
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembly and multifunctionality of peptide organogels: oil spill recovery, dye absorption and synthesis of conducting biomaterials†
Monikha Chetia‡
,
Swapna Debnath‡,
Sumit Chowdhury and
Sunanda Chatterjee *
*
Department of Chemistry, Indian Institute of Technology, Guwahati, Guwahati, Assam, India 781039. E-mail: sunanda.c@iitg.ac.in; Tel: +91-361-2583310
First published on 31st January 2020
Abstract
The self-assembly of a series of low molecular weight gelator dipeptides containing para amino benzoic acid has been studied in mechanistic detail. All four dipeptides form phase selective, thermoreversible, rigid gels in a large range of organic solvents and fuels such as petrol, diesel, and kerosene. The mechanism of self-assembly has been dissected in detail using several experimental techniques. Self-assembly is driven mainly by aromatic and hydrophobic interactions. Hydrogen bonding groups, though present, seem to make a trivial contribution towards the self-assembly process. Phase selective gelation abilities in fuels in the presence of acidic, basic and saline conditions, together with the easy recovery of fuels from the organogels, render the peptides potential candidates for addressing oil-spill recovery. Being electron rich systems, these organogelators can absorb cationic dyes with >90% efficiency from wastewater. Finally, conducting biomaterials have been synthesized by the insertion of reduced graphene oxide into the organogels. Such small peptide based gelator molecules, being economically viable and easy to prepare, in addition to being multifunctional, are a hot area of research in the field of materials chemistry.
Introduction
Low molecular weight supramolecular gels (LMWGs) are an important class of materials that can encapsulate large amounts of solvents in mesh-like entangled structures that are formed via the ordered hierarchical arrangement of individual molecules.1–4 These assemblies are driven by non-covalent forces such as hydrogen bonding, π–π interactions, hydrophobic interactions, van der Waals forces, charge transfer interactions, etc.,5–9 and can be controlled by various stimuli such as concentration, pH, UV radiation, temperature, solvent polarity and by salts.10–24 Various small molecules such as amides, peptides, ureas, peptoids, nucleobases, etc. have shown spectacular abilities to self-assemble. Supramolecular gels have been used in various applications such as in water purification, through the removal of organic dyes, oils and metal ions;25–38 as templates for nanoparticle fabrication;39–44 and in cosmetics, catalysts,45,46 dye sensitized solar cells,47 drug delivery systems,48–54 etc. Although the self-assembly of peptides was achieved serendipitously previously, extensive research over the last couple of decades has established that, by proper choice of the individual building blocks that are capable of promoting various non-covalent interactions, smart molecules with predictable and tunable self-assembling properties can be designed. However, even now the design of LMWGs is an important field of research due to their reduced expenditure, easy synthesis, biocompatibility and multifunctionality.Water pollution is one of the fundamental environmental problems faced by humanity at present. Marine oil spills due to the leakage of crude oil and petroleum products (i.e. fuels) are a major source of water pollution that creates a great environmental hazard for marine ecosystems.55 Different methods such as bioremediation,56,57 dispersants,58 solidifiers,59–61 sorbents62–64 and skimmers65–67 have been developed to tackle the problem. However, each of these approaches have their own shortcomings.68 Hydro/organogel materials have also been used in the recovery of oil spills.26,68–71 Another critical source of water pollution is the organic dyes that are routinely used in several industries.72 Dye effluents and their degradation products are carcinogenic, and have a negative impact on the reproductive and immune systems.73–77 Conventional methods of treating dye effluents such as incineration, biological treatment, absorption upon solid matrices such as activated carbon, chemical precipitation, electrochemical techniques, ion exchange, and others, have their own limitations due to their low sensitivity, incomplete removal, high energy requirements, and production of toxic sludge.78 Hydro/organogel-based soft materials28–38 offer an appealing alternative for the removal of dyes from contaminated water, due to their high water permeability, large surface area for adsorption and simplicity of use, along with their reusability and good biodegradability.
The supramolecular assembly of bioconjugates appended to optoelectronically active π-conjugated chromophores in organic solvents has already been reported.79–84 Hybrid materials using graphene and its derivatives with inorganic/polymeric materials are highly conducting and have been used in interesting applications including renewable energy conversion/storage devices, supercapacitors, electroresponsive systems, electrocatalysis, optoelectronic devices, electromagnetic interference shielding etc.85–96 In a number of studies, graphene and its derivatives have also been incorporated into peptide organogels. Peptide-based hybrid materials become moderately conducting in nature upon insertion of graphene or its derivates.97–100
In this study, four dipeptides, P1–P4, which are rich in aromatic moieties were synthesized in order to study their self-assembly and to generate materials with multiple applications. The building blocks of P1–P4 were chosen so that their self-assembly would be guided by aromatic and hydrophobic interactions. The mechanism of self-assembly of P1–P4 was established using various spectroscopic and microscopic experiments. Interestingly, all the peptides were capable of forming gels in a variety of organic solvents and in fuels such as petrol, diesel and kerosene, and this made them ideal materials to be used for oil spill recovery. All the organogels studied here were excellent adsorbents for cationic dyes. RGO was incorporated into the peptide organogels to generate conducting biomaterials. Generating environmentally friendly materials that have multiple applications, using directed self-assembly methods, is of considerable interest and was the main focus of our study.
Materials and methods
Materials
All amino acids, L-Phe, L-Trp, PABA, L-Phg, L-HomoPhe, all solvents, di-t-butyl dicarbonate, N,N′-dicyclohexylcarbodiimide (DCC), sodium hydroxide, 1-hydroxybenzotriazole (HOBt), and thionyl chloride were purchased from Spectrochem, Merck and Rankem. CDCl3 and DMSO-d6 solvents were obtained from Sigma Aldrich. The dyes crystal violet (CV) and rhodamine B (RB) were purchased from Spectrochem. Fuels such as kerosene, diesel and petrol were obtained from Bharat Petroleum Gas Station. Graphite powder was purchased from Asbury Carbons.Methods
Yield: P1, 78%; P2, 80%; P3, 82%; P4, 86%.
All peptides were characterized using HPLC (Fig. S1–S4†), ESI-MS (Fig. S5–S8†) and 1H NMR (Fig. S9–S12†).
ESI-MS. ESI-MS of P1. Mass calc. for P1: (M + H)+ = 438.208 Da; mass obs.: (M + H)+ = 438.2092 Da.
ESI-MS of P2. Mass calc. for P2: (M + H)+ = 413.208 Da; mass obs.: (M + H)+ = 413.2020 Da.
ESI-MS of P3. Mass calc. for P3: (M + H)+ = 385.178 Da; mass obs.: (M + H)+ = 385.1934 Da.
ESI-MS of P4. Mass calc. for P4: (M + H)+ = 399.188 Da; mass obs.: (M + H)+ = 399.1927 Da.
1H NMR. P1 (Fig. S9†): 1H NMR (400 MHz, DMSO-d6) δ 10.82 (s, 1H, NH of indole ring), 10.40 (s, 1H, NH of backbone), 7.92 (d, J = 8.5 Hz, 2H, aromatic ring Hs), 7.75 (d, J = 8.6 Hz, 2H, aromatic ring Hs), 7.65 (d, J = 7.9 Hz, 1H, aromatic ring H), 7.32 (d, J = 8.1 Hz, 1H, NH of Boc group), 7.18 (s, 1H, H of aromatic ring), 7.05 (t, J = 7.7 Hz, 2H, aromatic ring Hs), 6.97 (t, J = 7.4 Hz, 1H, H of aromatic ring), 4.39 (d, J = 6.7 Hz, 1H, H of alpha –CH), 3.82 (s, 3H, Hs of –CH3 group), 3.12 (dd, J = 14.6, 5.3 Hz, 1H, H of beta –CH2), 3.00 (dd, J = 14.5, 9.0 Hz, 1H, H of beta –CH2), 1.33 (s, 9H, Hs of Boc group).
P2 (Fig. S10†): 1H NMR (600 MHz, DMSO-d6) δ 10.36 (s, 1H, NH of backbone), 7.93–7.89 (m, 2H, Hs of aromatic ring), 7.74 (d, J = 8.8 Hz, 2H, Hs of aromatic ring), 7.32 (d, J = 7.6 Hz, 1H, NH of Boc group), 7.27 (t, J = 7.5 Hz, 2H, Hs of aromatic ring), 7.24–7.19 (m, 2H, Hs of aromatic ring), 7.19–7.15 (m, 1H, H of aromatic ring), 4.10 (ddd, J = 9.5, 7.5, 4.7 Hz, 1H, H of alpha –CH group), 3.82 (s, 3H, Hs of –CH3 group), 2.71 (ddd, J = 13.3, 10.8, 5.0 Hz, 1H, H of beta –CH2 group), 2.58 (ddd, J = 13.5, 10.8, 5.9 Hz, 1H, H of beta CH2), 1.97–1.83 (m, 2H, Hs of gamma –CH2 group), 1.40 (s, 9H, Hs of Boc group).
P3 (Fig. S11†): 1H NMR (400 MHz, DMSO-d6) δ 10.57 (s, 1H, H of backbone NH), 7.93–7.88 (m, 2H, Hs of aromatic ring), 7.74–7.68 (m, 2H, Hs of aromatic ring), 7.59 (d, J = 8.1 Hz, 1H, NH of Boc group), 7.52–7.47 (m, 2H, Hs of aromatic ring), 7.38–7.27 (m, 3H, Hs of aromatic ring), 5.36 (d, J = 8.1 Hz, 1H, H of alpha –CH), 3.81 (s, 3H, Hs of –CH3), 1.39 (s, 9H, Hs of Boc group).
P4 (Fig. S12†): 1H NMR (400 MHz, DMSO-d6) δ 10.39 (s, 1H, NH of backbone), 7.96–7.90 (m, 2H, Hs of aromatic ring), 7.77–7.69 (m, 2H, Hs of aromatic ring), 7.29 (dt, J = 14.8, 7.5 Hz, 4H, Hs of aromatic ring and NH of Boc group), 7.20 (d, J = 7.7 Hz, 2H, Hs of aromatic ring), 4.44–4.16 (m, 1H, H of alpha –CH), 3.82 (s, 3H, Hs of –CH3 group), 2.99 (dd, J = 13.7, 4.7 Hz, 1H, H of beta –CH2), 2.84 (dd, J = 13.7, 10.1 Hz, 1H, H of beta –CH2), 1.32 (s, 9H, Hs of Boc group).
To make the RGO incorporated P1 organogel (RGO-P1), the freshly prepared RGO (1 mg, 0.2 wt%) was added to 500 μL 1,2-DCB solution of peptide P1 at its MGC. The mixture was sonicated until a homogeneous suspension of RGO in the peptide was obtained. This was further heated at 100 °C for 15 min, followed by vortexing for 10 s. Upon cooling the mixture, a black colored organogel was formed within a further 5–10 min. Hybrid organogels were formed by varying the amount of RGO.
| Dye loaded = initial dye − unabsorbed dye, |
| loading efficiency = (dye loaded/initial dye) × 100 |
Results and discussion
We synthesized four dipeptides, Boc-Trp-PABA-OMe (P1), Boc-homoPhe-PABA-OMe (P2), Boc-Phg-PABA-OMe (P3) and Boc-Phe-PABA-OMe (P4) (Fig. 1), using a standard solution based peptide synthesis strategy, purified them using column chromatography and characterized them using analytical HPLC, 1H NMR and ESI-MS (Fig. S1–S12†). All four dipeptides were terminally protected and contained an aromatic delta amino acid residue, PABA. The second amino acid residue (Trp, Homo-Phe, Phg or Phe) was aromatic in nature. The design of the building blocks intended to facilitate self-assembly using aromatic interactions and hydrophobic interactions. Homologs of Phe, namely Phg and Homo-Phe, were used to understand the role of the length of the side chain on the assembly of the peptides.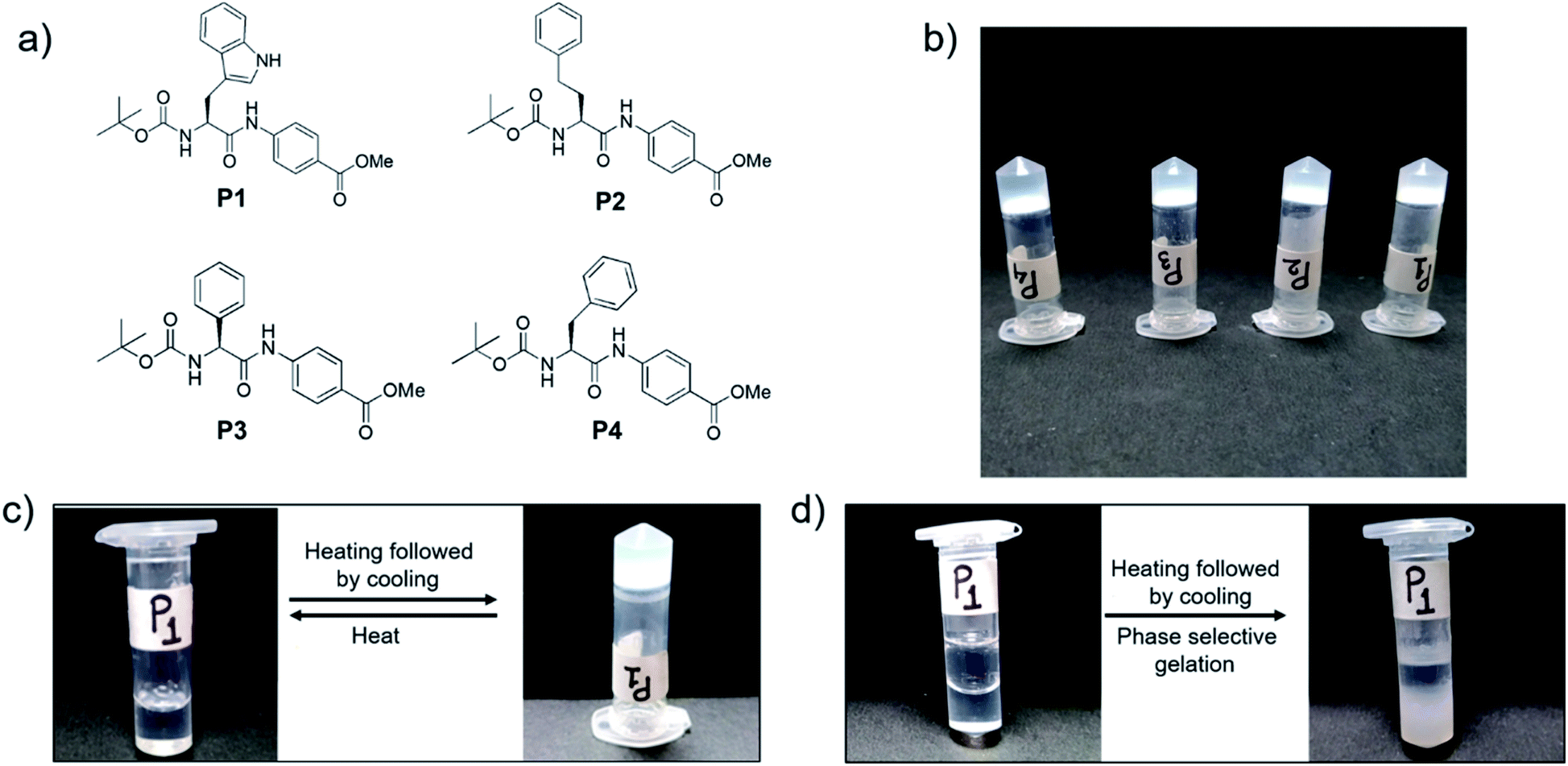 | ||
| Fig. 1 (a) Chemical structures of LMWG P1–P4, (b) gels formed in 1,2-DCB by P1–P4 (c) thermoreversibility of the organogel formed by P1 in 1,2-DCB and (d) phase selective gelation of P1 in 1,2-DCB in the presence of water. | ||
All of the dipeptides readily formed phase selective, thermoreversible gels in various organic solvents such as chloroform, THF, 1,2-dichlorobenzene, etc. (Fig. 1 and Table S1†) and in fuels such as kerosene, diesel and petrol, upon heating and subsequent cooling. The minimum gelation concentration of the peptides depended on the solvents, but it was generally between 0.2–0.4 w/v% (Table S1†). The thermal stability of the peptides was studied from the Tgel value, which was in the range of 30–80 °C. As the gels were translucent and were most thermostable in 1,2-DCB, further characterization of the organogels was carried out in this solvent.
The self-assembled morphologies of P1–P4 in 1,2-DCB in both solution and gel states were studied using Field Emission Scanning Microscopy (FESEM) (Fig. 2 and S13†). It was clear that the morphology of P1 in solution (Fig. S13†) and in the gel state (Fig. 2) were different. The morphology of P1 was nano fibrous in the solution state, while it appeared as a densely packed continuous matrix in the gel state. The morphologies of the peptides P2 and P3 in solution and in the gel state were the same, but with slight variations. Peptide P4 formed a fibrous entangled morphology both in solution and in the gel state, with a denser mesh in the latter. The subtle differences in the morphologies of P1–P4 can be attributed to the varying chains of the amino acids present in them.
 | ||
| Fig. 2 FESEM images of the organogels obtained in 1,2-DCB for (a) P1, (b) P2, (c) P3 and (d) P4. | ||
Rheology
The mechanical strength and the stability of the organogels derived from self-assembly of the individual peptides were studied by rheology. The storage modulus (G′) and the loss modulus (G′′) were measured as functions of different parameters such as angular frequency and the strain sweep. For this study, gels were formed from P1–P4 in 1,2-DCB at 1.0% w/v. In the angular frequency sweep experiment that was performed at a constant strain of 0.1% at 25 °C, for all four organogels, the G′ value was found to dominate over the G′′ value, until about 100 rad s−1 (Fig. 3a and S14a†). In addition, G′ and G′′ were found to be independent of angular frequency in the region of 1–100 rad s−1, indicating the formation of stable organogels. The storage moduli of all the organogels were of the order of ≥104 Pa in the frequency sweep experiment, indicating considerable mechanical strength of all the organogels. In the strain sweep experiment, where the storage moduli (G′) and the loss moduli (G′′) for the four organogels were plotted as a function of % strain (0.1–100%) (Fig. 3b and S14b†), it was found that G′ was higher than G′′(of the order of 104 Pa) until a particular strain (linear viscoelastic region (LVR), γ = 0.1% for P1–P4).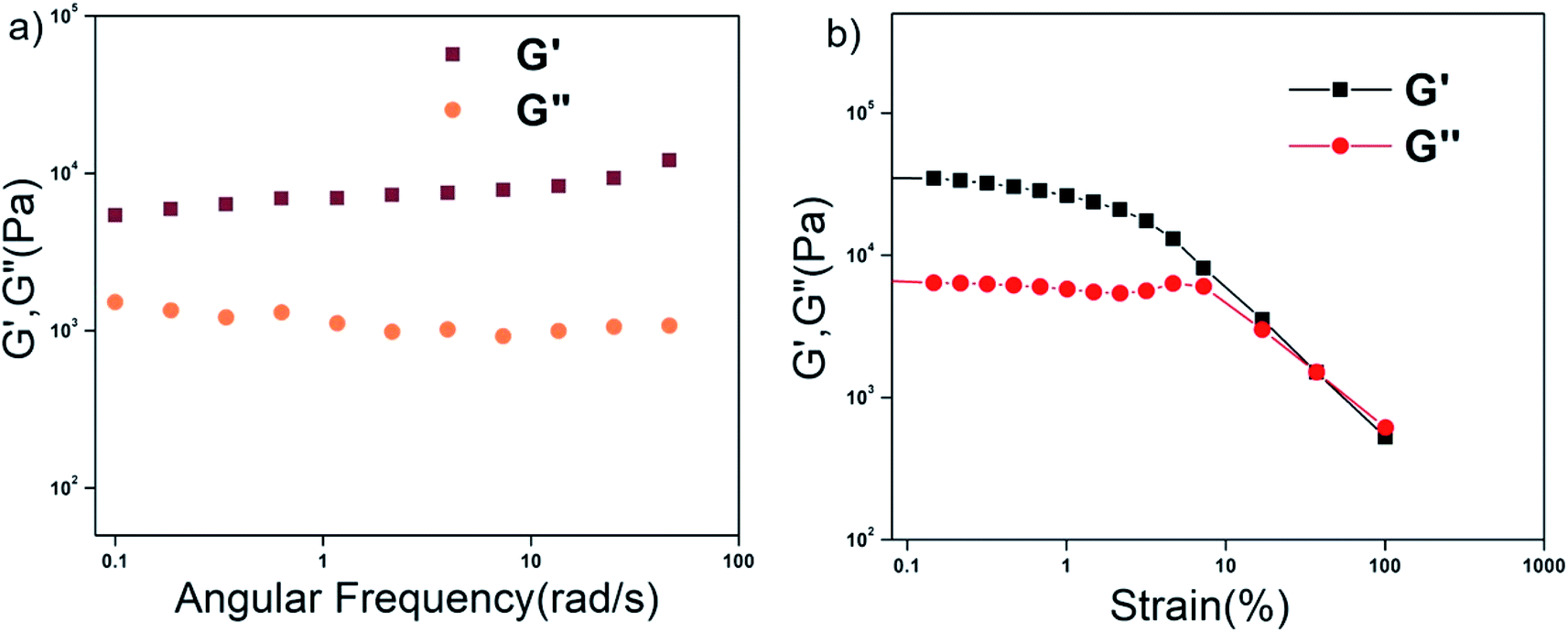 | ||
| Fig. 3 Rheology of the P1 xerogel. (a) Frequency dependence and (b) strain dependence of the dynamic storage modulus (G′) and the loss modulus (G′′) of the organogel from P1 in 1,2-DCB at 2% w/v. | ||
Non-covalent interactions in promoting self-assembly
The peptides were designed to be hydrophobic and rich in aromatic moieties. Thus, aromatic pi–pi interactions and hydrophobic interactions were expected to be the key non-covalent forces driving self-assembly. Peptide backbones are rich in hydrogen bond donor and acceptor groups and hence hydrogen bonding might be another important factor aiding self-assembly. The following experiments were performed to identify the key interactions driving the assembly process.Fluorescence studies
The intrinsic fluorescence of the tryptophan residue was used to study the role of aromatic π–π stacking interactions in the self-assembly of peptide P1. Fig. 4a shows the fluorescence emission spectra of P1 in THF at various concentrations (0.1–7 mM). The emission maxima of tryptophan at 325 nm was steadily quenched upon increasing the concentration of the peptide. This could be attributed to the stacking of the tryptophan moieties with progressive self-assembly upon increasing the concentration of the peptide. A prominent excimer peak was observed at 455 nm, and this was quenched upon increasing the concentration of the peptide. Usually, increasing the peptide concentration leads to more excimer formation, which is accompanied by an increase in the intensity of the excimer fluorescence peak. P1 contains two types of aromatic moieties, the indole ring from Trp and the aromatic backbone of the PABA amino acid. If the self-assembly of P1 leads to stacking of these two different moieties, then an exciplex is formed instead of an excimer. The fluorescence intensity for the exciplex may be quenched with the increase in the concentration of the exciplex.101 Thus, this experiment clearly proves that in the case of P1, aromatic π–π stacking interactions play an important role and suggests that stacking occurs between the Trp side chains and the aromatic ring of PABA.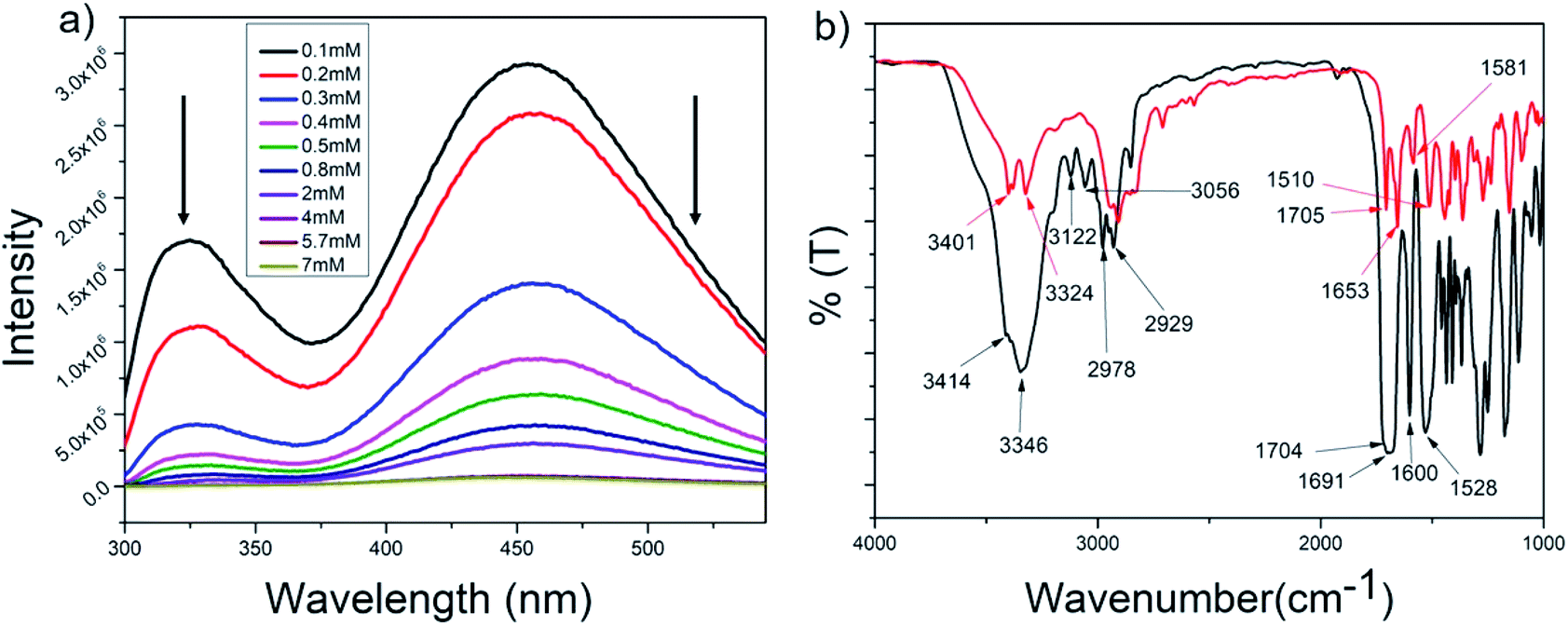 | ||
| Fig. 4 (a) Concentration dependent tryptophan fluorescence spectra for P1 in THF (λex at 280 nm) and (b) superimposed FTIR spectra of P1 in the powdered (black) and xerogel (red) states. | ||
As peptides P2–P4 are structurally very similar to P1, it can be assumed that aromatic stacking also plays an important role in their self-assembly.
FT-IR
Fig. 4b shows the superposition of the FT-IR spectra recorded for the powder and xerogel samples in 1,2-DCB for P1. There are two distinct peaks observed at 3414 and 3346 cm−1 in the powdered form of P1 for non-hydrogen bonded and hydrogen bonded NHs, respectively, and these shift to 3401 and 3324 cm−1, respectively, in the xerogel, indicating increased hydrogen bonding in the xerogel state. However, the presence of two peaks indicates both the existence of hydrogen bonded and non-hydrogen bonded NHs. The CO stretching frequency shifts from 1691 cm−1 in the powdered form to 1653 cm−1 in the xerogel form, suggesting that the backbone conformation of the P1 is not beta sheet-like. In the case of P2–P4 (Fig. 5a and S15†), a broad peak is observed in the NH stretching region at around 3420–3430 cm−1 for the xerogel, indicating the presence of non-hydrogen bonded backbone NHs. The lack of any peak at around 3200 cm−1 suggests the absence of hydrogen bonded amide protons in these systems in the self-assembled state. The amide I peaks in P2–P4 do not indicate a typical β-sheet type of conformation. The presence of the hydrogen bonding status of the amide protons was further studied using NMR spectroscopy.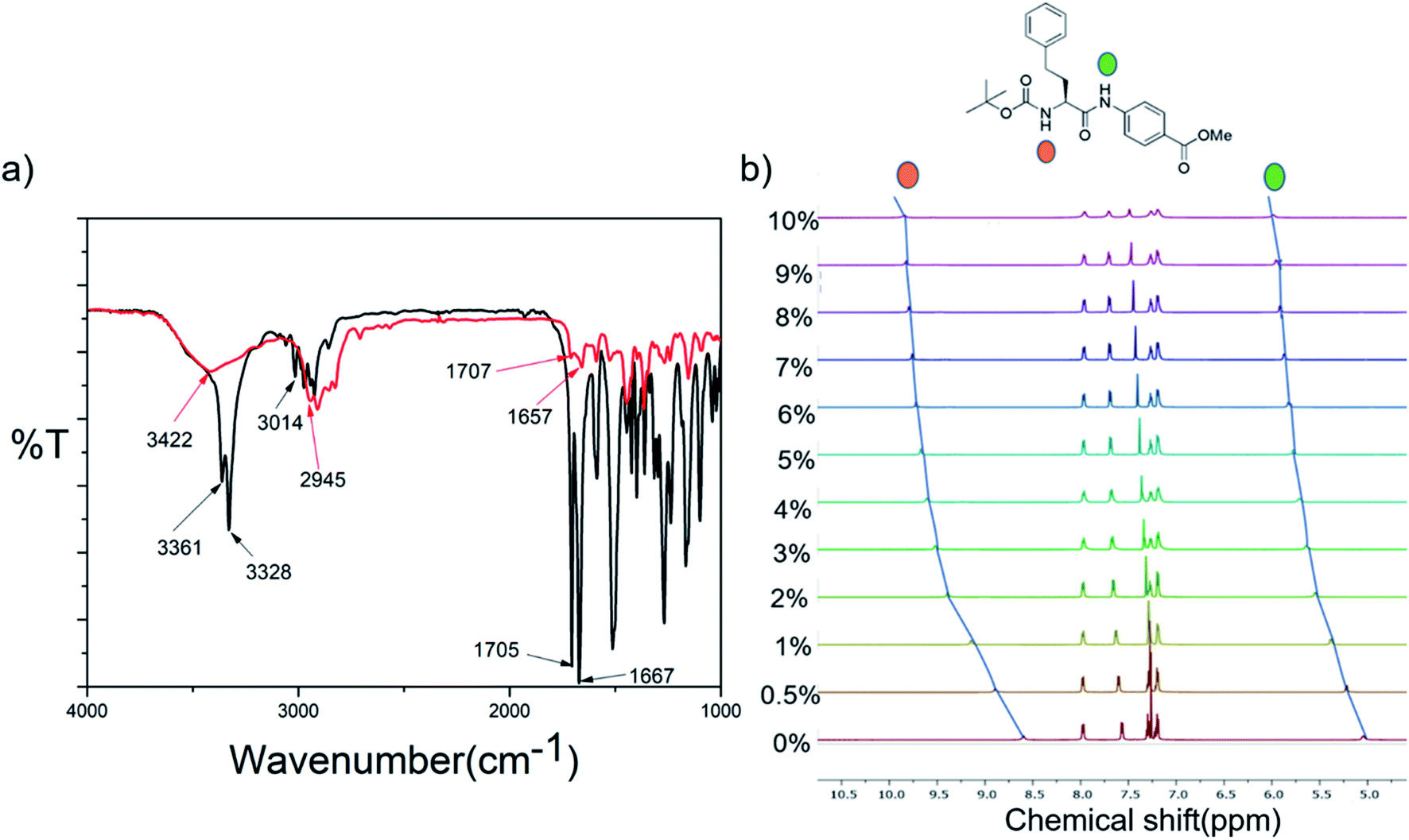 | ||
| Fig. 5 (a) Superimposed FT-IR spectra of P2 in powder (black) and xerogel (red) states. (b) Stacked NH region of the 1H NMR spectra obtained upon addition of increasing amounts of DMSO-d6 to the CDCl3 solution of P2. | ||
NMR
The role of intermolecular hydrogen bonds in driving self-assembly was probed by monitoring the NH chemical shifts of P1 and P2 in CDCl3 at concentrations below, above and at their minimum gelation concentration (Fig. S16a and b†). For both peptides, none of the NH protons showed any change in their chemical shifts over the entire concentration range, indicating that there was no change in the hydrogen bonded state of the backbone amide protons over the concentration range in which the study was performed. Furthermore, in order to understand the hydrogen bonded state of the amide protons, DMSO titration experiments of P1 and P2 were performed (Fig. S17† and 5b). For P1, it was seen that the NH of tryptophan did not show any change in the chemical shift value, suggesting it to be hydrogen bonded, while the NH of the PABA residue and the indole NH showed considerable changes in their chemical shift values, suggesting that they were solvent exposed or non-hydrogen bonded. This corroborates the observations from FT-IR experiments of P1 in the xerogel state, where there was the presence of both hydrogen bonded, as well as non-hydrogen bonded, NHs. Aromatic protons show a striking upfield shift upon the addition of DMSO-d6 to the CDCl3 solution of P1. The addition of increasing amounts of DMSO-d6 disrupts the self-assembly, thus changing the electronic environment of the aromatic protons, which become shielded. This is an indirect proof of the fact that stacking of aromatic moieties plays an important role in the self-assembly of P1. In the case of P2, NHs of both the amino acid residues were solvent exposed or were non-hydrogen bonded, supporting the FT-IR data where only a broad peak corresponding to non-hydrogen bonded NHs was observed.Thus, in summary, the hydrogen bonding pattern in P1 is slightly different from that in P2–P4. Hydrogen bonding does not seem to play a crucial role in the self-assembly process of P2–P4, unlike it does in P1.
PXRD
Information on the molecular packing of peptides P1–P4 in the gel state was obtained upon performing PXRD experiments (Fig. S18†). The PXRD pattern for the P1 xerogel is ordered, unlike that for its powdered form (Fig. S19†). The wide angle PXRD patterns for the P1–P4 xerogels showed similar periodic diffraction patterns, indicating the presence of a similar ordered arrangement in all the xerogels. In the peptide xerogels of P1–P4, peaks were observed at d values of 6.25 Å (2θ = 14.12), 5.19 Å (2θ = 17.04), 4.75 Å (2θ = 18.64) and 4.10 Å (2θ = 21.60) (Table S2†). The ideal centroid to centroid distance in the π–π stacked aromatic systems is about 4 Å. The distance of 4.20 Å that was present in all the xerogels may be attributed to the distance in between the aromatic moieties of the gelator molecules present in the system. This again indicates that aromatic pi–pi interactions are crucial for self-assembly, and that they are present in all the xerogel systems. The peak at around 4 Å is absent in the PXRD pattern of the powdered samples of P1, P3 and P4, indicating the lack of aromatic pi–pi interactions in the powdered form of the peptides.Crystal structures of P2 and P4
The structures of peptides P2 and P4 were elucidated from the single crystal X-ray analysis. Crystals were grown from the MeOH–H2O system by slow evaporation of solvent at room temperature (Table S3†). The backbone of both peptides P2 and P4 are kinked at the Cα carbon of the alpha amino acid residue (Fig. 6, S20 and Table S4†). There are no intramolecular hydrogen bonds present in either of the peptides. We could not obtain crystals for peptides P1 or P3 but, as the backbone of all the molecules is the same, we can assume that a similar backbone conformation is also present in P1 and P3. P2 and P4 do not have a beta sheet-like conformation, and this explains the absence of any characteristic peak in the amide I region in the FTIR spectra of the xerogels. Fig. S21 and S22† show the packing in crystals of P2 and P4, respectively. The packing in both the systems is fairly similar. For both the peptides, pairs of molecules pack together and such pairs stack to form column-like structures. In the case of P2, Boc groups of the two individual molecules in a pair face each other. The aromatic rings of PABA come close together, while the aromatic groups of Homo-Phe stick out in opposite directions. In P4, the packing of a pair of molecules is a bit different, with the Boc groups of one molecule pointing away from the other and the Phe side chain of one molecule stacking with the phenyl ring in the backbone of PABA of the other molecule. In both P2 and P4, pairs of molecules self-assemble to form columnar structures, primarily through aromatic stacking interactions, hydrophobic interactions and weak hydrogen bonding interactions (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H–N bond distance of about 2.2 Å, Table S5†).
O⋯H–N bond distance of about 2.2 Å, Table S5†).
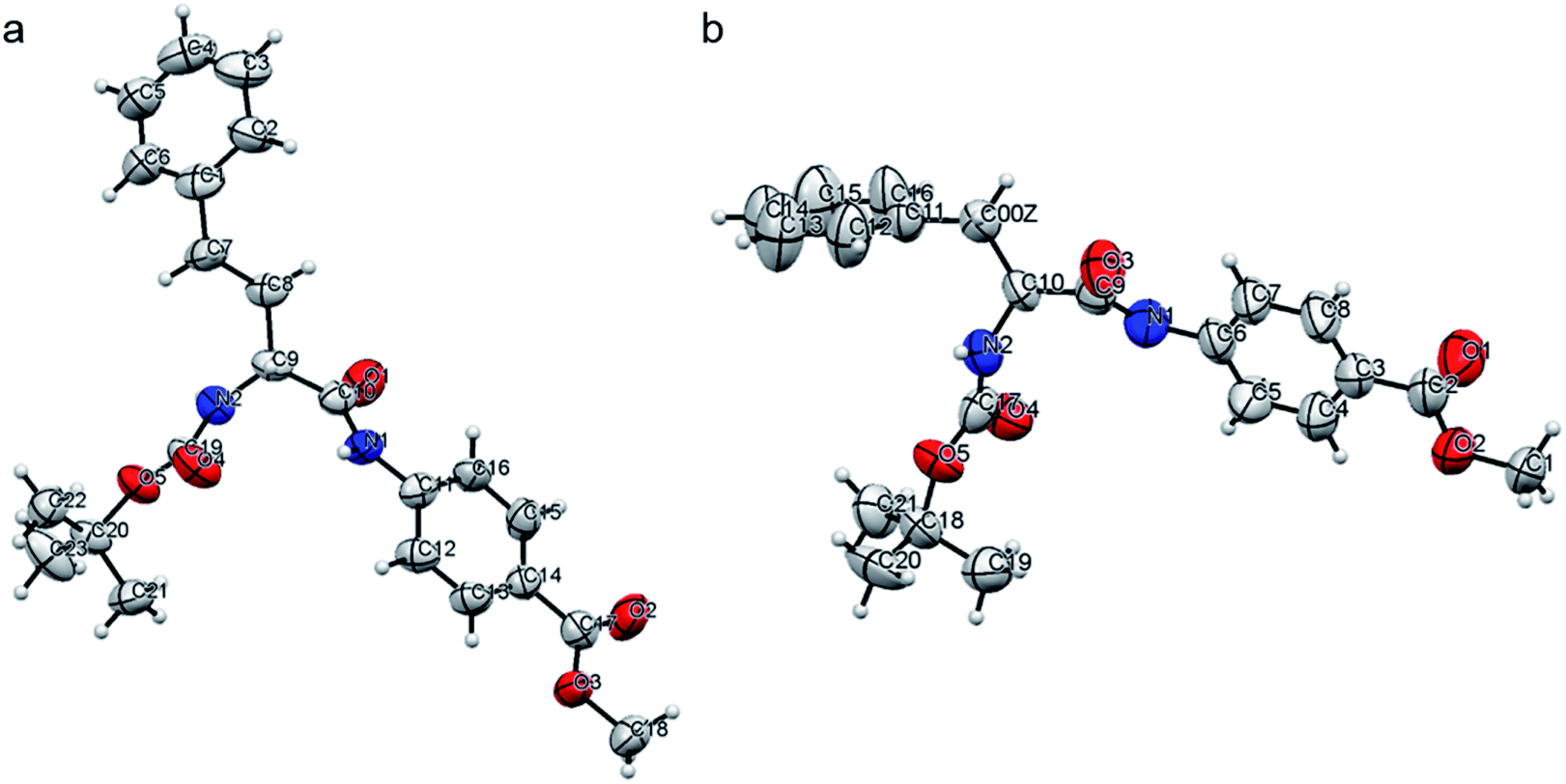 | ||
| Fig. 6 An ORTEP diagram for peptides (a) P2 and (b) P4, with an ellipsoid of 50% probability, as obtained in the crystals grown from the slow evaporation of MeOH–H2O. | ||
Mechanism of self-assembly
Fig. 7 summarizes the mechanism of self-assembly of LMWG P1–P4. The molecules assemble primarily through aromatic pi–pi stacking and hydrophobic interactions. Pi–pi stacking might occur in between the same or different aromatic moieties. Although all the molecules contain ample hydrogen bond donors and acceptors in the backbone, there seems to be an absence of hydrogen bonding in the self-assembled system, as evidenced from the FT-IR and NMR studies. While P2–P4 completely lack hydrogen bonding, P1 contains both hydrogen bonded and non-hydrogen bonded NHs. In the crystals of P2 and P4, weak hydrogen bonds (2.2 Å) are present, which help with crystal packing. A closer approach of the molecules to yield stronger hydrogen bonding might be absent in the crystals to minimize associated unfavorable van der Waals interactions. In the supramolecular packing arrangement in the organogels, the presence of solvent molecules further weakens the hydrogen bonds to the limit of non-existence. Thus, it is clear that the mere presence of hydrogen bonding groups does not ensure hydrogen bonding to be one of the driving forces in the self-assembly of the system. Instead, the assembly of molecules exploits only those parameters that are sufficient, to give rise to a thermodynamically favored system. In this case, the aromatic pi–pi stacking interactions and the hydrophobic interactions were sufficient enough to create and sustain thermodynamically favorable self-assembly. There might be subtle differences in the self-assembly of the molecules of P1–P4, depending on the side chains of the first amino acid residue. However, the global arrangement of the molecules is similar, as evidenced from the PXRD experiments. The backbone conformation of the peptides is kinked at the Cα carbon atom, preventing the formation of beta sheets, as is evidenced by the FT-IR studies. The molecules stack on top of each other, initiating unidirectional self-assembly, giving rise to column like structures with fibrous morphology. Many of these thin fibers assemble together due to favorable hydrophobic interactions, forming thicker fibers that are seen in the FESEM experiments.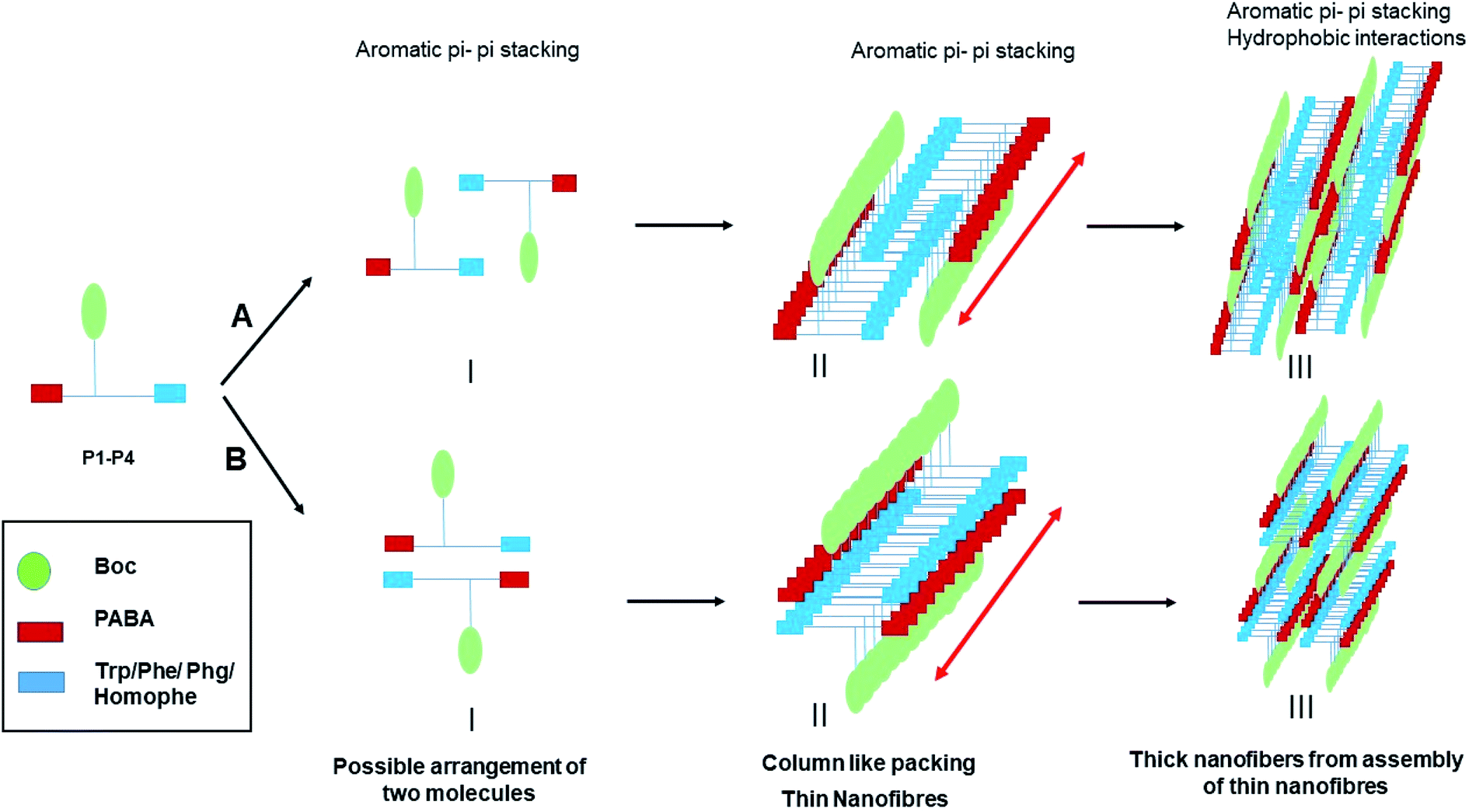 | ||
| Fig. 7 Mechanism of the self-assembly of P1–P4. | ||
Careful observation indicates that P1–P4 form gels in several less polar organic solvents such as in toluene, chloroform, dichlorobenzene etc., in contrast to more polar organic solvents such as ethanol, DMSO etc. This seems to be logical, as the non-polar solvents are better accommodated in the entangled network formed by the hydrophobic peptides P1–P4. As the solvents are non-polar and there is no evidence of hydrogen bonding obtained from either FTIR or NMR, it can be concluded that the only interaction of the peptides with the solvents is of hydrophobic origin.
Oil spill recovery
Peptides P1–P4 phase selectively formed gels in fuels such as kerosene, petrol and diesel in the presence of acidic, basic and saline aqueous media upon being heated and subsequently cooled (Fig. 8, S23b and Table S6†). The gels had a fibrous morphology, as seen from FESEM results (Fig. 8b) and they were mechanically robust (Fig. 8c). In the frequency sweep experiment, the G′ value was greater than the G′′ value throughout the entire range of the experiment. G′ had a value in the order of 104 to 105, and this suggested that the gels were robust in nature (Fig. 8c). The backbone conformation of the peptide in the xerogel state was probed using FT-IR (Fig. 8d). Although the amide I peak in the xerogel of P1 in kerosene at 1636 cm−1 was in the antiparallel beta sheet region, the observation of the peak at 3445 cm−1 indicated the presence of non-hydrogen bonded amide protons in the system, and this counters the presence of the beta sheet-like conformation. PXRD of the P1 xerogel in kerosene showed a similar peak pattern and hence interplanar distances as observed for the xerogels of P1 obtained from other organic solvents (Fig. S23a†). This indicated identical arrangement of the molecules in both cases. Summarizing all of the above, the mode of self-assembly of P1 in kerosene seems to be identical to that in other organic solvents. About 70% of the kerosene could be easily recovered upon vacuum distillation of the P1 organogel in kerosene. Facile recovery of the fuel in addition to the gelation ability of the peptides in the presence of variable pH and salt conditions renders the peptides potential candidates for addressing oil spill recovery.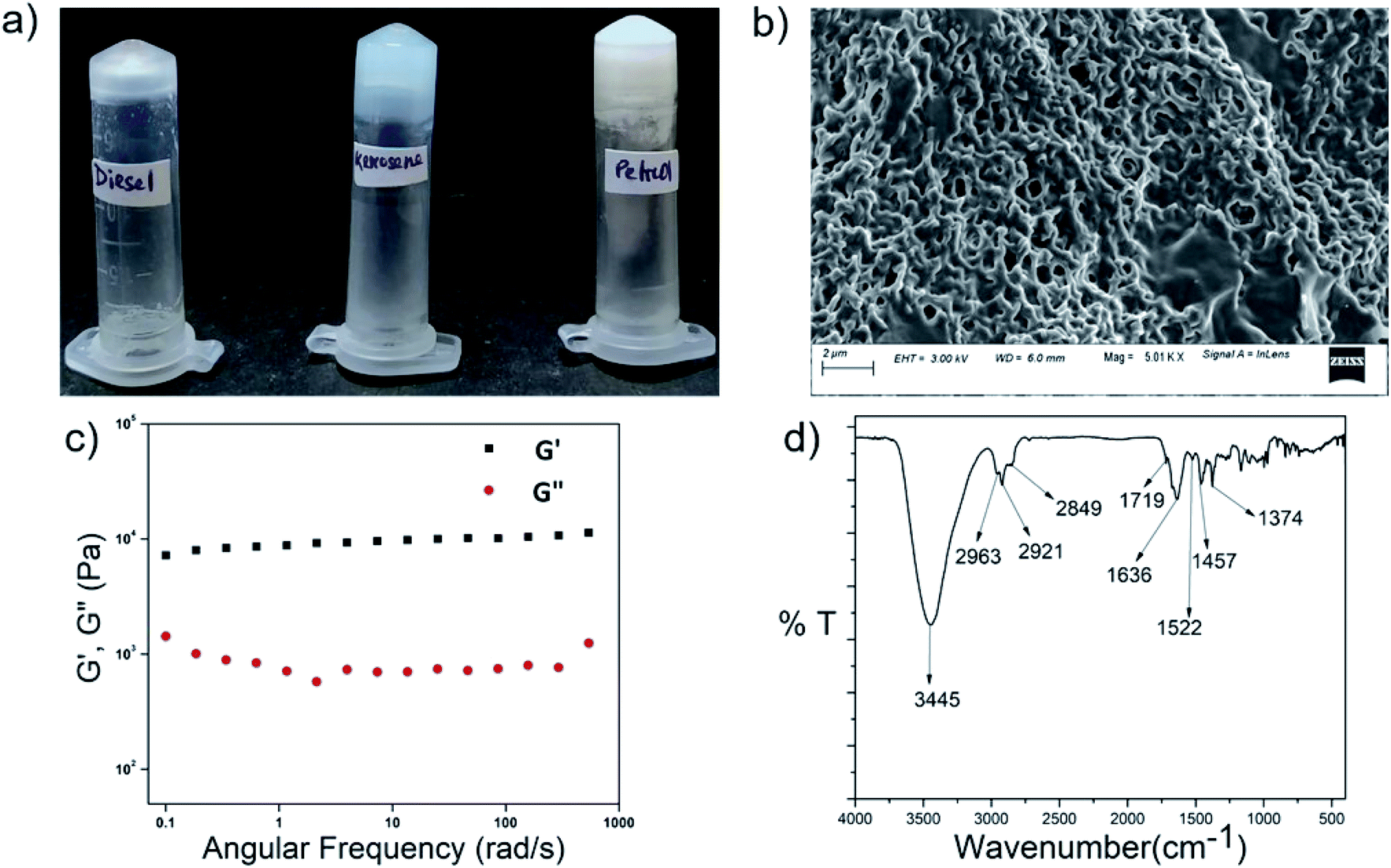 | ||
| Fig. 8 (a) Gels formed by P1 in kerosene, diesel and petrol, (b) FESEM image of the P1 organogel in kerosene, (c) frequency dependence of the dynamic storage modulus (G′) and the loss modulus (G′′) of the organogel from P1 in kerosene at 1% w/v. (d) FTIR spectrum of the xerogel of P1 in kerosene. | ||
Dye absorption
The presence of organic dye pollutants in water bodies is one of the major concerns for modern civilization. There have been previous reports in the literature on peptide-based dye absorbing materials.28–38 The organogels obtained from P1–P4 in the present study were checked for their dye absorption abilities. As the peptides were rich in aromatic moieties and hence rich in electrons, they might be good candidates for the absorption of cationic dyes (Fig. S24 and S25†). Most of the organogels showed high efficiency (≥90%) of absorption of the representative cationic dyes CV and RB (Fig. 9a and Table S7†). Organogel P4 showed relatively less efficiency in the absorption of RB.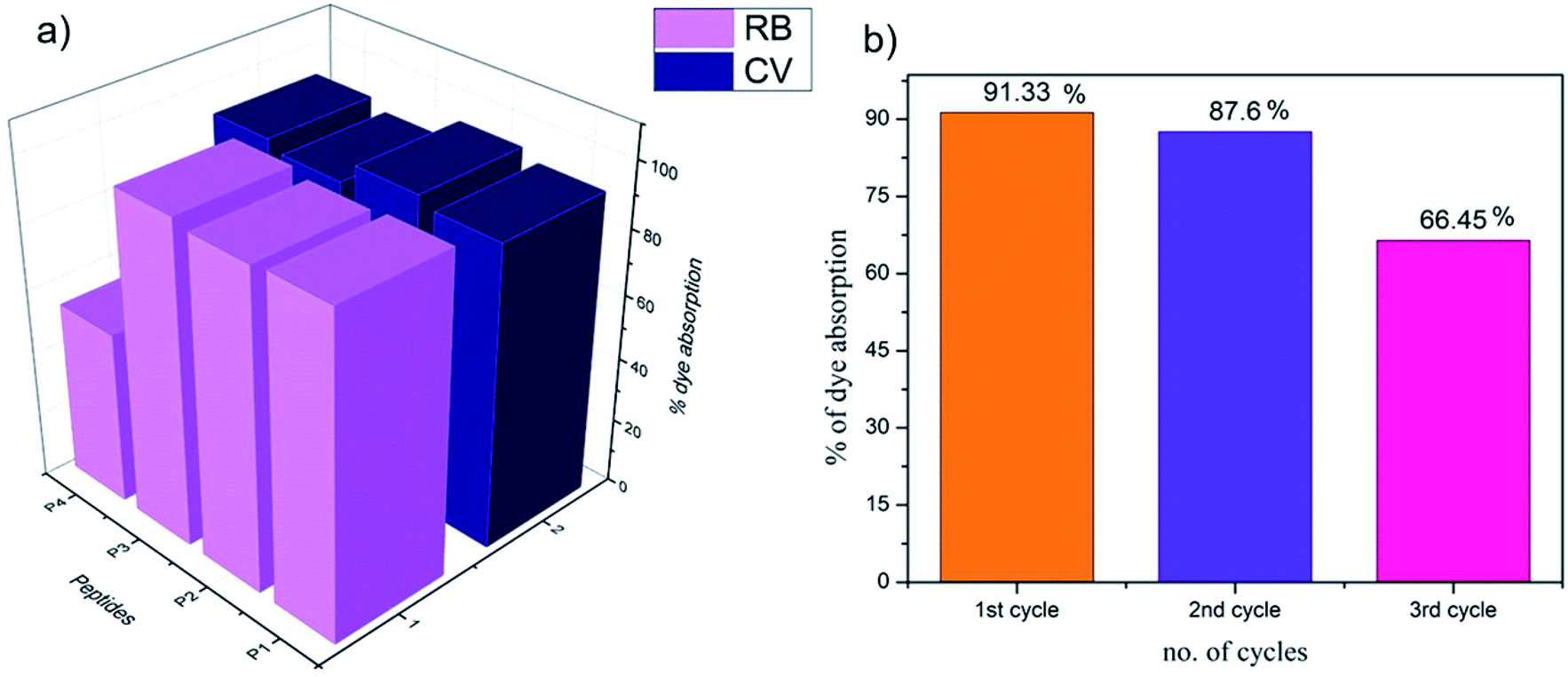 | ||
| Fig. 9 Dye absorption by the organogels P1–P4 in 1,2-DCB. A bar diagram representing the efficiency of (a) the absorption of cationic dyes CV and RB by organogels P1–P4, and (b) CV absorption in the subsequent cycles by the organogels from P1. | ||
Reusability of the organogel for dye absorption
For commercial use as dye absorbents in the purification of water, organogels should be reusable. The dyes could be released in diethyl ether medium over 24 hours and the original organogel recovered. The organogels from P1–P4 were tested for three consecutive cycles. Fig. 9b indicates the decreasing loading capacity of CV by the P1 organogel in three subsequent cycles.RGO incorporated organogels
In order to fabricate conducting organogels, we incorporated RGO into organogels from P1–P4. Reduced graphene oxide is conducting in nature, unlike graphene oxide. As the peptides P1–P4 were rich in aromatic amino acid residues, the incorporation of RGO into the gel was anticipated to be facilitated by aromatic pi–pi stacking interactions. All the peptides formed organogels in the presence of RGO at their MGC in 1,2-DCB (Fig. 10a). The RGO containing peptide organogels were thermoreversible in nature. The RGO doped organogels were thermostable in nature and their Tgel values were in the same range as those of the native gels. Different amounts of RGO were incorporated into the P1 organogel. In the present study, we incorporated a maximum of 1.2. wt% of RGO into the hybrid gels without destroying the gelation ability of the peptide gelator molecules. Attempts to load higher percentages of RGO in these organogels led to the precipitation of RGO and the formation of inhomogeneous material.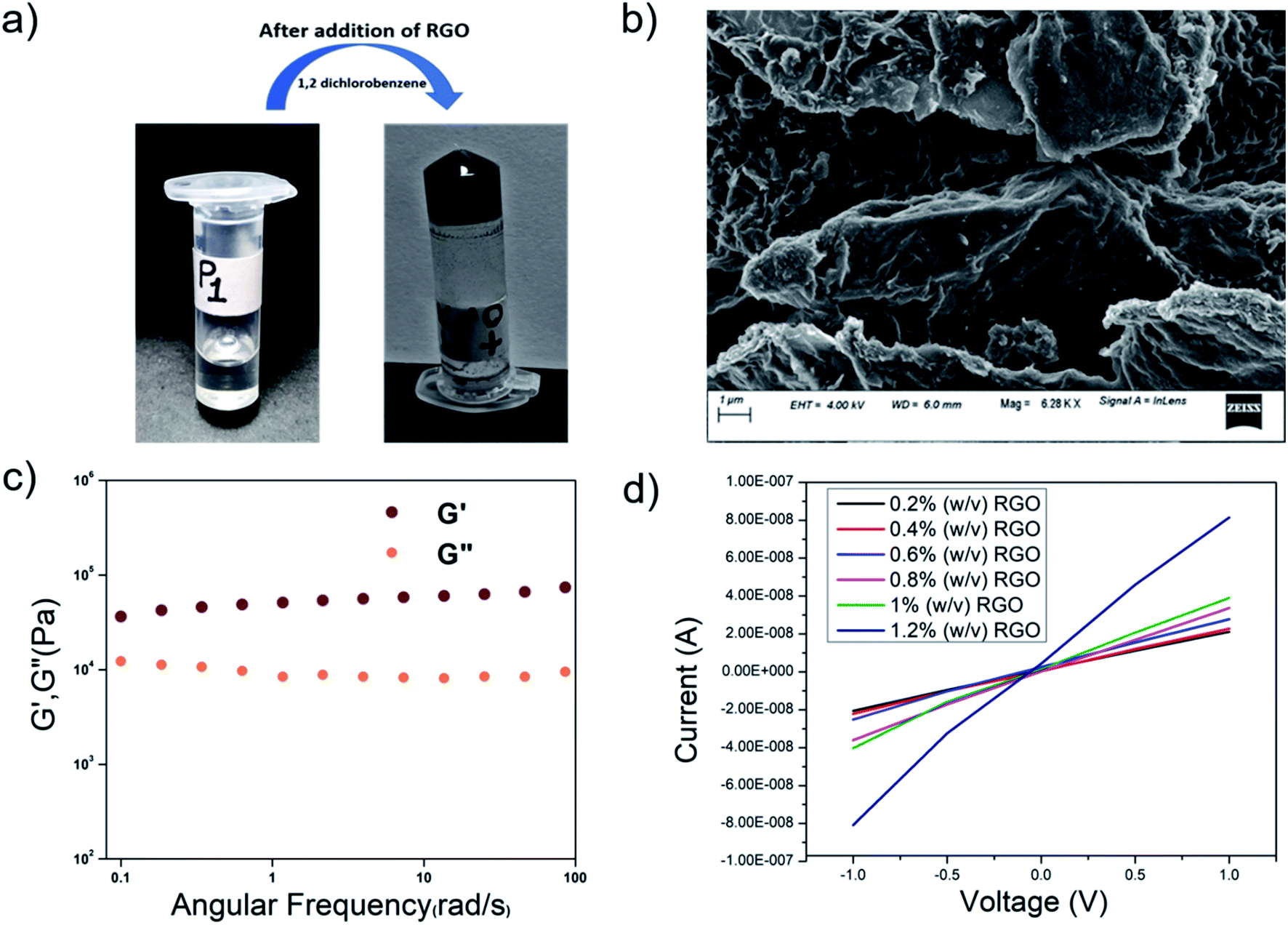 | ||
| Fig. 10 (a) The reduced graphene oxide incorporated hybrid organogel RGO-P1. (b) An FESEM image of the RGO-P1 hybrid organogel. (c) Frequency dependence of the dynamic storage modulus (G′) and the loss modulus (G′′) of the RGO-P1 hybrid organogel (1 wt%) in 1,2-DCB. (d) I–V curve showing the conductivity of the hybrid organogels upon RGO incorporation. | ||
To probe the morphology, FESEM was performed on the P1-RGO organogel. Fig. 10b shows the presence of both the RGO nanosheets and the peptide fibers in the hybrid RGO-P1 organogels containing 1 wt% of RGO. The RGO nanosheets are clearly visible with the peptide fibrous network dispersed on its surface, indicating good interaction in between the components of the hybrid organogel system. As the peptides are hydrophobic and contain a considerable amount of aromatic moieties, a good interaction with the RGO, rich in aromatic components, might be anticipated in the hybrid organogel.
To understand the backbone conformation of the peptide molecules in the hybrid organogel RGO-P1, FTIR was performed on the xerogel containing 1 wt% RGO (Fig. S26a†). The NH stretching frequency was observed at 3421 cm−1, indicating the presence of non-hydrogen bonded NHs in the hybrid xerogel, much like in the scenario of the pristine xerogels made from peptides alone. The amide I stretching peak was observed at 1667 cm−1, similar to that obtained from the xerogel containing the peptide alone. Thus, the backbone conformation of the peptides is identical both in the pristine and in the RGO-P1 xerogel. PXRD data obtained from the RGO-P1 xerogel is similar to that obtained from the pristine xerogel, indicating that the solid-state arrangement of the molecules is the same in both the hybrid and in the pristine organogel systems.
The viscoelastic properties of the RGO-P1 xerogel were probed by rheological studies. Studies were performed on the RGO-P1 hybrid organogels containing 1 wt% of RGO (Fig. 10c). In the frequency sweep experiment, the value of G′ was greater than that of G′′ throughout the experimental limits, indicating that the hybrid organogels were “solid-like”, which is similar to the native gel. Interestingly, the values of G′ and G′′ increased 10-fold in the case of RGO-P1 containing 1 wt% of RGO, in comparison to the native gel. This means that the incorporation of RGO enhances the rigidity of the gel. This could be attributed to the favorable interaction between the peptide nanofibers and the RGO sheets.
In summary, the insertion of RGO into the peptide organogel did not disrupt the self-assembly pattern that was present in the peptide-only system. The backbone conformation, solid-state packing etc. remained unaltered upon insertion of RGO. From the FESEM images, it was evident that both RGO and the peptides retained their distinct morphologies. Hence, the mode of co-assembly may be considered as “orthogonal” in this case. Favorable aromatic interactions between the peptides and the RGO might result in increased rigidity in the system.
Graphene is known for its highly conducting nature. The oxidation of graphite to graphene oxide changes some carbons to sp3, thus destroying the conductivity of the material. RGO, on the other hand, once again becomes conducting in nature, though it is not as good as graphene. Upon incorporation of RGO into the organogels, the hybrid materials became conducting in nature. There have been lots of studies in the literature where hybrid materials containing graphene and its derivatives have been used for energy conversion and storage.85–89 Most of these hybrid materials are of an inorganic nature and have high conductivity. There are fewer examples of peptide-based materials containing graphene and its derivatives.90,91 Although peptide-based hybrid materials are less conducting in nature compared to their inorganic counterparts, they are interesting as they might find applications in biomedical applications/devices, due to their biocompatibility. Fig. 10d shows the I–V curves obtained from the hybrid organogel RGO-P1 containing different amounts of RGO. The conductivity of the RGO-doped organogels increased proportionately to the amount of RGO in them. Homogeneous gels could not be prepared beyond the insertion of 1.2 w/v% of RGO, hence they were not considered for conductivity studies.
Conclusions
Small gelator peptides that are rich in aromatic moieties were designed in this work, and they readily formed phase selective, thermoreversible, mechanically robust organogels in a large variety of organic solvents and in fuels such as kerosene, diesel and petrol. The phase selective gelation ability of P1–P4 in petroleum fuels in various pH and saline conditions renders these organogels as potential materials for controlling marine oil spills. Aromatic pi–pi stacking and hydrophobic interactions were the driving forces for the assembly process, while hydrogen bonding played a trivial role in some systems and was completely absent in others. All four organogels, obtained from P1–P4, were excellent dye absorbents and are potential materials to be used in water purification. RGO was incorporated into the peptide organogels to yield hybrid organogels that were conducting in nature. The hybrid organogels were more rigid than the native peptide gels and their conductivity was proportional to the RGO content of the organogel. Hence, with rational design of the structural components in the LMWG, self-assembly can be directed to yield materials that are economically viable, recyclable and that can be utilized for multiple applications.Abbreviations
| DCU | Dicyclohexyl urea |
| Boc | Tertiary butyloxycarbonyl |
| FESEM | Field emission scanning electron microscopy |
| NMR | Nuclear magnetic resonance |
| FTIR | Fourier-transform infrared spectroscopy |
| PXRD | Powder X-ray diffraction |
| LMWG | Low molecular weight gelators |
| 1,2 DCB | 1,2 Dichlorobenzene |
| CV | Crystal violet |
| RB | Rhodamine B |
| MGC | Minimum gelation concentration |
| Trp | Tryptophan |
| Phe | Phenylalanine |
| Phg | Phenyl glycine |
| Homo-Phe | Homo phenylalanine |
| PABA | para aminobenzoic acid |
| RT | Room temperature |
| THF | Tetrahydrofuran |
| CDCl3 | Chloroform |
| DMSO-d6 | Dimethyl sulfoxide |
| GO | Graphene oxide |
| RGO | Reduced graphene oxide |
Author contributions
S. Chatterjee designed the project, M. C. and S. D. performed most of the experiments. S. C. performed the rheological experiments. S. Chatterjee analyzed the data and wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Chatterjee would like to acknowledge the Department of Science and Technology, India (Fast Track Project No. SB/FT/CS-070/2014) for financial support, and the COE FAST File No. 22-3/2016-TS.II/TC for use of the NMR instrument required in this research. S. Chatterjee acknowledges Arindam Bikash Neog for assistance in RGO synthesis. M. C., S. D. and S. C. acknowledge the IIT Guwahati for Scholarship, and the Central Instruments Facility, IITG for use of the FESEM instrument facility.References
- P. Dastidar, Chem. Soc. Rev., 2008, 37, 2699 RSC.
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821 RSC.
- G. Yu, X. Yan, C. Han and F. Huang, Chem. Soc. Rev., 2013, 42, 6697 RSC.
- S. Bera and D. Haldar, J. Mater. Chem. A, 2016, 4, 6933 RSC.
- A. Harada, R. Kobayashi, Y. Takashima, A. Hashidzume and H. Yamaguchi, Nat. Chem., 2011, 3, 34 CrossRef CAS PubMed.
- T. Park and S. C. Zimmerman, J. Am. Chem. Soc., 2006, 128, 11582 CrossRef CAS PubMed.
- A. Ajayaghosh and V. K. Praveen, Acc. Chem. Res., 2007, 40, 644 CrossRef CAS PubMed.
- S. Tanaka, M. Shirakawa, K. Kaneko, M. Takeuchi and S. Shinkai, Langmuir, 2005, 21, 2163 CrossRef CAS PubMed.
- T. Tu, W. Fang, X. Bao, X. Li and K. H. Dotz, Angew. Chem., Int. Ed., 2011, 50, 6601 CrossRef CAS PubMed.
- N. Fujita, Y. Sakamoto, M. Shirakawa, M. Ojima, A. Fujii, M. Ozaki and S. Shinkai, J. Am. Chem. Soc., 2007, 129, 4134 CrossRef CAS PubMed.
- S. Xiao, Y. Zou, M. Yu, T. Yi, Y. Zhou, F. Li and C. A. Huang, Chem. Commun., 2007, 4758 RSC.
- J. J. D. Jong, L. N. Lucas, R. M. Kellogg and J. H. V. Esch, Science, 2004, 304, 278 CrossRef PubMed.
- W. Cai, G. T. Wang, Y. X. Xu, K. Jiang and Z. T. Li, J. Am. Chem. Soc., 2008, 130, 6936 CrossRef CAS PubMed.
- O. J. Dautel, M. Robitzer, J. P. L. Porte, F. S. Spirau and J. J. E. Moreau, J. Am. Chem. Soc., 2006, 128, 16213 CrossRef CAS PubMed.
- P. Xue, R. Lu, G. Chen, Y. Zhang, H. Nomoto, M. Takafuji and H. Ihara, Chem.–Eur. J., 2007, 13, 8231 CrossRef CAS PubMed.
- D. Bardelang, Soft Matter, 2009, 5, 1969 RSC.
- Y. Li, T. Wang and M. Liu, Tetrahedron, 2007, 63, 7468 CrossRef CAS.
- G. Cravotto and P. Cintas, Chem. Soc. Rev., 2009, 38, 2684 RSC.
- C. Dou, D. Li, H. Gao, C. Wang, H. Zhang and Y. Wang, Langmuir, 2010, 26, 2113 CrossRef CAS PubMed.
- T. Naota and H. Koori, J. Am. Chem. Soc., 2005, 127, 9324 CrossRef CAS PubMed.
- K. Isozaki, H. Takaya and T. Naota, Angew. Chem., Int. Ed., 2007, 46, 2855 CrossRef CAS PubMed.
- H. Maeda, Chem.–Eur. J., 2008, 14, 11274 CrossRef CAS PubMed.
- A. Ajayaghosh, P. Chithra and R. Varghese, Angew. Chem., Int. Ed., 2007, 46, 230 CrossRef CAS PubMed.
- H. J. Kim, J. H. Lee and M. Lee, Angew. Chem., Int. Ed., 2005, 44, 5810 CrossRef CAS PubMed.
- K. Basu, N. Nandi, B. Mondal, A. Dehsorkhi, I. W. Hamley and A. Bannerjee, Interface Focus, 2017, 7, 1 CrossRef PubMed.
- M. Konda, I. Maity, D. B. Rasale and A. K. Das, ChemPlusChem, 2014, 79, 1482 CrossRef CAS.
- S. Basak, J. Nanda and A. Banerjee, J. Mater. Chem., 2012, 22, 11658 RSC.
- D. M. Wood, B. W. Greenland, A. L. Acton, F. Rodríguez-Llansola, C. A. Murray, C. J. Cardin, J. F. Miravet, B. Escuder, I. W. Hamley and W. Hayes, Chem.–Eur. J., 2012, 18, 2692 CrossRef CAS PubMed.
- F. Rodríguez-Llansola, B. Escuder, J. F. Miravet, D. Hermida-Merino, I. W. Hamley, C. J. Cardin and W. Hayes, Chem. Commun., 2010, 46, 7960 RSC.
- S. Basak, N. Nandi, S. Paul, I. W. Hamley and A. Banerjee, Chem. Commun., 2017, 53, 5910 RSC.
- B. Adhikari, G. Palui and A. Banerjee, Soft Matter, 2009, 5, 3452 RSC.
- S. Ray, A. K. Das and A. Banerjee, Chem. Mater., 2007, 19, 1633 CrossRef CAS.
- B. O. Okesola and D. K. Smith, Chem. Commun., 2013, 49, 11164 RSC.
- P. Chakraborty, B. Roy, P. Bairi and A. K. Nandi, J. Mater. Chem., 2012, 22, 20291 RSC.
- S. Sengupta and R. Mondal, J. Mater. Chem. A, 2014, 2, 16373 RSC.
- A. Chakrabarty, U. Maitra and A. D. Das, J. Mater. Chem., 2012, 22, 18268 RSC.
- N. Cheng, Q. Hu, Y. Guo, Y. Wang and L. Yu, ACS Appl. Mater. Interfaces, 2015, 7, 10258 CrossRef CAS PubMed.
- S. Debnath, A. Shome, S. Dutta S and P. K. Das, Chem.–Eur. J., 2008, 14, 6870 CrossRef CAS PubMed.
- J. H. Jung, H. Kobayashi, M. Masuda, T. Shimizu and S. Shinkai, J. Am. Chem. Soc., 2001, 123, 8785 CrossRef CAS PubMed.
- E. D. Sone, E. R. Zubarev and S. I. Stupp, Angew. Chem., Int. Ed., 2002, 41, 1705 CrossRef CAS.
- S. Kobayashi, N. Hamasaki, M. Suzuki, M. Kimura, H. Shinkai and K. Hanabusa, J. Am. Chem. Soc., 2002, 124, 6550 CrossRef CAS PubMed.
- C. Zhan, J. Wang, J. Yuan, H. Gong, Y. Liu and M. Liu, Langmuir, 2003, 19, 9440 CrossRef CAS.
- S. Kobayashi, K. Hanabusa, N. Hamasaki, M. Kimura, H. Shirai and S. Shinkai, Chem. Mater., 2000, 12, 1523 CrossRef CAS.
- P. Xue, R. Lu, Y. Huang, M. Jin, C. Tan, C. Bao, Z. Wang and Y. Zhao, Langmuir, 2004, 20, 6470 CrossRef CAS.
- M. Tena-Solsona, J. Nanda, S. Díaz-Oltra, A. Chotera, G. Ashkenasy and B. Escuder, Chem.–Eur. J., 2016, 22, 6687 CrossRef CAS.
- B. Escuder, F. Rodríguez-Llansola and J. F. Miravet, New J. Chem., 2010, 34, 1044 RSC.
- W. Kubo, T. Kitamura, K. Hanabusa, Y. Wada and S. Yanagida, Chem. Commun., 2002, 374 RSC.
- Y. Yamada and J. P. Schneider, Biomacromolecules, 2016, 17, 2634 CrossRef CAS PubMed.
- S. R. Jadhav, B. S. Chiou, D. F. Wood, G. DeGrande Hoffman, G. M. Glenn and G. John, Soft Matter, 2011, 7, 864 RSC.
- K. Basu, A. Baral, S. Basak, A. Dehsorkhi, J. Nanda, D. Bhunia, S. Ghosh, V. Castelletto, I. W. Hamley and A. Banerjee, Chem. Commun., 2016, 52, 5045 RSC.
- J. E. P. Sun, B. Stewart, A. Litan, S. J. Lee, J. P. Schneider, S. A. Langhans and D. J. Pochan, Biomater. Sci., 2016, 4, 839 RSC.
- V. Castelletto, A. Kaur, I. W. Hamley, R. H. Barnes, K. A. Karatzas, D. Hermida-Merino, S. Swioklo, C. J. Connon, J. Stasiak, M. Reza and J. Ruokolainen, RSC Adv., 2017, 7, 8366 RSC.
- S. Ray, A. K. Das and A. Banerjee, Chem. Mater., 2007, 19, 1633 CrossRef CAS.
- J. Nanda and A. Banerjee, Soft Matter, 2012, 8, 3380 RSC.
- M. Schrope, Nature, 2011, 472, 152 CrossRef CAS PubMed.
- B. Wang, W. Liang, Z. Guo and W. Liub, Chem. Soc. Rev., 2015, 44, 336 RSC.
- S. Venkatanarasimhan and D. Raghavachari, J. Mater. Chem. A, 2013, 1, 868 RSC.
- R. J. Fiocco and A. Lewis, Pure Appl. Chem., 1999, 71, 27 CAS.
- E. Pelletier and R. Siron, Environ. Toxicol. Chem., 1999, 18, 813 CrossRef CAS.
- F. L. Motta, S. R. Stoyanov and J. B. P. Soares, Chemosphere, 2018, 194, 837 CrossRef CAS PubMed.
- D. Sundaravadivelu, M. T. Suidan, A. D. Venosa and P. I. Rosales, Chemosphere, 2016, 144, 1490 CrossRef CAS PubMed.
- A. Tripathi, G. N. Parsons, O. J. Rojas and S. A. Khan, ACS Omega, 2017, 2, 4297 CrossRef CAS PubMed.
- H. M. Choi, Environ. Sci. Technol., 1992, 26, 772 CrossRef CAS.
- V. Singh, S. Jinka, K. Hake, S. Parameswaran, R. J. Kendall and S. Ramkumar, Ind. Eng. Chem. Res., 2014, 53, 11954 CrossRef CAS.
- N. Widiaksana, A. A. Yudiana and Y. S. Nugroho, IOP Conf. Ser. Earth Environ. Sci., 2018, 105, 1755 Search PubMed.
- S. Khandakar, M. N. Islam, R. I. Rubel and S. S. Yusuf, Bitlis Eren Univ. J. Sci. Technol., 2017, 7, 115 CrossRef.
- A. T. Hoang, V. V. Pham and D. N. Nguyen, Int. J. Appl. Eng. Res., 2018, 13, 4915 Search PubMed.
- S. Mondal, P. Bairi, S. Das and A. K. Nandi, J. Mater. Chem. A, 2019, 7, 381 RSC.
- S. Bhattacharya and Y. Krishnan-Ghosh, Chem. Commun., 2001, 185 RSC.
- M. Konda, I. Maity, D. B. Rasale and A. K. Das, ChemPlusChem, 2014, 79, 1482 CrossRef CAS.
- K. Basu, N. Nandi, B. Mondal, A. Dehsorkhi, I. W. Hamley and A. Banerjee, Interface Focus, 2017, 7, 1 CrossRef PubMed.
- N. D. Lourenco, J. M. Novais and H. M. Pinheiro, J. Biotechnol., 2001, 89, 163 CrossRef CAS PubMed.
- A. H. Gemeay, I. A. Mansour, R. G. El-Sharkawy and A. B. Zaki, J. Mol. Catal. A: Chem., 2003, 193, 109 CrossRef CAS.
- N. Daneshvar, H. Ashassi-Sorkhabi and A. Tizpar, Sep. Purif. Technol., 2003, 31, 153 CrossRef CAS.
- E. Rindle and W. J. Troll, J. Natl. Cancer Inst., 1975, 55, 181 CrossRef.
- D. Solpan and O. Guven, Radiat. Phys. Chem., 2002, 65, 549 CrossRef CAS.
- S. Kalyuzhnyi and V. Sklyar, Water Sci. Technol., 2000, 12, 23 CrossRef.
- F. I. Hai, K. Yamamoto and K. Fukushi, Crit. Rev. Environ. Sci. Technol., 2007, 37, 315 CrossRef CAS.
- E. K. Schillinger, E. Mena-Osteritz, J. Hentschel, H. G. Börner and P. Bäuerle, Adv. Mater., 2009, 21, 1562 CrossRef CAS.
- E. Jahnke, J. Weiss, S. Neuhaus, T. N. Hoheisel and H. Frauenrath, Chem.–Eur. J., 2009, 15, 388 CrossRef CAS PubMed.
- S. Bai, S. Debnath, N. Javid, P. W. J. M. Frederix, S. Fleming, C. Pappas and R. V. Ulijn, Langmuir, 2014, 30, 7576 CrossRef CAS PubMed.
- H. Xu, A. K. Das, M. Horie, M. S. Shaik, A. M. Smith, Y. Luo, X. Lu, R. Collins, S. Y. Liem, A. Song, P. L. A. Popelier, M. L. Turner, P. Xiao, I. A. Kinloch and R. V. Ulijn, Nanoscale, 2010, 2, 960 RSC.
- E. Torres, J. Puigmarti-Luis, A. Perez del Pino, R. M. Ortuno and D. B. Amabilino, Org. Biomol. Chem., 2010, 8, 1661 RSC.
- S. K. M. Nalluri, N. Shivarova, A. L. Kanibolotsky, M. Zelzer, S. Gupta, P. W. J. M. Frederix, P. J. Skabara, H. Gleskova and R. V. Ulijn, Langmuir, 2014, 30, 12429 CrossRef CAS PubMed.
- X. Huang, X. Qi, F. Boey and H. Zhang, Chem. Soc. Rev., 2012, 41, 666 RSC.
- U. N. Maiti, J. Lim, K. E. Lee, W. J. Lee and S. O. Kim, Adv. Mater., 2014, 26, 615 CrossRef CAS PubMed.
- C. Yang, Z. Liu, C. Chen, K. Shi, L. Zhang, X.-J. Ju, W. Wang, R. Xie and L.-Y. Chu, ACS Appl. Mater. Interfaces, 2017, 9, 15758 CrossRef CAS.
- R. Kumar, R. K. Singh, A. V. Alaferdov and S. A. Moshkalev, Electrochim. Acta, 2018, 281, 78e87 CrossRef.
- R. Kumar, E. T. S. G. da Silva, R. K. Singh, R. Savu, A. V. Alaferdov, L. C. Fonseca, L. C. Carossi, A. Singh, S. Khandka, K. K. Kar, O. L. Alves, L. T. Kubota and S. A. Moshkalev, J. Colloid Interface Sci., 2018, 515, 160 CrossRef CAS PubMed.
- R. Kumar, R. K. Singh, D. P. Singh, A. R. Vaz, R. R. Yadav, C. S. Rout and S. A. Moshkalev, Mater. Des., 2017, 122, 110 CrossRef CAS.
- R. Kumar, S. Sahoo, E. Joanni, R. K. Singh, W. K. Tan, K. K. Kar and A. Matsuda, Prog. Energy Combust. Sci., 2019, 75, 100786 CrossRef.
- R. Kumara, E. Joanni, R. K. Singh, D. P. Singh and S. A. Moshkalev, Prog. Energy Combust. Sci., 2018, 67, 115 CrossRef.
- B. Vedhanarayanan, B. Babu, M. M. Shaijumon and A. Ajayaghosh, ACS Appl. Mater. Interfaces, 2017, 9, 19417 CrossRef CAS PubMed.
- R. Y. Kuwahara, T. Oi, K. Hashimoto, S. Tamesue, T. Yamauchi and N. Tsubokawa, Colloid Polym. Sci., 2015, 293, 1635 CrossRef CAS.
- B. Adhikari, A. Biswas and A. Banerjee, ACS Appl. Mater. Interfaces, 2012, 4, 5472 CrossRef CAS PubMed.
- R. Kumar, W. C. Macedo Jr, R. K. Singh, V. S. Tiwari, C. J. L. Constantino, A. Matsuda and S. A. Moshkalev, ACS Appl. Nano Mater., 2019, 2, 4626 CrossRef CAS.
- B. Adhikari, J. Nanda and A. Banerjee, Chem.–Eur. J., 2011, 17, 11488 CrossRef CAS.
- B. Adhikari and A. Banerjee, Soft Matter, 2011, 7, 9259 RSC.
- T. H. Han, W. J. Lee, D. H. Lee, J. E. Kim, E.-Y. Choi and S. O. Kim, Adv. Mater., 2010, 22, 2060 CrossRef CAS.
- S. Roy, A. Baral and A. Banerjee, Chem.–Eur. J., 2013, 19, 14950 CrossRef CAS PubMed.
- J. X. Liang, Q. L. Nguyen and S. Matsika, Photochem. Photobiol. Sci., 2013, 12, 1387 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1961450 and 1961451. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra10395c |
| ‡ These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |