
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIlluminating glycoscience: synthetic strategies for FRET-enabled carbohydrate active enzyme probes
Meenakshi
Singh
a,
Michael
Watkinson
 a,
Eoin M.
Scanlan
b and
Gavin J.
Miller
*a
a,
Eoin M.
Scanlan
b and
Gavin J.
Miller
*a
aLennard-Jones Laboratories, School of Chemical and Physical Sciences, Keele University, Staffordshire, ST5 5BG, UK. E-mail: g.j.miller@keele.ac.uk
bSchool of Chemistry and Trinity Biomedical Sciences Institute, Trinity College Dublin, Dublin 2, Ireland
First published on 17th September 2020
Abstract
Carbohydrates are an essential class of biomolecule and carbohydrate active enzymes (CAZys) catalyse their synthesis, refinement, and degradation, hence contributing an overall regulatory capacity to their underpinning physiological roles. As such, there is a considerable current requirement to be able to monitor, quantify and inhibit CAZy activity. It is here that Förster resonance energy transfer (FRET) is emerging as a powerful tool in enabling this, through synthetic conjugation of appropriate fluorogenic partners into a particular CAZy substrate. In this review we survey recent accomplishments in synthetic methodology for accessing defined carbohydrate structures, suitably equipped with FRET probe capability, followed by their utilisation in studying particular classes of CAZy. The majority of examples concern endo-acting glycosidases, but emergent probes for exo-glycosidases, glycosyl transferases and catalytic antibodies are also examined.
 Meenakshi Singh | Meenakshi Singh obtained her MSc in Analytical chemistry from India and worked on kinetic studies of organoselenium compounds using NMR spectroscopy. Following her MSc, she joined IISER Bhopal and worked on synthetic strategies for bioactive molecules. Subsequently, she moved to IOCB Prague and worked on the total synthesis of bioactive prenylated indole alkaloids. Then she pursued her PhD at the University of Milan as a part of GLYCOVAX network under Marie Skłodowska-Curie actions and developed a synthetic scheme for GBS Type II Oligosaccharide for vaccines. Meenakshi is currently a research associate with Dr Gavin Miller at Keele, focusing on carbohydrate chemical biology. |
 Michael Watkinson | Mike Watkinson graduated from the University of St. Andrews in 1991 before completing his PhD at UMIST in 1994. After a year as a Royal Society Postdoctoral Fellow at the University of Santiago de Compostela, Spain in 1995 he returned to UMIST before his appointment at QMUL in 1998 where he remained until joining Keele University as the Head of School of Physical and Chemical Sciences in 2018. His current research focuses on the development of small molecule fluorescent probes. |
 Eoin M. Scanlan | Eoin Scanlan completed his PhD at the University of St. Andrews (2004). Following postdoctoral work at the University of Bern, Switzerland and at the University of Oxford, UK he joined the School of Chemistry in Trinity College Dublin in 2008 where he is Associate Professor of Organic and Medicinal Chemistry and a PI in the Trinity Biomedical Sciences Institute. He leads an international research team in Trinity College with a focus on new synthetic methods and the discovery of novel therapeutics, diagnostics and biomaterials. He is co-founder and CSO of Glycome Biopharma, a biotech start-up company based in Dublin. |
 Gavin J. Miller | Gavin read Chemistry with Medicinal Chemistry at UMIST where he was awarded an MChem. Continuing his studies at the University of Manchester, he completed a PhD in synthetic carbohydrate chemistry followed by a PDRA at St. Andrews University. After a time in industry he returned to the University of Manchester as a PDRA at the Manchester Institute of Biotechnology. He joined Keele in 2016 and is currently Senior Lecturer in Organic Chemistry. His group focus on the chemical synthesis of carbohydrates, including sugar nucleotides, glycosyl 1-phosphates, oligosaccharides and nucleoside analogues. |
1. Introduction
Glycosylation is ubiquitous in nature and carbohydrates play pivotal roles in a diverse range of complex biological processes including fertilisation, neuronal development, cell–cell interactions, hormone activity, inflammatory responses, and infection.1–3 Metabolism of glycans is therefore critical to the regulated biological function of an organism. Carbohydrates are composed of individual monosaccharides covalently linked through a diverse array of glycosidic bonds. Degradation of carbohydrates is challenging due to the relative stability of these glycosidic linkages, however, their hydrolysis is catalysed by enzymes known as glycosidases, which typically increase the rate of this hydrolysis by 1017 fold, compared to spontaneous hydrolysis events.4 This remarkable acceleration in rate identifies glycosidases as highly efficient catalysts for this process and although they catalyse similar reactions, they also exhibit exquisite substrate selectivity.5The large number of glycosidases found in nature (e.g. approximately 3% of the human genome is dedicated to the encoding of glycosidases) has led to their classification into various sub-categories based on criteria such as catalytic mechanism, substrate specificity, site of glycosidic bond cleavage, and amino acid sequence.6,7exo-Glycosidases act specifically on the termini of polysaccharides, while endo-glycosidases promote internal hydrolysis of glycosidic linkages. Hydrolysis can occur via one of two widely accepted mechanisms; proceeding with either retention or inversion of stereochemistry at the anomeric position, as outlined by Koshland in 1953.8 In both mechanisms, oxacarbenium ion intermediates and two amino acids (Asp or Glu) are involved and several studies outlining the development of mechanistic details of glycosidase activity on various carbohydrates have been published.5,9,10 More recently, evidence for a novel epoxide intermediate in glycosidase catalysis has also been reported.11 Glycosidases have been organised into more than 120 different families based on amino acid sequence similarity and this classification has led to the generation of the CAZy database (Carbohydrate Active Enzyme, available online at http://www.cazy.org).12–14 Glycosidases grouped within a particular family share structural similarities and often their hydrolytic mechanisms are identical.
The emergence of glycosidases’ dominant role in many biological and industrial processes has spurred interest into the development of tools to accurately detect and profile these enzymes so as to improve our biological understanding of their function, particularly within the context of human health and disease.15 Glycosidase enzyme deficiencies are associated with several pathological conditions including Gaucher disease, Pompe disease, Chron's disease, and cancer.16,17 Schindler and Fabry diseases arise due to incomplete degradation of carbohydrates with terminal α-N-acetylgalactosamine and α-galactose, respectively.18 Heparanase, a heparan sulfate degrading endoglycosidase, is overexpressed in almost all cancer types where it is closely associated with tumorogenesis and metastasis.19 Furthermore, glycosidases have been reported to function as important gene markers, for example, the gene encoding β-galactosidase, lacZ, is extensively used as a reporter gene in animals and yeast.20 Glycosidases also play a critical role in industry with xylanases and cellulases of particular interest for the bulk production of bio-ethanol.21
This review aims to survey the development and application of fluorescent probes to detect this important class of enzyme using appropriately labelled carbohydrate substrates (from 2010 onwards). Focus is paid to the design and synthesis of chemical probes whereby a glycosidic linkage is hydrolysed by the enzyme under study and a reporter molecule is released, enabling a ratiometric monitoring of activity using Förster Resonance Energy Transfer (FRET). The review will not cover other families of fluorescent probes or activity-based glycosidase probes,22 which have been comprehensively reviewed elsewhere.23 A brief overview of FRET is presented first, followed by consideration of probes for endo and exo-acting glycosidases and ending with recent examples beyond this classification, for example, glycosyl transferases and antibodies with innate glycosidase activity.
2. Förster resonance energy transfer (FRET)
FRET, often also referred to as fluorescence resonance energy transfer, is the process by which energy is transferred in a non-radiative manner from an excited state donor molecular chromophore (D) to another acceptor chromophore (A), instead of being emitted from the donor as a photon i.e. a conventional fluorescence response, and is ratiometric in nature. There are a number of requirements for FRET to occur and the underlying theory has been reviewed extensively elsewhere and is therefore not covered here.24,25The phenomenon is highly distance dependent and decreases exponentially with distance between donor and acceptor molecule and is generally only considered to be effective over distances below 10 nm in biological systems. As this is also the length scale of proteins, it paves a way to extremely widespread utility in probing protein–protein interactions, modifications, conformational changes, and a plethora of biochemical signalling events and processes,26 and has often been described as a ‘molecular ruler’. In addition to this close proximity, there must also be adequate overlap between the fluorescence spectrum of D and the absorption spectrum of A along with the sufficiently high quantum yield of D (fD) and the high absorption coefficient of A (eA) (Fig. 1).
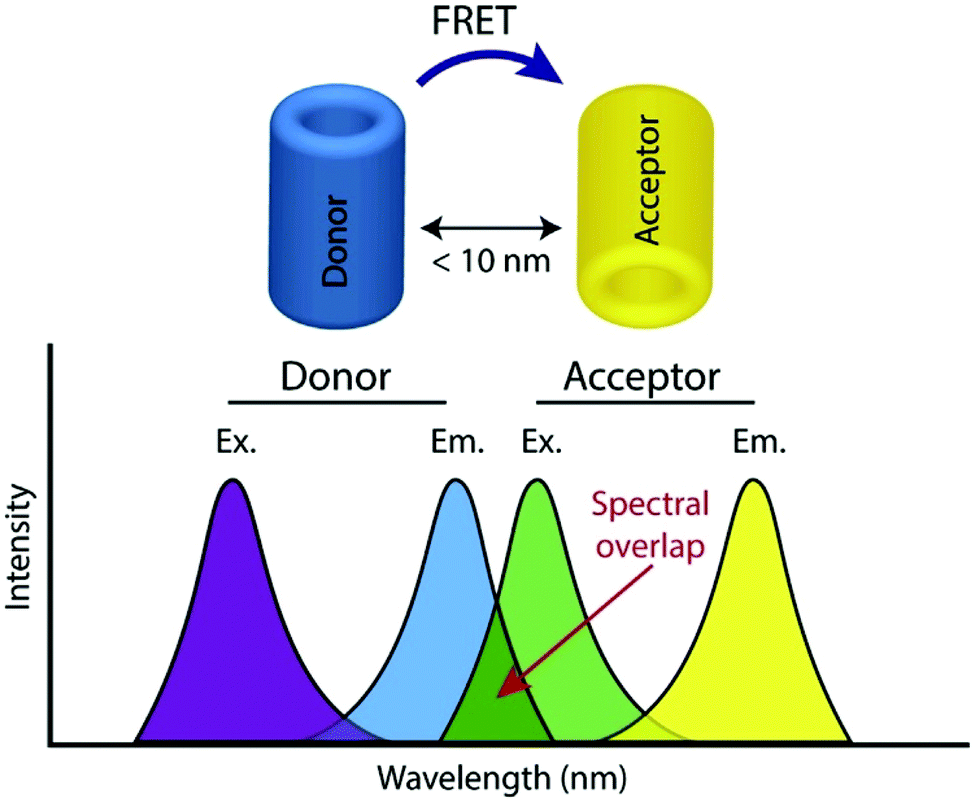 | ||
| Fig. 1 Illustration of donor and acceptor partners required for FRET, including the spectral overlap region of component fluorophores, adapted with permission from ref. 26. | ||
A further requirement for FRET to occur is that the transition dipoles of both D and A must be appropriately oriented or at least one (or both) must have a level of rapid rotational freedom, otherwise distance estimations can suffer large errors; this condition is usually met for chromophores attached to biomolecules. FRET is also sensitive to the local environment (e.g. solvent, pH, polarity and viscosity) and these elements need to be considered when interpreting data.
A wide range of chromophores have been employed in FRET systems and whilst genetically encoded biosensors utilising fluorescent proteins have found considerable applications in the life sciences,27 a number of other chromophores have also been exploited including organic dyes, quantum dots, and lanthanide complexes.28 Organic fluorophores are particularly useful as they can readily be attached covalently to a range of substrates such as amino acid residues, nucleic acids, and carbohydrates; large arrays of such labels are synthetically accessible or commercially available with desirable photochemical properties.29 Quantum dots are also increasingly used and are appealing chromophores due to their highly tunable photochemical properties, broad absorption and narrow emission bands. In addition, they generally display excellent chemical and thermal stability and there are increasingly reliable and variable methods available to link them to the analyte in question.27 Lanthanide complexes (principally of Tb3+ and Eu3+) are also increasingly popular donors as although they have generally narrower absorption profiles they are excellent donors, due to their long photoluminescent decays.
In this review the focus is on FRET systems that exploit enzymatic cleavage to elicit a change in the fluorescence response. This has been widely utilised in a number of biosensors to detect enzymatic activity in which the biosensor design includes a specific cleavage sequence for the enzyme of interest.26 Prior to enzymatic action, the FRET donor and acceptor are located closely enough for efficient energy transfer to occur (FRET quenching). However, upon cleavage of the biomolecule by the enzyme, they move apart resulting in a reduction of the FRET response and a concomitant increase in the fluorescent response of the donor fluorophore allowing analyte quantitation.
3. FRET probes for endo-acting glycosyl hydrolases
In the following Sections 3–6 we survey recent synthetic developments for the synthesis of FRET-enabled carbohydrate probes. Generally, the design of such systems centres around covalently equipping the carbohydrate target with organic fluorophores at the reducing and non-reducing termini of the glycan (or close thereto); although examples of both components being incorporated at the reducing end have also been developed (Section 4.1). From a synthetic perspective, this usually requires the installation of an appropriate functional tether at the reducing anomeric position and also at a ring position on the non-reducing sugar. Alternatively, reactive functional groups within the carbohydrate structure can be targeted directly (Section 3.5). Typically, the carbohydrate motif of interest is synthesised first using established step-wise glycosylation methodologies, but incorporating benign tethers for later functionalisation, and is completed by final stage deprotections and covalent attachment of the required organic dyes.3.1. Maltoligosaccharide probes for α-glucosidase
In 2012 Matsuoka and co-workers synthesised a series of FRET oligosaccharide probes for α-amylase,30 an enzyme responsible for the cleavage of α-(1,4)-glycosidic bonds in large, polysaccharide architectures, including starch and glycogen. Links between α-amylase isozyme activity and diseases such as myeloma and diabetes have been reported and synthetic probes that could ratiometrically determine α-amylase activity are therefore important.31 Building upon their previous work in this area,31 the group synthesised a small library of maltoligosaccharides with terminal FRET partners (a naphthylmethyl, NAP, donor and dansyl, DANSYL, acceptor), varying the length of the repeating α-(1,4)-D-Glc sugar chain from trisaccharide up to heptasaccharide.Peracetylated maltoligosaccharide starting materials were sourced from maltodextrin or through acetolysis of cyclodextrin components and model work using maltose was first undertaken to establish an efficient route for fluorescent label incorporation (Scheme 1).
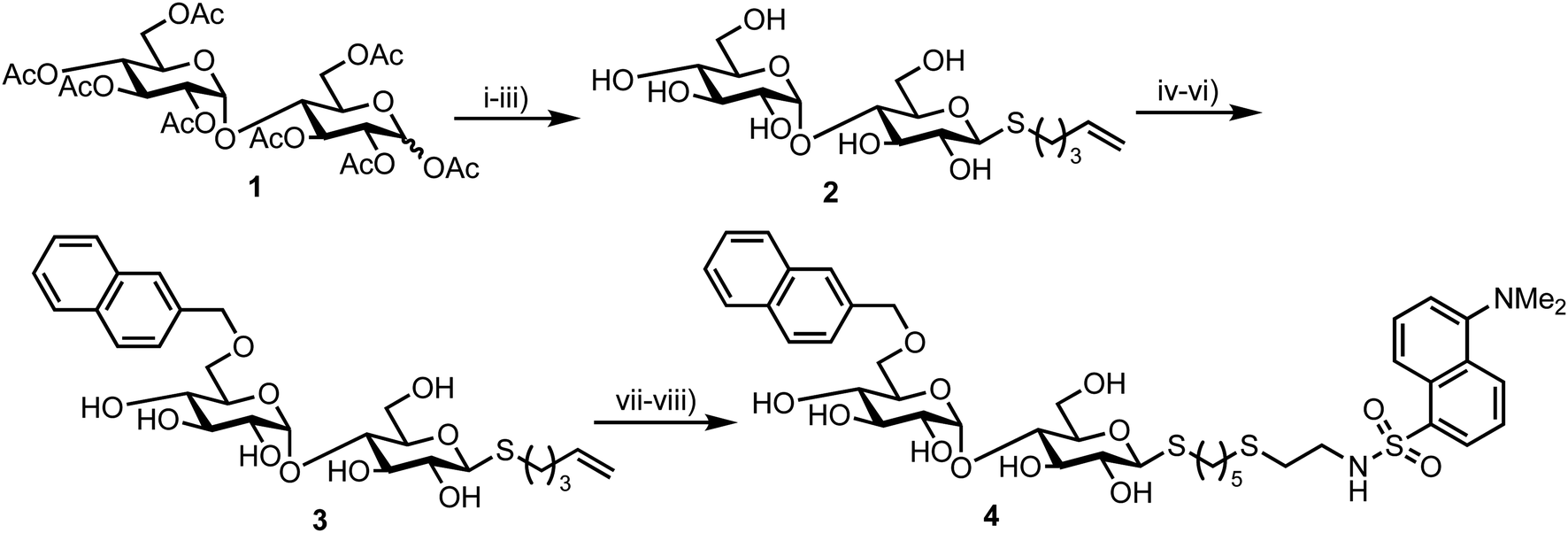 | ||
| Scheme 1 Reagents and conditions: (i) HBr/AcOH, Ac2O, AcOH, then AcSK, DMF, 72% two steps (ii) 5-bromopent-1-ene, DMF, diethylamine, 91%; (iii) NaOMe, MeOH, quant.; (iv) 2-naphthaldehyde di-ibutyl acetal, CSA, then Ac2O-pyr, 45%; (v) BH3·NMe3, AlCl3, MS 4 Å, THF, 74%; (vi) NaOMe, MeOH, quant.; (vii) HS(CH2)2NH2·HCl, MeOH, H2O, UV irradiation, quant.; (viii) dansyl chloride, Et3N, MeOH, quant. | ||
Accordingly, conversion of anomeric acetate mixture 1 to an anomeric thioacetate was accomplished via an SN2 reaction of the derived α-bromo glycoside with KSAc. Subsequent chemoselective S-deacetylation using diethylamine was followed by installation of an S-pentenyl glycoside at the reducing end in 72% yield over two steps. Following this, Zémplen deacetylation afforded S-pentenyl maltoside 2 in quantitative yield. A 4,6-naphthylidine acetal was next installed at the non-reducing end (followed by re-acetylation of the remaining hydroxyl groups) and opened regioselectively using BH3·NMe3–AlCl3 in 74% yield to incorporate the NAP-donor at the non-reducing end 6-position. Another Zémplen deacetylation step gave 6′-O-NAP-S-pentenyl maltoside 3. With the non-reducing component in place, radical mediated anti-Markovnikov addition of 2-mercaptoethyl amine to the S-pentenyl group and subsequent reaction of the free amine with dansyl chloride afforded model probe 4, with both of these steps reported in quantitative yield. Solvent choice was important for effecting the radical functionalisation reaction with MeOH/H2O found to provide an optimal solubility of both starting materials.
This successful synthetic route was then adapted for longer maltoligosaccharide sequences to afford a small matrix of compounds, 5–10, with the inter-chain length varying between one and five sugar units (Fig. 2a). When completing this synthesis, the authors noted a need to increase the reaction time for standard Zémplen transesterification on longer substrates and also that the previously successful non-reducing-end 4,6-naphthylidine acetal installation was not regioselective, requiring careful chromatographic separation of mono-, di- and tri-O-naphthylmethylidenated materials.
 | ||
| Fig. 2 (a) Library of non-reducing end NAP and reducing-end DANSYL functionalised maltoligosaccharide probes 5–10, the α-amylase cleavage point is shown in green (b) time course for hydrolysis of maltoligosaccharide probes by α-amylase using fluorescence detection at 333 nm, following excitation at 290 nm, adapted with permission from ref. 30. Orange and blue curves represent hexa- (compound 9) and heptasaccharide (compound 10) substrates, illustrating higher fluorescence intensities of NAP emission upon enzymatic cleavage. | ||
The installed NAP and DANSYL fluorogenic partners enabled a continuous assay to be established for each of substrates 5–10 using human saliva α-amylase (Fig. 2b). For sequences longer than pentasaccharide, a significant increase in relative fluorescence intensity at 333 nm (NAP emission) was observed (corrected for the initial FRET dansyl intensity), indicating that chain length correlated to enzymatic capability and that larger sequences may be better substrates for α-amylase. The results obtained by Matsuoka correlated to those reported previously using a 14C-labelled maltoligosaccharide.32
3.2. Chitooligosaccharides for an endo-β-glycosidase
In 2013, Halila and colleagues developed a FRET probe of chitin,33 a linear polysaccharide composed of β-(1,4)-linked N-acetylglucosamine (GlcNAc) units. Chitin is a natural polysaccharide with valuable biological and biomedical properties and methods to access and functionalise these materials in homogeneous form is an area of active research. Utilising a reducing end, hemiacetal/aldehyde modification with aniline catalysed amine ligation followed by reductive amination, the group installed a click-ready tether at this terminus (Scheme 2). Starting from tetra-N-acetyl-chitopentaose 11 an optimised ligation method was developed to deliver propargyl modified oligosaccharide 12. By using an excess of propargyl amine ([propargylamine]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[11]:[aniline] stoichiometry = 5:1:0.5) a yield of 70% for 12 was obtained. The authors proposed that the reaction sequence proceeded via rapid formation of an intermediate aniline-iminium ion which then underwent transimination with propargyl amine followed by reduction with NaCNBH4 to 12.
:[11]:[aniline] stoichiometry = 5:1:0.5) a yield of 70% for 12 was obtained. The authors proposed that the reaction sequence proceeded via rapid formation of an intermediate aniline-iminium ion which then underwent transimination with propargyl amine followed by reduction with NaCNBH4 to 12.
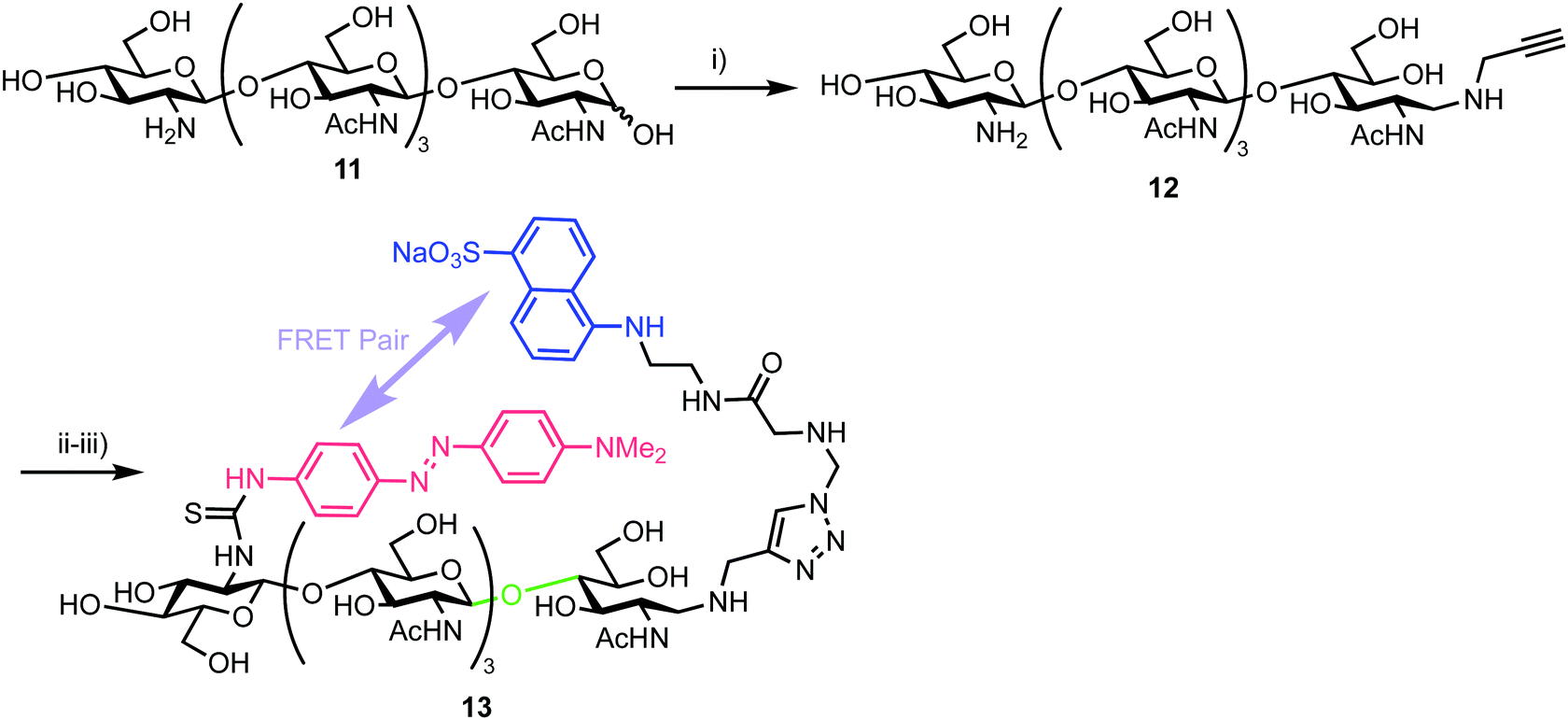 | ||
| Scheme 2 Reagents and conditions: (i) NaCNBH4, AcONH4, aniline, propargyl amine, 70%; (ii) N3-EDANS, CuSO4, sodium ascorbate; (iii) DABITC, NaHCO3, 48% two steps. | ||
With this oligosaccharide in hand, a FRET pair [5-(2-aminoethyl)amino-1-naphthalene-sulfonic acid, EDANS and dimethylaminophenylazophenyl, DAB] was installed utilising a method developed by Cottaz (Scheme 2).34 This introduced the fluorescent EDANS component at the open reducing-end sugar and the DAB acceptor through the amine of the non-reducing GlcNAc residue. Copper catalysed azide–alkyne cylcoaddition (CuAAC) ‘click’ chemistry was used to incorporate EDANS followed by free amine reaction with the isocyanate form of DAB (4-dimethylaminoazobenzene-4′-isothiocyanate, DABITC) in 48% yield over the two steps to deliver the final probe 13. This fluorescence quenched material was shown to be an active substrate for Chitinase A1 from Bacillus cirulans, with a time dependent increase in fluorescence at 490 nm corresponding to EDANS emission upon cleavage of the oligosaccharide by the enzyme and separation of the FRET-quenched pair.
3.3. Fluorogenic probe for endo-β-acetylglucosiminidases
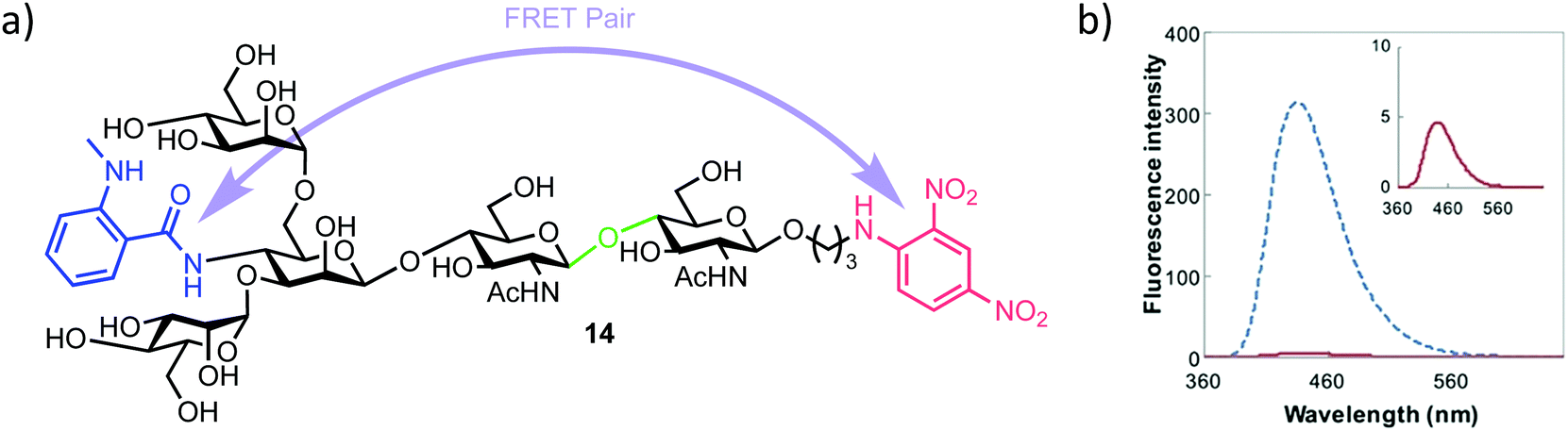 | ||
| Fig. 3 (a) Pentasaccharide ENGase FRET probe 14, green highlights the ENGase cleavage point, blue the NMA fluorescent component and red the DNP acceptor (b) fluorescence emission spectrum of 14 (inset red) alongside the NMA cleavage product (blue), following excitation and probe cleavage at 340 nm, concentration of 14 = 40 μm, adapted with permission from ref. 35. | ||
The required probe 14 was first shown to be 98% quenched (donor NMA fluorescence quenched by the DNP acceptor) by association of the appended intramolecular components (Fig. 3b, inset) followed by confirmed hydrolysis of 14 by commercial ENGase (Endo-M) from Mucor hiemalis. The process was monitored by HPLC and the characteristic retention times for enzymatic hydrolysis products confirmed using comparison to authentic standards.
Following this initial study, a real-time activity monitoring of the hydrolysis reaction was tested against five different ENGases from different species and activity confirmed for human and mouse ENGases, Endo-OM and Endo-CC, but no activity was observed against Endo-H. This was rationalised by Endo-H not generally being considered able to cleave core N-glycan regions, instead preferring high mannose type ligands. Finally, the authors adapted their probe to a high throughput screen (HTS) format. Detection of the hydrolysis of 14 was possible down to 0.5 μM and, importantly for HTS development, the inclusion of up to 20% DMSO in the reaction medium had no effect on probe activity. Using either thiazoline or Rabeprazole sodium inhibitors of Endo-M, IC50 values for these compounds were obtained as 626 μm and 40 μm respectively, indicating the applicability of this probe to screen libraries of potential ENGase inhibitors.
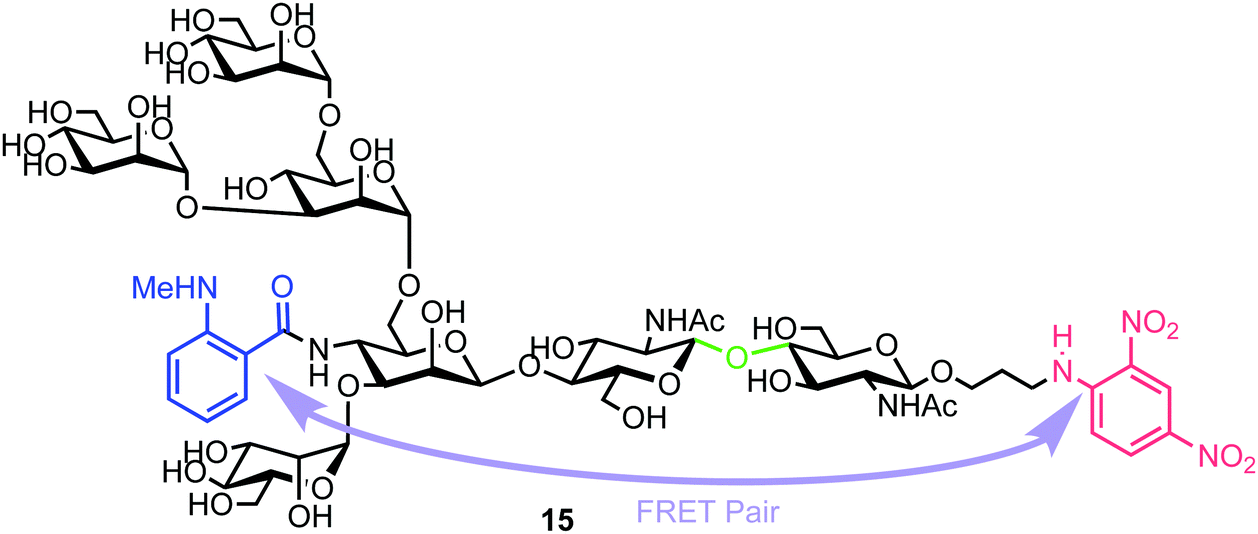 | ||
| Fig. 4 Heptasaccharide Endo-H FRET probe 15, green region highlights the Endo-H cleavage point, blue the NMA fluorescent component and red the DNP acceptor. | ||
Chemical synthesis of the probe started with galactosylchitobiose 16 as an acceptor and mannose thioglycoside 17 as donor (Scheme 3). The C-3 hydroxyl group in the galactose component of 16 had been shown to have enhanced nucleophilicity,38 and coupling with 17 was therefore completed using an unprotected galactosyl component within 16, providing tetrasaccharide 18 in 14% yield. Tetrasaccharide 18 was next reacted with a mannotriose donor 21, synthesized from the glycosylation of mannosyl acceptor 19 with chloride donor 20 using silver triflate (AgOTf) as a promoter. A trifluoromethanesulfonic anhydride (Tf2O) promoted coupling of donor 21 with tetrasaccharide 18 provided C-6 galactose-linked heptasaccharide 22 in 25% yield. Following this, triflation of the C-2 and C-4 hydroxyl groups in 22 was completed to give 23.
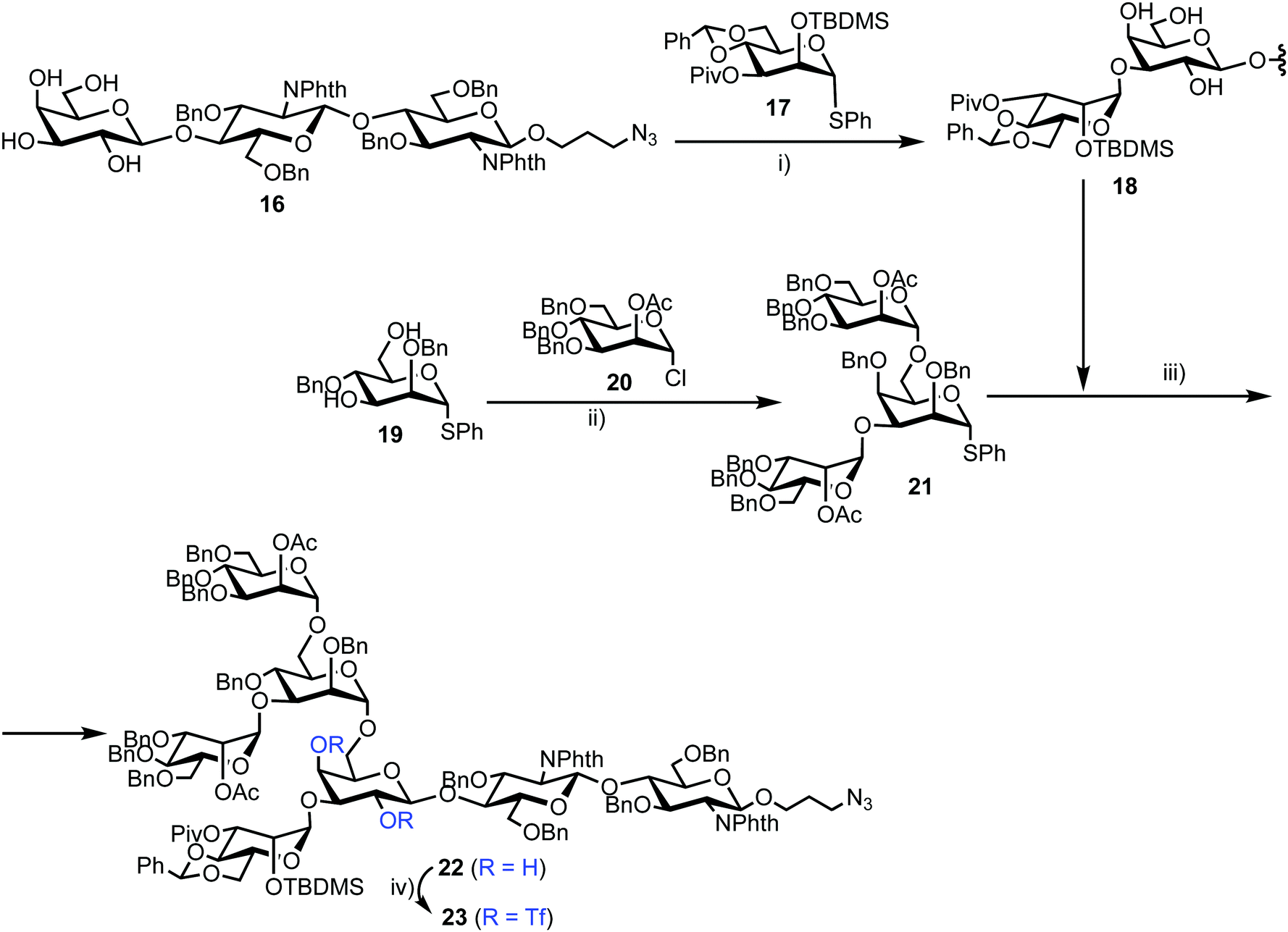 | ||
| Scheme 3 Reagents and conditions: (i) NIS, TfOH, MS 4 Å, CH2Cl2, −78 °C, 14%; (ii) AgOTf, MS 4 Å, CH2Cl2, −40 °C, 92%; (iii) NIS, TfOH, MS 4 Å, CH2Cl2, −78 °C, 25%; (iv) Tf2O, pyridine, CH2Cl2, −10 °C. | ||
Successful triflation then enabled a regioselective nucleophilic substitution of the C-4 triflate with azide using TBAN3 (Scheme 4). A second nucleophilic substitution at C-2 with CsOAc using 18-crown-6 in toluene under ultrasonication yielded heptasaccharide derivative 24. This material then underwent a series of protecting group manipulations, developed previously by Hindsgaul,39 followed by deprotection. Exploiting a difference in reactivity between the two free amino groups, a DNP group was selectively introduced at the reducing end amino propyl group followed by non-reducing end labelling with N-methylanthranilonic acid (MANT) to afford the final FRET-enabled heptasaccharide probe 15.
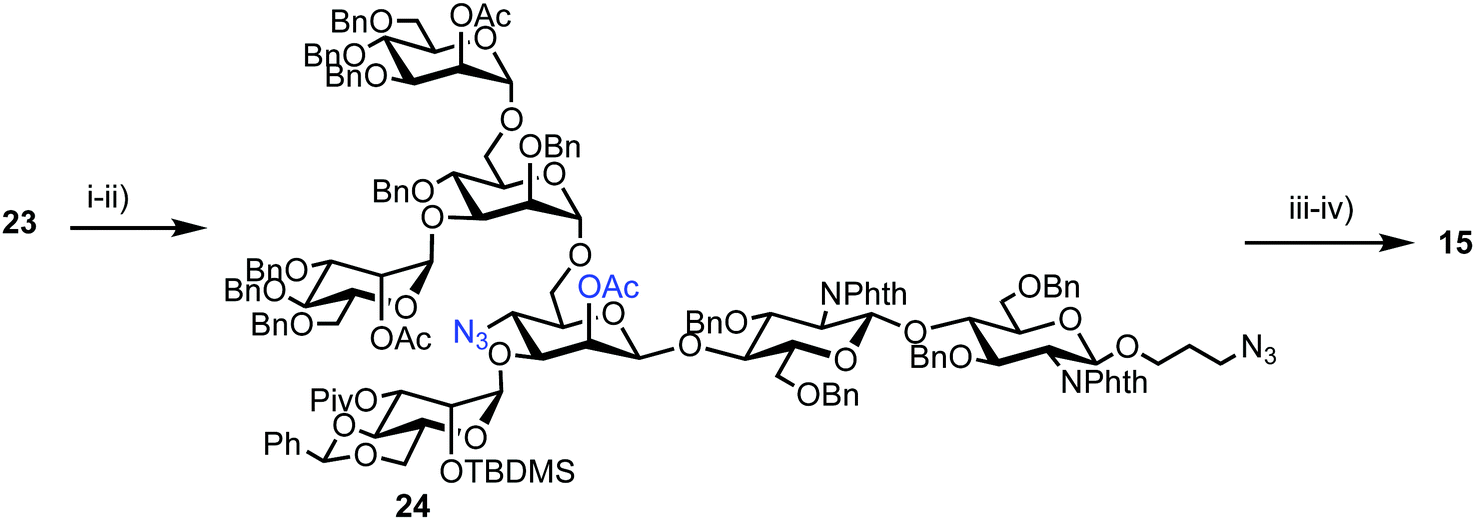 | ||
| Scheme 4 Reagents and conditions: (i) TBAN3, toluene; (ii) CsOAc, 18-crown-6, toluene, ultrasonication, 40 °C, 65% (3 steps from 22); (iii) (a) TBAF, THF, 40 °C, (b) ethylenediamine, n-BuOH, 90 °C, (c) Ac2O, pyridine, 40 °C, (d) NaOMe, MeOH, THF, 30 °C, (e) H2, Pd(OH)2, THF, H2O, 40 °C, 57% (5 steps); (iv) (a) 2,4-dinitrophenylfluoride, NaHCO3, MeOH, H2O, (b) N-methylanthranilic acid, HATU, DMAP, DMSO, 31% (2 steps). | ||
The NMA acceptor within heptasaccharide 15 was confirmed as 96% quenched by the DNP acceptor and this material was then incubated with Endo-H and the hydrolysis reaction monitored by HPLC using 360 nm (UV) and fluorescence detection (440 nm, excitation at 430 nm). The results observed from the fluorescence monitoring showed formation of hexasaccharide and monosaccharide products, indicative of cleavage, alongside an increase in fluorescence with increasing concentrations of Endo-H. Comparatively, a pentasaccharide with no branching mannose residues (MM3D) was not hydrolysed by Endo-H (Fig. 5). To demonstrate broader application for the probe, the team performed substrate specificity studies using Endo-M, belonging to GH family 85, comparing the hydrolysis of 15 and MM3D. They observed that both probes exhibited activity but that activity was higher for the MM3D ligand, confirming previous substrate specificity for Endo-M.
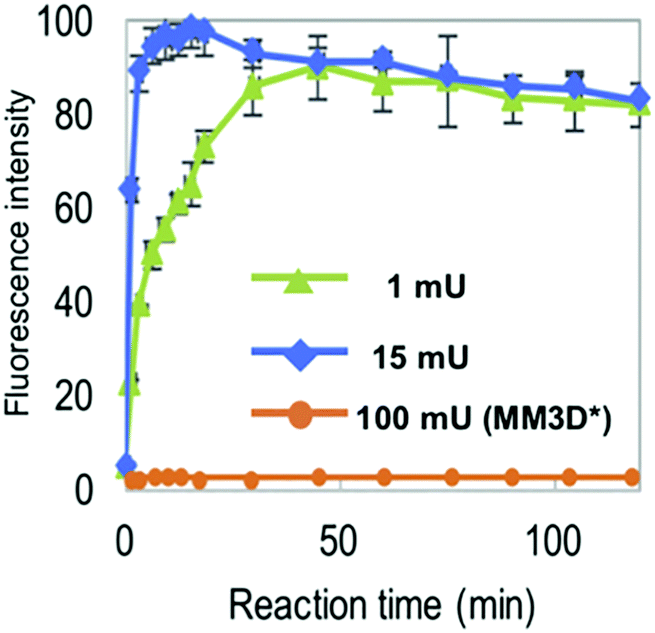 | ||
| Fig. 5 Monitoring hydrolysis of probe 15 by Endo-H at different substrate concentrations (green and blue traces) compared to pentasaccharide probe MM3 (orange trace), adapted with permission from ref. 37. | ||
3.4. A FRET assay for monitoring Golgi endo-α-mannosidase activity
Matsuo et al. also reported the synthesis of a tetrasaccharide probe for quantitative detection of hydrolytic activity for a Golgi endo-α-mannosidase (G-EM) using a fluorescence quenching assay.40 G-EM catalyses deglycosylation of N-glycans and plays crucial roles in the post-endoplasmic reticulum quality control pathway.41 Radiolabelled N-glycans have previously been used for measuring G-EM activity however these techniques are faced with limitations, such as challenging chromatographic separation of radioisotopically labeled compounds, poor reproducibility, and lack of immediate access to real-time monitoring. Furthermore, fluorogenic probes require an enzyme inhibitor to be included in the assay setup to avoid removal of the glucose residue by ER glucosidase II,42 and can therefore complicate the process. To overcome these limitations, the group synthesized tetrasaccharide probe 25 to provide accurate measurement of G-EM activity based on FRET quenching and which was not susceptible to glucosidase activity. The tetrasaccharide probe was labelled with MANT as the donor dye at the non-reducing end and DNP group as the acceptor at the reducing end (Fig. 6).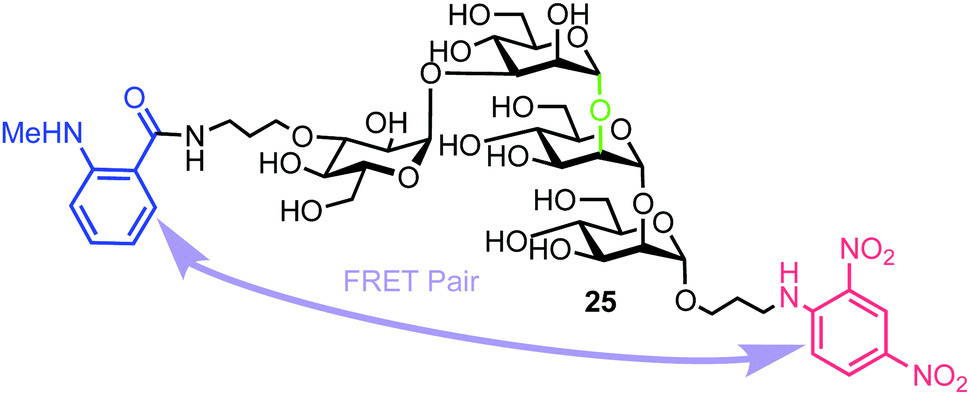 | ||
| Fig. 6 Structure of fluorogenic probe 25 containing a high-mannose type oligosaccharide labelled with MANT and DNP components. The enzyme cleavage position is shown in green. | ||
The synthesis of 25 utilised mannotriose 26, conveniently prepared from a common thioglycoside precursor, with a pivaloyl (Piv) group at the non-reducing terminal C-3 position to provide selectivity in introducing the glucose component (Scheme 5). tert-Butyldimethylsilyl (TBS) deprotection and subsequent selective removal of the Piv group afforded 27 which was then acetylated to obtain acceptor 28. Glycosylation of this acceptor was performed using NIS/AgOTf as promoter and donor 29 (obtained in two steps from a benzylidene glucose derivative) to afford tetrasaccharide 30. This reaction afforded both α- and β-anomers of 30, which were separated by silica gel column chromatography followed by final deprotection and introduction of the MANT and DNP fluorescent dyes, which was carried out in a stepwise manner to yield doubly labelled α-tetrasaccharide probe 25.
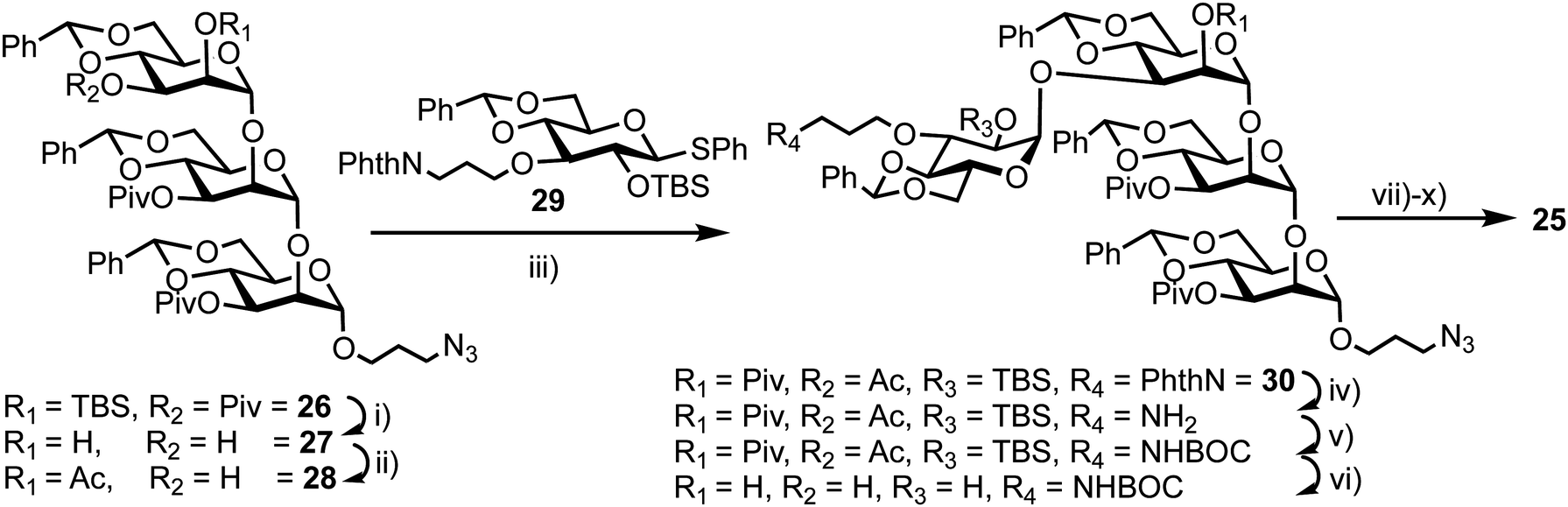 | ||
| Scheme 5 Reagents and conditions: (i) (a) TBAF, THF, (b) NaOMe, THF, MeOH, 40 °C, 83%, 2 steps; (ii) (a) trimethylorthoacetate, CSA, CH3CN (b) HCl, EtOAc, 61%, 2 steps; (iii) NIS, AgOTf, MS 4 Å, CH2Cl2, −20 °C, 29, 63%; (iv) (a) TBAF, THF, 40 °C, (b) ethylenediamine, n-BuOH, 90 °C, 60%, 2 steps; (v) Boc2O, THF; (vi) NaOMe, THF, MeOH, 40 °C, 74%, 2 steps; (vii) Pd(OH)2/C, t-BuOH, H2O, 40 °C, 97%; (viii) 1-fluoro-2,4-dinitrobenzene, NaHCO3, MeOH, H2O, 61%; (ix) TFA, 0 °C; (x) (a) N-methylanthranilic anhydride, NaHCO3, 1,4-dioxane, (b) NaOMe, MeOH, 61%, 3 steps. | ||
The photophysical properties of probe 25 were investigated by comparing the fluorescence intensity and decay of 25 and a MANT labelled disaccharide 31 (the expected product of hydrolysis of 25 by G-EM). Fluorescence intensity of the latter was found to be 53 times higher than for probe 25, indicating the MANT group to be 95% quenched by the DNP acceptor.
To test the biological capability of 25, hydrolysis experiments were performed with recombinant human G-EM,43 and the reaction was monitored by HPLC with UV detection. The results indicated that G-EM cleaved the central glycosidic linkage of 25 to give glucomannose 31 and mannobiose derivative 32 (Fig. 7).
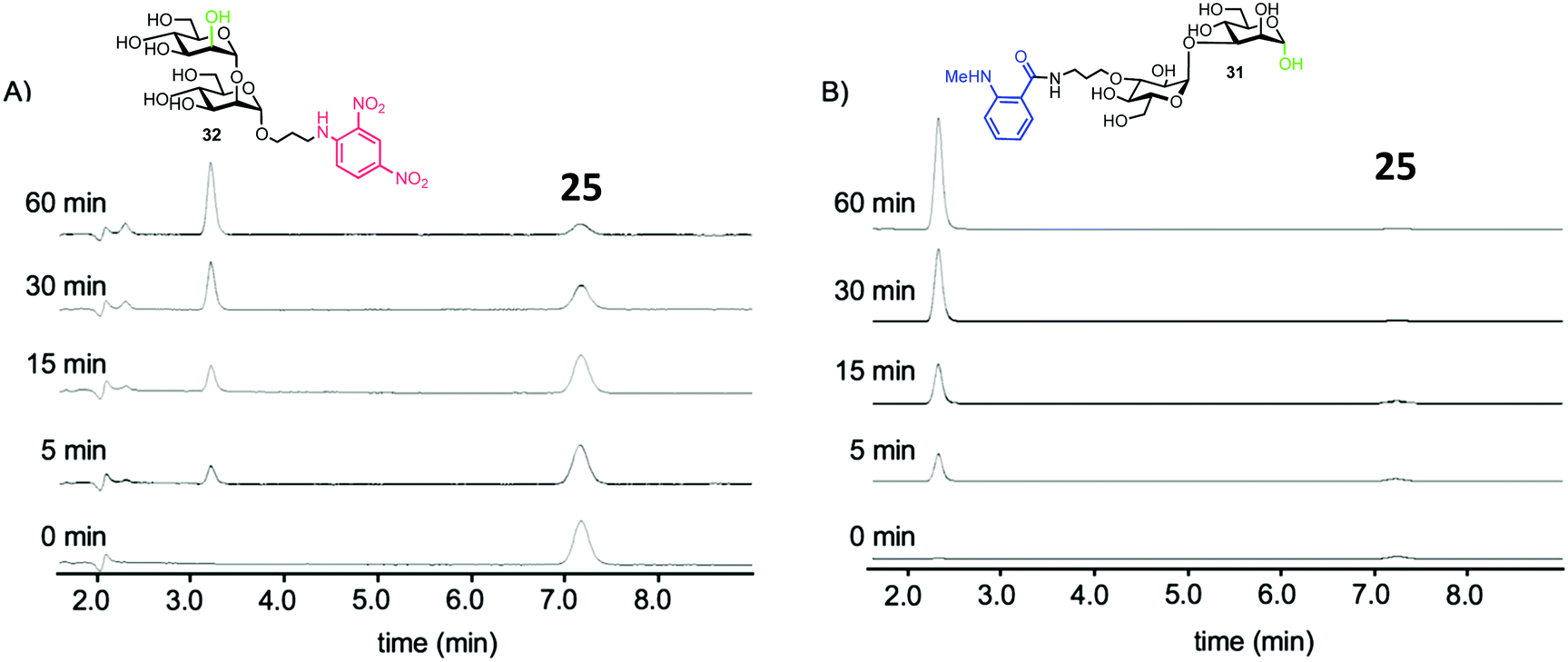 | ||
| Fig. 7 HPLC traces for cleavage of probe 15 by human G-EM. Disaccharide products 31 and 32 were detected at 360 nm and 440 nm for 32 and 31 respectively, adapted with permission from ref. 40. | ||
The group also investigated hydrolysis of probe 25 by G-EM in real time using a microplate reader. Differing concentrations of G-EM were incubated with 5 μM of 25 and the observed fluorescence intensity increased rapidly with increasing concentration of enzyme. Notably, the fluorescence intensity remained unchanged when the β-Glc configured stereoisomer of 25 was used, confirming specificity of G-EM for Glc-1,3-Man α-linkages. Determination of the Michaelis constant (Km) for 25 with G-EM (Km = 19 μM) and for a tetrasaccharide derivative without the MANT donor (Km = 15 μM) indicated no significant difference with regard to enzyme recognition. The resistance of fluorogenic probe 25 to glucosidase was also confirmed by incubating 25 with glucosidase II and no reaction was observed. Importantly this research described the first example of a FRET probe for detecting G-EM activity without the need for a glucosidase inhibitor to be included in the assay setup.
3.5. Fluorescent heparin to monitor endo-β-D-glucuronidase activity
Human heparanase (Hpa) is an endo-β-D-glucuronidase that cleaves linkages between glucuronic acid (GlcA) and glucosamine (GlcN) components in heparin (H) and heparan sulfate (HS) chains (Fig. 8). These polymeric chains are present on cell surfaces and in the extracellular matrix, playing a crucial role in cellular adhesion, tumour angiogenesis, and metastasis.44,45 Due to the pathophysiological ubiquity of Hpa, several biochemical and biophysical methods have been developed to detect its activity.46–48 Initially, these methods focused on quantifying levels of H/HS degradation products e.g. GlcA-terminated reducing sugars, and more recently, activity-based probes for Hpa have been established.49 In 2019 Desai and colleagues reported the synthesis of fluorescently labelled H as an efficient FRET probe for quantifying human Hpa activity and to screen for potential inhibitors.50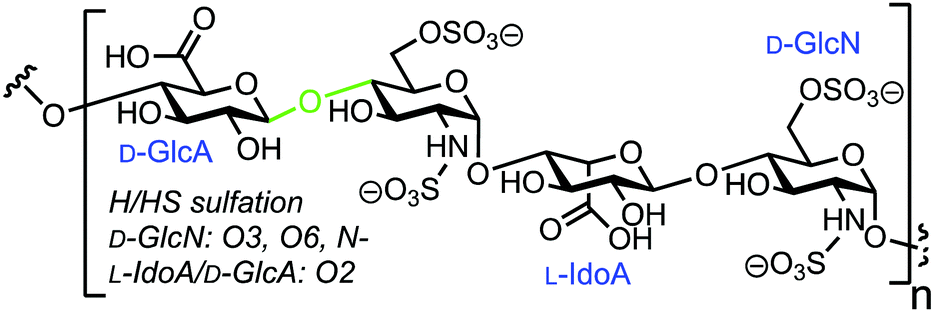 | ||
| Fig. 8 Chemical structure of heparin (H)/heparan sulfate (HS), Hpa cleavage site shown in green. | ||
The team utilised heterogeneous heparin (containing repeat GlcA–GlcNS6S units) and conjugated with 4′-dimethylaminoazobenzene-4-carboxylate (DABCYL) and EDANS as the FRET pair. This was completed using conventional carbodiimide coupling of uronic acid carboxylates within the carbohydrate polymer to amine-terminated fluorescent labels. As such, either probe could theoretically be introduced anywhere along the chain and the team prepared four heparin–FRET samples using varying equivalents of EDANS and DABCYL (1:1, 1:2, 2:2, and 2:1) per average chain of unfractionated heparin. 1H NMR spectroscopy was used to determine the stoichiometries of the two FRET labels in the final product. The cleavage of these substrates by Hpa and consequent FRET signal enhancement was evaluated and the modified polymer with singular incorporation of both EDANS and DABCYL was observed to show the highest increase in fluorescent intensity.
Next, the team investigated the kinetic properties of the substrate to obtain optimal conditions for a Hpa activity assay. A Km was calculated as 46 ± 12 μM and inhibition experiments of Hpa by suramin,51 using the FRET probe, showed it to offer sufficiently sensitive measurement capability; an IC50 for suramin was established as 330 ± 2.5 μM. Furthermore, the group implemented the FRET assay to monitor the activity of Hpa secreted by cancer cells. The human breast cancer cell line MCF7 was grown in both normoxic and hypoxic conditions with varying amounts of fetal bovine serum (FBS, 0%, 2%, and 10%) to mimic the in vivo conditions. Hpa activity was found to be almost 10% higher under hypoxic conditions (compared to normoxic) for cells grown in FBS-free media.52 Cells grown in 2% FBS did not show much difference in activity between hypoxic and normoxic conditions and 10% FBS media, interestingly, repressed the Hpa activity under both conditions.
The establishment of this rapid and simple to use assay for heparanase activity is important as the field progresses to identify potential inhibitors and undertake monitoring of enzyme activity in cells. Importantly, the authors identified caution that must be taken using this approach with labeled heterogeneous heparin samples and the requirement to analytical reproducibility in terms of future commercial viability.
3.6. A multi-purpose FRET probe for ganglioside-processing enzyme activity
In 2015, Withers et al. reported the synthesis and application of a fluorescent probe to detect and quantify ganglioside degrading enzymes in cell lysates, as well as in living cells.53 Gangliosides, a group of glycosphingolipids containing one or more terminal sialic acid residues, are found in the cell membrane and play crucial roles in biochemical signalling, pathogen entry, membrane transport, and intracellular protein sorting.54The design of the probe featured 7-hydroxycoumarin and BODIPY as FRET donor and acceptor components, attached to either end of a ganglioside GM3 derivative (33, Fig. 9). This was proposed to enable recognition and cleavage by different ganglioside-degrading enzymes, namely sphingolipid ceramide N-deacylase (Enz1), endoglycoceramidase (Enz2), and neuraminidase (Enz3). Cleavage of the probe by any of these enzymes would yield a large decrease in FRET efficiency, providing a detectable ratiometric fluorescent signal for enzyme activity.
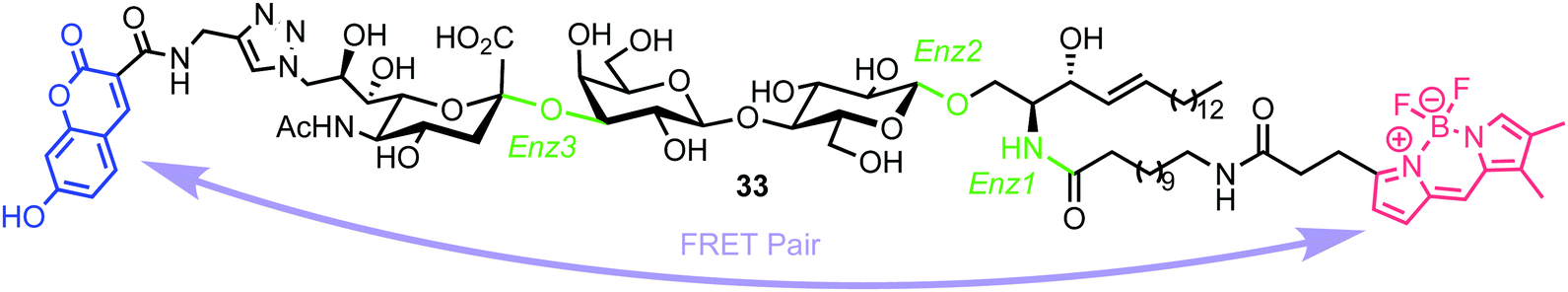 | ||
| Fig. 9 Chemical structure of ganglioside FRET probe 33 with cleavage points by ganglioside processing enzymes highlighted in green. | ||
The synthesis of the target probe was completed chemoenzymatically, initially using enzymatic methods previously established by the group (Scheme 6).55,56 Glycosylation of D-erythro-sphingosine with lactosyl fluoride 34 was completed using EGCase glycosynthase to yield lactosyl sphingosine 35 followed by coupling of 9-azido sialic acid using the sialyltransferase Cst-I from C. jejuni to obtain trisaccharide 36. Next, a coupling of coumarin derivative 37 with 36 using CuAAC ‘click’ chemistry resulted in a fluorescent lyso-GM3 derivative, which was followed by enzymatic condensation of with BODIPY-coupled lauric acid 38 to afford the target FRET-enabled probe 33.
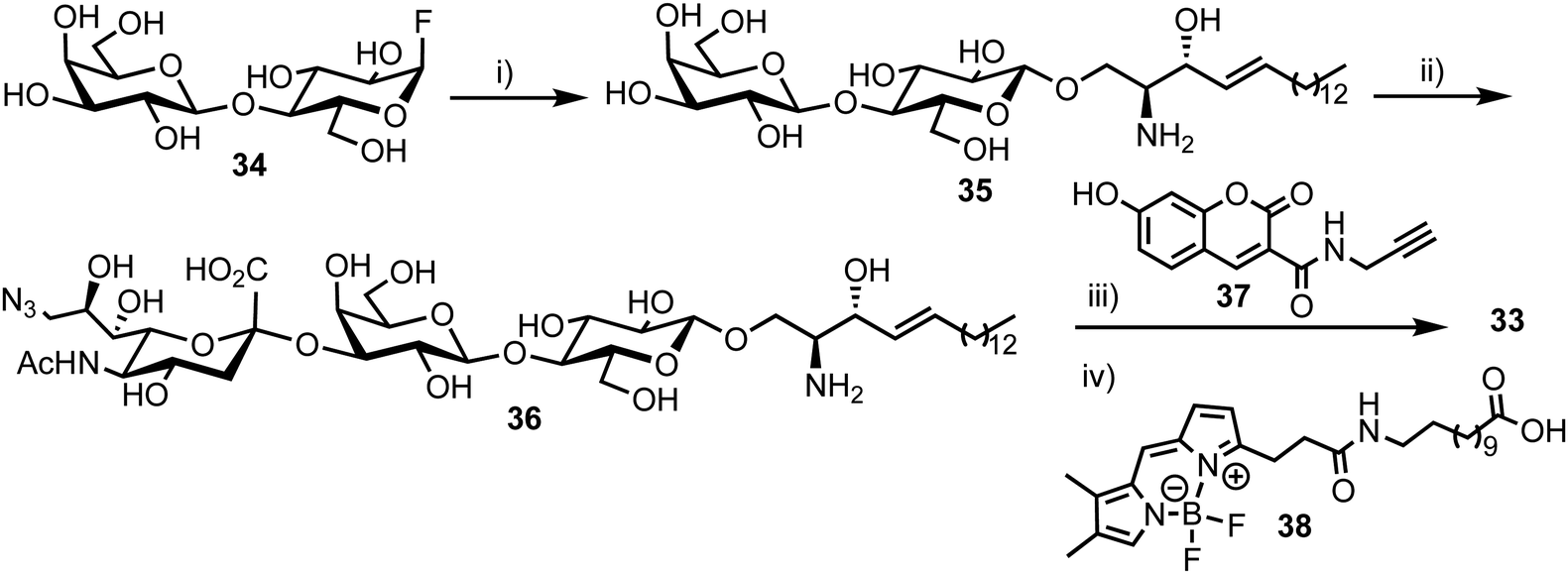 | ||
| Scheme 6 Reagents and conditions: (i) EGCase glycosynthase, D-erythro-sphingosine, NaOAc buffer pH 5.3, 10% dimethoxyethane, 37 °C; (ii) CMP-9-azido-9-deoxy-sialic acid, α-2,3-sialyltransferase, alkaline phosphatase, Tris buffer (pH 7.5), MgCl2, RT; (iii) 37, BTTES buffer, sodium ascorbate, CuSO4, DMF, 35 °C; (iv) 38, SCDase glycosphingolipid N-deacylase, HEPES buffer (pH 7.5), 10% dimethoxyethane, 35 °C. | ||
Probe 33 formed micelles in aqueous solution affecting its fluorescent properties significantly, so the team used Triton X-100 as a spacer surfactant to reduce the self-quenching that was initially observed. The emission of 7-hydroxycoumarin at 450 nm was barely observed when excitation was completed at 360 nm, while a strong emission from BODIPY was evident at 518 nm, with a FRET efficiency of >95%.
Next, the individual addition of ganglioside degrading enzymes (Enz1 from Shewanella alga G8, Enz2 from Rhodococcus strain M-777 or Enz3 from Micromonospora viridifaciens) increased the fluorescence emission at 450 nm significantly, accompanied by a decrease in emission at 518 nm. The products obtained after the cleavage of 33 by Enz1, Enz2 and Enz3 were visualized by TLC and a markedly different pattern was observed in each case, making 33 suitable for simultaneous detection in a multi-enzyme system (Fig. 10). The changes in ratio of emission (λem 450 nm and λem 518 nm) for the FRET components were up to 70-fold for Enz2 and Enz3. However, with Enz1 the change was significantly smaller (7.3-fold) since an equilibrium is established between hydrolysis and forward synthesis.
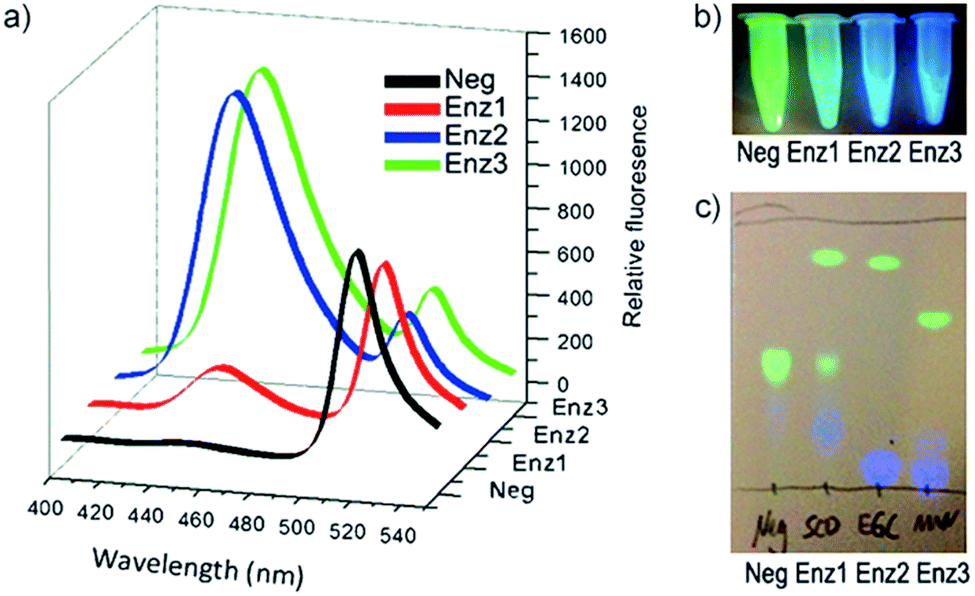 | ||
| Fig. 10 Hydrolysis of probe 33 by sphingolipid ceramide N-deacylase (Enz1), endoglycoceramidase (Enz2), or neuraminidase (Enz3), coumarin fluorescence (blue) and BODIPY fluorescence (green). (a) Fluorescence spectra before and after 30 min incubation with the different enzymes. (b) Visual detection of the reactions under a UV lamp. (c) Different hydrolysis patterns of probe 33 after hydrolysis by Enz1, Enz2, and Enz3, adapted with permission from ref. 53. | ||
The ability of 33 to insert into the membrane of human lymphoma cells was investigated next and its rapid hydrolysis was observed upon simple addition to the cell media due to neuraminidase activity. To diminish this effect, a microinjector was utilised to add a small amount of 33 in the vicinity of cells, during imaging. From the observed BODIPY emission, it was clear that staining of the cells occurred primarily at the plasma membrane. The addition of 33 strongly suppressed the decrease in fluorescence signal in all three channels, suggesting that the decrease monitored was primarily due to neuraminidase activity. The background-corrected FRET signal from cells labelled with 33 showed large differences in their rate of decay in the FRET channel in the presence versus the absence of 2,3-didehydro-N-acetyl-neuraminic acid (DANA), a neuraminidase inhibitor used to block the action of these enzymes.
The team concluded that 33 inserted into the plasma membrane where it is acted on as a substrate of human neuraminidases, although they could not rule out that other enzymes or physical processes were contributing to the observed emission changes. The experiments suggested no background hydrolysis of the probe in lysates of control E. coli BL21 cells, even after 12 h. However, on addition of lysate from E. coli cells expressing a control neuraminidase, the changes in fluorescence were observed to be dependent on the lysate dose. The synthesized probe 33 is ideal for the high-throughput assay of ganglioside degrading enzymes, enabling a highly sensitive real time observation of the process and compliments previously developed multienzyme detection probes.57
4. FRET probes for exo-acting glycosyl hydrolases
4.1. Bis-acetal based substrates for probing exo-glycosidase activity
In 2017, Vocadlo and Cecioni developed bis-acetal based substrates (BABS) as fluorescence quenched probes to monitor exo-glycosidase activity.58exo-Glycosidases cleave internal glycosidic linkages which are usually more difficult to target with fluorescence equipped probes as the binding sites are often sterically encumbered and pocket shaped. This class of CAZy have emerging roles in human health and systems that utilise covalent attachment have previously been developed in this area.59 The team proposed to position the fluorescent components outside of the enzyme–substrate binding region, thereby preserving the essential carbohydrate recognition motif.Accordingly, a fluorescence quenched FRET pair attached to a glycoside substrate at the anomeric position (as a bis-acetal) was envisaged, with subsequent action of a glycosidase releasing the carbohydrate and FRET component as hemi-acetals, the latter then breaking down to release the FRET pair and fluorogenic readout garnered from the partners no longer being in close proximity. The known stability of anomeric acetals (through endocyclic oxygen) compared to regular acetals was suggested to enable stability across a physiological pH gradient.
The chemical synthesis of two BABS derivatives started from conveniently prepared D-GlcNAc derivative 39 (Scheme 7).60 Firstly, a regio- and stereoselective bromoalkoxylation of 39 was completed using N-bromosuccinimide (NBS) and 2-(2-azidoethoxy)ethanol followed by a three-step one-pot functional group manipulation to deliver carboxylic acid 40. An EDANS donor fluorophore was then installed using amide coupling with carboxylate 40 followed by addition of a DABCYL quencher through the pendant azide. Final deprotection furnished GlcNAc-Br-BABS probe 41. For an alternative GlcNAc-OH-BABS system, the alkene in 39 first underwent stereoselective epoxidation to 42 followed by regioselective acid mediated ring opening with 2-(2-azidoethoxy)ethanol and functional group manipulation to deliver 43, which was then labelled in a similar manner to afford 44.
 | ||
| Scheme 7 Reagents and conditions: (i) 2-(2-azidoethoxy)ethanol, NBS, DCM, 57%; (ii) (a) LiOH, H2O, THF (b) Ac2O, pyr. (c) THF, H2O, 57–62% three steps; (iii) EDANS-NH2, HBTU, DIPEA, DMF; (iv) DABCYL-alkyne, Cu(MeCN)4PF6, DIPEA, DCM, 64%, two steps (v) NaOMe, MeOH; (vi) mCPBA, Na2SO4, toluene, 67%; (vii) 2-(2-azidoethoxy)ethanol, CSA, DCM, 66%; (viii) (a) LiOH, H2O, THF (b) Ac2O, pyr. (c) THF, H2O, 57–62% three steps; (ix) EDANS-NH2, HBTU, DIPEA, DMF; (x) DABCYL-alkyne, Cu(MeCN)4PF6, DIPEA, DCM, 81%, two steps; (xi) NaOMe, MeOH. | ||
With fluorescence quenched probes 41 and 44 in hand, the group first confirmed their stability across a pH range from 2 to 10 and then examined hydrolytic susceptibility using the exo-acting human O-GlcNAcase (hOGA), which cleaves O-linked β-N-GlcNAc units from serine and threonine residues. hOGA successfully catalysed the hydrolysis of 41 and 44 revealing kcat/Km values comparable to the native substrate (263 and 519 M−1 s−1, respectively) (Fig. 11).
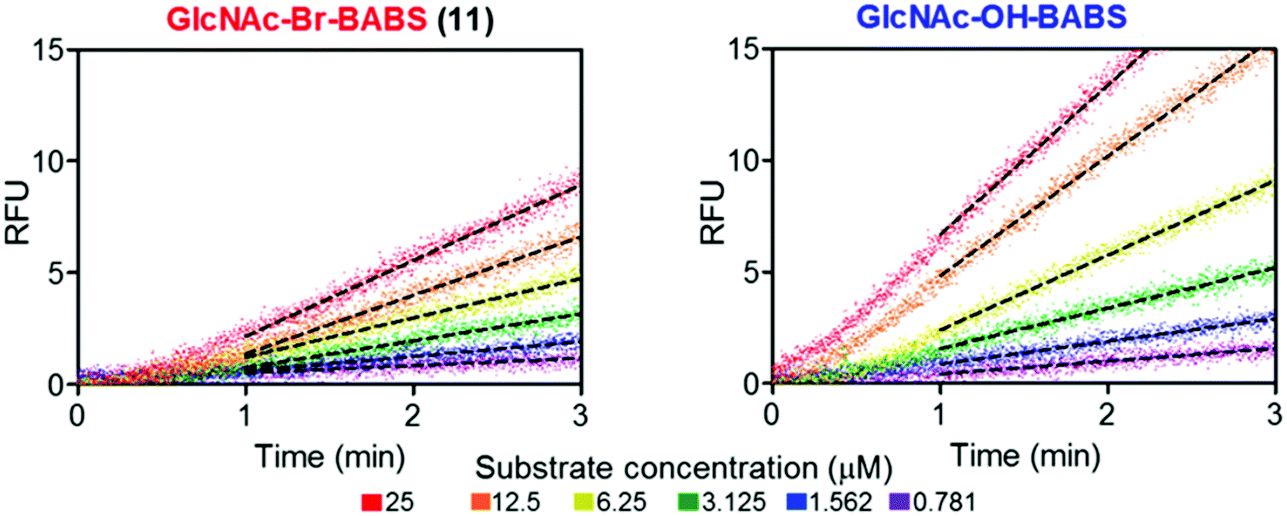 | ||
| Fig. 11 BABS probes 41 and 44 are turned over by hOGA. Evolution of fluorescence (RFU) for different concentrations of 41 and 44 in the presence of hOGA. Dotted lines represent the linear rates reached at steady state, adapted with permission from ref. 58. | ||
During these experiments, the authors noticed a short lag phase (approx. 1 minute) before linear activity rates were achieved which was proposed to be from an accumulation of the hydrolysed hemi-acetal intermediate containing both FRET components. To explore this effect, the enzymatic activity was stopped after 1 minute by addition of a tight binding hOGA inhibitor (Thimaet-G, 100 μM, Ki = 2 nM), which enabled monitoring of the breakdown of hemi-acetal generated before this point. Hemi-acetal from 44 broke down 4.5 times faster than 41, supporting a stabilising effect of the alpha bromine in reducing hemi-acetal decomposition.
Finally, turnover of 41 was monitored in SK-N-SH cell lysate with increasing concentrations of hOGA and processing rates similar to those in buffer were observed. Overall, the glyco-BABS probes provided proof of concept for monitoring exo-glycosidase activity in vitro and in cells and could be used in harmony with inhibitor development programs or to compliment recent capabilities in detecting post-translational glycan modification and regulation.61
5. FRET probes for glycosyl transferases
5.1. Monitoring α-1,3-fucosyltransferase IX activity
Fucosyltransferases catalyse the transfer of L-fucose from a donor molecule (a sugar nucleotide, NDP-sugar) to an acceptor. Within this classification, human fucosyltransferase IX (hFucT IX) belongs to the α-1,3-family, primarily transferring L-fucose with inversion of the anomeric configuration to terminal N-acetyllactosamine units. This process completes the synthesis of Lewis X (Lex), an epitope of many glyconconjugates. Lex is involved in cell–cell interaction and adhesion processes in both healthy and pathogenic pathways. The ability to accurately monitor hFucT IX catalysed activity in vitro would enable the elucidation of vital structure–function information offering the potential to then exploit the pathways it regulates to be uncovered, for example through the development of inhibitors.In 2013, Meir and Hahn described the synthesis of a fluorescently labelled GDP-β-L-fucose and used this to form a FRET-enabled Lex conjugate, catalysed by hFucT IX, thus establishing the capability to monitor the transformation.62 The group selected two commercial ATTO dyes as the FRET pair (donor ATTO550 and acceptor ATTO647N) and first completed a synthesis of ATTO 550 labelled GDP-β-L-fucose 48 (Scheme 8).
 | ||
| Scheme 8
Reagents and conditions: (i) (a) DMF, 4 Å MS; (b) CH3OH/H2O/Et3N (7:3:1), 41% two steps; (ii) H2, Pd/C, CH3OH, 90%; (iii) ATTO 550-NHS, NaHCO3, DMSO, buffer (pH = 8.3), 73%. | ||
The chemical synthesis of NDP-sugars is an historically challenging process, often associated with low yields and long reactions times.63,64 The same group have recently developed cycloSal-nucleotides,65 which serve as active phosphate ester building blocks for combination with an anomerically pure glycosyl 1-phosphate to reliably access NDP-sugars in high yield. Accordingly, 5-nitro-cycloSal-N2-acetyl-2′,3′-di-O-acetylguanosine monophosphate 45 and 2,3,4-tri-O-acetyl-6-azido-6-deoxy-α-L-galactopyranosyl phosphate 46 were coupled together in 41% yield (following deacetylation) to afford GDP-sugar 47. This material was then reduced at the L-Gal C-6 position using standard conditions in very good yield (90%), followed by facile coupling with the NHS-ester form of ATTO 550, to give fluorescent NDP-sugar 48 in 73% yield. The authors observed formation of hydrolysed dye during this final coupling step, but were able to separate this material from the required compound using silica gel and gel filtration purification techniques. HRMS confirmed the molecular mass of 48 (the structure of ATTO 550 is currently not disclosed but is related to other rhodamine dyes) and the conjugate exhibited identical absorbance and fluorescence maxima, compared to the NHS dye.
To access a fluorescently labelled acceptor partner for 48, N-acetylactosamines 52 and 53 were synthesised from lactal 49 (Scheme 9). Iodoacetoxylation of 49 followed by treatment of the resultant 2-iodo anomeric acetate with TMSN3/TMSOTf furnished an α-glycosyl azide. Subsequently, an amido glycosylation approach developed by Lafont was used to prepare β-glycosides of N-acetyl lactosaminide using either allyl alcohol or 4-penten-1-ol.66 Under these conditions the starting α-glycosyl azide forms an α-iminophosphorane through reaction with PPh3. This intermediate underwent intramolecular reaction to eliminate the C2 iodide and form a bridging C1–C2 aziridinium ion which was subsequently opened by the incoming alcohol nucleophile. Following basic workup, lactosaminides 50 were isolated in good yields. Finally, Zémplen deprotection, alkene functionalisation with cysteamine gave 51, and reaction with the NHS-ester form of ATTO 647N was completed. This afforded acceptor components 52 and 53 with an ATTO 467N acceptor dye at the reducing terminal, distanced from the anomeric centre by either propyl or pentyl hydrocarbon spacers and cysteamine.
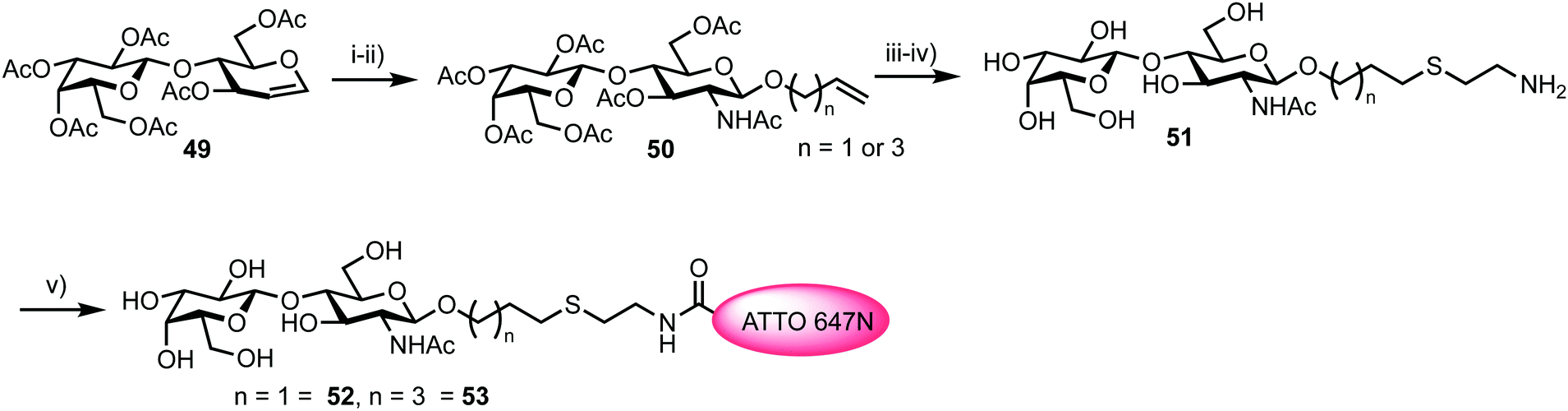 | ||
| Scheme 9 Reagents and conditions: (i) (a) TMSN3, TMSOTf; (ii) (a) ROH (allyl alcohol or 4-penten-1-ol), Ph3P, CH2Cl2; (b) Dowex OH−, EtOH, 65% two steps; (iii) NaOMe, MeOH, 94%; (iv) HSCH2CH2NH2·HCl, hν = 254 nm, H2O, 79% and 71%; (v) ATTO 647N-NHS, DMSO. | ||
The turnover of fluorescently labelled substrates 48 and 52/53 by hFucT IX was then analysed by high-performance thin layer chromatography coupled with mass spectrometry and dual colour fluorescence cross correlation spectroscopy. These studies revealed that both the sugar nucleotide donor 48 and disaccharide acceptor substrates 52/53 were accepted by recombinant hFucT IX, establishing a quick and versatile method for monitoring progress of this enzymatic process. Evidence for a FRET effect between the two dyes incorporated within the newly formed trisaccharide 54 was investigated (Fig. 12). Following successful glycosylation, a decrease in fluorescence intensity for ATT550 would be expected (FRET quenching). This was indeed observed by the authors, however, the additional expected increase in the fluorescence emission of the ATTO647N acceptor was not observed, suggesting a distance of >10 nm between the pair in 54. This work provides an exciting precedent for utilising labelled donor and acceptor components to monitor glycosyl transferase activity and also highlights the important molecular distance relationships required to capitalise on FRET effects.
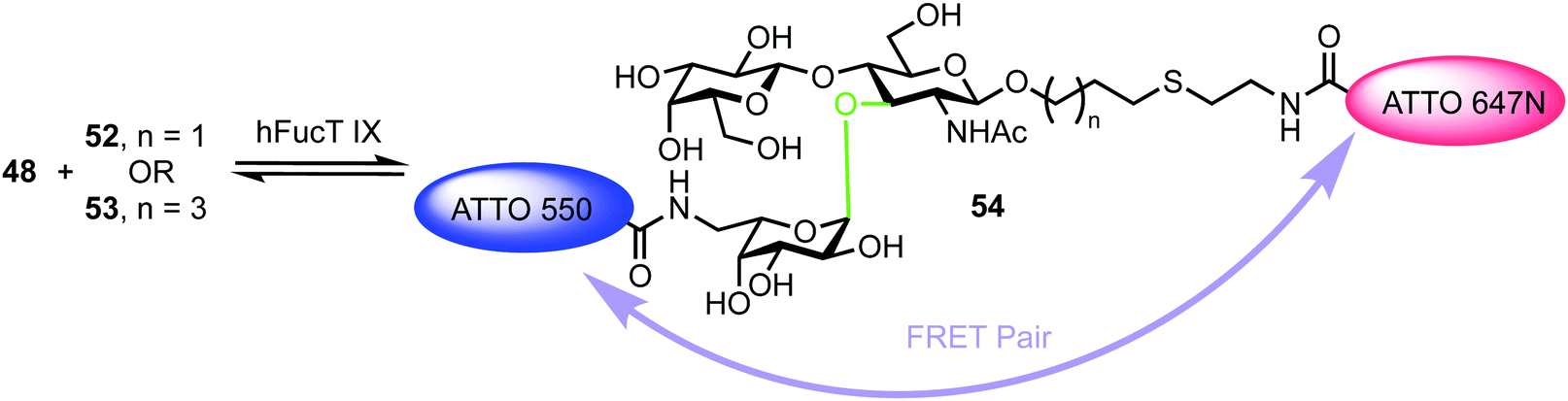 | ||
| Fig. 12 hFucT IX synthesis of Lex derivative 54 containing FRET enabled ATTO dyes. The α-fucosyltransferase catalysed bond formed is shown in green. | ||
6. Screening catalytic antibody activity
6.1. FRET probes for screening catalytic monoclonal antibody activity
Recently, Casadevall and Oscarson reported the design and synthesis of oligosaccharide FRET probes to characterise the hydrolytic activity of four catalytic monoclonal antibodies, effectively demonstrating their innate glycosidase activity.67 This was undertaken as part of a strategy targeting glycoconjugate vaccine candidates against the opportunistic fungi Cryptococcus neoformans, where the capsule polysaccharide glucuronoxylomannan (GXM) dominates. Based on their prior work developing synthetic glycan arrays of GXM structures,68 the group synthesised GXM decasaccharide targets 55 and 56, equipped with FRET capability, using a coumarin/DNP pairing (Fig. 13).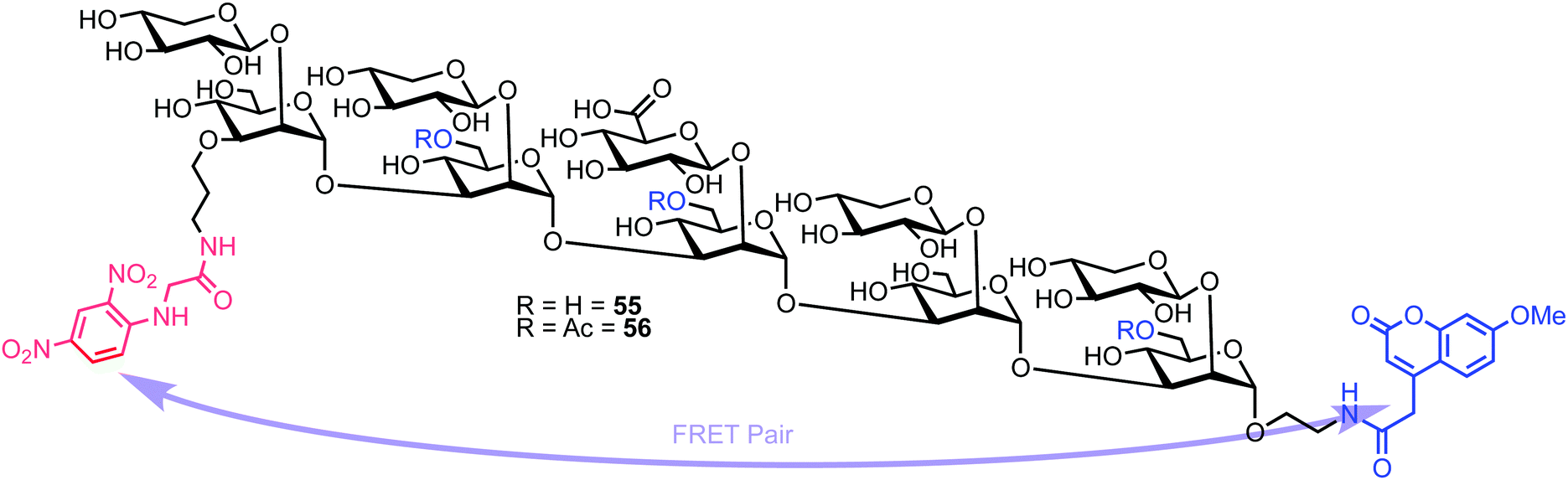 | ||
| Fig. 13 Decasaccharide GXM FRET probes for screening catalytic antibody activity. | ||
The stepwise synthesis and amalgamation of building blocks was carried out using methods previously developed by the group.69Scheme 10 highlights the synthesis, starting from tetrasaccharide 57, which underwent iterative coupling/deprotection sequences with glycosyl donors 58 and 59, to reveal a protected decasaccharide 60. This material was then globally deprotected using a pre-treated palladium catalyst,70 and consecutively conjugated with FRET donor and acceptor components to reveal probe 56. The non-acetylated probe 55 was accessed by removal of the acetyl groups from 56.
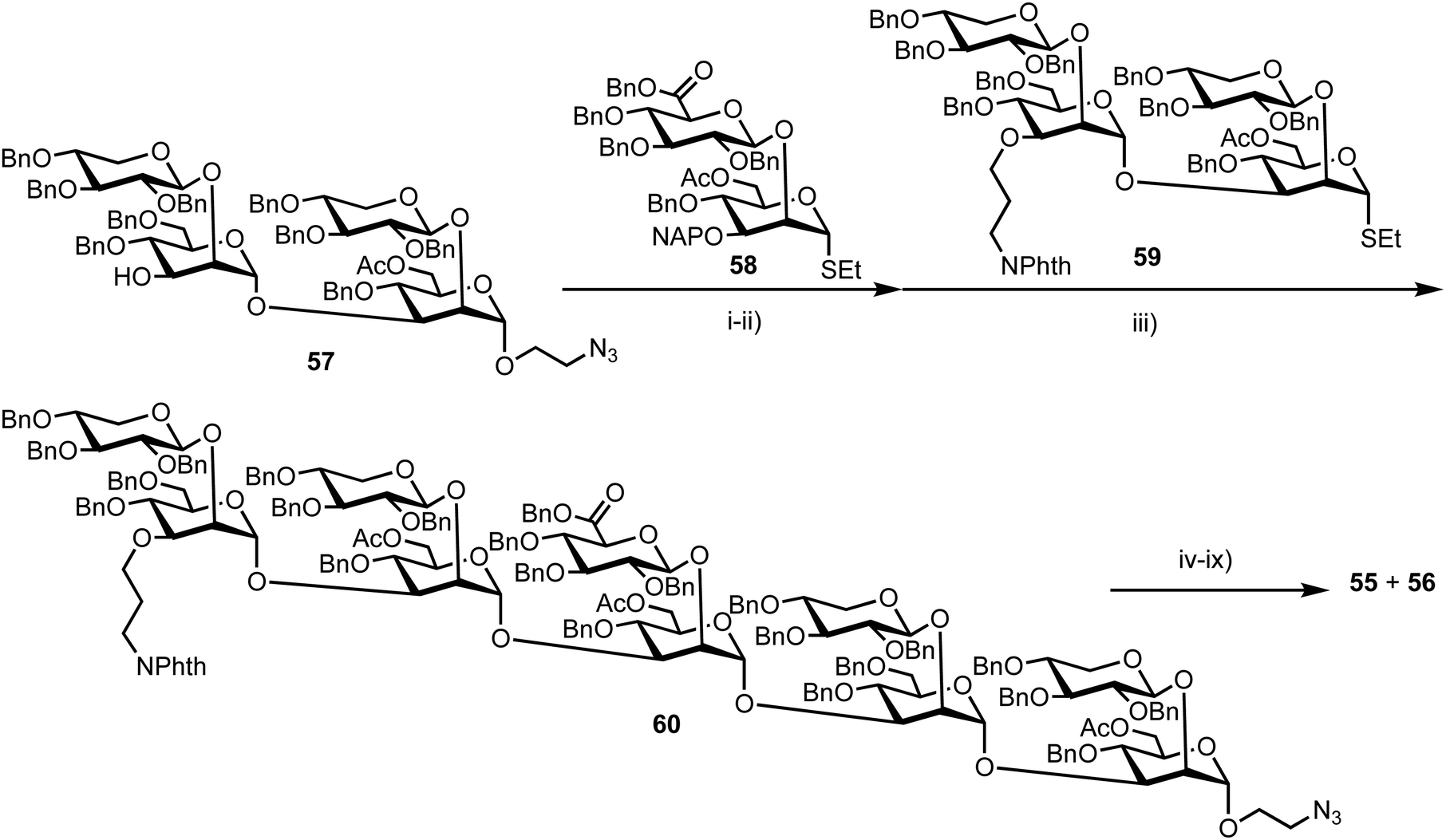 | ||
| Scheme 10 Reagents and conditions: (i) DMTST, Et2O, 4 Å MS, 0 °C 65%: (ii) DDQ, CH2Cl2, PBS pH 7.5, 0 °C, 70%; (iii) DMTST, Et2O, 4 Å MS, 0 °C, 85%; (iv) ethylene diamine, n-BuOH, 90 °C; (v) Boc2O, THF, H2O, 57%, 2 steps; (vi) Pd/C (pre-treated catalyst), H2, THF, t-BuOH, PBS (pH 5), 66%; (vii) 7-methoxycoumarin-4-acetic acid-N-succinimidyl ester, Et3N, DMSO; (viii) HCl, MeOH, H2O; (ix) N-(2,4-dinitrophenyl) glycine-N-succinimidyl ester, Et3N, DMSO. | ||
Next, the team implemented acylated and non-acylated FRET probes 55 and 56 to detect the catalytic activity of four murine monoclonal antibodies (mAbs): 18B7 (IgG1), 2H1 (IgG1), and two isotypes of 3E5, an IgG1 and IgG3. Highlights from this study showed the IgG3 isotype of 3E5 differed in specificity towards the two FRET probes when compared to 3E5 IgG1, despite these mAbs having the same variable sequence. The IgG3 displayed no activity towards 55, but the second highest activity against the acetylated ligand 56.
An evaluation of the Michaelis–Menten kinetics of mAb 2H1 against both FRET probes was also performed. Probe 56 had a Km of 2.12 × 10−4 mM, while 55 had a Km of 1.8 × 10−4 mM indicating marginally higher affinity of 2H1 for 55. However, determination of kcat (6.16 × 10−3 s−1 for 55 and 1.3 × 10−2 s−1 for 56) suggested 2H1 to be twice as efficient in hydrolysing 55. The kcat/Km values showed mAb 2H1 to be three times more efficient at hydrolysis of 55 than 18B7, suggesting 2H1 to be much more efficient at catalysing the hydrolysis of GXM oligosaccharides.
Overall this study gathered important new understanding regarding the role of oligosaccharide acetylation in antibody recognition and catalysis, with the inclusion of appropriate FRET components enabling kinetic parameters to be evaluated and the catalytic activity observable on the native capsule of heat-killed C. neoformans cells.
7. Summary of common FRET pairings used in oligosaccharide probe design
In Table 1, we summarise the common FRET pairings used to equip synthetic glycans for probing CAZy activity. Conventionally, these organic dyes are attached at the reducing and non-reducing ends of oligosaccharides at a late stage in the synthesis of linear sequences (ranging in length from tetrasaccharide up to polymeric systems). Parings have also been accomplished at only the reducing end of monosaccharides and at branching units in non-linear systems.| FRET pair | Spectral properties | ||
|---|---|---|---|
| Donor | Quencher | Donor (λabs/λem-max) (nm) | Quencher (λabs-max) (nm) |
| NAP | DANSYL | 290/333 | 333 |
| EDANS | DAB | 340/490 | 430 |
| NMA (MANT) | DNP | 340/440 | 360 |
| ATTO550 | ATTO647N | 554/576 | 646 |
| EDANS | DABCYL | 340/490 | 470 |
| 7-Hydroxycoumarin | BODIPY | 325/450 | 520 |
| 7-Methoxycoumarin | DNP | 325/410 | 360 |
8. Conclusion and outlook
Carbohydrate active enzymes control the production, processing and degradation of glycans which are underpinning to fundamental biology. Non-destructive analytical tools that can be utilised to monitor their function are therefore important and highly desirable. FRET-enabled carbohydrate probes are rapidly emerging to fulfil this requirement as important enabling tools. Their conceptualisation broadly involves synthetically installing the required fluorescent components (acceptor and donor) within a structurally defined carbohydrate enzyme substrate, thus enabling monitoring of catalytic function as the enzyme acts on the probe. Ratiometric monitoring enables alterations in fluorogenic readout to be captured as FRET quenching is removed and this provides direct access to important kinetic parameters regarding enzymatic activity and opens a pathway to screen for inhibitors. Nonetheless, judicious choices must still be made when designing probes for FRET experiments, such as instrumentation, choice of donor/acceptor parings and control experiments to unambiguously demonstrate FRET.71Exciting achievements in the past ten years have largely focused on endo-acting glycosyl hydrolases (enzymes that cleave internal glycosidic linkages in a given oligosaccharide chain) and as this field continues to evolve, methodologies to address a broader range of enzyme classes are required. From a synthetic chemistry perspective there will always be a design challenge to functionalise a substrate with FRET components without compromising target binding and this will need to be addressed as more challenging enzyme–substrate binding environments are considered, for example, glycosyl transferases or exo-acting hydrolases. Additionally, limiting factors from the perspective of requirements for FRET function (distance between fluorophores) will need to be addressed as capability extends carbohydrate chain lengths into polysaccharide space. Flexibility in the attachment position along the carbohydrate chain and the rigidity of the fluorophore need to be key focus points here.
Finally, the transition of these probe systems, which have to date largely been utilised in vitro, to live biological scenarios for direct monitoring of specific enzymatic activity is required. This is particularly important in the established context of upregulated CAZy activity as a marker in disease diagnosis and in addressing clinically relevant questions. Advancement here will expand the field of FRET-carbohydrate probes to capability levels apparent for application of this sensing platform within other biological macromolecule classes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Engineering and Physical Sciences Research Council (EPSRC, EP/T007397/1) are thanked for project grant funding.References
- C. R. Bertozzi and L. L. Kiessling, Science, 2001, 291, 2357–2364 Search PubMed.
- P. M. Rudd, T. Elliott, P. Cresswell, I. A. Wilson and R. A. Dwek, Science, 2001, 291, 2370–2376 Search PubMed.
- A. Varki, Glycobiology, 1993, 3, 97–130 Search PubMed.
- R. Wolfenden, X. Lu and G. Young, J. Am. Chem. Soc., 1998, 120, 6814–6815 Search PubMed.
- D. L. Zechel and S. G. Withers, Acc. Chem. Res., 2000, 33, 11–18 Search PubMed.
- G. J. Davies and M. L. Sinnott, Biochemistry, 2008, 30, 26–32 Search PubMed.
- P. M. Coutinho, E. Deleury and B. Henrissat, J. Appl. Glycosci., 2003, 50, 241–244 Search PubMed.
- D. E. Koshland, Biol. Rev. Camb. Philos. Soc., 1953, 28, 416–436 Search PubMed.
- J. D. McCarter and G. S. Withers, Curr. Opin. Struct. Biol., 1994, 4, 885–892 Search PubMed.
- C. S. Rye and S. G. Withers, Curr. Opin. Chem. Biol., 2000, 4, 573–580 Search PubMed.
- L. F. Sobala, G. Speciale, S. Zhu, L. Raich, N. Sannikova, A. J. Thompson, Z. Hakki, D. Lu, S. S. K. Abadi, A. R. Lewis, V. Rojas-Cervellera, G. Bernardo-Seisdedos, Y. Zhang, O. Millet, J. Jiménez-Barbero, A. J. Bennet, M. Sollogoub, C. Rovira, G. J. Davies and S. J. Williams, ACS Cent. Sci., 2020, 6, 760–770 Search PubMed.
- V. Lombard, H. G. Ramulu, E. Drula, P. M. Coutinho and B. Henrissat, Nucleic Acids Res., 2013, 42, D490–D495 Search PubMed.
- K. A. Stubbs, Carbohydr. Res., 2014, 390, 9–19 Search PubMed.
- B. Henrissat and G. Davies, Curr. Opin. Struct. Biol., 1997, 7, 637–644 Search PubMed.
- H. M. Burke, T. Gunnlaugsson and E. M. Scanlan, Chem. Commun., 2015, 51, 10576–10588 Search PubMed.
- M. C. Patterson, Swaiman's Pediatric Neurology, Elsevier, 6th edn, 2017 Search PubMed.
- P. S. Kishnani and Y.-T. Chen, Emery and Rimoin's Principles and Practice of Medical Genetics, 6th edn, 2013 Search PubMed.
- I. B. Tomasic, M. C. Metcalf, A. I. Guce, N. E. Clark and S. C. Garman, J. Biol. Chem., 2010, 285, 21560–21566 Search PubMed.
- I. Vlodavsky, P. Singh, I. Boyango, L. Gutter-Kapon, M. Elkin, R. D. Sanderson and N. Ilan, Drug Resist. Updates, 2016, 29, 54–75 Search PubMed.
- L. Plegt and R. J. Bino, Mol. Gen. Genet., 1989, 216, 321–327 Search PubMed.
- D. Kalyani, K.-M. Lee, T.-S. Kim, J. Li, S. S. Dhiman, Y. C. Kang and J.-K. Lee, Fuel, 2013, 107, 815–822 Search PubMed.
- K. Kalidasan, Y. Su, X. Wu, S. Q. Yao and M. Uttamchandani, Chem. Commun., 2013, 49, 7237–7239 Search PubMed.
- L. I. Willems, J. Jiang, K.-Y. Li, M. D. Witte, W. W. Kallemeijn, T. J. N. Beenakker, S. P. Schröder, J. M. F. G. Aerts, G. A. van der Marel, J. D. C. Codée and H. S. Overkleeft, Chem. – Eur. J., 2014, 20, 10864–10872 Search PubMed.
- R. M. Clegg, Curr. Opin. Biotechnol., 1995, 6, 103–110 Search PubMed.
- G. Bunt and F. S. Wouters, Biophys. Rev., 2017, 9, 119–129 Search PubMed.
- J. A. Broussard and K. J. Green, J. Invest. Dermatol., 2017, 137, e185–e191 Search PubMed.
- B. T. Bajar, E. S. Wang, S. Zhang, M. Z. Lin and J. Chu, Sensors, 2016, 16, 1488 Search PubMed.
- A. Kaur and S. Dhakal, Trends Anal. Chem., 2020, 123, 115777 Search PubMed.
- K. E. Sapsford, L. Berti and I. L. Medintz, Angew. Chem., Int. Ed., 2006, 45, 4562–4589 Search PubMed.
- H. Oka, T. Koyama, K. Hatano and K. Matsuoka, Bioorg. Med. Chem., 2012, 20, 435–445 Search PubMed.
- H. Oka, T. Koyama, K. Hatano, D. Terunuma and K. Matsuoka, Bioorg. Med. Chem. Lett., 2010, 20, 1969–1971 Search PubMed.
- T. Suganuma, R. Matsuno, M. Ohnishi and K. Hiromi, J. Biochem., 1978, 84, 293–316 Search PubMed.
- A. Guerry, J. Bernard, E. Samain, E. Fleury, S. Cottaz and S. Halila, Bioconjugate Chem., 2013, 24, 544–549 Search PubMed.
- S. Cottaz, B. Brasme and H. Driguez, Eur. J. Biochem., 2000, 267, 5593–5600 Search PubMed.
- N. Ishii, C. Sunaga, K. Sano, C. Huang, K. Iino, Y. Matsuzaki, T. Suzuki and I. Matsuo, ChemBioChem, 2018, 19, 660–663 Search PubMed.
- I. Matsuo, M. Isomura and K. Ajisaka, J. Carbohydr. Chem., 1999, 18, 841–850 Search PubMed.
- N. Ishii, K. Sano and I. Matsuo, Bioorg. Med. Chem. Lett., 2019, 29, 1643–1646 Search PubMed.
- I. Matsuo, M. Isomura, T. Miyazaki, T. Sakakibara and K. Ajisaka, Carbohydr. Res., 1997, 305, 401–413 Search PubMed.
- O. Kanie, S. C. Crawley, M. M. Palcic and O. Hindsgaul, Carbohydr. Res., 1993, 243, 139–164 Search PubMed.
- K. Sano, T. Kuribara, N. Ishii, A. Kuroiwa, T. Yoshihara, S. Tobita, K. Totani and I. Matsuo, Chem. – Asian J., 2019, 14, 1965–1969 Search PubMed.
- R. G. Spiro, Cell. Mol. Life Sci., 2004, 61, 1025–1041 Search PubMed.
- W. A. Lubas and R. G. Spiro, J. Biol. Chem., 1987, 262, 3775–3781 Search PubMed.
- S. Iwamoto, Y. Kasahara, Y. Yoshimura, A. Seko, Y. Takeda, Y. Ito, K. Totani and I. Matsuo, ChemBioChem, 2017, 18, 1376–1378 Search PubMed.
- R. D. Sanderson, M. Elkin, A. C. Rapraeger, N. Ilan and I. Vlodavsky, FEBS J., 2016, 284, 42–55 Search PubMed.
- N. Takemoto, T. Suehara, H. L. Frisco, S. Sato, T. Sezaki, K. Kusamori, Y. Kawazoe, S. M. Park, S. Yamazoe, Y. Mizuhata, R. Inoue, G. J. Miller, S. U. Hansen, G. C. Jayson, J. M. Gardiner, T. Kanaya, N. Tokitoh, K. Ueda, Y. Takakura, N. Kioka, M. Nishikawa and M. Uesugi, J. Am. Chem. Soc., 2013, 135, 11032–11039 Search PubMed.
- S. Tsuchida, K. A. Podyma-Inoue and M. Yanagishita, Anal. Biochem., 2004, 331, 147–152 Search PubMed.
- E. Hammond, C. P. Li and V. Ferro, Anal. Biochem., 2009, 396, 112–116 Search PubMed.
- C. M. Melo, I. L. S. Tersariol, H. B. Nader, M. A. S. Pinhal and M. A. Lima, Carbohydr. Res., 2015, 412, 66–70 Search PubMed.
- L. Wu, J. Jiang, Y. Jin, W. W. Kallemeijn, C.-L. Kuo, M. Artola, W. Dai, C. van Elk, M. van Eijk, G. A. van der Marel, J. D. C. Codée, B. I. Florea, J. M. F. G. Aerts, H. S. Overkleeft and G. J. Davies, Nat. Chem. Biol., 2017, 13, 867–873 Search PubMed.
- J. C. Sistla, S. Morla, A.-H. B. Alabbas, R. C. Kalathur, C. Sharon, B. B. Patel and U. R. Desai, Carbohydr. Polym., 2019, 205, 385–391 Search PubMed.
- H. Li, H. Li, H. Qu, M. Zhao, B. Yuan, M. Cao and J. Cui, Cancer Cell Int., 2015, 15, 52 Search PubMed.
- N. Poupard, P. Badarou, F. Fasani, H. Groult, N. Bridiau, F. Sannier, S. Bordenave-Juchereau, C. Kieda, J.-M. Piot, C. Grillon, I. Fruitier-Arnaudin and T. Maugard, Mar. Drugs, 2017, 15, 134 Search PubMed.
- G.-Y. Yang, C. Li, M. Fischer, C. W. Cairo, Y. Feng and S. G. Withers, Angew. Chem., Int. Ed., 2015, 54, 5389–5393 Search PubMed.
- K. Sandhoff and K. Harzer, J. Neurosci., 2013, 33, 10195–10208 Search PubMed.
- J. R. Rich and S. G. Withers, Angew. Chem., Int. Ed., 2012, 51, 8640–8643 Search PubMed.
- S. M. Hancock, J. R. Rich, M. E. C. Caines, N. C. J. Strynadka and S. G. Withers, Nat. Chem. Biol., 2009, 5, 508–514 Search PubMed.
- Y. Li, H. Wang, J. Li, J. Zheng, X. Xu and R. Yang, Anal. Chem., 2011, 83, 1268–1274 Search PubMed.
- S. Cecioni and D. J. Vocadlo, J. Am. Chem. Soc., 2017, 139, 8392–8395 Search PubMed.
- M. D. Witte, W. W. Kallemeijn, J. Aten, K.-Y. Li, A. Strijland, W. E. Donker-Koopman, A. M. C. H. van den Nieuwendijk, B. Bleijlevens, G. Kramer, B. I. Florea, B. Hooibrink, C. E. M. Hollak, R. Ottenhoff, R. G. Boot, G. A. van der Marel, H. S. Overkleeft and J. M. F. G. Aerts, Nat. Chem. Biol., 2010, 6, 907–913 Search PubMed.
- D. S. Larsen and R. J. Stoodley, J. Chem. Soc., Perkin Trans. 1, 1989, 1841–1852 Search PubMed.
- R. B. Cejas, V. Lorenz, Y. C. Garay and F. J. Irazoqui, J. Biol. Chem., 2019, 294, 2997–3011 Search PubMed.
- N. Lunau, K. Seelhorst, S. Kahl, K. Tscherch, C. Stacke, S. Rohn, J. Thiem, U. Hahn and C. Meier, Chem. – Eur. J., 2013, 19, 17379–17390 Search PubMed.
- S. Ahmadipour and G. J. Miller, Carbohydr. Res., 2017, 451, 95–109 Search PubMed.
- S. Ahmadipour, L. Beswick and G. J. Miller, Carbohydr. Res., 2018, 469, 38–47 Search PubMed.
- S. Wolf, T. Zismann, N. Lunau and C. Meier, Chem. – Eur. J., 2009, 15, 7656–7664 Search PubMed.
- D. Lafont, P. Boullanger, F. Carvalho and P. Vottero, Carbohydr. Res., 1997, 297, 117–126 Search PubMed.
- C. Crawford, M. P. Wear, D. F. Q. Smith, C. d’Errico, A. Casadevall and S. Oscarson, ChemRxiv DOI:10.26434/chemrxiv.12144699.v1.
- L. Guazzelli, R. Ulc, A. Bowen, C. Crawford, O. McCabe, A. Jedlicka, M. Wear, A. Casadevall and S. Oscarson, ChemRxiv DOI:10.26434/chemrxiv.11914905.v1.
- L. Guazzelli, R. Ulc, L. Rydner and S. Oscarson, Org. Biomol. Chem., 2015, 13, 6598–6610 Search PubMed.
- C. Crawford and S. Oscarson, Eur. J. Org. Chem., 2020, 3332–3337 Search PubMed.
- W. R. Algar, N. Hildebrandt, S. S. Vogel and I. L. Medintz, Nat. Methods, 2019, 16, 815–829 Search PubMed.
| This journal is © The Royal Society of Chemistry 2020 |