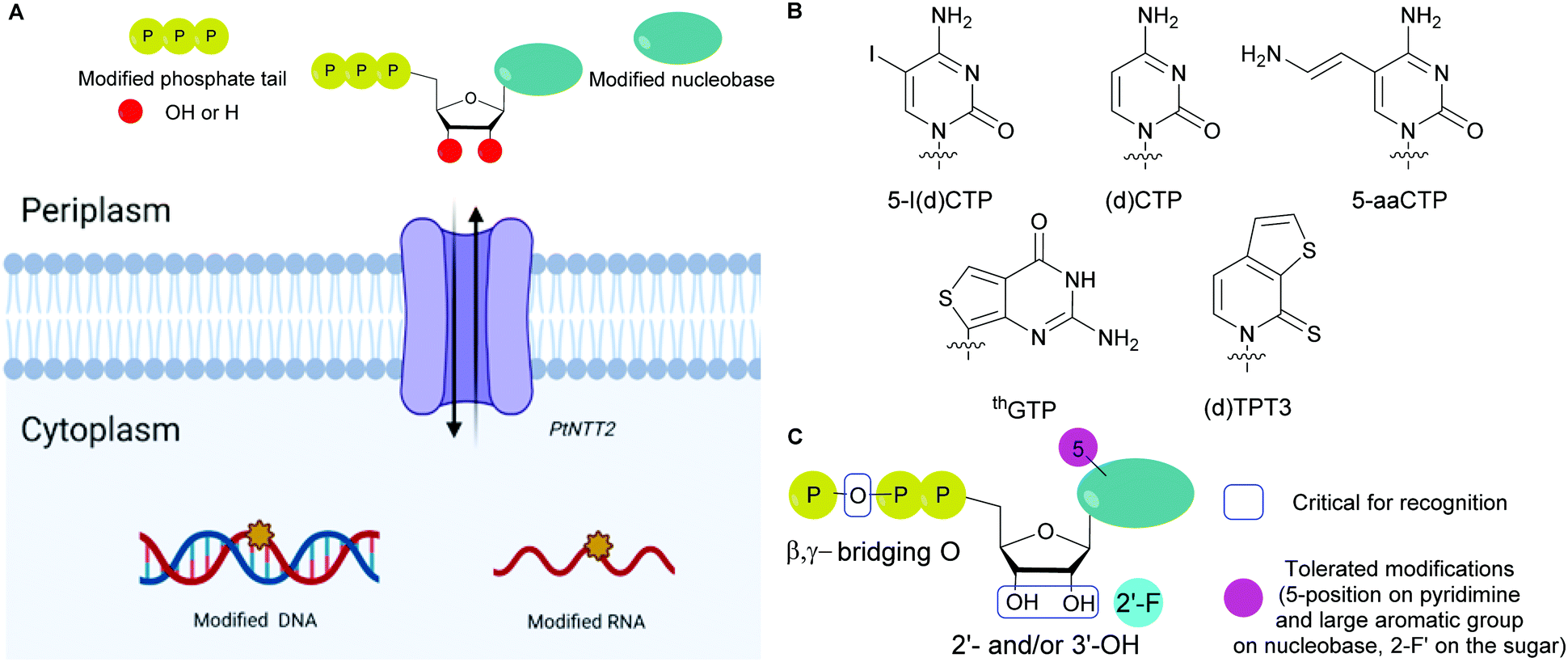
DOI:
10.1039/D0CB00078G
(Review Article)
RSC Chem. Biol., 2020,
1, 333-351
Modified nucleoside triphosphates in bacterial research for in vitro and live-cell applications
Received
27th May 2020
, Accepted 21st August 2020
First published on 14th September 2020
Abstract
Modified nucleoside triphosphates (NTPs) are invaluable tools to probe bacterial enzymatic mechanisms, develop novel genetic material, and engineer drugs and proteins with new functionalities. Although the impact of nucleobase alterations has predominantly been studied due to their importance for protein recognition, sugar and phosphate modifications have also been investigated. However, NTPs are cell impermeable due to their negatively charged phosphate tail, a major hurdle to achieving live bacterial studies. Herein, we review the recent advances made to investigate and evolve bacteria and their processes with the use of modified NTPs by exploring alterations in one of the three moieties: the nucleobase, the sugar and the phosphate tail. We also present the innovative methods that have been devised to internalize NTPs into bacteria for in vivo applications.
1. Introduction
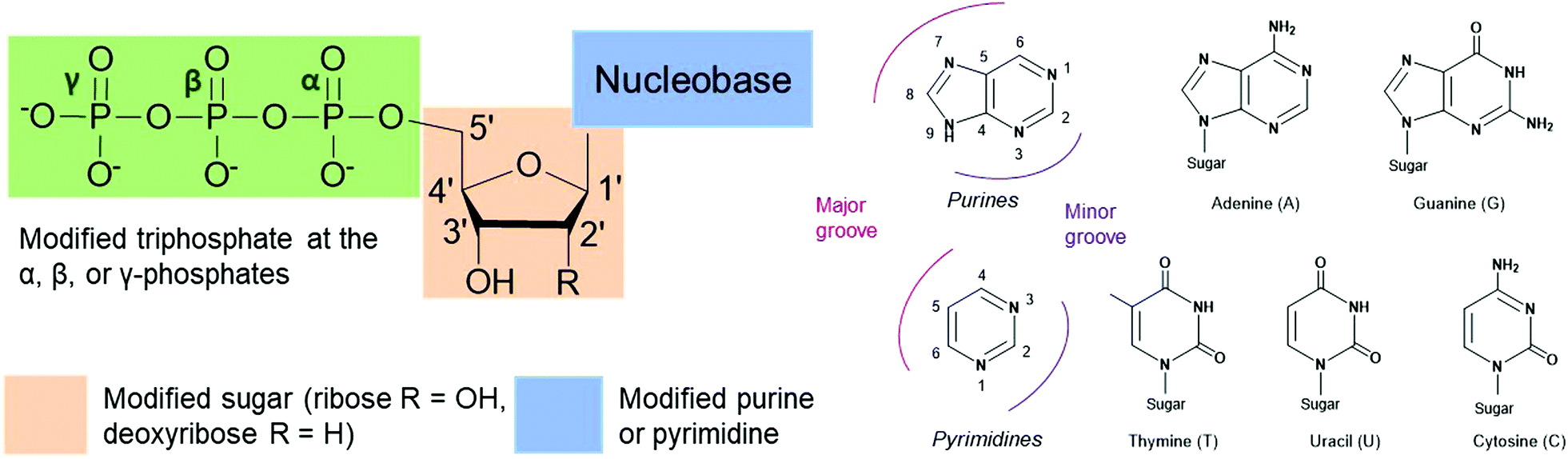
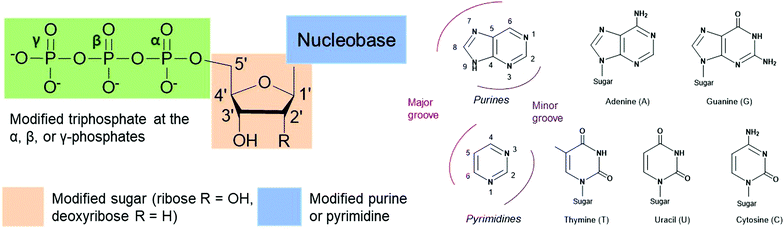
Nucleoside triphosphates (NTPs) are involved in numerous fundamental biological systems and are essential molecules for all living organisms, including bacteria. NTPs consist of a ribose or deoxyribose sugar core connected to a nucleobase (a purine or a pyrimidine) by a glycosylic bond at the 1′-carbon and a tail of three phosphates through the 5′-carbon (Fig. 1). These molecules are the building blocks of nucleic acids and are incorporated by polymerases to achieve many cellular processes. For instance, ribonucleic acid (RNA) is assembled by RNA polymerases from NTPs whereas deoxyribonucleic acid (DNA) is built from deoxyribonucleoside triphosphates (dNTPs), which are building blocks that contain a hydrogen atom on the 2′-carbon of the ribose instead of a hydroxyl group. The NTP adenosine triphosphate (ATP) is the energy currency as hydrolysis of the terminal ATP phosphoanhydride bond releases energy and actively powers many enzymes. ATP is also involved in signal transduction and gene regulation.1,2 Modified NTPs have long been exploited to study and evolve complex bacterial enzymatic mechanisms such as transcription, translation and protein activity. They are critical to advance our fundamental knowledge of bacterial protein machinery. Nevertheless, NTPs are cell impermeable due to their negatively charged phosphate tail, which creates challenges for their internalization and in vivo studies. Active work is currently underway to resolve this long-standing issue. In this review, we present recent advances in the exploration and alteration of bacterial functions using modified NTPs to decipher NTP-related bacterial mechanisms. We also introduce the innovative methods that have been developed to achieve their internalization into cells for live studies with the ultimate goal of uncovering novel bacterial functions and pathways.
 |
| | Fig. 1 Possible sites of modification on nucleoside triphosphates (NTPs) to study bacterial cellular processes. Modifications are found on the nucleobase (purines A and G or pyrimidines T, U and C), the sugar moiety (ribose or deoxyribose) or the triphosphate tail (α-, β- or γ-position). | |
2. Modifications on the nucleobase of NTPs
The nucleobases adenine (A), guanine (G), cytosine (C), and thymine (T) or uracil (U), used in DNA and RNA respectively, play a crucial role in biorecognition during DNA and RNA synthesis, replication and translation (Fig. 1). The high level of fidelity conferred by DNA/RNA polymerases and ribosomal proteins results from recognition of the base pair shape in the minor groove of nucleic acids and hydrogen bonding between the nucleobase pairs A and T/U or G and C.3 Nevertheless, protein plasticity has been observed resulting in the accommodation of modified NTPs within the DNA and RNA structure. Structural modifications of these nucleobases have been exploited to better understand DNA and RNA polymerase activity and their recognition mechanisms of nucleic acids, to expand the genetic alphabet, and to evolve enzymes.
2.1. Alterations impacting the major groove of DNA
DNA polymerases control insertion of the correct dNTPs into DNA through proof-reading and repair mechanisms tightly correlated with the three-dimensional structure of the DNA strand template and the dNTP building blocks.4,5 Most alterations of the dNTPs are located on the nucleobase at the 7- and 8-positions of the purines and the 5-position of the pyrimidines and are oriented towards the major groove of DNA (Fig. 1). These modifications result in minimal interferences with the activity of DNA polymerases and with hydrogen bonding between the base partners.6,7
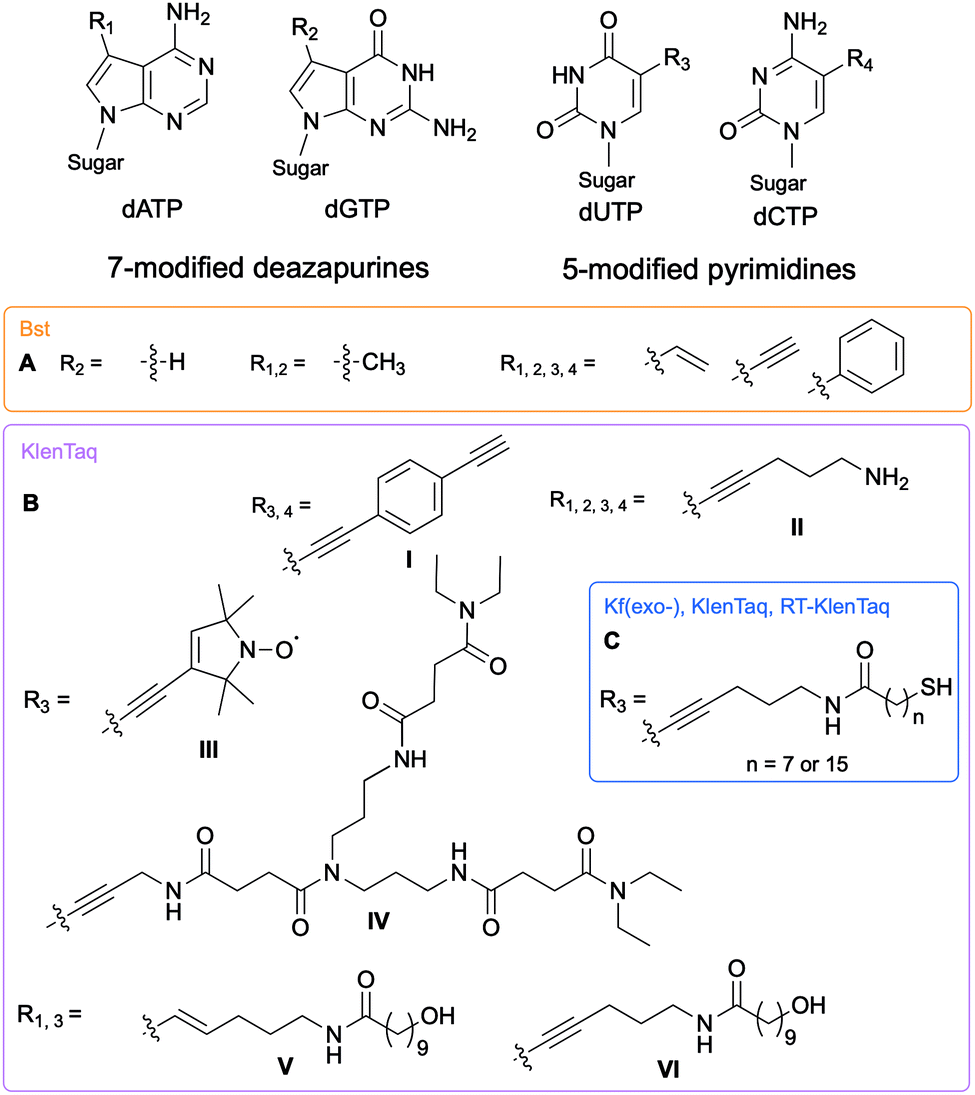
The Hocek and Marx research groups have thoroughly explored nucleobase-modified dNTPs with alterations oriented towards the major groove to establish the key parameters regulating their incorporation into DNA duplexes by bacterial polymerases. Kinetic and competition assays between altered and endogenous dNTPs, along with modeling studies, were implemented by Hocek and co-workers to understand the molecular basis for DNA polymerase selectivity with bacterial family A and archaeal family B polymerases [Bst large fragment from Geobacillus stearothermophilus, KOD XL a recombinant form of KOD1 from Thermococcus kodakaraensis, Pwo from Pyrococcus woesei, and Vent(exo-) from T. litoralis]. Using 7-substituted 7-deazaadenine and 7-deazaguanine in conjunction with 5-substituted pyrimidines containing moieties of increasing bulkiness (H, methyl, vinyl, ethynyl and phenyl), the studies demonstrated that cation–π interactions with an arginine residue within the Bst polymerase active site were crucial to the superior affinity of phenyl-modified deazapurine dNTPs with this enzyme and successful DNA extension (Fig. 2A).8 The critical role of the arginine residue was also established by Marx and co-workers who conducted a series of molecular and structural studies on Klenow fragment of Taq (KTq) DNA polymerase from the bacterium Thermus aquaticus, a truncated version of Taq polymerase (Fig. 2BI, II, III, IV).7,9,10 Hydrogen bonding is another important parameter involved in DNA polymerase-modified dNTPs recognition. Increased incorporation efficiency of the dendrimer IV compared to the spin label III was rationalized by the stabilization effects brought by hydrogen bonding between the dendrimer and the active site. Only modification III interactions with arginine played a stabilizing role. They furthered their understanding of the enzyme plasticity by employing modified dNTPs on the C5 position of pyrimidines and C7 position of 7-deazapurines bearing alkyne and alkene linkers (Fig. 2BV, VI)11 in addition to several consecutive modifications, including enrichment handles (Fig. 2BI).12,13 The authors showed that dNTPs bearing an alkene (V) or an alkyne (VI) were incorporated into DNA by KTq with a higher efficiency than their native counterparts except for dUTP modified with VI. The structural studies could not explain this observation. Furthermore, the presence of an amide moiety increased stabilization of the modified dNTPs in the active site by establishing an additional interactions with the O-helix or primer backbone.11 Aromatic, rigid and non-polar modifications (I) yielded additional cation–π interactions as previously discussed. Taken together, these results indicate that bacterial DNA polymerases can accommodate large and rigid groups as long as key interactions are present.12,13 Marx and co-workers demonstrated that DNA polymerases could tolerate extremely large modifications such as proteins of size comparable to the polymerases.14,15 For instance, they conjugated a mouse IgG1-Apoliprotein A1 monoclonal antibody to a dCTP at the 5-position of the nucleobase that was successfully incorporated by archaeal and bacterial DNA polymerases (KOD, Thermococcus sp. 9°N, and KTq) opposite to a guanine in a primer extension experiment.14 Similarly, Baccaro et al. showed that dTTPs conjugated to oligodeoxynucleotides, about 40 times bulkier than native NTPs, could be incorporated into DNA by KTq and Therminator (engineered from Thermococcus sp. 9°N-7) but at a lower efficiency than the natural dTTP.16 Interestingly, some modified dNTPs had a higher affinity for the DNA polymerase active site than their natural counter-parts due to newly formed interactions.17 Hocek and coworkers demonstrated that 7-aryl-7-deaza-modified dATPs had tighter affinities, improved incorporation into DNA and increased primer extension in comparison to their natural counter-part for the following bacterial DNA polymerases: Taq, DNA polymerase I large (Klenow) fragment (Kf) from Escherichia coli and Bst from G. stearothermophilus.17
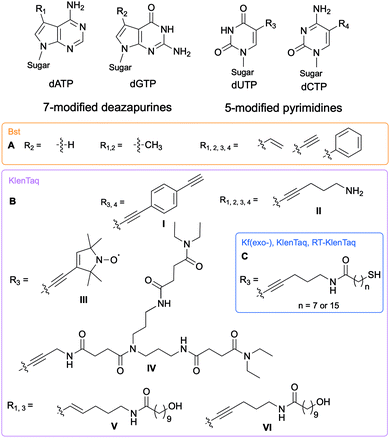 |
| | Fig. 2 Structures of nucleobase alterations of nucleobase-modified dNTPs oriented towards the major groove of DNA for investigating bacterial polymerase activity. | |
2.2. Alterations impacting the minor groove of DNA
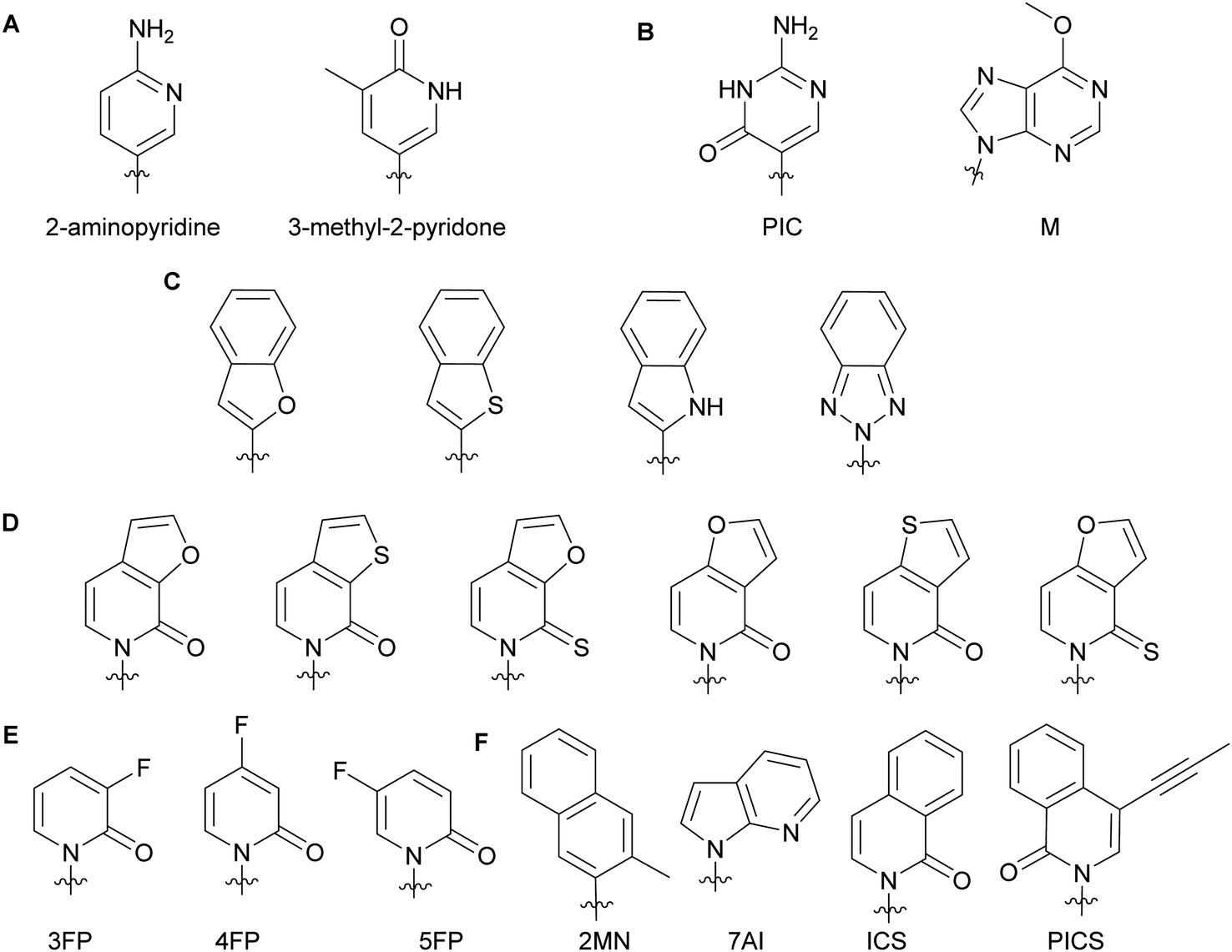
Minor groove modifications are more challenging to implement since they directly impact hydrogen bonding between base pairs, DNA helix integrity and DNA–protein interactions. Nevertheless, they are critical for investigating DNA-related proteins and for expanding the genetic alphabet.18 The first use of minor groove modifications on the 2-position of dNTPs for catalysis of DNA synthesis by polymerases was implemented by Matyašovský et al. They assessed dATPs that had been modified at the 2-position with a series of groups of increasing bulkiness (chloro, amino, methyl, vinyl, ethynyl and phenyl) for primer extension and post-DNA synthesis labeling. Due to its large size, 2-phenyl dATP was not accepted by DNA polymerases [Bst, KOD XL and Vent(exo-)]. All other modified dATPs were successfully incorporated into DNA. Furthermore, DNA containing vinyl- or ethynyl-groups were successfully conjugated to a fluorescent thiol- or azide-containing molecules respectively, demonstrating the possibility of using minor groove modifications as chemical tools to study DNA-related questions.18 Also looking at the modification of natural nucleobases, McLaughlin and co-workers showed that removal of the 2-oxo group from pyrimidines inhibited DNA polymerase (Taq and Kf) activity (Fig. 3A). They hypothesized that the absence of a critical hydrogen bond between the 2-oxo group of pyrimidines and the active site of the polymerases prevented an essential conformational change for the enzyme to achieve dNTP addition and/or resulted in a misfolding of the polymerase active site.19 The impact of modifications at the 4-position have also been evaluated with Taq, Kf(exo-) and Bsm from B. smithii by Meškys and co-workers. Various large acyl groups at the N4-position of dCTPs, including benzophenone for photo-crosslinking applications, were successfully used for primer extension with slight differences in incorporation efficiency for bulky modifications. Interestingly, N4-modified dCTPs could be recognized as TTPs and added opposite to adenine.6 Remarkably, modifications in the minor groove have led to natural pair rewiring. The addition of pseudoisocytidine and 6-methoxypurine dNTPs into a DNA fragment by Kf(exo-) followed by DNA amplification lead to interconversion of the G–C and A–T base pairs and, thus, created a point mutation (Fig. 3B).20 Examples of poor bacterial DNA polymerase substrates included imidazole-based nucleobases for Kf(exo-)21 and 4-hydroxy-2-mercaptobenzimidazole for Kf(exo-), Taq and Tfl from T. flavus.22
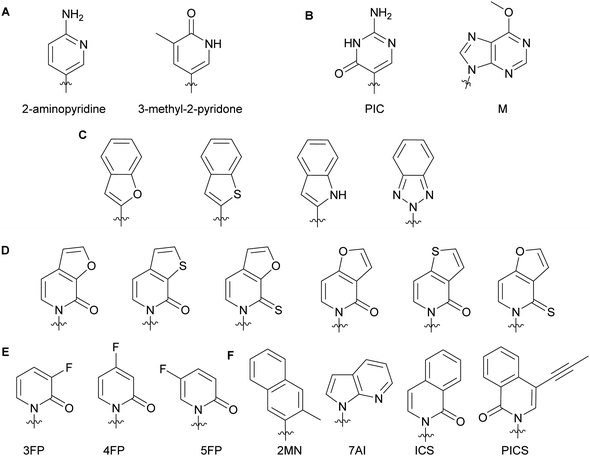 |
| | Fig. 3 Examples of dNTP unnatural nucleobases utilized to evaluate minor groove modifications on DNA stability and related enzymes (A–C). Modified natural nucleobases for DNA stability and natural base pair rewiring studies (B). Unnatural nucleobases for understanding the impact of hydrophobicity, polarity, polarizability and size on DNA pairing, synthesis and replication (D–F). | |
In the context of expanding the genetic alphabet and developing semi-synthetic organisms capable of functioning with non-canonical nucleic acids, Romesberg and co-workers have extensively studied base-pairing and polymerase activity with nucleobases that are structurally different from the natural ones. Through kinetics studies and crystallographic analysis, they investigated the impact of minor groove modifications on DNA polymerization with nucleotides displaying benzofuran, benzothiophene, indole and benzotriazole nucleobases using Kf(exo-) (Fig. 3C). While self-pairing was mostly independent of the type of unnatural nucleobase incorporated and destabilization of the DNA duplex was not observed, DNA elongation was impacted by the nature of the unnatural nucleobase. They attributed this result to the orientation of the unnatural nucleobase 3′-OH involved in the nucleophilic attack of the incoming NTP, which was dependent on the nature of the atom oriented towards the minor groove of the DNA polymerase.23 Other work with dNTPs containing heteroatom derivatization and fluoro-modified NTPs demonstrated that DNA duplex stability and replication do not solely rely on hydrogen bonding interactions but also on hydrophobic interactions.23–25 Polarity, shape and polarizability (destabilization of self-pairing when an oxygen atom is replaced by a sulfur atom on the exocyclic double-bond) are key factors (Fig. 3D).24 Moreover, fluorine derivatization and minor groove hydrogen-bond acceptors tend to positively support the extension step during replication (Fig. 3E).25 Examples of dNTPs with non-natural hydrophobic nucleobases were incorporated opposite of other non-natural ones into DNA with similar or higher efficiency than natural dNTPs opposite of their complementary bases (Fig. 3F). Moreover, unnatural dNTPs were orthogonal to natural dNTPs. These results established that hydrophobic packing forces could replace DNA hydrogen-bonding interactions and drive base pairing and replication.26 In a recent account, Hottin and Marx summarized the structural parameters governing the processing of nucleobase-modified dNTPs by family A polymerases. Furthermore, they highlighted the key differences between family A and family B polymerases by explaining how nucleobase-modified dNTPs were better accommodated by family B.7
2.3. Impact of unnatural nucleobases on DNA and RNA repair mechanisms
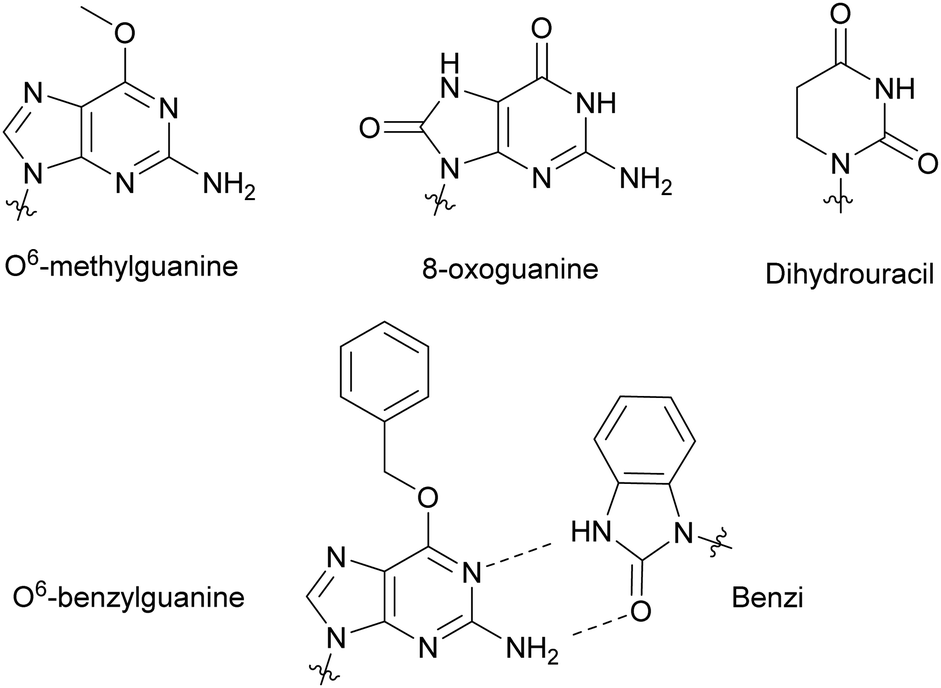
Modified NTPs have been investigated to understand DNA repair mechanisms and propagation of DNA mutations, from environmental or spontaneous sources, leading to altered protein synthesis.27 For instance, DNA templates bearing non-bulky nucleotide damaged products, such as O6-methylguanine or 8-oxoguanine, at specific locations led to mutagenic base insertion and decreased transcription when placed near the promoter region (Fig. 4).28 Similarly, introduction of dihydrouracil, the product resulting from the effects of ionizing radiation on cytosine present in DNA or RNA, caused an adenine insertion (Fig. 4). This suggested that mutagenic effects of this modification occurred in both replication and transcription.29 To detect non-natural adducts in DNA caused by alkylating agents found in chemotherapy treatment or in the environment, Wyss et al. found that a mutated KTq DNA polymerase could recognize O6-benzylguanine. The mutated polymerase could specifically incorporate a benzimidazole-derived dNTP (Benzi) opposite to it and catalyze the formation of full-length DNA products (Fig. 4).30,31 Walsh and Beuning reviewed a series of modified NTPs used as a means to investigate DNA polymerase specificity and to understand how DNA polymerases cope with non-natural NTPs in the context of mutagenesis and repair mechanisms.32
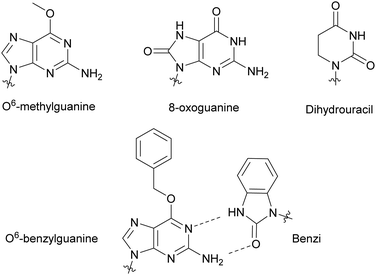 |
| | Fig. 4 Nucleobase modifications relevant to DNA damage present in NTPs, utilized to study DNA and RNA repair mechanisms and DNA adduct detection. | |
2.4. Impact of unnatural nucleobases on bacterial RNA polymerases
Bacteria possess only one type of RNA polymerase (RNAP), an enzyme central to transcription and gene expression. The core protein complexed with one of the specificity factors σ utilizes dNTPs to generate a nascent RNA from a DNA template.33 As for DNA polymerases, bacterial RNAP activity and the transcription step have been probed with nucleobase-modified dNTPs.34 Raindlová et al. inserted dNTPs with 5-substituted pyrimidines and 7-substituted 7-deazapurines with various alkyl chains into DNA templates to investigate the impact of these modifications on B. subtilus and E. coli transcription mechanisms (Fig. 2A).35 Although bulkier modifications had a dramatic effect on the inhibition of transcription compared to smaller ones, the correct RNA sequences were observed when transcription proceeded. This indicated that only the initial transcription was affected by major groove DNA modifications. This knowledge is a key step to control RNAP initiation. Remarkably, the replacement of N7 by a carbon atom in modified adenine and guanine present in DNA did not affect RNAP transcription activity, pinpointing the lack of hydrogen bonding interactions between the nitrogen and the RNAP active site. Janoušková et al. demonstrated that transcription could be modulated using groups acting as epigenetic marker mimics. The 5-(hydroxymethyl) (5-hm) modification on uracil and cytosine acted as an epigenetic epitope and altered E. coli RNAP activity as a function of the promoter sequence. An increase in transcription was observed when a promoter called Pveg from B. subtilus, recognized by E. coli RNAP, was utilized.36 In an effort to control transcription, VanÍková et al. used the photocleavable group nitrobenzyl (NB) to protect 5-hm-uracil or cytosine. NB acted as an epigenetic marker mimic for E. coli RNAP. Transcription of the DNA strand was halted in the presence of NB, whereas transcription resumed once NB was removed (Fig. 5).37 Photocaging as a means of regulating transcription has been thoroughly investigated by Hocek and co-workers.37,38
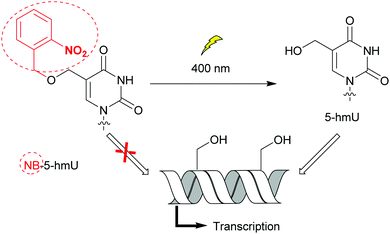 |
| | Fig. 5 The nitrobenzyl (NB) group operates as an epigenetic marker mimetic. The presence of the NB moiety on 5-hydroxymethyluracil (5-hmU) prevented DNA transcription by E. coli RNAP. Photocleavage of NB controlled transcription. | |
2.5. Probing restriction endonucleases with modified dNTPs
In bacteria, restriction endonucleases (REs) are a defense mechanism against foreign DNA.39 Due to their ability to recognize highly specific sequences, REs are common molecular biological tools for routine DNA manipulation that enable facile introduction of a small segment of DNA into plasmids or genomic DNA. Numerous studies have introduced modified dNTPs into DNA fragments to evaluate the impact of modified pyrimidines or purines on a series of RE activities based on their location within the DNA segment and the type of modifications (halogenated atoms, bulky and electron withdrawing groups, etc.). Their goal was to better understand DNA–protein affinity requirements for the development of biological tools.40–46 The Hocek research group has thoroughly evaluated the impact of major groove nucleobase modifications on REs, and a few examples are presented here. Macíčková-Cahová et al. demonstrated that 5-modified cytosine with moieties such as alkyl, phenyl or 3-nitrophenyl were not recognized by any of the bacterial type II REs tested including Afel, EcoRI and PvuII. However, ScaI was able to cleave DNA with 5-vinylcytosine demonstrating promiscuity for substrate affinity. In contrast, the presence of small substituents at the 5-position of uracil did not affect the activity of most tested REs (ScaI, PvuII, RsaI, KpnI, SacI, SphI, AflII, ApaLI, BglII, AfeI and PspGI). AfeI and PspGI were the most tolerant REs, accepting 5-phenyl and 5-nitrophenol on uracil. These results indicated that major groove interactions between DNA and some REs are critical for recognition especially for the G–C pair.42,44 Similarly, 8-bromo and 8-methyl adenine derivatives were well accepted by type II REs unlike some 7-modified-7-deazaadenine with halogens, aromatic or alkyl groups.41 Additional work on 7-modified-7-deaza dGTPs confirmed the importance of the G:C pairs for type II RE recognition, as most of the modified restriction sequences were not cleaved except the ones containing 7-acetyl-7-deaza dGTP by Afel from Alcaligenes faecalis. The presence of a H-bond acceptor, in this case the acetyl group, was critical for Afel activity.43 A first account of the impact of minor groove modifications on RE activity has recently been reported by Matyašovský et al. No global trend could be drawn from the results, which were dependent on the combination of enzymes and modifications tested. Nevertheless, minor groove alterations had an impact on RE activity, sequence recognition of DNA occurred at the DNA major groove.47
2.6. Selected applications of nucleobase-modified dNTPs
2.6.1. Development of chemical biology tools with nucleobase-modified dNTPs.
NTP nucleobase alterations have been instrumental in the development of chemical biology applications such as biomolecular fluorescence visualization, conjugation methods, and spectroscopy.10,18 The promiscuity of polymerases has been utilized to develop a myriad of chemical biology tools. For instance, Marx and co-workers demonstrated that KTq polymerase and the exonuclease deficient Kf(exo-) of E. coli could handle much larger modifications on altered dNTPs. Using this knowledge, they developed a naked-eye detection system using horseradish peroxidase for visualizing single nucleotide incorporation. DeoxyTTP connected to a horseradish peroxide, a 40 kDa glycoprotein, at the C5 position was successfully incorporated into DNA with KTq and Kf polymerases and into RNA using RT-KTq DNA polymerase on an E. coli 16S ribosomal RNA as the template. The presence of a thiol on the modified dTTP was designed to react with a maleimide conjugated to a horseradish peroxidase (Fig. 2C).15 In light of therapeutic development and drug target testing, Srivatsan and Tor synthesized a fluorescent bacterial A site, the aminoglycoside 16S ribosomal RNA target, using a furan-modified UTP. Binding of aminoglycosides to the A-site active site resulted in a change in fluorescence and a binding affinity that could be measured.48 Sandin et al. demonstrated the incorporation of fluorescent dCTP analogs in DNA by E. coli Kf, enabling direct visualization of the polymerase activity and investigation of polymerase flexibility towards dNTPs with large and structurally distinct nucleobase.49 Similarly, Caton-Williams et al. determined that 4-seleno-dTTP could provide a direct and visible readout of polymerase activity. Replacement of the oxygen atom by the selenium did not impact polymerase activity by promoting native hydrogen bonding between Se and the NH group from the polymerase. This 4-seleno-dTTP could be applied to nucleic acid structure determination by X-ray crystallography using multiwavelength anomalous dispersion (MAD) phasing.50
The incorporation of bioorthogonal handles into NTPs for copper-catalyzed alkyne-azide cycloaddition (CuAAC) and copper-free strain-promoted azide–alkyne cycloaddition (SPAAC) is beneficial for visualizing and investigating enzyme activity with minimal disruption due to their small size.18,51–53 For example, Zheng et al. developed 7-ethynyl-8-aza-7-deaza ATP (7-EAATP) to localize and track events related to poly(A) polymerase, a bacterial protein that catalyzes the addition of adenine nucleotides to the end of mRNA during mRNA maturation. Incorporation of 7-EAATP at the 3′-end of RNA was observed via CuAAC with biotin-azide but at a lower level than native ATP.54 An interesting earlier example of photoactivable ATP derivative, 8-[(4-azidophenacyl)thio] ATP (8-APAS-ATP), for exploring the interactions between an incoming NTP into RNAP through crosslinking, was devised by Costas et al. Although 8-APAS-ATP could not be used as an initiation substrate for transcription, this analog could be used as an elongation substrate with the simultaneous addition of a small amount of ATP. Molecular modeling studies revealed that 8-APAS-AMP rotated from a syn to an anti conformation when incorporated into the RNA transcript. The fact that free 8-APAS-AMP preferred a syn conformation may have contributed to its inability to act as an initiation substrate for RNA polymerase.55
2.6.2. Non-natural enzymes and aptamer design for in vitro selection applications.
To generate DNAzymes (single-stranded DNA with novel catalytic activity) or aptamers (single-stranded oligonucleotide chains which can bind to a specific target) for in vitro selection applications, such as Systematic Evolution of Ligands by EXponential enrichment (SELEX), nucleobase-modified dNTPs have been evaluated with a myriad of DNA polymerases for DNA incorporation and primer extension.56 In this context, Perrin and co-workers synthesized 5-aminomethyl dUTP derivatives further functionalized on the amine handle with aromatic and hydrophobic groups and evaluated their incorporation into DNA fragments with a series of bacterial, archaeal and bacteriophagic DNA polymerases (Fig. 6A). Archael Vent(exo-) DNA polymerase was found to be superior to the other enzymes tested as it accepted all derivatives evaluated to successfully carry out full length DNA elongation.57 Furthermore, this enzyme was used to incorporate a 5-(4-hydroxybenzamide)-modified dUTP instead of dTTP into DNA aptamers (Fig. 6B). Ultimately, using the SELEX methodology, an aptamer acting like an antibody was selected to detect the non-pathogenic E. coli strain DH5α.58 A series of polymerases from bacterial family A (Taq and Tth) and archaeal family B [Pfu, Pwo, and Vent(exo-)] were evaluated for their efficiency at producing PCR products with dUTPs and dCTPs containing a flexible and hydrophilic 7-amino-2,5-dioxaheptyl linker located at the C5 position on the nucleobase (Fig. 6C). Kuwahara et al. determined that the levels of incorporation of altered dCTPs and dUTPs and subsequent amplification of the corresponding DNA fragments correlated with the nature of the C5 group on the pyrimidines. While the long flexible 7-amino-2,5-dioxaheptyl modification was only accepted by members of the family B polymerases, shorter and more rigid groups (propynyl and methyl groups) except for 5-propynyl dCTP were utilized by the bacterial and archaeal polymerases. These results indicated that steric hindrance partially dictates selectivity.59 Numerous studies have been performed for the systematic evaluation of modified NTPs for generating novel nucleic acids towards in vitro selection applications. In a review focused on in vitro selection techniques, Dellafiore et al. described in detail the use of modified NTPs in the context of SELEX, aptamer, DNAzymes and ribozyme design. They focused on the elaboration of novel mutant enzymes for the accommodation of modified NTPs.60 A complementary review by Hollenstein encompassed the synthesis of modified dNTPs and their applications for SELEX, along with the design of DNAzymes and aptamers.1
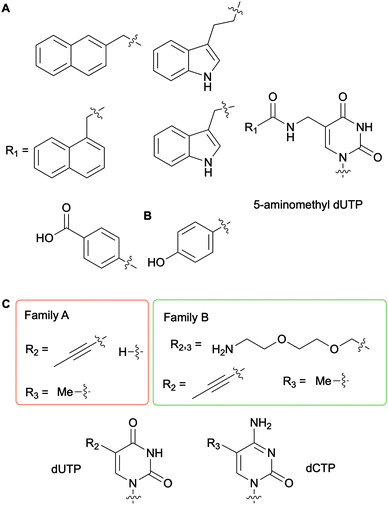 |
| | Fig. 6 Examples of nucleobase-modified dUTPs and dCTPs used in the context of in vitro selection applications. | |
2.6.3.
In vivo applications of nucleobase-modified dNTPs.
Numerous studies involving modified dNTPs have resulted in the expansion of the genetic alphabet and its use in living organisms. Understanding how organismal machinery can handle non-natural NTPs and function in the presence of alterations is the key for establishing an autonomous entity relying on non-native nucleic acids.61–64 Marlière et al. studied the impact of nucleobase-modified dNTPs on mechanisms such as NTP metabolism and bacterial adaptation and survival. They devised the first evolved E. coli capable of survival on three canonical bases A, C and G and one non-canonical base 5-chlorouracil resulting in the replacement of 90% of all thymidine with chlorodeoxyuridine in its DNA.65 Similarly, Schultz and coworkers successfully replaced 75% of thymidine in an autonomous E. coli with 5-hm-deoxyuridine after phosphorylation to the corresponding dUTP, exploiting the endogenous bacterial machinery and plasmids encoding for a hydroxymethylase from SPO1 bacillus phage gp29 and a mononucleotide kinase from T4 bacteriophage gp1.66 Additionally, 5-hm-cytidine was successfully incorporated into the E. coli genome instead of 2′-deoxycytidine up to 63%. These results demonstrated that an organism could survive with a non-canonical base.67 Romesberg and co-workers have designed numerous unnatural base pairs for expanding the genetic alphabet (see Section 2.2 of this review). They found that hydrophobicity and packing forces were important characteristics of modified nucleobases for successful DNA incorporation, primer extension and transcription.61,68 They showed primer extension in the presence of the unnatural base pair dTPT3–dNaM and PCR-amplification of the template containing the dTPT3–dNaM pair by Taq with high fidelity and efficiency.68,69 A different unnatural pair d5SICS–dNaM was incorporated into different plasmids which was in turn transformed into a semi-synthetic E. coli (Fig. 7A). This plasmid was subsequently replicated by the endogenous bacterial machinery using the unnatural dNTPs imported via NTP importers called PtNTT2 encoded in the organism (see Section 5.2 of this review).70,71 This was the first reported organism to function with an unnatural base pair. Optimization of this semi-synthetic organism yielded improved retention of the unnatural bases in the DNA sequences.71,72 Using the unnatural pair dNaM–dTPT3 and the semi-synthetic organism E. coli, Zhang et al. demonstrated for the first time the expression of a superfolder green fluorescent protein containing non-canonical amino acids at specific locations after successful DNA incorporation of unnatural dNTPs, transcription and translation.73 Herdewijn and co-workers have shown that the presence of non-canonical bases in a plasmid coding gene did not prevent E. coli from successfully recognizing the morphed gene, replicating it utilizing natural NTPs, and expressing the encoded protein. E. coli is sensitive to trimethoprim through the inhibition of its dihydrofolate reductase (DHFR). As a proof-of-concept, they created a morphed gene coding for a structurally different DHFR to the native E. coli DHFR, R67DHFR, but that was not inhibited by trimethoprim. After transformation of the plasmids containing the morphed gene, the bacterial machinery successfully replicated the plasmids and expressed the functional R67DHFR protein, conferring E. coli with resistance to trimethoprim (Fig. 7B).74,75 All of these studies have paved the way to the design of autonomous bacteria that live with modified genetic codes.
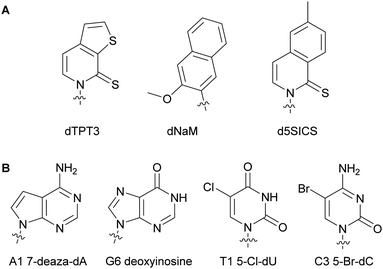 |
| | Fig. 7 Examples of structures of nucleobase-modified dNTPs successfully used in an autonomous E. coli. | |
3. Modifications on the sugar group of NTPs
NTPs have been modified to bear a sugar distinct from the natural ribose or deoxyribose to enrich the chemical and functional diversity of DNA and RNA segments. Similar to the nucleobase alterations, the principal goals of the sugar modifications are the expansion of the genetic code, the generation of novel enzymes and therapeutics, and the investigation of the rules of NTP enzymatic incorporation during transcription and translation. These sugar-modified NTPs are the basis for xeno-nucleic acids (XNAs), which are nucleic acid analogs that have a different sugar backbone and overall structure than canonical nucleic acids. XNAs can store information, manipulate polymerase activities and be used to evolve bacterial polymerases.76–78
3.1. Sugar-modified NTPs for the study of bacterial polymerases
The substrate specificity of DNA polymerases relies on the evaluation of the incoming NTP 2′- and 3′-positions by the enzyme active site.79 For instance, the exclusion of RNA NTPs from bacterial DNA polymerases is achieved by a steric gate that prevents NTPs with a 2′-OH from accessing the catalytic domain. The polymerase family determines whether the steric gate is composed of a tyrosine, phenylalanine or glutamic acid.80,81 Overall, DNA polymerases are less amenable to sugar and phosphate-modified NTPs than nucleobase-modified NTPs.7 Nevertheless, numerous NTPs with structurally modified sugars have been evaluated as polymerase substrates (Fig. 8).82 One method to study the impact of sugar-based modifications on the activity of bacterial polymerases is to explore sugar size and ring enlargement. Herdewijn and co-workers assessed whether DNA segments containing cyclohexenyl (CE), a 6-membered ring sugar, as backbone could be recognized and replicated by different polymerases, including bacterial Taq. Their goal was to develop an autonomous in vitro replicating system where polymerases were evolved to solely recognize a CE-sugar backbone. Although a CE-based DNA strand could be successfully copied by Taq and other polymerases, the incorporation of CE-dNTPs into a DNA strand was limited and was not observed opposite of a CE-based DNA strand for Taq. The authors attributed these observations to the larger size of the CE ring compared to the natural ribose, which caused steric hindrance in the polymerase active site or the incapacity of the CE to undergo the conformational changes required for the polymerase to catalyze the reaction (Fig. 8A).83 Similarly, Pochet et al. evaluated hexitol-based dTTP for the development of a novel nucleic acid due to its 3D-structural similarity to ribose. Primer extension from a hexitol-based DNA template by Kf(exo-) and Taq was observed for a maximum of 4 nucleotides, and a hexitol-modified DNA template could be used to consecutively polymerize a maximum of three hexitol-modified dTTPs (Fig. 8A). Ultimately, they recovered thymidylate synthase activity encoded in an E. coli plasmid, which had been inactivated due to the deletion of 2 codons within this gene, by replacing it with the corresponding hexameric hexitol template. These studies demonstrated that hexitol-based nucleic acids could transmit genetic information within a natural environment.84
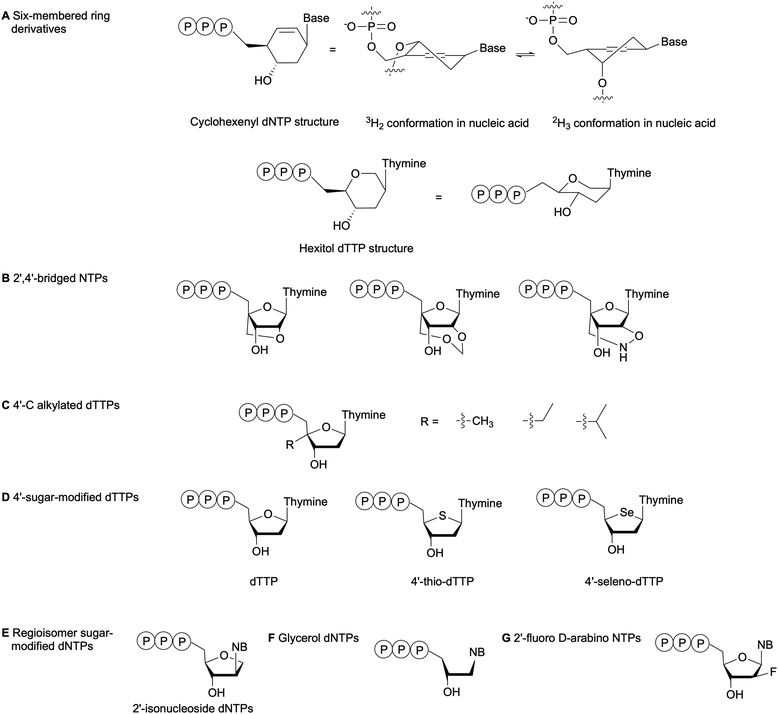 |
| | Fig. 8 Examples of sugar-modified NTPs evaluated to understand DNA polymerase sensitivity to sugar modifications. NB: nucleobase. | |
Kuwahara et al. evaluated the effects of 2′,4′-bridged NTPs that have been incorporated into a DNA template with various spacing for primer elongation using a series of DNA polymerases, including family A Taq and family B Vent(exo-) and KOD Dash (Fig. 8B). Full-length products were observed in high yields with Taq only when the sugar-modified nucleotides were spaced every 2 or 3 nucleotides. They also showed that none of the tested polymerases could perform complete primer elongation on a DNA template containing seven consecutive 2′,4′-bridged NTPs at the elongation site. Therefore, utilizing this type of modified NTPs for in vitro applications such as SELEX would be challenging.85 Marx and co-workers have explored the incorporation of 4′-modified dTTPs into DNA by E. coli DNA polymerases (Fig. 8C). These modifications located in the minor groove of DNA resulted in increased steric hindrance and limited the flexibility of the incoming modified dTTP within the narrow binding pocket of the DNA polymerase. These studies highlighted the importance of the interactions between the sugar backbone and the enzyme. Moreover, the active site tightness and steric constraints were found to promote dNTP selectivity and, thus, DNA polymerase fidelity.86–89 Tarashima et al. synthesized 4′-thio-dTTP (STTP) and 4′-seleno-dTTP (SeTTP) for primer extension by bacterial and archaeal DNA polymerases (Fig. 8D). Both Kf(exo-) and Taq showed limited primer extension, whereas extension was observed with archaeal polymerases. Kinetic analysis revealed that SeTTP and STTP were poorly recognized by Kf(exo-).90 These observations were attributed to the loss of interactions between the modified dNTP and the enzyme, resulting from the high steric barrier for conformational changes when the oxygen atom in the carbohydrate ring was replaced by a selenium or sulfur atom.90 Ogino et al. evaluated the incorporation of 2′-isonucleoside dNTPs (Fig. 8E), regioisomers of dNTPs, into DNA by family A and B polymerases as potential oligonucleotide therapeutics. Their studies attributed poor primer extension by Kf(exo-) or family A polymerases compared to family B polymerases to differences in the recognition mechanism. Family A polymerases identify the shape of the incoming dNTP, whereas family B polymerases recognize the DNA template-primer complex.91,92 Glycerol-based dNTPs (gNTPs) have also been evaluated as DNA polymerase substrates since glycerol-based nucleic acids (GNAs) form stable duplexes constructed on Watson–Crick base-pairing (Fig. 8F). Chen et al. showed that primer extension with gNTPs by bacterial polymerases Taq and Bst on a native DNA template was limited to only one or two additions, compared to up to five additions with therminator polymerase. Moreover, none of the tested polymerases could synthesize GNAs in the presence of a GNA template with gNTPs. This result was attributed to a conformational difference between the DNA duplex A-form and the GNA duplex structure.93 With the goal of designing and synthesizing aptamers for mRNA gene silencing, Peng et al. tested several bacterial family A and family B DNA polymerases for full length DNA synthesis using 2′-fluoro D-arabino-NTPs (Fig. 8G). While family B polymerases successfully incorporated and generated full-length aptamers, bacterial polymerases produced only truncated products. This indicated that great structural differences exist between the active sites of these enzyme families.94
This section reviewed sugar-modified NTPs used in the context of bacteria. A review from Dellafiore et al. includes other sugar-modified NTPs developed for additional systems.60
3.2. Evolution of bacterial polymerases to achieve sugar-modified NTP recognition
Sugar modifications have a greater impact than nucleobase modifications on bacterial polymerase enzymatic activity and, as a result, sugar-modified NTP recognition by these polymerases is limited.7 Consequently, focus has turned to the evolution of these enzymes to achieve recognition of these artificial substrates.95 Most of the work performed to date has been with archaeal polymerases such as Tgo from T. gorgonarius, which has been evolved to accommodate sugar modifications and XNA.77 Nevertheless, examples of evolved bacterial polymerases have also been reported.95,96 Leconte and coworkers studied the incorporation of 2′-sugar-modified NTPs by evolved Taq polymerase mutants and evaluated the structural components that yielded the observed results. They investigated the impact of a fluorine atom, an azido, a methoxy or an amino group on nucleotide recognition in mutants AA40, SFR3 and SFM19 previously designed to use 2′-modified NTPs. They found that SFM19 was able to recognize and incorporate the largest range of 2′-modified NTPs due to two mutations. First, the replacement of a neutral isoleucine with glutamic acid suggested that the presence of a negative charge affected recognition. Second, the replacement of a glutamate residue with glycine eliminated the steric gate, which enabled the enzyme to accommodate the presence of a group at the 2′-position.97 Modifications of the 2′-position of NTPs with a methoxy group or a fluorine atom are of interest as they make oligonucleotides containing these modifications resistant to nucleases, which is crucial to the design of stable aptamers for therapeutic purposes.98 In this context, Chen et al. have reported an improved method for the design of mutated polymerases, based on the structure of the Stoffel fragment (SF) of Taq polymerase, that are capable of recognizing and incorporating 2′-methoxy or 2′-fluoro NTPs into nucleic strands for transcription and reverse transcription as well as PCR applications.98 This method was initially devised for evolving SF as an RNAP.99 The polymerase SFM4-3 was the first example of an evolved enzyme capable of PCR amplification of oligonucleotide strands containing any type of 2′-modifications.98 In the context of SELEX and aptamer design, Lauridsen et al. have published a thorough review about sugar-modified NTPs and RNA and DNA polymerase recognition mechanisms by natural and evolved polymerases from diverse organisms.100
3.3. Chemical biology applications of sugar-modified NTPs
Visualization of enzyme activity is one of the applications where modifications on the NTP sugar are beneficial. The addition of affinity or fluorescent tags to RNA by CuAAC and SPAAC is the method of choice for RNA visualization and investigation of RNA-related enzymatic mechanisms.101 To develop a RNA functionalization technique for visualization purposes, Jäschke and co-workers inserted an azide at various positions on NTPs to study their incorporation into RNA by a series of nucleotidyl transferases, including E. coli poly(A) polymerase (PAP). The bacterial enzyme successfully added 2′-azido-2′-dNTP to the 3′-end of RNA, and the RNA was subsequently conjugated to a fluorophore by CuAAC and SPAAC. Polyadenylation of an RNA template containing a 2′-azido-modification at the 3′-end proceeded almost quantitatively with E. coli PAP. Moreover, kinetic control of the PAP step permitted the addition of only one modified NTP site-specifically.102
A unique bacterial DNA polymerase of particular interest is Bst from G. stearothermophilus, an enzyme capable of performing reverse transcription activity on native and non-native RNA containing sugar-modified nucleic acids (i.e., glycerol, 2′-fluoro-D-arabino). To gain insight into the molecular mechanisms that control the accommodation of sugar-modified NTPs, Chaput and coworkers obtained crystal structures of Bst in the presence of sugar-modified nucleic acids during post-translocation of the DNA products from XNA templates [threose-based nucleic acid (TNA) and 2′-fluoro D-arabino-based nucleic acid]. They found that the Bst active site was highly flexible compared to other family A DNA polymerases such as Taq. This allowed the sugar-modified XNA to establish extra points of contact with the enzyme, demonstrating that its activity is driven by template recognition.5 They also combined the threose modification with a fluorescent phenothiazine as a nucleobase to devise a novel threose-based CTP (tCfTP). Successful transcription of tCfTP-containing TNA by KOD-RI, a mutant of archaeal T. kodakarensis, followed by reverse transcription with Bst DNA polymerase yielded a high-fidelity rate of 98.4%. This validated tCfTP as a suitable replacement for threose-based CTP. Moreover, this analog presented additional beneficial properties, including improved hydrophobicity and base stacking character that was not observed with canonical bases. This base could be used as an internal visualization system for polymerase activity.103 A similar approach was used to design fluorescent TNA with a 7-deaza-7-modified threose-based GTP.104 DNA/RNA sequencing have benefited from the use of modified NTPs. Initially, Sanger sequencing relied on dideoxyribo NTPs as strand terminators and the Kf from E. coli.105 The development of fluorescent dideoxyribo NTPs, as well as novel detection methods resulted in the automation of Sanger sequencing.105 To reduce the cost of Sanger sequencing, Martinez et al. evaluated several alternatives to dideoxyribo NTPs including both allylic/acyclic and acyclic NTPs as DNA polymerization terminator with Taq and other DNA polymerases. Nevertheless, limited incorporation of these modified NTPs by DNA polymerases was observed.106,107 Reversible terminators modified at the 3′-position with a hydroxylamine have shown potential for next-generation sequencing.108 In this context, Chen et al. discusses the use of NTPs containing reversible terminators with diverse DNA polymerases for sequencing by synthesis.109
4. Modifications on the phosphate tail of NTPs
A myriad of NTPs with modified triphosphate tails have been generated to study critical bacterial processes such as metabolism, signal transduction, and energy transfer. Modifications are located at various positions on the phosphate tail depending on the intended application, since the triphosphate moiety can be hydrolyzed at differing phosphodiester bonds to power enzymatic activity. For instance, during the synthesis of nucleic acids, DNA and RNA polymerases catalyze transfer of the α-phosphate at the 3′-end of the primer by releasing a pyrophosphate group.110 In two-component systems (TCSs), which are the major bacterial signaling pathways, information is transferred inside of the cell by shuttling the γ-phosphate of ATP between several catalytic residues.111
Radioactive NTP isotopes are commonly used to evaluate phosphate-related processes. They are structurally “equivalent” to natural NTPs and, thus, are recognized by the enzymes as near-native substrates. Moreover, radioactive assays provide a direct readout of the enzyme activity and are typically more sensitive than fluorescent-based assays. Consequently, this is a method of choice for mechanistic studies under native conditions and for determining enzymatic kinetic parameters.112 For instance, radioactive [α-32P]dNTPs and [α-32P]NTPs have been utilized to investigate DNA110,113 and RNA114,115 polymerases, respectively. Radioactive [γ-32P]ATP and less energetic [γ-33P]ATP have been utilized to investigate the phosphorylation of numerous TCSs, as well as histidine kinase (HK) phosphatase activity since the γ-phosphate is transferred between proteins.116–121 Nevertheless, the safety hazards inherent to the use of radioactive materials, as well as their limited scope for use in whole cells, has resulted in the development of modified NTPs with various non-radioactive tags on their phosphate tail.
4.1. Modifications at the α-position of the phosphate tail
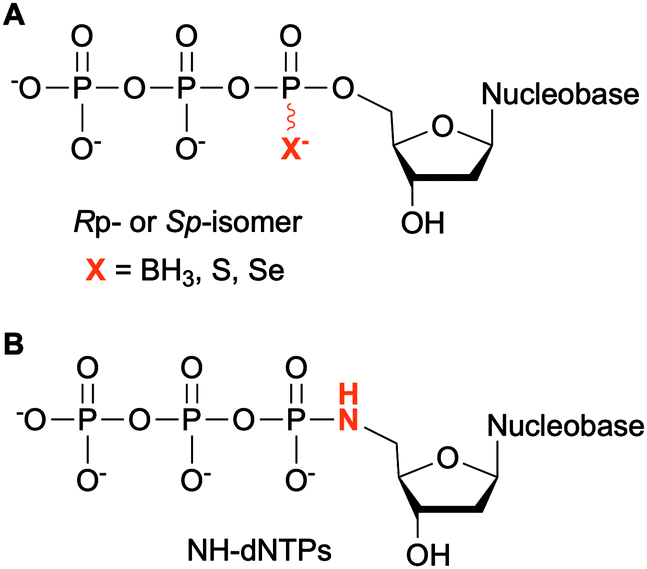
Modifications at the α-phosphate position have been investigated in the context of nucleic acids. For example, replacement of the α-non-bridging oxygen atom by a sulfur atom was implemented by Seo et al. to assess the incorporation of α-thiophosphate dNTP derivatives by DNA polymerases into DNA, creating a phosphorothioate backbone. These modified dNTPs caused minimal disturbance to Kf activity. Efficient incorporation into DNA segments was observed as well as their subsequent amplification with high fidelity and efficiency. Moreover, the sulfur atom conferred the newly formed modified DNA with a chemical handle, enabling the installation of a reporter group for analytical purposes.122 The phosphorothioate DNA backbone also provides stability towards nucleases, an important feature for therapeutic applications.123 Dobrikov et al. evaluated the incorporation of α-P-thio- and α-P-borano-dNTPs by Kf and Taq polymerases (Fig. 9A). Two stereoisomers of these modified dNTPs were formed, the Rp- and the Sp-isomers. They found that Sp-dNTPαBs were not suitable as substrates of bacterial DNA polymerases. Kf and Taq incorporated Rp-dNTPαB into DNA on average 3 and 5 times less efficiently, respectively, than natural dNTPs. For Kf, Sp-dNTPαSs were better substrates than the stereochemically equivalent Rp-dNTPαBs, but not to the level of native dNTPs. These results were explained by the lower electronegativity of the sulfur and the boron atoms compared to the oxygen atom, decreasing the rate of nucleophilic attack by the 3′-hydroxyl group of the DNA strand.124 The presence of boron in the backbone of DNA at the 3′-end increased resistance to ExoIII exonuclease from E. coli, especially if this modification was coupled with an alkyl group at the 5-position of the dCTP.125 To facilitate the structural determination of nucleic acids in complexes with polymerases using X-ray crystallography with MAD phasing contrast, Carrasco et al. substituted the non-bringing α-oxygen atom of dTTP with a selenium atom and showed quantitative incorporation of both stereoisomers into a DNA strand by Kf (Fig. 9A). The presence of selenium in the DNA backbone had an inhibitory effect against E. coli ExoIII.126 In contrast, Hu et al. found that only Sp-dNTPαSe were recognized by DNA polymerase I and Kf from E. coli, and Bst from B. stearothermophilus for primer extension. PCR amplification of seleno-based DNA (SNA) with Sp-dNTPαSe followed by sequencing demonstrated sequence integrity indicating that SNA and Sp-dNTPαSe could be used instead of native DNA and dNTPs. Interestingly, the use of dNTPαSe compared to native dNTPs suppressed non-specific DNA polymerization.127
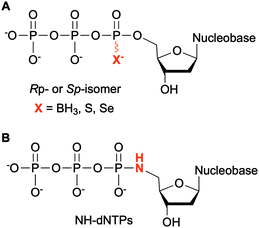 |
| | Fig. 9 Modifications at the α-phosphate of non-natural NTPs. | |
NTPs bearing a 5′-amino group have also been evaluated for DNA incorporation due to their enhanced reactivity compared to the native hydroxyl group and potential use for genome sequencing.128 Wolfe et al. demonstrated that all four 5′-amino dNTPs (NH-dNTPs, Fig. 9B) were easily incorporated by Kf of DNA polymerase I. They noted that the polymerization occurred more efficiently at basic pH. Indeed, the phosphoramidate bond is known to be unstable at acidic and neutral pH values. The authors utilized this property to develop sequence ladders.129 With the intent of engineering biosafe genetically modified organisms using novel and degradable genetic material, Nguyen et al. investigated NH-dCTP for the synthesis of phosphoramidate-based DNA that was susceptible to degradation under mildly acidic conditions (Fig. 9B). Although primer extension by Taq and Kf of DNA polymerase I from E. coli using NH-dCTP as a substrate was successful, PCR amplification of NH-dCTP-containing DNA by Taq was not. Nevertheless, an alternative cloning method based on the assembly of single-stranded phosphoramidate-containing DNA fragments was used to construct a coding region in a plasmid for trimethoprim (Tmp) resistance using NH-dCTPs. Successful transformation into E. coli and expression of the Tmp gene demonstrated the possibility of using phosphoramidate-modified dNTPs as a novel and degradable alphabet.76,130 Propagation of XNAs in the environment could lead to gene transfer and adaptative mechanisms especially in bacteria. Therefore, precautions should be taken in parallel to the development of XNAs and autonomous organisms living on XNAs. Phosphoramidate-based XNA provides a key advantage as it is easily degraded and its spreading between organisms would be limited.
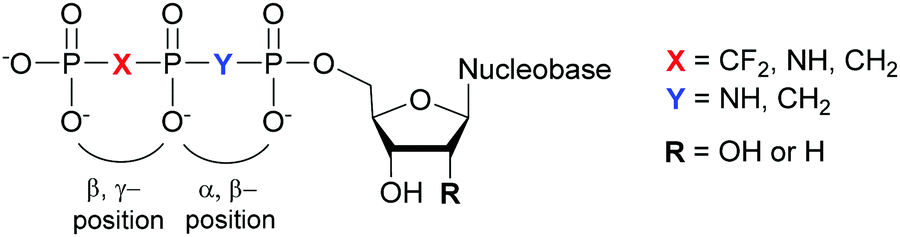
4.2. Modifications at the α,β- and β,γ-bridging atoms of the NTP phosphate tail
Structural investigation of enzymes by nuclear magnetic resonance (NMR) or X-ray crystallography benefits from the use of non-hydrolyzable NTP analogues to visualize the enzyme–NTP complex without substrate turnover. For example, an ATP analog, α,β-methylene-ATP (AMP-CPP), has been utilized to determine the catalytic mechanism of E. coli MutT pyrophosphohydrolase by NMR (Fig. 10).131 Barabás et al. crystallized an E. coli dUTPase in the presence of α,β-imido-dUTP to investigate the removal of dUTP from DNA to maintain its integrity (Fig. 10).132 Marina and coworkers employed β,γ-imido-ATP (AMP-PNP) to explore the binding of ATP in the ATP-binding pocket of bacterial HKs, the enzymatic portion of the TCSs, during phosphotransfer to the catalytic histidine (Fig. 10). They successfully deciphered the stepwise HK autophosphorylation mechanism.133 AMP-PNP and another ATP-analog, β,γ-methylene-ATP (AMP-PCP), have been exploited to study enzymatic-mediated histidine mechanisms either by X-ray crystallography134,135 or by biochemical assays (Fig. 10).136 Other NTP analogs found include α,β-methylene-CTP (CMP-CPP) with E. coli RNAP137 for biochemical assays, and β,γ-difluoromethylene-ATP (AMP-PCF2P) with biotin carboxylase from E. coli,138 β,γ-imido-GTP (GMP-PNP) with the GTPase domain of Ffh from T. aquaticus,139 and α,β-imido-dGTP (dGMP-NPP) with KTq from T. aquaticus140 in X-ray crystallography studies (Fig. 10).
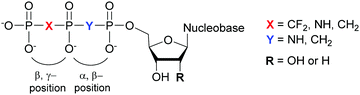 |
| | Fig. 10 Possible bridging atom modifications present in NTP analogs developed to study bacterial enzymatic processes. | |
4.3. Modifications at the γ-position of the NTP phosphate tail
NTPs modified at the γ-position are commonly employed to study the activity and catalytic mechanisms of enzymes such as kinases.
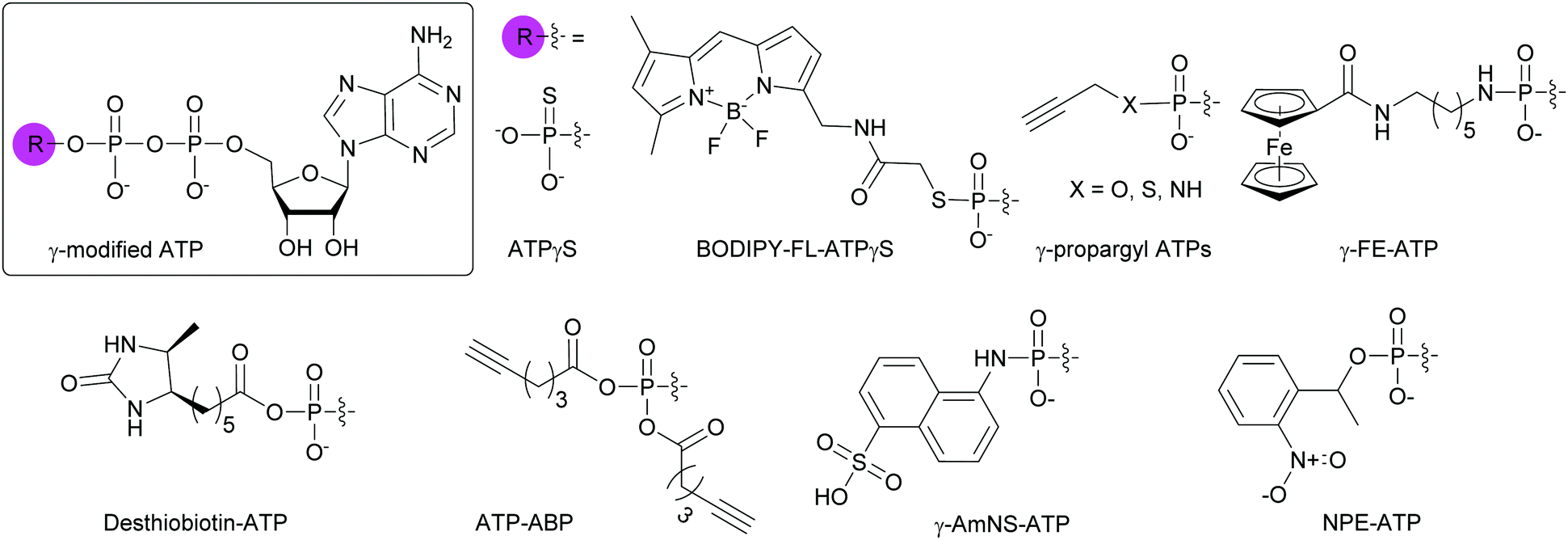
4.3.1. ATP molecules with a γ-modified phosphate.
Due to the abundance of ATP-binding proteins in bacteria including HKs and ATP-binding cassette transporters, numerous γ-modified ATP analogs have been investigated. Some of these proteins are critical to bacterial survival and pathogenesis.141,142 Our research group and others have developed and utilized γ-modified ATP molecules as a means to globally investigate bacterial TCS signaling. One of the major hurdles for the study of HKs is the instability of the phosphohistidine species to hydrolysis at the phosphoramidate bond between the nitrogen atom of the catalytic histidine and the phosphorus atom of the γ-phosphate. To circumvent this issue, ATPγS, an ATP analog in which one of the terminal oxygen atoms had been replaced by a sulfur atom, was shown to form thiophosphohistidine (Fig. 11). The presence of a sulfur conferred increased stability of this species due to the lower electronegativity of the sulfur atom compared to oxygen.143 A biochemical assay using ATPγS was developed by Marletta and co-workers to detect phosphorylated HKs and their cognate response regulators (RR). After phosphorylation of the histidine or aspartate residues, the thiophosphate was further alkylated using para-nitrobenzylmesylate. These thiophosphate esters could then be immunodetected.144 BODIPY-FL-ATPγS (B-FL-ATPγS), a fluorescent ATP analog, was the first non-radioactive ATP probe employed for evaluating HK autophosphorylation activity through direct readout (Fig. 11). Using the HK/RR pair HK853/RR468 from Thermotoga maritima, we demonstrated that transfer of the modified γ-phosphate from the HK to the conserved aspartate residue of the cognate RR was possible. Typically, this transfer event is fast (seconds to minutes) but the presence of a sulfur slowed the phosphotransfer event, enabling visualization over a few hours.119 Further mechanistic studies using radioactive [γ-33P]ATP and its analog [γ-35S]ATP, as well as B-FL-ATPγS were implemented to understand the slow kinetics observed with B-FL-ATPγS. We found that its large fluorescent group impacted the autophosphorylation kinetics much more than the decreased electronegativity of the sulfur atom, resulting in a reduction in catalytic efficiency by two orders of magnitude compared to native ATP.116 To overcome this challenge, we pursued ATP analogs with decreased bulk at the γ-position by replacing the B-FL moiety with a propargyl group for versatile purposes through conjugation to chemical handles using CuAAC. Moreover, these derivatives presented a γ-amino-, γ-thio-, or γ-hydroxyphosphate (Fig. 11). By combined experimental and computational studies, we sought to rationalize the effects of atom size and electronegativity, as well as alkyl group on HK autophosphorylation and phosphohistidine hydrolysis. We found that reaction sterics and recognition of the ATP analog by the HK drove histidine phosphorylation kinetics and that the free energy of the hydrolysis products determined phosphohistidine stability. Alkylation provided increased stability only under very acidic conditions (pH = 1).120 Kraatz and coworkers developed a 5′-γ-ferrocenyl-ATP (γ-FE-ATP) containing a γ-aminophosphate for monitoring bacterial HK activity electrochemically and by immunodetection (Fig. 11). Interestingly, they found that the γ-FE-ATP concentration affected autophosphorylation efficiency. They explained this observation by the fact that at certain concentrations of Fe2+ cations or ferrocenyl groups, these moieties may have an inhibitory effect on the proteins by interacting with their sensory domain.145 Despite these advances, radioactive ATP derivatives remain the most widely implemented method for the study of HK and RR activity.144,146,147
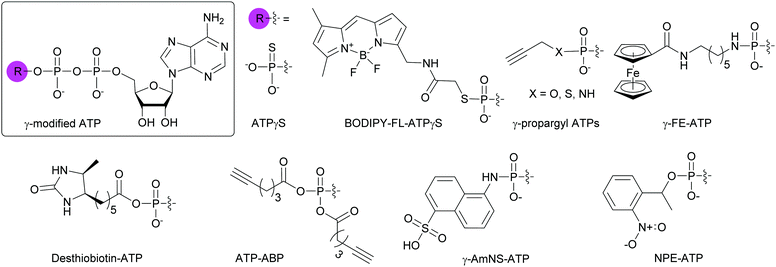 |
| | Fig. 11 Structures of γ-modified ATP analogs for the study of bacterial ATP-binding proteins. | |
To globally map the Mycobacterium tuberculosis ATP-binding proteome and identify potential ATP-related targets for the development of novel antibacterial drugs, Wolfe et al. used desthiobiotin-ATP, a lysine reactive acyl ATP, in a chemoproteomic approach (Fig. 11).148 In the case of acyl phosphate probes, only the acyl group present on the modified γ-phosphate is transferred to the protein, more specifically, to a lysine in the active site of ATP-binding proteins. Similarly, Wright and co-workers used an alkyne-based probe called ATP-ABP to validate over 250 proteins and identify 72 hypothetical proteins as ATP-binding enzymes, with some involved in the pathogenesis of M. tuberculosis (Fig. 11).149 A fluorescent γ-modified ATP-analog, γ-(sulfo-1-naphthyl)amide ATP (γ-AmNS-ATP) was found to be a substrate for RNAP and valyl-tRNA to a lower extent, but not for alkaline phosphatase, adenyl kinase and acetate synthetase kinase from E. coli (Fig. 11).150 This analog was used as a sensitive tool to measure RNA transcription by E. coli RNAP.151 γ-AmNS-ATP has also been utilized to evaluate nucleotide binding to an intrinsically fluorescent protein called isocitrate dehydrogenase kinase/phosphatase from E. coli and to elucidate the catalytic mechanism through changes in protein or γ-AmNS-ATP fluorescence upon binding to the active site.152
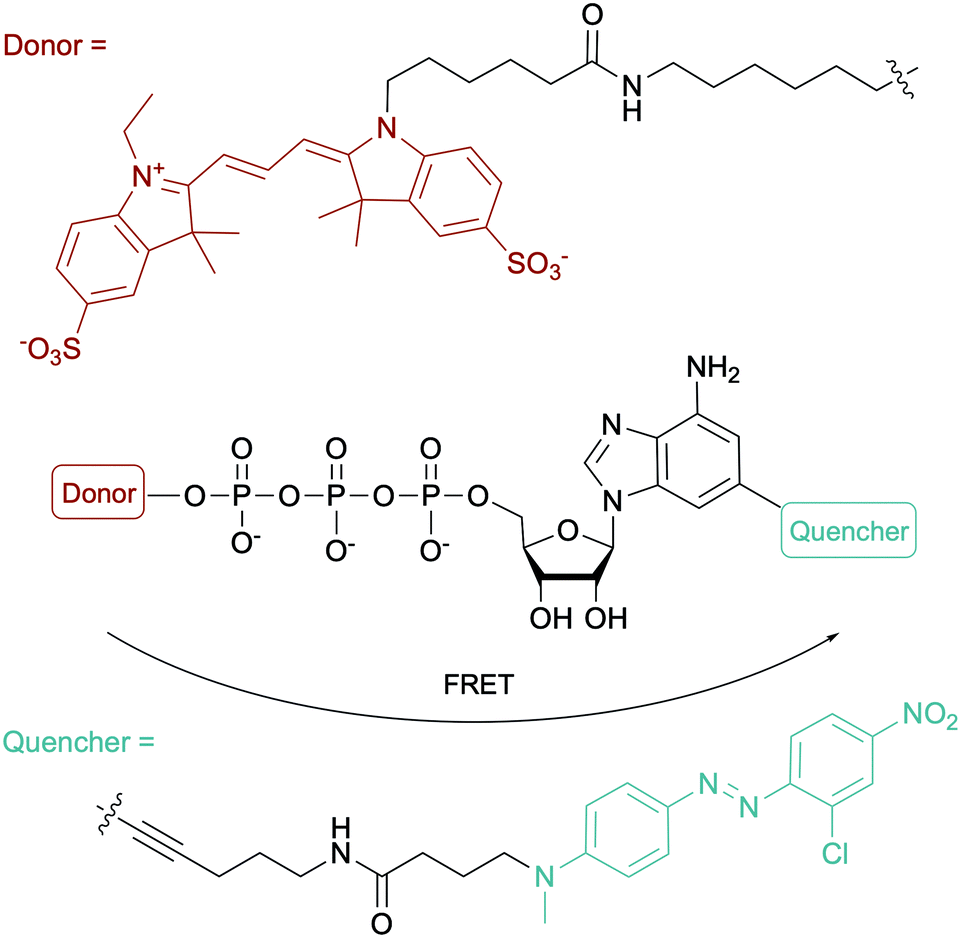
To evaluate the ATP-dependent carbonylation reaction used by Desulfococcus biacutus to degrade acetone, Marx and co-workers devised a Förster-resonance energy transfer (FRET)-based assay using a dual-modified ATP with groups on the γ-phosphate (FRET donor) and the 2-position of the adenine base (FRET quencher). They determined that thiamine pyrophosphate was involved in the carbonylation reaction (Fig. 12).153 For more ATP-related molecules used not only in bacterial systems, Pflum and co-workers reviewed ATP analogs modified at the nucleobase, sugar or phosphate position in the context of probing eukaryotic kinase proteins.154
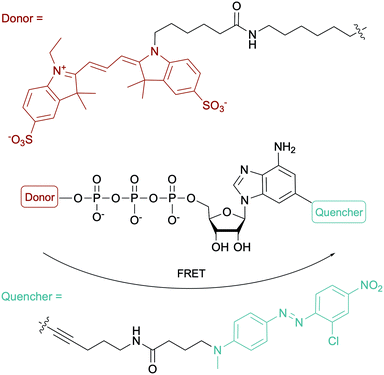 |
| | Fig. 12 Structure of the fluorogenic probe used in a FRET-based assay for monitoring an ATP-dependent carbonylation reaction in D. biacutus. | |
4.3.2. Other NTP molecules with a γ-modified phosphate.
In addition to the large body of literature related to γ-modified ATPs, other γ-modified NTPs have been investigated. For instance, Serdjukow et al. described the preparation of dNTPs and NTPs modified at the γ-position with fluorescent groups. They were synthesized through CuAAC by coupling the aminobutynyl group on the dNTP γ-phosphate with a fluorescent azide. Their incorporation into full DNA strands by DNA polymerase Kf(exo-) was quantified and full primer extension was observed with all dye-labeled dNTP analogs tested, except fluorescein-dTTP. The presence of an alkyne group provided flexibility for accessing various fluorophore-γ-labeled NTPs, a strategy that could be further employed in a situation-dependent manner.155
4.4. Chemical biology applications of phosphate-modified NTPs
A variety of chemical biology applications have utilized phosphate-modified NTPs. Here, we present a small collection to show the breath of possible usages. Fluorescence based-assays are commonly implemented for the high-throughput screening (HTS) of compounds against bacterial targets.156,157 For instance, Bhat et al. put to use γ-AmNS-UTP in a HTS to assess the potency of 670![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compounds against RNAP from E. coli as a mimic of RNAP from M. tuberculosis due to safety considerations. The hits were validated via a complementary radioactive assay using mycobacterial RNAP, with a 20% rate of false positives, possibly explained by the fact that the RNAP from E. coli was used instead of that from Mycobacteria in the initial HTS.158 To prevent nuclease-based degradation of therapeutic aptamers, Gorestein and co-workers designed a library of thioaptamers using dATPαS and dCTPαS in combination with dTTP and dGTP, using Kf polymerase for aptamer replication and Taq polymerase for aptamer PCR amplification. They successfully identified a candidate for inhibiting growth factor-β1, a protein associated with immune system dysregulation and tumor mediation.159 Photocaged molecules are compounds that lose their protecting group (the “cage”) upon controlled irradiation, releasing the molecule of interest. Fendler and co-workers applied this technique with P3-1-(2-nitro)phenylethyl-ATP (NPE-ATP) to study the translocation mechanism of the Na+ active transporter of Ilyobacter tartaricus that is powered by hydrolysis or formation of ATP (Fig. 11). They found that an electrogenic reaction occurred during Na+ transport.160 Likewise, NPE-GTP was used to probe the interactions between two members of the E. coli divisome (FtsZ and FtsA), demonstrating that FtsZ modulated FtsA interactions with the bacterial membrane.161 For more applications of phosphate-modified nucleotides, Hacker and co-workers published a review focusing on protein activity investigation with various analytical techniques in multiple biological systems including the use of nucleotides with longer phosphate chains to study polymerase activity and for next-generation sequencing.162
000 compounds against RNAP from E. coli as a mimic of RNAP from M. tuberculosis due to safety considerations. The hits were validated via a complementary radioactive assay using mycobacterial RNAP, with a 20% rate of false positives, possibly explained by the fact that the RNAP from E. coli was used instead of that from Mycobacteria in the initial HTS.158 To prevent nuclease-based degradation of therapeutic aptamers, Gorestein and co-workers designed a library of thioaptamers using dATPαS and dCTPαS in combination with dTTP and dGTP, using Kf polymerase for aptamer replication and Taq polymerase for aptamer PCR amplification. They successfully identified a candidate for inhibiting growth factor-β1, a protein associated with immune system dysregulation and tumor mediation.159 Photocaged molecules are compounds that lose their protecting group (the “cage”) upon controlled irradiation, releasing the molecule of interest. Fendler and co-workers applied this technique with P3-1-(2-nitro)phenylethyl-ATP (NPE-ATP) to study the translocation mechanism of the Na+ active transporter of Ilyobacter tartaricus that is powered by hydrolysis or formation of ATP (Fig. 11). They found that an electrogenic reaction occurred during Na+ transport.160 Likewise, NPE-GTP was used to probe the interactions between two members of the E. coli divisome (FtsZ and FtsA), demonstrating that FtsZ modulated FtsA interactions with the bacterial membrane.161 For more applications of phosphate-modified nucleotides, Hacker and co-workers published a review focusing on protein activity investigation with various analytical techniques in multiple biological systems including the use of nucleotides with longer phosphate chains to study polymerase activity and for next-generation sequencing.162
5. Challenges and methods for the internalization of NTPs into bacteria
Bacteria cell envelopes are composed of several layers including a peptidoglycan layer and phospholipid membranes. The two major classes of bacteria, Gram-positive and Gram-negative, are differentiated based on the structure of their cell envelopes. The Gram-positive bacteria cell envelope is comprised of a cytoplasmic cell membrane of phospholipids and several peptidoglycan layers containing teichoic acids. In Gram-negative bacteria, the phospholipid membrane is also present but does not contain teichoic acids. The rest of the envelope consists of a thin peptidoglycan layer with an additional outer membrane of lipopolysaccharides and surface proteins.163,164 Negatively charged molecules present in the cell envelope (lipopolysaccharides in Gram-negative bacteria and teichoic acids in Gram-positive bacteria) prevent NTPs from being imported inside most bacterial cells due to negative charge repulsion.165 The biological importance of NTPs for the study of bacterial molecular mechanisms in vivo has led to the development of strategies to facilitate their internalization into cells.
5.1. Natural bacterial NTP transporters
Natural transport of NTPs into bacteria has been reported and was found in organisms that scavenge NTPs from their host due to their inability to synthesize the corresponding nucleotides. Winkler and co-workers reported a carrier mediated transport system called Tcl1 specific for ATP and ADP as energy sources in Rickettsia prowazekii, a Gram-negative bacterium that grows in the cytoplasm of eukaryotic cells and is responsible for epidemic typhus.166,167 This mechanism was coined as “energy parasitism.”168 Other translocases, Tlc 4 and 5, were reported to participate in the import of ribonucleotides for nucleic acid biosynthesis in R. prowazekii.169 These translocases are transmembrane proteins bound to the cytoplasmic membrane of the bacteria.170 Native bacterial ATP transporters, nucleoside phosphate transporters 1 and 2 (Npt1Ct and Npt2Ct), have also been characterized in Chlamydia trachomatis. After successful heterologous expression in E. coli, using radiolabeled NTPs, Npt1Ct was found to catalyze the transport of ATP/ADP (energy transport) whereas Npt2Ct specifically imported ribonucleotides via a proton motive force.171 Similarly, Neuhaus and coworkers studied NTP carriers in Protochlamydia amoebophila related to the Chlamydiaceae family and found related importers (PamNTT1, PamNTT2, PamNTT3, and PamNTT5). PamNTT1 has an identical function to Tcl1 and Npc1Ct. PamNTT2 transports all RNA NTPs such as Npt2Ct. PamNTT3 and PamNTT5 import UTP and GTP/ATP, respectively, using a proton motive force.172
5.2. Expression of non-native bacterial NTP transporters for the internalization of modified NTPs
The absence of NTP importers in most bacteria has been a long-standing hurdle to studying NTP functions in vivo. Recently, with the goal of expanding the genetic alphabet, Romesberg and co-workers engineered semi-synthetic organisms to internalize non-natural NTPs by expressing exogenous importers from other species.70,71,73,173 The importer system was designed from a NTP transporter, PtNTT2, from the algae Phaeodactylum tricornutum that was incorporated into E. coli.73,174 For example, a semi-synthetic E. coli successfully incorporated non-natural NTPs into DNA and transcribed the information, which resulted in the production of a superfolder green fluorescent protein containing non-canonical amino acids.73 Using an uptake inhibition assay with radiolabeled [α-32P]ATP and a LC-MS/MS method, they determined the NTP uptake rates into the cells by PtNTT2 and explored the impact of modifications on the three core moieties of NTPs on their import. They found that groups at the 5-position on the pyrimidine nucleobases and large non-native aromatic nucleobases did not prevent internalization. They also determined that 2′- and 3′-hydroxyl groups were important for recognition by the translocase. Finally, the presence of a β,γ-bridging oxygen in the phosphate tail was deemed more significant than the α- or γ-nonbridging oxygen for internalization, which is rationalized by the fact that this enzyme is inherently specific to NTPs (Fig. 13). Using this importer, [α-32P]-labeled and 2′-fluorinated NTPs were successfully incorporated into RNA and DNA.174 Using a similar approach, Marlière and co-workers designed an E. coli organism that relies on the uptake of dTTP for survival with an exogeneous nucleoside triphosphate transporter (NTT) from Thalassiosira pseudonana for the uptake of dNTPs. Preference for dCTP import was observed over other dNTPs (in order of decreasing affinity dATP, dGTP and dTTP). Import of sugar-modified NTPs (dedeoxyNTPs and gemcitabine triphosphate) or a nucleobase-modified dCTP analog (5-aza dCTP with a nitrogen instead of a carbon at the 5-position of the pyrimidine) resulted in poor survival and increased DNA mutations.175
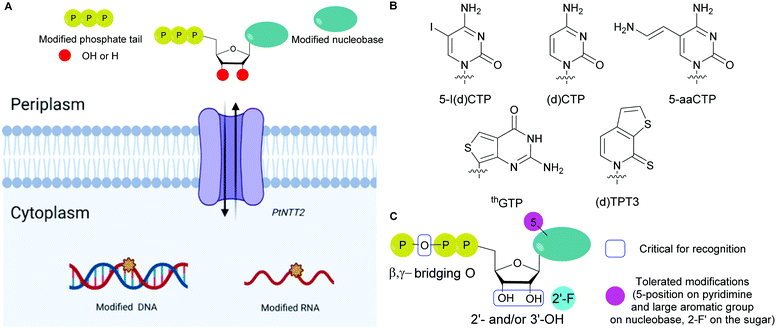 |
| | Fig. 13 (A) General scheme for the internalization of modified NTPs into E. coli using the exogenous PtNTT2 from Phaeodactylum tricornutum followed by their incorporation into DNA and RNA. (B) Structures of modified NTP nucleobases internalized in E. coli through PtNTT2. (C) Critical features in modified NTPs required for internalization and tolerated modifications for internalization. | |
5.3. Non-natural transporters for the internalization of modified NTPs
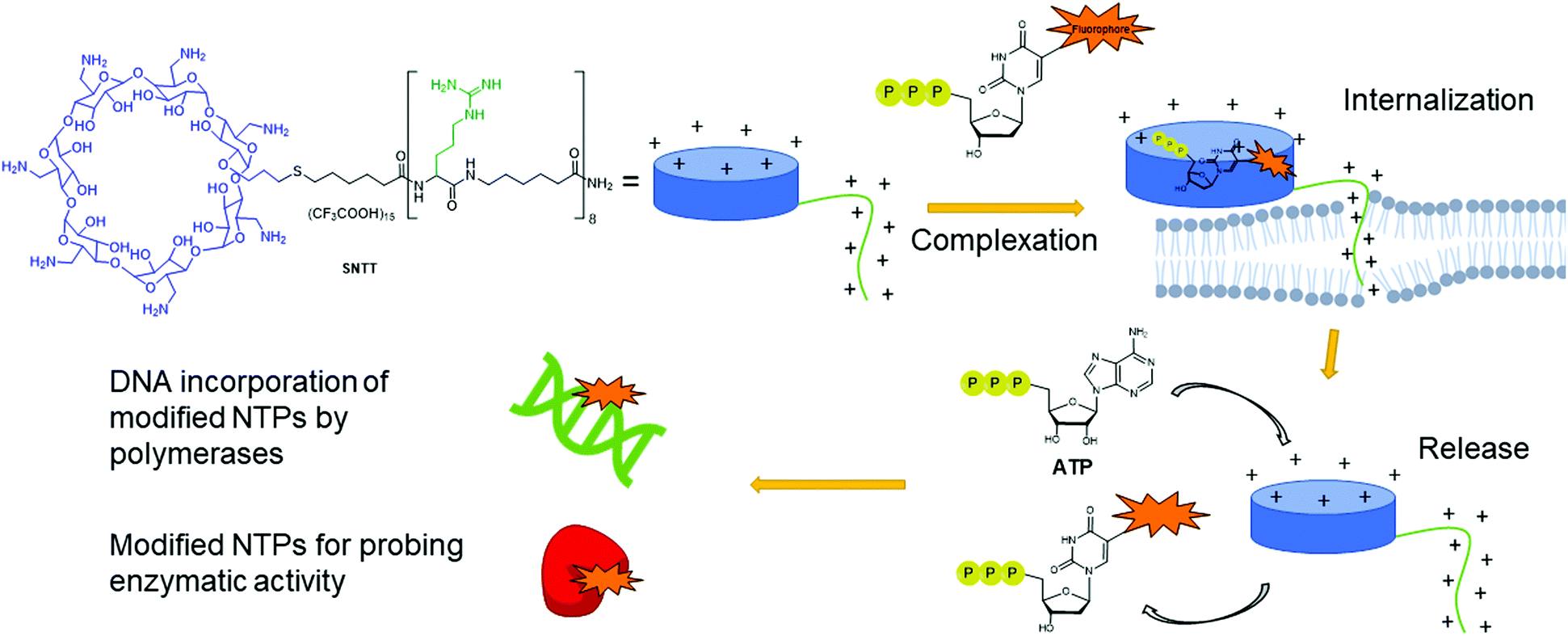
Despite having multiple applications, the scope of expressed importers is limited. They are restricted to organisms capable of heterogeneously expressing the NTT importer from the corresponding plasmid, typically E. coli. Aiming to develop a universal and facile internalization method that could be applied to multiple organisms, Kraus and co-workers designed a cyclodextrin derivative called synthetic nucleoside triphosphate transporter (SNTT; Fig. 14).176 This molecule contains a per-6-amino-β-cyclodextrin and an octa-aminocaproic acid/arginine tail. At neutral pH, the positively charged amines complex with the negatively charged phosphate tail of the NTPs. Furthermore, the positively charged arginine tail acts as a cell-penetrating agent by disrupting the phospholipid membrane, allowing the whole complex to enter the cells. The complexed NTP is released from the SNTT through displacement by the presence of the large excess of natural ATP and other NTPs present in the cytoplasm. Internalization of fluorescent NTPs was demonstrated in eukaryotic and bacterial cells by live-cell imaging. For instance, successful delivery of Cy3-dUTP was observed in E. coli and M. smegmatis.176
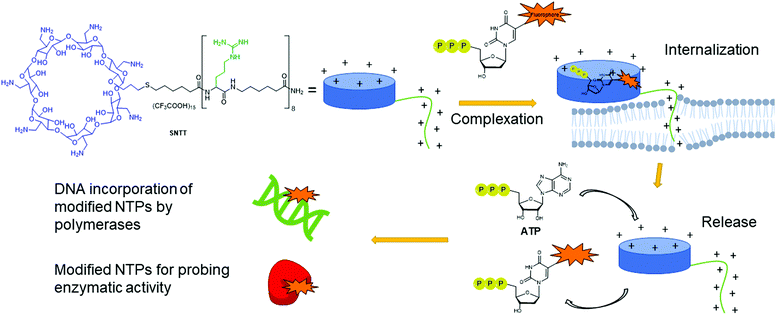 |
| | Fig. 14 Delivery mechanism of modified NTPs into cells using SNTT. A complex is formed between the modified NTP and the cyclodextrin ring. The positively charged arginine tail acts as a cell penetrating agent, pulling the complex into the cells. ATP present in large quantities in the cells displaces the modified NTPs from SNTT, releasing it into the cytoplasm of the cell. Modified NTPs are now available for incorporation into DNA or for probing enzymatic activity. | |
5.4. Examples of strategies to deliver non-natural NTPs or derivatives into cells with potential for bacteria
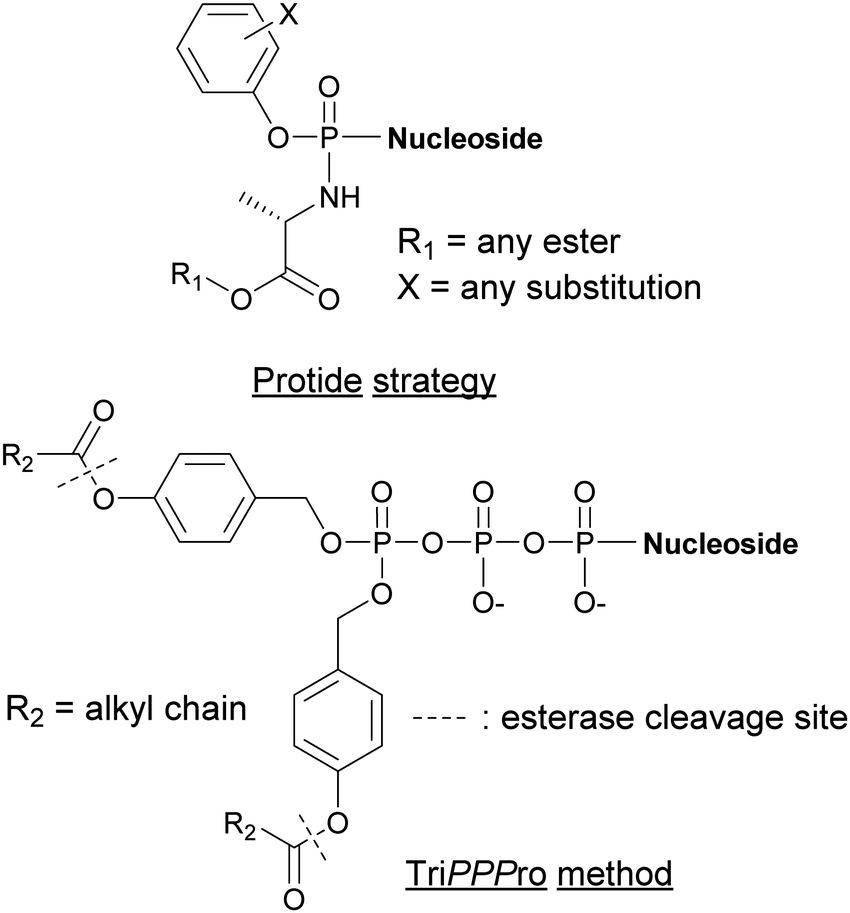
Alternative strategies such as the PROdrug + nucleoTIDE (ProTide) and the delivery of NTPs through a prodrug (TriPPPro) approach have been devised and are based on alterations of nucleoside derivatives to internalize into cells (Fig. 15).177
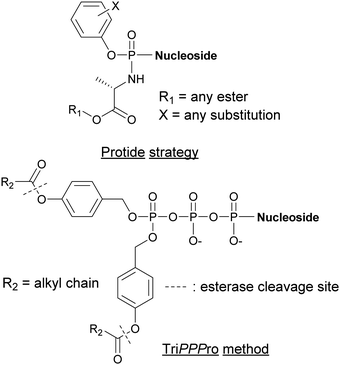 |
| | Fig. 15 Structures of nucleotide derivatives utilized for the Protide and TiPPPro strategies. | |
In the ProTide approach developed by McGuigan and co-workers, the negatively charged oxygen atoms present on the phosphate of the nucleoside monophosphate (NMP) are masked, forming a neutral compound that can be internalized by the cells. Most nucleotide analogs suffer from poor cell permeability.178 Once inside the cells, the ProTide is metabolized into the NMP, which can be further phosphorylated to the corresponding nucleoside di- or triphosphates.179 This method has been applied to the delivery of protides of N-(3-(5-(2′-deoxyuridine-5′-monophosphate))prop-2-ynyl)octanamide into M. tuberculosis, a potent inhibitor of flavin-dependent thymidylate synthase X (ThyX), which is essential to its survival. The enhanced lipophilicity of the phosphoramidate-protected derivatives improved internalization through the cell wall.180 Consequently, we can foresee the application of the ProTide strategy to other nucleotide-based inhibitors of bacterial targets to promote the development of effective antibiotics. A well-known example of a nucleotide-based prodrug is Sofosbuvir, an anti-viral drug used in combination therapy to treat hepatitis C. Once internalized into the virus, the compound is deprotected and metabolized to form the active metabolite, the 5′-triphosphosphate derivative. This metabolite inhibits the viral NS5B polymerase and, consequently, causes termination of viral RNA synthesis by RNA chain termination.181
The TriPPPro approach designed by Meier and co-workers relies on the addition of a lipophilic acyloxybenzyl moiety to two of the nucleophilic oxygen atoms of the NTP γ-phosphate for successful internalization into cells.182–184 The presence of remaining charged oxygen atoms on the α- and β-phosphates is balanced by adjusting the lipophilicity of the acyloxybenzyl moiety. The advantage of this strategy is that only one hydrolysis step is required by the cells to release the active NTP, bypassing the phosphorylation metabolism steps.185,186 Although the TriPPPro method has only been utilized in viruses, there are promising signs for its future use in bacteria.
6. Conclusion and outlook
Modified nucleoside triphosphates have been exploited to explore bacterial enzymatic mechanisms, expand the genetic alphabet, and develop novel proteins and therapeutics. Numerous studies have focused on nucleobase alterations due to their critical importance for protein recognition and potential for generating proteins or aptamers with novel functionalities. Studies that evaluated sugar-modified NTPs demonstrated that these molecules are in general poor substrates for bacterial enzymes. As a result, bacterial proteins have been evolved to accommodate modified NTPs, enabling a better understanding of the enzymatic structural features guiding NTP recognition and selectivity. Modifications of the NTP phosphate moiety are of particular interest since various enzymatic mechanisms rely on phosphate hydrolysis. Therefore, specific phosphate modifications are excellent tools for probing enzymatic activity. Nevertheless, the presence of a negatively charged phosphate tail prevents NTPs from being internalized into bacteria. This issue had been a long-standing hurdle for utilizing modified NTPs for in vivo applications. Recently, several methods have been established to facilitate uptake of NTPs into bacteria. These strategies were driven by different applications, such as the creation of autonomous organisms that rely on non-canonical nucleic acid, the internalization of NTP-based probes, and the delivery of therapeutics to cells. These novel methods pave the way for the exploration of a myriad of NTP-related proteins in their native environment and for creating novel organisms. Ultimately, these chemical tools will facilitate future studies that deepen our understanding of fundamental bacterial processes.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank J. Shirley for helpful suggestions. This work was supported by the University of Minnesota (UMN) Department of Chemistry, the UMN NIH Biotechnology Training Grant (5T32GM008347, A. E.), the UMN Undergraduate Research Opportunities Program (A. A. C.), a Minnesota Turkey Research and Promotion Council Research grant, and NIH R01GM134538-01A1. Fig. 13 and 14 were created using BioRender.
References
- M. Hollenstein, Molecules, 2012, 17, 13569–13591 CrossRef CAS
 .
.
- A. J. Cozzone, J. Cell. Biochem., 1993, 51, 7–13 CrossRef CAS .
- E. Westhof, M. Yusupov and G. Yusupova, F1000Prime Rep., 2014, 6, 19 Search PubMed .
- W. A. Beard, D. D. Shock, B. J. Vande Berg and S. H. Wilson, J. Biol. Chem., 2002, 277, 47393–47398 CrossRef CAS .
- L. N. Jackson, N. Chim, C. Shi and J. C. Chaput, Nucleic Acids Res., 2019, 47, 6973–6983 CrossRef CAS .
- J. Jakubovska, D. Tauraite, L. Birštonas and R. Meškys, Nucleic Acids Res., 2018, 46, 5911–5923 CrossRef CAS .
- A. Hottin and A. Marx, Acc. Chem. Res., 2016, 49, 418–427 CrossRef CAS .
- H. Cahová, A. Panattoni, P. Kielkowski, J. Fanfrlík and M. Hocek, ACS Chem. Biol., 2016, 11, 3165–3171 CrossRef .
- K. Bergen, A. L. Steck, S. Strutt, A. Baccaro, W. Welte, K. Diederichs and A. Marx, J. Am. Chem. Soc., 2012, 134, 11840–11843 CrossRef CAS .
- S. Obeid, A. Baccaro, W. Welte, K. Diederichs and A. Marx, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21327–21331 CrossRef CAS .
- A. Hottin, K. Betz, K. Diederichs and A. Marx, Chem. – Eur. J., 2017, 23, 2109–2118 CrossRef CAS .
- S. Obeid, H. Bußkamp, W. Welte, K. Diederichs and A. Marx, J. Am. Chem. Soc., 2013, 135, 15667–15669 CrossRef CAS .
- S. Obeid, H. Bußkamp, W. Welte, K. Diederichs and A. Marx, Chem. Commun., 2012, 48, 8320–8322 RSC .
- J. Balintová, M. Welter and A. Marx, Chem. Sci., 2018, 9, 7122–7125 RSC .
- M. Welter, D. Verga and A. Marx, Angew. Chem., Int. Ed., 2016, 55, 10131–10135 CrossRef CAS .
- A. Baccaro, A.-L. Steck and A. Marx, Angew. Chem., Int. Ed., 2012, 51, 254–257 CrossRef CAS .
- P. Kielkowski, J. Fanfrlík and M. Hocek, Angew. Chem., Int. Ed., 2014, 53, 7552–7555 CrossRef CAS .
- J. Matyašovský, P. Perlíková, V. Malnuit, R. Pohl and M. Hocek, Angew. Chem., Int. Ed., 2016, 55, 15856–15859 CrossRef .
- M.-J. Guo, M. J. Waring, S. Hildbrand, C. J. Leumann and L. W. McLaughlin, Nucleic Acids Res., 1998, 26, 1863–1869 CrossRef CAS .
- I. Hirao, M. Kimoto, S.-i. Yamakage, M. Ishikawa, J. Kikuchi and S. Yokoyama, Bioorg. Med. Chem. Lett., 2002, 12, 1391–1393 CrossRef CAS .
- S. Vichier-Guerre, L. Dugué and S. Pochet, Org. Biomol. Chem., 2019, 17, 290–301 RSC .
- K. Morihiro, H. Hoshino, O. Hasegawa, Y. Kasahara, K. Nakajima, M. Kuwahara, S.-i. Tsunoda and S. Obika, Bioorg. Med. Chem. Lett., 2015, 25, 2888–2891 CrossRef CAS .
- S. Matsuda, A. A. Henry, P. G. Schultz and F. E. Romesberg, J. Am. Chem. Soc., 2003, 125, 6134–6139 CrossRef CAS .
- A. A. Henry, C. Yu and F. E. Romesberg, J. Am. Chem. Soc., 2003, 125, 9638–9646 CrossRef CAS .
- G. T. Hwang, A. M. Leconte and F. E. Romesberg, ChemBioChem, 2007, 8, 1606–1611 CrossRef CAS .
- A. K. Ogawa, Y. Wu, D. L. McMinn, J. Liu, P. G. Schultz and F. E. Romesberg, J. Am. Chem. Soc., 2000, 122, 3274–3287 CrossRef CAS .
- S. Imoto, J. N. Patro, Y. L. Jiang, N. Oka and M. M. Greenberg, J. Am. Chem. Soc., 2006, 128, 14606–14611 CrossRef CAS .
- A. Viswanathan and P. W. Doetsch, J. Biol. Chem., 1998, 273, 21276–21281 CrossRef CAS .
- J. Liu and P. W. Doetsch, Nucleic Acids Res., 1998, 26, 1707–1712 CrossRef CAS .
- L. A. Wyss, A. Nilforoushan, F. Eichenseher, U. Suter, N. Blatter, A. Marx and S. J. Sturla, J. Am. Chem. Soc., 2015, 137, 30–33 CrossRef CAS .
- K. Betz, A. Nilforoushan, L. A. Wyss, K. Diederichs, S. J. Sturla and A. Marx, Chem. Commun., 2017, 53, 12704–12707 RSC .
- J. M. Walsh and P. J. Beuning, J. Nucleic Acids, 2012, 2012, 17 Search PubMed .
- J. Lee and S. Borukhov, Front. Mol. Biosci., 2016, 3, 73 Search PubMed .
- M. Hocek, Acc. Chem. Res., 2019, 52, 1730–1737 CrossRef CAS .
- V. Raindlová, M. Janoušková, M. Slavíčková, P. Perlíková, S. Boháčová, N. Milisavljevič, H. Šanderová, M. Benda, I. Barvík, L. Krásný and M. Hocek, Nucleic Acids Res., 2016, 44, 3000–3012 CrossRef .
- M. Janoušková, Z. Vaníková, F. Nici, S. Boháčová, D. Vítovská, H. Šanderová, M. Hocek and L. Krásný, Chem. Commun., 2017, 53, 13253–13255 RSC .
- Z. Vaníková, M. Janoušková, M. Kambová, L. Krásný and M. Hocek, Chem. Sci., 2019, 10, 3937–3942 RSC .
- S. Boháčová, Z. Vaníková, L. Poštová Slavětínská and M. Hocek, Org. Biomol. Chem., 2018, 16, 5427–5432 RSC .
- M. Saravanan, K. Vasu and V. Nagaraja, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10344–10347 CrossRef CAS .
- V. Valinluck, W. Wu, P. Liu, J. W. Neidigh and L. C. Sowers, Chem. Res. Toxicol., 2006, 19, 556–562 Search PubMed .
- H. Macíčková-Cahová and M. Hocek, Nucleic Acids Res., 2009, 37, 7612–7622 CrossRef .
- H. Macíčková-Cahová, R. Pohl and M. Hocek, ChemBioChem, 2011, 12, 431–438 CrossRef .
- M. Mackova, S. Bohacova, P. Perlikova, L. Postova Slavetinska and M. Hocek, ChemBioChem, 2015, 16, 2225–2236 CrossRef CAS .
- M. Mackova, R. Pohl and M. Hocek, ChemBioChem, 2014, 15, 2306–2312 CrossRef CAS .
- P. Kielkowski, N. L. Brock, J. S. Dickschat and M. Hocek, ChemBioChem, 2013, 14, 801–804 CrossRef CAS .
- J. Jakubovska, D. Tauraitė and R. Meškys, ChemBioChem, 2019, 20, 2504–2512 CrossRef CAS .
- J. Matyašovský and M. Hocek, Org. Biomol. Chem., 2020, 18, 255–262 RSC .
- S. G. Srivatsan and Y. Tor, J. Am. Chem. Soc., 2007, 129, 2044–2053 CrossRef CAS .
- P. Sandin, G. Stengel, T. Ljungdahl, K. Börjesson, B. Macao and L. M. Wilhelmsson, Nucleic Acids Res., 2009, 37, 3924–3933 CrossRef CAS .
- J. Caton-Williams and Z. Huang, Angew. Chem., Int. Ed., 2008, 47, 1723–1725 CrossRef CAS .
- U. Reisacher, D. Ploschik, F. Rönicke, G. B. Cserép, P. Kele and H.-A. Wagenknecht, Chem. Sci., 2019, 10, 4032–4037 RSC .
- M. Merkel, S. Arndt, D. Ploschik, G. B. Cserép, U. Wenge, P. Kele and H.-A. Wagenknecht, J. Org. Chem., 2016, 81, 7527–7538 CrossRef CAS .
- X. Ren, A. H. El-Sagheer and T. Brown, Analyst, 2015, 140, 2671–2678 RSC .
- Y. Zheng and P. A. Beal, Bioorg. Med. Chem. Lett., 2016, 26, 1799–1802 CrossRef CAS .
- C. Costas, E. Yuriev, K. L. Meyer, T. S. Guion and M. M. Hanna, Nucleic Acids Res., 2000, 28, 1849–1858 CrossRef CAS .
- D. Morrison, M. Rothenbroker and Y. Li, Small Methods, 2018, 2, 1700319 CrossRef .
- E. Liu, C. H. Lam and D. M. Perrin, Molecules, 2015, 20, 13591–13602 CrossRef .
- M. Renders, E. Miller, C. H. Lam and D. M. Perrin, Org. Biomol. Chem., 2017, 15, 1980–1989 RSC .
- M. Kuwahara, Y. Takahata, A. Shoji, A. N. Ozaki, H. Ozaki and H. Sawai, Bioorg. Med. Chem. Lett., 2003, 13, 3735–3738 CrossRef CAS .
- M. A. Dellafiore, J. M. Montserrat and A. M. Iribarren, Front. Chem., 2016, 4, 18 Search PubMed .
- V. T. Dien, M. Holcomb, A. W. Feldman, E. C. Fischer, T. J. Dwyer and F. E. Romesberg, J. Am. Chem. Soc., 2018, 140, 16115–16123 CrossRef CAS .
- Y. Zhang and F. E. Romesberg, Biochemistry, 2018, 57, 2177–2178 CrossRef CAS .
- K. H. Lee, K. Hamashima, M. Kimoto and I. Hirao, Curr. Opin. Biotechnol., 2018, 51, 8–15 CrossRef CAS .
- V. T. Dien, M. Holcomb and F. E. Romesberg, Biochemistry, 2019, 58, 2581–2583 CrossRef CAS .
- P. Marlière, J. Patrouix, V. Döring, P. Herdewijn, S. Tricot, S. Cruveiller, M. Bouzon and R. Mutzel, Angew. Chem., Int. Ed., 2011, 50, 7109–7114 CrossRef .
- A. P. Mehta, H. Li, S. A. Reed, L. Supekova, T. Javahishvili and P. G. Schultz, J. Am. Chem. Soc., 2016, 138, 7272–7275 CrossRef CAS .
- A. P. Mehta, H. Li, S. A. Reed, L. Supekova, T. Javahishvili and P. G. Schultz, J. Am. Chem. Soc., 2016, 138, 14230–14233 CrossRef CAS .
- L. Li, M. Degardin, T. Lavergne, D. A. Malyshev, K. Dhami, P. Ordoukhanian and F. E. Romesberg, J. Am. Chem. Soc., 2014, 136, 826–829 CrossRef CAS .
-
M. P. Ledbetter, D. A. Malyshev and F. E. Romesberg, in Non-Natural Nucleic Acids: Methods and Protocols, ed. N. Shank, Springer New York, New York, NY, 2019, pp. 193–212 DOI:10.1007/978-1-4939-9216-4_13 .
- D. A. Malyshev, K. Dhami, T. Lavergne, T. Chen, N. Dai, J. M. Foster, I. R. Corrêa, Jr. and F. E. Romesberg, Nature, 2014, 509, 385–388 CrossRef CAS .
- Y. Zhang, B. M. Lamb, A. W. Feldman, A. X. Zhou, T. Lavergne, L. Li and F. E. Romesberg, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1317–1322 CrossRef CAS .
- M. P. Ledbetter, R. J. Karadeema and F. E. Romesberg, J. Am. Chem. Soc., 2018, 140, 758–765 CrossRef CAS .
- Y. Zhang, J. L. Ptacin, E. C. Fischer, H. R. Aerni, C. E. Caffaro, K. San Jose, A. W. Feldman, C. R. Turner and F. E. Romesberg, Nature, 2017, 551, 644–647 CrossRef CAS .
- E. Eremeeva, M. Abramov, L. Margamuljana, J. Rozenski, V. Pezo, P. Marlière and P. Herdewijn, Angew. Chem., Int. Ed., 2016, 55, 7515–7519 CrossRef CAS .
- E. Eremeeva, M. Abramov, L. Margamuljana and P. Herdewijn, Chem. – Eur. J., 2017, 23, 9560–9576 CrossRef CAS .
- P. Herdewijn and P. Marlière, Chem. Biodiversity, 2009, 6, 791–808 CrossRef CAS .
- V. B. Pinheiro, A. I. Taylor, C. Cozens, M. Abramov, M. Renders, S. Zhang, J. C. Chaput, J. Wengel, S.-Y. Peak-Chew, S. H. McLaughlin, P. Herdewijn and P. Holliger, Science, 2012, 336, 341–344 CrossRef CAS .
- V. B. Pinheiro and P. Holliger, Curr. Opin. Chem. Biol., 2012, 16, 245–252 CrossRef CAS .
- C. M. Joyce, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 1619–1622 CrossRef CAS .
- J. W. Schroeder, J. R. Randall, L. A. Matthews and L. A. Simmons, Crit. Rev. Biochem. Mol. Biol., 2015, 50, 181–193 CrossRef CAS .
- J. A. Brown and Z. Suo, Biochemistry, 2011, 50, 1135–1142 CrossRef CAS .
- E. Eremeeva and P. Herdewijn, Curr. Opin. Biotechnol., 2019, 57, 25–33 CrossRef CAS .
- V. Kempeneers, M. Renders, M. Froeyen and P. Herdewijn, Nucleic Acids Res., 2005, 33, 3828–3836 CrossRef CAS .
- S. Pochet, P. A. Kaminski, A. Van Aerschot, P. Herdewijn and P. Marlière, C. R. Biol., 2003, 326, 1175–1184 CrossRef CAS .
- M. Kuwahara, S. Obika, J.-i. Nagashima, Y. Ohta, Y. Suto, H. Ozaki, H. Sawai and T. Imanishi, Nucleic Acids Res., 2008, 36, 4257–4265 CrossRef CAS .
- D. Summerer and A. Marx, J. Am. Chem. Soc., 2002, 124, 910–911 CrossRef CAS .
- M. Strerath, D. Summerer and A. Marx, ChemBioChem, 2002, 3, 578–580 CrossRef CAS .
- J. Cramer, G. Rangam, A. Marx and T. Restle, ChemBioChem, 2008, 9, 1243–1250 CrossRef CAS .
- D. Summerer and A. Marx, Synlett, 2004, 0217–0224 CAS .
- N. Tarashima, T. Sumitomo, H. Ando, K. Furukawa, T. Ishida and N. Minakawa, Org. Biomol. Chem., 2015, 13, 6949–6952 RSC .
- T. Ogino, K. Sato and A. Matsuda, ChemBioChem, 2010, 11, 2597–2605 CrossRef CAS .
- T. Ogino, N. Inoue, K. Sato and A. Matsuda, Nucleic Acids Symp. Ser., 2007, 51, 355–356 CrossRef .
- J. J. Chen, C.-H. Tsai, X. Cai, A. T. Horhota, L. W. McLaughlin and J. W. Szostak, PLoS One, 2009, 4, e4949–e4949 CrossRef .
- C. G. Peng and M. J. Damha, J. Am. Chem. Soc., 2007, 129, 5310–5311 CrossRef CAS .
- G. Houlihan, S. Arangundy-Franklin and P. Holliger, Acc. Chem. Res., 2017, 50, 1079–1087 CrossRef CAS .
- A. I. Taylor, V. B. Pinheiro, M. J. Smola, A. S. Morgunov, S. Peak-Chew, C. Cozens, K. M. Weeks, P. Herdewijn and P. Holliger, Nature, 2015, 518, 427–430 CrossRef CAS .
- H. J. Schultz, A. M. Gochi, H. E. Chia, A. L. Ogonowsky, S. Chiang, N. Filipovic, A. G. Weiden, E. E. Hadley, S. E. Gabriel and A. M. Leconte, Biochemistry, 2015, 54, 5999–6008 CrossRef CAS .
- T. Chen, N. Hongdilokkul, Z. Liu, R. Adhikary, S. S. Tsuen and F. E. Romesberg, Nat. Chem., 2016, 8, 556–562 CrossRef CAS .
- G. Xia, L. Chen, T. Sera, M. Fa, P. G. Schultz and F. E. Romesberg, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 6597–6602 CrossRef CAS .
- L. H. Lauridsen, J. A. Rothnagel and R. N. Veedu, ChemBioChem, 2012, 13, 19–25 CrossRef .
- J. M. Holstein and A. Rentmeister, Methods, 2016, 98, 18–25 CrossRef CAS .
- M.-L. Winz, A. Samanta, D. Benzinger and A. Jäschke, Nucleic Acids Res., 2012, 40, e78–e78 CrossRef CAS .
- H. Mei, C. Shi, R. M. Jimenez, Y. Wang, M. Kardouh and J. C. Chaput, Nucleic Acids Res., 2017, 45, 5629–5638 CrossRef CAS .
- H. Mei, J.-Y. Liao, R. M. Jimenez, Y. Wang, S. Bala, C. McCloskey, C. Switzer and J. C. Chaput, J. Am. Chem. Soc., 2018, 140, 5706–5713 CrossRef CAS .
- C.-Y. Chen, Front. Microbiol., 2014, 5, 305 Search PubMed .
- C. I. Martinez, L. H. Thoresen, K. Burgess and R. A. Gibbs, Nucleic Acids Res., 1999, 27, 1271–1274 CrossRef CAS .
- C. I. Martinez, M. Ali Ansari, R. Gibbs and K. Burgess, Bioorg. Med. Chem. Lett., 1997, 7, 3013–3016 CrossRef CAS .
- D. Hutter, M.-J. Kim, N. Karalkar, N. A. Leal, F. Chen, E. Guggenheim, V. Visalakshi, J. Olejnik, S. Gordon and S. A. Benner, Nucleosides, Nucleotides Nucleic Acids, 2010, 29, 879–895 CrossRef CAS .
- F. Chen, M. Dong, M. Ge, L. Zhu, L. Ren, G. Liu and R. Mu, Genomics, Proteomics Bioinf., 2013, 11, 34–40 CrossRef CAS .
- C. R. Burke and A. Lupták, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 980–985 CrossRef CAS .
- M. P. Bhate, K. S. Molnar, M. Goulian and W. F. DeGrado, Structure, 2015, 23, 981–994 CrossRef CAS .
- C. J. Hastie, H. J. McLauchlan and P. Cohen, Nat. Protoc., 2006, 1, 968–971 CrossRef CAS .
- Z. Huang and J. W. Szostak, Nucleic Acids Res., 1996, 24, 4360–4361 CrossRef CAS .
- V. Svetlov, D. G. Vassylyev and I. Artsimovitch, J. Biol. Chem., 2004, 279, 38087–38090 CrossRef CAS .
- K. Sanders, C.-L. Lin, A. J. Smith, N. Cronin, G. Fisher, V. Eftychidis, P. McGlynn, N. J. Savery, D. B. Wigley and M. S. Dillingham, Nucleic Acids Res., 2017, 45, 3875–3887 CrossRef CAS .
- O. M. Chase, A. Espinasse, K. E. Wilke and E. E. Carlson, Biochemistry, 2018, 57, 4368–4373 CrossRef CAS .
- J. Fischer, R. A. Johnson and E. Boon, J. Visualized Exp., 2017, 55129, DOI:10.3791/55129 .
- E. Hentschel, C. Mack, C. Gätgens, M. Bott, M. Brocker and J. Frunzke, Mol. Microbiol., 2014, 92, 1326–1342 CrossRef CAS .
- K. E. Wilke, S. Francis and E. E. Carlson, J. Am. Chem. Soc., 2012, 134, 9150–9153 CrossRef CAS .
- A. Espinasse, X. Wen, J. D. Goodpaster and E. E. Carlson, ACS Chem. Biol., 2020, 15, 1252–1260 CrossRef CAS .
- M. Khodai-Kalaki, D. F. Aubert and M. A. Valvano, J. Biol. Chem., 2013, 288, 30473–30484 CrossRef CAS .
- Y. J. Seo, D. A. Malyshev, T. Lavergne, P. Ordoukhanian and F. E. Romesberg, J. Am. Chem. Soc., 2011, 133, 19878–19888 CrossRef CAS .
- L. Wang, S. Jiang, Z. Deng, P. C. Dedon and S. Chen, FEMS Microbiol. Rev., 2018, 43, 109–122 CrossRef .
- M. I. Dobrikov, K. M. Grady and B. R. Shaw, Nucleosides, Nucleotides Nucleic Acids, 2003, 22, 275–282 CrossRef CAS .
- K. He, K. W. Porter, A. Hasan, J. D. Briley and B. R. Shaw, Nucleic Acids Res., 1999, 27, 1788–1794 CrossRef CAS .
- N. Carrasco and Z. Huang, J. Am. Chem. Soc., 2004, 126, 448–449 CrossRef CAS .
- B. Hu, Y. Wang, S. Sun, W. Yan, C. Zhang, D. Luo, H. Deng, L. R. Hu and Z. Huang, Angew. Chem., Int. Ed., 2019, 58, 7835–7839 CrossRef CAS .
- R. L. Letsinger, J. S. Wilkes and L. B. Dumas, Biochemistry, 1976, 15, 2810–2816 CrossRef CAS .
- J. L. Wolfe, T. Kawate, A. Belenky and V. Stanton Jr, Nucleic Acids Res., 2002, 30, 3739–3747 CrossRef CAS .
- H. Nguyen, M. Abramov, E. Eremeeva and P. Herdewijn, ChemBioChem, 2020, 21, 272–278 CrossRef CAS .
- J. Lin, C. Abeygunawardana, D. N. Frick, M. J. Bessman and A. S. Mildvan, Biochemistry, 1997, 36, 1199–1211 CrossRef CAS .
- O. Barabás, V. Pongrácz, J. Kovári, M. Wilmanns and B. G. Vértessy, J. Biol. Chem., 2004, 279, 42907–42915 CrossRef .
- P. Casino, L. Miguel-Romero and A. Marina, Nat. Commun., 2014, 5, 3258 CrossRef .
- G. Rivera-Cancel, W.-h. Ko, D. R. Tomchick, F. Correa and K. H. Gardner, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 17839–17844 CrossRef CAS .
- D. Albanesi, M. Martín, F. Trajtenberg, M. C. Mansilla, A. Haouz, P. M. Alzari, D. de Mendoza and A. Buschiazzo, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 16185–16190 CrossRef CAS .
- R. G. Yount, D. Babcock, W. Ballantyne and D. Ojala, Biochemistry, 1971, 10, 2484–2489 CrossRef CAS .
- A. Lass-Napiorkowska and T. Heyduk, Biochemistry, 2016, 55, 647–658 CrossRef CAS .
- I. Mochalkin, J. R. Miller, A. Evdokimov, S. Lightle, C. Yan, C. K. Stover and G. L. Waldrop, Protein Sci., 2008, 17, 1706–1718 CrossRef CAS .
- S. Padmanabhan and D. M. Freymann, Structure, 2001, 9, 859–867 CrossRef CAS .
- H. M. Kropp, S. L. Dürr, C. Peter, K. Diederichs and A. Marx, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 9992–9997 CrossRef CAS .
- K. J. Tanaka, S. Song, K. Mason and H. W. Pinkett, Biochim. Biophys. Acta, Biomembr., 2018, 1860, 868–877 CrossRef CAS .
- C. Cousin, A. Derouiche, L. Shi, Y. Pagot, S. Poncet and I. Mijakovic, FEMS Microbiol. Lett., 2013, 346, 11–19 CrossRef CAS .
- M. Lasker, C. D. Bui, P. G. Besant, K. Sugawara, P. Thai, G. Medzihradszky and C. W. Turck, Protein Sci., 1999, 8, 2177–2185 CrossRef CAS .
- H. K. Carlson, L. Plate, M. S. Price, J. J. Allen, K. M. Shokat and M. A. Marletta, Anal. Biochem., 2010, 397, 139–143 CrossRef CAS .
- N. Wang, Z. She, Z. Ingar, S. Martic and H.-B. Kraatz, Chem. – Eur. J., 2017, 23, 3152–3158 CrossRef CAS .
- G. D. Sankhe, N. M. Dixit and D. K. Saini, mSphere, 2018, 3, e00111 CrossRef CAS .
- A. E. Dago, A. Schug, A. Procaccini, J. A. Hoch, M. Weigt and H. Szurmant, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, E1733–E1742 CrossRef CAS .
- L. M. Wolfe, U. Veeraraghavan, S. Idicula-Thomas, S. Schürer, K. Wennerberg, R. Reynolds, G. S. Besra and K. M. Dobos, Mol. Cell. Proteomics, 2013, 12, 1644–1660 CrossRef CAS .
- C. Ansong, C. Ortega, S. H. Payne, D. H. Haft, L. M. Chauvignè-Hines, M. P. Lewis, A. R. Ollodart, S. O. Purvine, A. K. Shukla, S. Fortuin, R. D. Smith, J. N. Adkins, C. Grundner and A. T. Wright, Chem. Biol., 2013, 20, 123–133 CrossRef CAS .
- L. R. Yarbrough, Biochem. Biophys. Res. Commun., 1978, 81, 35–41 CrossRef CAS .
- J. G. Schlageck, M. Baughman and L. R. Yarbrough, J. Biol. Chem., 1979, 254, 12074–12077 CAS .
- K. Rittinger, D. Negre, G. Divita, M. Scarabel, C. Bonod-Bidaud, R. S. Goody, A. J. Cozzone and J.-C. Cortay, Eur. J. Biochem., 1996, 237, 247–254 CrossRef CAS .
- O. B. Gutiérrez Acosta, N. Hardt, S. M. Hacker, T. Strittmatter, B. Schink and A. Marx, ACS Chem. Biol., 2014, 9, 1263–1266 CrossRef .
-
T. M. Anthony, P. M. Dedigama-Arachchige, D. M. Embogama, T. R. Faner, A. E. Fouda and M. K. H. Pflum, in Kinomics, ed. H. B. Kraatz and S. Martic, 2015, pp. 137–168 Search PubMed .
- S. Serdjukow, F. Kink, B. Steigenberger, M. Tomás-Gamasa and T. Carell, Chem. Commun., 2014, 50, 1861–1863 RSC .
- K. E. Wilke, S. Francis and E. E. Carlson, ACS Chem. Biol., 2015, 10, 328–335 CrossRef CAS .
- K. E. Wilke, C. A. Fihn and E. E. Carlson, Bioorg. Med. Chem., 2018, 26, 5322–5326 CrossRef CAS .
- J. Bhat, R. Rane, S. M. Solapure, D. Sarkar, U. Sharma, M. N. Harish, S. Lamb, D. Plant, P. Alcock, S. Peters, S. Barde and R. K. Roy, J. Biomol. Screening, 2006, 11, 968–976 CrossRef CAS .
- J. Kang, M. S. Lee, J. A. Copland, B. A. Luxon and D. G. Gorenstein, Bioorg. Med. Chem. Lett., 2008, 18, 1835–1839 CrossRef CAS .
- C. Burzik, G. Kaim, P. Dimroth, E. Bamberg and K. Fendler, Biophys. J., 2003, 85, 2044–2054 CrossRef CAS .
- M. Jiménez, A. Martos, M. Vicente and G. Rivas, J. Biol. Chem., 2011, 286, 11236–11241 CrossRef .
- S. Ermert, A. Marx and S. M. Hacker, Top. Curr. Chem., 2017, 375, 28 CrossRef .
-
W. Levinson, Review of Medical Microbiology and Immunology, 14e, McGraw-Hill Education, New York, NY, 2016 Search PubMed .
- T. J. Silhavy, D. Kahne and S. Walker, Cold Spring Harbor Perspect. Biol., 2010, 2, a000414 CrossRef .
- G. K. Auer and D. B. Weibel, Biochemistry, 2017, 56, 3710–3724 CrossRef CAS .
- H. H. Winkler, J. Biol. Chem., 1976, 251, 389–396 CAS .
- H. H. Winkler and R. M. Daugherty, J. Bacteriol., 1984, 160, 76–79 CrossRef CAS .
- O. Trentmann, B. Jung, H. E. Neuhaus and I. Haferkamp, J. Biol. Chem., 2008, 283, 36486–36493 CrossRef CAS .
- J. P. Audia and H. H. Winkler, J. Bacteriol., 2006, 188, 6261–6268 CrossRef CAS .
- G. V. Plano and H. H. Winkler, J. Bacteriol., 1991, 173, 3389–3396 CrossRef CAS .
- J. Tjaden, H. H. Winkler, C. Schwoppe, M. Van Der Laan, T. Mohlmann and H. E. Neuhaus, J. Bacteriol., 1999, 181, 1196–1202 CrossRef CAS .
- I. Haferkamp, S. Schmitz-Esser, M. Wagner, N. Neigel, M. Horn and H. E. Neuhaus, Mol. Microbiol., 2006, 60, 1534–1545 CrossRef CAS .
- A. W. Feldman, V. T. Dien, R. J. Karadeema, E. C. Fischer, Y. You, B. A. Anderson, R. Krishnamurthy, J. S. Chen, L. Li and F. E. Romesberg, J. Am. Chem. Soc., 2019, 141, 10644–10653 CrossRef CAS .
- A. W. Feldman, E. C. Fischer, M. P. Ledbetter, J. Y. Liao, J. C. Chaput and F. E. Romesberg, J. Am. Chem. Soc., 2018, 140, 1447–1454 CrossRef CAS .
- V. Pezo, C. Hassan, D. Louis, B. Sargueil, P. Herdewijn and P. Marlière, ACS Synth. Biol., 2018, 7, 1565–1572 CrossRef CAS .
- Z. Zawada, A. Tatar, P. Mocilac, M. Buděšínský and T. Kraus, Angew. Chem., Int. Ed., 2018, 57, 9891–9895 CrossRef CAS .
- M.-J. Camarasa, ChemMedChem, 2018, 13, 1885–1889 CrossRef CAS .
- M. Serpi, V. Ferrari and F. Pertusati, J. Med. Chem., 2016, 59, 10343–10382 CrossRef CAS .
- Y. Mehellou, H. S. Rattan and J. Balzarini, J. Med. Chem., 2018, 61, 2211–2226 CrossRef CAS .
- C. McGuigan, M. Derudas, B. Gonczy, K. Hinsinger, S. Kandil, F. Pertusati, M. Serpi, R. Snoeck, G. Andrei, J. Balzarini, T. D. McHugh, A. Maitra, E. Akorli, D. Evangelopoulos and S. Bhakta, Bioorg. Med. Chem., 2014, 22, 2816–2824 CrossRef CAS .
- H. K. Bhatia, H. Singh, N. Grewal and N. K. Natt, J. Pharmacol. Pharmacother., 2014, 5, 278–284 CrossRef .
- C. Meier, Antiviral Chem. Chemother., 2017, 25, 69–82 CrossRef .
- X. Jia, D. Schols and C. Meier, J. Med. Chem., 2020, 63, 6003–6027 CrossRef CAS .
- C. Meier, C. Zhao, S. Weber, D. Schols and J. Balzarini, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202003073 .
- T. Gollnest, T. D. de Oliveira, D. Schols, J. Balzarini and C. Meier, Nat. Commun., 2015, 6, 8716 CrossRef CAS .
- S. Weising, V. Sterrenberg, D. Schols and C. Meier, ChemMedChem, 2018, 13, 1771–1778 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Hannah K.
Lembke
a,
Hannah K.
Lembke