
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Benzothiadiazole and its π-extended, heteroannulated derivatives: useful acceptor building blocks for high-performance donor–acceptor polymers in organic electronics
Yang
Wang
and
Tsuyoshi
Michinobu
*
Department of Organic and Polymeric Materials, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8552, Japan. E-mail: michinobu.t.aa@m.titech.ac.jp; Fax: +81-3-5734-3774; Tel: +81-3-5734-3774
First published on 1st June 2016
Abstract
The past five years have witnessed significant achievements in the field of flexible, stretchable, and printable organic electronics, especially polymer-based organic photovoltaics (OPVs) and organic field-effect transistors (OFETs), which have become competitive to their inorganic counterparts. One of the main driving forces attributed to this remarkable progress is the rapid development of semiconducting polymeric materials. Therefore, the design and synthesis of new building blocks for efficient polymer semiconductors have attracted increasing attention from both the academic and industrial communities. This review attempts to critically summarize the recent advances with respect to the electron-deficient building blocks based on benzothiadiazole and its π-extended, heteroannulated derivatives, which have been mostly developed over the past five years for constructing π-conjugated polymers, particularly donor–acceptor (D–A) polymers. Semiconducting polymers containing these building blocks have demonstrated interesting properties and promising performances as active layers in OPVs and OFETs. The structural implications related to the performances of organic electronic devices are discussed.
 Yang Wang | Yang Wang was born in Zhejiang Province, People’s Republic of China. He obtained his MS degree from the University of Chinese Academy of Sciences in 2013. Currently, he is a PhD candidate at the Tokyo Institute of Technology under the supervision of Prof. Tsuyoshi Michinobu. His doctoral research is focusing on the design and synthesis of conjugated polymers for organic thin film transistor and organic thermoelectric applications. His research interests also include the synthesis of small molecules, organic/inorganic hybrid materials, and related optoelectronic devices. |
 Tsuyoshi Michinobu | Tsuyoshi Michinobu was born in Hiroshima, Japan (1976). He received his PhD degree in chemistry from Waseda University, Japan (2003). After working as a postdoctoral fellow at ETH Zurich and National Institute for Materials Science (2003–2006), he started his academic career at the Tokyo University of Agriculture and Technology as Research Associate in 2006. He then moved to the Tokyo Institute of Technology as Assistant Professor in 2008 and was promoted to Associate Professor in 2012. |
1. Introduction
Heralded as the “Third Generation” semiconducting polymers, donor–acceptor (D–A) polymers are worthy of the reputation of “synthetic metals”.1 Typical D–A polymers are π-conjugated semiconducting polymers with alternating π-electron-rich (donor)2 and π-electron-deficient (acceptor)3 building blocks arrayed along the polymer backbones. Such polymers are usually synthesized by the palladium-catalyzed polycondensation between bifunctional donor and acceptor monomers.4 Therefore, the development of novel donor and acceptor building blocks has become one of the main driving forces to spur the advancement of semiconducting polymers.5 By utilizing D–A polymers as the active layers, organic photovoltaics (OPVs) with power conversion efficiencies (PCEs) beyond 10%6 as well as organic field-effect transistors (OFETs) with mobilities above 10 cm2 V−1 s−1 have been achieved.7D–A polymers could be traced back to the 1980s when Wudl and his co-workers developed the first narrow bandgap polymer, poly(isathianapthene).8 It is postulated that the modulation of the bandgap can be achieved when the polymer backbone consists of an alternating sequence of aromatic and quinoid units.9 Another related concept is the combination of electron-releasing (push) and electron-withdrawing (pull) units to modify the bandgap of the polymer, which was initially proposed by Yamamoto et al.10 and effectively demonstrated by Havinga et al.11 However, many recent research studies have substantiated that the above two concepts are intimately connected, and that the design of D–A polymers probably requires a combination of both concepts.12 Besides the designed conceptual development previously mentioned, techniques for synthesizing D–A polymers have also undergone significant advancements.13 Modern D–A polymers are synthesized by palladium-catalyzed cross coupling polycondensations, such as the Sonogashira,14 Heck,15 Suzuki-Miyaura,16 and Stille coupling polycondensations,17 catalyst-transfer condensation polymerization,18 and the state-of-the-art direct arylation polycondensation (DArP).19
With the development of high performance D–A polymers, electron-rich donors have been extensively studied and fully reported in many reviews,20 including specific references reporting the correlations between chemical structures and device performances.21 However, much less attention has been focused on the acceptor structures, which affect the material properties and hence the device performances.22 Among the few acceptors, benzo[c][2,1,3]thiadiazole (BT, Chart 1) is one of the most widely studied units.23 However, based on the development of new synthetic protocols,24 novel π-extended and heteroannulated benzothiadiazole derivatives have recently emerged, and a new series of D–A polymers based on these new acceptor units were synthesized and used in organic electronics.25 Therefore, it is an appropriate time to summarize BT and its π-extended, heteroannulated derivatives, which are used to produce high performance D–A polymers. Based on the complexity of their chemical structures, we classified them into four categories. The first category is BT and its heteroatom substituted derivatives, such as fluorinated benzothiadiazole (FBT and 2FBT, Chart 1). Second, the BT ring is extended to the triple fused ring structures, such as benzobisthiadiazole (BBT, Chart 1) and its heteroatom substituted analogues. Third, BT is further extended to the tetracyclic structures, such as naphthobisthiadiazole (NTz, Chart 1). Finally, other large π-extended benzothiadiazole derivatives with much more complicated structures are described. We note that the research on designing and synthesizing new acceptors for D–A polymers has made a significant contribution to the rapid advances in OPVs and OFETs26 and will make further contributions to other emerging devices, such as ternary blend OPVs,27 fullerene-free OPVs,28 and n-type organic thermoelectronics.29 We hope to encourage both academia and industry to direct much more attention towards this exciting and fruitful research field.
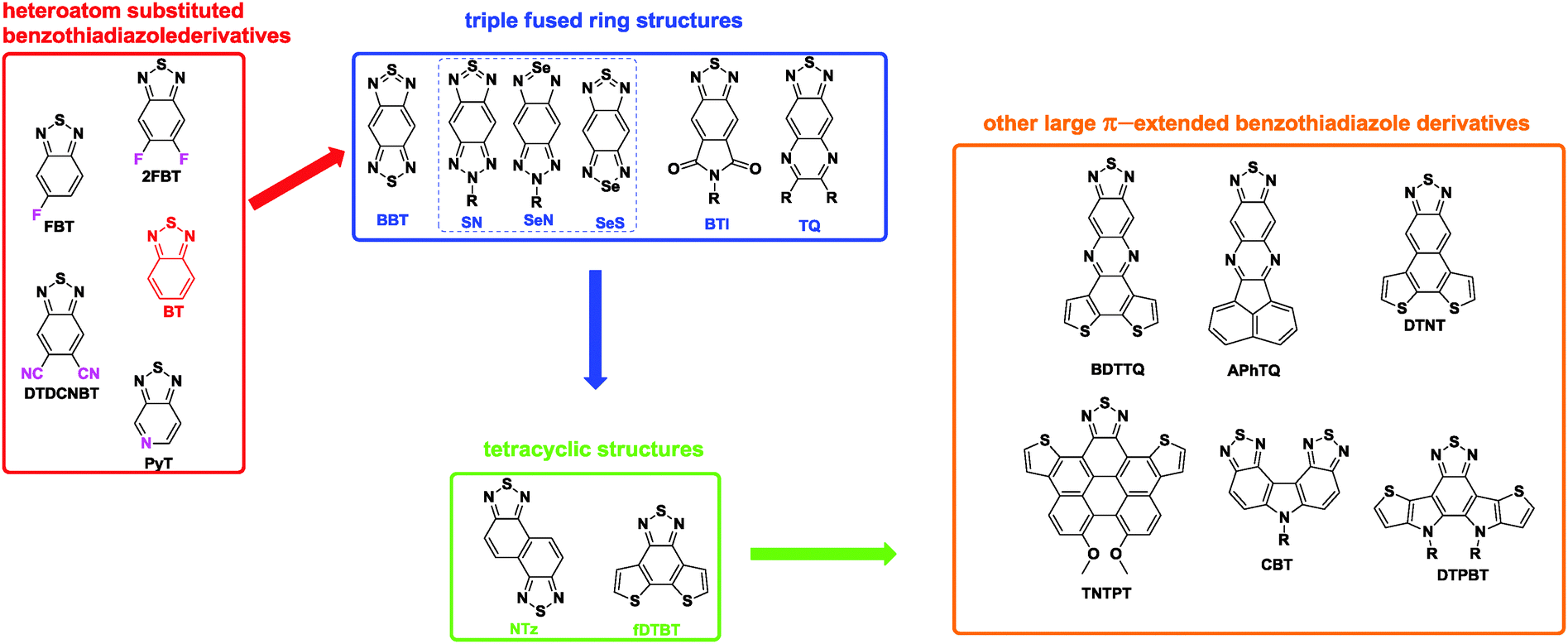 | ||
| Chart 1 The chemical structures of benzothiadiazole and its π-extended, heteroannulated derivatives. | ||
2. Benzo[c][2,1,3]thiadiazole and its derivatives
Brominated benzo[c][2,1,3]thiadiazole was first synthesized by Pilgram et al. in the 1970s.30 However, the first alternating donor–acceptor (D–A) copolymer (BT-Py, Chart 2) based on BT was synthesized by Meijer et al. in 1996.31 Since then, the BT unit has been frequently used to construct high performance D–A copolymers for organic electronics, such as OFETs and OPVs.32 For example, poly(N-9′-heptadecanyl)-2,7-carbazole-alt-5,5-(4′,7′-di-2-thienyl-2′,1′,3′-benzothiadiazole) (PCDTBT, Chart 2) has been developed for high performance BHJ solar cells with a high open-circuit voltage (Voc ∼0.90 V) and a power conversion efficiency (PCE) of 6.1%.33 So far, PCDTBT, surpassing the PCEs of the devices based on the most studied semiconducting polymer of P3HT, has been regarded as one of the benchmark polymers (such as PTB7) for the development of more efficient BHJ solar cells.34 In the research field of OFETs, BT is also frequently integrated into efficient hole transporting polymeric materials. For example, PIDT-BT with indacenodithiophene (IDT) as the donor and BT as the acceptor unit (Chart 2) has been recognized as one of the first successful BT based D–A copolymers with a mobility exceeding 1 cm2 V−1 s−1.35 Although the thin film of PIDT-BT exhibited low crystallinity with a large π-stacking distance of 4.1 Å and face-on orientation, a high hole mobility up to 1.25 cm2 V−1 s−1 could be achieved in bottom-contact/top-gate (BC/TG) transistors with CYTOP as the gate dielectric.35 Subsequently, Müllen and his co-workers tried to increase the crystallinity by increasing the molecular weight (Mn) of the BT-based polymers. They successfully synthesized the cyclopentadithiophene-alt-BT polymer (CDT-BTZ-C16, Chart 2) with the high Mn of 51 kg mol−1, which showed a good crystalline order and mobilities as high as 3.3 cm2 V−1 s−1.36 Soon afterwards, a much higher mobility of 5.5 cm2 V−1 s−1 based on CDT-BTZ-C16 was realized by the pronounced alignment of the polymer chains in a single fiber using the solvent vapor enhanced drop casting (SVED) method (Fig. 1).37 Very recently, CDT-BTZ-C20 (Chart 2) exceeded the hole mobility of 10 cm2 V−1 s−1 through mechanical compression of polymer chains at the surface of an ionic liquid.38 Interestingly, the charge transport behavior at near room temperature was band-like as the mobility increased with decreasing temperature.38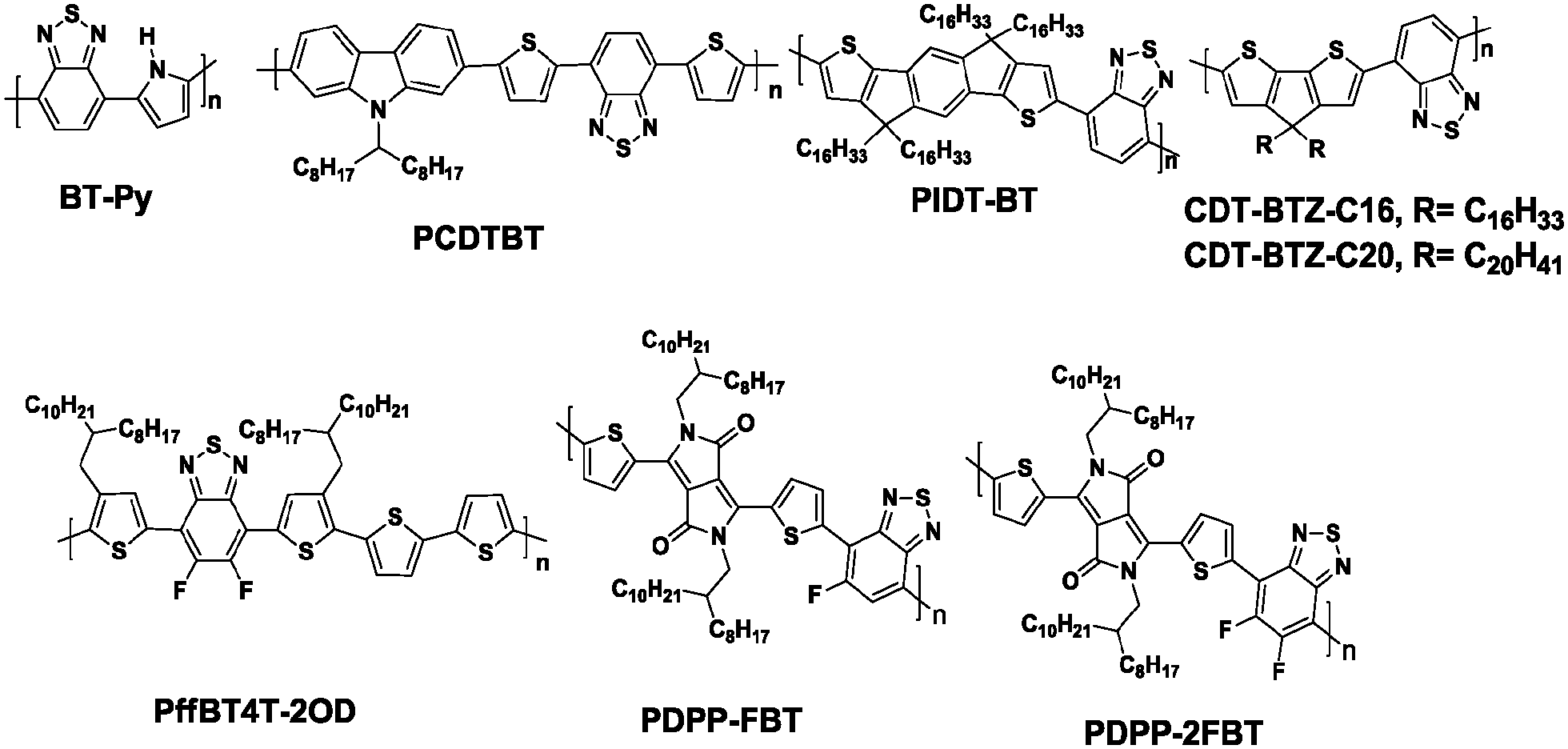 | ||
| Chart 2 Representative benzothiadiazole-based polymers. | ||
 | ||
| Fig. 1 (a) SEM and (b) SAED of CDT-BTz-C16. (c) Schematic illustration of the polymer organization in the SVED fiber. Reprinted with permission from the publisher.37 | ||
Further chemical modification of the BT core produced the fluorinated BT units (either single- or double-fluorinated BT, namely FBT and 2FBT, respectively, Chart 1). Fluorination of the molecules is a simple yet effective method of finely tuning the Frontier energy levels and other physiochemical properties.39 Thus, the fluorinated semiconducting polymers generally show better device performances because of their lower highest occupied molecular orbital (HOMO) levels, more planar backbones, and higher internal dipole moments than their unsubstituted counterparts.40 Over the past five years, the development of FBT- and 2FBT-based polymers has been regarded as one of the most remarkable advances in the field of PSCs and OFETs. For example, poly[(5,6-difluoro-2,1,3-benzothiadiazol-4,7-diyl)-alt-(3,3′′′-di(2-octyldodecyl)-2,2′;5′,2′′;5′′,2′′′-quaterthiophen-5,5′′′-diyl)] PffBT4T-2OD (Chart 2), also known as PCE11, has recently become one the most efficient photovoltaic polymers. PffBT4T-2OD was able to achieve a PCE of 10.8% and enabled six cases of high-efficiency BHJ PSCs (9.6–10.8%) in combination with the traditional PC61BM, PC71BM, and other fullerene derivatives.41 Very recently, PffBT4T-C9C13 with the same polymer backbone as PffBT4T-2OD but different alkyl chains (Fig. 2) displayed the remarkably high PCE of 11.7%.42 The authors attributed the excellent performance of PffBT4T-C9C13 to its significant temperature-dependent aggregation behavior caused by the alkyl side chains. Particularly interesting was that PffBT4T-C9C13 could be processed from a non-halogenated solvent (trimethylbenzene, TMB, Fig. 2a) with non-halogenated additives (phenylnaphthalene, PN, Fig. 2a), which paved the way towards highly efficient and environmentally friendly BHJ solar cells.42
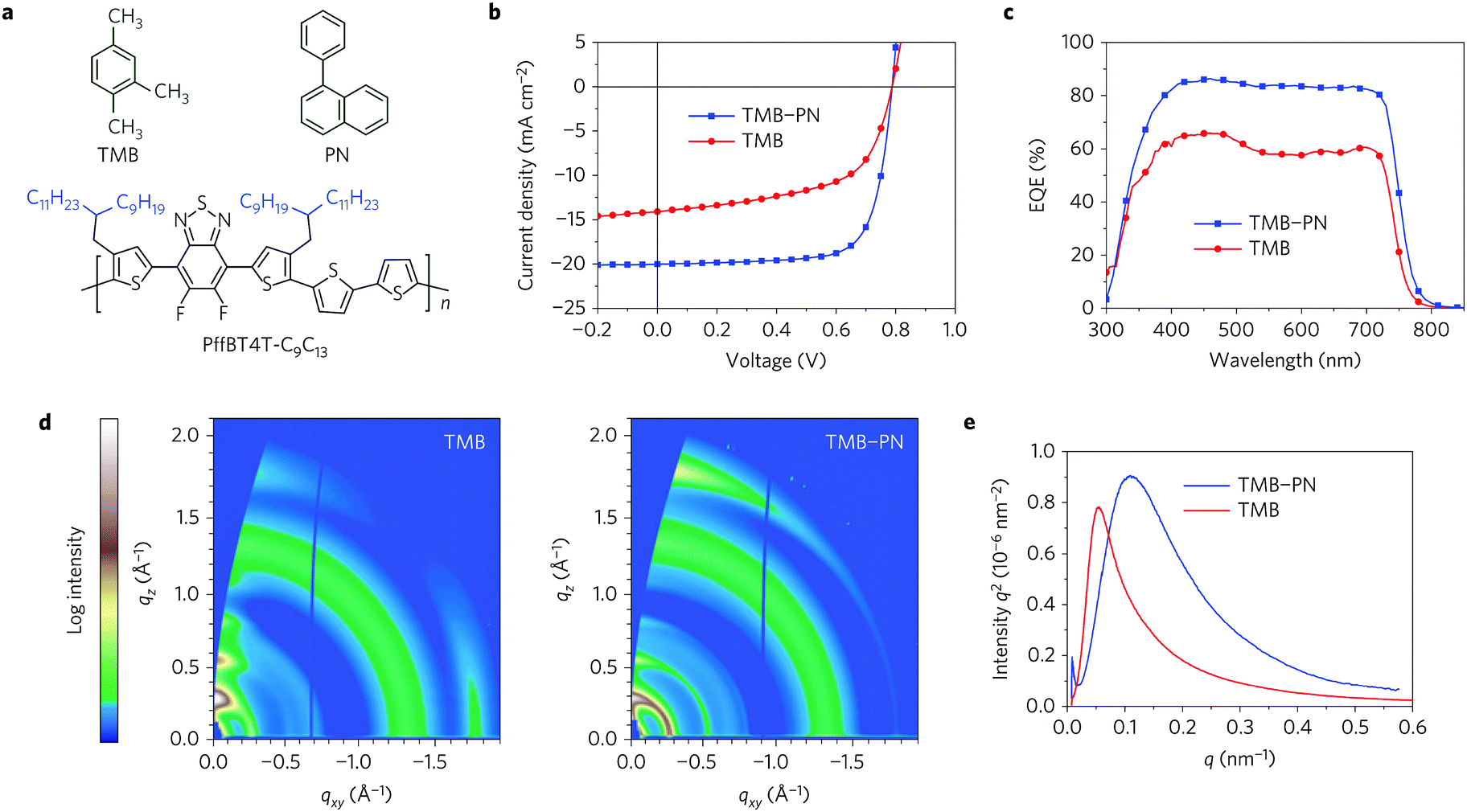 | ||
| Fig. 2 (a) Chemical structures of TMB, PN, and PffBT4T-C9C13, (b) current density (J)–voltage (V) curves, (c) external quantum efficiency (EQE) curves, (d) two-dimensional GIXD images, (e) RSoXS plots of PffBT4T-C9C13:PC71BM-based films processed from TMB or TMB–PN. Reprinted with permission from the publisher.42 | ||
Although dramatic advancements have been achieved in the research field of p-type organic semiconductors,43 n-type materials generally lag in terms of their mobility, chemical stability, and building block availability.44 Therefore, by employing the strong electron-accepting diketopyrrolopyrrole (DPP) and fluorinated-BT units, Yang et al. developed two new polymers (PDPP-FBT and PDPP-2FBT, Chart 2) to investigate the influence of the F atoms on the electron-transport characteristics of the OFETs.45 PDPP-FBT and PDPP-2FBT exhibited the maximum electron mobilities of 0.42 and 0.30 cm2 V−1 s−1, respectively, while they maintained the hole mobilities higher than 0.1 cm2 V−1 s−1. These high mobilities were realized by the cooperative effects of the strong intermolecular noncovalent interactions and highly electron-accepting π-systems, both of which were somewhat induced by the F substituents. This result indicated the high utility of the fluorinated BT building blocks in the semiconducting polymers, particularly when developing high performance ambipolar OFETs.
In order to develop unipolar n-type polymers based on BT units, more electron-deficient substituents should be attached to the BT core to lower the lowest unoccupied molecular orbital (LUMO) levels of the polymers. This is also expected to promote the air-stability and efficient electron injection from common electrodes.46 Therefore, 4,7-di(thiophen-2-yl)-5,6-dicyano-2,1,3-benzothiadiazole (DTDCNBT, Chart 1) was developed by Heeney and his coworkers.47 In order to investigate the potential of this new acceptor unit in semiconducting polymers, alkylated indacenodithiophene (IDT) and dithienogermole (DTG) derivatives were copolymerized with DTDCNBT, producing P(Ge-DTDCNBT) and P(IDT-DTDCNBT), respectively (Table 1). For the sake of a quantitative comparison, they also synthesized the corresponding fluorinated polymers, namely P(Ge-DTDFBT) and P(IDT-DTDFBT) (Table 1).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Polymers | M n (kg mol−1) | PDIa | Stille polymerization (%) | E HOMO (eV) | E LUMO (eV) | Device structure | μ h (cm2 V−1 s−1) | μ e (cm2 V−1 s−1) |
| a Determined by GPC in chlorobenzene using polystyrene standards at 80 °C. b Energy levels were estimated from the onsets of the first oxidation and reduction peaks, while the potentials were determined using ferrocene (Fc) as standard using empirical formulae. | ||||||||
| P(Ge-DTDCNBT) | 20.5 | 2.17 | 63 | −5.14 | −3.77 | TG/BC | N/A | 2.8 × 10−3 |
| P(Ge-DTDFBT) | 40.0 | 2.30 | N/A | −5.07 | N/A | TG/BC | 6.2 × 10−2 | N/A |
| P(IDT-DTDCNBT) | 47.3 | 1.72 | 90 | −5.34 | −3.67 | BG/TC | N/A | 4.9 × 10−4 |
| P(IDT-DTDFBT) | 51.9 | 1.43 | 80 | −5.31 | −3.39 | BG/TC | 6.1 × 10−2 | N/A |
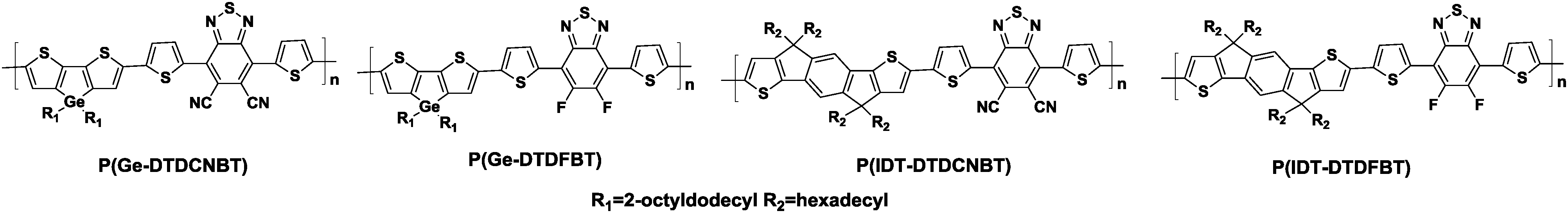
As a consequence, the DTDCNBT unit was found to be a much stronger electron acceptor than the 2FBT unit due to the increase in the electron affinity by around 0.4 eV. The charge mobilities of these polymers were evaluated in OFET devices. It was found that replacing the fluorine substituents with the cyano groups led to the dramatic change in the charge transporting feature from p-type to n-type in the OFET devices as shown in Table 1. It is remarkable that switching the type of charge transport could be achieved through a subtle alternation of the substituents.47
Another chemical modification of the BT unit is replacing its benzene ring with a π-electron-deficient pyridine unit, which forms the thiadiazolo[3,4-c]pyridine unit (PyT, Chart 1). Pyt was found to be a very useful building block to construct regio-regular oligomers and polymers for high performance OPVs and OFETs.48 For example, Perez et al. reported regio-irregular (RI) and regio-regular (RR) donor–acceptor conjugated copolymers based on cyclopenta[2,1-b:3,4-b′]dithiophene (CDT) and PyT (Fig. 3).49 Compared to its RI counterpart, the RR polymer exhibited a 2-order of magnitude higher hole mobility. Based on the grazing incidence X-ray diffraction (GIXD) measurements (Fig. 3), the RI polymer adopted the edge-on orientation throughout the film. However, in the RR film, the crystallites adopted a mixed edge-on and face-on texture in the bulk and a slight preponderance of edge-on at the surface, which eventually afforded better charge transport properties due to 3-D conduction channels.
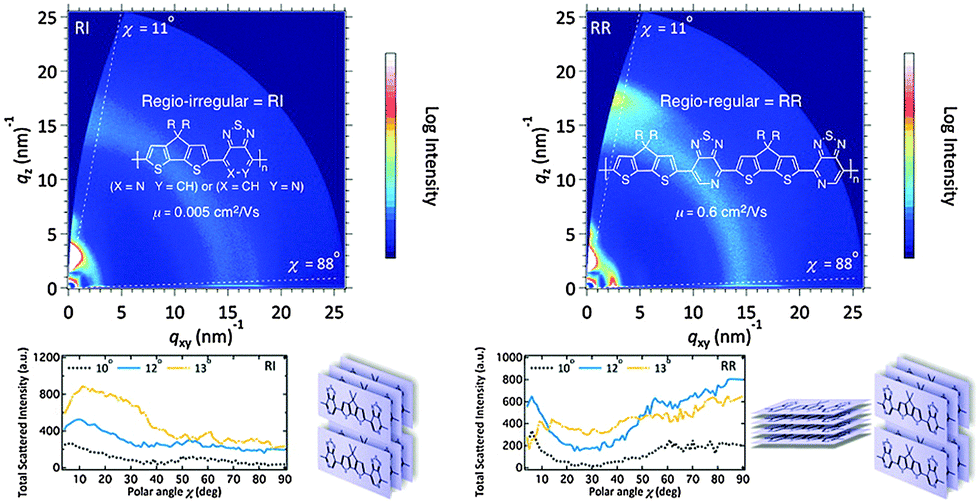 | ||
| Fig. 3 GIXD patterns of regio-irregular (RI) and regio-regular (RR) Pyt-based polymers. Reprinted with permission from the publisher.49 | ||
3. π-Extended benzothiadiazole derivatives with triply fused ring structure
Benzobisthiadiazole (BBT) is one of the most studied π-extended benzothiadiazole derivatives with tricyclic structures. The brominated BBT unit is first synthesized by Yamashita et al. with good planarity, extremely high electron affinity originating from its tetravalent sulfur atom, and strong interactions induced by short intermolecular contacts of the BBT molecules.50 While BBT was originally synthesized via a complicated route starting with the brominated BT unit, a more facile one-pot synthetic methodology was recently developed.51Unlike the state-of-the-art DPP units, the solubilizing side-groups (such as 2-octyldodecyl chain) have to be attached to the flanking thiophene rings in order to synthesize solution-processable semiconducting polymers based on the BBT unit. The solubility problem is the main reason why the applications of BBT-based polymers to PSCs and OFETs were not reported before 2011. Despite the variable mobilities, BBT-based polymers are quite unique amongst the acceptor building blocks discussed in this review due to their strong ambipolarity.52 For example, Wudl et al. developed a family of four new D–A polymers based on the BBT unit, namely, PBBTCD, PBBTPD, PBBTSiD, and PBBTFL (Chart 3) and reported their ambipolar thin film transistor (TFT) performances in 2011. All the polymers in the family showed low-lying LUMO energy levels ranging from −3.9 to −4.2 eV as well as HOMO energy levels around −4.8 eV. Integrating the polymers into the bottom gate/top contact TFTs resulted in balanced ambipolar performances with the highest μh and μe values of 0.13 and 0.10 cm2 V−1 s−1, respectively.53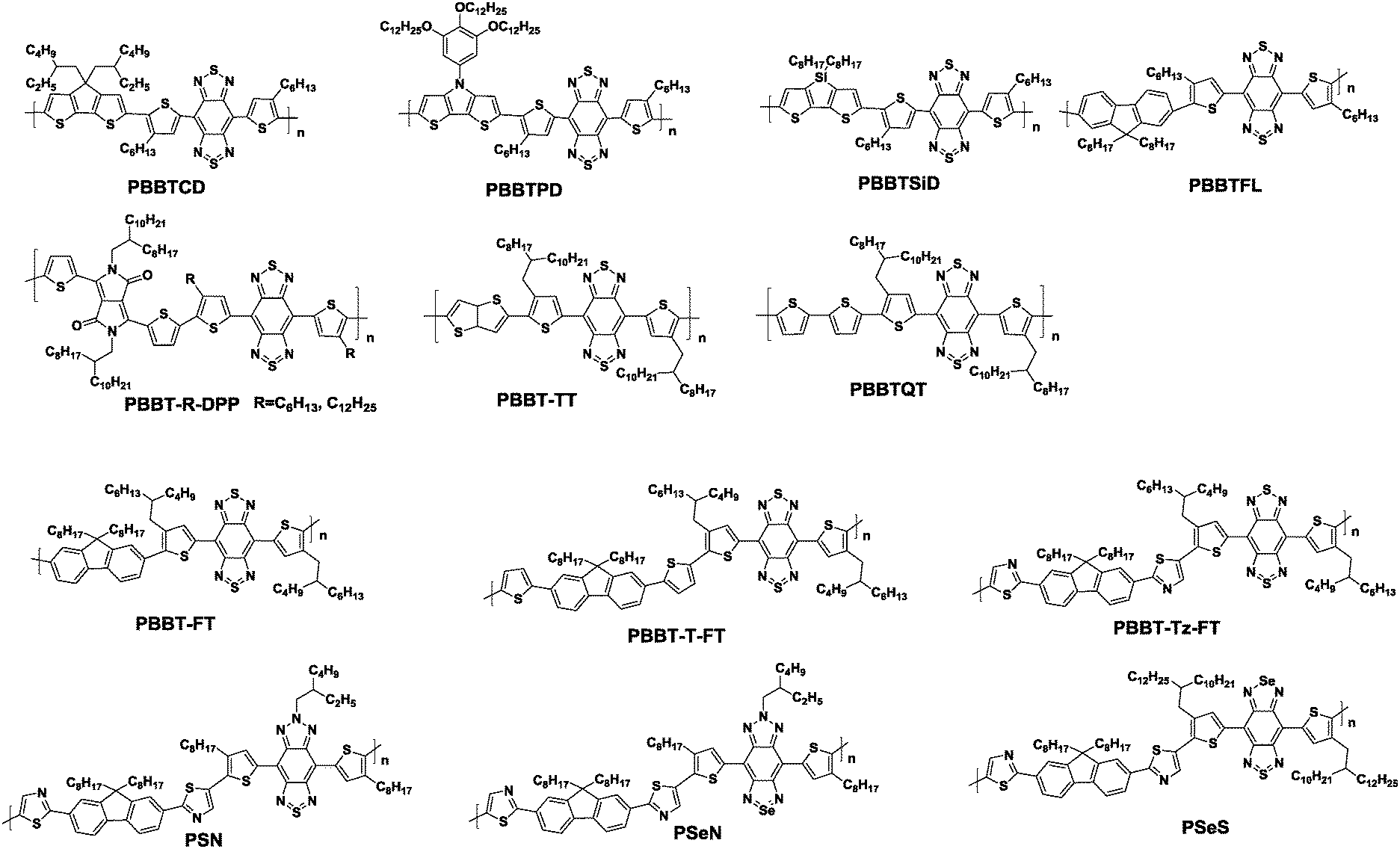 | ||
| Chart 3 Chemical structures of high performance semiconducting polymers based on BBT and its analogues. | ||
Shortly thereafter, the same group designed and synthesized a series of high performance BBT-based copolymers, namely, PBBT-R-DPP, PBBTTT, and PBBTQT (Chart 3). When BBT is copolymerized with the DPP units, the resulting polymer, namely PBBT-R-DPP, showed significant ambipolar charge tansport properties with the electron mobility of 1.32 cm2 V−1 s−1 and the hole mobility of 1.17 cm2 V−1 s−1.54 Also, the copolymer with thienothiophene (TT), PBBT-TT, displayed a high ambipolarity with the hole mobility exceeding 1 cm2 V−1 s−1 and electron mobility reaching 0.7 cm2 V−1 s−1.55 In contrast, when coupled with bithiophene, PBBTQT exhibited typical p-channel TFT characteristics with the high carrier mobility of up to 2.5 cm2 V−1 s−1 due to the shallow LUMO level and a very tight packing pattern with the short π–π stacking distance of 3.5 Å.56
However, there are three remaining problems regarding these high-performance BBT-based polymers. First of all, the high-lying HOMO energy levels (higher than −5.0 eV) limit their long-term air-stability. It should be noted that their excellent TFT performances were most likely achieved by utilizing strict device fabrication conditions under a nitrogen atmosphere. Second, most BBT-based polymers reported so far possessed low crystallinity as demonstrated by their X-ray diffraction patterns.53–56 Therefore, in order to develop more promising BBT-based polymers, one should try to enhance crystallinity via molecular engineering, such as synthesizing a more planar backbone, side-chain engineering, and heteroatom substitution. Third, BBT-based polymers usually exhibit ambipolar charge transport properties with a high off-current and low on/off ratio, thus resticting the range of their applications.53–56 Accordingly, developing unipolar p-type and/or n-type polymers based on the BBT units with a low off-current and high on/off ratio are highly in demand.
Recently, our group has successfully increased the air-stability of the BBT-based semiconducting polymers by inserting the appropriate π-conjugated bridges into the polymer backbone to lower the HOMO energy levels (Fig. 4). We designed and synthesized three new D–A conjugated polymers, namely, PBBT-FT, PBBT-T-FT, and PBBT-Tz-FT, with different π-conjugated bridges (as shown in Chart 3).57 All three polymers showed similar optical properties with a broad absorption from 300 to 1200 nm in the UV-vis-NIR range. However, electrochemical measurements suggested that the HOMO energy levels significantly varied from −5.05 to −5.42 eV. Most importantly, the TFTs demonstrated well-defined differences in their air-stability and ambipolar transport properties. p-Type dominant TFT performances with the μh value reaching 0.13 cm2 V−1 s−1 were observed for PBBT-FT without additional π-conjugated bridges. However, the TFT based on PBBT-T-FT with thiophene bridges showed lower, but more balanced μh and μe values of 6.5 × 10−3 and 1.2 × 10−3 cm2 V−1 s−1, respectively. Moreover, replacement of the thiophene bridges with the electron-accepting thiazole units further enhanced this trend. Thus, the TFT based on PBBT-Tz-FT with the thiazole bridges exhibited a μh value of 6.8 × 10−3 cm2 V−1 s−1 and a significantly enhanced μe value of 1.5 × 10−2 cm2 V−1 s−1. It is worth noting that the PBBT-FT-based devices demonstrated a very good stability under ambient conditions with a μh of 0.1 cm2 V−1 s−1 and a μe of 0.002 cm2 V−1 s−1 even when stored in air for over one month (Fig. 4).
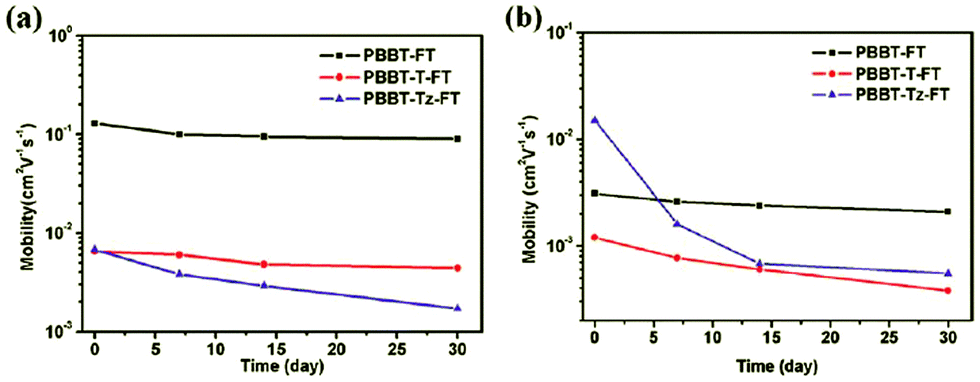 | ||
| Fig. 4 Time-dependent performance changes in (a) μh and (b) μe of the polymer TFTs under ambient conditions. Reprinted with permission from the publisher.57 | ||
Although the air-stability has been achieved for the above-mentioned polymers, unipolar p-type and/or unipolar n-type polymers based on the BBT-units were still elusive. In order to realize p-type and/or n-type unipolar polymers, our next approach was the simple, yet effective, heteroatom substitution on the BBT core.58 In this context, new semiconducting polymers based on thiadiazolobenzotriazole (SN), selenadiazolobenzotriazole (SeN), and selenadiazolobenzothiadiazole (SeS) with different heteroatoms on the BBT core were synthesized (Chart 3). The effects of heteroatom substitution were clearly observed on the energy levels of these polymers, determined from cyclic voltammetry (CV) measurements.58 Accordingly, TFTs based on these polymers showed different charge transport properties. For example, the p-type unipolar performances with the hole mobility as high as 0.65 cm2 V−1 s−1 was achieved for the device based on PSN. In contrast, the device based on PSeS displayed n-type dominant charge transport properties with the electron mobility up to 0.087 cm2 V−1 s−1. Most intruguingly, during the course of the GIXD study, it was found that the polymer orientation significantly changed from the edge-on dominant to face-on dominant packing orientations on a Si substrate by simply substituting the heteroatoms of the BBT core. For example, the substitution of sulfur by the selenium atom led to a face-on orientation texture, while replacing the sulfur atom with the nitrogen atom resulting in a molecular packing ordering with more preference to the edge-on orientation (Fig. 5). In addition, there was a clear correlation between the polymer orientation and TFT performances, indicating that the polymers with greater edge-on orientations tended to show higher mobilities.
 | ||
| Fig. 5 2D-GIXD patterns of the thin films of (a) PSN, (b) PSeN, and (c) PSeS after the treatment of optimized annealing conditions. Reprinted with permission from the publisher.58 | ||
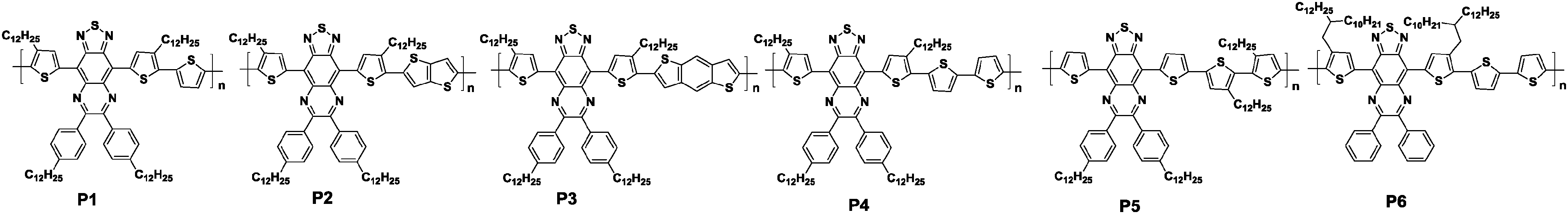
Another class of π-extended benzothiadiazole derivatives with triply fused ring structures is thiadiazoloquinoxalines (TQ, Chart 1). TQ-based oligomers and polymers possess various unique properties, such as high extinction coefficients and intensive broad absorptions reaching the NIR range.59 However, TQ-based polymers usually show low carrier mobilities due to their disordered thin film structures.60 Therefore, Baumgarten and his co-workers systematically studied the correlation between the structure and charge transport properties of the TQ-based polymers in order to identify the essential parameters for achieving highly-ordered microstructures (Table 2).61 First, different donors without any alkyl chains, namely, thiophene, thienothiophene, benzodithiophene, and bithiophene, were copolymerized with TQ, producing the corresponding polymers P1, P2, P3, and P4 (see Table 2). These donor units were expected to decrease the torsion angles along the polymer backbone, increasing the backbone coplanarity of the copolymers and potentially producing ordered structures in the thin film states. The two-dimensional wide-angle X-ray scattering measurements revealed that P4 had a higher crystallinity because its coherence length (5.8 nm) was 2 to 3 times greater than those of the other three polymers (1.7–2.9 nm). This then led to the relatively high field-effect hole mobility of 0.1 cm2 V−1 s−1 among the four polymers (Table 2). Second, by varying the substitution positions and density of the alkyl chains, P5 and P6 were synthesized to improve the solubility. However, P5 showed a lower hole mobility of merely 0.007 cm2 V−1 s−1 due to the sacrifice for the coplanarity of the polymer backbone (Table 2). Taking this result into account, P6 with a pair of 2-decyl-tetradecyl alkyl chains attached to the flanking thiophene units was designed in reference to P4, and this polymer demonstrated the highest hole mobility of up to 0.24 cm2 V−1 s−1 among the six polymers (Table 2).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Polymers | M n (kg mol−1) | PDIa | Stille polymerization (%) |
E
optg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b (eV) b (eV) |
IPc (eV) | EAc (eV) | μ h,max,avg (cm2 V−1 s−1) | I on/off |
| a Determined by GPC in 1,2,4-trichlorobenzene using polystyrene standards at 135 °C. b Drop-cast from toluene solution (2 mg mL−1), and Eopt = 1240 nm/λonset. c IP and EA values were estimated from the onsets of the first oxidation and reduction peaks, while the potentials were determined using ferrocene (Fc) as standard using empirical formulae IP/EA = −(EOx/Red onset − EFc/Fc+(1/2) + 4.8) eV, wherein EFc/Fc+(1/2) = 0.40 eV. | ||||||||
| P1 | 7.0 | 2.68 | 81 | 0.75 | −4.95 | −3.92 | 0.063, 0.046 | 10 |
| P2 | 5.4 | 2.15 | 67 | 0.81 | −5.04 | −3.99 | 0.081, 0.066 | 50–100 |
| P3 | 8.0 | 2.34 | 86 | 0.96 | −5.28 | −3.93 | 0.042, 0.033 | 100–150 |
| P4 | 9.1 | 2.33 | 57 | 0.94 | −5.19 | −3.94 | 0.10, 0.097 | 100–150 |
| P5 | 12.7 | 4.05 | 85 | 0.91 | −5.21 | −3.98 | 0.012, 0.007 | 100–150 |
| P6 | 18.8 | 3.48 | 71 | 0.95 | −5.23 | −3.99 | 0.24, 0.19 | ∼104 |
In addition, the TQ-based polymers can also be applied to electrochromic devices62 and polymer solar cells.63 For example, Andersson and his coworkers reported two electrochromic polymers based on the TQ unit (P1 and P2, Fig. 6). Even without doping, these polymers showed a λmax farthest into the NIR at 1410 (for P1) and 1476 nm (for P2) with the very low band gaps of <0.7 eV estimated from the onset wavelengths (Fig. 6). Most interestingly, these polymers exhibited a well-defined bleaching of their neutral absorptions in the NIR region and had an electrochromic contrast of up to 30% at the switching speed of 3 s.62
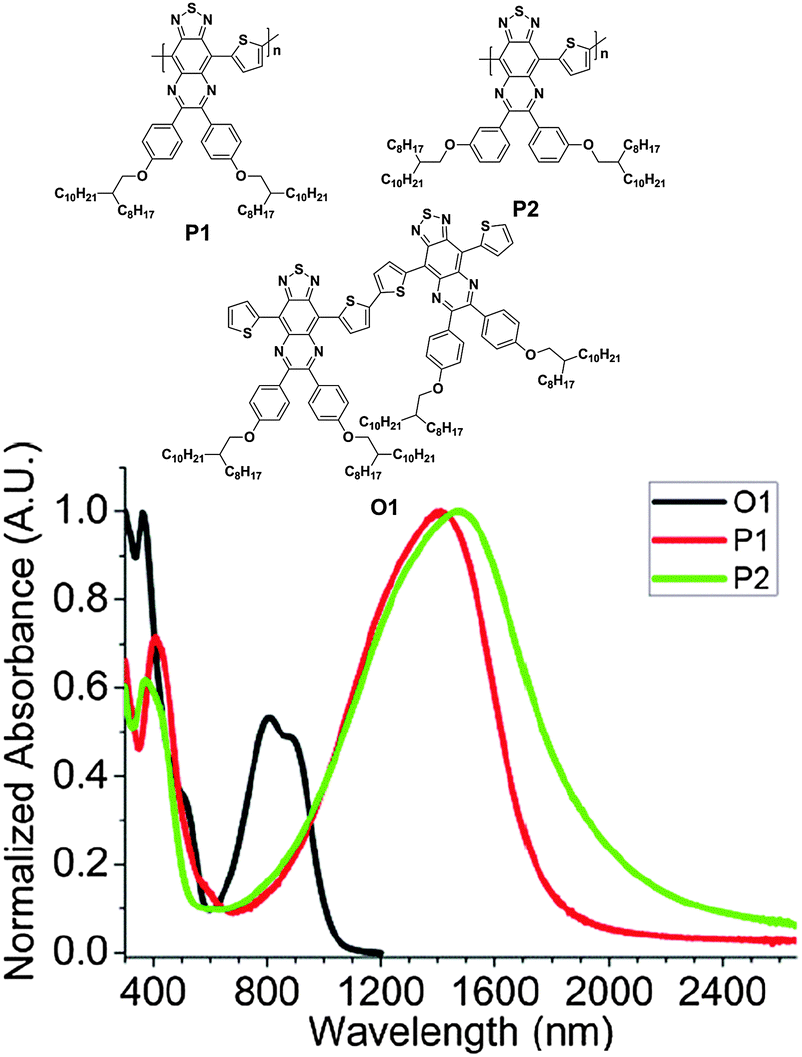 | ||
| Fig. 6 Chemical structures and UV-vis-NIR absorption spectra of thin films of O1, P1, and P2. Reprinted with permission from the publisher.62 | ||
Besides the TQ units, another important class of BT derivatives with triply fused ring structures is the 2,1,3-benzothiadiazole-5,6-dicarboxylic imide (BTI, Chart 1) derivatives, which are promising acceptor units for high-performance photovoltaic polymers. The BTI unit possessed a significantly lower LUMO level as compared to the fluorinated BT unit, indicating that the cyclic imide attached to the BT unit served as an effective electron-accepting substituent. The concurrent lowering of the HOMO level resulted in an increase in the Voc as compared to the conventional photoactive polymers incorporating the fluorinated BT unit. In addition, various alkyl chains could be introduced into the dicarboxylic imide building block for tuning the polymer solubility. Therefore, high-performance photovoltaic polymers based on the BTI unit could be developed.64,65 For example, two D–A copolymers using BTI as the acceptor unit, namely PDI-BDTT and PDI-BDTO, were synthesized and applied to the PSCs (Table 3).64 PDI-BDTT showed a pronounced bathochromically shifted absorption with a broad range from 300 to 800 nm. The electrochemical measurements suggested that the HOMO level of PDI-BDTT was as deep as −5.51 eV, which is beneficial for achieving a high Voc. Therefore, PSCs based on PDI-BDTT displayed PCEs as high as 5.19% with a high Voc of 0.91 V (Table 3). BBTI-1 and BBTI-2 reported by McCulloch et al. are more promising BTI-based polymers.65 BBTI-1 could generate an excellent blend morphology without the addition of solvent additives or thermal and/or solvent-annealing steps during the device fabrication. As a consequence, the blend films of BBTI-1 with PC71BM could be simply cast from a single solvent system to reproducibly afford PSCs with the PCEs of greater than 8% (Table 3).
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Polymers | M n (kg mol−1) | PDIa | Stille polymerization (%) | E HOMO (eV) | E LUMO (eV) | DIOd (v%) | J sc (mA cm−2) | V oc (V) | FF (%) | PCEe (%) |
| a The number-average molecular weights measured by GPC using polystyrene as standard, for PDI-BDTT and PDI-BDTO using THF as eluent; for BDTI-1 and BDTI-2 using chlorobenzene at 80 °C. b Fractionation by means of preparative size exclusion chromatography in chlorobenzene at 80 °C. c From the onset of CV in thin film. d DIO is short for 1,8-diiodooctane. e For PDI-BDTT and PDI-BDTO, the solar cells were fabricated with a conventional device configuration using PC71BM as the electron acceptor materials; for BDTI-1 and BDTI-2, the solar cells were fabricated with an inverted OPV device architecture using PC71BM as the electron acceptor materials. | ||||||||||
| PDI-BDTT | 29 | 4.8 | 58 | −5.51 | −3.76 | 2.5 | 11.09 | 0.91 | 49.2 | 5.19 |
| PDI-BDTO | 44 | 3.2 | 86 | −5.44 | −3.74 | 2.5 | 5.53 | 0.80 | 47.1 | 2.10 |
| BDTI-1 | 64 | 1.99 | 41b | −5.2 | N/A | 0 | 16.45 | 0.80 | 63 | 8.3 |
| BDTI-2 | 75 | 2.11 | 34b | −5.3 | N/A | 3 | 11.00 | 0.81 | 67 | 6.0 |
4. π-Extended benzothiadiazole derivatives with tetracyclic structures
Naphtho[1,2-c:5,6-c′]bis[2,1,3]thiadiazole (NTz), which can be regarded as a doubly BT-fused ring, is thought to be one of the most widely studied benzothiadiazole derivatives with tetracyclic structures. Thanks to its high planarity and high electron deficiency,66 NTz is a fascinating acceptor unit for high-performance D–A polymers used in OPVs67–70 and OFETs.68 Except for a few reports from other groups,70 Takimiya et al. have intensively investigated the NTz-based semiconducting polymers. For example, when NTz was copolymerized with naphtho[1,2-b:5,6-b′]dithiophene (NDT3), the resulting copolymer, PNDT3NTz-DT (Table 4), displayed very close π–π stacking distances of 3.5 Å in the thin film state. This remarkable close π stacking distance would originate from the extremely planar polymer backbone (π-extended four-ring-fused heteroarenes both as the donor and acceptor units).68 Due to the suitable energy levels, large absorption range, high crystallinity, and favorable molecular orientations, PNDT3NTz-DT was found to be one of the most versatile polymers that exhibit both the high carrier mobility of ∼0.5 cm2 V−1 s−1 in OFETs and the high PCE of 4.9% in OPVs.68 It was later revealed that by attaching a linear dodecyl (–C12H25) chain at the 5,10-positions of the NDT3, the new polymer, namely PNNT-12HD (Table 4), exhibited a highly-ordered face-on orientation on a substrate, giving rise to the very high PCEs of over 8%.71 As shown in Fig. 7a, the original polymer (PNDT3NTz-DT, also called PNNT-DT) was oriented in an edge-on manner, as the diffractions corresponding to the lamellar structure and the π–π stacking predominantly appeared along the qz and qxy axes, respectively. However, in sharp contrast to this polymer, PNNT-12HD preferentially adopted the face-on orientation as the π–π stacking diffraction appeared along the qz axis (Fig. 7b). After blending with PC61BM, the positions and distances of both lamellar structures and π–π stacking did not change, indicating the preservation of the polymer packing structures. The face-on orientation and the crystalline π–π stacking of PNNT-12HD were consistent with the low in-plane mobility but, on the other hand, the high out-of-plane mobility compared to PNDT3NTz-DT, which would be the cause of the significantly enhanced FF of the PSCs especially when the thicker active layers were fabricated. In the conventional PSCs, the optimized thickness of the active layers is usually 80–120 nm, because as the thickness increases, the FF significantly decreases. However, in the case of PNNT-12HD with the highly crystalline face-on packing structure, the FF was still as high as 0.64 when the thickness reached 300 nm. Since thick films can harvest more solar photons, a higher Jsc is also expected. Therefore, PSCs based on PNNT-12HD eventually achieved the high PCE of 8.2% at the thickness of 300 nm, while PSCs based on PNDT3NTz-DT showed the maximum PCE of 5.5% at the optimized thickness of 170 nm.|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Polymers | M n (kg mol−1) | PDI | Stille polymerrization (%) | E HOMO (eV) | E LUMO (eV) | J sc (mA cm−2) | V oc (V) | FF (%) | PCE (%) | μ FET (cm2 V−1 s−1) |
| a Devices with the conventional structure: PEDOT/PSS/Polymer:PC61BM/LiF/Al. b The active layer thickness is around 300 nm. c Devices with inverted structures: ITO/ZnO/Polymer:PC61BM/MoOx/Ag. d Devices with inverted structures: ITO/ZnO/Polymer:PC71BM/MoOx/Ag. | ||||||||||
| PNDT3NTz-DTa | 31.0 | 10.0 | 95 | −5.25 | N/A | 11.3 | 0.83 | 52 | 4.9 | 0.54 |
| PNNT-12HDa | 36.0 | 2.5 | 85 | −5.22 | N/A | 15.6 | 0.82 | 64 | 8.2 | 0.1 |
| PNNT-12DTa | 46.1 | 3.0 | 99 | −5.22 | N/A | 7.3 | 0.86 | 65 | 4.1 | 0.01 |
| PNTz4Ta | 52.6 | 2.4 | 94 | −5.16 | −3.77 | 12.0 | 0.76 | 69 | 6.3 | 0.56 |
| PNTz4Tb | 52.6 | 2.4 | 94 | −5.16 | −3.77 | 17.7 | 0.74 | 67 | 8.7 | 0.56 |
| PNTz4Tc | 52.6 | 2.4 | 94 | −5.16 | −3.77 | 18.2 | 0.73 | 74 | 9.8 | 0.56 |
| PNTz4Td | 52.6 | 2.4 | 94 | −5.16 | −3.77 | 19.4 | 0.71 | 73 | 10.1 | 0.56 |
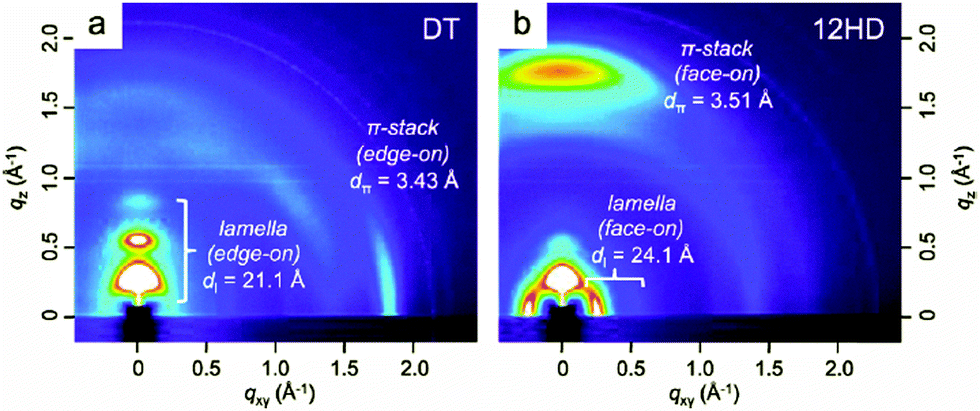 | ||
| Fig. 7 2D-GIXD images of as-spun thin films of (a) PNDT3NTz-DT (also called PNNT-DT) and (b) PNNT-12HD. Reprinted with permission from the publisher.71 | ||
In order to acquire higher PCEs, a new D–A polymer with quaterthiophene and NTz was developed (PNTz4T, Table 4).72 Thanks to its broad absorption in the range of 300–800 nm and a relatively deep HOMO level of −5.16 eV, PNTz4T showed the high PCE of 6.3% in single-junction PSCs blended with PC61BM. Another intriguing feature of PNTz4T was that it forms a crystalline structure with a lamellar motif and with a π–π stacking distance of ∼3.5 Å in the thin film. This π–π stacking distance is remarkably short as compared to that of typical high-performance D–A polymers employed in PSCs.73 In addition, PNTz4T formed a face-on orientation in the BHJ blend film, which was favorable for PSCs, but formed an edge-on orientation in the polymer-only film which was suitable for OFETs (Fig. 8). Recently, PCE values reaching 10% were reported for the single-junction PSCs with an inverted architecture using PNTz4T as the p-type material and PC71BM as the n-type material.74 The high efficiency was attributed to the highly crystalline structure with the short π–π stacking distance and favorable face-on orientation. Furthermore, it was found that the polymer crystallites with the face-on orientation were present in greater abundance on the ZnO surface than on the PEDOT:PSS surface. Also, the face-on orientation was enriched in the bulk and at the top contact, while the edge-on orientation was enriched at the bottom contact (Fig. 9a–d). These unique characteristics of the backbone orientation facilitated the charge transport and effectively reduced the charge recombination, particularly in the inverted device architecture (Fig. 9e and f), resulting in higher Jsc and FF values. As a consequence, the PNTz4T/PC61BM-based inverted device achieved the PCE of 9.8%, whereas the conventional device finally reached the efficiency of 8.7% (Table 4).
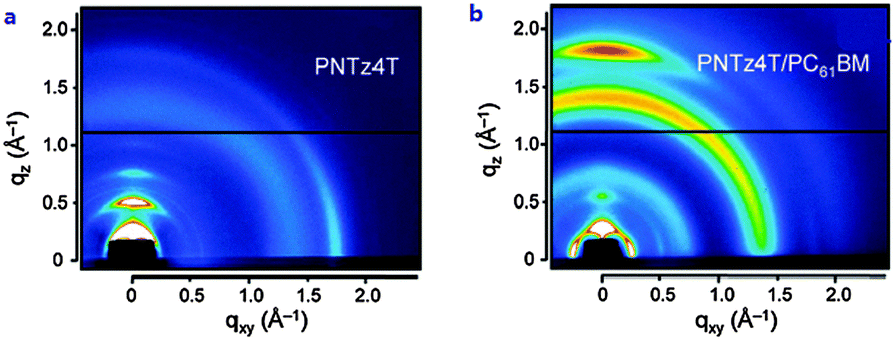 | ||
| Fig. 8 2D-GIXD images of (a) PNTz4T-only film and (b) PNTz4T/PC61BM blend film. Reprinted with permission from the publisher.72 | ||
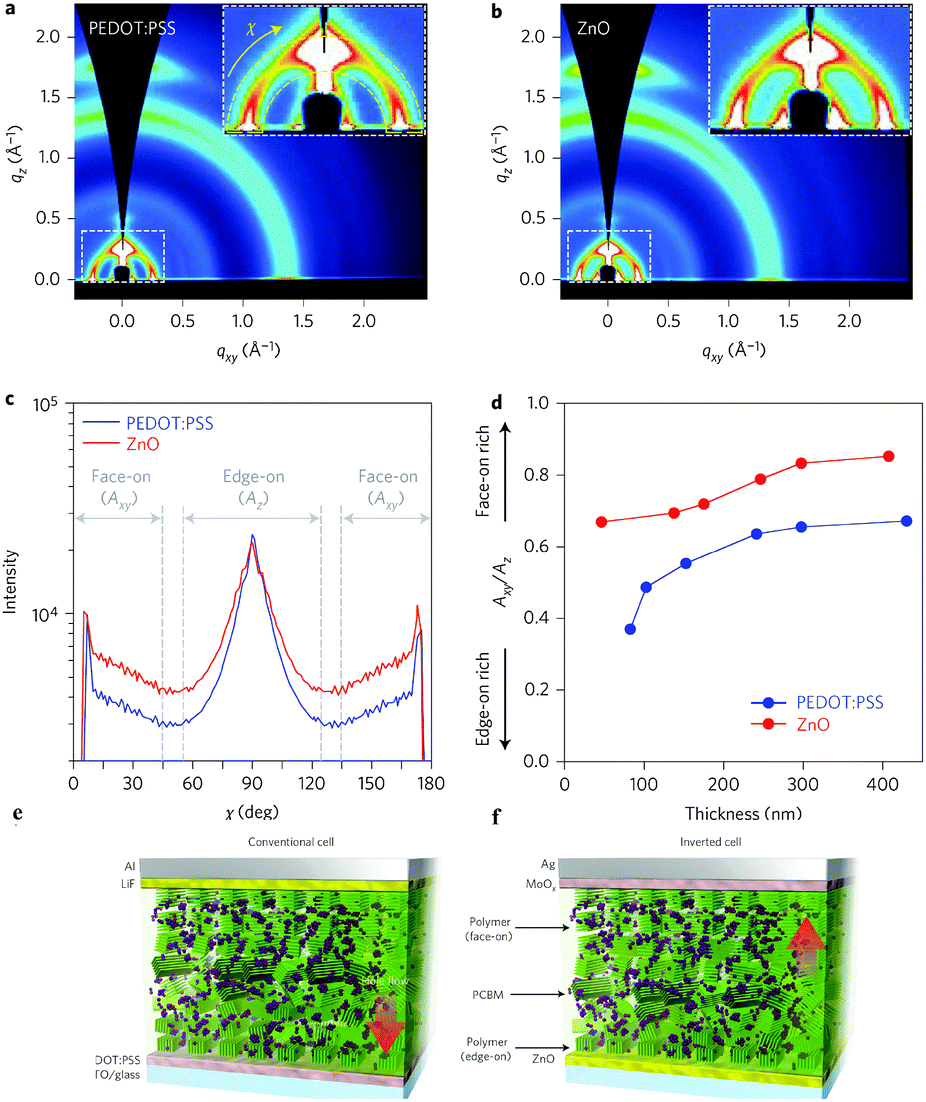 | ||
| Fig. 9 (a) 2D GIXD image of the blended film on the ITO/PEDOT:PSS substrate, (b) 2D GIXD image of the blended film on the ITO/ZnO substrate, (c) pole figures extracted from the lamellar diffraction for the blended films, (d) dependence of Axy/Az on film thickness, where Axy/Az is the ratio of the face-on to edge-on orientation, (e) conventional device structure with PEDOT:PSS as the bottom and LiF as the top interlayer, and (f) inverted device structure with ZnO as the bottom and MoOx as the top interlayer. Reprinted with permission from the publisher.74 | ||
Besides the NTz unit, dithienobenzothiadiazole (fDTBT) is another class of novel benzothiadiazole derivatives with tetracyclic structures (Fig. 10).75 Compared to 4,7-dithienyl-2,1,3-benzothiadazole (DTBT), the chemical structure of fDTBT is based on the direct fusion of the two end thienyl units into the central benzothiadiazole. Therefore, it can suppress the inter-annular rotation, allow a better π-electron delocalization, enhance the intermolecular interactions, and improve the properties of light absorption and charge transportation.76 When it was copolymerized with the benzo[1,2-b:4,5-b′]dithiophene (BDT) unit, a series of D–A copolymers, P(BDTn-fDTBT), with different alkyl chains attached to the BDT unit were produced.77 P(BDT8-fDTBT), P(BDT10-fDTBT), and P(BDT12-fDTBT) with short alkyl side chains did not dissolve in chloroform, chlorobenzene (CB), or o-dichlorobenzene (o-DCB) even when heated. In contrast, P(BDT16-fDTBT) with 2-hexyldecyl side chains was soluble in hot CB and ODCB, and P(BDT20-fDTBT) in chlorinated solvents, such as chloroform, at room temperature. Surprisingly, P(BDT24-fDTBT) has a very good solubility even in hexane. In the PSCs, P(BDT16-fDTBT) showed the highest PCE of 4.5%, obviously outperforming P(BDT20-DTBT) with the PCE of 3.98% (Table 5). Very interestingly, the solubility was in the order of P(BDT16-fDTBT) < P(BDT20-fDTBT) < P(BDT24-fDTBT). However, it was found that the order of the PCEs was opposite: P(BDT16-fDTBT) > P(BDT20-fDTBT) > P(BDT24-fDTBT).77 This trend could be attributed to the bulkiness of the alkyl chains. Generally speaking, on the premise of a sufficient solubility, the smaller the alkyl chain, the better the OPV performance.78
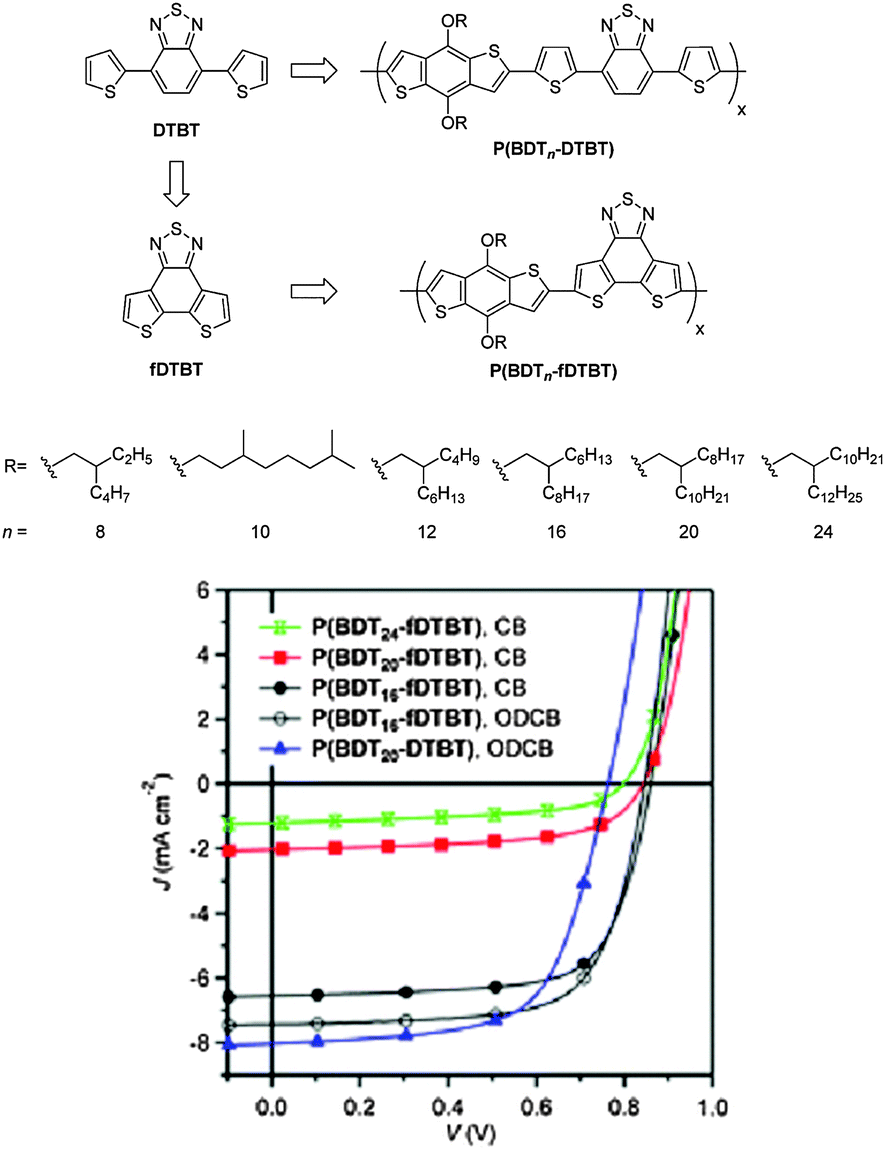 | ||
| Fig. 10 Chemical structures of DTBT and fDTBT as well as the copolymers, P(BDTn-DTBT) and P(BDTn-fDTBT), and their OPV performaces. Reprinted with permission from the publisher.77 | ||
| Polymers | M n (kg mol−1) | PDI | Stille polymerization (%) | E HOMO (eV) | E LUMO (eV) | J sc (mA cm−2) | V oc (V) | FF (%) | PCEe (%) | μ h (cm2 V−1 s−1) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Determined by GPC in 1,3,5-trichlorobenzene at 150 °C using polystyrene standards. b Determined by GPC in chloroform at room temperature using polystyrene standards. c Estimated from the onsets of the first oxidation peaks. d The values in parentheses are the calculated JSC from EQE spectra. e The average values, while those in parentheses are the best value among the measured devices. | ||||||||||
| P(BDT16-fDTBT) | 29.7a | 2.33 | 80–90 | −5.39 | N/A | 7.44 (6.43) | 0.85 | 69.0 | 4.36 (4.50) | 1.09 × 10−4 |
| P(BDT20-fDTBT) | 23.1b | 1.65 | 80–90 | −5.50 | N/A | 2.04 (1.73) | 0.84 | 60.6 | 1.04 (1.07) | 6.85 × 10−5 |
| P(BDT24-fDTBT) | 27.1b | 1.94 | 80–90 | −5.52 | N/A | 1.16 (0.96) | 0.79 | 54.0 | 0.49 (0.56) | 4.74 × 10−5 |
| P(BDT20-DTBT) | 31.7a | 1.84 | 80–90 | −5.26 | N/A | 8.02 (6.85) | 0.76 | 64.4 | 3.93 (3.98) | 1.25 × 10−4 |
Recently, Cao et al. employed the fDTBT unit to synthesize a copolymer with the terthiophene unit, producing a new D–A copolymer, namely DTfBT-Th3 (Fig. 11a).79 Due to strong interchain aggregation, DTfBT-Th3 could not easily dissolve in CB and DCB at room temperature, but this polymer could be processed using hot CB and o-DCB solutions at ∼100 °C. By using the technique of hot spin-coating,80 DTfBT-Th3 displayed an OFET hole mobility as high as 0.55 cm2 V−1 s−1. This is the first report of the OFET mobility for fDTBT-based semiconducting polymers. Moreover, a high PCE of 6.67% was achieved for the PSCs with the active layers deposited from the hot CB solution. These results suggested that fDTBT is a versatile building block to construct high performance semiconducting polymers for high-mobility OFETs and efficient PSCs.81
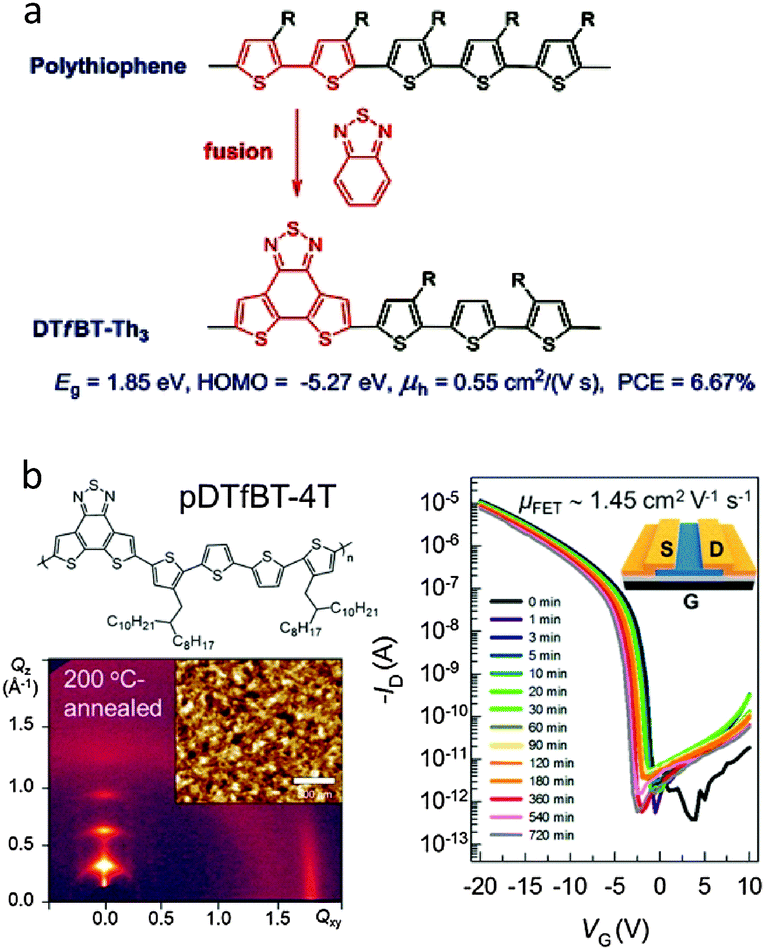 | ||
| Fig. 11 (a) Core extension design of DTfBT-Th3 and (b) polymer structure, thin film morphology, and OFET performances of pDTfBT-4T. Reprinted with permission from the publishers.79,82 | ||
Very recently, Pei et al. synthesized a high-molecular-weight (Mn = 276 kg mol−1) fDTBT-based copolymer, namely pDTfBT-4T, by incorporating a relatively more flexible tetrathiophene (4T) instead of terthiophene (Fig. 11b).82 The polymer, pDTfBT-4T, could be dissolved in warm CB (60 °C), which was spin-cast to form homogeneous thin films as the semiconducting channel layer in the OFETs. After annealing at various temperatures for 10 min, a phase transition behavior from one-dimensional (1D) nanorods to two-dimensional (2D) nanosheets was clearly observed. By utilizing the optimized crystalline structures, the OFETs based on pDTfBT-4T exhibited the excellent hole mobility of 1.45 cm2 V−1 s−1 with negligible hysteresis (Fig. 11b).
5. Other benzothiadiazole derivatives with large π-extended cyclic systems
In order to achieve better π-electron delocalization and stronger intermolecular interactions, some research groups have tried to covalently fasten the BT unit with donor structures (or vice versa) to produce more π-extended fused cyclic entities. For example, two BT rings were fused through a pyrrole ring to afford a new acceptor unit, namely carbazole-bisthiadiazole (CBT, Chart 1).83 Although the PCEs based on the CBT-containing polymer (PCBTBDT, Fig. 12) were not noticeable (2.0%), the authors claimed that developing new organic chromophores is the innovative approach to next-generation D–A copolymers.83 That is to say, they created a new concept based on the D–A integrated structure. When CBT is looked at from another point of view, it can also be regarded as a dithiadiazole-containing carbazole. Thus, the two electron-withdrawing thiadiazole subunits interact with the electron-donating carbazole, leading to the smaller HOMO–LUMO bandgap. Compared to the traditional D–A linkage, the directly fused D–A structures are expected to have more efficient intramolecular charge transfer (ICT) interactions.83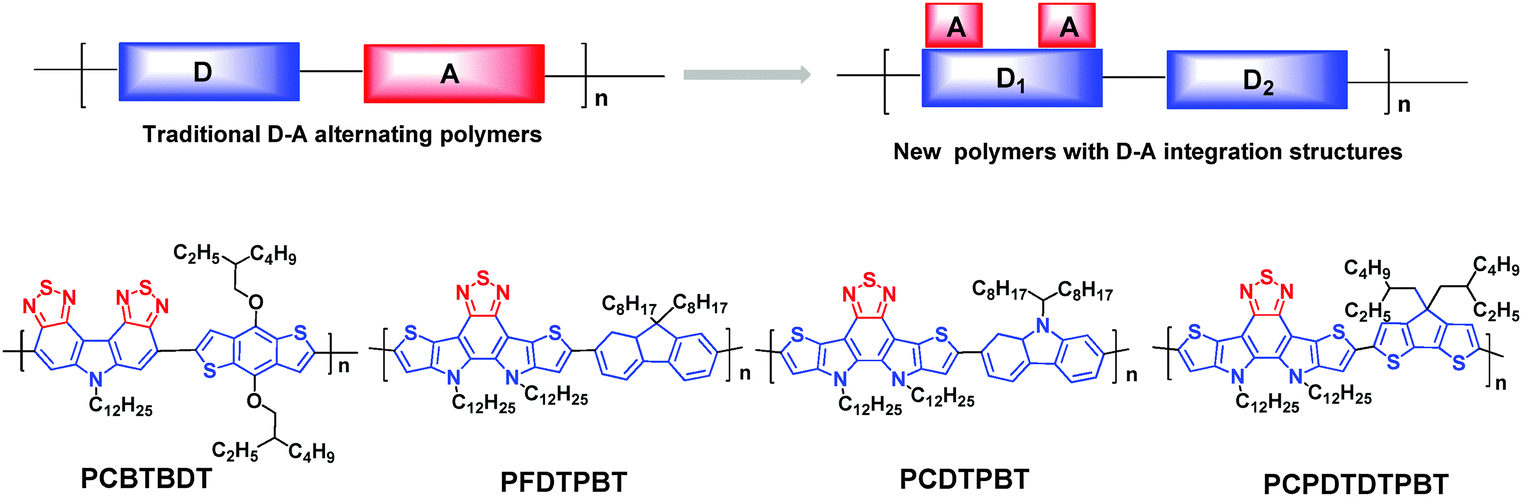 | ||
| Fig. 12 The traditional D–A copolymers and the novel D–A copolymers based on the D–A integrated structure.83,84 | ||
In the meantime, Cheng et al. successfully developed a nitrogen-bridged D–A–D multifused building block, dithienopyrrolo-benzothiadiazole (DTPBT), in which BT was fused with two thieno[3,2-b]pyrrole moieties (Chart 1).84 DTPBT can indeed be classified as the D–A integrated structure, where a thiadiazole unit is integrated into a pentacyclic structure. DTPBT was then copolymerized with the fluorene (F), carbazole (C), and cyclopentadithiophene (CPDT) units via the Suzuki or Stille coupling polycondensations to afford three new copolymers, namely PFDTPBT, PCDTPBT, and PCPDTDTPBT, respectively (Fig. 12). Although the PSC performances were moderate with the PCE of 3.11% for the device based on PFDTPBT, the authors found an enhanced light-absorption ability in the UV region by ICT transitions upon photoexcitation.84
In order to further reduce the bandgap for absorbing more sunlight in OPV applications, a conjugated biphenyl unit was fused with DTBT to afford a perylene-embedded benzothiazolyl unit (namely TNTPT, Fig. 13). This unit showed both higher HOMO and LUMO levels, but a smaller bandgap than DTBT.85 TNTPT was then copolymerized with benzo[1,2-b:4,5-b′]dithiophene (BDTT) to give a new polymer, PTNTPT-BDTT, for PSC applications. Although the device based on PTNTPT-BDTT displayed a moderate PCE of 3.22%, the efficient sun light absorption significantly contributed to this performance. It is noteworthy that TNTPT had a very high molar extinction coefficient of 9.5 × 104 M−1 cm−1, and that of PTNTPT-BDTT (ε = 6.6 × 105 M−1 cm−1) was even higher. The authors claimed that these ε values are some of the highest for OPV donor materials.86
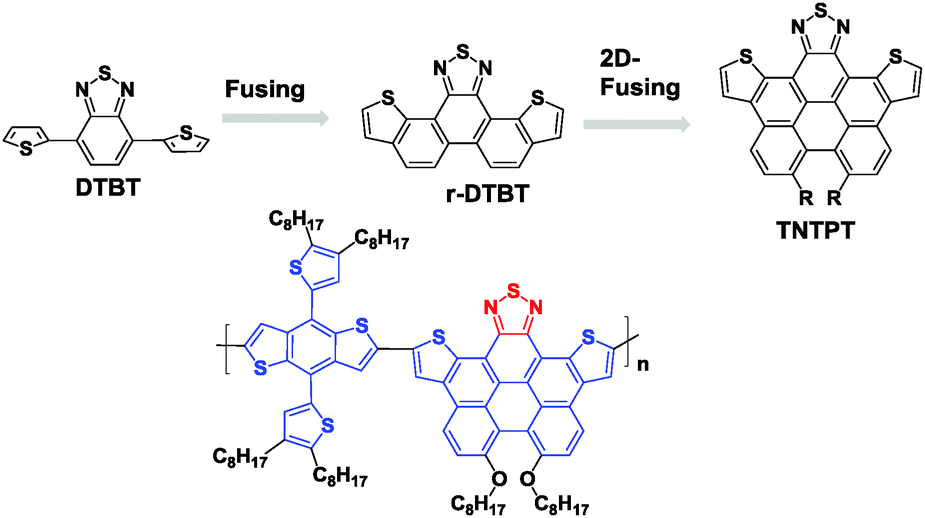 | ||
| Fig. 13 Chemical structures of TNTPT and its copolymers.85 | ||
As discussed in Section 4, dithienobenzothiadiazole (fDTBT) is a versatile unit to construct high performance semiconducting polymers used in both OPVs and OFETs. In order to further broaden the absorption range for a higher Jac87 and obtain a narrower bandgap with a lower HOMO level for a higher Voc,88 Nakanishi et al. designed a T-shaped monomer, dithieno-naphthothiadiazole (DTNT), consisting of two thiophene donors and a thiadiazole acceptor fused through a naphthalene ring (Chart 4).89 Copolymerizing with bithiophene (BTh) and 3,6-bis(2-thienyl)pyrrolo[3,4-c]pyrrole-1,4-dione (DPP) with different alkyl chains yielded four new polymers, namely PDTNTBThEH, PDTNTBThOD, PDTNTDPPEH, and PDTNTDPPOD (Chart 4). Among these polymers, the best PCE of 2.76% was obtained for the device based on PDTNTBThEH/PC71BM. The devices based on PDTNTBThOD and PDTNTDPPOD showed even poorer performances. The authors claimed that one of the main problems of the DTNT-based polymers is the synthetic difficulty in the conversion from DTNT to the boric ester or tin derivatives for the Suzuki or Stille coupling. Thus, C–H activated direct arylation polycondensation was adopted to synthesize these polymers. However, the emerging polymerization technique unfortunately produced low molecular weight polymers (∼5000 g mol−1), which also resulted in limited crystallinity and photovoltaic properties.
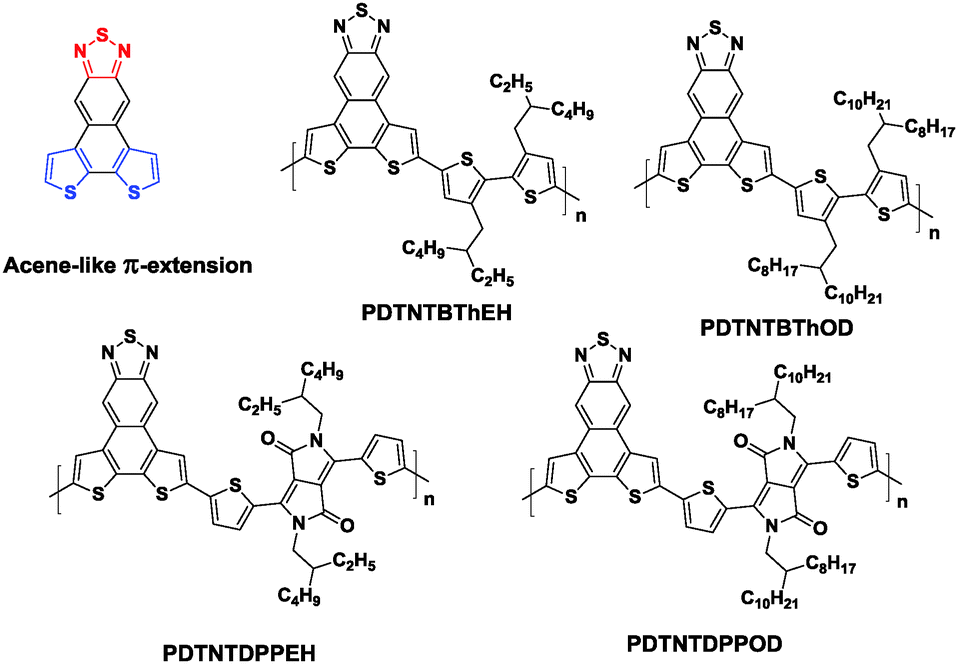 | ||
| Chart 4 Chemicals structures of DTNT and its copolymers.87 | ||
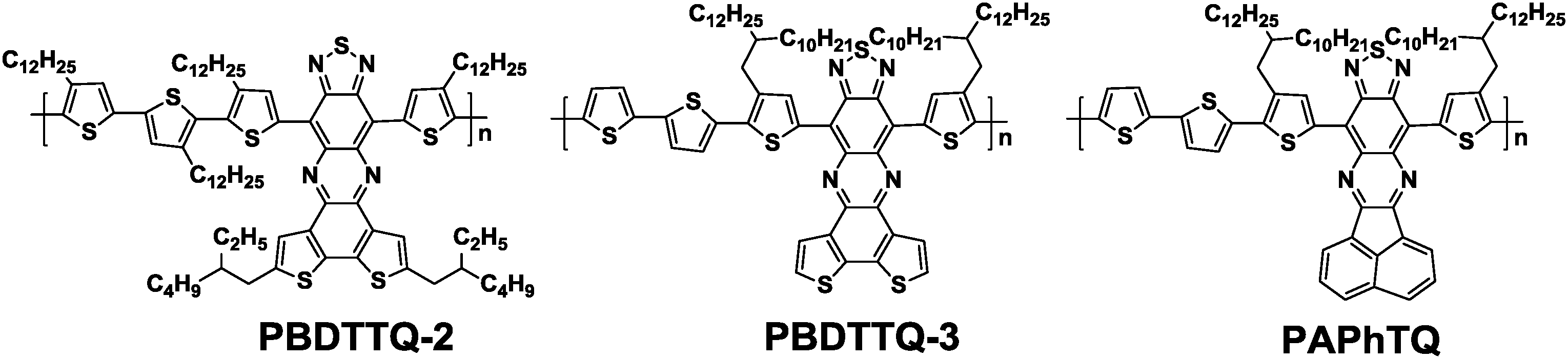
As previously discussed in Section 3, thiadiazoloquinoxaline (TQ) derivatives are much stronger electron acceptors (LUMO: −3.43 to −3.83 eV)90 than the DPP unit (LUMO: −3.13 eV).91 A few copolymers based on TQs showed decent hole transporting properties, but no ambipolar behavior was observed. Therefore, in order to achieve a lower LUMO level and enhance the electron-accepting power of the TQ, extended fused π-frameworks were designed.92 For example, An et al. developed a new acceptor, benzodithiophene-thiadiazoloquinoxaline (BDTTQ), by fusing benzodithiophene with TQ (Chart 1).93 By copolymerizing with bithiophene, the resulting polymer (PBDTTQ-2, Table 6) showed the narrow bandgap of 1.03 eV and a relatively low LUMO level of −4.01 eV while maintaining the deep HOMO level of −5.50 eV. The OFETs based on PBDTTQ-2 showed a balanced ambipolar transport with the hole mobility of 1.2 × 10−3 cm2 V−1 s−1 and the electron mobility of 6.0 × 10−4 cm2 V−1 s−1. The relatively low mobilities were mainly due to the lack of π-stacking ordering induced by too many alkyl chains. Subsequently, on premise of ensuring solubility, An et al. deliberately reduced the number of alkyl chains attached to PBDTTQ-2, thus producing PBDTTQ-3 (Table 6).94 As a consequence, the optimized ambipolar OFET performances with the hole mobility of up to 0.22 cm2 V−1 s−1 and the electron mobility of up to 0.21 cm2 V−1 s−1 resulted. Changing the electron-donating fused aromatic system from benzodithiophene to acenaphthylene furnished a new acceptor, namely APhTQ (Chart 1). The APhTQ-based new polymer, namely PAPhTQ, exhibited a shallower LUMO level of −3.94 eV as compared to that of PBDTTQ-3 (−4.08 eV), whereas the HOMO levels of the two polymers were nearly identical, suggesting the dominant contribution of the tetrathiophene unit to the HOMO of both polymers. Consequently, PAPhTQ was also an ambipolar semiconducting polymer with the slightly lower hole mobility of 0.11 cm2 V−1 s−1 and the electron mobility of 0.02 cm2 V−1 s−1 (Table 6).
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Polymers | M n (kg mol−1) | PDI | Stille polymerization (%) |
E
optgb (eV) |
E HOMO (eV) | E LUMO (eV) | μ h (cm2 V−1 s−1) | μ e (cm2 V−1 s−1) |
| a Determined by GPC in THF using polystyrene standards. b Drop-cast from choloform solution. c HOMO and LUMO levels are estimated from the onsets of the first oxidation and reduction peaks, respectively. | ||||||||
| PBDTTQ-2 | 11.8 | 1.7 | 63 | 1.03 | −5.48 | −4.01 | 0.0006 | 0.0012 |
| PBDTTQ-3 | 76.3 | 3.6 | 76 | 0.76 | −5.05 | −4.08 | 0.22 | 0.21 |
| PAPhTQ | 100.9 | 6.5 | 72 | 0.99 | −5.10 | −3.94 | 0.11 | 0.02 |
6. Conclusion and outlook
The previous sections presented the state-of-the-art benzothiadiazole and its π-extended, heteroannulated derivatives, which were developed for producing the semiconducting polymers for use in OPVs and OFETs. We believe that the rational design of these acceptor building blocks, in addition to the donor units, is one of the key factors for obtaining remarkable advances in the organic electronics field. The benzothiadiazole and its π-extended, heteroannulated derivatives discussed in this review are only a small part of the library among the entire acceptor building blocks. However, we hope that this information would be useful for the readers and spur the exploration and development of new acceptors for semiconducting polymers.Among the presented semiconducting polymers, deep EHOMO levels can be found in BTI- and fDTBT-based polymers. For example, PDI-BDTT (Table 3) possessed a deep EHOMO of −5.51 eV. On the other hand, deep ELUMO levels can be obtained in BBT-based polymers, as represented by the ELUMO of −4.20 eV for PBBTPD (Chart 3). The difference between the EHOMO and ELUMO levels, called an electrochemical band gap, has a correlation with the end absorption of the UV-vis-NIR spectrum. A remarkably narrow band gap of 0.64 eV was observed for one of the TQ-based polymers (P2, Fig. 6). It was explicitly shown that the use of these polymers in OPV and PFET devices is very promising. 2FBT-based polymers usually displayed excellent PCEs in BHJ solar cells. For instance, PffBT4T-C9C13 (Fig. 2) achieved a remarkably high PCE of 11.7% even when spin-coated from non-chlorinated solvents. In OFETs, the highest hole mobility exceeding 10 cm2 V−1 s−1 was reported for CDT-BTZ-C20 (Chart 2), while the highest electron mobility exceeding 1 cm2 V−1 s−1 for the BBT-based polymers (PBBT-R-DPP, Chart 3).
Although the semiconducting polymers based on benzothiadiazole and its derivatives have mainly been used in the OPV and OFET devices up to now, the new applications of these semiconducting polymers will be pursued in the near future. In general, narrow band gap semiconducting polymers have not been fully investigated in the fields of near-infrared (NIR) polymer light-emitting diodes (PLEDs),95 biosensors,96 organic/inorganic hybrid devices,97 and sorting of semiconducting carbon nanotubes.98 It is therefore interesting to examine new polymers for these applications.
Acknowledgements
Financial support from the Support for Tokyotech Advanced Researchers (STAR) is acknowledged. Y. W. is grateful to the Japanese government (MEXT: Monbukagakusho) scholarship.Notes and references
- A. J. Heeger, Chem. Soc. Rev., 2010, 39, 2354 RSC.
- S. D. Oosterhout, N. Kopidakis, Z. R. Owczarczyk, W. A. Braunecker, R. E. Larsen, E. L. Ratcliff and D. C. Olson, J. Mater. Chem. A, 2015, 3, 9777 CAS.
- J. Huang, Y. Ie, M. Karakawa and Y. Aso, J. Mater. Chem. A, 2013, 1, 15000 CAS.
- A. Marrocchi, A. Facchetti, D. Lanari, C. Petrucci and L. Vaccaro, Energy Environ. Sci., 2016, 9, 763 CAS.
- (a) J. Mei, Y. Diao, A. L. Appleton, L. Fang and Z. Bao, J. Am. Chem. Soc., 2013, 135, 6724 CrossRef CAS PubMed; (b) Y. Wang, Y. Liu, S. Chen, R. Peng and Z. Ge, Chem. Mater., 2013, 25, 3196 CrossRef CAS; (c) Y. Wang, F. Yang, Y. Liu, R. Peng, S. Chen and Z. Ge, Macromolecules, 2013, 46, 1368 CrossRef CAS; (d) Q. Liu, Y. Liu, Y. Wang, L. Ai, X. Ouyang, L. Han and Z. Ge, New J. Chem., 2013, 37, 3627 RSC; (e) G. Kim, S.-J. Kang, G. K. Dutta, Y.-K. Han, T. J. Shin, Y.-Y. Noh and C. Yang, J. Am. Chem. Soc., 2014, 136, 9477 CrossRef CAS PubMed; (f) L. Fan, R. Cui, X. Guo, D. Qian, B. Qiu, J. Yuan, Y. Li, W. Huang, J. Yang, W. Liu, X. Xu, L. Li and Y. Zou, J. Mater. Chem. C, 2014, 2, 5651 RSC; (g) Y. Liu, Y. Wang, L. Ai, Z. Liu, X. Ouyang and Z. Ge, Dyes Pigm., 2015, 121, 363 CrossRef CAS; (h) Y. Wang, M. F. G. Klein, J. Hiyoshi, S. Kawauchi, W. W. H. Wong and T. Michinobu, J. Photopolym. Sci. Technol., 2015, 28, 385 CrossRef CAS; (i) Y. M. Yang, W. Chen, L. Dou, W.-H. Chang, H.-S. Duan, B. Bob, G. Li and Y. Yang, Nat. Photonics, 2015, 9, 190 CrossRef CAS; (j) W. Hong, C. Guo, B. Sun and Y. Li, J. Mater. Chem. C, 2015, 3, 4464 RSC.
- (a) J. You, L. Dou, K. Yoshimura, T. Kato, K. Ohya, T. Moriarty, K. Emery, C.-C. Chen, J. Gao, G. Li and Y. Yang, Nat. Commun., 2013, 4, 1446 CrossRef PubMed; (b) Z. He, B. Xiao, F. Liu, H. Wu, Y. Yang, S. Xiao, C. Wang, T. P. Russell and Y. Cao, Nat. Photonics, 2015, 9, 174 CrossRef CAS; (c) H. Hu, K. Jiang, G. Yang, J. Liu, Z. Li, H. Lin, Y. Liu, J. Zhao, J. Zhang, F. Huang, Y. Qu, W. Ma and H. Yan, J. Am. Chem. Soc., 2015, 137, 14149 CrossRef CAS PubMed.
- (a) I. Kang, H.-J. Yun, D. S. Chung, S.-K. Kwon and Y.-H. Kim, J. Am. Chem. Soc., 2013, 135, 14896 CrossRef CAS PubMed; (b) G. Kim, S. J. Kang, G. K. Dutta, Y. K. Han, T. J. Shin, Y. Y. Noh and C. Yang, J. Am. Chem. Soc., 2014, 136, 9477 CrossRef CAS PubMed.
- F. Wudl, M. Kobayashi and A. J. Heeger, J. Org. Chem., 1984, 49, 3382 CrossRef CAS.
- J. Kuerti, P. R. Surjan and M. Kertesz, J. Am. Chem. Soc., 1991, 113, 9865 CrossRef CAS.
- Z. H. Zhou, T. Maruyama, T. Kanbara, T. Ikeda, K. Ichimura, T. Yamamoto and K. Tokuda, J. Chem. Soc., Chem. Commun., 1991, 27, 1210 RSC.
- E. E. Havinga, W. ten Hoeve and H. Wynberg, Synth. Met., 1993, 55, 299 CrossRef CAS.
- G. L. Gibson, T. M. McCormick and D. S. Seferos, J. Am. Chem. Soc., 2011, 134, 539 CrossRef PubMed.
- P. Bujak, I. Kulszewicz-Bajer, M. Zagorska, V. Maurel, I. Wielgus and A. Pron, Chem. Soc. Rev., 2013, 42, 8895 RSC.
- (a) T. Matsumoto, K. Tanaka and Y. Chujo, Macromolecules, 2015, 48, 1343 CrossRef CAS; (b) Y. Feng, C. Dai, J. Lei, H. Ju and Y. Cheng, Anal. Chem., 2016, 88, 845 CrossRef PubMed.
- Y. Lee, Y. Liang and L. Yu, Synlett, 2006, 2879 CAS.
- B. He, A. B. Pun, D. Zherebetskyy, Y. Liu, F. Liu, L. M. Klivansky, A. M. McGough, B. A. Zhang, K. Lo, T. P. Russell, L. Wang and Y. Liu, J. Am. Chem. Soc., 2014, 136, 15093 CrossRef CAS PubMed.
- B. Carsten, F. He, H. J. Son, T. Xu and L. Yu, Chem. Rev., 2011, 111, 1493 CrossRef CAS PubMed.
- (a) T. Yokozawa, Y. Nanashima and Y. Ohta, ACS Macro Lett., 2012, 1, 862 CrossRef CAS; (b) G. C. Schmidt, D. Höft, K. Haase, A. C. Hübler, E. Karpov, R. Tkachov, M. Stamm, A. Kiriy, F. Haidu, D. R. T. Zahn, H. Yan and A. Facchetti, J. Mater. Chem. C, 2014, 2, 5149 RSC; (c) T. Yokozawa and Y. Ohta, Chem. Rev., 2016, 116, 1950 CrossRef CAS PubMed.
- R. Matsidik, H. Komber, A. Luzio, M. Caironi and M. Sommer, J. Am. Chem. Soc., 2015, 137, 6705 CrossRef CAS PubMed.
- (a) I. McCulloch, R. S. Ashraf, L. Biniek, H. Bronstein, C. Combe, J. E. Donaghey, D. I. James, C. B. Nielsen, B. C. Schroeder and W. Zhang, Acc. Chem. Res., 2012, 45, 714 CrossRef CAS PubMed; (b) C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208 CrossRef CAS PubMed; (c) L. Ye, S. Zhang, L. Huo, M. Zhang and J. Hou, Acc. Chem. Res., 2014, 47, 1595 CrossRef CAS PubMed.
- (a) X. Guo, M. Zhang, L. Huo, F. Xu, Y. Wu and J. Hou, J. Mater. Chem., 2012, 22, 21024 RSC; (b) L. Dou, J. Gao, E. Richard, J. You, C.-C. Chen, K. C. Cha, Y. He, G. Li and Y. Yang, J. Am. Chem. Soc., 2012, 134, 10071 CrossRef CAS PubMed; (c) S. Zhang, L. Ye, W. Zhao, D. Liu, H. Yao and J. Hou, Macromolecules, 2014, 47, 4653 CrossRef CAS; (d) S. Shi, X. Xie, C. Gao, K. Shi, S. Chen, G. Yu, L. Guo, X. Li and H. Wang, Macromolecules, 2014, 47, 616 CrossRef CAS; (e) H. Yao, H. Zhang, L. Ye, W. Zhao, S. Zhang and J. Hou, ACS Appl. Mater. Interfaces, 2016, 8, 3575 CrossRef CAS PubMed.
- (a) Y. Zhao, Y. Guo and Y. Liu, Adv. Mater., 2013, 25, 5372 CrossRef CAS PubMed; (b) J. H. Park, E. H. Jung, J. W. Jung and W. H. Jo, Adv. Mater., 2013, 25, 2583 CrossRef CAS PubMed; (c) O. O. Adegoke, I. H. Jung, M. Orr, L. Yu and T. Goodson, J. Am. Chem. Soc., 2015, 137, 5759 CrossRef CAS PubMed.
- (a) C. Edder, P. B. Armstrong, K. B. Prado and J. M. J. Fréchet, Chem. Commun., 2006, 1965 RSC; (b) J. Lu, F. Liang, N. Drolet, J. Ding, Y. Tao and R. Movileanu, Chem. Commun., 2008, 5315 RSC; (c) Z. Mohsan, A. L. Kanibolotsky, A. J. Stewart, A. R. Inigo, L. Dennany and P. J. Skabara, J. Mater. Chem. C, 2015, 3, 1166 RSC.
- T. Mori, T. Nishimura, T. Yamamoto, I. Doi, E. Miyazaki, I. Osaka and K. Takimiya, J. Am. Chem. Soc., 2013, 135, 13900 CrossRef CAS PubMed.
- T. C. Parker, D. G. Patel, K. Moudgil, S. Barlow, C. Risko, J. Brédas, J. R. Reynolds and S. R. Marder, Mater. Horiz., 2015, 2, 22 RSC.
- Y. Lin and X. Zhan, Mater. Horiz., 2014, 1, 470 RSC.
- N. Felekidis, E. Wang and M. Kemerink, Energy Environ. Sci., 2016, 9, 257 CAS.
- W. Ni, M. Li, B. Kan, F. Liu, X. Wan, Q. Zhang, H. Zhang, T. P. Russell and Y. Chen, Chem. Commun., 2016, 52, 465 RSC.
- K. Shi, F. Zhang, C.-A. Di, T.-W. Yan, Y. Zou, X. Zhou, D. Zhu, J.-Y. Wang and J. Pei, J. Am. Chem. Soc., 2015, 137, 6979 CrossRef CAS PubMed.
- K. Pilgram, M. Zupan and R. Skiles, J. Heterocycl. Chem., 1970, 7, 629 CrossRef CAS.
- H. A. M. van Mullekom, J. A. J. M. Venkemans and E. W. Meijer, Chem. Commun., 1996, 2163 RSC.
- (a) J. Chen and Y. Cao, Acc. Chem. Res., 2009, 42, 1709 CrossRef CAS PubMed; (b) P.-L. T. Boudreault, A. Najari and M. Leclerc, Chem. Mater., 2010, 23, 456 CrossRef.
- (a) N. Blouin, A. Michaud and M. Leclerc, Adv. Mater., 2007, 19, 2295 CrossRef CAS; (b) S. Wakim, S. Beaupré, N. Blouin, B. Aich, S. Rodman, R. Gaudiana, Y. Tao and M. Leclerc, J. Mater. Chem., 2009, 19, 5351 RSC; (c) H. Park, A. Roy, S. Beaupré, S. Cho, N. Coates, J. S. Moon, D. Moses, M. Leclerc, K. Lee and A. J. Heeger, Nat. Photonics, 2009, 3, 297 CrossRef.
- S. Beaupré and M. Leclerc, J. Mater. Chem. A, 2013, 1, 11097 Search PubMed.
- W. Zhang, J. Smith, S. E. Watkins, R. Gysel, M. McGehee, A. Salleo, J. Kirkpatrick, S. Ashraf, T. Anthopoulos, M. Heeney and I. McCulloch, J. Am. Chem. Soc., 2010, 132, 11437 CrossRef CAS PubMed.
- H. N. Tsao, D. M. Cho, I. Park, M. R. Hansen, A. Mavrinskiy, D. Y. Yoon, R. Graf, W. Pisula, H. W. Spiess and K. Müllen, J. Am. Chem. Soc., 2011, 133, 2605 CrossRef CAS PubMed.
- S. Wang, M. Kappl, I. Liebewirth, M. Muller, K. Kirchhoff, W. Pisula and K. Müllen, Adv. Mater., 2012, 24, 417 CrossRef CAS PubMed.
- Y. Yamashita, F. Hinkel, T. Marszalek, W. Zajaczkowski, W. Pisula, M. Baumgarten, H. Matsui, K. Müllen and J. Takeya, Chem. Mater., 2016, 28, 420 CrossRef CAS.
- (a) M. L. Tang and Z. Bao, Chem. Mater., 2011, 23, 446 CrossRef CAS; (b) Z. Xiao, J. Subbiah, K. Sun, D. J. Jones, A. B. Holmes and W. W. H. Wong, Polym. Chem., 2014, 5, 6710 RSC; (c) Z. Xiao, K. Sun, J. Subbiah, T. Qin, S. Lu, B. Purushothaman, D. J. Jones, A. B. Holmes and W. W. H. Wong, Polym. Chem., 2015, 6, 2312 RSC.
- (a) H. Bronstein, J. M. Frost, A. Hadipour, Y. Kim, C. B. Nielsen, R. S. Ashraf, B. P. Rand, S. Watkins and I. McCulloch, Chem. Mater., 2013, 25, 277 CrossRef CAS; (b) Y. Yang, R. Wu, X. Wang, X. Xu, Z. Li, K. Li and Q. Peng, Chem. Commun., 2014, 50, 439 RSC.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS PubMed.
- J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Nature Energy, 2016, 1, 15027 CrossRef.
- Y. Zhao, Y. Guo and Y. Liu, Adv. Mater., 2013, 25, 5372 CrossRef CAS PubMed.
- X. Gao and Y. Hu, J. Mater. Chem. C, 2014, 2, 3099 RSC.
- J. Lee, M. Jang, S. M. Lee, D. Yoo, T. J. Shin, J. H. Oh and C. Yang, ACS Appl. Mater. Interfaces, 2014, 6, 20390 CAS.
- (a) T. D. Anthopoulos, F. B. Kooistra, H. J. Wondergem, D. Kronholm, J. C. Hummelen and D. M. de Leeuw, Adv. Mater., 2006, 18, 1679 CrossRef CAS; (b) H. Usta, C. Risko, Z. Wang, H. Huang, M. K. Deliomeroglu, A. Zhukhovitskiy, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2009, 131, 5586 CrossRef CAS PubMed; (c) H. Zhong, J. Smith, S. Rossbauer, A. J. P. White, T. D. Anthopoulos and M. Heeney, Adv. Mater., 2012, 24, 3205 CrossRef CAS PubMed.
- A. Casey, Y. Han, Z. Fei, A. J. P. White, T. D. Anthopoulosc and M. Heeney, J. Mater. Chem. C, 2015, 3, 265 RSC.
- (a) L. F. Lai, J. A. Love, A. Sharenko, J. E. Coughlin, V. Gupta, S. Tretiak, T.-Q. Nguyen, W.-Y. Wong and G. C. Bazan, J. Am. Chem. Soc., 2014, 136, 5591 CrossRef CAS PubMed; (b) M. Wang, H. Wang, T. Yokoyama, X. Liu, Y. Huang, Y. Zhang, T.-Q. Nguyen, S. Aramaki and G. C. Bazan, J. Am. Chem. Soc., 2014, 136, 12576 CrossRef CAS PubMed; (c) W. W. H. Wong, J. Subbiah, S. R. Puniredd, W. Pisula, D. J. Jones and A. B. Holmes, Polym. Chem., 2014, 5, 1258 RSC.
- L. A. Perez, P. Zalar, L. Ying, K. Schmidt, M. F. Toney, T.-Q. Nguyen, G. C. Bazan and E. J. Kramer, Macromolecules, 2014, 47, 1403 CrossRef CAS.
- (a) Y. Yamashita, K. Ono, M. Tomura and S. Tanaka, Tetrahedron, 1997, 53, 10169 CrossRef CAS; (b) K. Ono, S. Tanaka and Y. Yamashita, Angew. Chem., Int. Ed., 1994, 33, 1977 CrossRef; (c) T. Kono, D. Kumaki, J. Nishida, S. Tokito and Y. Yamashita, Chem. Commun., 2010, 46, 3265 RSC; (d) J. L. Banal, J. Subbiah, H. Graham, J.-K. Lee, K. P. Ghiggino and W. W. H. Wong, Polym. Chem., 2013, 4, 1077 RSC.
- T. L. Tam, H. Li, F. Wei, K. J. Tan, C. Kloc, Y. M. Lam, S. G. Mhaisalkar and A. C. Grimsdale, Org. Lett., 2010, 12, 3340 CrossRef CAS PubMed.
- T. T. Steckler, X. Zhang, J. Hwang, R. Honeyager, S. Ohira, X. H. Zhang, A. Grant, S. Ellinger, S. A. Odom, D. Sweat, D. B. Tanner, A. G. Rinzler, S. Barlow, J. L. Bredas, B. Kippelen, S. R. Marder and J. R. Reynolds, J. Am. Chem. Soc., 2009, 131, 2824 CrossRef CAS PubMed.
- J. D. Yuen, R. Kumar, D. Zakhidov, J. Seifter, B. Lim, A. J. Heeger and F. Wudl, Adv. Mater., 2011, 23, 3780 CAS.
- J. D. Yuen, J. Fan, J. Seier, B. Lim, R. Hufschmid, A. J. Heeger and F. Wudl, J. Am. Chem. Soc., 2011, 133, 20799 CrossRef CAS PubMed.
- J. Fan, J. D. Yuen, M. Wang, J. Seifter, J. H. Seo, A. R. Mohebbi, D. Zakhidov, A. Heeger and F. Wudl, Adv. Mater., 2012, 24, 2186 CrossRef CAS PubMed.
- J. Fan, J. D. Yuen, W. Cui, J. Seifter, A. R. Mohebbi, M. Wang, H. Zhou, A. Heeger and F. Wudl, Adv. Mater., 2012, 24, 6164 CrossRef CAS PubMed.
- Y. Wang, T. Kadoya, L. Wang, T. Hayakawa, M. Tokita, T. Mori and T. Michinobu, J. Mater. Chem. C, 2015, 3, 1196 RSC.
- Y. Wang, H. Masunaga, T. Hikima, H. Matsumoto, T. Mori and T. Michinobu, Macromolecules, 2015, 48, 4012 CrossRef CAS.
- T. Dallos, D. Beckmann, G. Brunklaus and M. Baumgarten, J. Am. Chem. Soc., 2011, 133, 13898 CrossRef CAS PubMed.
- (a) C.-Y. Yu, C.-P. Chen, S.-H. Chan, G.-W. Hwang and C. Ting, Chem. Mater., 2009, 21, 3262 CrossRef CAS; (b) A. P. Zoombelt, M. Fonrodona, M. M. Wienk, A. B. Sieval, J. C. Hummelen and R. A. J. Janssen, Org. Lett., 2009, 11, 903 CrossRef CAS PubMed.
- C. An, M. Li, T. Marszalek, X. Guo, W. Pisula and M. Baumgarten, J. Mater. Chem. C, 2015, 3, 3876 RSC.
- T. T. Steckler, P. Henriksson, S. Mollinger, A. Lundin, A. Salleo and M. R. Andersson, J. Am. Chem. Soc., 2014, 136, 1190 CrossRef CAS PubMed.
- M. L. Keshtov, S. A. Kuklin, N. A. Radychev, A. Y. Nikolaev, E. N. Koukaras, A. Sharma and G. D. Sharma, RSC Adv., 2016, 6, 14893 RSC.
- L. Wang, D. Cai, Q. Zheng, C. Tang, S.-C. Chen and Z. Yin, ACS Macro Lett., 2013, 2, 605 CrossRef CAS.
- C. B. Nielsen, R. S. Ashraf, N. D. Treat, B. C. Schroeder, J. E. Donaghey, A. J. P. White, N. Stingelin and I. McCulloch, Adv. Mater., 2015, 27, 948 CrossRef CAS PubMed.
- S. Mataka, K. Takahashi, Y. Ikezaki, T. Hatta, A. Torii and M. Tashiro, Bull. Chem. Soc. Jpn., 1991, 64, 68 CrossRef CAS.
- M. Wang, X. Hu, P. Liu, W. Li, X. Gong, F. Huang and Y. Cao, J. Am. Chem. Soc., 2011, 133, 9638 CrossRef CAS PubMed.
- I. Osaka, T. Abe, M. Shimawaki, T. Koganezawa and K. Takimiya, ACS Macro Lett., 2012, 1, 437 CrossRef CAS.
- T. Yang, M. Wang, C. Duan, X. Hu, L. Huang, J. Peng, F. Huang and X. Gong, Energy Environ. Sci., 2012, 5, 8208 CAS.
- (a) M. Wang, X. Hu, L. Liu, C. Duan, P. Liu, L. Ying, F. Huang and Y. Cao, Macromolecules, 2013, 46, 3950 CrossRef CAS; (b) Y. Sun, J. Seifter, M. Wang, L. A. Perez, C. Luo, G. C. Bazan, F. Huang, Y. Cao and A. J. Heeger, Adv. Energy Mater., 2014, 4, 1301601 Search PubMed.
- I. Osaka, T. Kakara, N. Takemura, T. Koganezawa and K. Takimiya, J. Am. Chem. Soc., 2013, 135, 8834 CrossRef CAS PubMed.
- I. Osaka, M. Shimawaki, H. Mori, I. Doi, E. Miyazaki, T. Koganezawa and K. Takimiya, J. Am. Chem. Soc., 2012, 134, 3498 CrossRef CAS PubMed.
- J. M. Szarko, J. Guo, Y. Liang, B. Lee, B. S. Rolczynski, J. Strzalka, T. Xu, S. Loser, T. J. Marks, L. Yu and L. X. Che, Adv. Mater., 2010, 22, 5468 CrossRef CAS PubMed.
- V. Vohra, K. Kawashima, T. Kakara, T. Koganezawa, I. Osaka, K. Takimiya and H. Murata, Nat. Photonics, 2015, 9, 403 CrossRef CAS.
- F. A. Arroyave, C. A. Richard and J. R. Reynolds, Org. Lett., 2012, 14, 6138 CrossRef CAS PubMed.
- D. G. Patel, F. Feng, Y. Ohnishi, K. A. Abboud, S. Hirata, K. S. Schanze and J. R. Reynolds, J. Am. Chem. Soc., 2012, 134, 2599 CrossRef CAS PubMed.
- C.-Y. Mei, L. Liang, F.-G. Zhao, J.-T. Wang, L.-F. Yu, Y.-X. Li and W.-S. Li, Macromolecules, 2013, 46, 7920 CrossRef CAS.
- (a) J. Mei and Z. Bao, Chem. Mater., 2014, 26, 604 CrossRef CAS; (b) L. Chen, P. Shen, Z. Zhang and Y. Li, J. Mater. Chem. A, 2015, 3, 12005 RSC.
- J. Huang, Y. Zhu, J. Chen, L. Zhang, J. Peng and Y. Cao, Macromol. Rapid Commun., 2014, 35, 1960 CrossRef CAS PubMed.
- J. Zhao, Y. Li, A. Hunt, J. Zhang, H. Yao, Z. Li, J. Zhang, F. Huang, H. Ade and H. Yan, Adv. Mater., 2016, 28, 1868 CrossRef CAS PubMed.
- (a) A. Efrem, C.-J. Lim, Y. Lu and S.-C. Ng, Tetrahedron Lett., 2014, 55, 4849 CrossRef CAS; (b) A. Efrem, Y. Lei, B. Wu, M. Wang, S. C. Ng and B. S. Ong, Dyes Pigm., 2016, 129, 90 CrossRef CAS.
- M. Pei, J. Huang, M. Jang, J.-H. Kim, M. Lee, J. Chen, D. H. Hwang and H. Yang, J. Phys. Chem. C, 2016, 120, 903 CAS.
- S. Cai, L. Chen, D. Zha and Y. Chen, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 624 CrossRef CAS.
- Y.-J. Cheng, Y.-J. Ho, C.-H. Chen, W.-S. Kao, C.-E. Wu, S.-L. Hsu and C.-S. Hsu, Macromolecules, 2012, 45, 2690 CrossRef CAS.
- R. Chen, C. Zheng, C. Li, H. Li, Z. Wang, Y. Tang, H. Jiang, Z. Tan and W. Huang, Polym. Chem., 2016, 7, 780 RSC.
- W. Zhou, F. Jin, X. Huang, X.-M. Duan and X. Zhan, Macromolecules, 2012, 45, 7823 CrossRef CAS.
- H. Yao, H. Zhang, L. Ye, W. Zhao, S. Zhang and J. Hou, Macromolecules, 2015, 48, 3493 CrossRef CAS.
- T. Ma, K. Jiang, S. Chen, H. Hu, H. Lin, Z. Li, J. Zhao, Y. Liu, Y.-M. Chang, C.-C. Hsiao and H. Yan, Adv. Energy Mater., 2015, 5, 1501282 Search PubMed.
- T. Nakanishi, Y. Shirai and L. Han, J. Mater. Chem. A, 2015, 3, 4229 CAS.
- H. Li, T. L. Tam, Y. M. Lam, S. G. Mhaisalkar and A. C. Grimsdale, Org. Lett., 2011, 13, 46 CrossRef CAS PubMed.
- G. Qian, J. Qi, J. A. Davey, J. S. Wright and Z. Y. Wang, Chem. Mater., 2012, 24, 2364 CrossRef CAS.
- G. Qian, Z. Zhong, M. Luo, D. Yu, Z. Zhang, D. Ma and Z. Y. Wang, J. Phys. Chem. C, 2009, 113, 1589 CAS.
- C. An, S. R. Puniredd, X. Guo, T. Stelzig, Y. Zhao, W. Pisula and M. Baumgarten, Macromolecules, 2014, 47, 979 CrossRef CAS.
- C. An, M. Li, T. Marszalek, D. Li, R. Berger, W. Pisula and M. Baumgarten, Chem. Mater., 2014, 26, 5923 CrossRef CAS.
- T. T. Steckler, M. J. Lee, Z. Chen, O. Fenwick, M. R. Andersson, F. Cacialli and H. Sirringhaus, J. Mater. Chem. C, 2014, 2, 5133 RSC.
- X. Feng, L. Liu, S. Wang and D. Zhu, Chem. Soc. Rev., 2010, 39, 2411 RSC.
- Y. S. Kwon, J. Lim, H.-J. Yun, Y.-H. Kim and T. Park, Energy Environ. Sci., 2014, 7, 1454 CAS.
- H. Wang and Z. Bao, Nano Today, 2016, 10, 737 CrossRef.
| This journal is © The Royal Society of Chemistry 2016 |