimers. In vitro assays showed cleavage specificity of diUbs and branched triUbs generated using thiol–ene coupling for tested DUBs. Using this approach, the Strieter group also reported the generation of K6, K48, K63 Ub polymers up to seven subunits starting with specific Ub monomer in a single step TEC reaction.65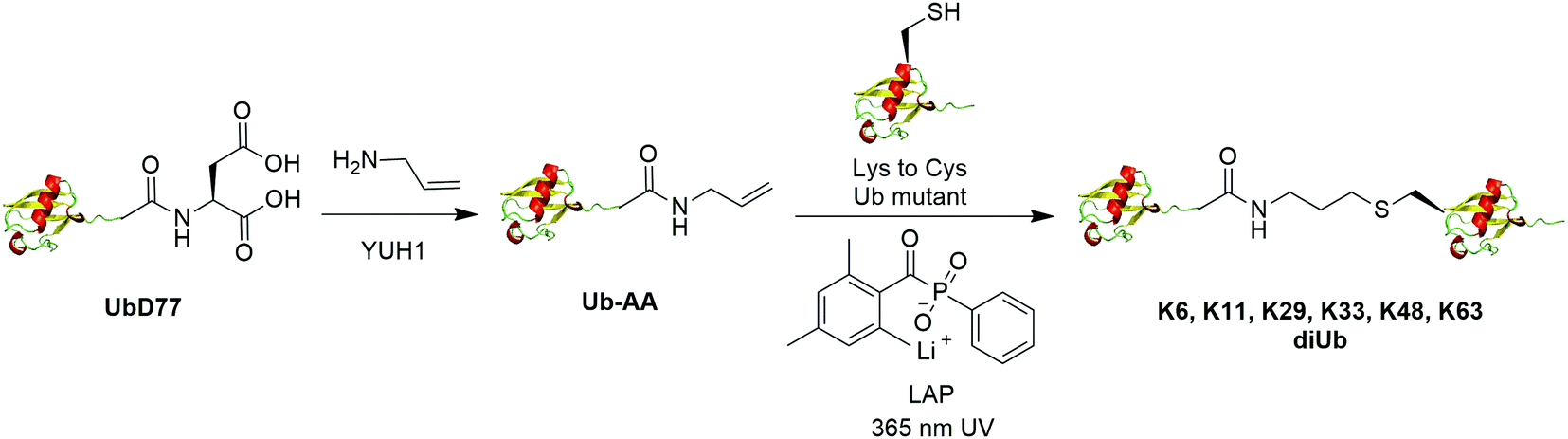 | ||
| Fig. 12 Chemical ubiquitination using thiol–ene reaction. | ||
3.5 Chemical ubiquitination through oxime and hydrazone formation
Oxime formation represents a fast chemoselective condensation reaction between aminoxy and aldehyde to form a stable linkage. In 2010, Shanmugham et al. reported the generation of ubiquitinated isopeptide using oxime formation that is resistant to DUBs cleavage (Fig. 13A).67 In this report, the authors used Ub-aldehyde that can be generated from Ub-acetal and subsequently ligated with an aminoxy-modified peptide to form a nonhydrolyzable ubiquitinated isopeptide as the product. Using this method, the authors successfully generated K48- and K63-linked ubiquitinated isopeptide mimicking diUb as well as ubiquitinated PCNA, H2A, H2B, FANCD2 isopeptides.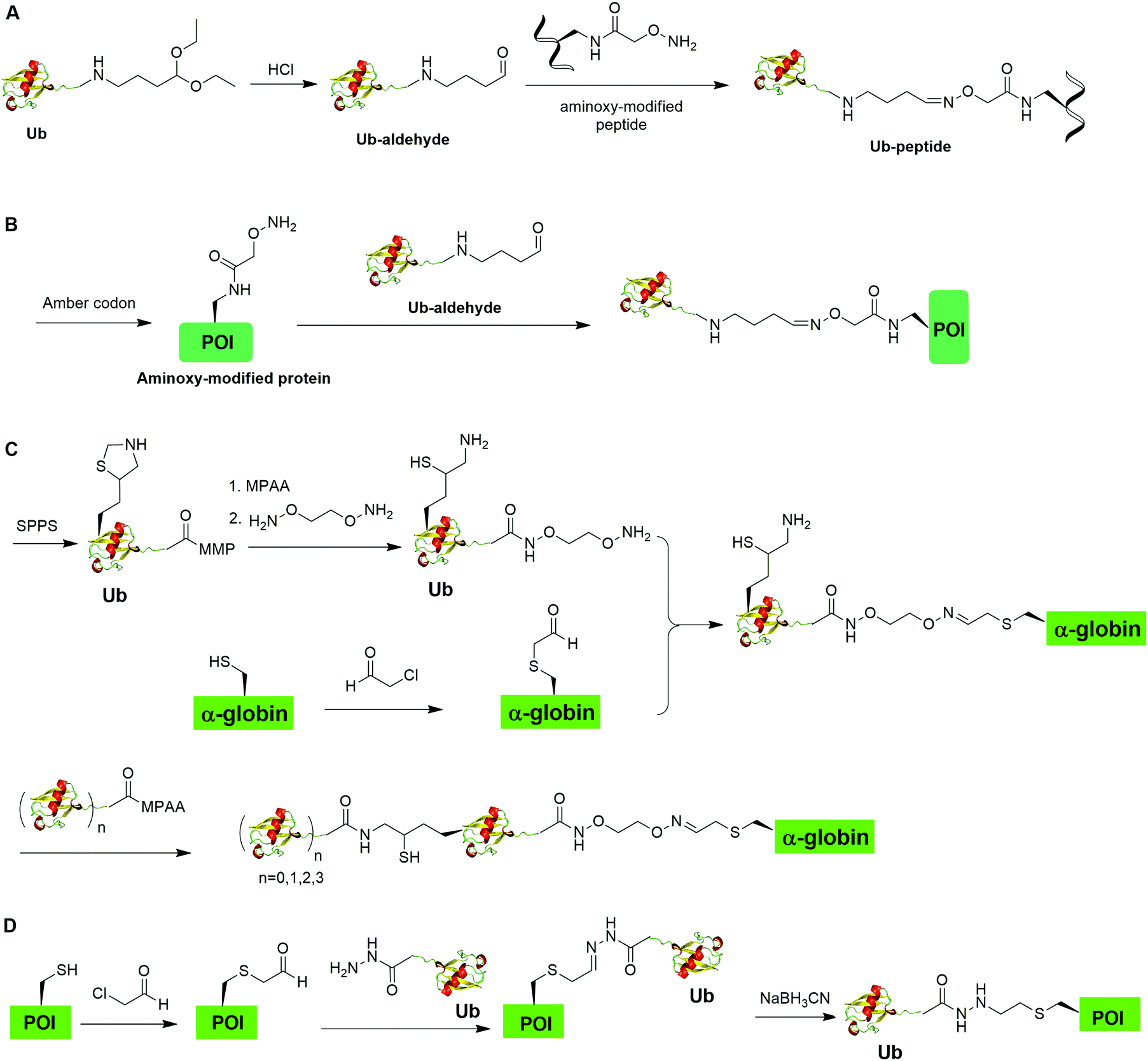 | ||
| Fig. 13 Chemical ubiquitination through oxime and hydrazone formation. (A) Generation of Ub-peptides through oxime formation. (B) Generation of Ub-POI through oxime formation. (C) Generation of polyUb-α-globin through oxime formation. (D) Generation of Ub-POI through hydrazone formation. | ||
Using a similar oxime formation chemistry, in 2016, Stanley et al. reported the generation of full length diUb (Fig. 13B).68 In this report, the authors used a pyrrolysyl-tRNA synthetase PylRS/tRNACUA pair to site-specifically incorporate the unnatural amino acid aminooxy-L-lysine into proximal Ub. Then, biorthogonal oxime ligation between aminooxy-lysine in the proximal Ub and distal Ub-aldehyde afforded a full-length diUb as the final product. Using this method, the authors successfully generated Ub chain and ubiquitinated SUMO.
Later in 2016, Singh et al. reported the generation of polyUb-α-globin utilizing oxime formation chemistry coupled with δ-mercaptolysine mediated chemical ubiquitination (Fig. 13C).69 In this report, the authors used SPPS to incorporate a protected δ-mercaptolysine into distal Ub. Following modification of the C-terminus of Ub and deprotection to free the δ-mercaptolysine, the Ub building block was ready to be ligated with chemically modified α-globin. Site-directed mutagenesis was used to mutate the designated Lys to Cys in α-globin. After the reaction of thiol in α-globin with chloroacetaldehyde, the α-globin was ready to be ligated with the oxime functional group in the Ub building block to form ubiquitinated α-globin. PolyUb α-globin was generated by NCL with Ubn-MPAA creating a stable linkage between Ub and α-globin resistant to DUB cleavage. Through incubation of the generated polyUb-α-globin with purified human 26S proteasome, the authors found that while the diUb- and triUb-α-globins are susceptible to trimming by proteasome-associated DUBs but not degradation, the tetraUb-α-globin are predominantly degraded by the proteasome.
In 2018, Bhat et al. reported a chemical ubiquitination method through hydrazone formation (Fig. 13D).70 In this report, the authors used site-directed mutagenesis to mutate the designated Lys to Cys in histone. Followed by thiol alkylation with chloroacetaldehyde, condensation with Ub-hydrazide, and reduction with sodium cyanoborohydride, ubiquitinated histone H2B and diUb mimics were generated successfully using this hydrazone formation method. Importantly, the authors showed that the hydrazone linkage of ubiquitinated POI can be cleaved by DUBs.
3.6 Chemical ubiquitination using conjugate addition
Chemical ubiquitination by exploiting the nucleophilicity of cysteine thiol that can readily react with a Ub C-terminal electrophile represents an efficient way of generating ubiquitinated POI. The first example is the chemical ubiquitination using thiol–Michael addition. The thiol-Michael addition reaction characterized by the conjugate addition of a thiol to a Michael acceptor has been widely used in bioconjugation and materials chemistry.117,118 In 2014, Hemantha et al. reported the generation of monoUb- and diUb-α-globin using thiol–Michael addition (Fig. 14A).60 The authors first used SPPS to generate monoUb- and diUb-hydrazide, followed the treatment with NaNO2 at pH 3 to convert the hydrazide group to acyl azide. Following the addition of an excess amount of commercially available N-(2-aminoethyl)-maleimide, mono- and di-Ub maleimide were generated. Finally, a thiol–Michael addition between the thiol in an α-globin mutant and the maleimide in Ub yielded monoUb-α-globin with good efficiency.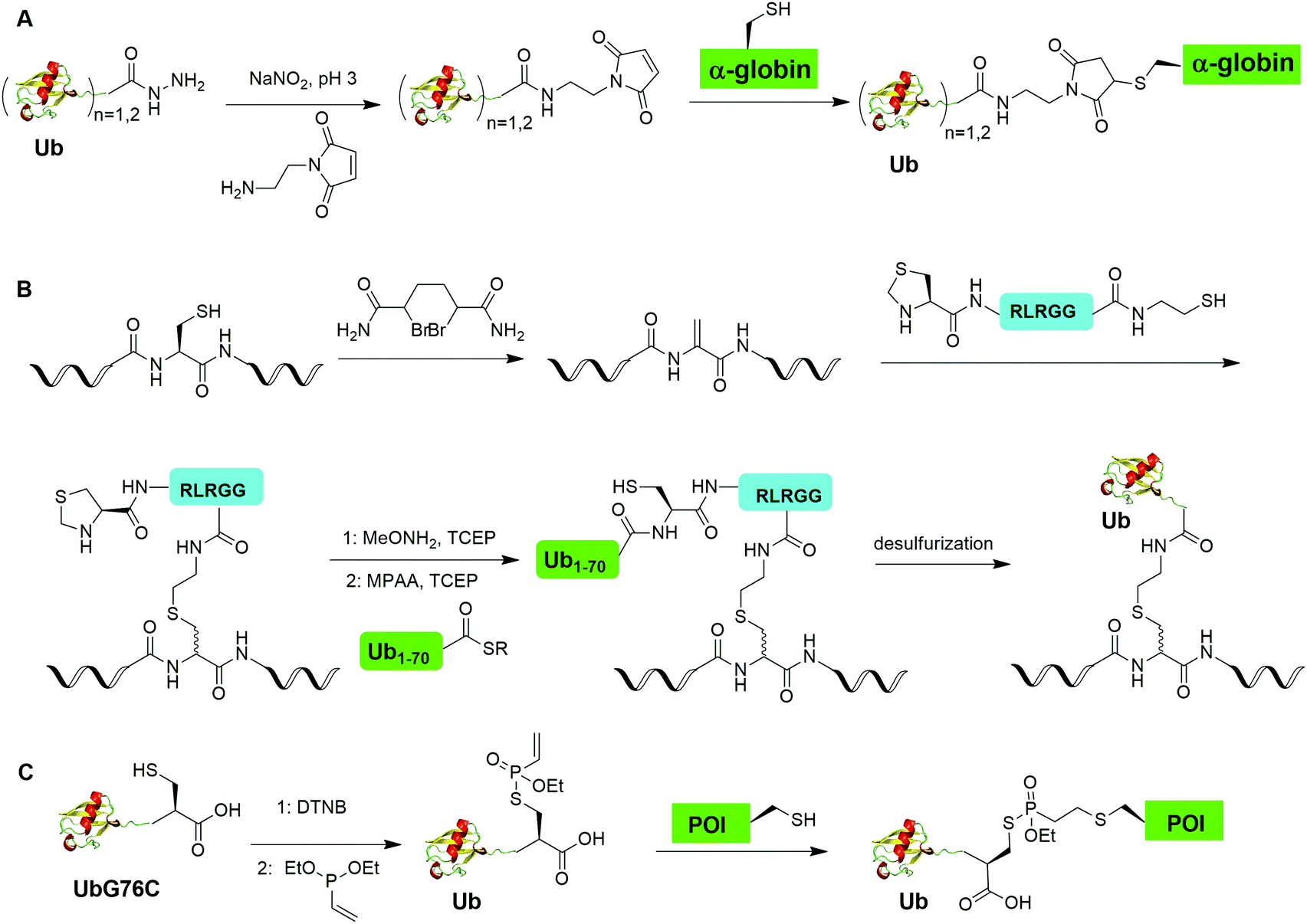 | ||
| Fig. 14 Chemical ubiquitination through conjugate addition. (A) Generation of Ub-α-globin through thiol maleimide conjugate addition. (B) Generation of Ub-POI through thiol dehydroalanine conjugate addition. (C) Generation of ubiquitinated POI through vinylphopshonite-mediated conjugate addition. | ||
Another strategy of chemical ubiquitination using thiol-Michael addition is through the generation of dehydroalanine (Dha) Michael acceptor in the protein backbone and ligation with thiol group introduced at the C-terminus of Ub. Dha-based chemistry has been proved as a versatile strategy for the preparation of proteins bearing PTM mimetics.119–122 In 2016, Meledin et al. reported a chemical protein ubiquitination method via a thiol dehydroalanine Michael addition reaction (Fig. 14B).72 In this report, the authors first used 1,4-dibromobutane to convert the cysteine residue in the substrate protein to Dha. Followed by a thiol-Michael addition with a hexapeptide bearing a thiol reactive handle derived from the C-terminal of Ub, a thioether linkage between the Ub C-terminal peptide and a target protein peptide was generated. Followed by extension of Ub chain by NCL with the complementary Ub polypeptide Ub(1–70)-thioester and desulfurization, a full-length ubiquitinated peptide was generated with good yield. Using this method, the authors also successfully generated K29- and K48-linked diUb and ubiquitinated α-globin.
Using a similar thiol dehydroalanine Michael addition strategy, Pan et al. reported the generation of K27-diUb and the crystal structure of a UCHL3–K27diUb complex.74 In this report, the authors first generated K27UbDha, followed by the ligation with an auxiliary molecule through thiol-Michael addition. Subsequent deprotection of the auxiliary group exposed the thiol group for NCL with distal Ub-hydrazide yielding a diUb intermediate. K27-Linked diUb was successfully generated by removal of the auxiliary group using trifluoroacetic acid. Importantly, the authors solved the crystal structure of UCHL3–K27diUb and found that binding of K27-diUb to UCHL3 changed the conformation of the ubiquitin dimer.
Recently, Baumann et al. reported a chemical ubiquitination method using vinylphosphonothiolate electrophiles for thiol–thiol bioconjugations (Fig. 14C).73 In this report, the authors first generated UbG76C dimer. Then, DTT was used to reduce the disulfide bond and DTNB to activate the thiol group generating a UbG76C-TNB intermediate. Followed by the addition of vinylphosphonite, the TNB group can be substituted by vinylphosphonite forming a UbG76C vinylphosphonothiolate intermediate. Finally, the cysteine residue in the POI can undergo a conjugate addition with the UbG76C vinylphosphonothiolate intermediate, yielding a ubiquitinated POI. Using this method, the authors successfully generated K48-linked diUb and K6-ubiquitinated α-synuclein.
3.7 Sortase-mediated ubiquitination
In 2019, Fottner et al. reported a sortase-mediated ubiquitination method (Fig. 15).75 SrtA can recognize peptide sequence LPXTG and cleave the peptide bond between T and G, forming a thioester intermediate between the sortase active site Cys and LPXT, which undergoes a specific transpeptidation with a peptide containing a N-terminal glycine residue. In this report, the authors modified the C-terminal peptide of Ub from its native sequence LRLRGG to LPLTGG, allowing for its recognition by SrtA and the subsequent formation of a thioester-linked Ub-SrtA intermediate. This intermediate can be nucleophilically attacked by an α-amino group of the terminal glycine of a GGK group introduced on POI, forming the ubiquitinated protein with a native isopeptide bond between the targeted lysine on the POI and the G76 of Ub. To introduce GGK in POI, the authors used genetic-code expansion to insert an unnatural amino acid AzGGK into a specific position of POI. Then, the azide group in the side chain of AzGGK was reduced to an amine through a bioorthogonal Staudinger reduction, generating a GGK amenable for sortase-mediated ubiquitination. Also, the authors showed that this method can also be applied to chemical SUMOylation of POI and successfully generated diUb, Ub-PCNA and SUMO-GFP. Importantly, this approach allows the site-specific ubiquitination and SUMOylation of non-refoldable, multidomain proteins in mammalian cells through a native isopeptide bond but resistant to DUB cleavage as the C-terminus of Ub was modified.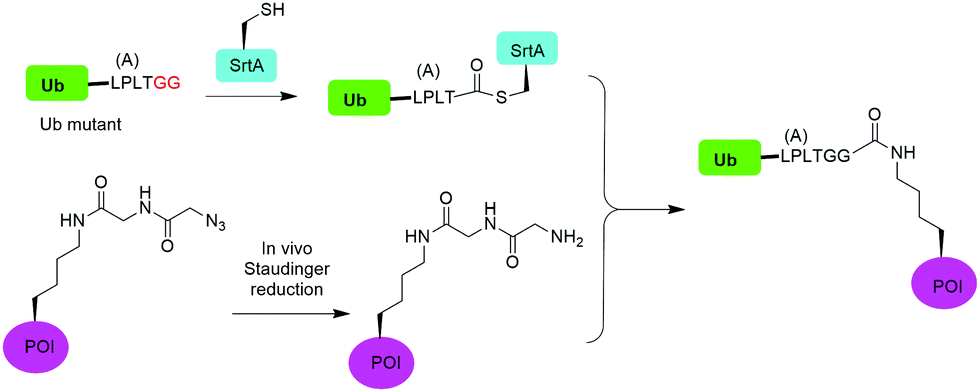 | ||
| Fig. 15 Sortase mediated protein ubiquitination. | ||
3.8 Cysteine-aminoethylation assisted chemical ubiquitination
In 2019, Chu et al. reported a CAACU (cysteine-aminoethylation assisted chemical ubiquitination) strategy for the efficient synthesis of ubiquitinated and SUMOylated histone analogs (Fig. 16A).76 In this report, the authors first mutated the target Lys in histones to Cys, followed by the installation of an auxiliary group as shown in Fig. 16A. Subsequent removal of the thiol-protecting acetamidomethyl (Acm) group enables the ligation with Ub hydrazide, yielding an amid linkage between Ub and histones. After removal of the auxiliary group, ubiquitinated histones were generated. Using this method, the authors also generated SUMOylated histones. Cleavage assay using DUBs showed that the thioether-containing isopeptide linkage was cleaved by DUBs as efficiently as a native isopeptide bond in Ub-histone.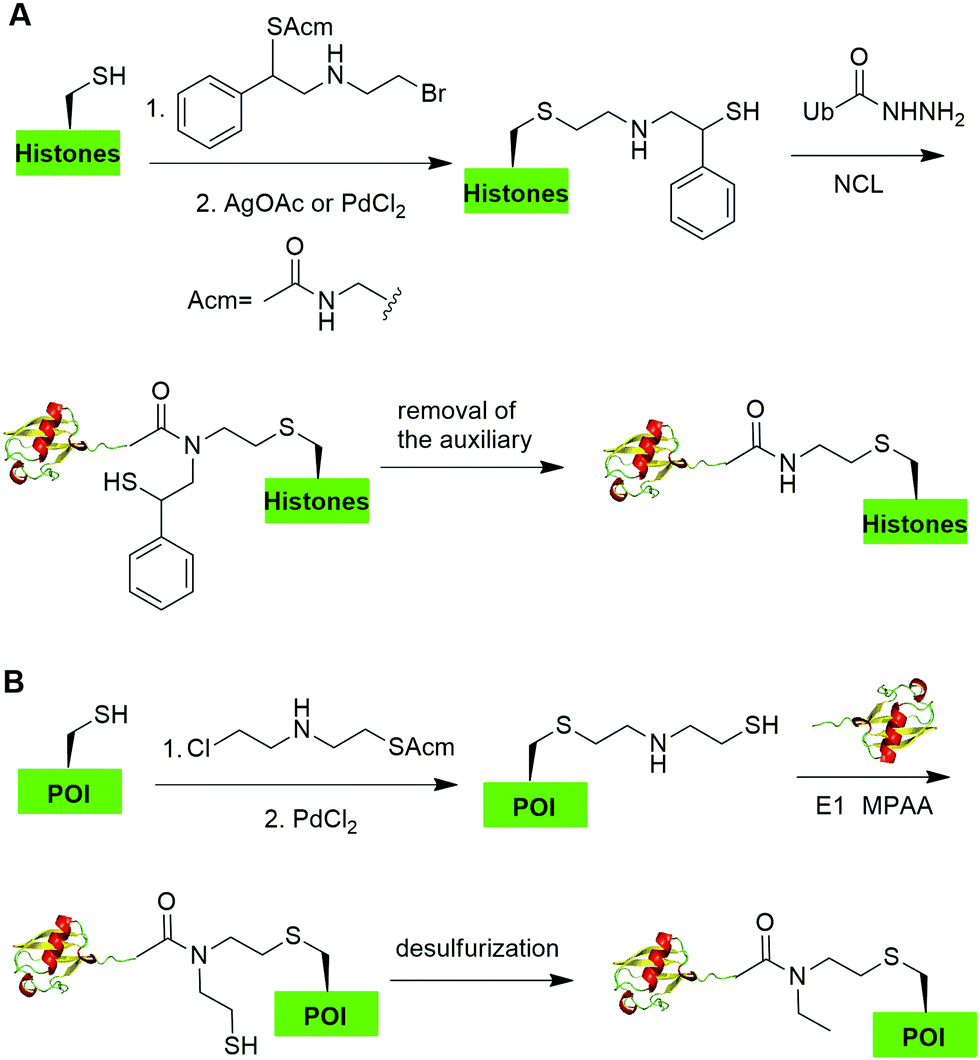 | ||
| Fig. 16 Cysteine-aminoethylation-assisted chemical ubiquitination. (A) Cysteine-aminoethylation-assisted chemical ubiquitination of histones. (B) Cysteine-aminoethylation-mediated NCL to generate isopeptide-N-ethylated diUb, ployUb, branched polyUb, and ubiquitinated POI. | ||
Recently, Zheng et al. reported a 2-((2-chloroethyl)amino)ethanethiol-mediated and E1-catalyzed strategy to generate an isopeptide-N-ethylated diUb which is resistant to DUB cleavage (Fig. 16B).77 In this report, the authors first synthesized a bifunctional small molecule 2-((2-chloroethyl)amino)ethanethiol protected with Acm. This small molecule can readily react with the thiol group in the POI. Subsequent removal of the ACM protection group exposed a β-mercaptoethylamin as a reactive handle for subsequent ligation with distal Ub-thioester generated by an E1 catalyzed reaction. Followed by desulfurization, an N-ethyl isopeptide linked Ub-POI was generated successfully.
4. Summary and perspective
Ubiquitination has attracted much attention since the discovery of its involvement in proteasomal protein degradation. The diversity of Ub chain linkages and length presents tremendous challenges to the investigation of protein ubiquitination and deubiquitination. In vitro enzymatic assembly of polyUb chains provided an initial method for biochemists to generate the Ub chain of different linkages, allowing for early studies of Ub modification. The more recent breakthrough in synthetic and semisynthetic methods in preparation of linkage and site-specific polyUb chains and ubiquitinated POI has greatly helped elucidating Ub chain assembly, recognition, and processing with molecular details. Chemical ubiquitination through a nonnative linkage also enabled structural investigations of Ub pathway enzymes (Ub ligases and DUBs) in complex with Ub chains and ubiquitinated proteins. Related chemical approaches were also successfully applied to generate ubiquitin-based probes.123–127 These efforts have facilitated the discovery of inhibitors that can modulate the enzymatic ubiquitination and deubiquitination processes.Since the introduction of chemical ubiquitination methods to the ubiquitin field, great achievement has been made towards the structural and functional understanding of Ub signaling at a molecular level. However, the development of more efficient methods for generating well-defined polyubiquitinated proteins remains central to our understanding of the complex mechanisms of Ub signaling mediated by different types of polyUb chains, particularly the noncanonical and branched Ub chains. The generation of proteins site-specifically modified by polyUb chain with defined linkage and length in sufficient quantity still represents a challenge especially when the target proteins are of larger size and prone to denaturation. Efficient chemoenzymatic methods that are generally applicable to a wide variety of proteins for polyubiquitination still need to be developed. In addition, introducing Ub branch and other post-translational modifications, such as phosphorylation,128 onto the linear polyUb chain adds further complexity. Generating non-lysine ubiquitinated proteins through chemical approaches will also allow a better understanding of this non-canonical ubiquitin modification.129,130 Lastly elucidation of Ub signaling in live cells is of great importance. Recently, several cell-permeable Ub probes have been reported allowing for intracellular profiling of DUBs.111,131–133 Achieving controlled polyubiquitination of a target protein in cells using bioorthogonal methods is still a challenge. To this end, new cell-compatible chemical reactions needs to be adopted for achieving chemical ubiquitination in live cells. The ability of generating well-defined polyubiquitinated proteins will help to identify reader proteins of a large number of ubiquitinated proteins and shed light on the molecular basis of Ub chain signaling in cells. In conjunction with activity-based Ub probes for Ub pathway enzymes, the chemical ubiquitination approaches will also play an indispensable role in uncovering new targets in the Ub proteasome system for therapeutic intervention of human diseases.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the US National Institutes of Health (GM129468) for supporting the work in the Zhuang laboratory.References
- D. Komander and M. Rape, Annu. Rev. Biochem., 2012, 81, 203–229 CrossRef CAS.
- M. J. Clague, C. Heride and S. Urbé, Trends Cell Biol., 2015, 25, 417–426 CrossRef CAS.
- R. Yau and M. Rape, Nat. Cell Biol., 2016, 18, 579–586 CrossRef CAS.
- K. N. Swatek and D. Komander, Cell Res., 2016, 26, 399–422 CrossRef CAS.
- A. Hershko and A. Ciechanover, Annu. Rev. Biochem., 1998, 67, 425–479 CrossRef CAS.
- A. L. Schwartz and A. Ciechanover, Annu. Rev. Pharmacol. Toxicol., 2009, 49, 73–96 CrossRef CAS.
- F. Ikeda, N. Crosetto and I. Dikic, Cell, 2010, 143, 677–681 CrossRef CAS.
- K. K. Deol, S. Lorenz and E. R. Strieter, Front. Physiol., 2019, 10 Search PubMed.
- C. M. Pickart, Annu. Rev. Biochem., 2001, 70, 503–533 CrossRef CAS.
- B. A. Schulman and J. W. Harper, Nat. Rev. Mol. Cell Biol., 2009, 10, 319–331 CrossRef CAS.
- Y. Ye and M. Rape, Nat. Rev. Mol. Cell Biol., 2009, 10, 755–764 CrossRef CAS.
- R. J. Deshaies and C. A. P. Joazeiro, Annu. Rev. Biochem., 2009, 78, 399–434 CrossRef CAS.
- C. E. Berndsen and C. Wolberger, Nat. Struct. Mol. Biol., 2014, 21, 301–307 CrossRef CAS.
- M. Groettrup, C. Pelzer, G. Schmidtke and K. Hofmann, Trends Biochem. Sci., 2008, 33, 230–237 CrossRef CAS.
- M. B. Metzger, V. A. Hristova and A. M. Weissman, J. Cell Sci., 2012, 125, 531–537 CrossRef CAS.
- V. Chau, J. W. Tobias, A. Bachmair, D. Marriott, D. J. Ecker, D. K. Gonda and A. Varshavsky, Science, 1989, 243, 1576–1583 CrossRef CAS.
- K. E. Wickliffe, A. Williamson, H.-J. Meyer, A. Kelly and M. Rape, Trends Cell Biol., 2011, 21, 656–663 CrossRef CAS.
- Z. J. Chen and L. J. Sun, Mol. Cell, 2009, 33, 275–286 CrossRef CAS.
- K. Iwai, H. Fujita and Y. Sasaki, Nat. Rev. Mol. Cell Biol., 2014, 15, 503–508 CrossRef CAS.
- Y. Kulathu and D. Komander, Nat. Rev. Mol. Cell Biol., 2012, 13, 508–523 CrossRef CAS.
- S. M. Nijman, M. P. Luna-Vargas, A. Velds, T. R. Brummelkamp, A. M. Dirac, T. K. Sixma and R. Bernards, Cell, 2005, 123, 773–786 CrossRef CAS.
- D. Komander, M. J. Clague and S. Urbé, Nat. Rev. Mol. Cell Biol., 2009, 10, 550–563 CrossRef CAS.
- K. Husnjak and I. Dikic, Annu. Rev. Biochem., 2012, 81, 291–322 CrossRef CAS.
- C. M. Pickart and S. Raasi, Methods Enzymol., 2005, 399, 21–36 CAS.
- S. Raasi and C. M. Pickart, Methods Mol. Biol., 2005, 301, 47–55 CAS.
- A. Bremm and D. Komander, Methods Mol. Biol., 2012, 832, 219–228 CrossRef CAS.
- M. K. Hospenthal, S. M. Freund and D. Komander, Nat. Struct. Mol. Biol., 2013, 20, 555–565 CrossRef CAS.
- M. A. Michel, P. R. Elliott, K. N. Swatek, M. Simicek, J. N. Pruneda, J. L. Wagstaff, S. M. Freund and D. Komander, Mol. Cell, 2015, 58, 95–109 CrossRef CAS.
- Y. A. Kristariyanto, S. A. Abdul Rehman, D. G. Campbell, N. A. Morrice, C. Johnson, R. Toth and Y. Kulathu, Mol. Cell, 2015, 58, 83–94 CrossRef CAS.
- R. Hofmann, G. Akimoto, T. G. Wucherpfennig, C. Zeymer and J. W. Bode, Nat. Chem., 2020, 12, 1008–1015 CrossRef CAS.
- S. M. Mali, S. K. Singh, E. Eid and A. Brik, J. Am. Chem. Soc., 2017, 139, 4971–4986 CrossRef CAS.
- R. Layfield, K. Franklin, M. Landon, G. Walker, P. Wang, R. Ramage, A. Brown, S. Love, K. Urquhart, T. Muir, R. Baker and R. J. Mayer, Anal. Biochem., 1999, 274, 40–49 CrossRef CAS.
- K. S. Kumar, L. Spasser, S. Ohayon, L. A. Erlich and A. Brik, Bioconjugate Chem., 2011, 22, 137–143 CrossRef CAS.
- S. Tang, L. J. Liang, Y. Y. Si, S. Gao, J. X. Wang, J. Liang, Z. Mei, J. S. Zheng and L. Liu, Angew. Chem., Int. Ed., 2017, 56, 13333–13337 CrossRef CAS.
- C. Chatterjee, R. K. McGinty, J. P. Pellois and T. W. Muir, Angew. Chem., Int. Ed., 2007, 46, 2814–2818 CrossRef CAS.
- R. K. McGinty, J. Kim, C. Chatterjee, R. G. Roeder and T. W. Muir, Nature, 2008, 453, 812–816 CrossRef CAS.
- R. Yang, K. K. Pasunooti, F. Li, X. W. Liu and C. F. Liu, J. Am. Chem. Soc., 2009, 131, 13592–13593 CrossRef CAS.
- K. S. Ajish Kumar, M. Haj-Yahya, D. Olschewski, H. A. Lashuel and A. Brik, Angew. Chem., Int. Ed., 2009, 48, 8090–8094 CrossRef CAS.
- R. Yang, K. K. Pasunooti, F. Li, X. W. Liu and C. F. Liu, Chem. Commun., 2010, 46, 7199–7201 RSC.
- M. Haj-Yahya, K. S. Ajish Kumar, L. A. Erlich and A. Brik, Biopolymers, 2010, 94, 504–510 CrossRef CAS.
- K. S. A. Kumar, L. Spasser, L. A. Erlich, S. N. Bavikar and A. Brik, Angew. Chem., Int. Ed., 2010, 49, 9126–9131 CrossRef CAS.
- K. S. A. Kumar and A. Brik, Isr. J. Chem., 2011, 51, 900–907 CrossRef CAS.
- S. Virdee, P. B. Kapadnis, T. Elliott, K. Lang, J. Madrzak, D. P. Nguyen, L. Riechmann and J. W. Chin, J. Am. Chem. Soc., 2011, 133, 10708–10711 CrossRef CAS.
- M. Hejjaoui, M. Haj-Yahya, K. S. Kumar, A. Brik and H. A. Lashuel, Angew. Chem., Int. Ed., 2011, 50, 405–409 CrossRef CAS.
- K. S. Kumar, S. N. Bavikar, L. Spasser, T. Moyal, S. Ohayon and A. Brik, Angew. Chem., Int. Ed., 2011, 50, 6137–6141 CrossRef CAS.
- M. Haj-Yahya, B. Fauvet, Y. Herman-Bachinsky, M. Hejjaoui, S. N. Bavikar, S. V. Karthikeyan, A. Ciechanover, H. A. Lashuel and A. Brik, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17726–17731 CrossRef CAS.
- C. E. Weller, W. Huang and C. Chatterjee, ChemBioChem, 2014, 15, 1263–1267 CrossRef CAS.
- C. E. Weller, A. Dhall, F. Ding, E. Linares, S. D. Whedon, N. A. Senger, E. L. Tyson, J. D. Bagert, X. Li, O. Augusto and C. Chatterjee, Nat. Commun., 2016, 7, 12979 CrossRef CAS.
- M. Pan, S. Gao, Y. Zheng, X. Tan, H. Lan, D. Sun, L. Lu, T. Wang, Q. Zheng, Y. Huang, J. Wang and L. Liu, J. Am. Chem. Soc., 2016, 138, 7429–7435 CrossRef CAS.
- H. Sun, S. M. Mali, S. K. Singh, R. Meledin, A. Brik, Y. T. Kwon, Y. Kravtsova-Ivantsiv, B. Bercovich and A. Ciechanover, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 7805–7812 CrossRef CAS.
- S. Virdee, Y. Ye, D. P. Nguyen, D. Komander and J. W. Chin, Nat. Chem. Biol., 2010, 6, 750–757 CrossRef CAS.
- C. Castañeda, J. Liu, A. Chaturvedi, U. Nowicka, T. A. Cropp and D. Fushman, J. Am. Chem. Soc., 2011, 133, 17855–17868 CrossRef.
- L. Yin, B. Krantz, N. S. Russell, S. Deshpande and K. D. Wilkinson, Biochemistry, 2000, 39, 10001–10010 CrossRef CAS.
- Y. E. Lewis, T. Abeywardana, Y. H. Lin, A. Galesic and M. R. Pratt, ACS Chem. Biol., 2016, 11, 931–942 CrossRef CAS.
- K. Yang, G. Li, P. Gong, W. Gui, L. Yuan and Z. Zhuang, ChemBioChem, 2016, 17, 995–998 CrossRef CAS.
- M. T. Morgan, M. Haj-Yahya, A. E. Ringel, P. Bandi, A. Brik and C. Wolberger, Science, 2016, 351, 725–728 CrossRef CAS.
- J. Chen, Y. Ai, J. Wang, L. Haracska and Z. Zhuang, Nat. Chem. Biol., 2010, 6, 270–272 CrossRef CAS.
- C. Chatterjee, R. K. McGinty, B. Fierz and T. W. Muir, Nat. Chem. Biol., 2010, 6, 267–269 CrossRef CAS.
- F. Meier, T. Abeywardana, A. Dhall, N. P. Marotta, J. Varkey, R. Langen, C. Chatterjee and M. R. Pratt, J. Am. Chem. Soc., 2012, 134, 5468–5471 CrossRef CAS.
- H. P. Hemantha, S. N. Bavikar, Y. Herman-Bachinsky, N. Haj-Yahya, S. Bondalapati, A. Ciechanover and A. Brik, J. Am. Chem. Soc., 2014, 136, 2665–2673 CrossRef CAS.
- K. Yang, P. Gong, P. Gokhale and Z. Zhuang, ACS Chem. Biol., 2014, 9, 1685–1691 CrossRef CAS.
- N. D. Weikart and H. D. Mootz, ChemBioChem, 2010, 11, 774–777 CrossRef CAS.
- S. Eger, M. Scheffner, A. Marx and M. Rubini, J. Am. Chem. Soc., 2010, 132, 16337–16339 CrossRef CAS.
- S. Eger, B. Castrec, U. Hubscher, M. Scheffner, M. Rubini and A. Marx, ChemBioChem, 2011, 12, 2807–2812 CrossRef CAS.
- V. H. Trang, E. M. Valkevich, S. Minami, Y. C. Chen, Y. Ge and E. R. Strieter, Angew. Chem., Int. Ed., 2012, 51, 13085–13088 CrossRef CAS.
- E. M. Valkevich, R. G. Guenette, N. A. Sanchez, Y. C. Chen, Y. Ge and E. R. Strieter, J. Am. Chem. Soc., 2012, 134, 6916–6919 CrossRef CAS.
- A. Shanmugham, A. Fish, M. P. Luna-Vargas, A. C. Faesen, F. El Oualid, T. K. Sixma and H. Ovaa, J. Am. Chem. Soc., 2010, 132, 8834–8835 CrossRef CAS.
- M. Stanley and S. Virdee, ChemBioChem, 2016, 17, 1472–1480 CrossRef CAS.
- S. K. Singh, I. Sahu, S. M. Mali, H. P. Hemantha, O. Kleifeld, M. H. Glickman and A. Brik, J. Am. Chem. Soc., 2016, 138, 16004–16015 CrossRef CAS.
- S. Bhat, Y. Hwang, M. D. Gibson, M. T. Morgan, S. D. Taverna, Y. Zhao, C. Wolberger, M. G. Poirier and P. A. Cole, J. Am. Chem. Soc., 2018, 140, 9478–9485 CrossRef CAS.
- E. K. Dixon, C. A. Castañeda, T. R. Kashyap, Y. Wang and D. Fushman, Bioorg. Med. Chem., 2013, 21, 3421–3429 CrossRef CAS.
- R. Meledin, S. M. Mali, S. K. Singh and A. Brik, Org. Biomol. Chem., 2016, 14, 4817–4823 RSC.
- A. L. Baumann, S. Schwagerus, K. Broi, K. Kemnitz-Hassanin, C. E. Stieger, N. Trieloff, P. Schmieder and C. P. R. Hackenberger, J. Am. Chem. Soc., 2020, 142, 9544–9552 CrossRef CAS.
- M. Pan, Q. Zheng, S. Ding, L. Zhang, Q. Qu, T. Wang, D. Hong, Y. Ren, L. Liang, C. Chen, Z. Mei and L. Liu, Angew. Chem., Int. Ed., 2019, 58, 2627–2631 CrossRef CAS.
- M. Fottner, A. D. Brunner, V. Bittl, D. Horn-Ghetko, A. Jussupow, V. R. I. Kaila, A. Bremm and K. Lang, Nat. Chem. Biol., 2019, 15, 276–284 CrossRef CAS.
- G.-C. Chu, M. Pan, J. Li, S. Liu, C. Zuo, Z.-B. Tong, J.-S. Bai, Q. Gong, H. Ai, J. Fan, X. Meng, Y.-C. Huang, J. Shi, H. Deng, C. Tian, Y.-M. Li and L. Liu, J. Am. Chem. Soc., 2019, 141, 3654–3663 CrossRef CAS.
- Q. Zheng, T. Wang, G. C. Chu, C. Zuo, R. Zhao, X. Sui, L. Ye, Y. Yu, J. Chen, X. Wu, W. Zhang, H. Deng, J. Shi, M. Pan, Y. M. Li and L. Liu, Angew. Chem., Int. Ed., 2020, 59, 13496–13501 CrossRef CAS.
- Z. Chen and C. M. Pickart, J. Biol. Chem., 1990, 265, 21835–21842 CrossRef CAS.
- R. M. Hofmann and C. M. Pickart, J. Biol. Chem., 2001, 276, 27936–27943 CrossRef CAS.
- W. J. Cook, L. C. Jeffrey, E. Kasperek and C. M. Pickart, J. Mol. Biol., 1994, 236, 601–609 CrossRef CAS.
- J. Piotrowski, R. Beal, L. Hoffman, K. D. Wilkinson, R. E. Cohen and C. M. Pickart, J. Biol. Chem., 1997, 272, 23712–23721 CrossRef CAS.
- R. Varadan, O. Walker, C. Pickart and D. Fushman, J. Mol. Biol., 2002, 324, 637–647 CrossRef CAS.
- C. Alfano, S. Faggiano and A. Pastore, Trends Biochem. Sci., 2016, 41, 371–385 CrossRef CAS.
- K. Kliza and K. Husnjak, Front. Mol. Biosci., 2020, 7, 21 CrossRef CAS.
- M. J. Clague, S. Urbé and D. Komander, Nat. Rev. Mol. Cell Biol., 2019, 20, 338–352 CrossRef CAS.
- M. van Huizen and M. Kikkert, Front. Cell Dev. Biol., 2020, 7, 392 CrossRef.
- M. A. Michel, D. Komander and P. R. Elliott, Methods Mol. Biol., 2018, 1844, 73–84 CrossRef CAS.
- B. Merrifield, Science, 1986, 232, 341–347 CrossRef CAS.
- R. Ramage, J. Green, T. W. Muir, O. M. Ogunjobi, S. Love and K. Shaw, Biochem. J., 1994, 299(Pt 1), 151–158 CrossRef CAS.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Science, 1994, 266, 776–779 CrossRef CAS.
- D. Bang, G. I. Makhatadze, V. Tereshko, A. A. Kossiakoff and S. B. Kent, Angew. Chem., Int. Ed., 2005, 44, 3852–3856 CrossRef CAS.
- M. Uckelmann and T. K. Sixma, DNA Repair, 2017, 56, 92–101 CrossRef CAS.
- J. A. Schmid, M. Berti, F. Walser, M. C. Raso, F. Schmid, J. Krietsch, H. Stoy, K. Zwicky, S. Ursich, R. Freire, M. Lopes and L. Penengo, Mol. Cell, 2018, 71, 897–910.e898 CrossRef CAS.
- H. Wang, L. Wang, H. Erdjument-Bromage, M. Vidal, P. Tempst, R. S. Jones and Y. Zhang, Nature, 2004, 431, 873–878 CrossRef CAS.
- R. Pavri, B. Zhu, G. Li, P. Trojer, S. Mandal, A. Shilatifard and D. Reinberg, Cell, 2006, 125, 703–717 CrossRef CAS.
- B. Zhu, Y. Zheng, A. D. Pham, S. S. Mandal, H. Erdjument-Bromage, P. Tempst and D. Reinberg, Mol. Cell, 2005, 20, 601–611 CrossRef CAS.
- L. Spasser and A. Brik, Angew. Chem., Int. Ed., 2012, 51, 6840–6862 CrossRef CAS.
- F. El Oualid, R. Merkx, R. Ekkebus, D. S. Hameed, J. J. Smit, A. de Jong, H. Hilkmann, T. K. Sixma and H. Ovaa, Angew. Chem., Int. Ed., 2010, 49, 10149–10153 CrossRef CAS.
- M. G. Spillantini, T. D. Bird and B. Ghetti, Brain Pathol., 1998, 8, 387–402 CrossRef CAS.
- W. P. Gai, J. H. Power, P. C. Blumbergs, J. G. Culvenor and P. H. Jensen, J. Neurochem., 1999, 73, 2093–2100 CAS.
- B. C. Campbell, C. A. McLean, J. G. Culvenor, W. P. Gai, P. C. Blumbergs, P. Jäkälä, K. Beyreuther, C. L. Masters and Q. X. Li, J. Neurochem., 2001, 76, 87–96 CrossRef CAS.
- L. Stefanis, E. Emmanouilidou, M. Pantazopoulou, D. Kirik, K. Vekrellis and G. K. Tofaris, J. Neurochem., 2019, 150, 577–590 CrossRef CAS.
- R. Rott, R. Szargel, J. Haskin, V. Shani, A. Shainskaya, I. Manov, E. Liani, E. Avraham and S. Engelender, J. Biol. Chem., 2008, 283, 3316–3328 CrossRef CAS.
- J. T. Lee, T. C. Wheeler, L. Li and L. S. Chin, Hum. Mol. Genet., 2008, 17, 906–917 CrossRef CAS.
- A. Bank and J. V. O'Donnell, Nature, 1969, 222, 295–296 CrossRef CAS.
- J. R. Shaeffer, J. Biol. Chem., 1983, 258, 13172–13177 CrossRef CAS.
- J. R. Shaeffer, J. Biol. Chem., 1994, 269, 22205–22210 CrossRef CAS.
- J. R. Shaeffer and M. A. Kania, Biochemistry, 1995, 34, 4015–4021 CrossRef CAS.
- G. Li, Q. Liang, P. Gong, A. H. Tencer and Z. Zhuang, Chem. Commun., 2014, 50, 216–218 RSC.
- P. Gong, G. A. Davidson, W. Gui, K. Yang, W. P. Bozza and Z. Zhuang, Chem. Sci., 2018, 9, 7859–7865 RSC.
- W. Gui, C. A. Ott, K. Yang, J. S. Chung, S. Shen and Z. Zhuang, J. Am. Chem. Soc., 2018, 140, 12424–12433 CrossRef CAS.
- F. Meng, W. E. Hennink and Z. Zhong, Biomaterials, 2009, 30, 2180–2198 CrossRef CAS.
- J. F. McGouran, S. R. Gaertner, M. Altun, H. B. Kramer and B. M. Kessler, Chem. Biol., 2013, 20, 1447–1455 CrossRef CAS.
- D. Flierman, G. J. van der Heden van Noort, R. Ekkebus, P. P. Geurink, T. E. Mevissen, M. K. Hospenthal, D. Komander and H. Ovaa, Cell Chem. Biol., 2016, 23, 472–482 CrossRef CAS.
- X. Zhang, A. H. Smits, G. B. van Tilburg, P. W. Jansen, M. M. Makowski, H. Ovaa and M. Vermeulen, Mol. Cell, 2017, 65, 941–955.e948 CrossRef CAS.
- X. Zhang, A. H. Smits, G. B. van Tilburg, H. Ovaa, W. Huber and M. Vermeulen, Nat. Protoc., 2018, 13, 530–550 CrossRef CAS.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- Y. Sun, H. Liu, L. Cheng, S. Zhu, C. Cai, T. Yang, L. Yang and P. Ding, Polym. Int., 2018, 67, 25–31 CrossRef CAS.
- J. Guo, J. Wang, J. S. Lee and P. G. Schultz, Angew. Chem., Int. Ed., 2008, 47, 6399–6401 CrossRef CAS.
- J. M. Chalker, S. B. Gunnoo, O. Boutureira, S. C. Gerstberger, M. Fernández-González, G. J. L. Bernardes, L. Griffin, H. Hailu, C. J. Schofield and B. G. Davis, Chem. Sci., 2011, 2, 1666–1676 RSC.
- J. M. Chalker, L. Lercher, N. R. Rose, C. J. Schofield and B. G. Davis, Angew. Chem., Int. Ed., 2012, 51, 1835–1839 CrossRef CAS.
- M. R. Vallée, M. W. Schombs, Z. J. Balaban, J. Colyer and B. G. Davis, Chem. Commun., 2016, 52, 3014–3017 RSC.
- R. Ekkebus, D. Flierman, P. P. Geurink and H. Ovaa, Curr. Opin. Chem. Biol., 2014, 23, 63–70 CrossRef CAS.
- D. S. Hewings, J. A. Flygare, M. Bogyo and I. E. Wertz, FEBS J., 2017, 284, 1555–1576 CrossRef CAS.
- D. Conole, M. Mondal, J. D. Majmudar and E. W. Tate, Front. Chem., 2019, 7, 876 CrossRef.
- W. Gui, P. Paudel and Z. Zhuang, Compr. Nat. Prod. Chem., 2020, 589–602 Search PubMed.
- X. Sui, Y. Wang, Y.-X. Du, L.-J. Liang, Q. Zheng, Y.-M. Li and L. Liu, Chem. Sci., 2020, 11, 12633–12646 RSC.
- S. Bondalapati, W. Mansour, M. A. Nakasone, S. K. Maity, M. H. Glickman and A. Brik, Chemistry, 2015, 21, 7360–7364 CrossRef CAS.
- H. Sun, R. Meledin, S. M. Mali and A. Brik, Chem. Sci., 2018, 9, 1661–1665 RSC.
- V. De Cesare, D. Carbajo Lopez, P. D. Mabbitt, A. J. Fletcher, M. Soetens, O. Antico, N. T. Wood and S. Virdee, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(4), e2006947118 CrossRef.
- D. S. Hameed, A. Sapmaz, L. Gjonaj, R. Merkx and H. Ovaa, ChemBioChem, 2018, 19, 2553–2557 CrossRef CAS.
- G. Mann, G. Satish, R. Meledin, G. B. Vamisetti and A. Brik, Angew. Chem., Int. Ed., 2019, 58, 13540–13549 CrossRef CAS.
- W. Gui, S. Shen and Z. Zhuang, J. Am. Chem. Soc., 2020, 142, 19493–19501 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |