
One-pot synthesis of gadolinium-doped carbon quantum dots for high-performance multimodal bioimaging†
Yi
Pan‡
ac,
Jun
Yang‡
ab,
Yaning
Fang‡
a,
Junhui
Zheng
b,
Rong
Song
a and
Changqing
Yi
*ac
aKey Laboratory of Sensing Technology and Biomedical Instruments (Guangdong Province), School of Engineering, Sun Yat-Sen University, Guangzhou, China. E-mail: yichq@mail.sysu.edu.cn; Fax: +86-20-39342380; Tel: +86-20-39342380
bGuangdong General Hospital, Guangzhou, China
cResearch Institute of Sun Yat-Sen University in Shenzhen, Shenzhen, China
First published on 15th November 2016
Abstract
In this study, an efficient and mild approach is reported for the facile synthesis of a carbon quantum dot (CQD)-based dual-modal fluorescence (FL)/magnetic resonance (MR) imaging probe by doping Gd(III) into CQDs through a one-pot pyrolysis process at low temperature. The as-prepared Gd-encapsulated CQDs (GCDs), which have an average diameter of ∼15 nm, are highly water-soluble. The GCDs are observed to have a high MR response with a longitudinal relaxation of 57.42 mM−1 s−1 and a strong fluorescence brightness with an absolute quantum yield of 40% while containing only 1.0% (w/w) of Gd3+ content. Stemming from the minimum Gd-doping and the inert carbon coating, GCDs exhibit excellent biocompatibility and blood compatibility. Dual-modality bioimaging applications of GCDs are successfully demonstrated by the use of HeLa cells and mice as models, revealing their great potential in fundamental biomedical research studies and even clinical fields such as fluorescence-guided surgery and dual-modal FL/MR imaging of blood vessels.
1. Introduction
The multimodal imaging probe, which synergistically combines several imaging modalities, has been proved to be a powerful tool for early diagnosis and disease treatment with high precision and accuracy. Of all the multimodal imaging probes, the fluorescence (FL)/magnetic resonance (MR) probe attracts significant attention in biomedical research and is successfully implemented in clinical practice, owing to its collective merits of high spatial and subcellular resolution, deep tissue penetration, and single-cell sensitivity. In particular, there has been widespread research interest in developing safer and more efficient nanoscale FL/MR dual-modal imaging probes which exhibit tunable physicochemical properties and favourable biodistribution.1–19 Recently, carbon quantum dots (CQDs) have become a superior scaffold to construct multi-modal imaging probes for in vivo investigation, owing to their facile synthesis and easy surface functionalization, high water-solubility and colloidal stability under physiological conditions, as well as excellent biocompatibility.Various synthesis strategies have been reported to prepare CQD-based multimodal imaging probes.8–20 One approach is to integrate magnetic Fe3O4 nanoparticles (NPs) into fluorescent CQDs by hydrothermal decomposition of organic precursors employing Fe3O4 NPs as seeds8–10 or by depositing Fe3O4 NPs on graphene oxide.11 However, magnetic NP-integrated CQDs suffer from the heavy accumulation in the reticuloendothelial organs mainly caused by their relatively large sizes.16
It has been reported that the incorporation of Gd into solid matrices can cause the slowing of tumbling of the Gd complex, thus significantly increasing r1 relaxivity.12 Therefore, combining Gd(III) and fluorescent CQDs should be a more promising approach for the preparation of CQD-based multi-modal imaging probes.12–19 The Gd-encapsulated CQDs (GCDs) can be prepared by simple pyrolysis in air of gadopentetate monomeglumine (GdPM),13 or gadopentetic acid (Gd-DTPA),16 or a mixture of gadopentetic acid, Tris base and betaine hydrochloride.12 Though these one-pot calcination methods are simple and time-saving, the poor hydrophilicity of the as-prepared GCDs caused by carbonization makes further functionalization of the probes difficult. And due to their relatively large sizes, the as-prepared GCDs might suffer from the non-negligible accumulation in the reticuloendothelial organs after systemic injection.20 Compared with the calcination in air at high temperature, preparation of the GCDs in aqueous solution under mild conditions, for example, by the hydrothermal approach with and without microwave assistance, should be more controllable and easy-to-accomplish, and the as-prepared GCDs exhibit high water solubility and colloidal stability under physiological conditions.14,15,19 The strategy of step-by-step surface modification in aqueous solution under mild conditions has also been demonstrated for the successful preparation of the GCDs, where Gd3+ ions are simply chelated onto the outer surface of the DTPA or cyclic DTPA dianhydride functionalized CQDs.17,18 Though these step-by-step surface modification methods can be used to prepare water soluble GCDs with relatively small sizes and good MR response, they still suffer from complicated and time-consuming multistep synthesis. Although various strategies have been successfully demonstrated for the synthesis of CQD-based FL/MR dual-modal imaging probes, their practical applications in biological process monitoring or medical diagnosis are still limited by either the low MR response or the weak fluorescence brightness. Therefore, continuous research efforts should be exerted to explore more efficient approaches for the synthesis of CQD-based multimodal probes with enhanced imaging performance.
An ideal FL/MR dual-modal imaging probe for future clinical translation should possess strong fluorescence brightness and a high MR response while containing as little Gd3+ content as possible.18 Due to the importance of the fluorescence intensity for bioimaging with high sensitivity and resolution, significant efforts have been devoted to the investigation of synthesis routes for improving the quantum yield (QY) of CQDs.21–29 Although the exact mechanism is still an open question, the photoluminescence (PL) properties of CQDs, such as QY, FL lifetime, as well as PL emission wavelength, strongly depend on the quantum-confinement effect, structural defects induced by surface modification and element doping. In particular, due to its good controllability, doping plays an important role in tuning the PL properties of CQDs. It has been reported that the carboxyl groups of critic acid (CA) and the amines of ethylenediamine are beneficial for condensation polymerization and further carbonization, thus greatly improving the QY of CQDs to 80% (quinine sulfate with QY = 0.54 as standard) via N-doping.29
Based on these understandings, an efficient and mild approach is reported in this study for the facile synthesis of biocompatible GCDs with high longitudinal relaxation and strong fluorescence brightness while containing as little Gd3+ content as possible. As illustrated in Fig. 1, CA monohydrate, branched polyethylenimine (BPEI) with an extremely high density of ethylene diamine units and Gd-DTPA were chosen as carbon, nitrogen and gadolinium sources, respectively. And the dual-modality applications of as-prepared GCDs are demonstrated by the use of HeLa cells and mice as models. Experimental results revealed that the GCDs are water soluble, biocompatible, and cell-membrane permeable. More importantly, the GCDs prepared in this study are observed to have a significantly higher MR response than commercially available contrast agents while still exhibiting strong fluorescence brightness with significantly improved QY, suggesting their potential for practical applications in fundamental biomedical research studies and even clinical fields.
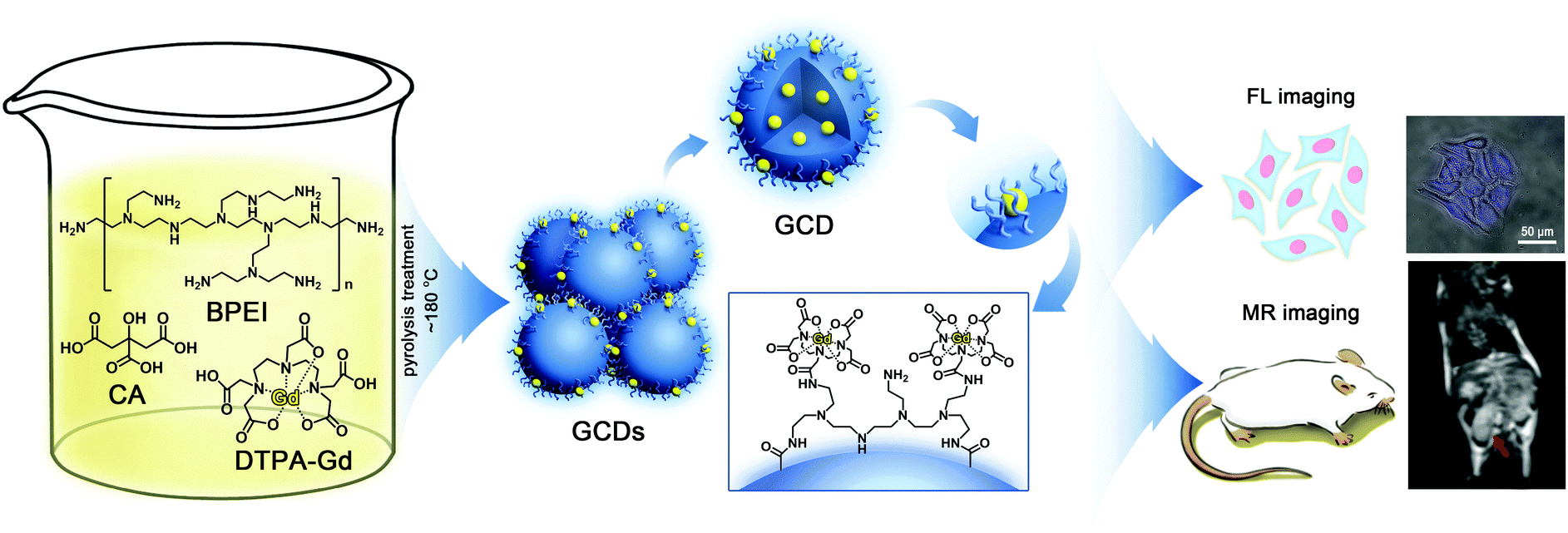 | ||
| Fig. 1 Schematic illustration for the synthesis and dual-modality imaging application of GCDs. | ||
2. Experimental section
2.1 Reagents and materials
All the chemical reagents are of analytical grade and used without further purification, including GdCl3·6H2O (Sigma), DTPA, branched polyethylenimine (BPEI, MW = 1800, Aladdin), CA, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide, dimethyl sulfoxide (DMSO), ninhydrin, L-threonine, xylenol orange, acetate buffer, and phosphate buffered saline (PBS). Ultrapure water was prepared with an A10 advantage deionized water system (Millipore). Reagents for cell culture include Fetal Bovine Serum (FBS) from Gibco, DMEM medium with high glucose (H-DMEM) from Hyclone, and penicillin–streptomycin from Hyclone.2.2 Synthesis of GCDs
GCDs were synthesized by the one-pot pyrolysis treatment of CA, BPEI, and Gd-DTPA at low temperature. Firstly, Gd-DTPA was freshly prepared by adding 5.0 mL of 0.09 g mL−1 DTPA solution to 5.0 mL of 0.06 g mL−1 GdCl3 solution under stirring for 3 h. Then, 5.0 mL of freshly prepared Gd-DTPA solution was mixed with a 5.0 mL solution containing 0.20 g mL−1 CA and 0.10 g mL−1 BPEI in a 50 mL beaker to form a homogenous solution. The mixture was heated moderately (∼180 °C) by oil heating until a uniform pale-yellow gel was obtained, followed by the addition of 1.0 mL of cold water under heating. The same procedure was repeated 10 times until the color of the gel turned orange. Finally, the products were successively dissolved in 10.0 mL of ultrapure water, centrifuged at 6000 rpm for 30 min and dialyzed (MWCO = 3500) against water for 3 days. The solid products were collected after freeze-drying and could be dissolved again for further characterization and experiments.2.3 Characterization
The physiochemical properties, including the morphology, structure, chemical composition, and optical properties, of the as-prepared GCDs were characterized using a comprehensive set of analytical techniques, such as high resolution transmission electron microscopy (HRTEM, JEM-2012GHR, JEOL), X-ray photoelectron spectroscopy (ESCALAB 250, Thermo-VG Scientific), X-ray diffraction (Empyrean, PANalytical), Fourier transform infrared spectroscopy (VERTEX 70, Bruker), UV-Vis spectrometry (DU730, Beckman) and fluorescence spectrometry (Fluoromax-4P, Horiba). The size distribution of the as-prepared GCDs was revealed by dynamic light scattering (DLS) measurements using a NanoZS90 instrument (Malvern). FL QY and the FL lifetime were measured on an FSP920-combined time resolved and steady state fluorescence spectrometer (Edinburgh Instruments) and a Fluorolog-3 fluorescence spectrometer (Horiba), respectively.The Gd content in GCDs was measured by a quadrupole-based iCAP Qc ICP-MS (ThermoFisher Scientific, Bremen, Germany) using the standard mode. The determination was carried out by external calibration in the range from 20 to 2000 ng L−1. In brief, 1.0 mL of nitric acid (65%) and 0.1 mg of GCDs were added to a centrifuge tube, and the mixture was allowed to react for 0.5 h. Then 1 mL of rhodium solution was added as an internal standard and diluted to a volume of 1.0 L. The final concentration of the internal standard was 1 μg L−1. The isotopes 103Rh, 158Gd and 160Gd were monitored with a dwell time of 0.1 s each. The MR property of GCDs was evaluated by capturing the T1-weighted MR images of GCDs in aqueous solution with a clinical 1.5T MRI instrument, where water protons in ultrapure water were used as the blank. T1-weighted MR imaging of GCDs in aqueous solution containing different Gd3+ concentrations (0.006, 0.013, 0.019, 0.032, 0.064 and 0.095 mM) was carried out, followed by the calculation of T1 and T2 relaxation rates.
2.4 Evaluation of the biocompatibility of GCDs
Since free Gd3+ is highly toxic, the possible Gd3+ leakage from GCDs into serum was firstly determined using xylenol orange as the indicator.30 In brief, 0.26 g of GCDs were dissolved in 3.0 mL of serum and transferred to a dialysis bag (MWCO = 1000). The dialysis bag was put into a centrifuge tube containing 27.0 mL of serum, followed by shaking the whole setup on a shaker at 100 rpm, 37 °C. At different time points (0, 1, 2, 4, 8, 12, 24 h), 200 μL of the sample was collected from the centrifuge tube. The acetic buffer solution of the xylenol orange dye was prepared by dissolving 3.0 mg of xylenol orange in 250 mL of acetate buffer (pH = 5.8). Then, 100 μL of the as-prepared xylenol orange solution and 50 μL of the sample were added to a 96-well microplate, followed by measuring the absorbance at 433 nm (Abs433) and 573 nm (Abs573) on a microplate reader. At each time point, three samples were parallelly measured. The concentration of free Gd3+ was proportional to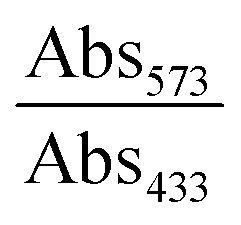 . The sample collected at 0 h was used as the blank where relative free Gd3+ concentration was regarded as 0%. The optical density of GdCl3 solution with a concentration of 0.145 mg L−1 was recorded to represent 100% relative free Gd3+ concentration.
. The sample collected at 0 h was used as the blank where relative free Gd3+ concentration was regarded as 0%. The optical density of GdCl3 solution with a concentration of 0.145 mg L−1 was recorded to represent 100% relative free Gd3+ concentration.
MTT assay was employed to evaluate the cytotoxicity of the as-prepared GCDs using HeLa cells (Cells Bank of the Chinese Academy of Science) which were incubated in H-DMEM supplemented with 10% FBS and 1% penicillin–streptomycin under the conditions of 5% CO2 at 37 °C. In brief, HeLa cells were seeded on a 96-well plate at an initial density of 5 × 103 cells per well and incubated for 24 h. After removing the culture medium, GCDs with concentrations ranging from 0 to 1.0 mg mL−1 were introduced and co-incubated with the HeLa cells for another 24 h. Six groups of concentration were examined and for each group the experiment was repeated three times. After incubation for 24 h, the cells were washed with PBS and then 100 μL of culture medium and 20 μL of MTT solution (dissolved in PBS, 5 mg mL−1) were added to each well, followed by incubation for another 4 h. All the incubation processes stated above were performed at 37 °C with 5% CO2. Finally, the supernatant was removed and 150 μL of DMSO was added to each well. The samples were shaken for 10 min before absorbance value measurement. The absorbance at 570 nm was measured on a microplate reader. The optical density of the control group which received no GCD treatment represented 100% growth of the HeLa cells.
A hemolysis assay was also carried out to evaluate the blood compatibility of GCDs.31,32 In brief, red blood cells were collected from human blood by centrifugation to remove the serum. Then, red blood cells were washed with PBS five times, followed by ten times dilution in PBS before usage. 0.1 mL of the diluted cell suspension was mixed with 1.5 mL of aqueous solution containing different concentrations of GCDs (0.65, 1.30, 1.94, 2.60 g L−1), and 1.5 mL of ultrapure water and PBS were used as positive and negative control, respectively. After mild shake and stew for 2 h at room temperature, the samples were centrifuged and the supernatants were collected for absorbance measurement which was performed on a UV-Vis spectrometer (DU730, Beckman) with a scanning range from 500 nm to 600 nm. Absorbance at 541 nm was specially recorded for the calculation of the hemolysis percentage using the following equation: Hemolysis (%) = (Abssample − Absnegative![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) control)/(Abspositivecontrol − Absnegative control) × 100%, where hemolysis for the positive control group and the negative control group were regarded as 100% and 0%, respectively.
control)/(Abspositivecontrol − Absnegative control) × 100%, where hemolysis for the positive control group and the negative control group were regarded as 100% and 0%, respectively.
2.5 In vitro dual-modal FL/MR imaging
HeLa cells were seeded on a 24-well plate at an initial density of 4 × 104 cells per well and incubated for 24 h mainly for cell adherence. The concentration of GCD solution for cellular imaging was 0.628 mg mL−1, which was determined on the basis of the MTT result. The cells were treated with the GCD solution for another 24 h, washed with PBS three times and fixed with 1 mL of 4% paraformaldehyde. The fluorescence images were acquired by a fluorescence microscope (Olympus, IX71) under UV excitation with a peak at ∼340 nm.MR imaging of HeLa cells was performed on a clinical 1.5T MRI instrument (Philip 1.5T Achieva), according to a previous study with some modification.16 Firstly, HeLa cells were cultured in 75 cm2 culture flasks. Upon reaching a confluence of 80%, 0.628 mg mL−1 GCDs and a culture medium without GCDs (as blank group) were added to two different culture flasks, respectively. After incubation for 24 h, the cells in both culture flasks were in turn washed with PBS, digested by trypsin, centrifuged and redispersed in PBS (pH = 7.4). Finally, the T1-weighted MR images of the HeLa cells were acquired using the following scanning parameters: TR/TE = 480/8.8 ms, 256 × 196 matrices, flip angle = 5°/26°, and slice thickness = 3 mm.
2.6 Animal models and in vivo MR imaging
In vivo MR imaging was conducted on C57BL mice (n = 6, male, gray hair, an average body weight of about 25 g), which were purchased from Laboratory Animal Center of Sun Yat-Sen University. All experiments were approved by the Animal Experimentation Ethics Committee of Sun Yat-sen University and conducted in accordance with guidelines for human care. Briefly, C57BL mice were anesthetized with 0.075 mL of chloral hydrate via intraperitoneal injection, and injected with 0.25 mL GCDs and Gd-DTPA (Magnevist, a commercial MR contrast) through the caudal vein at a Gd concentration of 0.01 M, respectively. The mice were then fixed on cardboard with medical tape and the T1-weighted MR images were obtained with a clinical MR instrument (Fig. S1, ESI† Philip 1.5T Achieva) using the scanning parameters and sequences listed in Table S1 (ESI†).3. Results and discussion
3.1 Synthesis and characterization of GCDs
As revealed by TEM images (Fig. 2A), the as-prepared GCDs exhibited a subround shape with a diameter of 15.0 ± 5.0 nm, correlating well with DLS measurements which revealed a size distribution between 10.0 and 20.0 nm (inset of Fig. 2A). As evidenced in HRTEM images, GCDs had an obvious lattice spacing of 0.279 nm (Fig. 2B), which is consistent with that of the (100) facet of graphite.33,34 This result confirmed that GCDs preserve the graphite crystal structure after the simultaneous doping of nitrogen and gadolinium elements, correlating with previous observations that the hydrothermal method typically yields crystal structures.14,15 while calcination always produces amorphous structures.12,16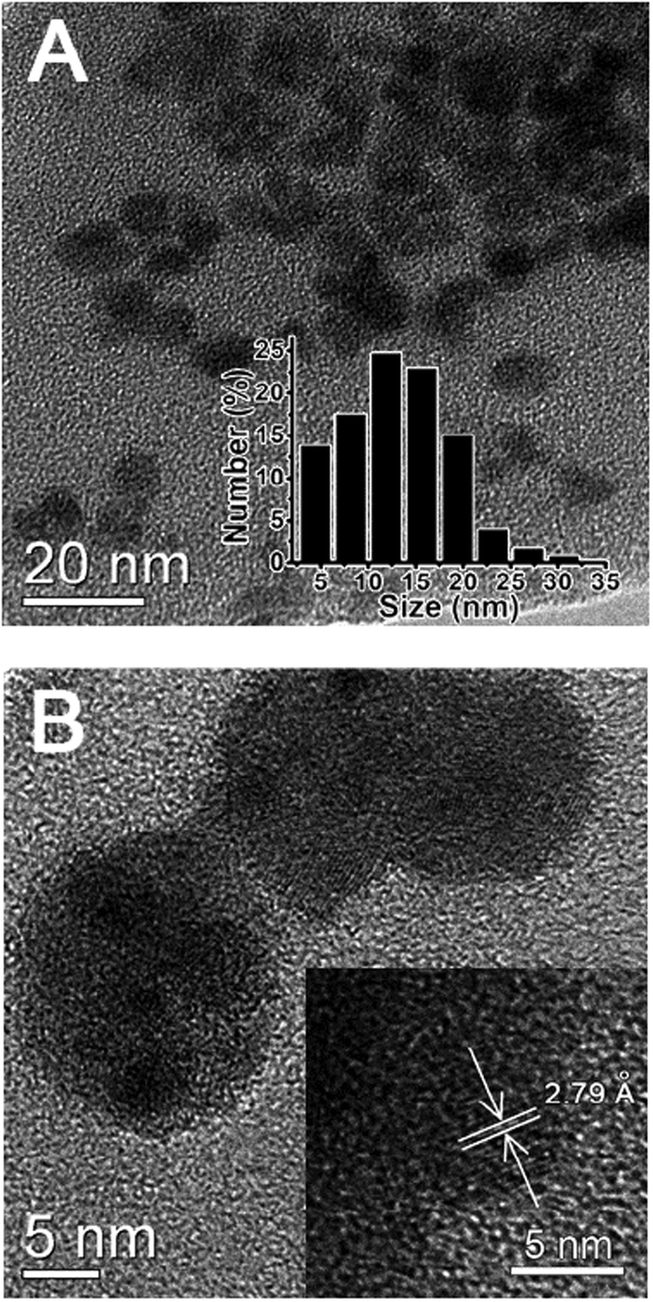 | ||
| Fig. 2 (A) TEM image of GCDs. Inset: the histogram of GCD size distribution obtained from DLS measurements. (B) HRTEM image of GCDs. Inset: the HRTEM image of GCDs with lattice spacing. | ||
XPS and FTIR was performed to further characterize the structure and components of the as-prepared GCDs. XPS analysis revealed that the characteristic peaks correspond to Gd 4d (141 eV), C 1s (285 eV), N 1s (398 eV), O 1s (530 eV), and Gd 3d (1187 eV) (Fig. 3A), confirming that the as-prepared GCDs were composed of 58.52% C, 25.34% O, 15.3% N and 0.58% Gd (Table S2, ESI†), and elemental mapping revealed that Gd was distributed evenly within GCDs (Fig. S2, ESI†). The high-resolution Gd 4d XPS spectra exhibited Gd 4d5/2 and Gd 4d3/2 lines at 141.0 and 147.2 eV, respectively (Fig. 3B). In comparison to the corresponding elemental Gd 3d5/2 (1186.0 eV), a higher binding energy of 1187.5 eV was observed for Gd 3d5/2 in GCDs (Fig. 3C), possibly due to the different coordination environments.15,35 The presence of both Gd 4d and Gd 3d peaks reveals that Gd still existed in the form of Gd3+ in GCDs,12–16 suggesting that the chemical environment and Gd coordination were not significantly altered by the synthesis process. Maintaining the oxidation state of Gd3+ is quite important, because MR imaging depends on its half-filled f orbital with seven unpaired electrons and its symmetrical s ground state.
 | ||
| Fig. 3 (A) XPS survey spectra, (B) Gd 4d spectra, (C) Gd 3d spectra, (D) C 1s spectra, (E) N 1s spectra, and (F) O 1s spectra of GCDs. | ||
The deconvoluted XPS C 1s spectra exhibited a dominant peak centered at 284.5 eV which is attributed to C–C, further validating the presence of a graphitic sp2 carbon structure in GCDs (Fig. 3D).36 The stretching vibration band of C–H at 2981.8 cm−1 (νC–H) and the bending vibration band of C–H at 1398.3 cm−1 (δC–H) in FTIR spectra also confirmed the carbon framework of GCDs (Fig. S3, ESI†). Two peaks centered at 398.4 and 400.1 eV presented on the deconvoluted XPS N 1s spectra are respectively attributed to C–N and C–N–H, confirming the successful doping of N elements in GCDs (Fig. 3E). FTIR analysis revealed the presence of surface amino groups on GCDs, as evidenced by the presence of the obvious stretching vibration band of N–H at 3423.5 cm−1 and the bending vibration band of N–H at 1591.2 cm−1, and the percentage of free amines on the surface of GCDs was determined to be 2.6% (w/w) using Kaiser tests (Fig. S4, ESI†).37
FTIR analysis also revealed peaks at 3568.2, 1706.9 and 1010.7 cm−1 that are, respectively, the characteristic stretching vibration bands of O–H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and C–O (Fig. S3, ESI†), indicating the presence of surface carboxyl groups which were either generated during the pyrolysis process and/or inherited from the DTPA precursors. This is also supported by the deconvoluted O 1s XPS spectra, finding a dominant peak centered at 530.1 eV and a shoulder peak centered at 531.3, eV which were attributed to C–O and CO, respectively (Fig. 3F). The surface carboxyl and amino groups endow GCDs with good colloidal stability and cell-membrane permeability.
O, and C–O (Fig. S3, ESI†), indicating the presence of surface carboxyl groups which were either generated during the pyrolysis process and/or inherited from the DTPA precursors. This is also supported by the deconvoluted O 1s XPS spectra, finding a dominant peak centered at 530.1 eV and a shoulder peak centered at 531.3, eV which were attributed to C–O and CO, respectively (Fig. 3F). The surface carboxyl and amino groups endow GCDs with good colloidal stability and cell-membrane permeability.
3.2 Optical and MR properties of GCDs
The as-prepared GCDs are highly dispersible in water, forming a transparent and homogeneous light-yellow aqueous solution without visible precipitation for months and emitting bright blue fluorescence under 365 nm excitation (inset of Fig. 4A). Fig. 4A shows the UV-vis absorption and fluorescence spectra of GCDs. The UV-vis spectra of GCDs exhibited a broad absorption band between 220 and 450 nm with a shoulder and an absorption peak appearing at ∼240 nm and 350 nm (curve a of Fig. 4A), which could be attributed to π → π* transition of CC and n → π* transition of CO, respectively. The absorption spectrum also validates the sp2 hybrid structure and carbonyl groups of GCDs, and resembles those reported previously for CQDs.14,34
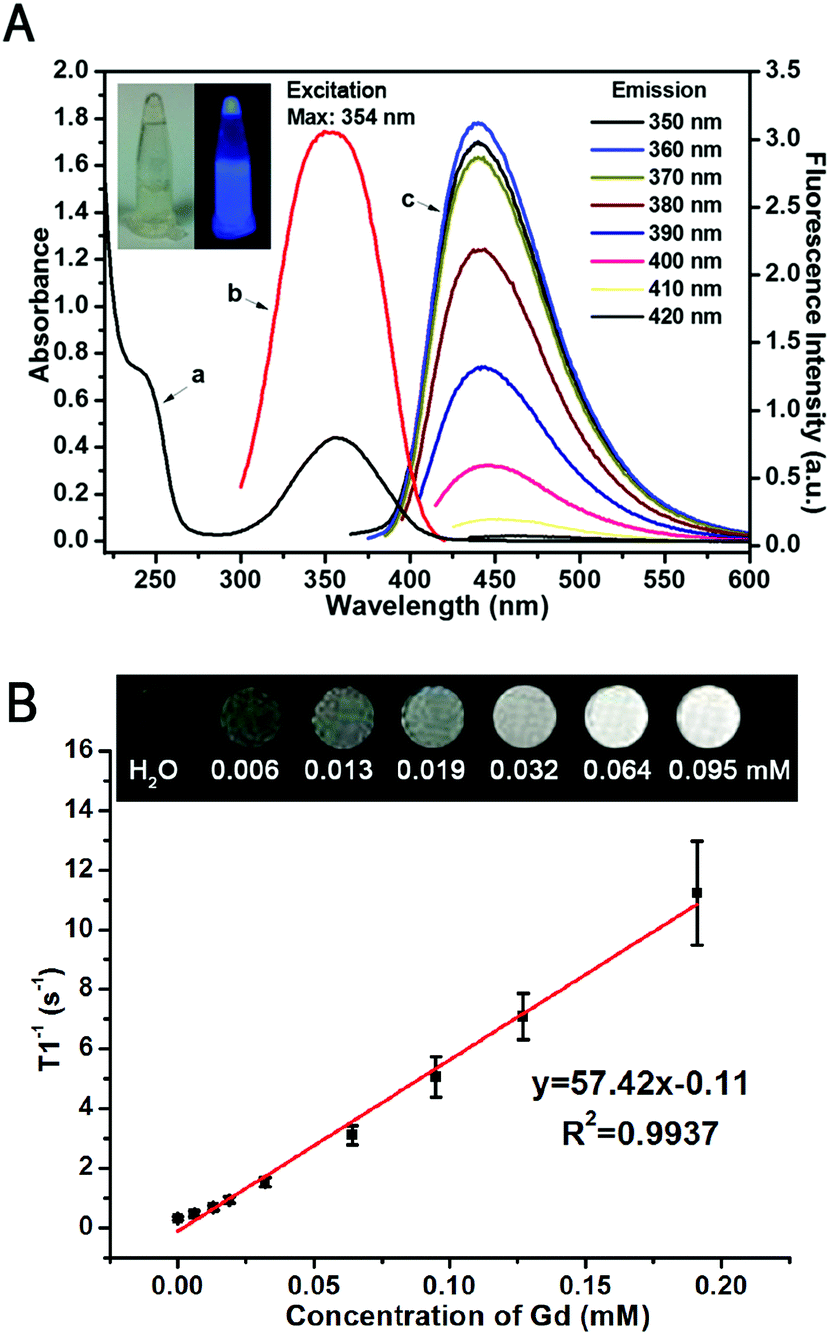 | ||
| Fig. 4 (A) The UV-Vis absorption spectrum (curve a), excitation spectrum (curve b) and emission spectra (curve c) of GCDs. The inset shows the photograph of GCDs under daylight and 365 nm UV light. (B) T1-Weighted MR images of GCDs with various concentrations (equivalent Gd concentration: 0, 0.006, 0.013, 0.019, 0.032, 0.064 and 0.095 mM), and a linear correlation between the longitudinal relaxivity (r1) and equivalent Gd concentration of GCDs. | ||
GCDs are highly fluorescent, and can emit strong PL upon excitation with light of a wide range of wavelengths (curve c of Fig. 4A). Unexpectedly, different from conventional CQDs, GCDs prepared in this study exhibited a wavelength-independent emission, where the fluorescence intensity increased to the maximum then decreased while the emission peak showed no obvious change upon excitation with light with wavelengths from 290 nm to 420 nm. The strongest emission peaks of GCDs which reflect their dominant energy gap are around 445 nm at the maximum excitation wavelength of 354 nm. It is possible that the fluorescence origin sites of GCDs were altered by simultaneous doping of N and Gd elements, resulting in a uniform chemical feature and the state of those sp2 clusters contained on the surface of GCDs.39–41 It has been reported previously that the N atom is the electron-acceptor in the heteroatom doped CQDs, showing obvious charge transfer and thus high QY.28 Therefore, as expected, the absolute QY of GCDs with heavy N-element doping was determined to be 40% which was significantly higher than previous studies,12–19 indicating the extra-high PL efficiency. As shown in Fig. 4A, the wavelength of the absorption peak of GCDs is almost the same as the maximum excitation wavelength of GCDs, indicating that most of the absorption is transformed into fluorescence and thus also possibly contributes to its high QY. The PL lifetime of GCDs was determined to be 9.65 ns (Fig. S5, ESI†), also reflecting its excellent optical properties and thus endowing GCDs with the ability to be applied in FL bioimaging especially for future FL-guided surgery and immune-fluorescence histology.
Doping Gd3+ into GCDs renders them the MRI modality. The T1 enhanced contrast capability of GCDs was thus evaluated on a clinical 1.5T MR instrument. As shown in Fig. 4B, the brightness of grey-scaled T1-weighted MR images enhanced with the increase of Gd3+ concentration, confirming the concentration-dependent positive contrast enhancement. The excellent MR response was further demonstrated by the large slope of the r1 relaxivity curve (Fig. 4B), from which the r1 relaxivity of GCDs was calculated to be 57.42 mM−1 s−1, significantly higher than that of the commercially available Gd contrast agents such as DTPA analogues Gadovist® (4.34 mM−1 s−1),42 Magnevists® (3.2 mM−1 s−1)/Omniscans® (3.3 mM−1 s−1),43 and ProHance®(4.3 mM−1 s−1).44 It has been well-established that the r2/r1 ratio of MR contrast agents is an important parameter to determine their classification and estimate their efficiency. The contrast agents favor positive contrast enhancement when the r2/r1 value is close to 1.0, otherwise they are considered as negative contrast agents when the ratio of r2/r1 is larger than 1.5.20 Based on the relaxivites of GCDs, the ratio of r2/r1 was calculated to be 1.14, indicating that GCDs is a T1-contrast agent. Clinically, a T1-contrast agent with high relaxivity is much more desirable than a T2 agent.45
Taking the high toxicity of Gd3+ into account, an ideal Gd-based MRI contrast agent should contain a minimum amount of Gd while exhibiting as high r1 relaxivity as possible. As quantitated by ICP-MS using HNO3 digestion, there is only 1.0% (w/w) Gd in GCDs, significantly lower than previous reports.12–17 Though the relationship between the Gd content in nanostructured MR contrast probes and their longitudinal relaxivity is still an open question, the incorporation of Gd into solid matrices can cause the slowing of tumbling of the Gd complex and maximize its function in curtailing the longitudinal relaxation time of hydrogen protons, thus significantly increasing r1 relaxivity,12,46,47 and the high local concentration of Gd3+ caused by the high specific surface ratios of GCDs might also contribute to its high r1 relaxivity.48 It is worth noting that the r1 relaxivity could achieve such a high level with only little amount of Gd, indicating that GCDs synthesized in this study is a promising T1-weighted MRI contrast agent for medical translation.
3.3 Biocompatibility of GCDs
Toxicity is one of the major concerns for the practical application of a bioimaging probe. Free Gd3+ can induce serious toxicity by changing intracellular reactive oxygen species (ROS) levels, inhibiting critical Ca2+ channels, and even causing cardiovascular and neurologic toxicity, and there is no biochemistry based on Gd3+ in human body.49–51 Therefore, examining the Gd3+ leakage from Gd-based MR contrast agents is of great importance. In this study, Gd3+ leakage from GCDs was detected using Xylenol Orange as the indicator. As shown in Fig. 5A, no noticeable amount of Gd3+ was released into serum for 24 h. It is possible that Gd3+ was stably trapped by and chelated with the carbon dot framework, effectively blocking the leakage of Gd3+ into the surroundings.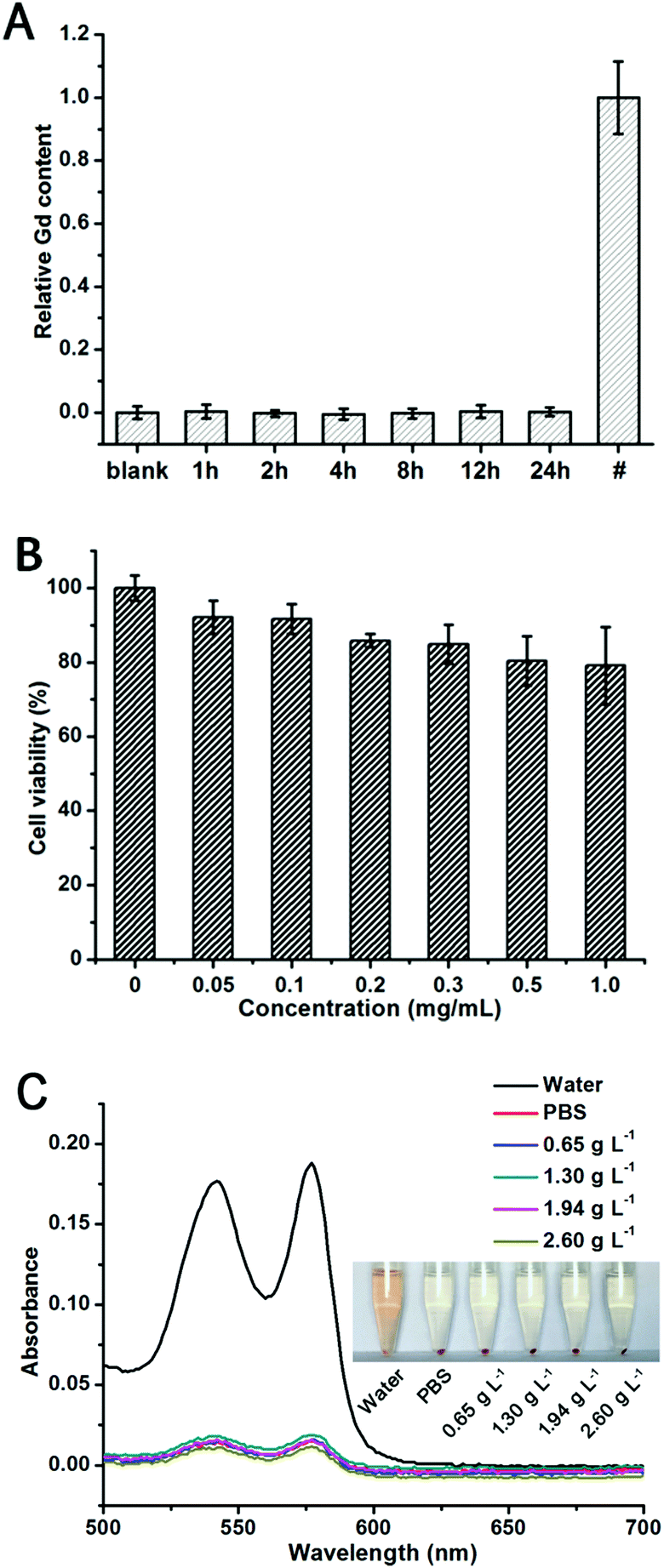 | ||
| Fig. 5 (A) Gd3+ release from GCDs over 24 h using xylenol orange (XO) as a Gd3+ indicator. GCDs were incubated in serum. # represents the overall Gd content in the GCDs used in the leakage test. (B) Viability of HeLa cells upon treatment of GCDs for 24 h. Results are mean ± SD of the triplicate experiments. (C) Hemolytic percentage of GCDs to human red blood cells. Inset shows the photographic images of the red blood cells treated with GCDs at various concentrations. | ||
Due to the intrinsic biocompatibility of CQDs and the negligible leakage of Gd3+, an outstanding biocompatibility is expected for GCDs. A typical MTT assay was carried out with HeLa cells to examine the comparative cell viability upon treatment with different concentrations of GCDs for 24 h. As shown in Fig. 5B, over 80% cell viability was observed even with the GCD concentration up to 1.0 mg mL−1 (equivalent Gd concentration is 0.06 mM), confirming the good biocompatibility of GCDs towards HeLa cells and emphasizing its potential application in bioimaging.
Because hemolysis in vivo can result in jaundice, anemia, and other pathological conditions, the hemolytic potential of GCDs is also evaluated.31,32 In this study, ultrapure water acted as a positive control while the PBS acted as a negative control. As shown in Fig. 5C, GCDs displayed the same phenomena and fitted similar absorbance curves as the negative control group among the concentration range from 0.65 g L−1 to 2.60 g L−1, validating their negligible hemolytic potential. All the above results confirm the excellent biocompatibility and blood compatibility of GCDs which guarantee its applicability in in vivo imaging.
3.4 FL and MR imaging of HeLa cells with GCDs as probes
A cell model was firstly employed to demonstrate the FL and MR dual response of GCDs. HeLa cells were incubated with 0.628 mg mL−1 GCDs and images were captured with a fluorescence microscope under bright field and UV light excitation (Fig. 6A–C). It can be seen from the bright-field image of Fig. 6A that the morphology of HeLa cells upon treatment with GCDs remained intact, further verifying their biocompatibility. GCDs produced a bright blue FL and can illuminate the HeLa cells, correlating well with their emission spectra. GCDs were observed to localize mainly in the cytoplasm, confirming that GCDs can cross the cell membrane barriers to enter the intracellular region. Previous reports have revealed that free amine groups could give rise to endosomolytic effects thus leading to cell membrane permeability.18,38,52 The hypothesis of GCD internalization through endocytosis is supported by the Kaiser test which confirmed the presence of 2.60% (w/w) free amine groups on the surface of GCDs. | ||
| Fig. 6 (A–C) The fluorescence microscopic images of HeLa cells treated with 0.628 mg mL−1 GCDs for 24 h. Images were taken under (A) bright field, and (B) UV light excitation (ranging from 335 to 380 nm with a peak at ∼340 nm). (C) Overlay of fluorescence and bright field images. (D and E) The photographic images (D) and T1-weighted MR images (E) of HeLa cells incubated with GCDs (left) of 0.628 mg mL−1. The blank group was incubated with the same volume of culture medium without GCDs (right). | ||
In addition, the grey-scaled T1-weighted images of HeLa cells upon treatment with 0.628 mg mL−1 GCDs were captured on a clinical MRI instrument. As shown in Fig. 6E, compared to the blank group, significantly enhanced signals were observed in the cells upon treatment with GCDs, indicating the obvious positive contrast enhancement. Cross-validation between the FL and MR results confirmed that GCDs were successfully internalized by HeLa cells and the FL and MR efficiency of GCDs was maintained, demonstrating the great potential of GCDs as in vivo FL and MR dual-response imaging probes.
3.5 In vivo MR imaging with GCDs as probes
In vivo MR imaging of GCDs was investigated with C57BL mice as models. In order to illustrate the clinical practicality of GCDs, the commercially available MR contrast agent Gd-DTPA (Magnevists®), which is widely used in clinical practice, is employed as the control in this study. Plain scan, contrast-enhanced scan, and dynamic contrast-enhanced (DCE) MR perfusion imaging including cardiac perfusion scan and aortic perfusion scan were performed to have a comprehensive assessment of the MR response of GCDs and Gd-DTPA (Magnevists®). As shown in Fig. 7A and E, both GCDs and Gd-DTPA entered the blood circulation of mice immediately after injection. The contrast-enhanced scan images (Fig. 7B) revealed an obviously positive contrast enhancement of GCDs, as compared to plain scan images (Fig. 7A). A similar phenomenon was observed for Gd-DTPA (Magnevists®) (Fig. 7E and F). The kidneys were highly visualized by the excellent contrast MR signal and were substantially distinguished from other organs (Fig. 7B). More importantly, a significantly stronger MR response was observed for GCDs than Gd-DTPA (Magnevists®), indicating its potential for future clinical translation (Fig. 7B and F).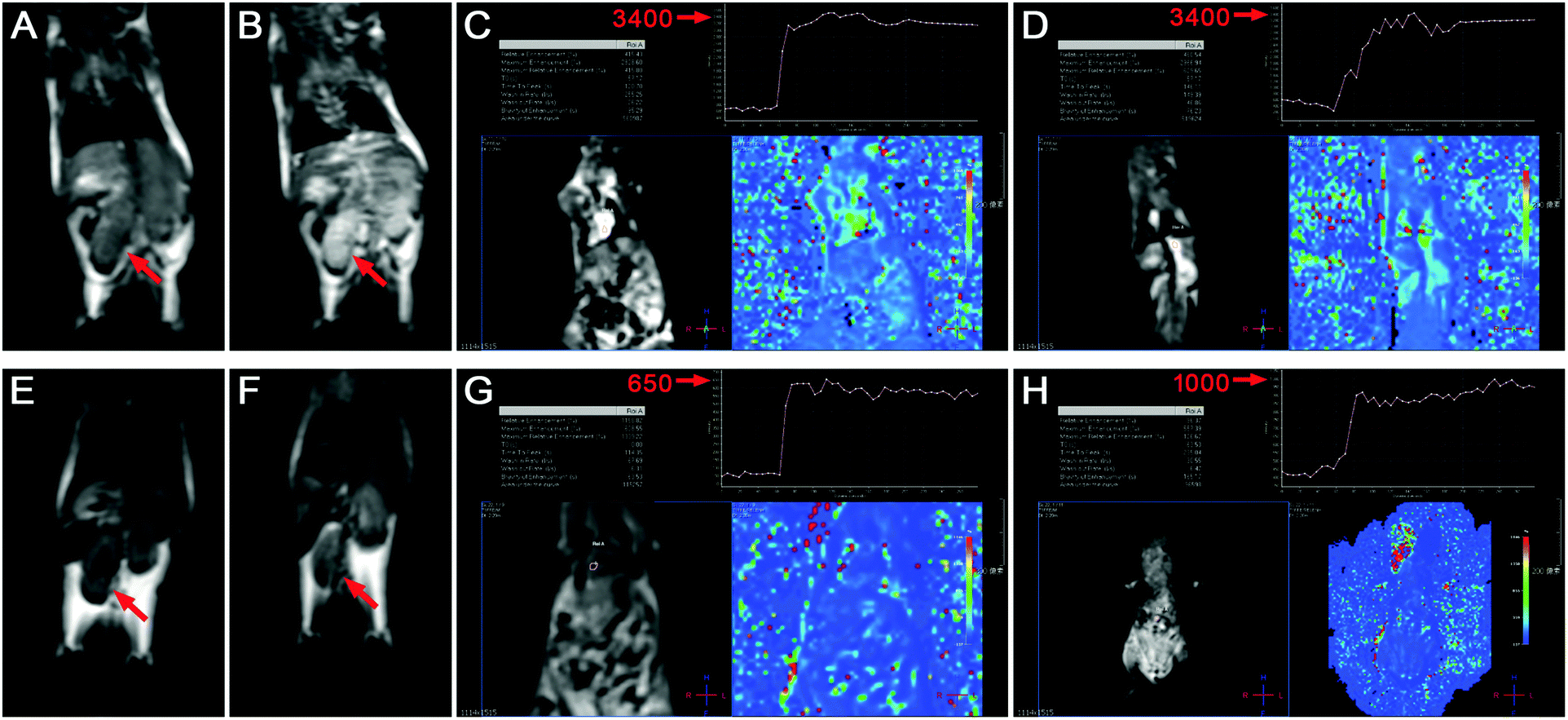 | ||
| Fig. 7 T 1-Weighted MR images of the mice injected with GCDs (A–D) and Gd-DTPA (Magnevists®) (E–H): (A and E) plain scan, (B and F) contrast-enhanced scan, (C and G) cardiac perfusion scan, and (D and H) aortic perfusion scan. | ||
Perfusion imaging is a kind of functional MR imaging technique which can reveal the blood perfusion state and provide information towards hemodynamics. Thanks to the high r1 relaxivity of GCDs, the peak intensity in cardiac and aortic perfusion scan reached 3400 in the mice injected with GCDs (Fig. 7C and D), whereas the values generated by Gd-DTPA (Magnevists®) were only 650 and 1000 (Fig. 7G and H), respectively. The significantly enhanced MR response by GCDs confirmed the great potential of GCDs in DCE perfusion MR imaging, especially in MR angiography.
4. Conclusions
In summary, a dual-modal FL/MR imaging probe is facilely synthesized via a straightforward low temperature pyrolysis process. A series of clear and accurate characterization techniques validated the graphite crystal structure of the as-prepared GCDs and the surface carboxyl and amino groups which afford GCDs good colloidal stability and cell-membrane permeability. Stemming from the minimum Gd-doping and the inert carbon coating, GCDs exhibit excellent biocompatibility and blood compatibility, guaranteeing their applicability for in vitro and in vivo imaging. GCDs afford not only a significantly improved r1 relaxivity (57.42 mM−1 s−1), but also strong fluorescence brightness with the absolute QY significantly improved to 40%, making them appealing as dual-modal FL/MR imaging probes which exhibit collective merits of high spatial and subcellular resolution, deep tissue penetration, and single-cell sensitivity. This potential is strengthened by the fact that a significantly stronger MR response was observed for GCDs than the commercially available MR contrast agent Gd-DTPA (Magnevists®). Overall, the GCDs synthesized in this study have shown great promise in clinical translation as safe and high-performance functional imaging probes.Acknowledgements
The financial support from Tip-top Scientific and Technical Innovative Youth Talents of Guangdong special support program (2014TQ01R417) and the Guangdong-HongKong Technology Cooperation Funding Scheme (2016A050503027) is gratefully acknowledged.References
- J. S. Kim, W. J. Rieter, K. M. L. Taylor, H. An, W. L. Lin and W. B. Lin, J. Am. Chem. Soc., 2007, 129, 8962–8963 CrossRef CAS PubMed.
- H. Gong, Z. L. Dong, Y. M. Liu, S. N. Yin, L. Cheng, W. Y. Xi, J. Xiang, K. Liu, Y. G. Li and Z. Liu, Adv. Funct. Mater., 2014, 24, 6492–6502 CrossRef CAS.
- E. S. Shibu, K. Ono, S. Sugino, A. Nishioka, A. Yasuda, Y. Shigeri, S. Wakida, M. Sawada and V. Biju, ACS Nano, 2013, 7, 9851–9859 CrossRef CAS PubMed.
- C. X. Li, D. M. Yang, P. A. Ma, Y. Y. Chen, Y. Wu, Z. Y. Hou, Y. L. Dai, J. H. Zhao, C. P. Sui and J. Lin, Small, 2013, 9, 4150–4159 CrossRef CAS PubMed.
- L. Cheng, C. Wang, X. X. Ma, Q. L. Wang, Y. Cheng, H. Wang, Y. G. Li and Z. Liu, Adv. Funct. Mater., 2013, 23, 272–280 CrossRef CAS.
- C. Y. Liu, Z. Y. Gao, J. F. Zeng, Y. Hou, F. Fang, Y. L. Li, R. R. Qiao, L. Shen, H. Lei, W. S. Yang and M. Y. Gao, ACS Nano, 2013, 7, 7227–7240 CrossRef CAS PubMed.
- Z. L. Zhao, H. H. Fan, G. F. Zhou, H. R. Bai, H. Liang, R. W. Wang, X. B. Zhang and W. H. Tan, J. Am. Chem. Soc., 2014, 136, 11220–11223 CrossRef CAS PubMed.
- S. Srivastava, R. Awasthi, D. Tripathi, M. K. Rai, V. Agarwal, V. Agrawal, N. S. Gajbhiye and R. K. Gupta, Small, 2012, 8, 961 CrossRef CAS.
- H. Wang, J. Shen, Y. Y. Li, Z. Y. Wei, G. X. Cao, Z. Gai, K. L. Hong, P. Banerjee and S. Q. Zhou, Biomater. Sci., 2014, 2, 915–923 RSC.
- S. Sahu, N. Sinha, S. K. Bhutia, M. Majhi and S. Mohapatra, J. Mater. Chem. B, 2014, 2, 3799–3808 RSC.
- R. Justin, K. Tao, S. Roman, D. X. Chen, Y. W. Xu, X. S. Geng, I. M. Ross, R. T. Grant, A. Pearson, G. D. Zhou, S. MacNeil, K. Sun and B. Q. Chen, Carbon, 2016, 97, 54–70 CrossRef CAS.
- A. B. Bourlinos, A. Bakandritsos, A. Kouloumpis, D. Gournis, M. Krysmann, E. P. Giannelis, K. Polakova, K. Safarova, K. Hola and R. Zboril, J. Mater. Chem., 2012, 22, 23327–23330 RSC.
- X. Y. Ren, L. H. Liu, Y. Li, Q. Dai, M. Zhang and X. L. Jing, J. Mater. Chem. B, 2014, 2, 5541–5549 RSC.
- Y. Xu, X. H. Jia, X. B. Yin, X. W. He and Y. K. Zhang, Anal. Chem., 2014, 86, 12122–12129 CrossRef CAS PubMed.
- N. Q. Gong, H. Wang, S. Li, Y. L. Deng, X. A. Chen, L. Ye and W. Gu, Langmuir, 2014, 30, 10933–10939 CrossRef CAS PubMed.
- H. M. Chen, G. D. Wang, W. Tang, T. Todd, Z. P. Zhen, C. Tsang, K. Hekmatyar, T. Cowger, R. B. Hubbard, W. Z. Zhang, J. Stickney, B. Z. Shen and J. Xie, Adv. Mater., 2014, 26, 6761–6766 CrossRef CAS PubMed.
- C. L. Huang, C. C. Huang, F. D. Mai, C. L. Yen, S. H. Tzing, H. T. Hsieh, Y. C. Lingd and J. Y. Chang, J. Mater. Chem. B, 2015, 3, 651–664 RSC.
- Y. P. Shi, Y. Pan, J. Zhong, J. Yang, J. H. Zheng, J. L. Cheng, R. Song and C. Q. Yi, Carbon, 2015, 93, 742–750 CrossRef CAS.
- H. Liao, Z. Y. Wang, S. Chen, H. Wu, X. J. Ma and M. Q. Tan, RSC Adv., 2015, 5, 66575–66581 RSC.
- C. C. Huang, C. H. Su, W. M. Li, T. Y. Liu, J. H. Chen and C. S. Yeh, Adv. Funct. Mater., 2009, 19, 249–258 CrossRef CAS.
- C. Q. Ding, A. W. Zhu and Y. Tian, Accounts Chem. Res., 2014, 47, 20–30 CrossRef CAS PubMed.
- Y. Q. Dong, H. C. Pang, H. B. Yang, C. X. Guo, J. W. Shao, Y. W. Chi, C. M. Li and T. Yu, Angew. Chem., Int. Ed., 2013, 52, 7800–7804 CrossRef CAS PubMed.
- X. L. Wang, Y. J. Long, Q. L. Wang, H. J. Zhang, X. X. Huang, R. Zhu, P. Teng, L. P. Liang and H. Z. Zheng, Carbon, 2013, 64, 499–506 CrossRef CAS.
- H. T. Li, X. D. He, Y. Liu, H. Huang, S. Y. Lian, S. T. Lee and Z. H. Kang, Carbon, 2011, 49, 605–609 CrossRef CAS.
- P. Anilkumar, X. Wang, L. Cao, S. Sahu, J. H. Liu, P. Wang, K. Korch, K. N. Tackett, A. Parenzan and Y. P. Sun, Nanoscale, 2011, 3, 2023–2027 RSC.
- X. Wang, L. Cao, S. T. Yang, F. S. Lu, M. J. Meziani, L. L. Tian, K. W. Sun, M. A. Bloodgood and Y. P. Sun, Angew. Chem., Int. Ed., 2010, 49, 5310–5314 CrossRef CAS PubMed.
- Z. M. Zhang, Y. Pan, Y. N. Fang, L. L. Zhang, J. Y. Chen and C. Q. Yi, Nanoscale, 2016, 8, 500–507 RSC.
- S. W. Yang, J. Sun, X. B. Li, W. Zhou, Z. Y. Wang, P. He, G. Q. Ding, X. M. Xie, Z. H. Kang and M. H. Jiang, J. Mater. Chem. A, 2014, 2, 8660–8667 CAS.
- S. J. Zhu, Q. N. Meng, L. Wang, J. H. Zhang, Y. B. Song, H. Jin, K. Zhang, H. C. Sun, H. Y. Wang and B. Yang, Angew. Chem., Int. Ed., 2013, 52, 3953–3957 CrossRef CAS PubMed.
- A. Barge, G. Cravotto, E. Gianolio and F. Fedeli, Contrast Media Mol. Imaging, 2006, 1, 184–188 CrossRef PubMed.
- M. A. Dobrovoiskaia, J. D. Clogston, B. W. Neun, J. B. Hall, A. K. Patri and S. E. McNeil, Nano Lett., 2008, 8, 2180–2187 CrossRef PubMed.
- R. C. Lv, P. P. Yang, F. He, S. L. Gai, C. X. Li, Y. L. Dai, G. X. Yang and J. Lin, ACS Nano, 2015, 9, 1630–1647 CrossRef CAS PubMed.
- S. N. Baker and G. A. Baker, Angew. Chem., Int. Ed., 2010, 49, 6726–6744 CrossRef CAS PubMed.
- Y. Q. Dong, R. X. Wang, H. Li, J. W. Shao, Y. W. Chi, X. M. Lin and G. N. Chen, Carbon, 2012, 50, 2810–2815 CrossRef CAS.
- M. Ahren, L. Selegard, F. Soderlind, M. Linares, J. Kauczor, P. Norman, P. O. Kall and K. Uvdal, J. Nanopart. Res., 2012, 14, 1006 CrossRef.
- Z. C. Yang, M. Wang, A. M. Yong, S. Y. Wong, X. H. Zhang, H. Tan, A. Y. Chang, X. Li and J. Wang, Chem. Commun., 2011, 47, 11615–11617 RSC.
- Z. M. Zhang, Y. P. Shi, Y. Pan, X. Cheng, L. L. Zhang, J. Y. Chen, M. J. Li and C. Q. Yi, J. Mater. Chem. B, 2014, 2, 5020–5027 RSC.
- O. Boussif, F. Lezoualc'h, M. A. Zanta, M. D. Mergny, D. Scherman, B. Demeneix and J. P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7297–7301 CrossRef CAS.
- Y. Q. Dong, J. W. Shao, C. Q. Chen, H. Li, R. X. Wang, Y. W. Chi, X. M. Lin and G. N. Chen, Carbon, 2012, 50, 4738–4743 CrossRef CAS.
- L. B. Tang, R. B. Ji, X. K. Cao, J. Y. Lin, H. X. Jiang, X. M. Li, K. S. Teng, C. M. Luk, S. J. Zeng, J. H. Hao and S. P. Lau, ACS Nano, 2012, 6, 5102–5110 CrossRef CAS PubMed.
- Y. Zhang and J. H. He, Phys. Chem. Chem. Phys., 2015, 17, 20154–20159 RSC.
- G. K. Das, B. C. Heng, S. C. Ng, T. White, J. S. C. Loo, L. D'Silva, P. Padmanabhan, K. K. Bhakoo, S. T. Selvan and T. T. Y. Tan, Langmuir, 2010, 26, 8959–8965 CrossRef CAS PubMed.
- F. Erogbogbo, C. W. Chang, J. L. May, L. W. Liu, R. Kumar, W. C. Law, H. Ding, K. T. Yong, I. Roy, M. Sheshadri, M. T. Swihart and P. N. Prasad, Nanoscale, 2012, 4, 5483–5489 RSC.
- R. D. Bolskar, A. F. Benedetto, L. O. Husebo, R. E. Price, E. F. Jackson, S. Wallace, L. J. Wilson and J. M. Alford, J. Am. Chem. Soc., 2003, 125, 5471–5478 CrossRef CAS PubMed.
- F. Chen, W. B. Bu, S. J. Zhang, J. N. Liu, W. P. Fan, L. P. Zhou, W. J. Peng and J. L. Shi, Adv. Funct. Mater., 2013, 23, 298–307 CrossRef CAS.
- H. B. Na and T. Hyeon, J. Mater. Chem., 2009, 19, 6267–6273 RSC.
- F. Q. Hu, H. M. Joshi, V. P. Dravid and T. J. Meade, Nanoscale, 2010, 2, 1884–1891 RSC.
- K. Malzahn, S. Ebert, I. Schlegel, O. Neudert, M. Wagner, G. Schutz, A. Ide, F. Roohi, K. Munnemann, D. Crespy and K. Landfester, Adv. Healthcare Mater., 2016, 5, 567–574 CrossRef CAS PubMed.
- P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293–2352 CrossRef CAS PubMed.
- S. Aime and P. Caravan, J. Magn. Reson. Imaging, 2009, 30, 1259–1267 CrossRef PubMed.
- X. M. Wu, Y. D. Zong, Z. Ye and Z. R. Lu, Pharm. Res-Dordr., 2010, 27, 1390–1397 CrossRef CAS PubMed.
- Y. P. Shi, Y. Pan, J. Y. Chen, C. M. Wong, H. Zhang, M. J. Li and C. Q. Yi, Mater. Sci. Eng., C, 2016, 69, 561–568 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6tb02115h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |