
MBenes: emerging 2D materials as efficient electrocatalysts for the nitrogen reduction reaction†
Xiaowei
Yang
,
Chanjuan
Shang
,
Si
Zhou
* and
Jijun
Zhao
Key Laboratory of Materials Modification by Laser, Ion and Electron Beams (Dalian University of Technology), Ministry of Education, Dalian 116024, China. E-mail: sizhou@dlut.edu.cn; Tel: +86-0411-84706100
First published on 15th May 2020
Abstract
MBenes, an emerging family of two-dimensional (2D) transition metal borides, have recently been synthesized in experiment and hold great promise for 2D ferromagnets and metal ion batteries. With a well-defined layered structure and outstanding electrical conductivity, MBenes provide an ideal platform for exploring the catalytic behavior of boride surfaces at the atomic level. Herein, we exploit ten kinds of MBenes for electrocatalysis of the N2 reduction reaction (NRR). By comprehensive first-principles calculations, we show that MBenes have high stability in aqueous environments and excellent selectivity toward the NRR against the hydrogen evolution reaction (HER). Intriguingly, both the surface boron and metal atoms of MBenes can serve as the active sites owing to the co-existence of occupied and unoccupied p (d) states for the boron (metal) atoms, while the boron reaction centers provide even superior activity compared to the metal atoms. Moreover, the activity of these 2D borides can be tuned by engineering their work function. Our theoretical results shine light on utilizing metal borides as a new category of non-precious catalysts for nitrogen fixation, and illuminate the fundamental principles for controlling their catalytic performance.
New conceptsInspired by the recent discovery of 2D transition metal borides, namely, MBenes, herein we proposed to use them for electrocatalysis of nitrogen reduction reaction (NRR). Compared to the conventional transition metal based catalysts, the competitive hydrogen evolution reaction (HER) is effectively suppressed on most MBenes. Superior activity can be achieved on the boron active surface by taking advantage of their partially occupied p orbitals and high adaptability for electron transfer with nitrogen intermediates, thus opening a new doorway for utilizing boron compounds on a large scale for practical electrocatalysis. We further illuminate the basic rule for control of the activity of MBenes by engineering their work function. This theoretical study provides important guidance for manipulating metal boride surfaces with atomic precision for various energy applications. |
1. Introduction
As the boron analog of MXenes,1 MBenes, namely, two-dimensional (2D) transition metal borides, were first predicted in theory2,3 and synthesized experimentally in the last two years.4–8 Similar to MXenes, monolayer or few-layer MBene sheets can be obtained by etching the A atomic layers (A = Al or In) from the synthetic crystals of MAB phases (namely, ternary layered transition metal borides, M for a transition metal). So far, a few kinds of MBenes including MoB,4 TiB5 and CrB6–8 have been prepared in the laboratory, which show metallic behavior. Ab initio calculations have not only confirmed the thermodynamic and dynamic stabilities of MBenes, but also proposed several fascinating physical and chemical properties, such as high electrical and thermal conductivities, robust ferromagnetism with a high Curie temperature, and superior electrochemical properties as the anode materials for Li/Na ion batteries and as catalysts for the CO2 reduction reaction.2,3,9 As emerging 2D materials, the fundamental properties and potential applications of MBenes remain largely unexplored.The renewable-electricity-driven N2 reduction reaction, using earth-abundant nitrogen as a feedstock, has been considered as one of the most compelling strategies for green energy storage and transportation via the N2/NH3 cycle.10–12 Metal catalysts such as Fe and Ru have been widely adopted for the N2 reduction reaction (NRR), but their efficiency is severely limited due to the highly competitive HER side reaction.13,14 Boron is an abundant non-metal element and has low valence, carrying both occupied and unoccupied p orbitals, endowing boron-based materials with the speciality of N2 fixation.15,16 Experiments showed that doping B in graphene induces electron redistribution, and the electron-deficient B centers exhibit enhanced adsorption strength with N2 and prohibit binding with the Lewis acid H+ for the HER, leading to greatly improved faradaic efficiency for the NRR.17 On the theoretical side, single B atoms anchored on substrates, such as 2D g-C3N4,18,19 C2N,20 and h-BN monolayers and nanoribbons,21,22 as well as various B-doped 2D materials,19 have been proposed as efficient catalysts for NRR. The activity is closely related to the charge transfer between the B atom and the surrounding environment.
Both experimental and theoretical studies have shown the great potential of boron for the NRR. However, how to balance the stability and activity of boron-based catalysts remains elusive. To utilize boron on a large scale for catalytic applications, boron-rich compound materials with a large number of surface active sites would be more desirable than dispersed or doped single B atom catalysts. Due to the electron-deficient character of boron, metal borides display exotic bonding states, giving them enormous varieties of geometrical and electronic structures.23 Therefore, it is imperative to achieve a comprehensive understanding of the principles for precisely manipulating the catalytic behavior of both metal and boron exposed surfaces for metal borides. MBenes, with diverse chemical compositions and well-defined surface structures having both exposed boron and metal sites, serve as ideal models for such mechanism study. Moreover, these novel 2D compound materials have unique advantages for electrocatalysis, including large surface area and excellent electrical conductivity.
Here, we exploited ten kinds of MBenes for electrocatalysis of the N2 reduction reaction. By comprehensive ab initio calculations, the stability of MBenes in an aqueous environment at ambient temperature has been characterized. The reaction pathway and activity for the NRR on different types of reaction sites, as well as the selectivity of the NRR over the HER, were fully addressed. More importantly, we elucidated the origin of the high activity of boron surfaces, and determined the key indicators—that are measureable in experiment—governing the NRR activity. These theoretical results open a doorway for rational design of low-dimensional transition metal borides for various energy applications.
2. Computational methods
Spin-polarized density functional theory (DFT) calculations were performed with the Vienna ab initio Simulation Package (VASP),24 using a plane wave basis set with an energy cutoff of 500 eV, projector augmented wave (PAW) potentials,25 and the generalized gradient approximation parameterized by Perdew, Burke and Ernzerhof (GGA-PBE) for the exchange and correlation functional.26 Grimme's semiempirical DFT-D3 scheme of dispersion correction was employed to describe the van der Waals (vdW) interactions.27Here we adopted a supercell consisting of 4 × 4 × 1 unit cells of MBenes, having lateral dimensions of 10.56–13.08 Å, and added a large vacuum space of 20 Å along the perpendicular orientation. The Brillouin zones were sampled by k-point grids of 11 × 11 × 1 for primitive unit cells and 3 × 3 × 1 for the supercells, respectively. All the structural models were fully optimized using convergence criteria of 10−4 eV for the total energy and 10−2 eV Å−1 for the forces on each atom, respectively. The on-site charges and bond order were further evaluated by Mulliken population analysis28 implemented in CASTEP using the PBE functional, a plane wave basis with an energy cutoff of 500 eV, and ultrasoft pseudopotentials.29
The adsorption energy ΔEads of a gas molecule involved in the NRR (N2 or NH3) on MBenes is defined as
| ΔEads = Etotal − EMBene − Egas | (1) |
The Gibbs free energy of formation (ΔG) for each elementary step of the NRR was computed as
| ΔG = ΔEDFT + ΔZPE − TΔS | (2) |
| ΔGH* =ΔEH* + ΔZPE − TΔS | (3) |
3. Results and discussion
3.1. Structures, electronic properties and stability of MBenes
Following previous theoretical calculations and experimental synthesis,2,6–8 we considered ten kinds of 2D MBenes as potential catalysts for the NRR, including FeB2, RuB2, OsB2, V3B4, Nb3B4, Ta3B4, CrB, MnB, ZrB and HfB, and their structures are displayed in Fig. 1. All the MBenes have bare surfaces, as there is no experimental evidence yet showing the presence of functional groups on MBenes.6 Previous theoretical studies have also demonstrated that these MBenes with bare surfaces have stable phonon dispersions without any imaginary bands, manifesting their dynamic stability.2,3 According to the ratio of metal and B atoms, these MBenes can be classified into three types: (I) FeB2, RuB2 and OsB2 with a thickness of 3.70–3.93 Å, in which a buckled layer of metal atoms is covered by two layers of B atoms on the outmost surfaces; (II) V3B4, Nb3B4 and Ta3B4, comprised of two layers of B atoms sandwiched by three layers of metal atoms with a thickness of 4.84–5.26 Å; and (III) CrB, MnB, ZrB and HfB having a bilayer buckled structure (a thickness of 2.23–2.85 Å) with metal atoms on the outmost surfaces. Thus, the active site of MBenes for adsorbing N2 molecules could be either the exposed B atoms (type I) or metal atoms (type II and III) on the upmost surface, which thereafter will be named the boron active surface (Fig. 1a) and the metal active surface (Fig. 1b and c), respectively. As summarized in Table 1, FeB2, RuB2, OsB2 and Nb3B4 are non-magnetic, while V3B4, Ta3B4, CrB, MnB, ZrB and HfB are ferromagnetic with a total magnetic moment of 0.78–4.49 μB per formula.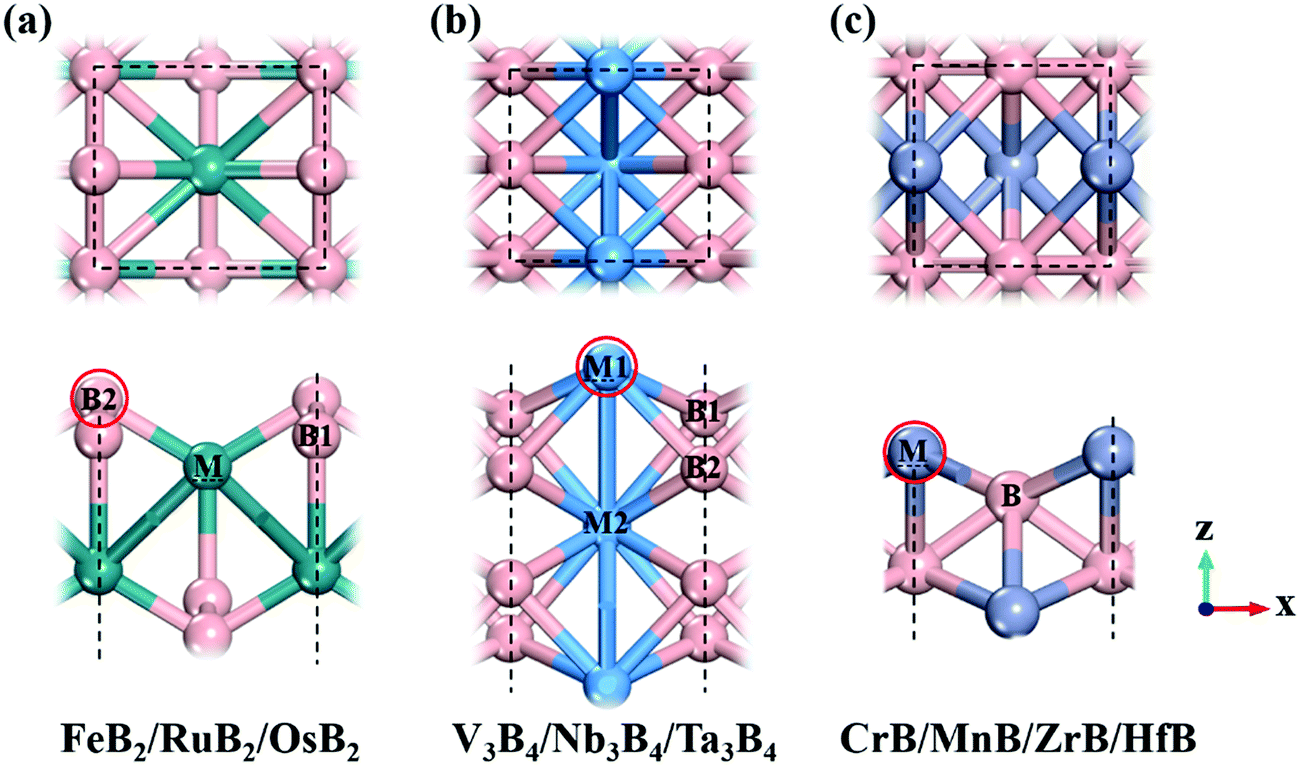 | ||
| Fig. 1 Top views (upper panel) and side views (lower panel) of different types of MBene structures: (a) FeB2, OsB2, and RuB2, (b) V3B4, Nb3B4, and Ta3B4, and (c) CrB, MnB, ZrB, and HfB. The B atom is shown in pink, and metal atoms are shown in turquoise, light blue, and purple colors. The dashed lines and boxes indicate the lateral dimension of the unit cell of MBene. The red circles indicate the active sites for N2 adsorption and reduction. | ||
| MBene | M tot (μB) | d M–B (Å) | BO | Φ (eV) | CT (e) | |||
|---|---|---|---|---|---|---|---|---|
| B1 | B2 | M1 | M2 | |||||
| FeB2 | 0.00 | 1.91 | 0.52 | 4.97 | 0.73 | 0.46 | −1.20 | — |
| RuB2 | 0.00 | 2.05 | 0.26 | 5.05 | 0.53 | 0.42 | −0.95 | — |
| OsB2 | 0.00 | 2.09 | 0.29 | 4.46 | 0.53 | 0.37 | −0.90 | — |
| V3B4 | 2.22 | 2.15 | 0.68 | 3.67 | 0.65 | 0.59 | −0.68 | −1.11 |
| Nb3B4 | 0.00 | 2.32 | 0.82 | 4.22 | 0.57 | 0.53 | −0.49 | −1.21 |
| Ta3B4 | 2.42 | 2.33 | 1.31 | 3.70 | 0.64 | 0.61 | −0.52 | −1.47 |
| CrB | 4.50 | 2.13 | 0.79 | 4.09 | 0.61 | — | −0.61 | — |
| MnB | 4.49 | 2.09 | 0.63 | 4.17 | 0.94 | — | −0.94 | — |
| ZrB | 0.78 | 2.43 | 0.69 | 3.44 | 0.61 | — | −0.61 | — |
| HfB | 1.13 | 2.41 | 0.98 | 3.38 | 0.70 | — | −0.70 | — |
The present MBenes have an M−B bond length of 1.91–2.43 Å with a bond order34 of 0.26–1.31, indicating strong covalent bonds between B and metal atoms, which are also consistent with the electron localization function in Fig. S1 of the ESI.† The B atoms gain a certain amount of electrons from the metal atoms in MBenes, which fill the B p orbitals. As revealed by the on-site charges in Table 1, the charge transfer to each B atom ranges from 0.37 e to 0.94 e, such that the B atoms (each originally having 3 valence electrons) in MBenes are still electron deficient, as also indicated by their partial charge densities near the Fermi level in Fig. S2 of the ESI.† In particular, the outmost B and metal atoms with lower coordination carry less electrons by 0.11–0.27 e and 0.43–0.95 e than the interior ones, respectively. Consequently, the p or d orbitals of boron or metal active surfaces of MBenes are partially filled. To some extent, the p orbitals of the active B atoms can mimic the metal d states, presenting both occupied and unoccupied states. For both B and metal sites, the unoccupied orbitals are able to accept the lone-pair electrons from the σ orbital of N2, while the occupied orbitals can back-donate electrons to the antibonding π* orbital of N2, endowing MBenes with activity for N2 fixation (see the schematic plot in Fig. 2). The work functions of MBenes range from 3.38 eV to 5.05 eV, which are noticeably lower than those of 2D MXenes (5.2–6.6 eV).35 Intriguingly, the work functions of MBene systems with boron active surfaces are larger than those with metal active surfaces, which is highly relevant for understanding their reactivity with N2 molecules, as will be discussed in the following content.
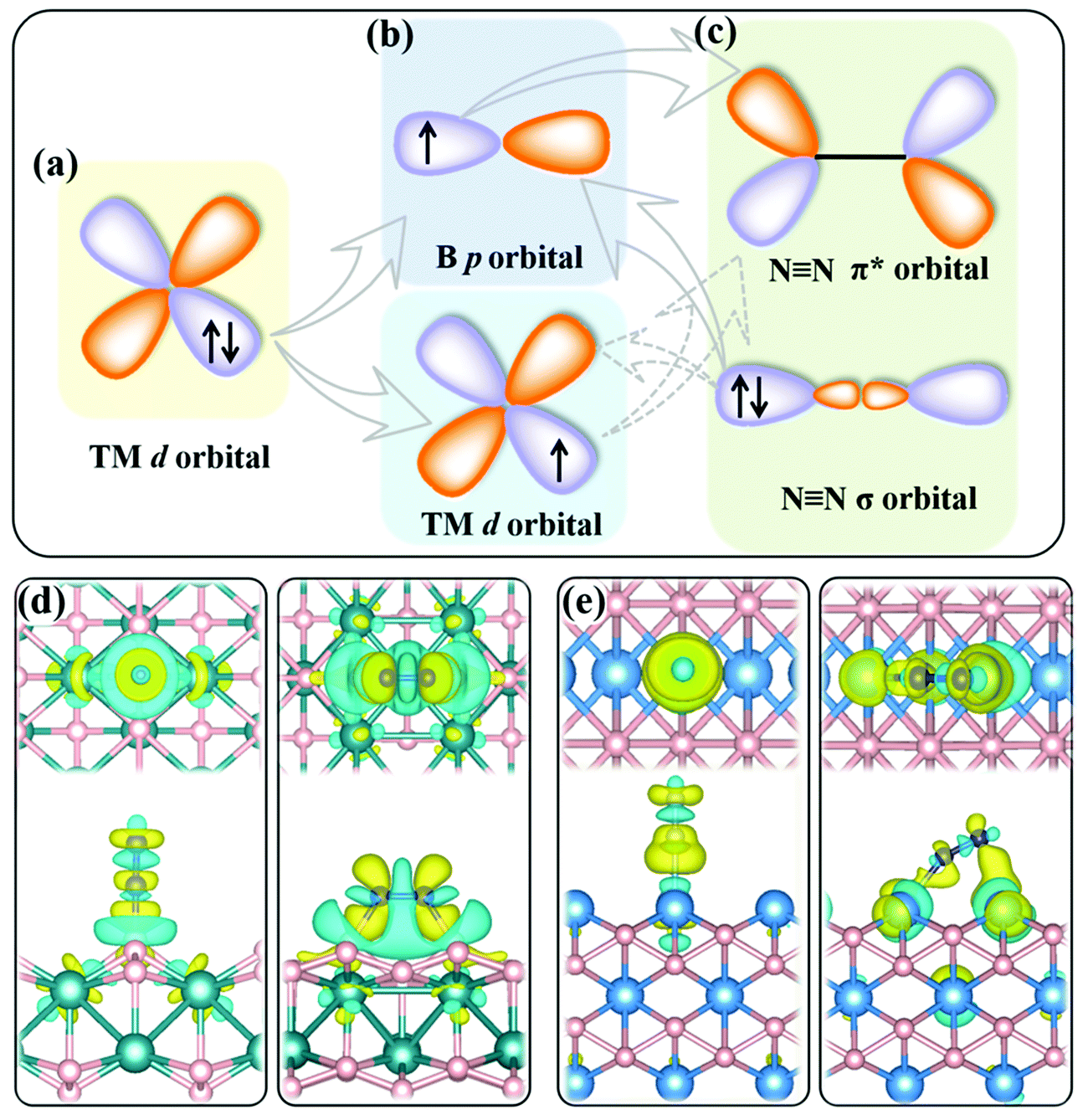 | ||
| Fig. 2 (a–c) Schematic illustration of the mechanism of activity of MBenes for N2 fixation. (d and e) Differential charge density distributions of N2 molecules absorbed on RuB2 and Ta3B4 surfaces, respectively, in the end-on configuration (left) and side-on configuration (right): top views (upper panels) and side views (bottom panels). The B, N, Ru and Ta atoms are shown in pink, blue, turquoise and light blue colors, respectively. The yellow and cyan colors represent electron accumulation and depletion regions, respectively, with an isosurface value of 0.0035 e Å−3. | ||
Electrochemical reactions usually occur in aqueous solutions.36 Thus, the stability of electrocatalysts in an aqueous environment, which can be reflected by the fluctuations of the current density in experiment,37 is an important concern for practical electrochemical processes. To assess the stability of MBenes, we performed ab initio molecular dynamics (AIMD) simulations on an MBene at a temperature of 300 K in a water environment,38,39 by taking RuB2 as a representative. The evolution of the energy and temperature with the simulation time is plotted in Fig. 3c. The mean square displacement (MSD) of atoms as a function of simulation time is given in Fig. S3 of the ESI.† After 10 ps, the structure of RuB2 remains intact in water without any noticeable deformation, and the MSD of B and metal atoms is in the range of 0.1–1.4 Å2. The deviation of the Ru–B bond length is below 0.2 Å with respect to that of the equilibrium structure (Fig. 3b), manifesting the outstanding stability of MBenes in aqueous solutions at ambient temperature. Therefore, MBenes may form a category of robust electrocatalysts for various energy applications.
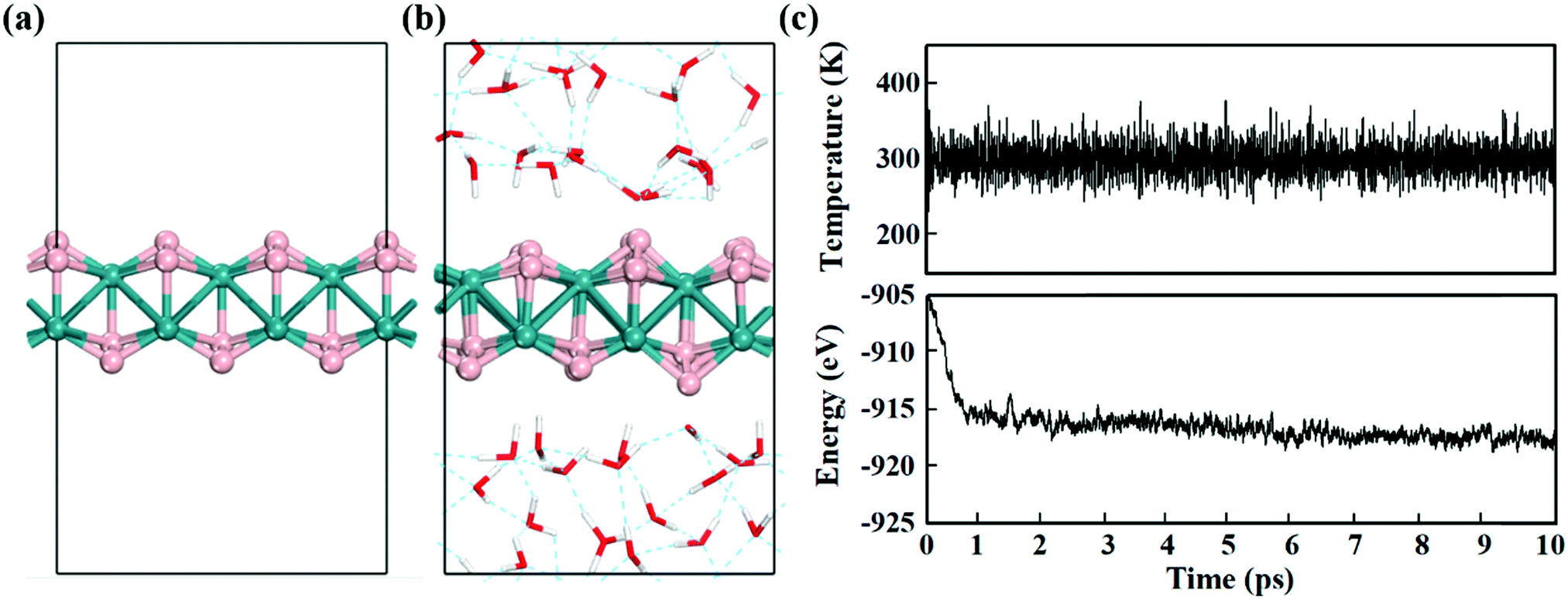 | ||
| Fig. 3 (a) Equilibrium structure of RuB2 in a vacuum after optimization. (b) Structure of RuB2 in a water environment after AIMD simulations at 300 K for 10 ps. (c) Variations of temperature and energy with the time of AIMD simulation for RuB2. The H, B, O and Ru atoms are shown in white, pink, red and turquoise, respectively. The boxes indicate the dimension of supercells for the calculations. | ||
3.2. N2 adsorption on MBenes
Effective adsorption of N2 on the catalyst is a prerequisite for the NRR. As shown in Fig. 4a–d, for both boron and metal active surfaces, a N2 molecule can chemisorb on MBenes via the end-on and side-on configurations. In the former case, only one N atom of N2 binds with the active site, while both N atoms form bonds with the underlying B or metal atoms in the latter case. The adsorption energies of N2 on various MBenes range from −0.47 eV to −1.75 eV as plotted in Fig. 4c, which are sufficiently strong to capture and activate the N2 molecule. For the B active sites, the adsorption strength of N2 in the end-on configuration is stronger than that of the side-on one by 0.28–0.45 eV in energy; for the side-on configuration, N2 adsorption along the armchair direction (on top of shorter B–B bonds) is usually more favorable than that along the zigzag direction (on top of longer B–B bonds) (Fig. S4 of the ESI†), while the opposite trend is observed for metal active sites, on which N2 adsorption in the side-on configuration is stronger by 0.03–1.05 eV. The only exception is MnB with weaker side-on N2 adsorption than the end-on one. Overall, the binding capability of these MBenes with N2 molecules falls in a moderate range that would be suitable for the NRR, as previous theoretical studies have shown that N2 adsorption energies in the range of −0.50 to −1.25 eV may be beneficial for achieving a low overpotential.21,40–43 The chemisorbed N2 molecule has an elongated N–N bond length, i.e. 1.13 Å for the end-on configuration, and 1.17–1.22 Å for the side-on one (1.11 Å for a free N2 molecule), accompanied by an amount of electron transfer of 0.01–0.24 e and 0.32–0.56 e to the N2 molecule, respectively.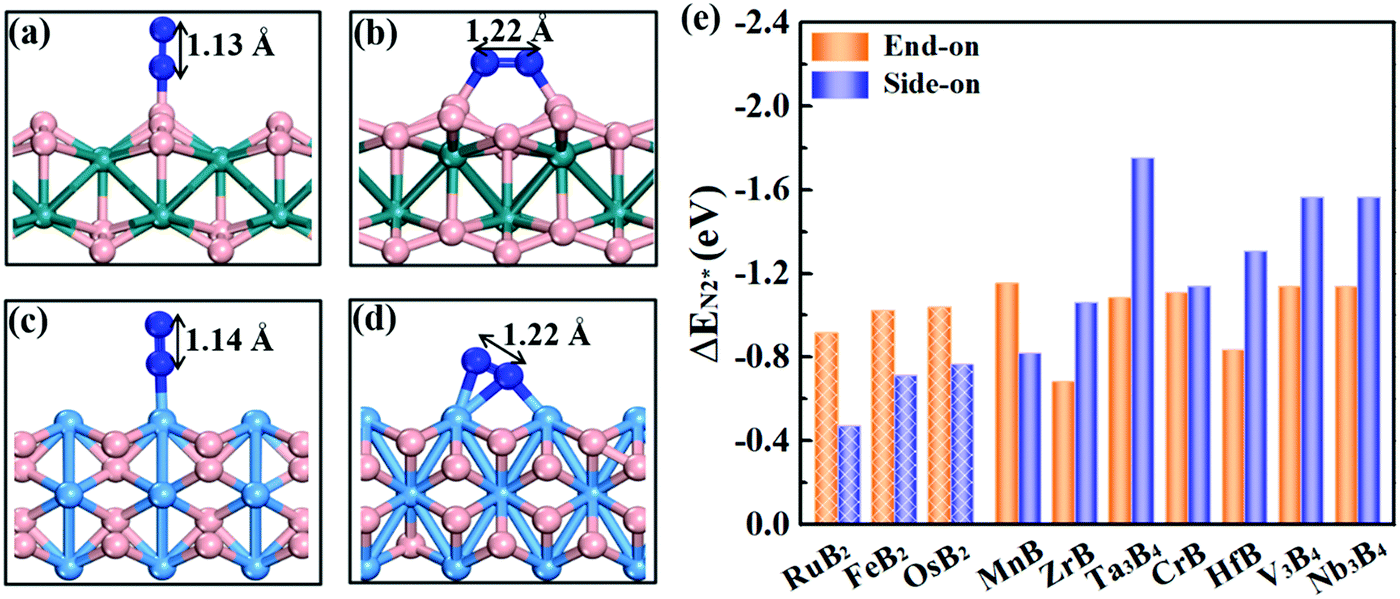 | ||
| Fig. 4 Side view of the structures of N2 molecules chemisorbed on RuB2 (a and b) and Ta3B4 (c and d) in the end-on and side-on configurations. The N–N bond length is given for each system. The B, N, Ru and Ta atoms are shown in pink, blue, turquoise and light blue colors, respectively. (e) Adsorption energies of N2 on various kinds of MBenes. Shaded (solid) histograms indicate MBenes with a B (metal) active surface. | ||
The interaction between N2 and MBenes with different surfaces can be better understood from the differential charge density (Δρ)44,45 displayed in Fig. 2d and e, which is defined as the charge density of an MBene with adsorption of a N2 molecule (ρN2@MBene) relative to that of the individual MBene (ρMBene) and N2 (ρN2):
| Δρ = ρN2@MBene − ρMBene − ρN2 | (4) |
It shows both electron accumulation (yellow color) and depletion (cyan color) on the N2 molecule as well as on the metal or boron active sites, corresponding to electron donation from the σ orbital of N2 to the MBene, and back-donation from the MBene to the antibonding π* orbital of N2. This is quite distinct from the unidirectional electron transfer for other gas molecules (such as H2, O2 and CO2) on catalyst surfaces, exhibiting electron accumulation on the gas molecule and depletion on the active sites.46–48 Moreover, for the boron active surface, N2 adsorption induces noticeable charge transfer between the active B atoms and the adjacent metal atoms. According to previous theoretical studies,49,50 the amount and direction of charge transfer between the nitrogen intermediates and the active site undergo substantial variation and fluctuation during the alternative protonation of nitrogen intermediates in the NRR. Such consecutive electron transfer processes would be favorable on MBenes with a boron active surface. Their outmost B and metal atoms can cooperatively serve as an electron reservoir with enhanced adaptability for accepting and donating various amounts of electrons with the nitrogen intermediates, which is thus beneficial for lowering the onset potential of the NRR (as will be shown later).
3.3. N2 reduction reaction on MBenes
To evaluate the catalytic performance of MBenes for N2 reduction, three typical reaction mechanisms have been considered, including the distal, alternating and enzymatic ones. Briefly speaking, the distal and alternating pathways start from the end-on configuration, while the enzymatic mechanism proceeds through the side-on configuration of N2 adsorption. All these three reaction pathways are completed by a six-electron reaction, i.e., N2 + 6H+ + 6e → 2NH3. For the distal pathway, the proton–electron pair (H+ + e) first reacts with one N atom in the adsorbed N2; whereas for the alternating and enzymatic mechanisms, the proton–electron pair attacks the two N atoms alternatively, toward the formation of two NH3 molecules.Under the above three mechanisms, we calculated the free energy diagrams of the NRR for all the MBene systems, as displayed in Fig. 5 and Fig. S5–S7 of the ESI.† For both boron and metal active surfaces of MBenes, the most efficient pathway that gives the lowest onset potential for the NRR is through the enzymatic mechanism. The only exception is Ta3B4, which prefers the alternating mechanism. Fig. 5a and b present the enzymatic pathway on RuB2 as a representative. The adsorbed *N–N species in the side-on configuration is protonated, and the N–N bond is broken during the formation of the *NH2–NH2 intermediate. The largest step ΔGmax in the free energy diagram ranges from 0.03 eV to 0.92 eV. Especially, the MBenes with boron active surfaces, i.e. FeB2, RuB2 and OsB2, exhibit the highest activity with ΔGmax = 0.26, 0.25 and 0.03 eV, respectively. The rate-determining step is the protonation of *NH–NH to yield the *NH–NH2 intermediate. On the other hand, CrB, MnB and ZrB with metal active surfaces have larger ΔGmax = 0.16, 0.43 and 0.92 eV, respectively. The rate-determining step is the first protonation step for the former two systems, and the protonation of *NH–NH2 to yield *NH2–NH2 for ZrB. For V3B4, Nb3B4, Ta3B4 and HfB, the rate-determining step is the formation of *NH3, associated with ΔGmax = 0.53–0.92 eV.
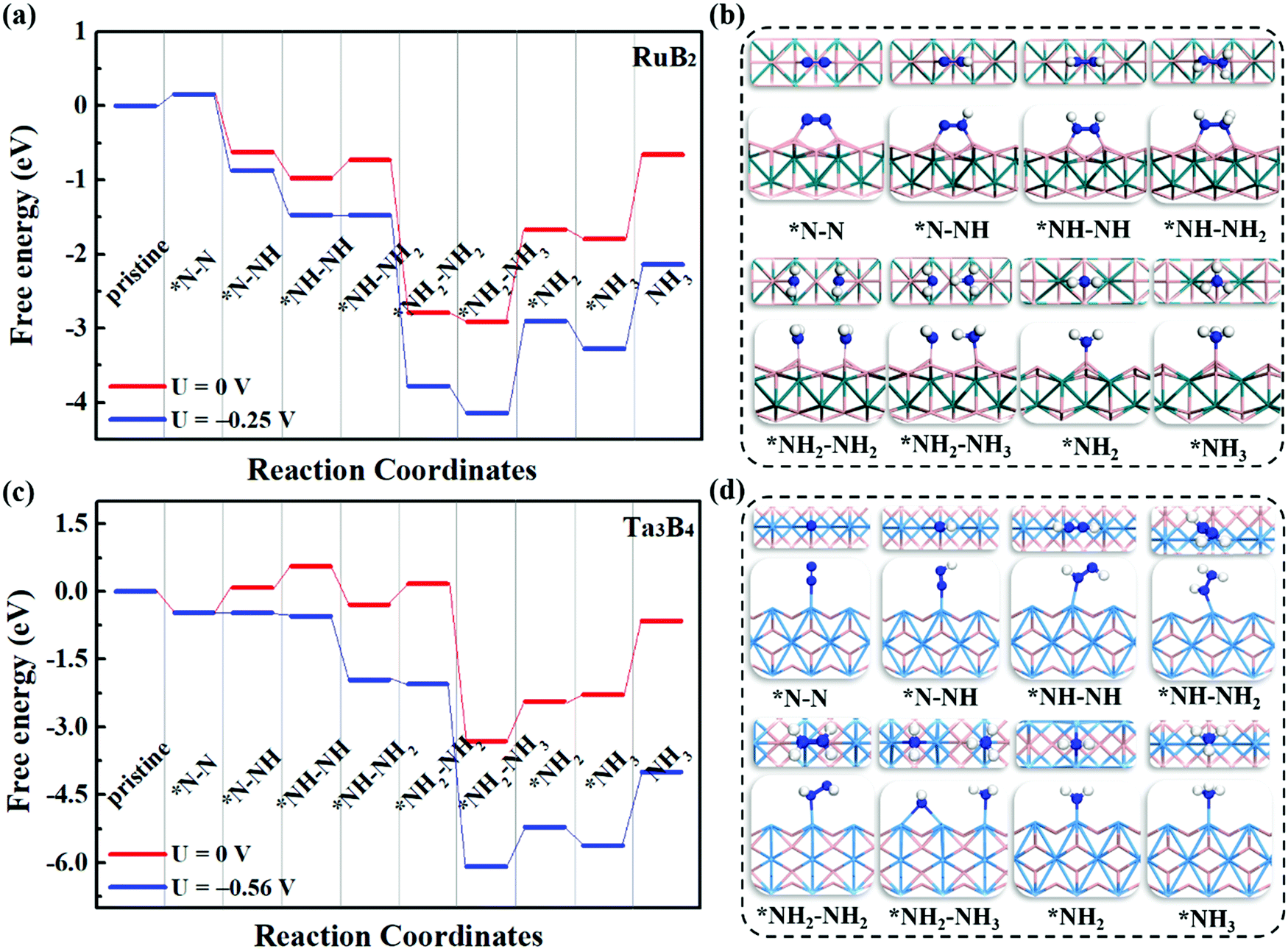 | ||
| Fig. 5 Free energy diagrams of the N2 reduction reaction on (a) RuB2via the enzymatic mechanism, and (c) Ta3B4via the alternative mechanism under different applied potentials. The symbol * denotes an intermediate adsorbed on the MBene. The structures (top view and side view) of the corresponding reaction intermediates on RuB2 and Ta3B4 are displayed in (b and d), respectively. The H, B, N, Ru and Ta atoms are shown in white, pink, blue, turquoise and light blue colors, respectively. | ||
Fig. 5c and d illustrate the alternating pathway on Ta3B4 as a representative. Protonation proceeds on the adsorbed *N–N species in the end-on configuration, and the breaking of the N–N bond occurs during the formation of the *NH2–NH3 intermediate. The rate-determining step is the first reaction step from *N–N to *N–NH for all the MBenes, involving ΔGmax = 0.56–1.40 eV (lowest value of 0.56 eV for Ta3B4). The distal reaction pathway requires ΔGmax above 0.84 eV, and thus is less efficient than the other two mechanisms. Note that desorption of NH3* from MBenes into the gas phase involves a Gibbs free energy of formation of −0.45 to −1.29 eV, which are in the range of the reported values for many other NRR electrocatalysts, such as Fe and Ru metals (<−0.8 eV)51–53 and low-dimensional catalysts (−1.21 to −1.79 eV).18,21,41,54,55 In practice, using acid electrolytes can facilitate the dissolution of NH3* species,56 making NH3 gas readily desorb from these catalysts. Therefore, it would be the protonation steps instead of NH3 desorption that determine the NRR activity of MBenes.
Fig. 6a plots the onset potentials U of all the MBene systems. MBenes with boron active surfaces have U as low as −0.03 to −0.26 V, while the ones with metal active surfaces show larger U of −0.16 to −0.92 V. In particular, FeB2, RuB2, OsB2, CrB and MnB have U below −0.50 V, highly competitive with the benchmark metal catalysts for the NRR, such as the Re(111) surface (−0.5 V),14 and early transition metals Sc, Y, Ti and Zr (−1.0 to −1.5 V),33 as well as the theoretically proposed single atom catalysts such as Mo atoms anchored on an h-BN monolayer (−0.35 V),49 B atoms immobilized on phosphorene (−0.20 V)57 or the edge of h-BN sheets (−0.29 V).21 In the electrochemical N2 reduction reaction, the HER is a competing side reaction and usually results in a low Faraday efficiency for the NRR for many transition metal catalysts.14,58,59 To evaluate the HER activity in MBenes, we calculated the free adsorption energy for H* species ΔGH*, which can be regarded as the overpotential for the HER. The corresponding atomic structures with adsorbates are displayed in Fig. S8 of the ESI.† Except V3B4, MnB, CrB and ZrB, the majority of the considered MBenes present larger overpotentials for the HER than the NRR (Fig. 6d); hence the NRR should overwhelm the HER in these MBenes. Therefore, the superiority of MBenes lies in not only the low NRR onset potentials, but also high selectivity against HER, outstanding stability in aqueous solutions, and excellent electrical conductivity, all of which are highly desirable for practical implementation of these electrocatalysts.
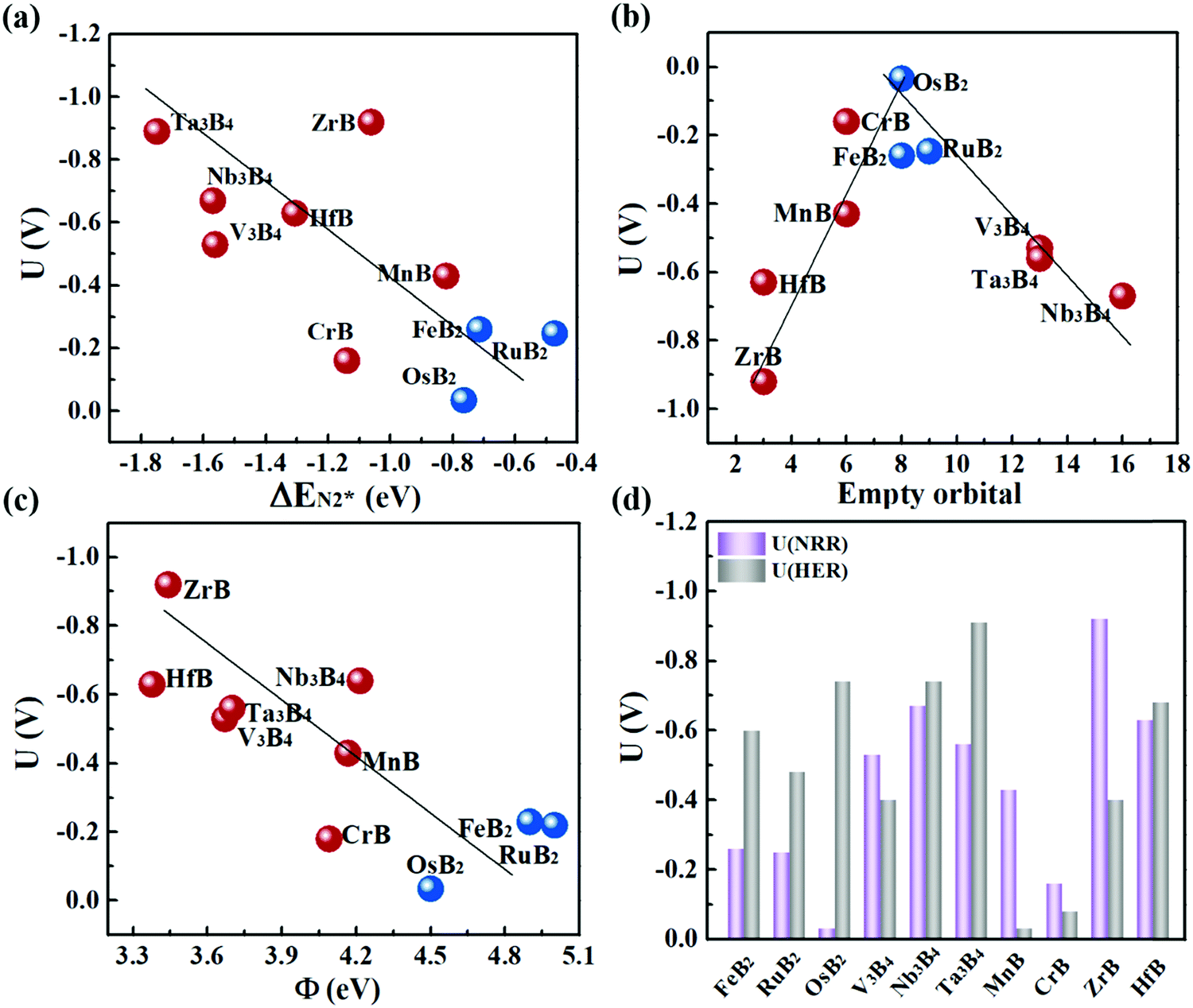 | ||
| Fig. 6 NRR onset potential vs. (a) N2 adsorption energy, (b) total number of empty p orbitals of B atoms and empty d orbitals of metal atoms in MBenes, and (c) work function of MBenes. (d) Onset potential of the NRR and ΔGH* for the HER on various MBenes. | ||
3.4. Band structure–activity relationship of MBenes
To explain the high catalytic activity of MBenes, we explored the underlying correlations between the onset potential, adsorption property and electronic band structure. As revealed by Fig. 6a, the NRR onset potential follows an approximately linear relation with the adsorption strength of N2 on MBenes – weaker binding of the N2 molecule leads to a lower NRR onset potential. This trend of activity is in contrast to that of some reported boron based electrocatalysts for the NRR.21,60–62 In MBenes, breaking of the N–N bond occurs during the protonation of NH2–NH to NH2–NH2 under the enzymatic pathway, and is thermodynamically favorable for all the systems. These MBenes may not have the same rate-determining step, and thus a system with stronger binding capability does not necessarily have higher activity. For instance, V3B4, Nb3B4, HfB and Ta3B4 with metal exposed surfaces provide stronger N2 binding than that of the other systems. However, they encounter a higher potential step during the protonation of NH2* to NH3*, and thus have lower activity. RuB2, OsB2 and FeB2 with boron active surfaces exhibit weaker adsorption strength with N2 and higher activity, with the NRR limited by the protonation of *NH–NH to *NH2–NH.The binding capability and onset potential of MBenes are correlated to the occupancy and energy levels of the frontier orbitals. During the NRR process, the B and metal atoms in MBenes cooperatively act as an electron reservoir for accepting electrons from the nitrogen intermediates or donating electrons to them. Fig. 6b plots the total number of empty p orbitals of B atoms and empty d orbitals of metal atoms in MBenes, which exhibits a volcano relation with the onset potential. Apparently, moderate orbital filling of B and metal atoms endows MBenes with optimal adaptability for electron transfer with the reaction intermediates and hence results in the highest activity.
Deeper insights can be gained by examining the electronic density of states (DOS) of MBenes. As shown in Fig. 7a and Fig. S9 of the ESI,† all the MBenes are metallic with prominent states at the Fermi level. Especially, for the MBenes with boron active surfaces, the exposed B atoms make a substantial contribution to the DOS in the vicinity of the Fermi level (Fig. 7b). Here we reference all the orbital levels to a vacuum; thus the absolute value of the Fermi energy equals the work function. Taking RuB2 as an example, due to the relatively large work function, its Fermi level (−5.05 eV) is located far below the 2π* antibonding orbital of the N2 molecule (−2.14 eV), resulting in a relatively weak interaction between N2 and RuB2. Nevertheless, backdonation from N2 to RuB2 is still possible, as demonstrated by the differential charge density in Fig. 2d. The electrons from the p orbitals of the active B atoms in RuB2 transfer to the 2π* orbital of N2, which is then lowered to −0.17 eV (Fig. 7a), consistent with the population analysis (0.29 e gained by N2). Intuitively, an MBene system with the Fermi level closer to the 2π* orbital of N2 would interact more favorably with the N2 molecule.63
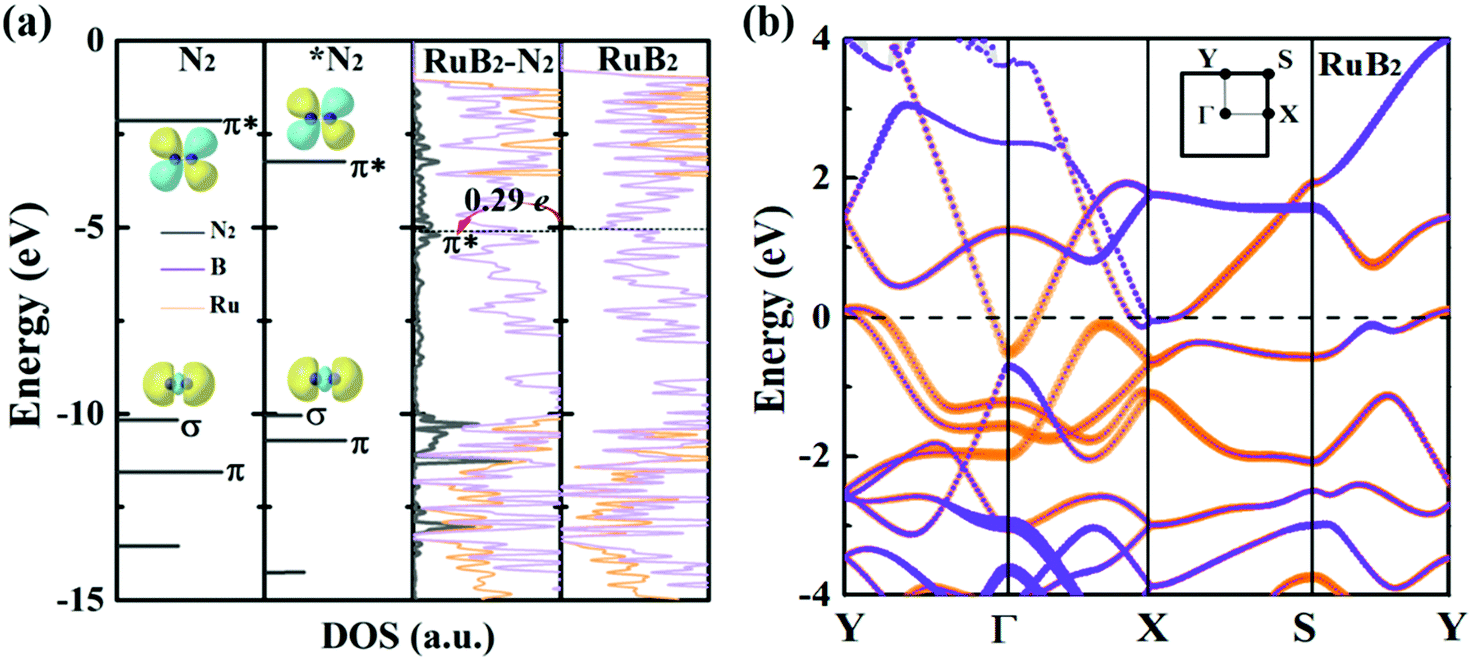 | ||
| Fig. 7 (a) From left to right: molecular orbital levels of a free N2 molecule and adsorbed *N2, and local density of states (LDOS) of RuB2 with and without adsorption of a N2 molecule. The dashed lines in the right two panels indicate the Fermi level, and the energies are relative to a vacuum. (b) Electronic band structure of RuB2. The purple and orange colors indicate the contributions of the bands from B and metal atoms, respectively, and the line width is proportional to weight. The Fermi level (dashed line) is set to zero. The inset shows the first Brillouin zone of the RuB2 unit cell. | ||
Indeed, such a correlation between the onset potential (or N2 adsorption energies) and work functions of MBenes is found in Fig. 6c and Fig. S10 of the ESI.† Overall, MBenes with larger work functions exhibit weaker binding capability and lower NRR onset potentials. Noticeably, the three MBene systems with boron active surfaces have larger work functions than those with metal active surfaces. They provide relatively weaker but suitable adsorption strength with N2, and hence show higher activity for the NRR. The data points in Fig. 6c have some deviations from the linear relation, indicating that there may be other factors that also influence the catalytic properties of MBenes. Previous studies have shown that the p electrons of non-metal elements can be activated by strategies like doping, nanostructuring, and hybridizing with metals or metal compounds.35,64,65 The activity of oxides, nitrides, carbon and silicon based materials has been found to be closely related to the p band center. Here we plotted the p band center of exposed B atoms and d band center of surface metal atoms in MBenes as a function of ΔEN2*, which, however, does not follow any clear relation (Fig. S11 of the ESI†). Although the catalytic performance of MBenes may simultaneously involve multiple descriptors, our analysis shows that the work function is the most relevant physical parameter to the NRR activity, which can be directly measured and controlled in experiment and hence should arouse attention for catalyst design.
4. Conclusion
To summarize, the recently discovered 2D transition metal borides, namely, MBenes with ten compositions and three types of atomic structures, have been computationally explored for electrocatalysis of the N2 reduction reaction. Our ab initio calculations show that MBenes possess outstanding stability in aqueous solution under ambient conditions and remarkable selectivity for the NRR against the HER side reaction. With a rich variety of chemical compositions and geometrical structures, MBenes can provide diverse active sites for N2 fixation and reduction. Intriguingly, the MBenes (FeB2, RuB2 and OsB2) with surface B atoms as the active sites exhibit superior activity for the NRR than those with metal active sites, as signified by the low onset potentials of −0.03 to −0.26 V. Such excellency can be ascribed to the partially occupied p orbitals, advantageous adsorption properties, and large adaptability as electron reservoirs of the surface B atoms. Moreover, the work function of MBenes is relevant to the N2 binding strength and onset potential, and thus is an important parameter that can help modulate the activity of MBenes. These exciting results shine light on utilizing transition metal borides as a novel family of catalysts for energy applications, and provide vital guidance for manipulating their activity for various chemical processes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (11974068), the Fundamental Research Funds for the Central Universities of China (DUT20LAB110), and the Supercomputing Center of Dalian University of Technology.References
- M. Naguib, V. N. Mochalin, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2014, 26, 992–1005 CrossRef CAS PubMed.
- Z. Jiang, P. Wang, X. Jiang and J. Zhao, Nanoscale Horiz., 2018, 3, 335–341 RSC.
- Z. Guo, J. Zhou and Z. Sun, J. Mater. Chem. A, 2017, 5, 23530–23535 RSC.
- L. T. Alameda, P. Moradifar, Z. P. Metzger, N. Alem and R. E. Schaak, J. Am. Chem. Soc., 2018, 140, 8833–8840 CrossRef CAS PubMed.
- J. Wang, T. N. Ye, Y. Gong, J. Wu, N. Miao, T. Tada and H. Hosono, Nat. Commun., 2019, 10, 2284 CrossRef PubMed.
- H. Zhang, H. Xiang, F.-Z. Dai, Z. Zhang and Y. Zhou, J. Mater. Sci. Technol., 2018, 34, 2022–2026 CrossRef.
- S. Kota, W. Wang, J. Lu, V. Natu, C. Opagiste, G. Ying, L. Hultman, S. J. May and M. W. Barsoum, J. Alloys Compd., 2018, 767, 474–482 CrossRef CAS.
- H. Zhang, F.-Z. Dai, H. Xiang, X. Wang, Z. Zhang and Y. Zhou, J. Mater. Sci. Technol., 2019, 35, 1593–1600 CrossRef.
- H. Yuan, Z. Li and J. Yang, J. Phys. Chem. C, 2019, 123, 16294–16299 CrossRef CAS.
- S. Giddey, S. P. S. Badwal and A. Kulkarni, Int. J. Hydrogen Energy, 2013, 38, 14576–14594 CrossRef CAS.
- J. M. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS.
- V. Rosca, M. Duca, M. T. de Groot and M. T. M. Koper, Chem. Rev., 2009, 109, 2209–2244 CrossRef CAS PubMed.
- Q. Li, L. He, C. Sun and X. Zhang, J. Phys. Chem. C, 2017, 121, 27563–27568 CrossRef CAS.
- J. H. Montoya, C. Tsai, A. Vojvodic and J. K. Norskov, ChemSusChem, 2015, 8, 2180–2186 CrossRef CAS PubMed.
- M.-A. Légaré, G. B. Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–900 CrossRef PubMed.
- M.-A. Légaré, M. Rang, G. B. Chabot, J. I. Schweizer, I. Krummenacher, R. Bertermann, M. Arrowsmith, M. C. Holthausen and H. Braunschweig, Science, 2019, 363, 1329–1332 CrossRef PubMed.
- X. Yu, P. Han, Z. Wei, L. Huang, Z. Gu, S. Peng, J. Ma and G. Zheng, Joule, 2018, 2, 1610–1622 CrossRef CAS.
- C. Ling, X. Niu, Q. Li, A. Du and J. Wang, J. Am. Chem. Soc., 2018, 140, 14161–14168 CrossRef CAS PubMed.
- C. Liu, Q. Li, C. Wu, J. Zhang, Y. Jin, D. R. MacFarlane and C. Sun, J. Am. Chem. Soc., 2019, 141, 2884–2888 CrossRef CAS PubMed.
- K. Bhattacharyya and A. Datta, Phys. Chem. Chem. Phys., 2019, 21, 12346–12352 RSC.
- X. Mao, S. Zhou, C. Yan, Z. Zhu and A. Du, Phys. Chem. Chem. Phys., 2019, 21, 1110–1116 RSC.
- J. Lee and J. Kang, J. Catal., 2019, 375, 68–75 CrossRef CAS.
- B. Albert and K. Hofmann, Handbook of Solid State Chemistry, Materials and Structure of Solids, 2017, ch. 10, vol. 1 Search PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- R. S. Mulliken, J. Chem. Phys., 1955, 23, 1841 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr. –Cryst. Mater., 2005, 220, 567–570 CAS.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1131–1135 RSC.
- K. Reuter and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 65, 035406 CrossRef.
- M. W. Chase, NIST-JANAF Thermochemical Tables, American Institute of Physics, New York, 1998 Search PubMed.
- E. Skulason, T. Bligaard, S. Gudmundsdottir, F. Studt, J. Rossmeisl, F. Abild-Pedersen, T. Vegge, H. Jonsson and J. K. Norskov, Phys. Chem. Chem. Phys., 2012, 14, 1235–1245 RSC.
- I. Mayer, Int. J. Quantum Chem., 1986, 29, 477–483 CrossRef CAS.
- S. Zhou, X. Yang, W. Pei, N. Liu and J. Zhao, Nanoscale, 2018, 10, 10876–10883 RSC.
- X. Cui, C. Tang and Q. Zhang, Adv. Energy Mater., 2018, 8, 1800369 CrossRef.
- W. Qiu, X. Xie, J. Qiu, W.-H. Fang, R. Liang, X. Ren, X. Ji, G. Cui, A. M. Asiri, G. Cui, B. Tang and X. Sun, Nat. Commun., 2018, 9, 3485 CrossRef PubMed.
- X. Guo, H. Du, F. Qu and J. Li, J. Mater. Chem. A, 2019, 7, 3531–3543 RSC.
- J. Jiao, R. Lin, S. Liu, W. Cheong, C. Zhang, Z. Chen, Y. Pan, J. Tang, K. Wu, S. Hung, H. Chen, L. Zheng, Q. Lu, X. Yang, B. Xu, H. Xiao, J. Li, D. Wang, Q. Peng, C. Chen and Y. Li, Nat. Chem., 2019, 11, 222–228 CrossRef CAS PubMed.
- C. Ling, Y. Ouyang, Q. Li, X. Bai, X. Mao, A. Du and J. Wang, Small Methods, 2019, 3, 1800376 CrossRef.
- Z. Chen, J. Zhao, L. Yin and Z. Chen, J. Mater. Chem. A, 2019, 7, 13284–13292 RSC.
- L. Yang, T. Wu, R. Zhang, H. Zhou, L. Xia, X. Shi, H. Zheng, Y. Zhang and X. Sun, Nanoscale, 2019, 11, 1555–1562 RSC.
- Q. Qin, T. Heil, M. Antonietti and M. Oschatz, Small Methods, 2018, 2, 1800202 CrossRef.
- W. Zhao, P. Wei, Q. Zhang, H. Peng, W. Zhu, D. Tang, J. Yu, H. Zhou, Z. Liu, X. Mu, D. He, J. Li, C. Wang, X. Tang and J. Yang, Nat. Commun., 2015, 6, 1–7 Search PubMed.
- L. Mo, Y. Wang, Y. Bai, Q. Xiang, Q. Li, W. Yao, J. Wang, K. Ibrahim, H. Wang, C. Wan and J. Cao, Sci. Rep., 2015, 5, 1–7 Search PubMed.
- X. Yang, N. Gao, S. Zhou and J. Zhao, Phys. Chem. Chem. Phys., 2018, 20, 19390–19397 RSC.
- Y. Zhao, N. Liu, S. Zhou and J. Zhao, J. Mater. Chem. A, 2019, 7, 16294–16303 RSC.
- S. Zhou, X. Yang, Y. Shen, R. B. King and J. Zhao, J. Alloys Compd., 2019, 806, 698–704 CrossRef CAS.
- J. Zhao and Z. Chen, J. Am. Chem. Soc., 2017, 139, 12480–12487 CrossRef CAS PubMed.
- X. Ma, J. Liu, H. Xiao and J. Li, J. Am. Chem. Soc., 2018, 140, 46–49 CrossRef CAS PubMed.
- X. Guo and S. Huang, Electrochim. Acta, 2018, 284, 392–399 CrossRef CAS.
- S. Maheshwari, G. Rostamikia and M. J. Janik, J. Chem. Phys., 2019, 150, 041708 CrossRef PubMed.
- M. Zafari, D. Kumar, M. Umer and K. S. Kim, J. Mater. Chem. A, 2020, 8, 5209–5216 RSC.
- T. He, S. K. Matta and A. Du, Phys. Chem. Chem. Phys., 2019, 21, 1546–1551 RSC.
- H. Zhu, Y. Hu, S. Wei and D. Hua, J. Phys. Chem. C, 2019, 123, 4274–4281 CrossRef CAS.
- H. Chun, V. Apaja, A. Clayborne, K. Honkala and J. Greeley, ACS Catal., 2017, 7, 3869–3882 CrossRef CAS.
- L. Shi, Q. Li, C. Ling, Y. Zhang, Y. Ouyang, X. Bai and J. Wang, J. Mater. Chem. A, 2019, 7, 4865–4871 RSC.
- F. Zhou, L. M. Azofra, M. Ali, M. Kar, C. McDonnell-Worth, C. Sun, X. Zhang and D. R. MacFarlane, Energy Environ. Sci., 2017, 10, 2516–2520 RSC.
- B. H. R. Suryanto, C. S. M. Kang, D. Wang, C. Xiao, F. Zhou, L. M. Azofra, L. Cavallo, X. Zhang and D. R. MacFarlane, ACS Energy Lett., 2018, 3, 1219–1224 CrossRef CAS.
- H. Yin, L. Gan and P. Wang, J. Mater. Chem. A, 2020, 8, 3910–3917 RSC.
- J. Zhang, Y. Zhao, Z. Wang, G. Yang, J. Tian, D. Ma and Y. Wang, New J. Chem., 2020, 44, 422–427 RSC.
- Y. Qian, Y. Liu, Y. Zhao, X. Zhang and G. Yu, EcoMaterials, 2020, 2, e12014 Search PubMed.
- J. C. Liu, X. L. Ma, Y. Li, Y. G. Wang, H. Xiao and J. Li, Nat. Commun., 2018, 9, 1610 CrossRef PubMed.
- S. Zhou, X. Yang, X. Xu, S. Dou, Y. Du and J. Zhao, J. Am. Chem. Soc., 2020, 142, 308–317 CrossRef CAS PubMed.
- W. Pei, S. Zhou, Y. Bai and J. Zhao, Carbon, 2018, 133, 260–266 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nh00242a |
| This journal is © The Royal Society of Chemistry 2020 |