
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Natural product scaffolds as inspiration for the design and synthesis of 20S human proteasome inhibitors
Grace E.
Hubbell
and
Jetze J.
Tepe
 *
*
Department of Chemistry, Michigan State University, East Lansing, MI 48823, USA. E-mail: tepe@chemistry.msu.edu
First published on 16th September 2020
Abstract
The 20S proteasome is a valuable target for the treatment of a number of diseases including cancer, neurodegenerative disease, and parasitic infection. In an effort to discover novel inhibitors of the 20S proteasome, many reseaarchers have looked to natural products as potential leads for drug discovery. The following review discusses the efforts made in the field to isolate and identify natural products as inhibitors of the proteasome. In addition, we describe some of the modifications made to natural products in order to discover more potent and selective inhibitors for potential disease treatment.
 Grace E. Hubbell | Grace Hubbell is currently working on her PhD in Chemistry at Michigan State University under the supervision of Dr Jetze Tepe. She received her BSc degrees in Biochemistry and Chemistry from Lake Superior State University (LSSU) in 2017. While at LSSU, Grace worked on the development of a methodology to access 1,2-disubstituted imidazolines and investigated their reactivity towards specific amino acids to further explore their potential biological activity. Her current research focuses are the total syntheses of marine alkaloids and the design and synthesis of novel small molecule inhibitors of the human proteasome. |
 Jetze J. Tepe | Prof. Jetze J. Tepe was born in Gouda, the Netherlands and moved to the US in 1988. He received his BS degree in 1992 from Jacksonville University, Florida. He received his PhD degree from the University of Virginia with Prof. Timothy L. Macdonald, after which he worked with Prof. Robert M. Williams at Colorado State University for his post-doctoral studies. He joint the faculty at Michigan State University in 2000, where he is currently a Professor of Chemistry. Research in the Tepe lab is focused on the synthesis and biological evaluation of small heterocyclic scaffolds and natural products, as part of an academic drug discovery program. |
Introduction
The ubiquitin-proteasome pathway plays an integral role in maintaining homeostasis in eukaryotic cells.1 The 26S proteasome is a 2.5 MDa threonine protease which is responsible for the degradation of redundant proteins in a cell into oligopeptides for further processing by other pathways (Fig. 1). Ubiquitin-proteasome-mediated degradation has been implicated in the regulation of many signalling proteins including cyclins (involved in cell-cycle progression), p53 (tumor suppressor),2 and IκBα (inhibitor of transcription factor NF-κB).3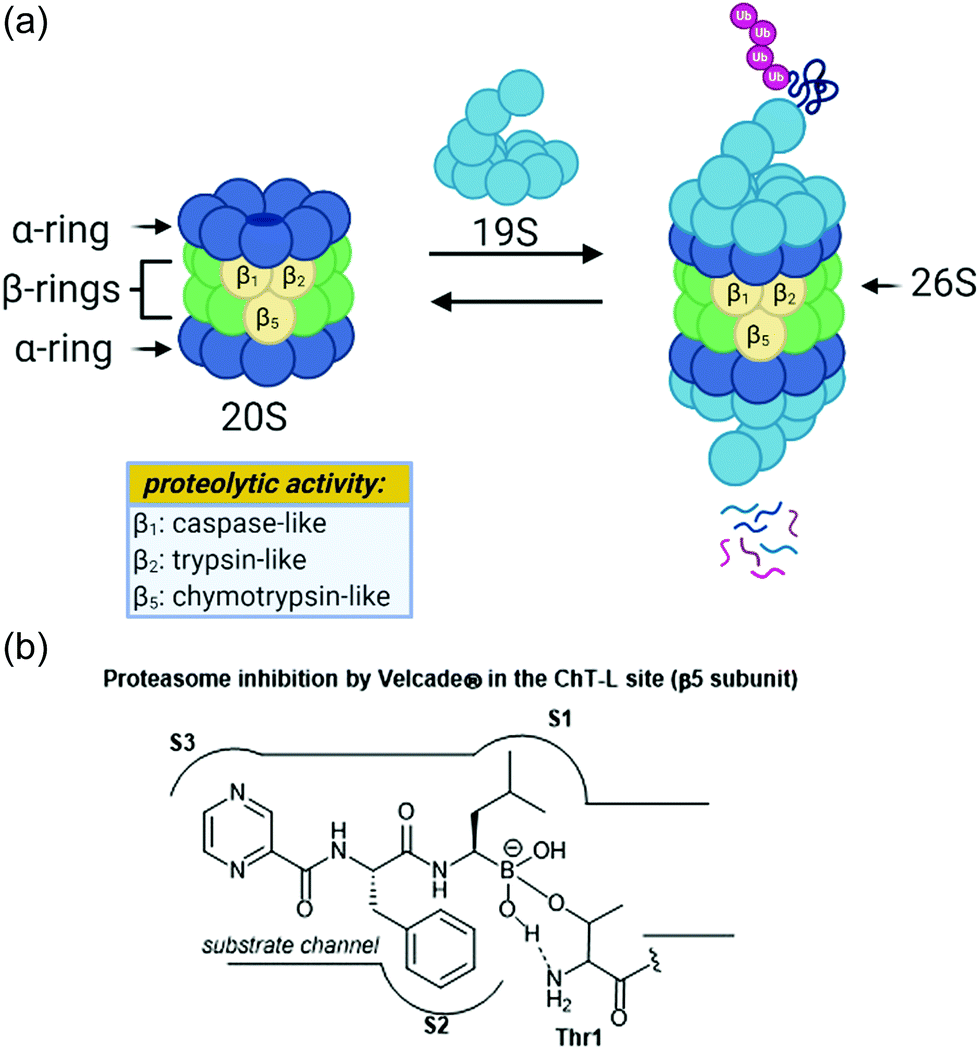 | ||
| Fig. 1 (a) The assembly of the 26S proteasome complex and depiction of proteolysis are shown above. The proteolytic activities of the beta subunits are also shown. (b) The mechanism of action of Velcade® towards the chymotrypsin-like site of the proteasome is shown. The boronic acid undergoes reversible nucleophilic attack by the Thr1 residue within the catalytic site. | ||
The 26S proteasome is comprised of one or two 19S regulatory particles (RP) which associate with the 700 kDA barrel-like 20S core particle (CP).4 The 20S core particle is composed of four stacked heptameric rings in an α1–7β1–7β1–7α1–7 arrangement.5 The outer α-rings are responsible for recognition of the regulatory particles, and the inner β-rings house the catalytic activity of the protease. The β-rings each consist of seven unique subunits, and three of these subunits (β1, β2, and β5) are responsible for the proteolytic activity of the proteasome. Binding of the 19S caps to the CP facilitates gate-opening, which in turn allows for the proteolytic degradation of polyubiquitinylated proteins. The 19S caps are responsible not only for this gate-opening,6 but also the recognition of polyubiquitinylated proteins, unravelling of these into linear peptides, and feeding the peptides into the catalytic chamber.7 Once fed into the chamber, unravelled proteins are cleaved into oligopeptides by the catalytic sites, which all proteolytically cleave the peptide chains with the aid of an N-terminal threonine residue (Thr1). Although all three catalytic sites use a nucleophilic threonine to carry out their activity, slight differences between their substrate channels allow for preference towards cleavage of specific peptide residues. The proteasome exhibits caspase-like (C-L, peptidyl-glutamidyl peptide hydrolysing), trypsin-like (T-L) and chymotrypsin-like (ChT-L) activities which are carried out by its β1, β2, and β5 subunits, respectively.8–10 The first 20S proteasome structure determined was that of the archaebacterium Thermoplasma acidophilum in 1995,5 and many eukaryotic proteasomes have since been characterized. Structural similarity between eukaryotic proteasomes is highly conserved, which has allowed for the use of models such as the yeast 20S proteasome11 and mammalian 20S proteasome12,13 for identification of molecules which exhibit modulatory activity against the proteasome. These models have been especially useful considering that while their X-ray crystal structures were solved throughout the 1990s and early 2000s, which has facilitated inhibitor design greatly, the X-ray crystal structure of the human 20S proteasome was only recently solved in 2015.14 Two other core particles have also been identified as the immunoproteasome15 and the thymoproteasome.16 The differences of selectivity between these are governed by slight differences in the topology within their substrate channels.17
Due to its integral role in maintaining cell homeostasis, modulation of the activity of the 20S proteasome has been considered as a potential way to treat several diseases including cancer, autoimmune disease, and neurodegeneration.18–20 Additionally, the proteasome of parasitic species has recently been targeted in the treatment of parasitic diseases.21 Inhibition of the proteasome has been implicated as a veritable strategy for the treatment of certain cancers, given that inhibition leads to ER stress and apoptosis.22 The invention of the proteasome-inhibitor Velcade® (bortezomib) is perhaps the best representation of how targeting the ubiquitin-proteasome pathway can lead to the treatment of some cancers. This dipeptide boronate was approved by the FDA in 2003 as a treatment for multiple myeloma, and its mechanism of action is through direct inhibition of the ChT-L activity of the proteasome.23 Activation of the proteasome has also been considered for the treatment of neurodegenerative diseases such as Parkinson's and Alzheimer's disease.24–26
Following the discovery of the 26S proteasome complex in the mid-1990s, researchers focused upon the development of novel inhibitors of the 20S proteasome. Most compounds which have been designed as inhibitors of the 20S proteasome are built upon peptide-based scaffolds. Peptide-based scaffolds contain optimized amino acid residues to mimic substrates of the 20S proteasome and improve recognition by the different catalytic subunits. The residues also participate in hydrogen-bonding and hydrophobic interactions to optimize interaction with the substrate channel. Specific design of the peptide chain has allowed for the generation of subsite-specific inhibitors of the proteasome. The design of such inhibitors is based upon the inherent preference of the C-L, T-L and ChT-L activities towards acidic, basic, and hydrophobic residues, respectively. Additionally, these compounds typically contain an electrophilic warhead at their C-terminus which interacts directly with the N-terminal threonine hydroxyl residue of proteolytic sites to block their catalytic activity. The first synthetic proteasome inhibitors were peptide aldehydes27 which had been shown to inhibit several types of proteases including serine and cysteine proteases. These peptide aldehydes act as reversible covalent inhibitors of the 20S proteasome, forming a hemiacetal with the nucleophilic threonine residue. Design of more selective inhibitors stemmed from alteration of the electrophilic warhead, leading to the eventual inclusion of boronic acids, vinyl sulfones,28 and α-ketoaldehydes29 within the scaffolds of synthetic inhibitors of the 20S proteasome. The interaction between covalent inhibitors and the catalytic sites is demonstrated in Fig. 1b with bortezomib. The substrate channel accommodates the peptide side chains of the molecule much like it would a peptide substrate. The electrophilic boronic acid moiety at the terminus of the molecule is susceptible to nucleophilic attack by the Thr1 residue of the catalytic site. This electrophile undergoes reversible nucleophilic addition by the amino acid, and the resulting charged species is stabilized through hydrophobic interactions and hydrogen-bonding interactions.30 Noncovalent inhibitors typically capitalize upon purely hydrophobic and hydrogen-bonding interactions to confer inhibition.
Nature is an attractive source for the identification of novel proteasome inhibitors: the inherent stereochemical complexity of many natural products may allow researchers insight into unique interactions relative to the established clinically available inhibitors. A variety of methods exist for mining nature for identification of novel proteasome inhibitors including the use of fluorogenic peptide assay, site-specific probes, pathway-specific accumulation assay and NMR spectroscopic assay.31 These methods have been especially helpful in regards to identification of natural product inhibitors, as some may be utilized with crude mixtures before isolation. Examples of the use of these methods are included in the review. Elucidation of the mechanisms of action by natural products through the methods of X-ray crystallography and computational docking allow for the design of more potent and selective inhibitors based upon the natural product scaffold. The focus of this review is to discuss several classes of natural products which have been identified for their inhibitory activity towards the 20S proteasome, their mechanism of action, and the efforts taken to optimize these scaffolds for potency and selectivity towards human 20S proteasome inhibition.
Natural product scaffolds that inhibit the 20S proteasome
Peptide-aldehydes
Several families of peptide aldehyde natural products have been identified for their inhibitory activity towards the 20S proteasome (Chart 1). The first established natural product-based peptide aldehyde inhibitors tyropeptins A and B are products of the actinomycete microbe Kitasatospora sp. MK993-dF2, a strain originally discovered in a soil sample from Kami-gun, Japan.32,33 After extensive spectroscopic analysis, the structures of the tyropeptins A and B were determined as isovaleryl-L-tyrosyl-L-valyl-DL-tyrosinal and n-butyryl-L-tyrosyl-L-leucyl-DL-tyrosinal, respectively. For confirmation of their structures, the total syntheses of the natural products were completed and compared with the isolated samples. The peptide backbone and reactive aldehyde moiety had already been established as a promising proteasome inhibiting scaffold,34 and therefore the potential biological activities of tyropeptins A and B were of interest to researchers. Tyropeptin A (1) competitively inhibits the ChT-L and T-L activities of the 20S proteasome with IC50 values of 0.20 μM and 2.9 μM, respectively. Tyropeptin B (2) is a weaker competitive inhibitor of the 20S proteasome, with IC50 values for the ChT-L and T-L sites of 0.39 μM and 7.8 μM, respectively. Tyropeptin A also exhibited cytotoxicity against HeLa S3 human cervical cells and HL-60 promyelocytic leukemia cells with IC50 values of 33.2 μM and 8.6 μM, respectively. Derivatives of tyropeptin A were designed by Momose et al. using computational methods.35,36 At the time, the crystal structure of the human 20S proteasome had not yet been solved; however, the crystal structure of the bovine 20S proteasome had been determined and was therefore used as a model.12,13 Tyropeptin A was believed to undergo nucleophilic attack by the Thr1Oγ residue of the active site to form a hemiacetal, and its side chains interact with the subpockets to form an antiparallel β-sheet. Introduction of bulky aromatic groups at the N-terminus enhanced inhibitory activity towards the ChT-L site of the proteasome. Optimization led to TP-110 (3), an N-terminal naphthyl derivative with methylated hydroxy positions (IC50: 0.027 μM). TP-110 exhibited improved growth inhibitory activity against cancer cells, likely due to increased hydrophobicity of the molecule to improve cell permeability. In subsequent in vitro studies, TP-110 also repressed the chymotrypsin-like activity of the proteasome in human prostate cancer PC-3 cells, inhibiting the proteasome activity by 56% with 0.16 μM.37 Furthermore, TP-110 induces apoptosis in the prostate cancer cell line. Substitution of the formyl group in TP-110 with boronates resulted in enhanced inhibitory potency towards the ChT-L site.38 Optimization of this scaffold led to boronate 4, which bore an N-terminal 3,6-dichloro-2-pyridyl substituent and exhibited comparable inhibitory potency towards the chymotrypsin-like activity of the proteasome (IC50: 0.053 μM) to that of bortezomib.39 Analog 4 also displayed good cytotoxic activity towards the RPMI8226 multiple myeloma cells, and was chosen for further in vitro and solid tumor studies.40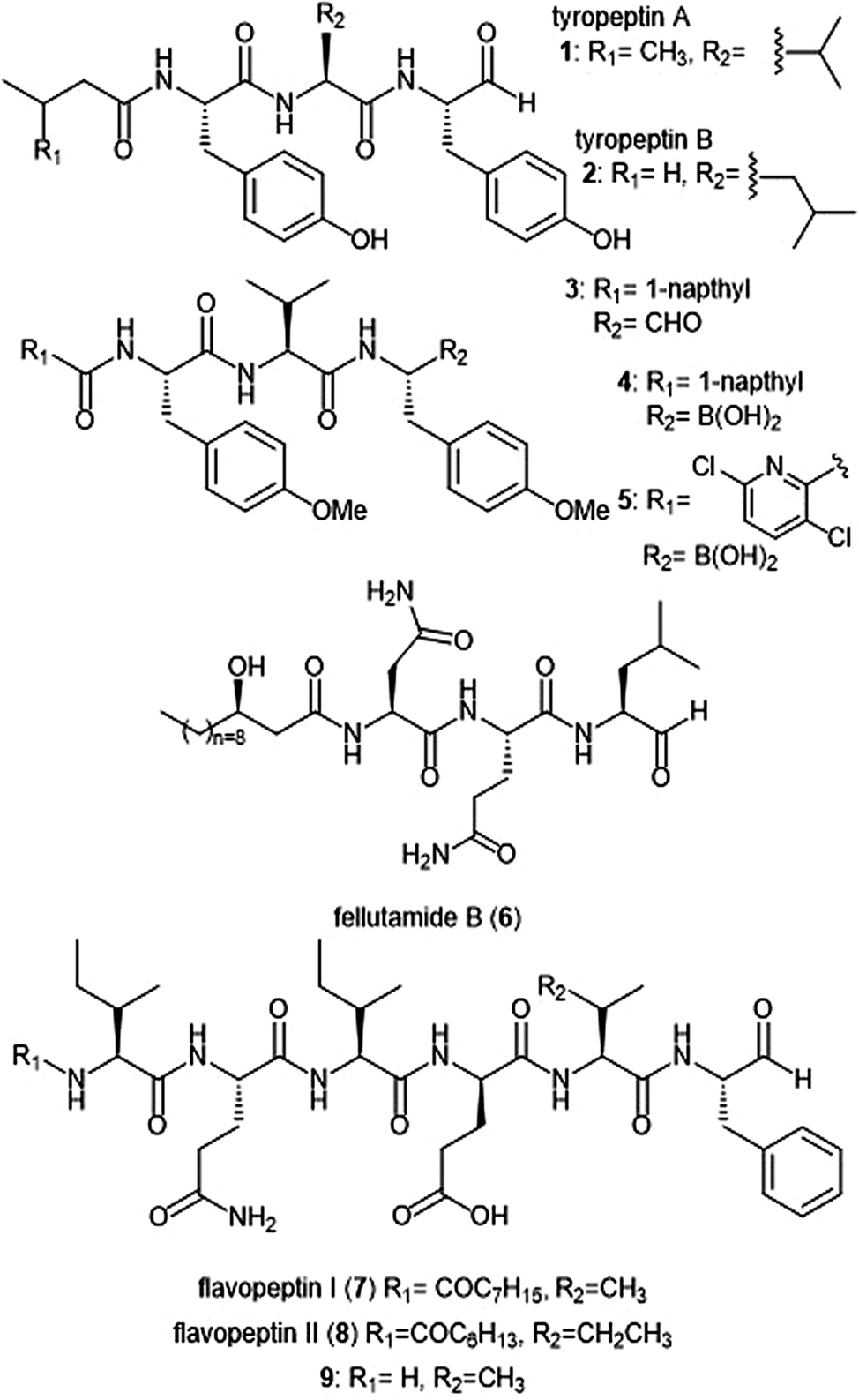 | ||
| Chart 1 Peptide aldehyde natural products and derivatives that inhibit the 20S proteasome are shown. | ||
Fellutamide B (5) was originally isolated from Penicillium fellutanium, a fungus which is found in the gastrointestinal tract of the marine fish Apogon endekataenia.41 Fellutamide B demonstrated potent cytotoxicity against murine leukemia P388 (IC50: 0.18 μM) and L1210 (IC50: 1.26 μM) cells, as well as human epidermoid carcinoma KB cells (IC50: 1.26 μM) in vitro, though the mechanism of cytotoxicity was not established. The total synthesis of fellutamide B was first achieved by the Crews group.42 Since then, other groups have also set out to achieve total syntheses of not only fellutamide B, but other members of the fellutamide family as well.43–45 Fellutamide B potently inhibits the chymotrypsin-like site (IC50: 9.4 ± 2.5 nM), but the trypsin-like and caspase-like sites to a lesser extent (IC50: 2.0 ± 0.4 μM and 1.2 ± 0.8 μM, respectively).46In vitro studies revealed the ability of the natural product to inhibit the proteasome within cells, as treatment of L–M mouse fibroblasts resulted in accumulation of ubiquitinated proteins.
The flavopeptin class of peptide aldehydes was discovered with the help of proteomics.47 The Kelleher group utilized the method of Proteomic Investigation of Secondary Metabolism (PrISM) to screen Streptomyces species for non-ribosomal peptide synthases (NRPS) responsible for secondary metabolite synthesis. Using the PrISM approach, the group was able to identify a novel NRPS gene cluster from Streptomyces sp. NRRL F-6652. With the help of bioinformatics analysis and metabolomics analysis, six flavopeptins were discovered as the products of this gene cluster. Flavopeptins I (6) and II (7) were synthesized using solid phase peptide synthesis (SPPS), along with the N-terminal amine derivative 8. Due to the similarity of the flavopeptins to other established inhibitors, the natural products were tested for their inhibitory activity towards the ChT-L and C-L sites of the 20S proteasome. Flavopeptins I and II are low micromolar inhibitors of both sites, and the terminal amine derivative 8 exhibits submicromolar IC50 values for both sites. The flavopeptins were also tested for their cytotoxicity against the multiple myeloma cell lines MM.M1S and FR4, as well as the histiocytic lymphoma cell line, U-937. Flavopeptin I displayed values of IC50: ∼35 and 13 μM for the MM cell lines and the lymphoma line, respectively. The N-terminal amine derivative 8 displayed no cytotoxic activity. The lack of activity is believed to be due to lower cell permeability and stability as compared to flavopeptins I and II.
Syrbactins
Several syrbactin natural products have also been identified for their proteasome inhibition (Chart 2). Syrbactins are divided into 3 sub-families: syringolins, glidobactins, and cepafungins. These compounds are products of plant pathogens, and their similarities include a macrocyclic lactam with an electrophilic α,β-unsaturated carboxamide. Glidobactins (A–H) are natural product syrbactins that are produced by the bacterial strain Polyangium brachysporum sp. nov. K481–B101.48–51 Glidobactins A–C are cytotoxic against melanotic melanoma B16 cells as well as human colon cancer HCT-116 cells, and exhibit potent in vivo antitumoral activity towards P388 leukemia in mice.48 Further modification of the glidobactin A structure by Oka et al. led to the preparation of several derivatives, which were also evaluated for their antitumoral activity.51 Glidobactin A (9) was later identified as a nanomolar inhibitor of the ChT-L site of the 20S proteasome, with a Ki value of 49 ± 5.4 nM.52 The compound also inhibited T-L activity of the 20S proteasome, albeit at considerably higher concentrations (2000 ± 600 nM, n = 6). The mechanism of inhibition by syrbactins was determined through elucidation of the X-ray crystal structure of the 20S proteasome in complex with glidobactin A and syringolin A. The α,β-unsaturated moiety of glidobactin A undergoes a nucleophilic Michael-type 1,4-addition by the Thr1Oγ of the chymotrypsin-like and trypsin-like sites of the proteasome. The lipophilic alkyl chain of glidobactin A contributes to its inhibitory activity and served as inspiration in the development of novel syrbactin proteasome inhibitors; the activity of these analogs is discussed in a later section.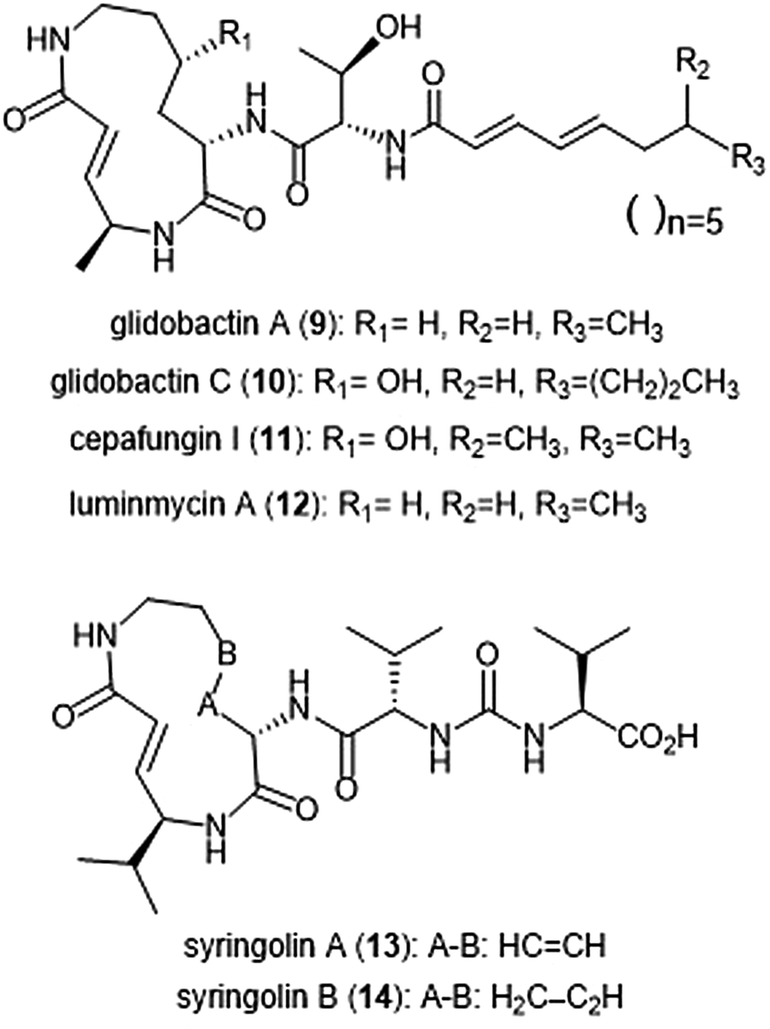 | ||
| Chart 2 Syrbactin natural products which inhibit the 20S proteasome. | ||
Glidobactin C (10) was recently identified as an inhibitor of the human constitutive and immunoproteasomes through a competitive metabolite profiling technique by the Böttcher group.53 Glidobactin C exhibited in vitro inhibitory activity towards the ChT-L and T-L sites of the human constitutive proteasome (IC50: 2.9 ± 2.2 nM and 2.4 ± 2.8 nM, respectively). The compound also inhibits the human immunoproteasome (ChT-L IC50: 7.1 ± 5.3 nM; T-L IC50: 2.5 ± 2.0 nM). The two additional methylene groups of 10 relative to 9 are thought to improve its ability to penetrate the cell membrane, resulting in greater activity in cell assays.
The cepafungins were observed as products of the bacteria Pseudomonas sp. CB-3 by Shoji et al.54 Cepafungins I, II, and III were identified as acylpeptide antibiotics which all contain a distinct 12-membered macrolactam ring.55 The macrolactam ring of all three compounds is comprised of the two amino acids γ-hydroxylysine and 4-amino-2-pentenoic acid, and the distinguishing feature among each is a variable fatty acyl tail at the N-terminus of the peptide. Based upon their analysis, the group concluded that the structure of cepafungin II is identical to that of known natural product glidobactin A. The complex of all three compounds displayed a moderate ability to prolong the survival period of mice which bear murine lymphatic leukemia P388 cells. Cepafungin I (11) was identified as a proteasome inhibitor in later studies: Stein et al. developed an NMR-based proteasome assay to identify natural product inhibitors of the yeast 20S proteasome.56 Cepafungin I potently inhibits the ChT-L activity of the proteasome (IC50: 4 nM), five times more potent than the known inhibitor glidobactin A (IC50: 19 nM). Cepafungin I also inhibits the T-L activity of the proteasome with an IC50 value of 24 nM. The improved inhibitory activity of cepafungin I as compared to glidobactin A is believed to be due to the increased stabilization of the fatty acyl tail through van der Waals interactions.
Luminmycin A (12) is another natural product of the syrbactin family which has recently gained attention as a potential proteasome inhibitor. A deoxy derivative of glidobactin A, luminmycin A contains the hallmark 12-membered macrolactam ring responsible for the inhibitory activity of the syrbactin family towards the human 20S proteasome. Luminmycin A was identified as a metabolite of a silenced gene cluster plu1881–plu1887 of Photorhabdus luminescens, and the associated gene cluster was utilized to produce luminmycin A using the method of heterologous expression.57 The natural product was also observed as a metabolite of crickets following their infection with Photorhabdus asymbiotica.58 Luminmycin A and other natural products belonging to the luminmycin class were successfully isolated following heterologous expression of the same gene cluster, using E. coli as host.59,60 Recently, luminmycin A was successfully synthesized in the laboratory by Servatius et al.61 Luminmycin A inhibits the constitutive proteasome (CP) as well as the immunoproteasome (IP), (ChT-L IC50: 0.039 ± 0.002 μM (CP), 0.016 ± 0.006 μM (IP); T-L 0.026 ± 0.008 μM (CP), 0.017 ± 0.0016 μM (IP)).53 The natural product also exhibits cytotoxic activity against human carcinoma HCT-116 cells (IC50: 91.8 nM).
The syringolin family of natural products were identified as secondary metabolites of the pathogen Pseudomonas syringae pv. syringae, a non-host pathogen of the Oryza sativa rice plants.62,63 Syringolin A was identified as an inhibitor of the human proteasome in 2008, thus prompting interest in the successful completion of its total synthesis by several research groups. The total syntheses of syringolins A and B were first completed in 2009, with many more syntheses to follow.64–66In vitro enzymatic assays revealed the inhibitory activities of syringolins A and B toward the 20S proteasome. Syringolin A (13) inhibits the ChT-L and C-L activities of the 20S proteasome (Ki: 1.1 ± 0.179 μM, and 10.3 ± 1.4 μM, respectively). The macrolactam binds covalently to the 20S proteasome by direct interaction with Thr1Oγ within the active sites.52,67 The α,β-unsaturated carboxamide of syringolin A is rendered especially electrophilic due to the ring strain of the macrocycle resulting from the presence of two trans alkenes; upon 1,4-Michael addition of Thr1Oγ, this ring strain is relieved. In comparison, syringolin B (14), which lacks one of the trans alkenes, is a significantly less potent inhibitor of the 20S proteasome (Ki: 7.7 ± 2.3 μM (ChT-L); 107.8 ± 39.2 μM (T-L).
Additional derivatization of the syringolin A scaffold has been pursued by several groups (Chart 3). Alteration of the N-terminus of syringolin A from a carboxylic acid to a lipophilic tail led to the production of SylA-LIP 15, which exhibits inhibitory activity towards all three catalytic sites of the proteasome (Ki: 8.65 ± 1.33 nM (ChT-L); 79.6 ± 29.3 nM (T-L); and 943 ± 100 nM (C-L)).68 TIR-199 (16) contains a slightly longer lipophilic tail than SylA-LIP and inhibits the ChT-L (Ki: 18 nM) and T-L (Ki: 194 nM) sites of the human proteasome in in vitro enzymatic assays.69 TIR-199 also inhibits the proteasome in multiple myeloma cell line MM1.RL and neuroblastoma cell line MYCN2 and has demonstrated exciting cytotoxic activity against bortezomib-resistant cell lines (MM1.S BzR and U266 BzR), indicating it can overcome bortezomib resistance in vivo.70 Clerc et al. further investigated the effects of substitution at the N-terminus through the synthesis of a syringolin A–glidobactin A hybrid 17.71 The inhibitory activity and subsite selectivity of the analogue was tested using competitive activity-based protein profiling (ABPP) in HEK cell lysates and living cells, compared to known syrbactin inhibitors. Further in vitro studies revealed that 17 inhibits all three catalytic sites of the 20S proteasome (Ki: 12.5 ± 1.5 nM (ChT-L), 136.9 ± 12.4 nM (T-L), 3.7 ± 1.2 μM (Casp-L)).72 Cell culture-based experiments from the same study revealed that the analogue carries out inhibition of the proteasome in several cancer cell lines. Introduction of hydrophobic sidechains allowed the group to target the S3 subpocket of the β5 subunit for inhibition. Compound 18, which contained a benzyl substituent and lipophilic side chain, displayed strong inhibitory activity against the ChT-L site of the proteasome (Ki′: 0.12 nM) and cytotoxicity towards human RPMI8226 multiple myeloma cells (IC50: 2.2 nM).66 Further optimization of 18 has focused on improving cytotoxicity.73
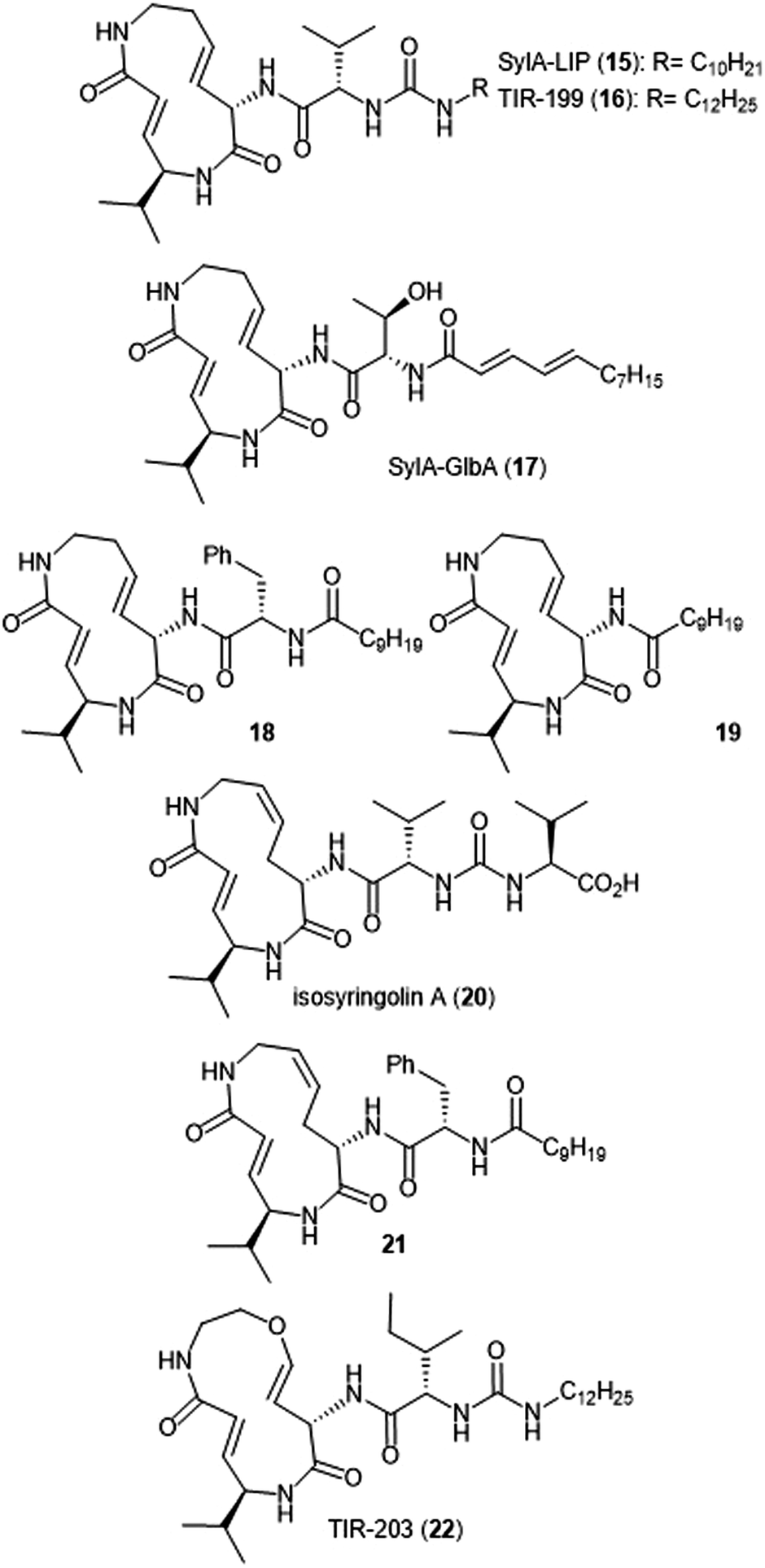 | ||
| Chart 3 Proteasome inhibitors based on the syringolin-A scaffold are shown. | ||
Simplification of the syringolin A scaffold has also been a focus of research by the Ichikawa group. One report focused on analogues that exhibited proteasome inhibition and cytotoxicity against cancer cells by targeting a different intermolecular H-bonding interaction.74 The N-terminal amide of analogue 18 is believed to participate in a hydrogen-bonding interaction with Asp114 (β6), similarly to its parent syringolin A. The group hypothesized that removal of the N-terminal amide group and replacement with a longer alkyl chain may allow for a switching in the hydrogen-bonding interactions within the β5 subunit and allow for a novel interaction between the substrate and Ala49. The hypothesis was verified through the synthesis of analogue 19, which displayed nanomolar inhibitory activity towards the ChT-L site of the proteasome (IC50: 107 nM). Additionally, analogue 19 exhibits nanomolar growth inhibitory activity against several cancer cell lines.
Alteration of the macrocycle structure was also explored. Researchers hypothesized that a more accessible alkene isomer with a similar three-dimensional structure might be also be able to carry out inhibitory activity. The group synthesized isosyringolin A (20) and its analogue 21, which both exhibited inhibition towards the ChT-L site (Ki = 590 nM; 1.53 nM, respectively).75 Compound 21 also exhibited potent cytotoxicity against OPM-2 human myeloma cancer cells (IC50: 6.7 nM) and bortezomib-resistant OPM-2 cells (IC50: 60 nM), displaying greater cytotoxicity as compared to bortezomib (IC50: 146 nM). Expansion of the syringolin A ring by Ibarra-Rivera et al. led to oxa-SylA-LIP (22), which displays potent cytotoxicity against the MYCN-2 cells (IC50: 0.4 μM) and also inhibits the proteasome in cell-based studies.76,77
Analogue development based upon the syringolin B scaffold has also been investigated by several research groups (Chart 4). For example, the SylB-LIP analogue (23) was developed as a direct comparison to SylA-LIP (15).76 Totaro et al. designed more potent syringolin B analogues with greater inhibition towards the β5 subunit based upon the synthesis of syringolin B by Pirrung et al.65,78 Introduction of aromatic groups at the P1 and P3 residues in the scaffold led to the discovery of potent inhibitor 24 (second order rate-constant (kin/Ki): 4305 M−1 s−1). The analogue also exhibits potent growth inhibition against various leukemia cell lines.
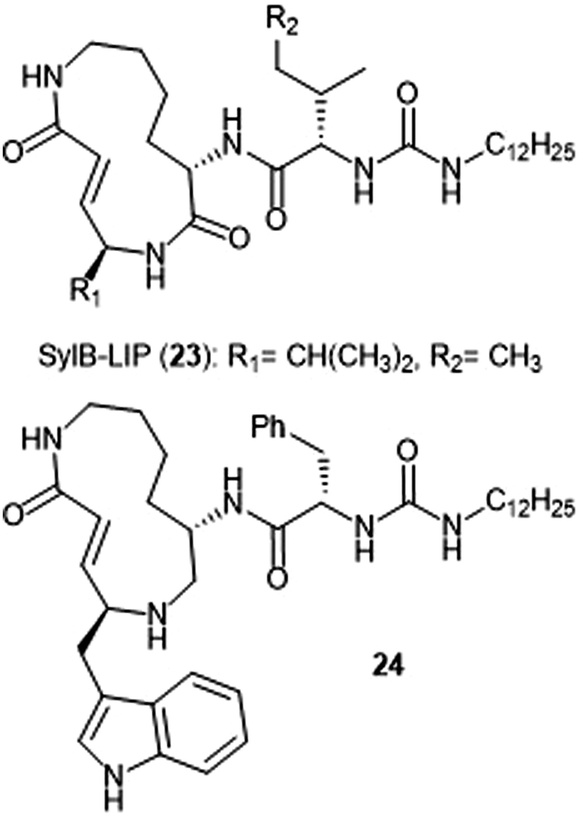 | ||
| Chart 4 Syringolin-B based inhibitors of the 20S proteasome are depicted. | ||
Thiasyrbactin analogues with a sulfur-for-carbon substitution within the macrolactam ring were synthesized with varying levels of sulfur oxidation. These analogues were reported for their activity towards both the constitutive and immunoproteasomes, more selectively inhibiting the T-L site of the immunoproteasome.79 The biological activity of the analogues was further examined through cytotoxicity experiments with numerous neuroblastoma cell lines. The thiasyrbactin scaffold has established itself as a drug-like starting point for inhibition of the immunoproteasome.
Macrocyclic peptides
Several classes of macrocyclic peptides have been identified for their inhibitory activity towards the proteasome. Perhaps the most explored class has been the TMC cyclic peptides (Chart 5). TMC-95 A, B, C, and D were isolated from the fermentation broth of Apiospora montagnei Sacc. TC 1093 by Koguchi et al. while the group was screening for 20S proteasome inhibitors.80,81 Among all of the TMC cyclic peptides, TMC-95 A (25) and B (26) were identified as specific inhibitors of the 20S proteasome, displaying activity against both the ChT-L (IC50: 5.4 nM (TMC-95 A) and 8.7 nM (TMC-95 B)) and T-L (IC50: 200 nM (TMC-95 A) and 490 nM (TMC-95 B)) subunits. TMC-95A further demonstrated cytotoxic activities against HCT-116 and HL-60 cells with IC50 values of 4.4 μM and 9.8 μM, respectively. The X-ray crystal structure of the yCP:TMC-95A complex was later solved by Groll et al. (2.9 Å resolution), revealing that the macrocycle reversibly inhibits the 20S CP through non-covalent interactions within the catalytic core.82 The specificity of the natural product for the chymotrypsin-like site is believed to be due to greater interaction with the S3 specificity pocket.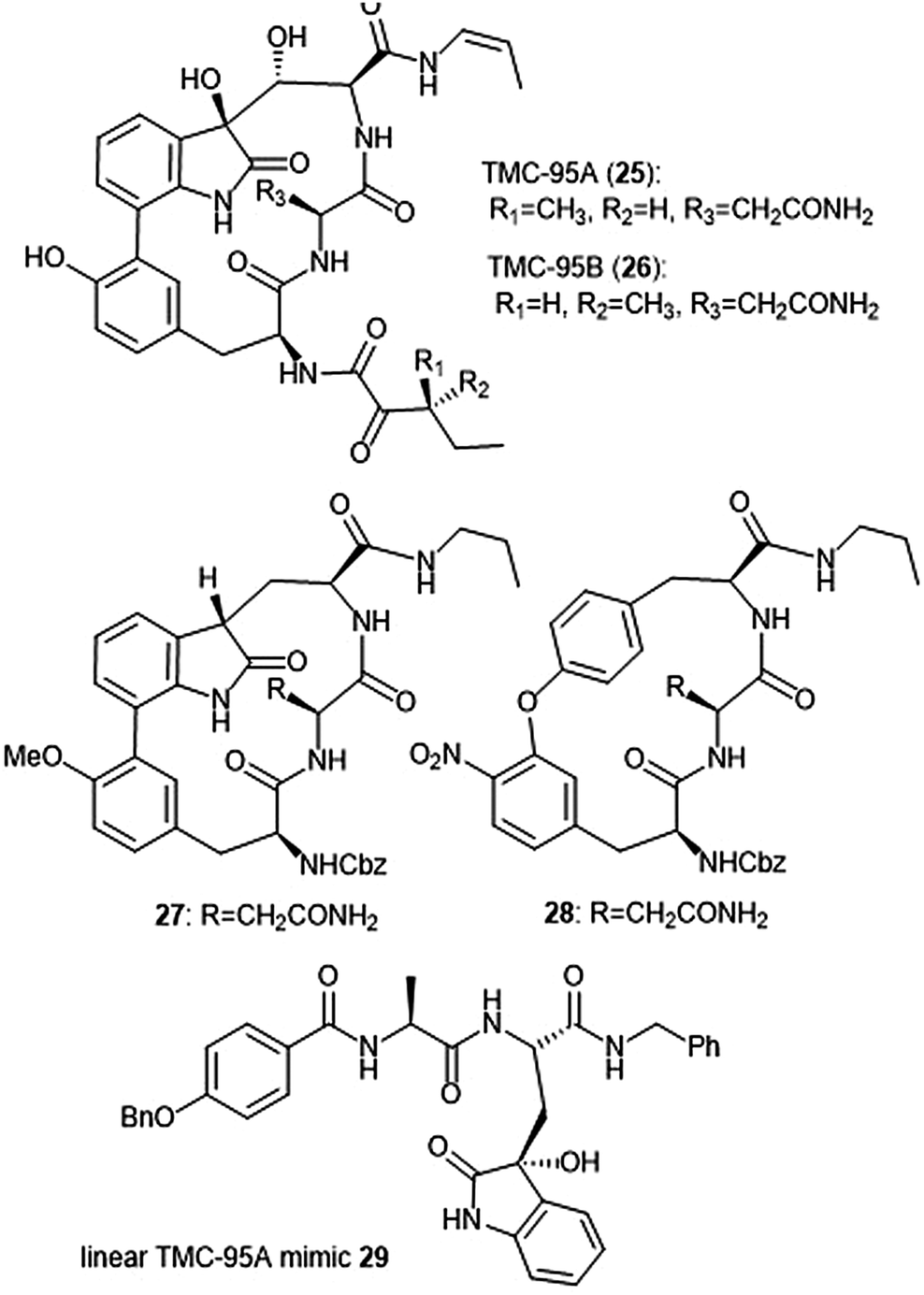 | ||
| Chart 5 The TMC-95 natural products and subsequent inhibitors developed based upon the scaffold are shown. | ||
Due to the biological activity and overall complexity of the TMC-95 natural products, many groups sought to undertake their synthesis.83–86 The first total syntheses of TMC-95A and B were achieved by Lin and Danishefsky in 2002.83 The presence of sensitive functional groups in addition to the difficulty in accessing the macrocycle has encouraged subsequent analogue development to be focused on the synthesis of simplified molecules. Kaiser et al. synthesized compound 27, a simplified analogue based upon the minimal requirements for binding as determined from the X-ray crystal structure of TMC-95A/proteasome complex.87 The analogue exhibited inhibitory activity against the ChT-L site of the 20S human proteasome (IC50: 1.9 μM) as compared to TMC-95A (IC50: 0.012 μM).88 The same researchers evaluated the effect of additional P′ residues at the C-terminus of TMC-95A in a separate study.89 The Danishefsky group established the importance of the enamide moiety at the C-8 position of the molecule;90 analogues lacking this displayed at least a 1000-fold less potent inhibition towards the ChT-L site as compared to their natural product parent molecule. Furthermore, alteration of the enamide sidechain also affected inhibitory activity; the presence of a propyl substituent in exchange for a propylene or allyl amide sidechain results in diminished activity. In previous studies, the biaryl moiety of TMC-95A was deemed responsible for the induction and stabilization of a β-type peptide backbone conformation while in complex with the 20S proteasome.82 Kaiser et al. implemented the substitution of the biaryl heterocycle using a more accessible biaryl ether moiety with the goal of retaining the similar β-type backbone conformation.91 The biaryl ether analogue 28 retains inhibitory potency towards the ChT-L site of the yCP (IC50: 5.5 μM). Later studies revealed that due to the lack of the oxindole, biaryl ether analogues do not adopt the desired stabilized β-type structure while in complex with the 20S proteasome.92 Wilson et al. later synthesized macrocyclic peptide aldehydes upon the previously established biaryl ether analogues which are nanomolar inhibitors of the ChT-L site.93 Further efforts to access simplified analogues of TMC-95A have eliminated synthesis of the challenging macrocycle altogether through the development of linear peptide mimics of the natural product. The Vidal group—in collaboration with many other research groups—has used this linear TMC-95A approach to synthesize effective inhibitors.94–99 In particular, the linear 3-hydroxyoxindole-containing derivatives exhibited subunit selectivity towards the C-L site).96 Optimization of the scaffold led to the discovery of potent inhibitor 29, which inhibited the ChT-L of the constitutive and immunoproteasomes (IC50: 7.1 ± 0.2 and 10.2 ± 0.1 nM, respectively).99 Dimerized linear TMC-95A mimics using the active derivative 29 scaffold have also been synthesized to evaluate their ability to inhibit multiple active sites at once. Using either PEG spacers100 or oligomers of aminohexanoic and adipic acid as spacers,101 researchers were able to achieve nanomolar inhibition of the ChT-L sites of the 20S proteasome.
Argyrins A–H were isolated from the culture broth of myxobacterium Archangium gephyra by the Hofle group in 2002.102,103 This group of cyclic octapeptides displayed growth inhibitory activity against a variety of mammalian cell lines,102 and became a focus of total synthesis for many groups.104–106 Argyrins A, B, C, D and F (30–34) (Chart 6) were later identified as low nanomolar inhibitors of the human proteasome.107,108 Bulow et al. established that the methoxy group in Trp2 as well as the exo-cyclic methylene group are necessary for the ability to inhibit the proteasome.108 The binding mode of the argyrins was later elucidated through NMR spectroscopy and molecular modeling.109 Computational inhibition studies using a humanized proteasome model indicated which interactions contributed to argyrin subunit specificity.110 Using molecular docking, researchers designed novel argyrin A analogues in silico which display increased specificity towards the caspase-like site of the 20S humanized proteasome model. The Loizidou group recently reported the specificity of argyrin B towards the immunoproteasome.111
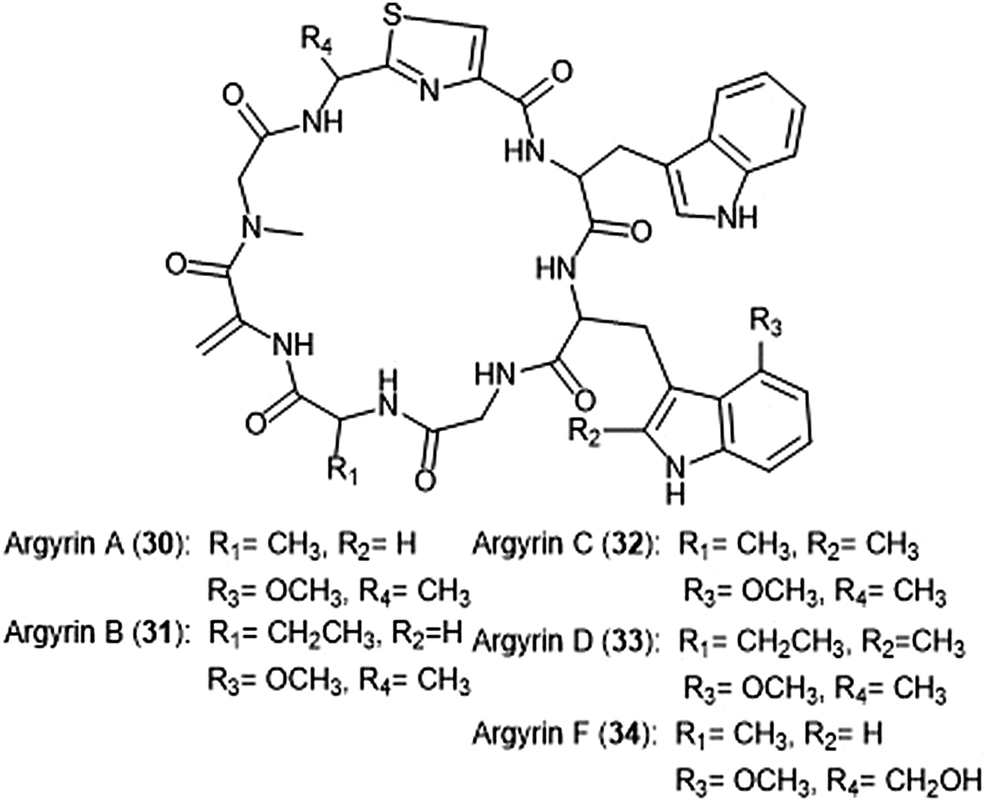 | ||
| Chart 6 The cyclic argyrin octapeptides which have been identified as inhibtors of the 20S proteasome are depicted. | ||
Macrocyclic thiopeptides have also been investigated as inhibitors of the human proteasome. Bhat et al. reported that thiopeptides thiostrepton (35) and siomycin A (36) exhibit inhibitory activity against the 20S proteasome, which in turn contributes to the effects observed on the oncogenic transcription factor Fox1 (Chart 7).112,113 The additional “B-ring” present in thiostrepton and siomycin A contributes to their inhibitory activity towards the 20S proteasome.114 One avenue of analogue development has focused on targeting the parasite P. falciparum, and again illustrated that maintenance of the “B-ring” confers inhibition.115 Zhang et al. employed the use of an S. laurentii tsrA mutant organism to produce Ala2 thiostrepton analogs and additionally evaluated them for their inhibitory activity towards the 20S proteasome.116
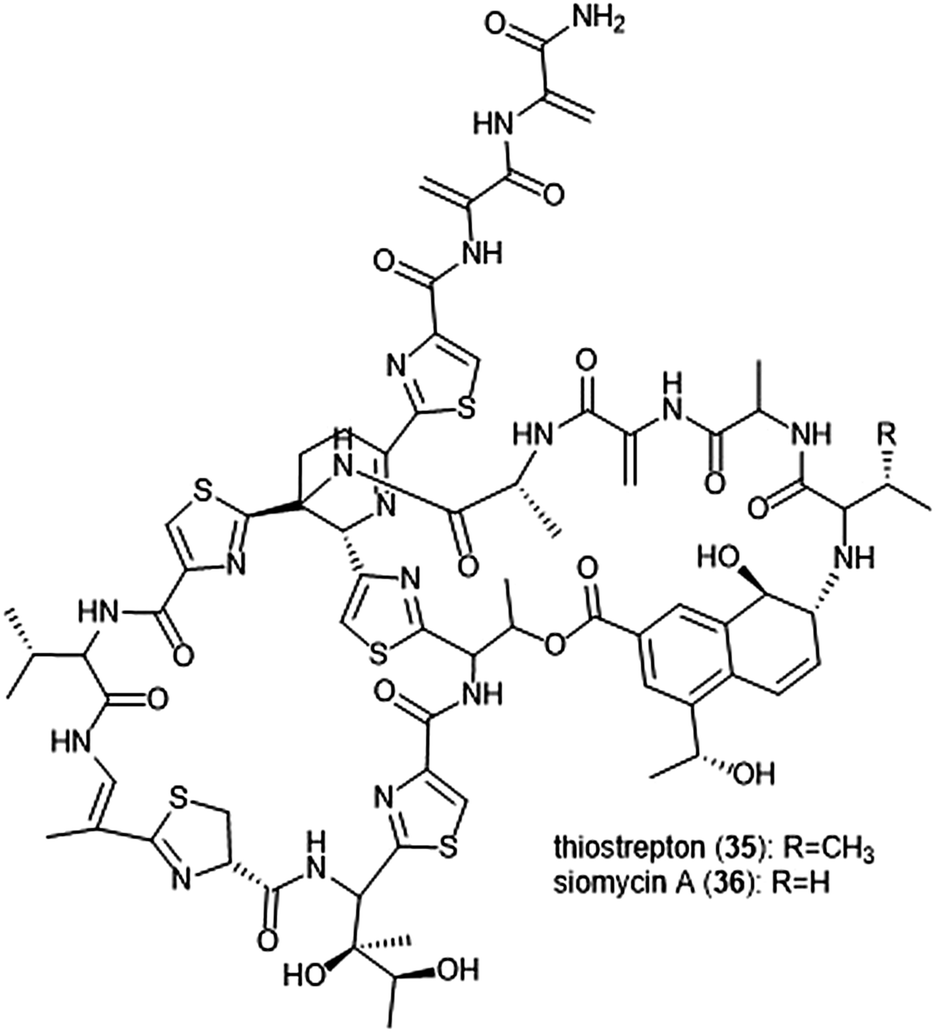 | ||
| Chart 7 The thiopeptide natural products thiostrepton and siomycin A are shown. The “B-ring” within both contributes to their ability to inhibit the 20S proteasome. | ||
Several macrocyclic peptide scaffolds that have been identified as inhibitors of the proteasome have yet to be further evaluated through SAR studies (Charts 8 and 9). Among these natural products are the phepropeptins. Phepropeptins A–D (37–40) are cyclic hexapeptides isolated by Sekizawa et al. from Streptomyces sp. MK600-cF7.117 All four natural products exhibit inhibitory activity against the β5 subunit (IC50: 30.8, 15.3, 17.9, and 10.7 μM, respectively). Because of their stability as cyclic peptides, these natural products were believed to interact with the proteasome through van der Waals and hydrophobic interactions rather than through covalent bond formation. Scytonemide A (41)was isolated from the cultured freshwater cyanobacterium Scytonema hofmannii (UTEX 1834).118 Scytonemide A potently inhibits the ChT-L site of the 20S proteasome (IC50: 96 nM). The presence of an imine moiety within the macrocycle of scytonemide A was hypothesized to be a potential site of attack by the nucleophilic Thr1Oγ of the active site; however, the X-ray crystal structure of this natural product in complex with the 20S proteasome to validate the hypothesis has not been reported. Recently, the cyclic hexapeptide baceridin (42) was isolated from the culture broth of an epiphytic Bacillus strain and underwent total synthesis by the Kalesse group.119 Baceridin inhibits all three catalytic sites of the 20S proteasome and inhibits proliferation in several tumor cell lines.
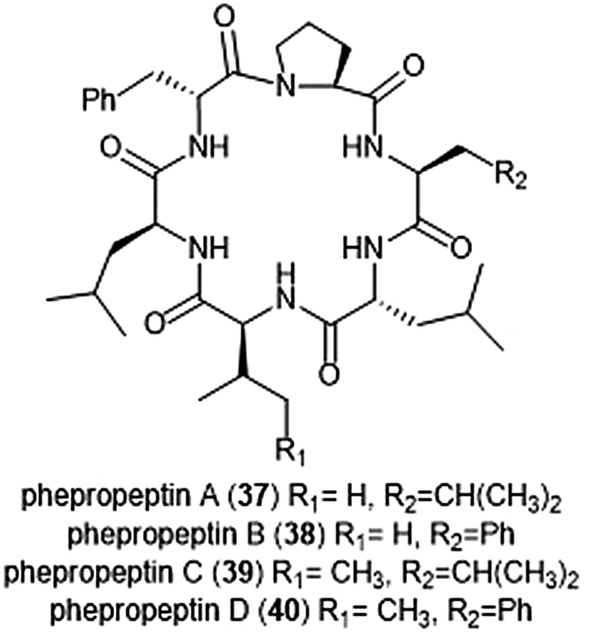 | ||
| Chart 8 Phepropeptins A–D are depicted; these cyclic peptides are believed to inhibit the proteasome through purely noncovalent interactions. | ||
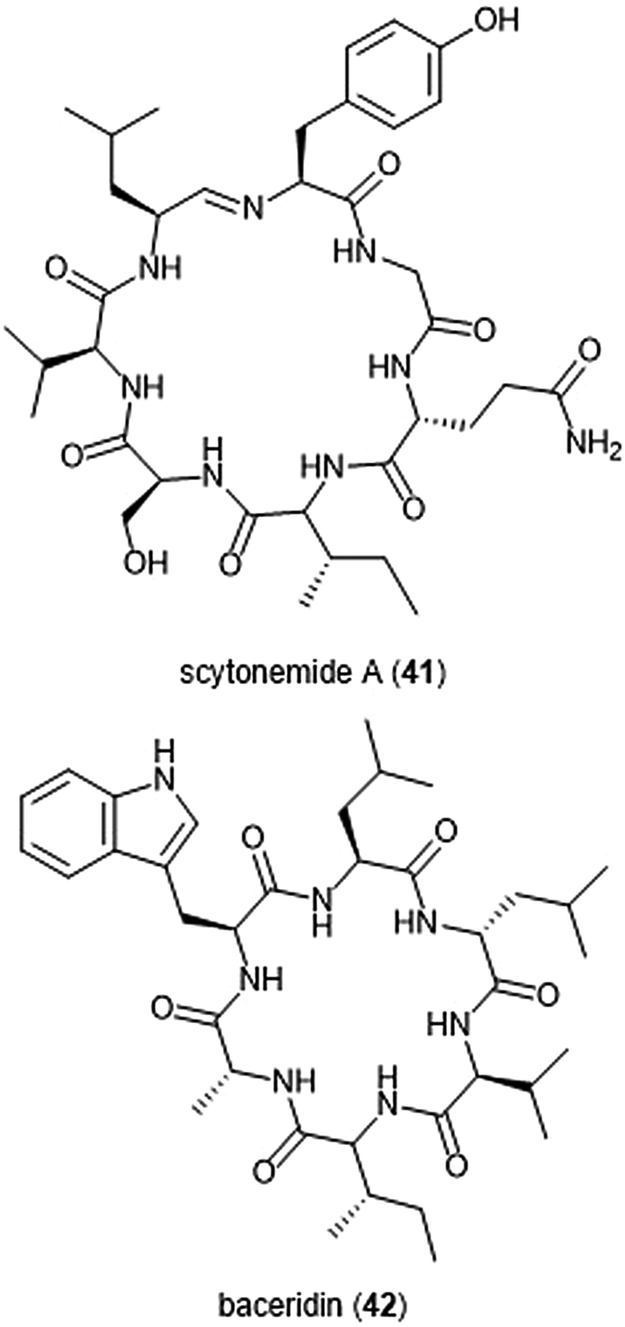 | ||
| Chart 9 Scytonemide A (41) and baceridin (42) are cyclic peptides which also inhibit the 20S proteasome. | ||
β-Lactones
The β-lactone natural products have also been heavily evaluated for their ability to inhibit the proteasome (Chart 10). Lactacystin (43) was isolated by Omura et al. from Streptomyces sp. OM-6519 and displayed biological activity in its initial discovery.120,121 In their 1994 study, Fenteany et al. researchers further evaluated lactacystin and analogues including omuralide for their biological activity. Results suggested the contribution of either the N-acetyl cysteine or β-lactone moieties towards their ability to affect cell cycle progression in Neuro 2A and MG-63 osteosarcoma cells, although the molecular target of these substrates was unidentified at the time.122 A subsequent study indicated that lactacystin inhibits the 20S proteasome.123 Researchers later determined that rather than acting as an inhibitor towards the 20S proteasome, lactacystin acts as a prodrug of its active β-lactone omuralide (44).124,125 The first total syntheses of lactacystin and omuralide were achieved by the Corey group, and many total syntheses and formal syntheses have been recorded since.126–128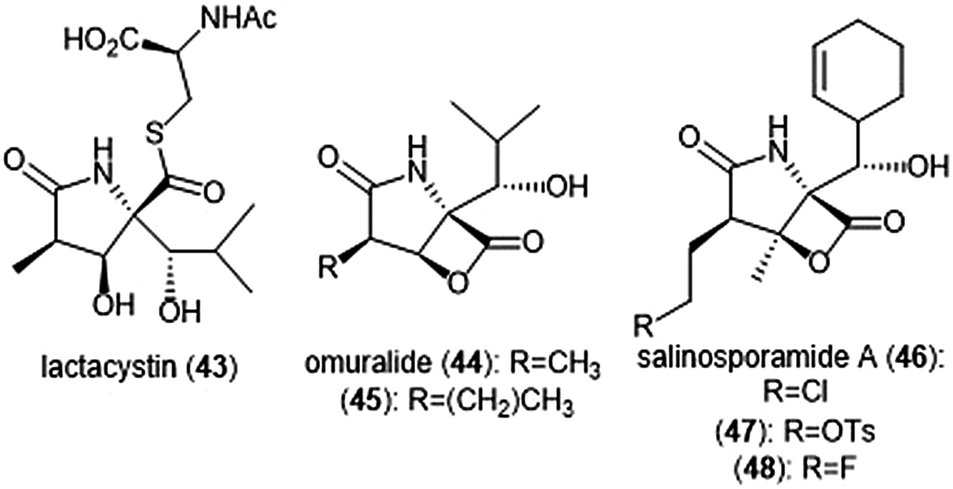 | ||
| Chart 10 Several natural products and derivatives which contain or form a reactive β-lactone ring and inhibit the proteasome are depicted. | ||
In an SAR study of the omuralide scaffold, the Corey group determined that while the hydroxy-isobutyl substituent at C9 is necessary for potent inhibition of 20S bovine proteasome, alteration of the methyl substituent at C7 with larger or bulky alkyl groups improves potency towards the target.129 Research conducted by Soucy et al. further complemented these results with derivative 45, which contains an n-propyl group in place of the methyl substituent at C7. Compound 45 as a Kobs/[I] which is greater than two-fold more potent than omuralide (Kobs/[I] = 46![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 M−1 s−1).130 The compact, densely functionalized omuralide scaffold indicated the importance of exploring non-peptidic natural products as potential inhibitors of the 20S proteasome.
500 M−1 s−1).130 The compact, densely functionalized omuralide scaffold indicated the importance of exploring non-peptidic natural products as potential inhibitors of the 20S proteasome.
Salinosporamide A was first isolated as a product of the ocean sediment-dwelling bacteria Salinospora strain CNB-392 and its structure was elucidated by Feling et al.131 The natural product shares its unique fused γ-lactam–β-lactone bicyclic ring scaffold with that of omuralide. Salinosporamide A was tested for its cytotoxicity against HCT-116 human colon carcinoma cells, displaying an IC50 value of 35.1 nM. Due to its structural similarity to omuralide, salinosporamide A (46) was also tested for its inhibitory activity towards 20S proteasome; it exhibits potent inhibitory activity towards the ChT-L site (IC50: 1.3 nM). Chauhan et al. further investigated the bioactivity of salinosporamide A in in vitro and in vivo studies.132 Salinosporamide A inhibits the ChT-L site with an EC50 value of 3.5 ± 0.3 nM as well as the C-L and T-L sites with EC50 values of 430 ± 34 nM and 28 ± 2 nM, respectively. Salinosporamide A also induced apoptosis in multiple myeloma cells which are resistant towards conventional and Bortezomib therapies.
Only a year after its discovery, the Corey group reported the first total synthesis of salinosporamide A;133 the molecule has since become an attractive target for total synthesis, and many syntheses of salinosporamide A have been reported.134–148 Exploration of the structure–activity relationship of the salinosporamide scaffold began with Macherla et al.,149 and has since been a focus of several research groups (Charts 10 and 11). Alteration from the cyclohexenyl ring to other substituted cyclohexanes resulted in lower inhibitory activity towards the 20S proteasome, as well as lower cytotoxicity against multiple myeloma RPMI 8226 cells. Replacement of the chlorine atom with a hydrogen resulted in a 10-fold reduction of its inhibitory activity towards the ChT-L site of the 20S proteasome, as well as a significant reduction of its cytotoxicity towards RPMI 8226 cells. Groll et al. later solved the crystal structure of the yeast proteasome core particle (yCP) in complex with salinosporamide A, implicating the importance of the chloroethyl group in the overall inhibitory activity towards the proteasome.150 Following transesterification of the β-lactone, the resulting hydroxyl group cyclizes upon the chloro substituent to form a tetrahydrofuran irreversibly.
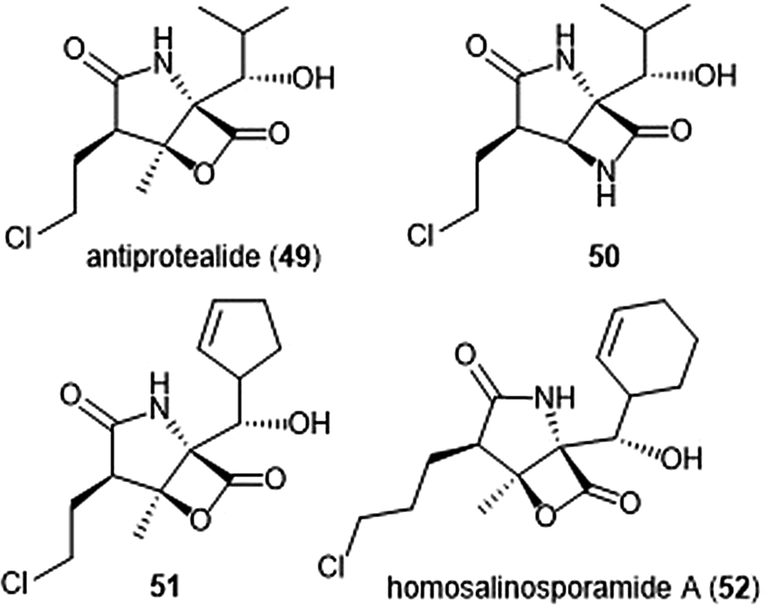 | ||
| Chart 11 Derivatives of β-lactone natural products are shown. | ||
Manam et al. synthesized salinosporamide A analogues which replaced the chloro substituents with functionalities of varying leaving group potentials to further probe the mechanism of inhibition.151 All analogues potently inhibit the ChT-L site of the 20S proteasome; tosyl analog 47 displays inhibitory activity towards the ChT-L site of the 20S proteasome with an IC50 value of 2.5 ± 0.4 nM. Qualitative dialysis experiments with the analogues indicated that the presence of a leaving group prolongs the duration of inhibition. The researchers also performed kinetics experiments with salinosporamide A to understand the mechanism of its proteasome inhibition, determining that the natural product is a slow tight binding inhibitor which confers its activity through a multi-step inhibition mechanism. Fluorosalinosporamide 48 was later synthesized by Eustáquio and Moore using a combination of genetic engineering and precursor-directed synthesis.152 Unlike salinosporamide A, 48 did not undergo displacement of its halogen to afford an irreversible adduct during inhibition, but rather acts as a reversible inhibitor towards the ChT-L activity of the 20S proteasome with two-fold reduced potency relative to salinosporamide A (IC50: 1.5 ± 0.05 nM). The ability of the fluorine to participate in hydrophobic interactions with the proteasome was cited as a justification of its inhibitory potency.
The Corey group further explored the salinosporamide A and omuralide scaffolds through the synthesis of a γ-lactam–β-lactone hybrid antiprotealide (49).153 Antiprotealide—later discovered as a natural product154—exhibited 2.5-fold more potent inhibitory activity towards the β5 subunit than omuralide, but was less potent than salinosporamide A. Synthesis of the γ-lactam–β-lactone congener (50) of antiprotealide was also successfully achieved by the Corey group. While the analogue displayed slower inactivation towards the 20S proteasome than salinosporamide A and omuralide, its improved stability under physiologic conditions was an advantageous feature for relevant drug development.
McGlinchey et al. further demonstrated that bulky groups at the C5 position were necessary for in vitro inhibition of the β5-subunit.155 Chemical synthesis and metabolic engineering were also used by this group to synthesize several novel salinosporamide derivatives with varying substituents at the P1 position.156 The analogue 51—bearing a cyclopentenyl substituent at the P1 position—exhibited equipotent inhibitory potency towards the chymotrypsin-like site of the proteasome in comparison to salinosporamide A, with an IC50 value of 2.2 ± 0.1 nM. Analog 51 also displays increased cytotoxicity against HCT-116 cells.
The length of the chloroalkyl chain at the C2 position of the salinosporamide scaffold is also an important feature with regards to its inhibitory potency. Nguyen et al. explored this in their development of an enantioselective route to (—)-salinosporamide A and derivative (—)-homosalinosporamide A.145 (—)-Homosalinosporamide A varies from (—)-salinosporamide A only through the length of its C2 sidechain; the compound contains an additional methylene carbon. The researchers proposed that the derivative would behave similarly to salinosporamide A in its mechanism of inhibition towards the human proteasome. (—)-homosalinosporamide A (52) displayed similar inhibitory potency towards the ChT-L activity of the 20S proteasome as compared to (—)-salinosporamide A, with an IC50 value of 0.7 ± 0.04 nM (versus 0.8 ± 0.08 nM). However, X-ray crystallographic evidence of the hCP: (—)-homosalinosporamide A complex illustrated that (—)-homosalinosporamide A does not form a tetrahydropyran during its inhibition of the proteasome.157
The structurally related cinnabaramides (Chart 12) were first isolated from the terrestrial Streptomycetes JS360 in 2007 by Stadler et al.158In vitro evaluation of the inhibitory potency of the cinnabaramides towards the human 20S proteasome mirrored results of previously established inhibitors of similar structure. For example, cinnabaramide A (53) (IC50: 1 nM) displayed similar potency as salinosporamide A. Cinnabaramide A is also cytotoxic against colon cancer cell line HCT-116 at a level relative to that of salinosporamide A. Cinnabaramides F (54) and G (55) are believed to act as a prodrug to form the reactive β-lactone in a manner reminiscent to lactacystin; these natural products display potent inhibition towards the human 20S proteasome with IC50 values of 6 nM and 0.6 nM, respectively. rac-Cinnabaramide A was later synthesized by the Romo group.137 In a subsequent study, Rachid et al. accessed chlorinated derivatives of cinnabaramides A–D using mutasynthetic methods.159 15-Chlorocinnabaramide A (56) exhibits greater inhibitory activity towards the β5 subunit of the 20S proteasome than its parent compound, with an IC50 value of 9.3 ± 5.9 nM (IC50 value of cinnabaramide A for this assay was 11.9 ± 7.4 nM). Derivative 56 also displays potent cytotoxicity against the HCT-116, RPMI8226, and SW840 cancer cell lines.
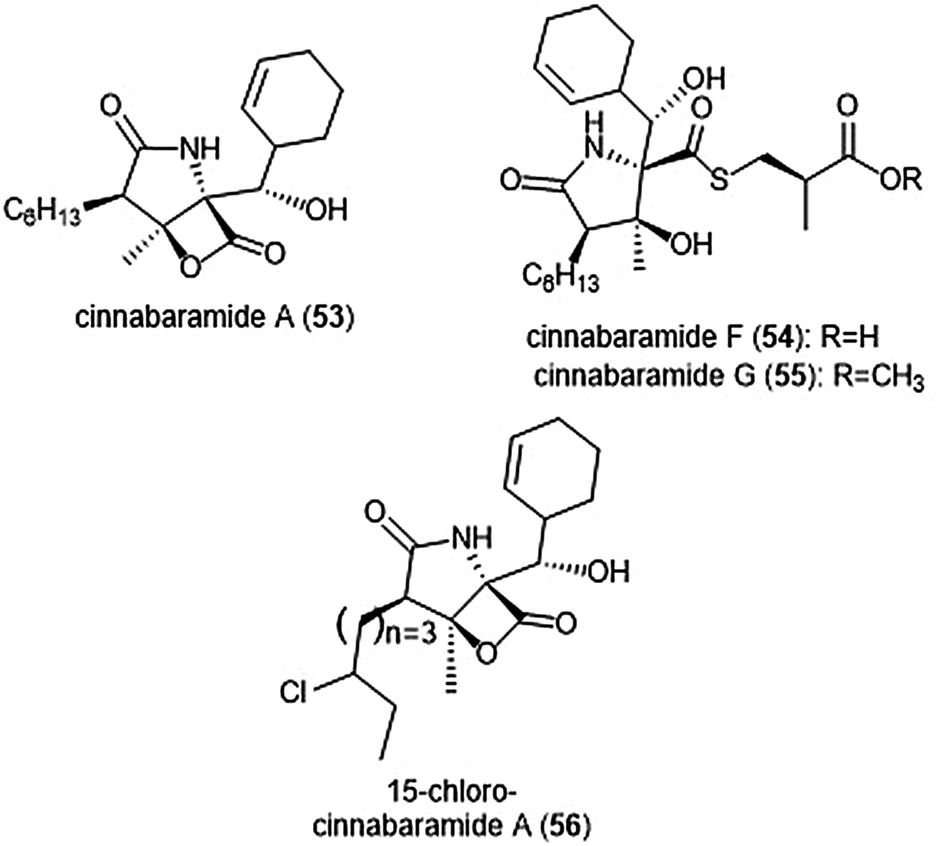 | ||
| Chart 12 The cinnabaramide natural products and their derivative (56) are depicted. | ||
Belactosins A and C (Chart 13) were first isolated from the culture broth of Streptomyces sp. KY11780 by Asai et al.160 The antiproliferative and antitumor activities of both natural products is attributed to a β-lactone moiety at their C-terminus. Belactosin A contains a cyclopropane ring within its backbone which is absent in that of belactosin C. Belactosin A and C both displayed in vitro antiproliferative activity against HeLa S3 cells with IC50 values of 51 μM and 200 μM, respectively. Belactosin A further demonstrated the ability to halt cell cycle progression in the G2/M stage in tumor cells. Akai et al. reported the molecular mechanism of action of belactosins A (57) and C (58): both demonstrate nanomolar inhibitory activity towards the ChT-L site of the rabbit 20S proteasome, both displaying IC50 values of 0.21 μM.161 Optimization of the belactosin A scaffold was further achieved by benzylation of it carboxylic acid. The first total synthesis of belactosin A was completed by Armstrong and Scutt in 2004.162 An enantioselective total synthesis of belactosins A, C and its homologue homobelactosin C (59) was reported shortly thereafter by Larionov and de Meijere.163 X-ray crystallographic evidence based upon the yCP:homobelactosin complex suggested that the β-lactone moiety undergoes nucleophilic attack by the Thr1Oγ residue within the catalytic site to form an ester through a covalent, irreversible bond.164
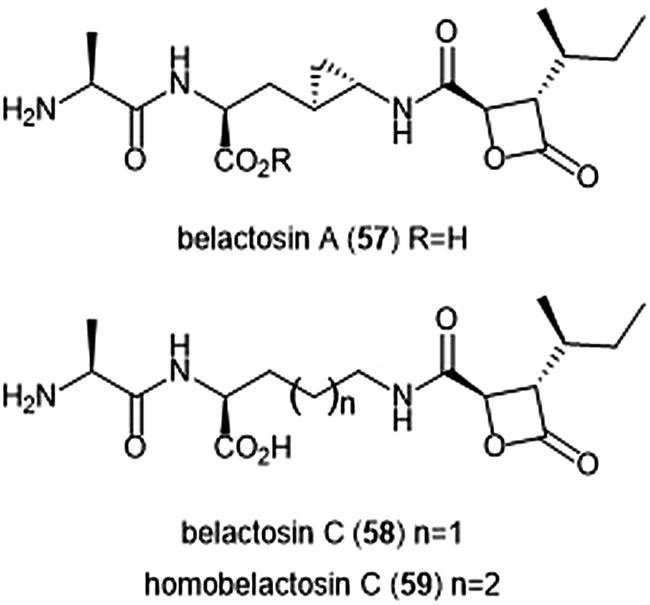 | ||
| Chart 13 The belactosin inhibitors are depicted. The β-lactone moeity contributes to their ability to inhibit the 20S proteasome. | ||
Exploration of the stereochemistry surrounding the belactosin A cyclopropyl ring indicated its contribution to biological activity (Chart 14).165–167 The (cis/L-anti) configuration demonstrated promise, as evidenced by compound 60. Compound 60 selectively inhibits the ChT-L site of the human 20S proteasome with an IC50 value of 5.7 ± 1.2 nM, comparable to that of bortezomib. Compound 60 also inhibits the growth of HCT116 cells with an IC50 value of 1.82 μM via the same mechanism as bortezomib (IC50 value: 0.01 μM). The X-ray crystal structure of yCP:60 complex (2.8 Å), provided researchers with key interactions that contribute to inhibition. Optimization of the transition-state conformation of 60 was subsequently reported.168 Installation of a methyl group at carbon C1′ adjacent to the cyclopropyl group led to analog 61, which is restricted into a predominant syn conformation. Derivative 61 exhibited lower inhibitory activity towards the ChT-L site relative to the parent compound with an IC50 value of 47 ± 2.9 nM. However, when evaluated for its growth inhibitory activity against several tumor cell lines (Hs-Sultan, KB, and HCT-116), compound 61 exhibits activity similar to its parent compound.
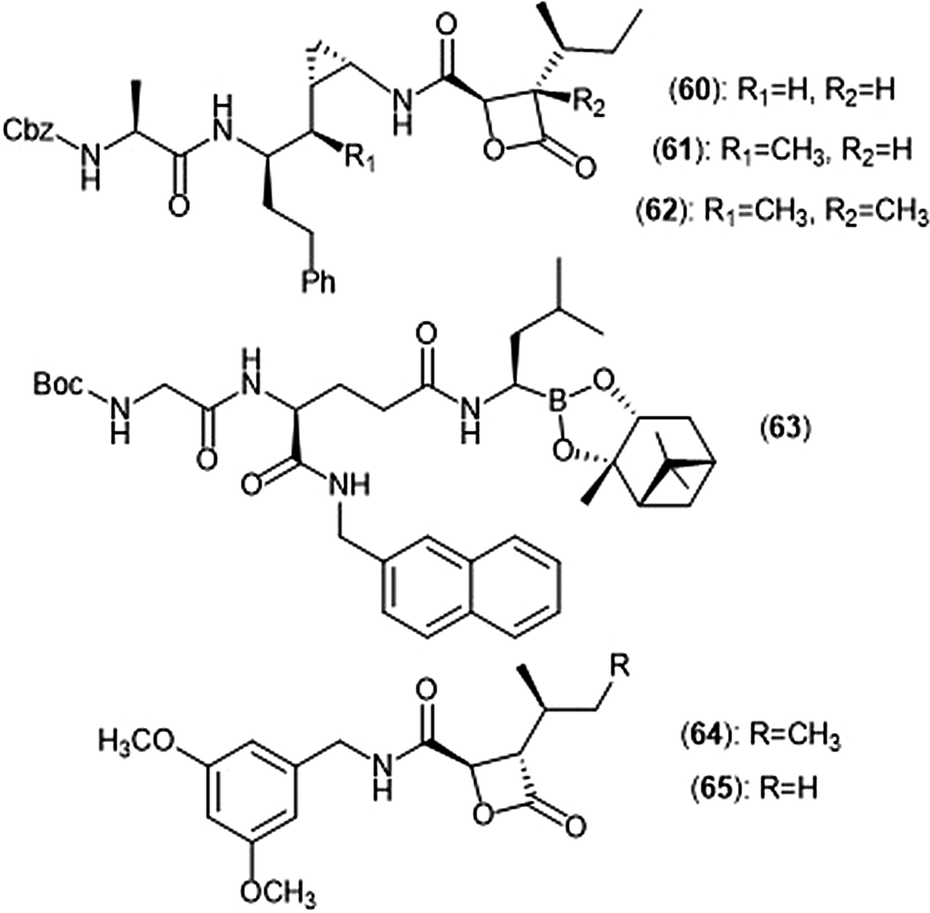 | ||
| Chart 14 Belactosin-based derivatives which exhibit inhibitory activity towards the proteasome are shown. | ||
Researchers hypothesized that poor performance of belactosin analogues under biological conditions and in cell studies may be due to chemical or enzymatic hydrolysis of the reactive β-lactone moiety,168 and thus sought to improve stability by increasing steric hindrance surrounding the β-lactone ring.169 Analogue 62 not only contains an additional methyl group at the α-position of the lactone carbonyl carbon, but also retains the same methyl group at the C1′ carbon adjacent to the cyclopropyl ring to lock it into the syn conformation. While analogue 62 displays lower activity towards the ChT-L site of the proteasome as compared to parent compound 60 (IC50: 1.3 μM and 0.0057 μM, respectively), 62 exhibited comparable growth inhibitory activity towards HCT-116 cells relative to the established inhibitor (IC50: 4.0 μM (62); 1.8 μM (60)). These results provided a novel belactosin analogue with improved biological stability.
Flexible achiral nonpeptidic analogues with a β-lactone moiety and aromatic tails were synthesized by Kawamura et al. using a topology-based scaffold hopping approach.170 These synthetically accessible inhibitors represent novel scaffolds with improved drug-likeness in comparison to past belactosin analogues. Simplified nonpeptidic belactosin scaffolds were further explored through synthesis of hybridized bortezomib and epoxomicin analogs.170,171 Divergence from the β-lactone moiety of the belactosin scaffold stemmed from an interest to incorporate more stable electrophilic warheads.
Nakamura et al. first sought to optimize the belactosin C scaffold in 2009 through the introduction of a boronic acid moiety like that of bortezomib.172 Compound 63 was identified as a potent inhibitor of the ChT-L (β5) site of the 20S proteasome (IC50: 0.28 ± 0.04 μM) and also exhibited submicromolar growth inhibitory activity against HeLa cells (IC50: 0.35 ± 0.02 μM) in an MTT assay. The de Meijere group expanded their previous synthetic endeavors of belactosin scaffolds through the synthesis of novel belactosin C-based proteasome inhibitors.173 A subsequent report by the same group elected to divert from the dipeptide scaffold of belactosin C to investigate more easily accessible analogues.174 Analogues containing the N-(3,5-dimethoxy)benzyl amido side chain displayed especially potent inhibitory activity towards the β5 subunit. This scaffold was later altered to produce a minimal β-lactone scaffold for selective inhibition of either the β5c or β5i of the human 20S proteasome.175 Analogue 64 contains a pseudo-isoleucine P1 side chain, whereas 65 contains a smaller pseudo-valine moiety. This difference in size of the P1 side chain resulted in selective inhibition. Compound 64 exhibited preference towards the β5i site (IC50: 14.37 nM) over the β5c (IC50: 21.35 nM), whereas 65 preferentially inhibits the β5c (IC50: 26.87 nM) over the β5i (IC50: 83.62 nM).
Another class of β-lactone containing natural products which display intriguing activity towards the proteasome are cystargolides A and B (Chart 15). Originally isolated by the Kerr group in 2015 from the actinomycete Kitasatospora cystarginea NRRLB16505, these natural products contain a dipeptide backbone with a β-lactone moiety.176 Cystargolides A (66) and B (67) exhibited inhibition towards the ChT-L site of the 20S proteasome with IC50 values of 0.36 ± 0.017 μM and 0.93 ± 0.032 μM, respectively. The total syntheses and absolute stereochemistry of cystargolides A and B were later successfully achieved by Tello-Aburto et al.177 Wolf et al. were also able to access the cystargolides and belactosins using biosynthetic methods. The stereochemistry of these natural products is integral to inhibitory activity: maintaining the (2R,3S) absolute stereochemistry contributes to potency of inhibitors.178 Benzylation of the N-terminus also improved inhibitory potency 100-fold, as evidenced by analogues 68 (IC50: 9.2 ± 0.59 nM) and 69 (IC50: 9.0 ± 1.4 nM).177 Subsequent optimization of the cystargolide scaffold was achieved in 2018: benzyl ether 70 inhibits the hβ5 subunit of Jurkat cell lysate with an IC50 value of 3.1 ± 0.2 nM as compared to cystargolide B (IC50: 0.90 ± 0.11 μM), and is also cytotoxic against MCF-7 and RPMI-8226 cells.179
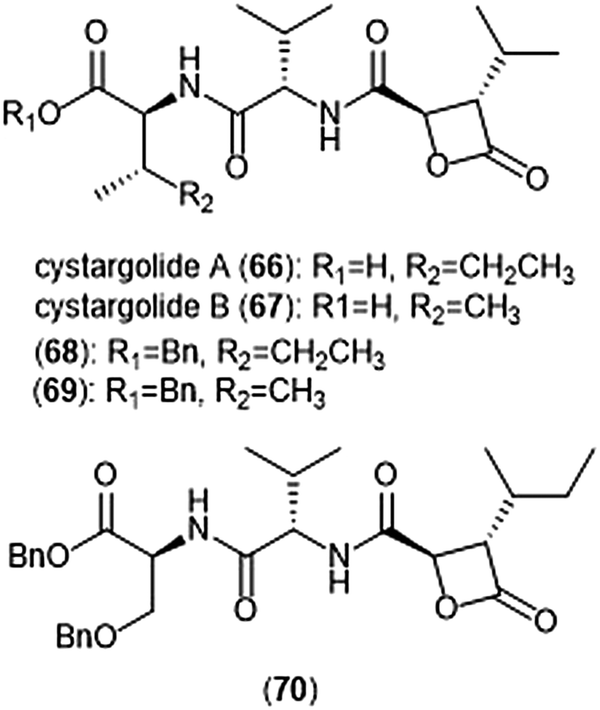 | ||
| Chart 15 The cystargolide natural products and derivatives are shown. | ||
α,β-Epoxyketones
The epoxy-ketone containing natural products have become a specific focus to researchers due to their ability to inhibit the 20S proteasome (Chart 16). Eponemycin is a dipeptide natural product which features a lipophilic tail at its N-terminus and an electrophilic α,β-epoxyketone moiety at its C-terminus. The natural product was isolated from the fermentation broth of the soil-dwelling Streptomyces hygroscopicus No. P247-71 (ATCC 53709).180 Eponemycin (71) and its derivative dihydroeponemycin (72) exhibit potent in vitro toxicity against B16–F10 and HCT-116 cells. The natural product also demonstrated itself as an inhibitor of angiogenesis.181 The cellular target of its antitumor activity was later identified by the Crews group as the proteasome with biotinylated eponemycin analogs.182 Dihydroeponemycin inhibits the proteasome in a competitive and irreversible manner, with the highest rate of inhibition towards the ChT-L site (kassoc: 66.4 ± 8.9 M−1 s−1). Kim et al. further explored the structure–activity relationship of dihydroeponemycin and analogues to improve understanding of subunit selectivity within the 20S proteasome.183 The ability of dihydroeponemycin to interact with the immunoproteasome was attributed to its lipophilic C-terminus.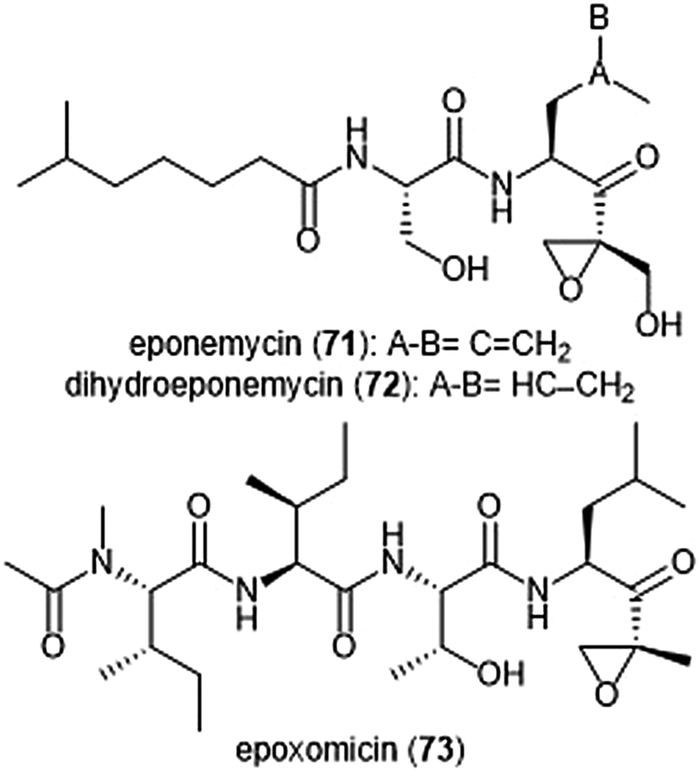 | ||
| Chart 16 The epoxy-ketone natural products eponemycin, dihydroeponemycin and epoxomicin are depicted. The electrophilic epoxy-ketone moeity within these scaffolds contributres directly to their ability to inhibit the 20S protesome. | ||
The natural product epoxomicin (73) was isolated from a soil-dwelling actinomycete no. Q996-17184 and is comprised of a tetrapeptide skeleton with a C-terminal α,β-epoxyketone. Epoxomicin exhibited strong in vitro cytotoxicities against various cancer cell lines including HCT-116 (IC50: 9.0 nM), and additionally exhibits antitumor activity against B16 melanoma. The total synthesis of epoxomicin was first reported by the Crews group in 1999.185 Due to previous reports that indicated peptide α,β-epoxyketones as inhibitors of the proteasome,186 epoxomicin was evaluated for this activity. Epoxomicin selectively inhibits the chymotrypsin-like site of the 20S proteasome (kassoc: 35400 M−1 s−1). The N-terminal P3 and P4 residues play a major role in the potency of inhibition towards the proteasome.183 As compared to dihydroeponemycin, epoxomicin and its analogues display greater inhibitory potency. The X-ray crystal structure of the yCP:epoxomicin complex was solved by Groll et al. (2.25 Å resolution),187 revealing the basis for its selectivity towards the ChT-L site. The α,β-epoxyketone was initially believed to undergo a reversible nucleophilic addition by the Thr1Oγ to produce a hemiacetal; subsequent attack of the epoxide by Thr1N forms a morpholino adduct irreversibly. Recently, the mechanism of proteasome inhibition by epoxyketones was revised based on X-ray crystallographic data by Schrader et al.188 Instead of forming a morpholino adduct, epoxyketones are believed to react with the Thr1 residue of the catalytic site to form a 1,4-oxepane ring.
Further optimization of the epoxomicin scaffold has been achieved by several groups (Chart 17). The Crews group set out to improve upon this natural product to access more potent and selective analogues, leading to the development of the tetrapeptide epoxyketone YU-101 (74).189In vitro studies indicate that YU-101 selectively inhibits the ChT-L site of the 20S proteasome with a kassoc value of 166000 (5–12 nM), greater than that of epoxomicin and bortezomib. Further optimization of this analogue led to carfilzomib (Kyprolis®, 75) by the biotech company Proteolix.190 Carfilzomib exhibits selective, potent inhibitory activity towards the ChT-L site of the proteasome, with an IC50 value of 6 nM; the compound also performed well in in vivo studies. In 2012 carfilzomib was approved by the FDA for the treatment of refractory multiple myeloma;191 further alteration of this compound led to the orally available oprozomib (76).192 Subsequent optimization of the epoxyketone scaffold has focused on developing analogues which can overcome bortezomib and carfilzomib-resistant multiple myeloma. Kim et al. recently reported a novel epoxyketone 77, which not only inhibits the ChT-L activity of proteasome from RPMI8226 cell lysates (IC50: 2.1 ± 0.9 nM), but also inhibits proteasome activity (ChT-L) nearly three-fold more potently in RPMI8226-Cfz resistant cells than carfilzomib itself (IC50: 106.2 ± 28.9 nM).193 Other recent advances of the epoxyketone moeity include the design of selective inhibitors for the β1i,194,195 β2i,196,197 and β5i subunits.198
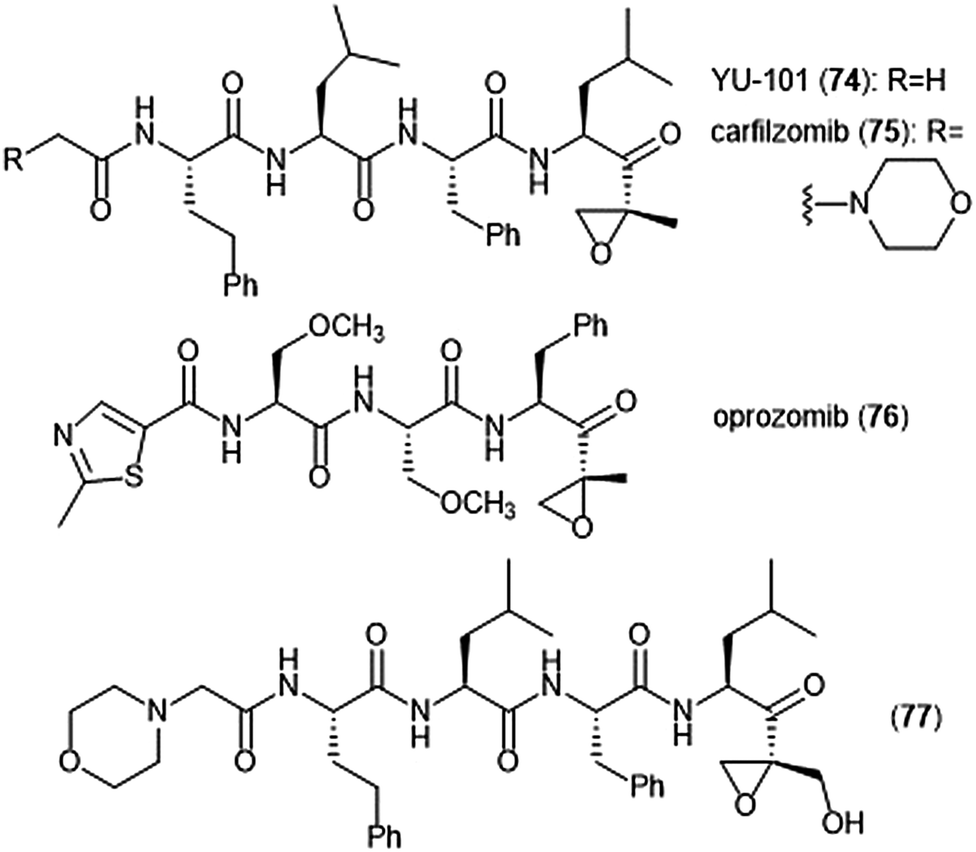 | ||
| Chart 17 Synthetic inhibitors of the 20S proteasome based upon epoxomicin are shown. | ||
The epoxyketone-containing natural products carmaphycin A and B were isolated from marine cyanobacterium Symploca sp.199 These tripeptides contain a lipophilic N-acylated tail at their N-terminus and a leucine-derived epoxyketone at their C-terminus. A scalable total synthesis of both carmaphycins was also conducted and both were evaluated for their ability to inhibit the yeast 20S proteasome. Carmaphycins A (78) and B (79) (Chart 18) exhibited potent inhibitory activity towards the ChT-L site with IC50 values of 2.5 ± 0.3 nM and 2.6 ± 0.9 nM, respectively. The natural products also displayed cytotoxic activity towards H-460 and HCT-116 cancer cell lines. Structural investigation implicated the importance of the sulfoxide/sulfone moieties in the methionine-derived residue in the interaction with the target; this interaction represents a distinctive binding mode for the carmaphycins relative to previously discovered α,β-epoxyketone inhibitors. Replacement of the α,β-epoxyketone moiety with an enone led to the identification of analogue 80, which is a selective nanomolar inhibitor towards the ChT-L site of the 20S yeast proteasome.200 Analogue 80 interacts through a unique two-step hydroamination mechanism with the catalytic site to form a morpholine adduct. Most recently, carmaphycin-based proteasome inhibitors have been designed for use as antibody drug conjugates as potential cancer treatments.201
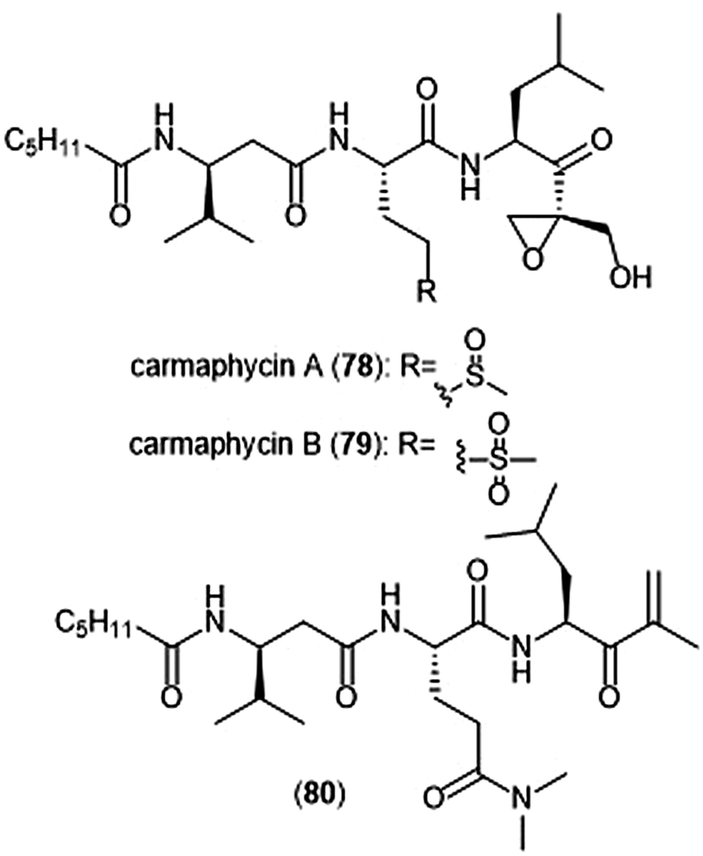 | ||
| Chart 18 Carmaphycins A and B, and the enone derivative (80) are shown. | ||
Several other epoxyketone natural products were also isolated and evaluated for their ability to inhibit the proteasome (Chart 19). TMC-86A (81) and B (82) are isolated from the fermentation broth of Streptomyces sp. TC 1084.202,203 Both display similar inhibitory potency towards the ChT-L site with IC50 values of 5.1 μM and 1.1 μM, respectively. TMC-86A and B also exhibited nanomolar cytotoxicity against various tumor cell lines. TMC-96 (83) was isolated from the fermentation broth of Saccharothrix sp. TC 1094,202,203 and contains a branched N-acylated terminus and a leucine-derived α,β-epoxyketone moiety similar to dihydroeponemycin. TMC-96 exhibits inhibitory activity towards the ChT-L and C-L sites of the 20S proteasome (IC50: 2.9 μM and 3.5 μM, respectively), and additionally displays nanomolar cytotoxic activity against a variety of tumor cell lines. TMC-89A (84) and B (85) are α,β-epoxyketone-containing tripeptides which were isolated from the fermentation broth of Streptomyces sp. TC 1087 by Koguchi et al. in their search for new proteasome inhibitors of bacterial origin.204 When tested for their proteasome inhibitory activity, both displayed equipotent micromolar activity towards the ChT-L site of the 20S proteasome, with IC50: 1.1 μM. TMC-89A and B also exhibit slight selectivity towards the T-L site of the 20S proteasome, with IC50 values of 390 nM and 510 nM, respectively. In vitro cytotoxicity studies reveal that in comparison to many other α,β-epoxyketones, TMC-89A and B are not remarkably cytotoxic against tumor cell lines. Subsequent optimization of these natural products has not been reported but may benefit from the addition of N-terminal lipophilic residues to improve cell permeability.
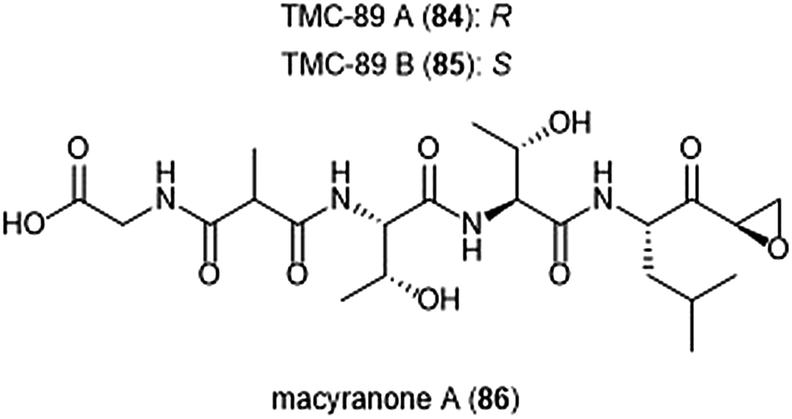 | ||
| Chart 19 Several epoxy-ketone containing peptide natural products have been identified as inhibitors of the 20S proteasome. | ||
The macyranones are a group of six linear peptides which were produced by the myxobacteria Cystobacter fuscus MCy9118.205 Macryanone A (86) contains an α,β-epoxyketone moiety which made it an intriguing compound to test for proteasome inhibition. Macyranone A inhibits the ChT-L activity of the yCP (IC50: 5.9 nM) and the human constitutive proteasome and immunoproteasome, with IC50 values of 21 nM and 15 nM, respectively. X-ray crystallographic analysis of the yCP: macyranone complex (2.8 Å resolution) revealed that it reacts irreversibly with the catalytic sites of the proteasome in a similar mechanism to epoxomicin. Despite its promising performance in in vitro enzymatic testing, macyranone A displayed poor cytotoxicity against mammalian cell lines including HCT-116, THP-1 and HL-60 cancer cell lines.
The most recently discovered epoxyketone natural product inhibitors were reported by Owen et al. The group utilized molecular evolutionary analyses of complex metagenomes to target and isolate the previously unidentified clarepoxcin and landepoxcin natural products (Chart 20).206 Clarepoxcins A–E (87–91) were produced by S. albus:AR456, while landepoxcins A (92) and B (93) were produced by S. albus:AR412. Clarepoxcins A–D are potent low nanomolar inhibitors of the human 20S proteasome, with an IC50 ∼ 6.9–15.1 nM (ChT-L). The clarepoxcins also exhibited potent cytotoxic activity against HCT-116 cells. While clarepoxcin E did not display inhibitory activity towards the 20S human proteasome, it did exhibit potent cytotoxicity against HCT-116. The authors proposed that in cells, clarepoxcin E acts as a prodrug to form the reactive α,β-epoxyketone moiety. Landepoxcins A and B displayed higher nanomolar inhibitory activity towards the human 20S proteasome, however landepoxcin A did exhibit impressive cytotoxicity against HCT-116 cells.
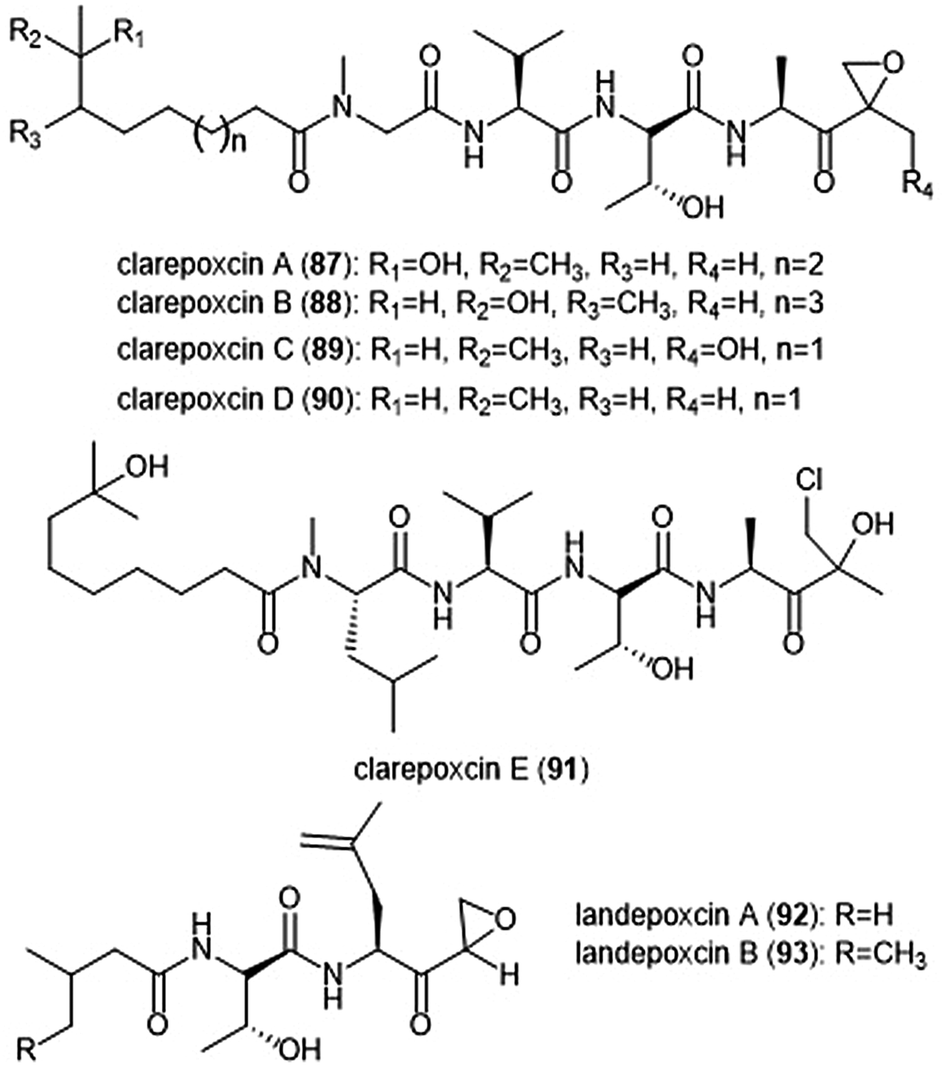 | ||
| Chart 20 The recently identified epoxy-ketone containing proteasome inhibitors clarepoxcins and landepoxcins are shown. Clarepoxcin E is believed to act as a prodrug of an epoxy ketone in cell studies. | ||
Terpenoids
Terpenoids represent a vast class of natural products that have been reported for their biological activities. For example, the agosterols were evaluated by the Tsukamoto group for their ability to inhibit the proteasome. Among the many tested, agosterol C demonstrated the most potent activity against the ChT-L site, with an IC50 value of 19.8 μM.207 Epoxyphomalins A and B were first isolated as products of the marine-derived fungus Phoma sp. by Mohamed et al.208 Epoxyphomalin A displayed cytotoxicity towards several human tumor cell lines. The intracellular target of epoxyphomalins A and B was later identified as the 20S proteasome;209 both epoxyphomalins A (94) and B (95) exhibit dose-dependent inhibition of the 20S human proteasome subunits (Chart 21). Epoxyphomalin A demonstrated equipotent inhibition against all three catalytic sites, whereas epoxyphomalin B preferentially inhibits the ChT-L site.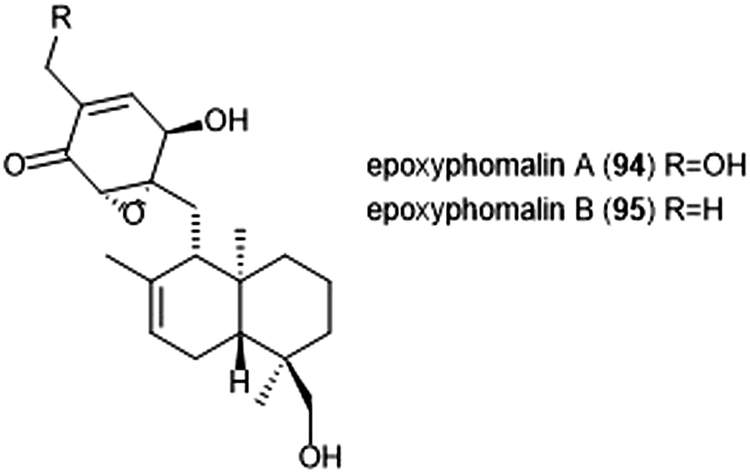 | ||
| Chart 21 The proteasome inhibitors epoxyphomalin A and B are shown. | ||
Noda et al. isolated a variety of stronglyphorines from marine sponge Petrosia corticata in 2015.210 The structure–activity relationship of the strongylophorines with their inhibitory activity revealed that the presence of a hemiacetal in addition to a hydroquinone moiety contributes significantly towards the potency of the proteasome inhibitors. Strongylophorines 13/14 (96) and strongylophorines 15/16 (97) exhibited the most potent inhibitory activity towards the ChT-L site of the 20S proteasome with an IC50 of 2.1 μM and 3.6 μM, respectively (Chart 22).
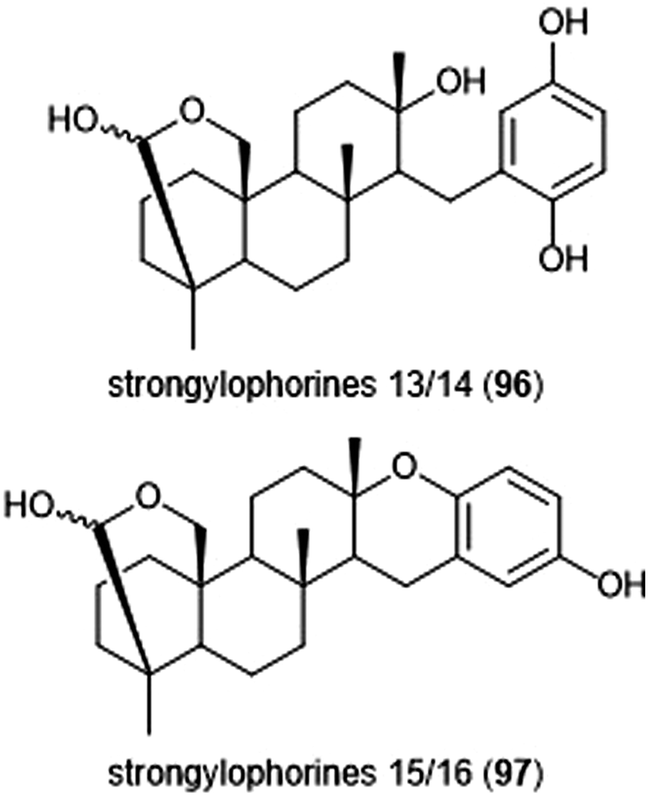 | ||
| Chart 22 The hemiacetal moieties in strongylophorines 13/14 and 15/16 contribute to their ability to inhibit the 20S proteasome. | ||
Petrosaspongiolide M (98) was isolated from the New Caledonian marine sponge Petrosaspongi nigra among the other petrosaspongiolides A–R. The petrosaspongiolides are sesterterpenes all containing a cheilantane skeleton, and were first identified as inhibitors of the preparation of phospholipase A2.211 Petrosaspongiolide M was later identified by Margarucci et al. as a proteasome inhibitor using chemical proteomics.212 The natural product inhibited the C-L and ChT-L activity of the proteasome in a cell-based assay with IC50 values of 0.85 ± 0.15 μM and 0.64 ± 0.15 μM, respectively. Petrosaspongiolide M was later reported to exhibit potent inhibitory activity towards the ChT-L and C-L activities of the proteasome with submicromolar values in an in vitro fluorometric enzyme assay.213 Optimization led to analogues 99 and 100, which both feature a benzothiophen-2-yl substituent at the C4 position of the butenolide ring. Analogue 99 exhibited potent inhibitory activity against the ChT-L and C-L site of the 20S proteasome, with IC50 values of 0.06 ± 0.009 and 0.22 ± 0.03 nM, respectively. The desbrominated analogue 100 also displayed potent inhibitory activity towards the ChT-L and C-L sites, with IC50 values of 0.07 ± 0.01 nM for both sites (Chart 23).
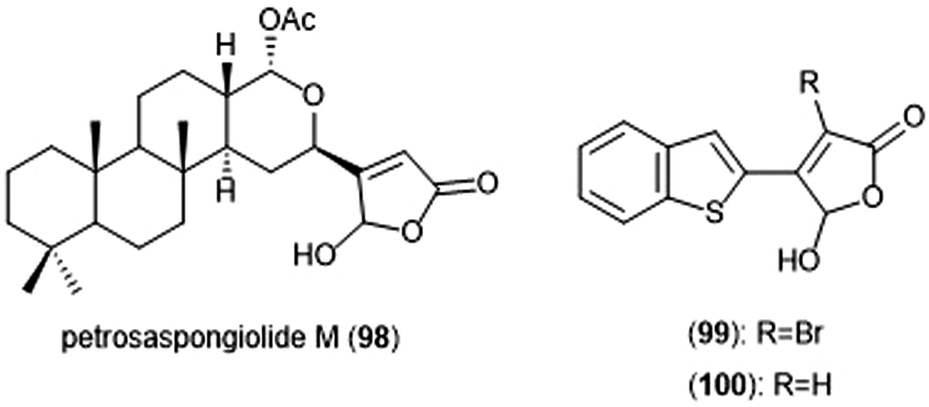 | ||
| Chart 23 Petrosaspongiolide M and its simplified analogues are depicted; all three inhibit the activity of the proteasome. | ||
Several other terpenoid natural products have also been identified as inhibitors of the 20S proteasome. The merosesterterpene acanthosulfate (101) was identified by West and Faulkner as a metabolite of the marine sponge Acanthodendrilla sp. in 2008.214 Acanthosulfate (Chart 24) exhibited inhibition towards the proteasome (IC50: 4.5 μM) but lacked selectivity and potency when tested for activity against the BMS Oncology Diverse Cell Panel (ODCA).
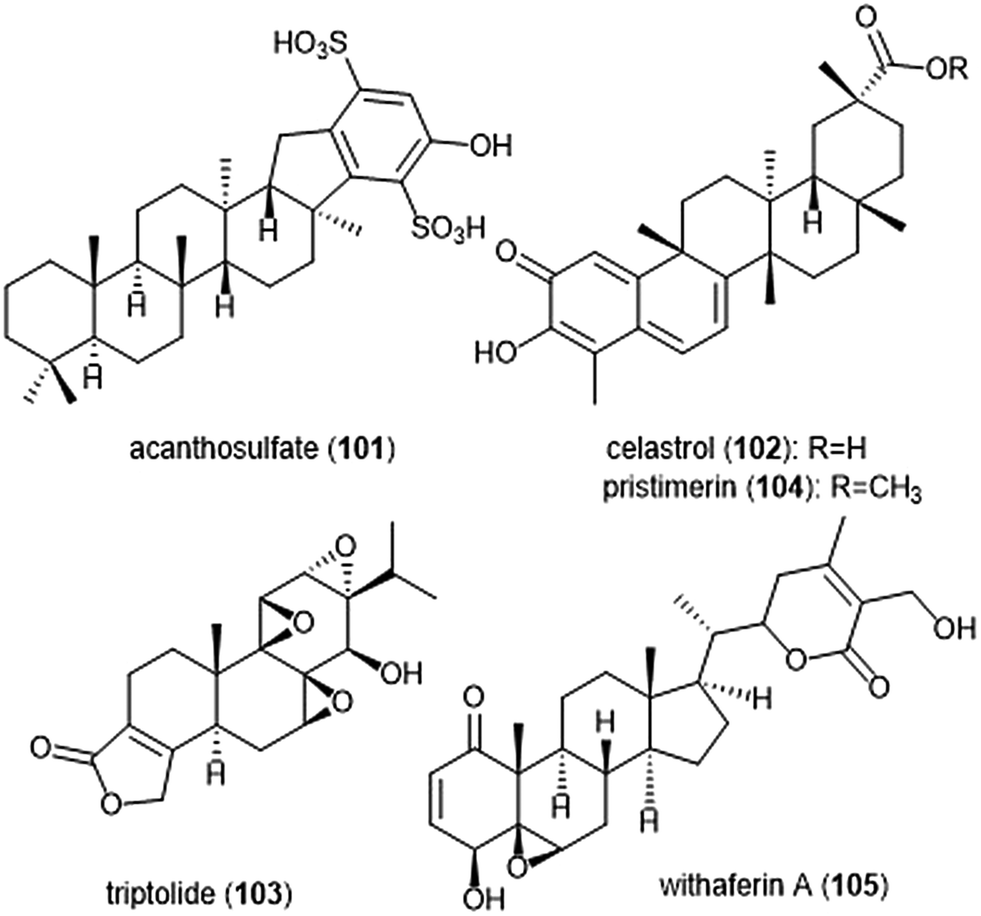 | ||
| Chart 24 Various terpenoids which have been identified as proteasome inhibitors are depicted. | ||
Celastrol (102) is a quinone methide triterpene which is extracted from the “Thunder God Vine” (Tripterygium wilfordii Hook F.) and has been recognized for its biological activity. Dou et al. discovered the inhibitory activity of celastrol towards the 20S proteasome in 2006.215 Celastrol selectively inhibits the ChT-L activity of the proteasome with an IC50 value of 2.5 μM. The compound also demonstrated the ability to inhibit the proteasome of prostate cancer cells in a cell-based assay at 1–5 μM. Celastrol further demonstrated antitumor activity in an in vivo study. Researchers reported that treatment of PC-3 containing mice with the natural product resulted in tumor reduction. Because celastrol has many intracellular targets,216 focus on optimization of the celastrol scaffold has involved improving its solubility and physiological properties. Recently, celastrol has been reported as cytotoxic against human multiple myeloma cells.217 Further investigation of compounds from the extract of the “Thunder God Vine” (Tripterygium wilfordii F. Hook) focused on the diterpene tri-epoxide lactone triptolide (103). Originally discovered in 1972, researchers reported its antileukemic activity thus providing impetus for further biological evaluaton.218,219 Although triptolide does not inhibit the ChT-L site of purified 20S proteasome, it was able to inhibit proteasomal activity within cancer cells. This interesting result suggests that triptolide acts as a prodrug, and one of its metabolites—perhaps an oxidized ketone—acts instead as the biologically active molecule.
Tiedemann et al. further demonstrated the inhibitory potential of the celastrol scaffold through the investigation of its natural product relative pristimerin and the mechanisms of its biological activity.220 Pristimerin (104) contains an identical scaffold to that of celastrol except for the carboxylic acid moiety, which is instead replaced by a methyl ester. Pristimerin selectively inhibits the ChT-L activity of the 20S proteasome with an EC50 value of less than 125 nM. Researchers further indicated in a cell-based experiment that pristimerin suppresses NF-KB activity through proteasome dependent and independent pathways. Pristimerin also demonstrated promising activities in cytotoxicity and in vivo studies. The steroidal lactone withaferin A (105) is a natural product which was isolated from the Indian Winter Cherry Withania somnifera Dunal, a plant which is popular for its use in Ayurvedic medicine.221 The steroid contains two conjugated ketones and bears structural similarity to celastrol. Withaferin A had previously been recognized for its various biological activities, though the mechanism of action by the natural product had not been elucidated. Dou et al. identified the 20S proteasome as an intracellular target of withaferin A.222 Withaferin A inhibits the ChT-L activity of the 20S proteasome with an IC50 value of 4.5 μM and also exhibits proteasomal inhibition within PC-3 cells. The natural product also performed well in in vivo tumor studies conducted on mice. In silico docking studies in addition to kinetic studies suggested that the conjugated ketone moieties of withaferin A may interact covalently with the Thr1Oγ within the β5 active site to confer inhibition of the 20S proteasome.
The neomacrophorins (Chart 25) were identified as products of soil-dwelling Trichoderma sp. 1212-03, and have recently been evaluated for their biological activities.223–225 Neomacrophorin I demonstrated cytotoxicity against human colorectal cancer COLO 201 cells, though its mode of action for this cytotoxicity was initially unclear. An MTT assay of the neomacrophorins with promyelocytic leukemia HL60 cells revealed that neomacrophorins I–VI (106–111) inhibit growth of the HL60 cells. The molecules were also responsible for inducing apoptotic cell death in HL60 cells, and further demonstrated in vitro inhibitory activity towards the 20S proteasome. Neomacrophorins I and IV demonstrated the most potent inhibitory activity towards the ChT-L site as compared to the other neomacrophorins, with IC50 values of 5.7 ± 1.0 and 5.3 ± 1.2 μM, respectively. The quinone moiety was deemed an important feature for the inhibitory proteasome activity and cytotoxicity.
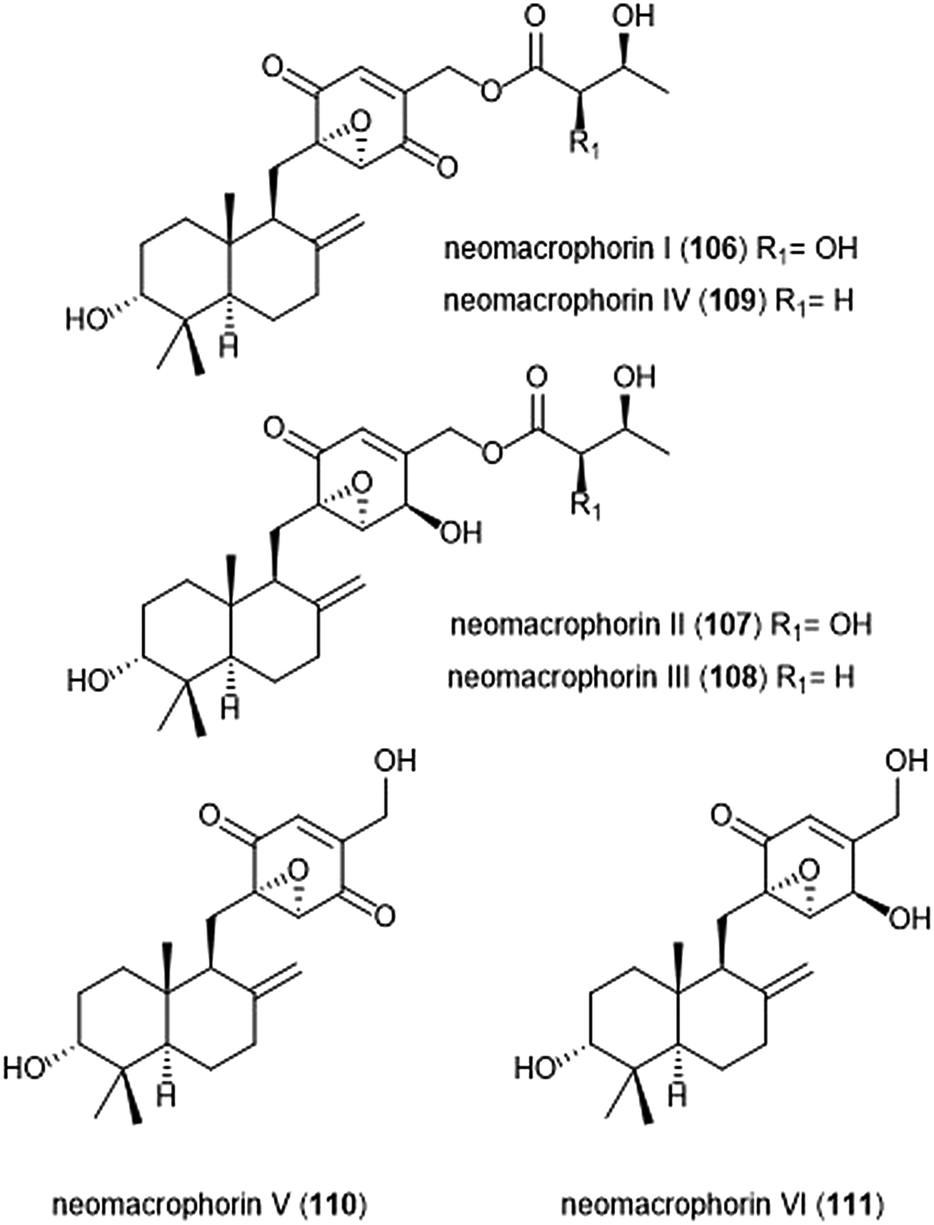 | ||
| Chart 25 The neomacrophorins I–VI are depicted. | ||
Alkaloids
Various alkaloids have also been identified as inhibitors of the proteasome. Tsukamoto et al. isolated the aaptamine natural products (Chart 26) from the marine sponge Aaptos suberitoides as part of their efforts to identify novel proteasome inhibitors from marine invertebrate extracts and marine-derived fungi cultures. Aaptamine (112), isoaaptamine (113) and demethylaaptamine (114) inhibit the ChT-L and C-L sites for both rat and human 20S proteasomes (IC50: 7.0–20.2 μM). When tested for cytotoxicity against HeLa cells, the aaptamines did not exhibit a correlation between cytotoxicity and proteasome inhibition.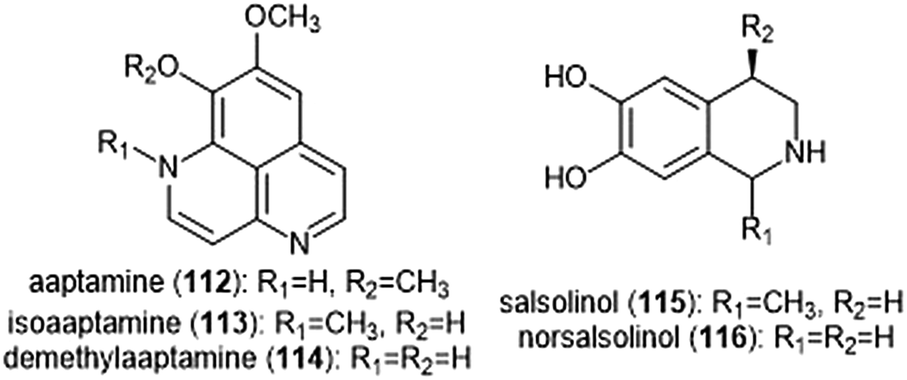 | ||
| Chart 26 The aaptamines and salsolinols all inhibit the 20S proteasome. | ||
The tetrahydroisoquinoline salsolinol alkaloids were later isolated from the marine sponge Xestospongia cf. vansoesti and evaluated for their proteasome inhibitory activity.226 Salsolinol (115) and its derivative norsalsolinol (116) inhibit the ChT-L site with IC50 values of 279.0 and 193.7 μM, respectively. Cytotoxicity studies indicated that salsolinol is cytotoxic against several cell lines; norsalsolinol displayed cytotoxicity against HeLa cells (IC50: 42.4 μM) but was not tested against other cell lines.
Specific manzamines have been identified as inhibitors of the 20S proteasome by Tsukamoto et al. (Chart 27).227,228 Following their isolation from the marine sponge Acanthostrongylophora ingens and structural identification, several were evaluated for their cytotoxicity and proteasome inhibitory activity. Manzamine A (117), neo-kauluamine (118) and pre-neo-kauluamine (119) exhibit potent inhibitory activity towards the ChT-L site of the 20S proteasome with IC50 values of 2.0, 0.13 and 0.34 μM, respectively. Acanthomanzamine D (120) also exhibits nanomolar inhibitory activity towards the chymotrypsin-like site of the 20S proteasome (IC50: 630 nM), albeit with less potency than the kauluamines. The presence of the eight-membered ring in addition to the β-carboline appeared to be necessary for proteasome inhibition.
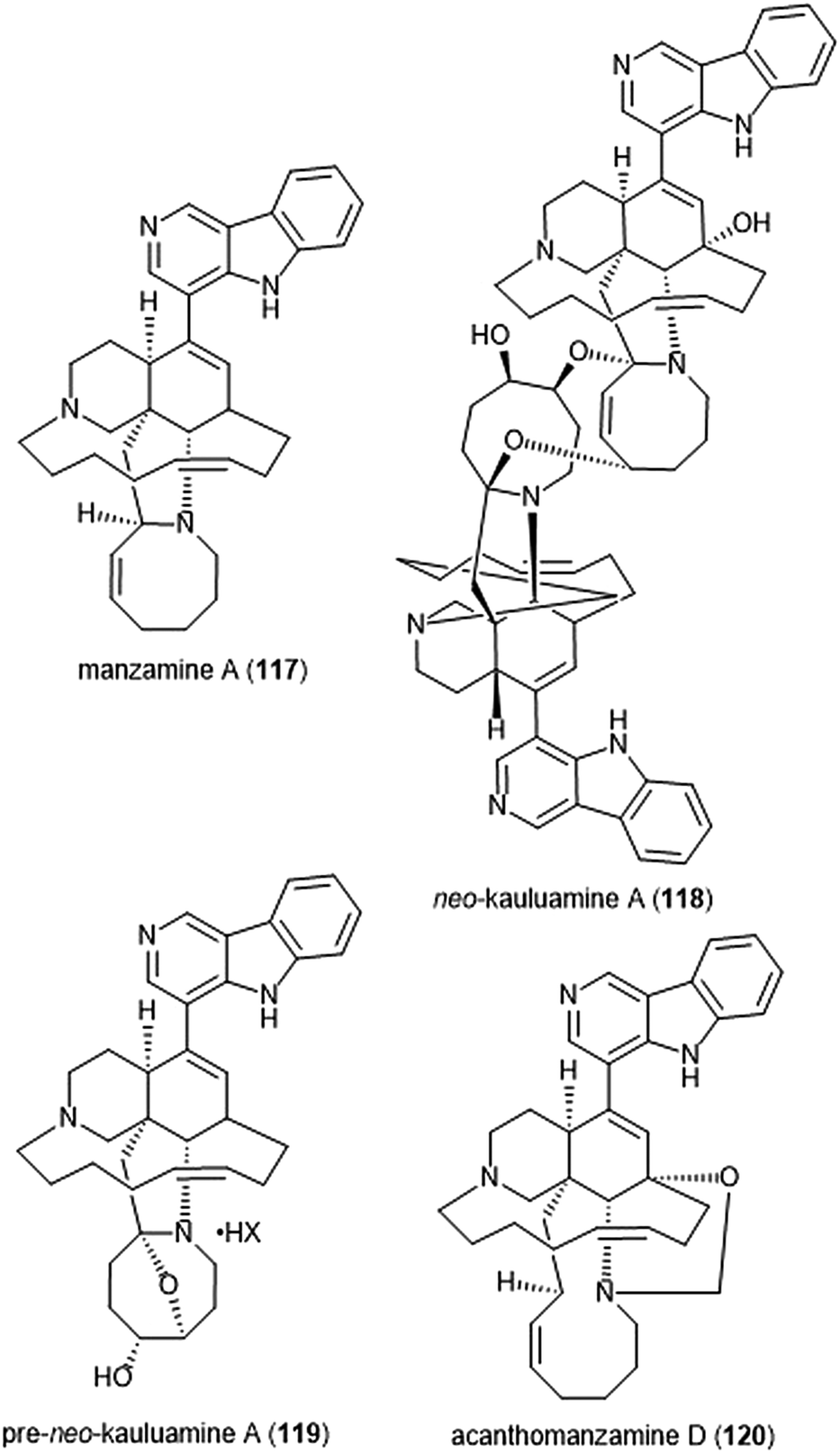 | ||
| Chart 27 Various manzamines have been identified for their ability to inhibit the 20S proteasome. The eight-membered ring has been credited as an integral contributor to inhibition. | ||
The Tsukamoto group also isolated two halicyclamines from Acanthostrongylophora ingens and evaluated them for their biological activity (Chart 28).229 Halicyclamine B (121) and tetradehydrohalicylamine B (122) differ only in their level of unsaturation; whereas the latter contains a pyridinium ring, the former contains a substituted tetrahydropyridine ring. The halicyclamines were tested for cytotoxic activities against HeLa cells which showed that tetradehydrohalicyclamine B exhibited low micromolar cytotoxicity (IC50: 12 μM), whereas halicyclamine B is not cytotoxic. Halicyclamine B and tetradehydrohalicyclamine B demonstrated inhibitory activity towards both the cCP and iCP.
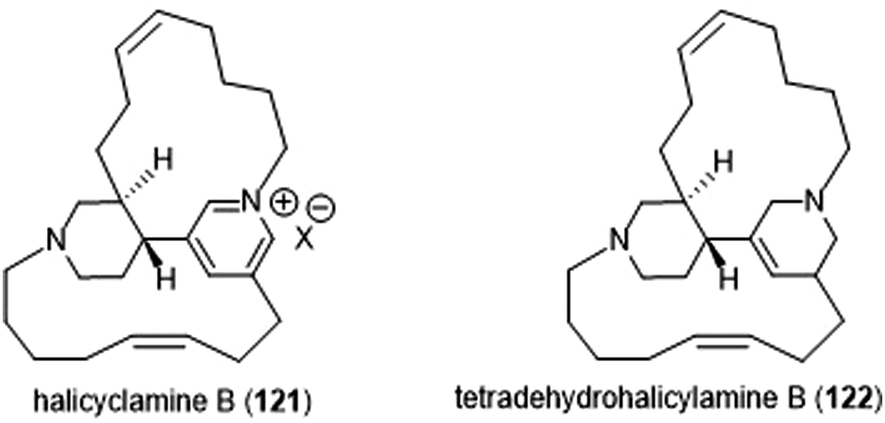 | ||
| Chart 28 Halicylamine B and tetradehydrohalicyclamine B are shown. | ||
Cerpegin (123) and its analogs (Chart 29) have been investigated as novel proteasome inhibitors in recent years. Cerpegin itself exhibits selective micromolar inhibition against the C-L activity of the 20S proteasome, with an IC50 value of 10.4 ± 0.5 μM.230 Optimization of the N5 position led to the identification of selective micromolar (IC50: ∼5 μM) inhibitors of the caspase-like activity of the 20S proteasome. In silico docking suggested an interaction of the N5 substituents with a Tyr residue (Tyr114, β2 subunit) for their selectivity. Introduction of a large, flexible hydrophobic residue at the C1 position led to the discovery of sixteen derivatives with micromolar (IC50: 2–5 μM) activity towards the β1 subunit.231In silico docking indicated that these hydrophobic moieties bind within the primed substrate binding channel of the β1 active site to confer selectivity. Replacement of the carbonyl at C4 with a benzylamino moiety further improved the inhibitory activity of the scaffold to lead to β1-selective nanomolar inhibitor (124) (IC50: 600 nM).232
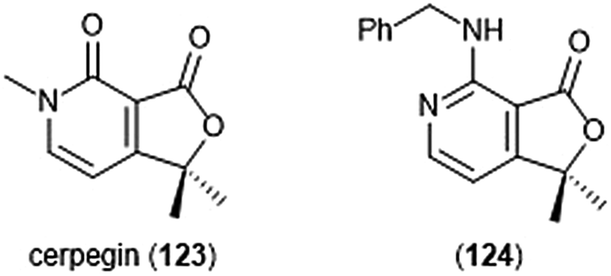 | ||
| Chart 29 The cerpegin scaffold has also been optimized for proteasome inhibition. Depicted are the natural product and its optimized benzylamino analog (124). | ||
Pyrrole–imidazole alkaloids have also been investigated for their ability to inhibit the proteasome (Chart 30). Pyrrole–imidazole alkaloids encompass a large group of heterocyclic natural products isolated from marine sponges.233 Perhaps one of the most famous pyrrole–imidazole alkaloids is palau ‘amine (125). Discovered by Scheuer et al. in 1993,234 researchers reported that palau ‘amine displayed cytotoxic activity. The first total synthesis of (±)-palau’amine was achieved by the Baran group in 2010,235 followed by an enantioselective synthesis of (—)-palau’amine shortly thereafter.236 Due to its reported cytotoxicity, palau’amine and its relatives (±)-dibromophakellin (126) and (±)-dibromophakellstatin (127) were evaluated as inhibitors of the ChT-L activity of the cCP and iCP.237 (—)-Palau’amine displayed 2-fold improved inhibitory activity towards the constitutive human 20S proteasome relative to its racemate (IC50: 2.5 (±0.7) vs. 5.5 (±1.5) μM, respectively), indicating that the (—) enantiomer is responsible for inhibitory activity towards the proteasome. The presence of a cyclic urea or guanidine ring is integral to inhibitory activity. Researchers synthesized several indole analogs238 as potential inhibitors based on the (±)-dibromophakellin scaffold using the same strategy as Hewlett and Tepe in their total synthesis.239 (±)-Indolophakellin analog 129 exhibited potent and specific inhibitory activity towards the β5c of the 20S proteasome (IC50: 3.5 ± 0.7 μM) at a comparable potency to (±)-palau’amine. An X-ray crystal structure of the yCP:129 complex (2.5 Å resolution) revealed that the analogue confers its inhibition through solely non-covalent interactions with the S3 subpocket of the β5 subunit, including H-bonding and halogen bonding interactions. Since this study, newly discovered pyrrole–imidazole alkaloids have frequently been tested for their proteasomal inhibitory activity. For example, 5-bromopalau’amine (128) was recently isolated among several bromopyrrole alkaloids from the Dictyonella sp. marine sponge and exhibited inhibition towards the ChT-L site of the 20S proteasome (IC50: 9.2 ± 3.2 μM).240
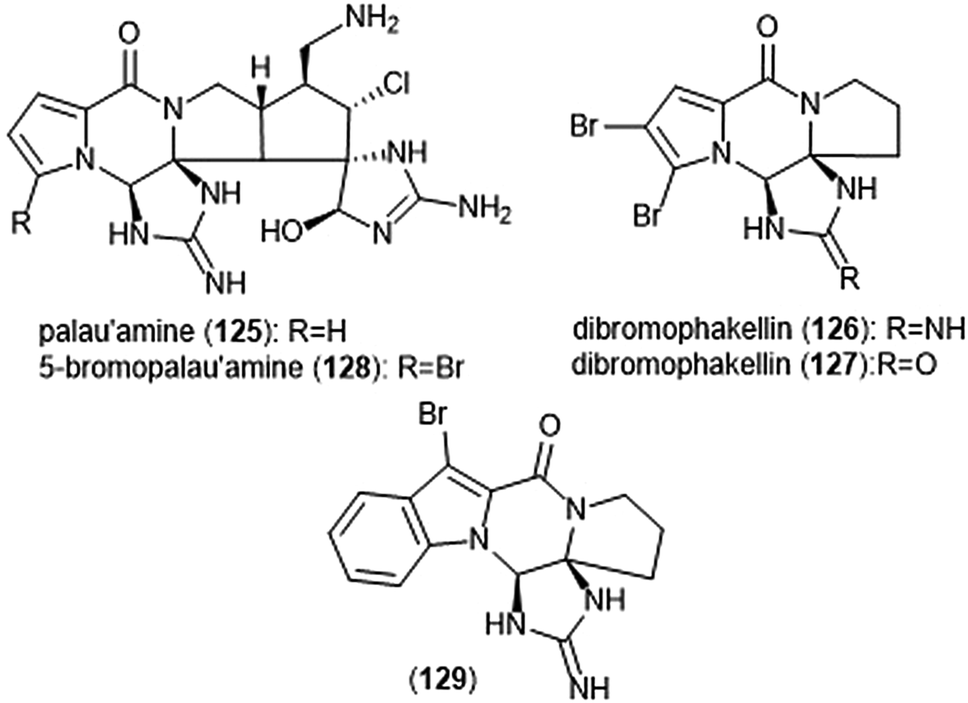 | ||
| Chart 30 Various pyrrole–imidazole alkaloids and analogs have been identified as inhibitors of the 20S proteasome; a selection are displayed. | ||
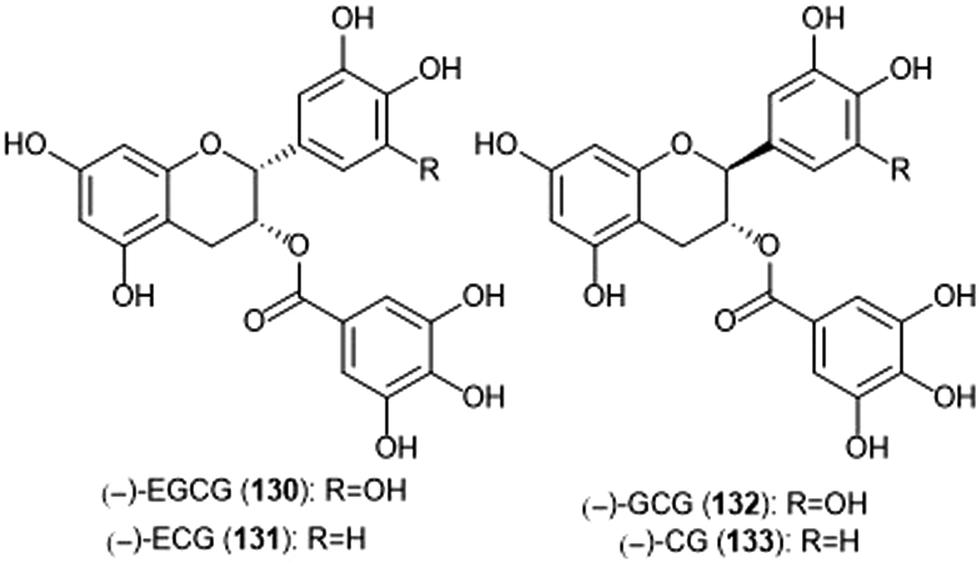 | ||
| Chart 31 Gallate-containing tea polyphenols which inhibit the 20S proteasome are shown. | ||
Flavonoids
Many flavonoids have been isolated and evaluated for their ability to inhibit the 20S proteasome based upon their intriguing bioactivities. The green tea-derived catechin polyphenols have been among these flavonoids of interest, and include epigallocatechin-3-gallate ((—)-EGCG), epicatechin ((—)-EC), epigallocatechin ((—)-EGC), gallocatechin-gallate ((—)-GCG), and catechin gallate. The ester moiety present in of some of these flavonoids was thought to contribute to proteasome inhibition, as the ester-containing catechins (—)-EGCG (130), (—)-ECG (131), (—)-GCG (132) and (—)-CG (133)(Chart 31) inhibit the chymotrypsin-like activity of the 20S proteasome in a range of IC50 values 86–194 nM, (—)-EGCG being the most potent among them.241 Catechins lacking the ester moiety do not display this same inhibitory potency. Subsequent cell-based studies using (—)-EGCG demonstrated the compound's ability to inhibit the 26S proteasome in Jurkat T cells; 10 μM (—)-EGCG inhibited ∼70% of the proteasomal ChT-L activity. EGCG also inhibits the ChT-L activity in breast (MCF-7) and prostate (PC-3 and LNCaP) cancer cells. Enantiomeric analogues of natural catechins, (+)-EGCG and (+)-GCG inhibit the ChT-L activity of the proteasome in both in vitro and in vivo studies to a similar potency of the natural catechins. However, global protection of the hydroxy groups of (+)-EGCG with benzyl moieties renders the compound inactive, implicating that the presence of at least one hydroxyl group is necessary for inhibitory activity.242 The mechanism of proteasome inhibition by catechins was later postulated by Smith et al.243 (—)-EGCG irreversibly inhibits the ChT-L activity of the 20S proteasome in a time-dependent manner, indicative of covalent bond formation within the active site. Docking studies carried out using AutoDock indicated this potential interaction. When docked with the ChT-L site, the ester carbonyl of (—)-EGCG is within 3.18 Å of Thr1Oγ for nucleophilic attack. The hydrophobic A ring of (—)-EGCG also sits within the S1 subpocket of the ChT-L site to confer selectivity between subunits. Due to instability of (—)-EGCG under biological conditions, subsequent studies of the tea polyphenols for proteasome inhibition focused upon improving analogue stability under physiologically-relevant conditions. Several studies by the Dou group have been conducted to generate bioactive analogues of (—)-EGCG: examples of these include peracetate esters244,245 and fluorinated analogues.246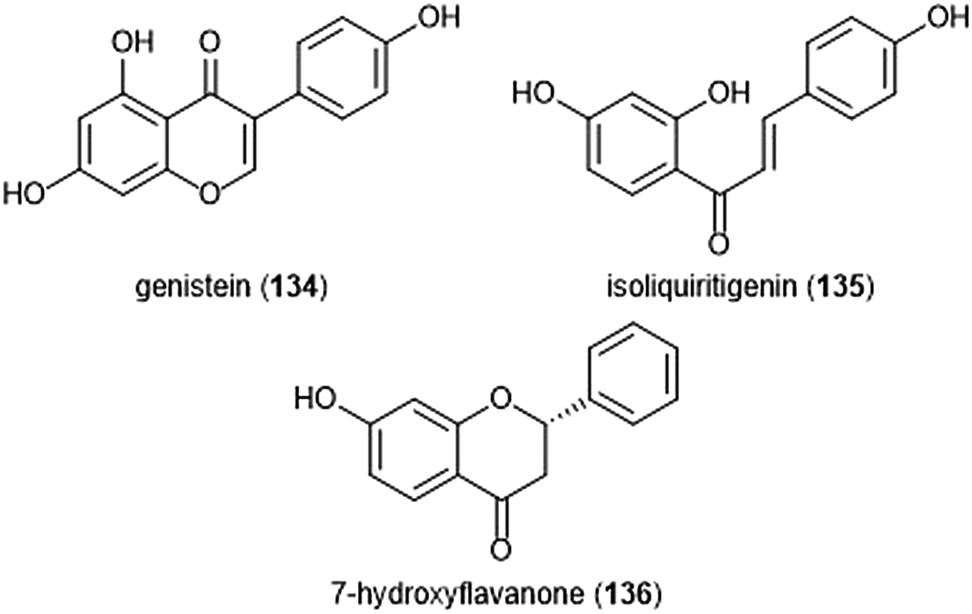 | ||
| Chart 32 The flavonoids genistein, isoliquiritigenin and 7-hydroxyflavanone are shown. | ||
In addition to the tea polyphenols, several other flavonoids have been identified for their ability to inhibit the proteasome (Charts 32–36). Specific features of the flavonoids contribute to the interactions between the compounds and target active sites. The soy isoflavone genistein (134) was first reported in 2003 by Kazi et al. for its inhibitory activity towards the ChT-L site of the 20S proteasome in a cell-free assay with an IC50 value of 26 μM.247 Docking studies suggested that unlike tea polyphenol (—)-EGCG, genistein does not covalently interact with the Thr1 residue of the active site. Genistein was also isolated in addition to proteasome inhibitors isoliquiritigenin (135) and 7-hydroxyflavanone (136) from the plant Spatholobus suberectus.248 The three compounds exhibited low micromolar inhibition towards the β5-site of the 20S proteasome, with IC50 values ranging from 4.88 (±1.55) to 9.26 (±1.2) μM. Apigenin (137), quercetin (138), kaempferol (139) and myricetin (140) were later evaluated by Chen et al. for their ability to inhibit the 20S proteasome.249 All four compounds inhibited the ChT-L site of the 20S proteasome in a cell-free assay, with apigenin displaying the most potent activity (IC50: 1.8 ± 0.03 μM). The compounds displayed similar inhibitory activity towards the 26S proteasome in intact Jurkat cells. Apigenin was still the most potent of the four compounds. In silico docking studies suggested that the carbonyl at C-4 is the site of nucleophilic attack by the Thr1Oγ residue of the active site. Additionally, the hydroxyl group at C-3 was believed to interfere with binding of the flavonoids to the active site. Further studies by the same group evaluated the structure–proteasome–inhibitory activity relationship of flavonoids chrysin (141), luteolin (142), naringenin (143), and eriodictyol (144) in comparison to apigenin.250 The 2,3 double-bond featured within the flavanones chrysin, apigenin and luteolin was deemed necessary for of inhibition towards the ChT-L site of the proteasome. A subsequent study by Wu and Fang reported that the inhibition of chrysin, apigenin and luteolin is selective for ChT-L and T-L catalytic activities of the proteasome within tumor cells.250
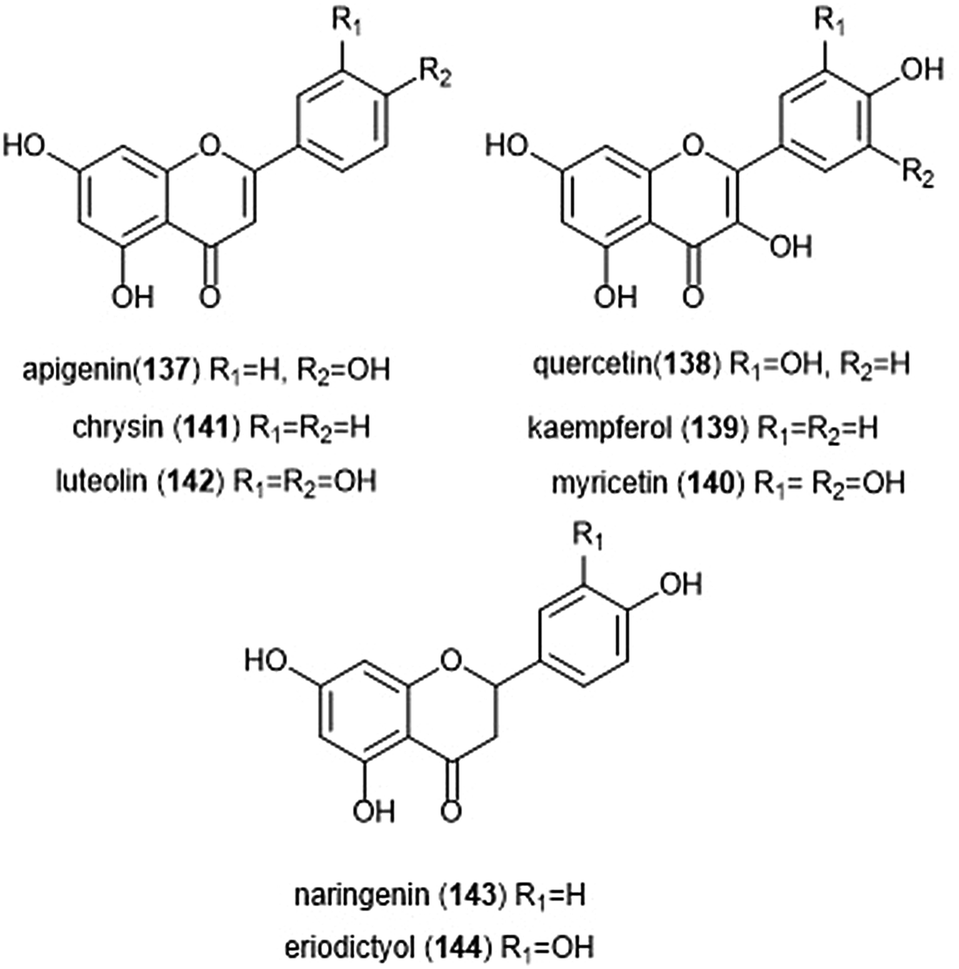 | ||
| Chart 33 Various flavonoids with flavone, flavanol and flavanone substructures inhibit the 20S proteasome, and are depicted. | ||
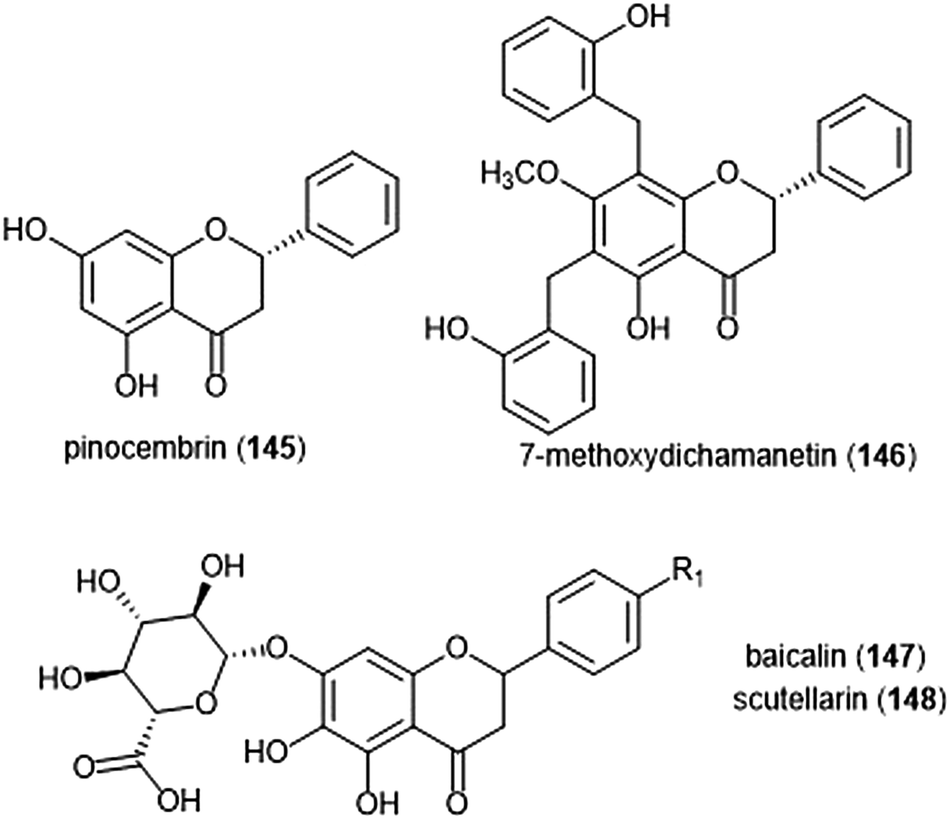 | ||
| Chart 34 Several flavanone-containing proteasome inhibitors have been discovered and are shown. | ||
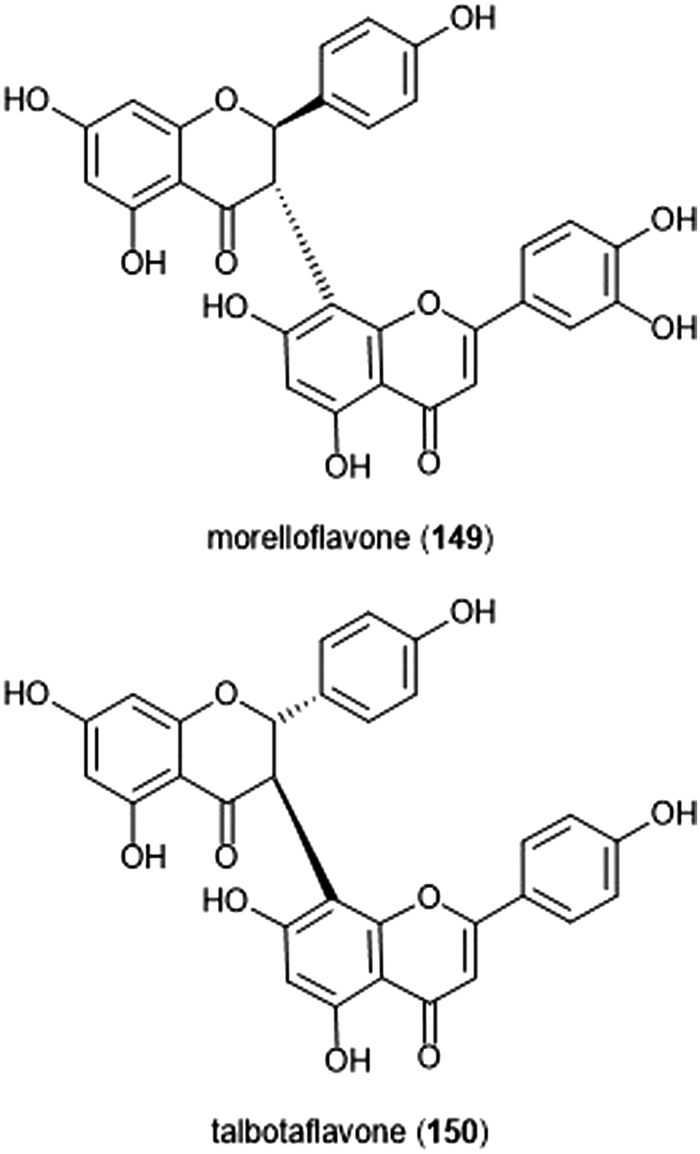 | ||
| Chart 35 The biflavonoid-containing proteasome inhibitors morelloflavone and talbotaflavone are shown. | ||
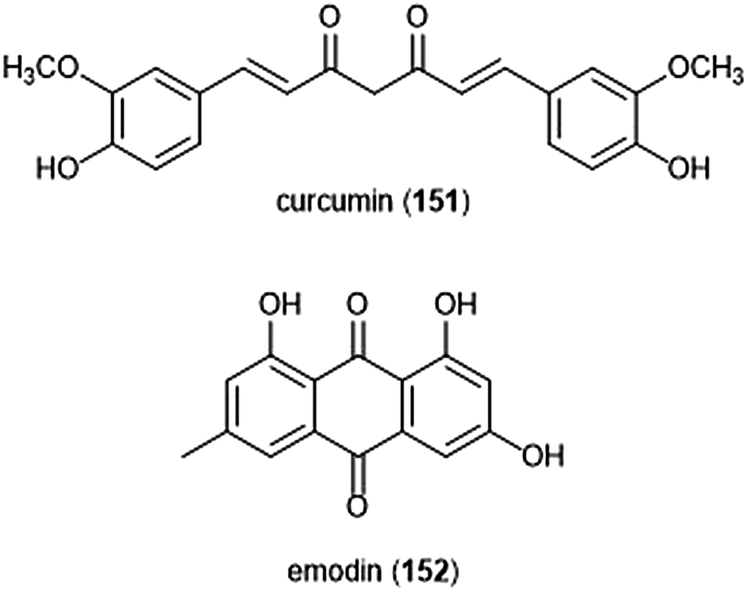 | ||
| Chart 36 The flavonoids curcumin and emodin are shown. | ||
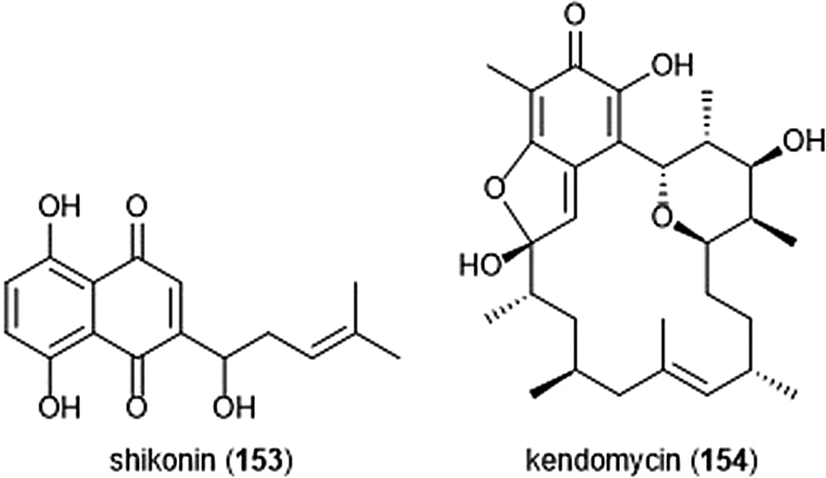 | ||
| Chart 37 The polyketides shikonin and kendomycin have also been identified as proteasome inhibitors. | ||
Pinocembrin (145) and 7-methoxydichamanetin (146) were recently isolated in a bioassay-guided fractionation of the Piper sarmentosum plant by Pan et al.251 These flavanones exhibited inhibitory activities toward the ChT-L site of the human 20S proteasome, with IC50 values of 2.87 ± 0.26 and 3.45 ± 0.18 μM, respectively. The flavonoid glycosides baicalin (147) and scutellarin (148) were isolated from the Chinese herbal medicines Scutellaria baicalensis and Erigeron breviscapus (Vant.) Hand-Mazz by Wu et al. These compounds exhibited the ability to preferentially inhibit ChT-L activity in A549 and HL60 cancer cells.252 The biflavonoids morelloflavone (149) and talbotaflavone (150) were isolated from the stem bark of Garcinia lateriflora by Ren et al., and exhibited inhibition against the ChT-L site of the 20S proteasome, with IC50 values of 1.3 μM and 4.4 μM, respectively.253 The prenylated flavonoid sanggenon C was also reported as an inhibitor of the 20S proteasome.254 Huang et al. indicated that the natural product inhibits the ChT-L activity of the 20S proteasome in enzymatic studies and also in H22 cell lysate in a dose-dependent manner.
The promiscuous active agent curcumin (151) is a symmetric polyphenol which was also reported as a proteasome inhibitor through in vitro and in vivo studies.255,256 Curcumin inhibits the ChT-L activity of the 20S mammalian proteasome with an IC50 value of 1.85 μM, demonstrates the ability to inhibit the 26S proteasome in human colon cancer HCT-116 and SW480 cell lines, and also induces apoptosis. In silico docking studies suggests that the ketone moieties are susceptible to nucleophilic attack by the Thr1Oγ of the chymotrypsin-like site. A recent study by Zhang et al. introduced an α-aminoboronic acid electrophile to the scaffold to improve potency.257 These compounds displayed impressive growth inhibitory activity against HCT-116 cells.
Emodin (152) was recently identified as an inhibitor of the 26S proteasome in the HEK293A-luciferase-cODC cell line. Emodin inhibited luciferase-ODC degradation with an EC50 of 6.33 μM and also exhibited inhibitory activity against the ChT-L and C-L sites of the proteasome, with IC50 values of 1.22 and 0.24 μM, respectively.258In silico docking with the catalytic sites of the proteasome indicated that the carbonyls are susceptible to nucleophilic attack by the Thr1Oγ much like the other carbonyl-containing flavonoids.
Polyketides
Polyketides represent another class of natural products which have been scrutinized for their biological activity (Chart 37). The napthoquinone shikonin (153) was isolated from the traditional Chinese medicine Zi Cao (gromwell) and reported as an inhibitor of ChT-L activity of 20S rabbit proteasome (IC50: 12.5 μM).259 The natural product also demonstrates inhibitory activity towards the 26S proteasome in cell studies (PC-3 and murine hepatoma H22). A later study by Wada et al. complemented these results, demonstrating the ability of shikonin to induce apoptosis in various multiple myeloma cells including bortezomib resistant cells KMS11/BTZ (SHK at 2.5–5 μM).260In silico docking studies suggest that the quinone carbonyls interact with the ChT-L site in such a way that they became highly susceptible to the catalytic site's nucleophilic Thr1 residue.Recently, the cytotoxic macrocycle kendomycin (154) was identified as a weak inhibitor of the proteasome.261 Kendomycin exhibited the ability to weakly inhibit the activity of the ChT-L site of the proteasome (IC50: 67.9 μM). X-ray crystallographic analysis of the yCP: kendomycin complex indicates that it does not interact with the inner chamber of the core particle, but rather covalently attaches to β2-H141Nγ along the outside of the 20S proteasome. Kendomycin sits within the surface-exposed pocket formed by the interface of the β2–β7′ subunits.
Macrolides
Seco-mycalolide A (155) was isolated alongside known mycalolide A (156) and 30-hydroxymycalolide A (157) from a marine sponge of the genus Mycale (Chart 38). The mycalolides had previously been reported for their cytotoxicity towards B-16 melanoma cells. In vitro studies indicated that these compounds inhibit the ChT-L activity of the proteasome, with IC50 values of 11.5, 33.0 and 49.4 μM, respectively.262 These results suggested that the intact macrocycle is not necessary for inhibitory activity, which could allow for the design of simplified analogues in future studies.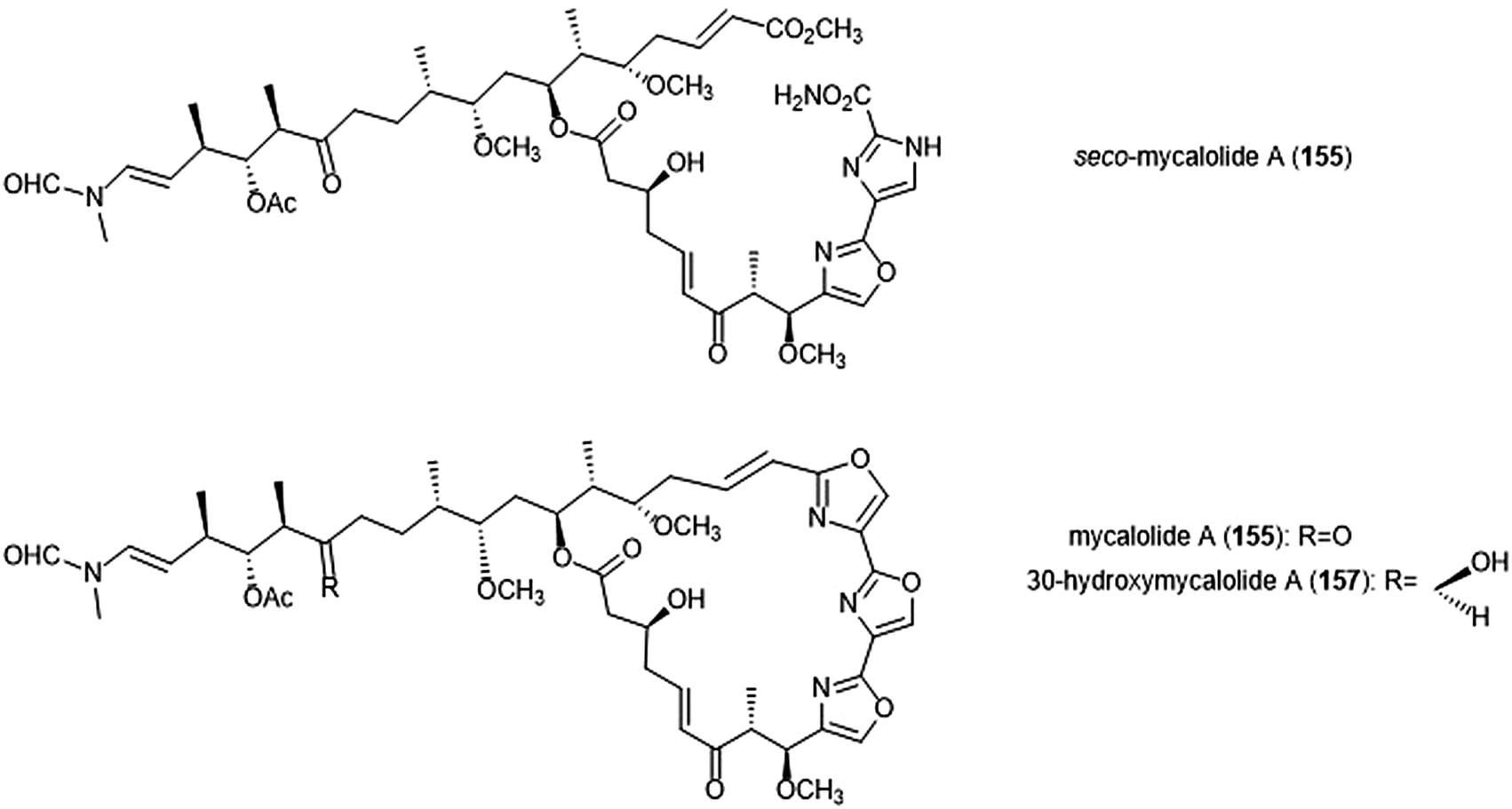 | ||
| Chart 38 Mycalolide B and its natural analogs seco-mycalolide A and 30-hydroxymycalolide A have been identified as inhibitors of the 20S proteasome, and their structures are displayed. | ||
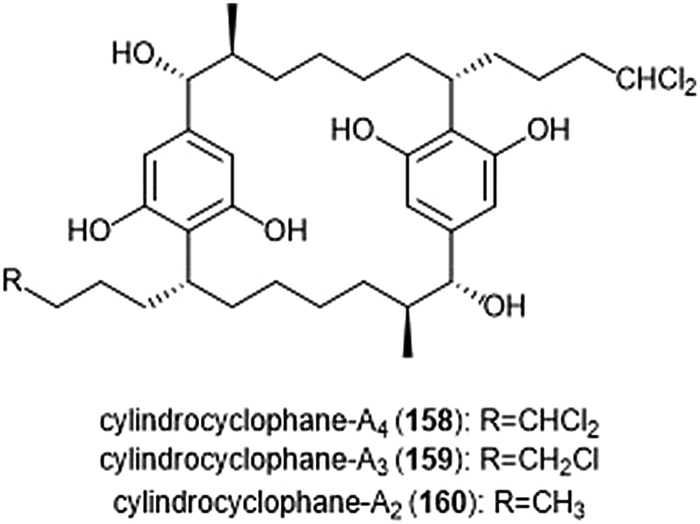 | ||
| Chart 39 The cylindrocyclophanes A2 and A4 are depicted. | ||
Several macrocyclic cylindrocyclophanes were also reported as proteasome inhibitors by Chlipala et al.263 The compounds were isolated from the extract of the terrestrial cyanobacteria Nostoc species (UIC 10022A) which was collected from a Chicago city parkway. The most potent cylindrocyclophanes A4–A2 (158–160) inhibit the ChT-L activity at low micromolar concentrations (IC50: 3.93 ± 0.18, 2.75 ± 0.31, and 2.55 ± 0.11 μM, respectively). Cylindrocyclophanes A4–A2 also display potent cytoxicity against HT-29 cells, with EC50 values of 2.0, 0.5 and 1.7 μM, respectively. Researchers suggested that in addition to a dichloromethyl moiety, the presence of hydroxyl group at C14 was also important for inhibitory activity. However, the inhibitory potency results did not correlate to cytotoxicity results, suggesting that the main reason for cytotoxicity was not due to proteasome inhibition (Chart 39).
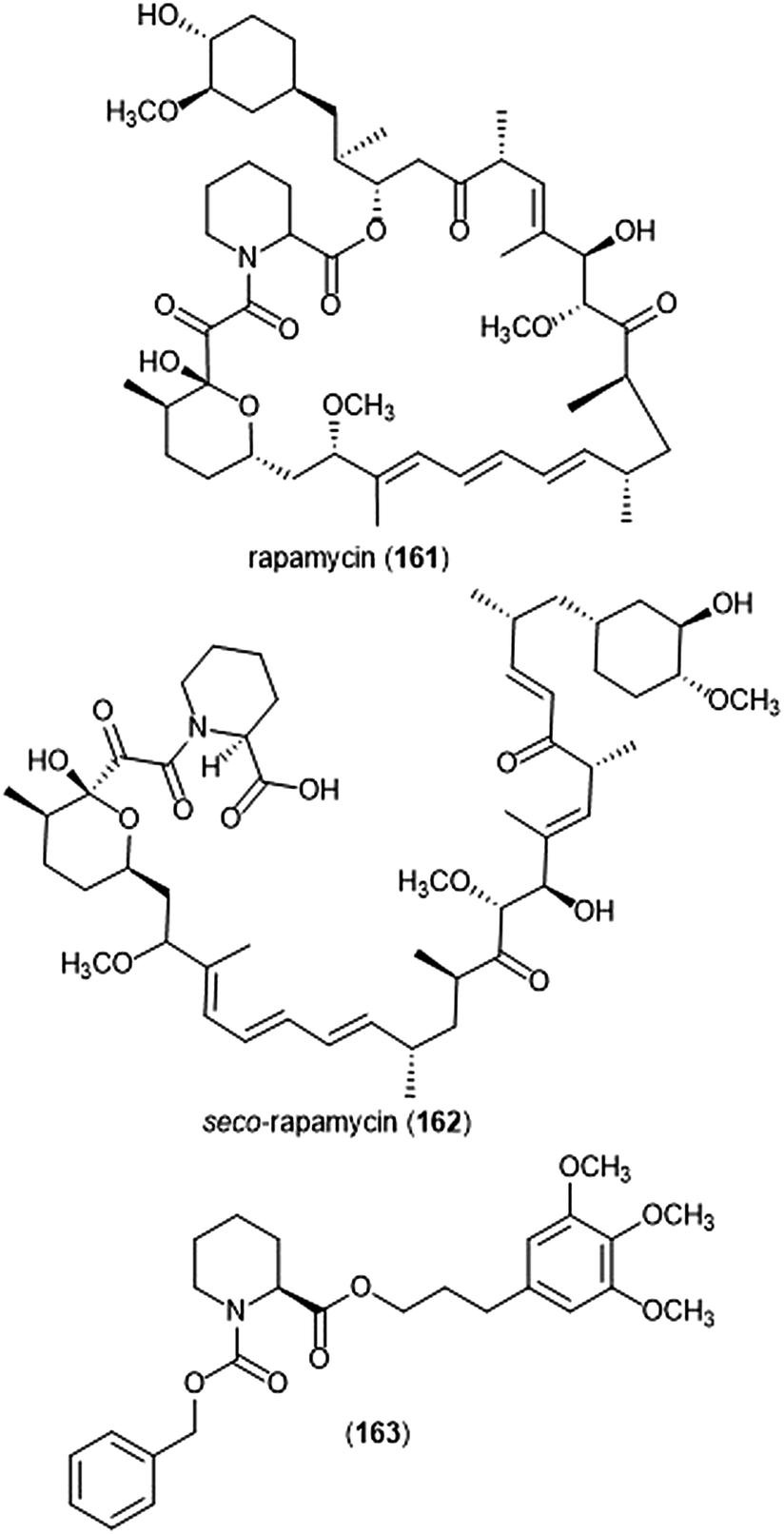 | ||
| Chart 40 Rapamycin and its natural and synthetic analogs are shown. | ||
The macrolide rapamycin (161), its analogues, and its acyclic analog seco-rapamycin (162) have also been identified as allosteric inhibitors of the 20S proteasome by Osmulski and Gaczynksa (Chart 40).264 Rather than bind competitively within the catalytic sites of the 20S proteasome, these compounds bind to specific grooves on the α-ring to confer inhibition. Rapamycin inhibits the ChT-L and T-L activities of the 20S proteasome in a reversible manner, with IC50 values of 1.9 and 0.4 μM, respectively. Identification of a minimal binding pharmacophore led researchers to the discovery of analog 163, a pipecolic ester carbamate which inhibits the ChT-L site of the 20S proteasome (IC50 value of 2.0 μM).265
Conclusions
Modulation of the 20S proteasome is a valuable strategy for the treatment of many diseases. Several natural product classes have been identified as proteasome inhibitors, making them intriguing starting points in the search of drug leads. Not only do these scaffolds provide opportunity for inhibition by allosteric and competitive modes, the complex structures have also facilitated extensive structure–activity relationship studies to better understand their mechanism of interaction with the 20S proteasome. Inherent challenges associated with the use of natural product-based inhibitors include product isolation from crude mixtures and subsequent synthesis of the complex substrates, as exemplified in the case of some natural product-based inhibitors. However, the use of X-ray crystallography and computational docking have been integral in the determination of their mechanisms of inhibition. These methods allow for the strategic synthesis of novel (often simplified) scaffolds, which retain the key components of their parent molecule. Modification of natural product scaffolds by researchers has led to more potent, physiologically relevant, and selective inhibitors as starting points for the treatment of disease. Thus far, the clinical impact of proteasome inhibitors has been significant with several agents currently clinically used to treat multiple myeloma and mantle cell lymphoma.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge financial support for this work from the National Institutes of Health 1R01 AG066223-01A1. Fig. 1B was created with http://BioRender.com.Notes and references
- A. Hershko and A. Ciechanover, Annu. Rev. Biochem., 1998, 67, 425–479 CrossRef.
- A. Ciechanover, J. A. DiGiuseppe, A. L. Schwartz and G. M. Brodeur, Prog. Clin. Biol. Res., 1991, 366, 37–43 Search PubMed.
- Z. Chen, J. Hagler, V. J. Palombella, F. Melandri, D. Scherer, D. Ballard and T. Maniatis, Genes Dev., 1995, 9, 1586–1597 CrossRef.
- J. M. Peters, Z. Cejka, J. R. Harris, J. A. Kleinschmidt and W. Baumeister, J. Mol. Biol., 1993, 234, 932–937 CrossRef.
- J. Lowe, D. Stock, B. Jap, P. Zwickl, W. Baumeister and R. Huber, Science, 1995, 268, 533–539 CrossRef.
- P. C. A. Da Fonseca and E. P. Morris, J. Biol. Chem., 2008, 283, 23305–23314 CrossRef.
- M. Orlowski and S. Wilk, Arch. Biochem. Biophys., 2000, 383, 1–16 CrossRef.
- K. Tanaka, T. Yoshimura, A. Kumatori, A. Ichihara, A. Ikai, M. Nishigai, K. Kameyama and T. Takagi, J. Biol. Chem., 1988, 263, 16209–16217 Search PubMed.
- M. Groll, W. Heinemeyer, S. Jäger, T. Ullrich, M. Bochtler, D. H. Wolf and R. Huber, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 10976–10983 CrossRef.
- M. Groll, M. Bochtler, H. Brandstetter, T. Clausen and R. Huber, ChemBioChem, 2005, 6, 222–256 CrossRef.
- M. Groll, L. Ditzel, J. Löwe, D. Stock, M. Bochtler, H. D. Bartunik and R. Huber, Nature, 1997, 386, 463–471 Search PubMed.
- M. Unno, T. Mizushima, Y. Morimoto, Y. Tomisugi, K. Tanaka, N. Yasuoka and T. Tsukihara, J. Biochem., 2002, 131, 171–173 CrossRef.
- M. Unno, T. Mizushima, Y. Morimoto, Y. Tomisugi, K. Tanaka, N. Yasuoka and T. Tsukihara, Structure, 2002, 10, 609–618 Search PubMed.
- W. D. Harshbarger, C. Miller, C. Diedrich and J. C. Sacchettini, Structure, 2015, 23, 418 CrossRef.
- M. Aki, N. Shimbara, M. Takashina, K. Akiyama, S. Kagawa, T. Tamura, N. Tanahashi, T. Yoshimura, K. Tanaka and A. Ichihara, J. Biochem., 1994, 115, 257–269 CrossRef.
- S. Murata, K. Sasaki, T. Kishimoto, S. I. Niwa, H. Hayashi, Y. Takahama and K. Tanaka, Science, 2007, 316, 1349–1353 CrossRef.
- E. M. Huber, M. Basler, R. Schwab, W. Heinemeyer, C. J. Kirk, M. Groettrup and M. Groll, Cell, 2012, 148, 727–738 CrossRef.
- E. M. Huber and M. Groll, Angew. Chem., Int. Ed., 2012, 51, 8708–8720 CrossRef.
- B. A. Teicher, G. Ara, R. Herbst, V. J. Palombella and J. Adams, Clin. Cancer Res., 1999, 5, 2638–2645 Search PubMed.
- S. Gillessen, M. Groettrup and T. Cerny, Onkologie, 2002, 25, 534–539 Search PubMed.
- S. C. Xie, L. R. Dick, A. Gould, S. Brand and L. Tilley, Expert Opin. Ther. Targets, 2019, 23, 903–914 Search PubMed.
- D. M. Pereira, P. Valentao, G. Correia-da-Silva, N. Teixeira and P. B. Andrade, Nat. Prod. Rep., 2015, 32, 705 RSC.
- R. C. Kane, P. F. Bross, A. T. Farrell and R. Pazdur, Oncologist, 2003, 8, 508–513 CrossRef.
- E. Njomen and J. J. Tepe, J. Med. Chem., 2019, 62, 6469–6481 CrossRef.
- C. L. Jones, E. Njomen, B. Sjogren, T. S. Dexheimer and J. J. Tepe, ACS Chem. Biol., 2017, 15, 2240–2247 CrossRef.
- C. L. Jones and J. J. Tepe, Molecules, 2019, 24, 2841 CrossRef.
- K. L. Rock, C. Gramm, L. Rothstein, K. Clark, R. Stein, L. Dick, D. Hwang and A. L. Goldberg, Cell, 1994, 78, 761–771 CrossRef.
- M. Bogyo, J. S. McMaster, M. Gaczynska, D. Tortorella, A. L. Goldberg and H. Ploegh, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 6629–6634 CrossRef.
- J. F. Lynas, P. Harriott, A. Healy, M. A. McKervey and B. Walker, Bioorg. Med. Chem. Lett., 1998, 8, 373–378 CrossRef.
- M. Groll, C. Berkers, H. Ploegh and H. Ovaa, Structure, 2006, 14, 451 CrossRef.
- M. L. Stein and M. Groll, Biochim. Biophys. Acta, Mol. Cell Res., 2014, 1843, 26–38 CrossRef.
- I. Momose, R. Sekizawa, H. Hashizume, N. Kinoshita, Y. Homma, M. Hamada, H. Iinuma and T. T. Takeuchi, J. Antibiot., 2001, 54, 997–1003 CAS.
- I. Momose, R. Sekizawa, S. Hirosawa, D. Ikeda, H. Naganawa, H. Inuma and T. Takeuchi, J. Antibiot., 2001, 54, 1004–1012 CAS.
- K. Shinohara, M. Tomioka, H. Nakano, S. Toné, H. Ito and S. Kawashima, Biochem. J., 1996, 317, 385–388 CAS.
- I. Momose, Y. Umezawa, S. Hirosawa, M. Iijima, H. Iinuma and D. Ikeda, Biosci., Biotechnol., Biochem., 2005, 69, 1733–1742 CrossRef CAS.
- I. Momose, Y. Umezawa, S. Hirosawa, H. Iinuma and D. Ikeda, Bioorg. Med. Chem. Lett., 2005, 15, 1867–1871 CrossRef CAS.
- I. Momose, M. Iijima, M. Kawada and D. Ikeda, Biosci., Biotechnol., Biochem., 2007, 71, 1036–1043 CAS.
- T. Watanabe, I. Momose, M. Abe, H. Abe, R. Sawa, Y. Umezawa, D. Ikeda, Y. Takahashi and Y. Akamatsu, Bioorg. Med. Chem. Lett., 2009, 19, 2343–2345 CAS.
- T. Watanabe, H. Abe, I. Momose, Y. Takahashi, D. Ikeda and Y. Akamatsu, Bioorg. Med. Chem. Lett., 2010, 20, 5839–5842 CAS.
- I. Momose, H. Abe, T. Watanabe, S. i. Ohba, K. Yamazaki, S. Dan, T. Yamori, T. Masuda and A. Nomoto, Cancer Sci., 2014, 105, 1609–1615 CrossRef CAS.
- H. Shigemori, S. Wakuri, K. Yazawa, T. Nakamura, T. Sasaki and J. i. Kobayashi, Tetrahedron, 1991, 47, 8529–8534 CAS.
- J. S. Schneekloth, J. L. Sanders, J. Hines and C. M. Crews, Bioorg. Med. Chem. Lett., 2006, 16, 3855–3858 CAS.
- A. M. Giltrap, K. M. Cergol, A. Pang, W. J. Britton and R. J. Payne, Mar. Drugs, 2013, 11, 2382–2397 Search PubMed.
- J. S. Yadav, S. S. Dachavaram, R. Grée and S. Das, Tetrahedron Lett., 2015, 56, 3999–4001 CrossRef CAS.
- M. C. Pirrung, F. Zhang, S. Ambadi and Y. Gangadhara Rao, Org. Biomol. Chem., 2016, 14, 8367–8375 RSC.
- J. Hines, M. Groll, M. Fahnestock and C. M. Crews, Chem. Biol., 2008, 15, 501–512 CAS.
- Y. Chen, R. A. McClure, Y. Zheng, R. J. Thomson and N. L. Kelleher, J. Am. Chem. Soc., 2013, 135, 10449–10456 CAS.
- M. Oka, Y. Nishiyama, S. Ohta, H. Kamei, M. Konishi, T. Miyaki, T. Oki and H. Kawaguchi, J. Antibiot., 1988, 41, 1331–1337 CAS.
- M. Oka, H. Ohkuma, H. Kamei, M. Konishi, T. Oki and H. Kawaguchi, J. Antibiot., 1988, 41, 1906–1909 CrossRef CAS.
- M. Oka, K. Yaginuma, K. Numata, M. Konishi, T. Oki and H. Kawaguchi, J. Antibiot., 1988, 41, 1338–1350 CrossRef CAS.
- M. Oka, K. i. Numata, Y. Nishiyama, H. Kamei, M. Konishi, T. Oki and H. Kawaguchi, J. Antibiot., 1988, 41, 1812–1822 CrossRef CAS.
- M. Groll, B. Schellenberg, A. S. Bachmann, C. R. Archer, R. Huber, T. K. Powell, S. Lindow, M. Kaiser and R. Dudler, Nature, 2008, 452, 755–758 CAS.
- A. Pawar, M. Basler, H. Goebel, G. O. Alvarez Salinas, M. Groettrup and T. Böttcher, ACS Cent. Sci., 2020, 6, 241–246 CAS.
- J. i. Shoji, H. Hinoo, T. Kato, T. Hattori, K. Hirooka, K. Tawara, O. Shiratori and Y. Terui, J. Antibiot., 1990, XLIII, 783–787 Search PubMed.
- Y. Terui, J. Nishikawa, H. Hinoo, T. Kato and J. i. Shoji, J. Antibiot., 1990, 43, 788–795 CAS.
- M. L. Stein, P. Beck, M. Kaiser, R. Dudler, C. F. W. Becker and M. Groll, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 18367–18371 CAS.
- J. Fu, X. Bian, S. Hu, H. Wang, F. Huang, P. M. Seibert, A. Plaza, L. Xia, R. Müller, A. F. Stewart and Y. Zhang, Nat. Biotechnol., 2012, 30, 440–446 CrossRef CAS.
- C. M. Theodore, J. B. King, J. You and R. H. Cichewicz, J. Nat. Prod., 2012, 75, 2007–2011 CrossRef CAS.
- X. Bian, A. Plaza, Y. Zhang and R. Müller, J. Nat. Prod., 2012, 75, 1652–1655 CAS.
- X. Bian, F. Huang, H. Wang, T. Klefisch, R. Müller and Y. Zhang, ChemBioChem, 2014, 15, 2221–2224 CrossRef CAS.
- P. Servatius, T. Stach and U. Kazmaier, Eur. J. Org. Chem., 2019, 3163–3168 CrossRef CAS.
- U. Wäspi, D. Blanc, T. Winkler, P. Rüedi and R. Dudler, Mol. Plant-Microbe Interact., 1998, 11, 727–733 CrossRef.
- U. Waspi, P. Hassa, A. A. Staempfli, L. P. Molleyres, T. Winkler and R. Dudler, Microbiol. Res., 1999, 154, 89–93 CrossRef CAS.
- C. Dai and C. R. J. Stephenson, Org. Lett., 2010, 12, 3453–3455 CrossRef CAS.
- M. C. Pirrung, G. Biswas and T. R. Ibarra-Rivera, Org. Lett., 2010, 12, 2402–2405 CrossRef CAS.
- T. Chiba, H. Hosono, K. Nakagawa, M. Asaka, H. Takeda, A. Matsuda and S. Ichikawa, Angew. Chem., Int. Ed., 2014, 53, 4836–4839 CrossRef CAS.
- J. Clerc, M. Groll, D. J. Illich, A. S. Bachmann, R. Huber, B. Schellenberg, R. Dudler and M. Kaiser, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6507–6512 CrossRef CAS.
- C. R. Archer, D. L. T. Koomoa, E. M. Mitsunaga, J. Clerc, M. Shimizu, M. Kaiser, B. Schellenberg, R. Dudler and A. S. Bachmann, Biochem. Pharmacol., 2010, 80, 170–178 CrossRef CAS.
- A. S. Bachmann, J. Opoku-Ansah, T. R. Ibarra-Rivera, L. P. Yco, S. Ambadi, C. C. Roberts, C. A. Chang and M. C. Pirrung, J. Biol. Chem., 2016, 291, 8350–8362 CrossRef CAS.
- M. R. Pierce, R. M. Robinson, T. R. Ibarra-Rivera, M. C. Pirrung, N. G. Dolloff and A. S. Bachmann, Leuk. Res., 2020, 88, 106271 CrossRef CAS.
- J. Clerc, N. Li, D. Krahn, M. Groll, A. S. Bachmann, B. I. Florea, H. S. Overkleeft and M. Kaiser, Chem. Commun., 2011, 47, 385–387 RSC.
- C. R. Archer, M. Groll, M. L. Stein, B. Schellenberg, J. Clerc, M. Kaiser, T. P. Kondratyuk, J. M. Pezzuto, R. Dudler and A. S. Bachmann, Biochemistry, 2012, 51, 6880–6888 CrossRef CAS.
- T. Chiba, A. Matsuda and S. Ichikawa, Bioorg. Med. Chem. Lett., 2015, 25, 4872–4877 CrossRef CAS.
- T. Chiba, S. Kitahata, A. Matsuda and S. Ichikawa, Chem. Pharm. Bull., 2016, 64, 811–816 CrossRef CAS.
- S. Kitahata, T. Chiba, T. Yoshida, M. Ri, S. Iida, A. Matsuda and S. Ichikawa, Org. Lett., 2016, 18, 2312–2315 CrossRef CAS.
- T. R. Ibarra-Rivera, J. Opoku-Ansah, S. Ambadi, A. S. Bachmann and M. C. Pirrung, Tetrahedron, 2011, 67, 9950–9956 CrossRef CAS.
- J. Opoku-Ansah, T. R. Ibarra-Rivera, M. C. Pirrung and A. S. Bachmann, Pharm. Biol., 2012, 50, 25–29 CrossRef CAS.
- K. A. Totaro, D. Barthelme, P. T. Simpson, R. T. Sauer and J. K. Sello, Bioorg. Med. Chem., 2015, 23, 6218–6222 CrossRef CAS.
- N. A. Bakas, C. R. Schultz, L. P. Yco, C. C. Roberts, C. e. A. Chang, A. S. Bachmann and M. C. Pirrung, Bioorg. Med. Chem., 2018, 26, 401–412 CrossRef CAS.
- Y. Koguchi, J. Kohno, M. Nishio, K. Takahashi, T. Okuda, T. Ohnuki and S. Komatsubara, J. Antibiot., 2000, 53, 105–109 CrossRef CAS.
- J. Kohno, Y. Koguchi, M. Nishio, K. Nakao, M. Kuroda, R. Shimizu, T. Ohnuki and S. Komatsubara, J. Org. Chem., 2000, 65, 990–995 CrossRef CAS.
- M. Groll, Y. Koguchi, R. Huber and J. Kohno, J. Mol. Biol., 2001, 311, 543–548 CrossRef CAS.
- S. Lin and S. J. Danishefsky, Angew. Chem., Int. Ed., 2002, 41, 512–515 CrossRef CAS.
- B. K. Albrecht and R. M. Williams, Org. Lett., 2003, 5, 197–200 CrossRef CAS.
- M. Inoue, H. Sakazaki, H. Furuyama and M. Hirama, Angew. Chem., Int. Ed., 2003, 42, 2654–2657 CrossRef CAS.
- S. Lin, Z. Q. Yang, B. H. B. Kwok, M. Koldobskiy, C. M. Crews and S. J. Danishefsky, J. Am. Chem. Soc., 2004, 126, 6347–6355 CrossRef CAS.
- M. Kaiser, M. Groll, C. Renner, R. Huber and L. Moroder, Angew. Chem., Int. Ed., 2002, 41, 780–783 CrossRef CAS.
- M. Kaiser, M. Groll, C. Siciliano, I. Assfalg-Machleidt, E. Weyher, J. Kohno, A. G. Milbradt, C. Renner, R. Huber and L. Moroder, ChemBioChem, 2004, 5, 1256–1266 CrossRef CAS.
- M. Kaiser, C. Siciliano, I. Assfalg-Machleidt, M. Groll, A. G. Milbradt and L. Moroder, Org. Lett., 2003, 5, 3435–3437 CrossRef CAS.
- Z. Q. Yang, B. H. B. Kwok, S. Lin, M. A. Koldobskiy, C. M. Crews and S. J. Danishefsky, ChemBioChem, 2003, 4, 508–513 CrossRef CAS.
- M. Kaiser, A. G. Milbradt, C. Siciliano, I. Assfalg-Machleidt, W. Machleidt, M. Groll, C. Renner and L. Moroder, Chem. Biodiversity, 2004, 1, 161–173 CrossRef CAS.
- M. Groll, M. Götz, M. Kaiser, E. Weyher and L. Moroder, Chem. Biol., 2006, 13, 607–614 CrossRef CAS.
- D. L. Wilson, I. Meininger, Z. Strater, S. Steiner, F. Tomlin, J. Wu, H. Jamali, D. Krappmann and M. G. Götz, ACS Med. Chem. Lett., 2016, 7, 250–255 CrossRef CAS.
- A. Berthelot, S. Piguel, G. Le Dour and J. Vidal, J. Org. Chem., 2003, 68, 9835–9838 CrossRef CAS.
- N. Basse, S. Piguel, D. Papapostolou, A. Ferrier-Berthelot, N. Richy, M. Pagano, P. Sarthou, J. Sobczak-Thépot, M. Reboud-Ravaux and J. Vidal, J. Med. Chem., 2007, 50, 2842–2850 CrossRef CAS.
- M. Groll, N. Gallastegui, X. Maréchal, V. Le Ravalec, N. Basse, N. Richy, E. Genin, R. Huber, L. Moroder, J. Vidal and M. Reboud-Ravaux, ChemMedChem, 2010, 5, 1701–1705 CrossRef CAS.
- A. Desvergne, Y. Cheng, S. Grosay-Gaudrel, X. Maréchal, M. Reboud-Ravaux, E. Genin and J. Vidal, J. Med. Chem., 2014, 57, 9211–9217 CrossRef CAS.
- K. Xu, K. Wang, Y. Yang, D. A. Yan, L. Huang, C. H. Chen and Z. Xiao, Eur. J. Med. Chem., 2015, 98, 61–68 CrossRef CAS.
- N. Richy, D. Sarraf, X. Maréchal, N. Janmamode, R. Le Guével, E. Genin, M. Reboud-Ravaux and J. Vidal, Eur. J. Med. Chem., 2018, 145, 570–587 CrossRef CAS.
- X. Maréchal, A. Pujol, N. Richy, E. Genin, N. Basse, M. Reboud-Ravaux and J. Vidal, Eur. J. Med. Chem., 2012, 52, 322–327 CrossRef.
- A. Desvergne, E. Genin, X. Maréchal, N. Gallastegui, L. Dufau, N. Richy, M. Groll, J. Vidal and M. Reboud-Ravaux, J. Med. Chem., 2013, 56, 3367–3378 CrossRef CAS.
- F. Sasse, H. Steinmetz, T. Schupp, F. Petersen, K. Memmert, H. Hofmann, C. Heusser, V. Brinkmann, P. von Matt, G. Höfle and H. Reichenbach, J. Antibiot., 2002, 55, 543–551 CrossRef CAS.
- L. Vollbrecht, H. Steinmetz and G. Höfle, J. Antibiot., 2002, 55, 715–721 CrossRef CAS.
- S. V. Ley, A. Prieur and C. Heusser, Org. Lett., 2002, 4, 711–714 CrossRef CAS.
- W. Wu, Z. Li, G. Zhou and S. Jiang, Tetrahedron Lett., 2011, 52, 2488–2491 CrossRef CAS.
- D. Pogorevc, Y. Tang, M. Hoffmann, G. Zipf, H. S. Bernauer, A. Popoff, H. Steinmetz and S. C. Wenzel, ACS Synth. Biol., 2019, 8, 1121–1133 CrossRef CAS.
- I. Nickeleit, S. Zender, F. Sasse, R. Geffers, G. Brandes, I. Sörensen, H. Steinmetz, S. Kubicka, T. Carlomagno, D. Menche, I. Gütgemann, J. Buer, A. Gossler, M. P. Manns, M. Kalesse, R. Frank and N. P. Malek, Cancer Cell, 2008, 14, 23–35 CrossRef CAS.
- L. Bülow, I. Nickeleit, A. K. Girbig, T. Brodmann, A. Rentsch, U. Eggert, F. Sasse, H. Steinmetz, R. Frank, T. Carlomagno, N. P. Malek and M. Kalesse, ChemMedChem, 2010, 5, 832–836 CrossRef.
- B. Stauch, B. Simon, T. Basile, G. Schneider, N. P. Malek, M. Kalesse and T. Carlomagno, Angew. Chem., Int. Ed., 2010, 49, 3934–3938 CrossRef CAS.
- E. Z. Loizidou and C. D. Zeinalipour-Yazdi, Chem. Biol. Drug Des., 2014, 84, 99–107 CrossRef CAS.
- D. J. Allardyce, C. M. Bell and E. Z. Loizidou, Chem. Biol. Drug Des., 2019, 94, 1556–1567 CAS.
- U. G. Bhat, P. A. Zipfel, D. S. Tyler and A. L. Gartel, Cell Cycle, 2008, 7, 1851–1855 CrossRef CAS.
- U. G. Bhat, M. Halasi and A. L. Gartel, PLoS One, 2009, 4, 1–5 CrossRef.
- B. Pandit, U. Bhat and A. L. Gartel, Cancer Biol. Ther., 2011, 11, 43–47 CrossRef CAS.
- S. Schoof, G. Pradel, M. N. Aminake, B. Ellinger, S. Baumann, M. Potowski, Y. Najajreh, M. Kirschner and H. D. Arndt, Angew. Chem., Int. Ed., 2010, 49, 3317–3321 CrossRef CAS.
- F. Zhang, C. Li and W. L. Kelly, ACS Chem. Biol., 2016, 11, 415–424 CrossRef CAS.
- R. Sekizawa, I. Momose, N. Kinoshita, H. Naganawa, M. Hamada, Y. Muraoka, H. Iinuma and T. Takeuchi, J. Antibiot., 2001, 54, 874–881 CrossRef CAS.
- A. Krunic, A. Vallat, S. Mo, D. D. Lantvit, S. M. Swanson and J. Orjala, J. Nat. Prod., 2010, 73, 1927–1932 CrossRef CAS.
- J. Niggemann, P. Bozko, N. Bruns, A. Wodtke, M. T. Gieseler, K. Thomas, C. Jahns, M. Nimtz, I. Reupke, T. Brüser, G. Auling, N. Malek and M. Kalesse, ChemBioChem, 2014, 15, 1021–1029 CrossRef CAS.
- S. Ōmura, K. Matsuzaki, T. Fujimoto, K. Kosuge, T. Furuya, S. Fujita and A. Nakagawa, J. Antibiot., 1991, 44, 117–118 CrossRef.
- S. Ōmura, T. Fujimoto, K. Otoguro, K. Matsuzaki, R. Moriguchi, H. Tanaka and Y. Sasaki, J. Antibiot., 1991, 44, 113–116 CrossRef.
- G. Fenteany, R. F. Standaert, G. A. Reichard, E. J. Corey and S. L. Schreiber, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 3358–3362 CrossRef CAS.
- G. Fenteany, R. F. Standaert, W. S. Lane, S. Choi, E. J. Corey and S. L. Schreiber, Science, 1995, 268, 726–731 CrossRef CAS.
- A. Craiu, M. Gaczynska, T. Akopian, C. F. Gramm, G. Fenteany, A. L. Goldberg and K. L. Rock, J. Biol. Chem., 1997, 272, 13437–13445 CrossRef CAS.
- L. R. Dick, A. A. Cruikshank, A. T. Destree, L. Grenier, T. A. McCormack, F. D. Melandri, S. L. Nunes, V. J. Palombella, L. A. Parent, L. Plamondon and R. L. Stein, J. Biol. Chem., 1997, 272, 182–188 CrossRef CAS.
- E. J. Corey and G. A. Reichard, J. Am. Chem. Soc., 1992, 114, 10677–10678 CrossRef CAS.
- E. J. Corey, W. Li and G. A. Reichard, J. Am. Chem. Soc., 1998, 120, 2330–2336 CrossRef CAS.
- E. J. Corey and W.-D. Z. Li, Chem. Pharm. Bull., 1999, 47, 1–10 CrossRef CAS.
- E. J. Corey, W. D. Z. Li, T. Nagamitsu and G. Fenteany, Tetrahedron, 1999, 55, 3305–3316 CrossRef CAS.
- F. Soucy, L. Grenier, M. L. Behnke, A. T. Destree, T. A. McCormack, J. Adams and L. Plamondon, J. Am. Chem. Soc., 1999, 121, 9967–9976 CrossRef CAS.
- R. H. Feling, G. O. Buchanan, T. J. Mincer, C. A. Kauffman, P. R. Jensen, W. Fenical and D. John, Angew. Chem., Int. Ed., 2003, 42, 355–357 CrossRef CAS.
- D. Chauhan, L. Catley, G. Li, K. Podar, T. Hideshima, M. Velankar, C. Mitsiades, N. Mitsiades, H. Yasui, A. Letai, H. Ovaa, C. Berkers, B. Nicholson, T. H. Chao, S. T. C. Neuteboom, P. Richardson, M. A. Palladino and K. C. Anderson, Cancer Cell, 2005, 8, 407–419 CrossRef CAS.
- L. R. Reddy, P. Saravanan and E. J. Corey, J. Am. Chem. Soc., 2004, 126, 6230–6231 CrossRef CAS.
- L. R. Reddy, J. F. Fournier, B. V. Subba Reddy and E. J. Corey, Org. Lett., 2005, 7, 2699–2701 CrossRef CAS.
- A. Endo and S. J. Danishefsky, J. Am. Chem. Soc., 2005, 127, 8298–8299 CrossRef CAS.
- N. P. Mulholland, G. Pattenden and I. A. S. Walters, Org. Biomol. Chem., 2006, 4, 2845–2846 RSC.
- Gil Ma, H. Nguyen and D. Romo, Org. Lett., 2007, 9, 2143–2146 CrossRef CAS.
- T. Ling, V. R. Macherla, R. R. Manam, K. A. McArthur and B. C. Potts, Org. Lett., 2007, 9, 2289–2292 CrossRef CAS.
- N. P. Mulholland, G. Pattenden and I. A. S. Walters, Org. Biomol. Chem., 2008, 6, 2782–2789 RSC.
- T. Fukuda, K. Sugiyama, S. Arima, Y. Harigaya, N. Tohru and S. Omura, Org. Lett., 2008, 10, 4239–4242 CAS.
- K. Takahashi, M. Midori, K. Kawano, J. Ishihara and S. Hatakeyame, Angew. Chem., Int. Ed., 2008, 47, 6244–6246 CrossRef CAS.
- R. A. Mosey and J. J. Tepe, Tetrahedron Lett., 2009, 50, 295–297 CrossRef CAS.
- H. Nguyen, G. Ma and D. Romo, Chem. Commun., 2010, 46, 4803–4805 RSC.
- Y. Kaiya, J.-i. Hasegawa, T. Momose, T. Sata and N. Chida, Chem. – Asian J., 2010, 6, 209–219 CrossRef.
- H. Nguyen, G. Ma, T. Gladysheva, T. Fremgen and D. Romo, J. Org. Chem., 2011, 76, 2–12 CrossRef CAS.
- N. Satoh, S. Yokoshima and T. Fukuyama, Org. Lett., 2011, 13, 3028–3031 CrossRef CAS.
- L. B. Marx and J. W. Burton, Chem. – Eur. J., 2018, 24, 6747–6754 CrossRef CAS.
- H. Gholami, A. Kulshrestha, O. K. Favor, R. J. Staples and B. Borhan, Angew. Chem., Int. Ed., 2019, 58, 10110–10113 CrossRef CAS.
- V. R. Macherla, S. S. Mitchell, R. R. Manam, K. A. Reed, T. H. Chao, B. Nicholson, G. Deyanat-Yazdi, B. Mai, P. R. Jensen, W. F. Fenical, S. T. C. Neuteboom, K. S. Lam, M. A. Palladino and B. C. M. Potts, J. Med. Chem., 2005, 48, 3684–3687 CrossRef CAS.
- M. Groll, R. Huber and B. C. M. Potts, J. Am. Chem. Soc., 2006, 128, 5136–5141 CrossRef CAS.
- R. R. Manam, K. A. McArthur, T. H. Chao, J. Weiss, J. A. Ali, V. J. Palombella, M. Groll, G. K. Lloyd, M. A. Palladino, S. T. C. Neuteboom, V. R. Macherla and B. C. M. Potts, J. Med. Chem., 2008, 51, 6711–6724 CrossRef CAS.
- A. S. Eustáquio and B. S. Moore, Angew. Chem., Int. Ed., 2008, 47, 3936–3938 CrossRef.
- L. R. Reddy, J. F. Fournier, B. V. S. Reddy and E. J. Corey, J. Am. Chem. Soc., 2005, 127, 8974–8976 CrossRef CAS.
- R. R. Manam, V. R. Macherla, G. Tsueng, C. W. Dring, J. Weiss, S. T. C. Neuteboom, K. S. Lam and B. C. Potts, J. Nat. Prod., 2009, 72, 295–297 CrossRef CAS.
- R. P. McGlinchey, M. Nett, A. S. Eustáquio, R. N. Asolkar, W. Fenical and B. S. Moore, J. Am. Chem. Soc., 2008, 130, 7822–7823 CrossRef CAS.
- M. Nett, T. A. M. Gulder, A. J. Kale, C. C. Hughes and B. S. Moore, J. Med. Chem., 2009, 52, 6163–6167 CrossRef CAS.
- M. Groll, H. Nguyen, S. Vellalath and D. Romo, Mar. Drugs, 2018, 16, 1–9 Search PubMed.
- M. Stadler, J. Bitzer, A. Mayer-Bartschmid, H. Müller, J. Benet-Buchholz, F. Gantner, H. V. Tichy, P. Reinemer and K. B. Bacon, J. Nat. Prod., 2007, 70, 246–252 CAS.
- S. Rachid, L. Huo, J. Herrmann, M. Stadler, B. Köpcke, J. Bitzer and R. Müller, ChemBioChem, 2011, 12, 922–931 CrossRef CAS.
- A. Asai, A. Hasegawa, K. Ochiai, Y. Yamashita and T. Mizukami, J. Antibiot., 2000, 53, 81–83 CrossRef CAS.
- A. Asai, T. Tsujita, S. V. Sharma, Y. Yamashita, S. Akinaga, M. Funakoshi, H. Kobayashi and T. Mizukami, Biochem. Pharmacol., 2004, 67, 227–234 CrossRef CAS.
- A. Armstrong and J. N. Scutt, Chem. Commun., 2004, 510–511 RSC.
- O. V. Larionov and A. De Meijere, Org. Lett., 2004, 6, 2153–2156 CrossRef CAS.
- M. Groll, O. V. Larionov, R. Huber and A. De Meijere, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4576–4579 CrossRef CAS.
- K. Yoshida, K. Yamaguchi, T. Sone, Y. Unno, A. Asai, H. Yokosawa, A. Matsuda, M. Arisawa and S. Shuto, Org. Lett., 2008, 10, 3571–3574 CrossRef CAS.
- K. Yoshida, K. Yamaguchi, A. Mizuno, Y. Unno, A. Asai, T. Sone, H. Yokosawa, A. Matsuda, M. Arisawa and S. Shuto, Org. Biomol. Chem., 2009, 7, 1868–1877 CAS.
- S. Kawamura, Y. Unno, A. List, A. Mizuno, M. Tanaka, T. Sasaki, M. Arisawa, A. Asai, M. Groll and S. Shuto, J. Med. Chem., 2013, 56, 3689–3700 CrossRef CAS.
- S. Kawamura, Y. Unno, M. Tanaka, T. Sasaki, A. Yamano, T. Hirokawa, T. Kameda, A. Asai, M. Arisawa and S. Shuto, J. Med. Chem., 2013, 56, 5829–5842 CrossRef CAS.
- S. Kawamura, Y. Unno, A. Asai, M. Arisawa and S. Shuto, Org. Biomol. Chem., 2013, 11, 6615–6622 RSC.
- S. Kawamura, Y. Unno, T. Hirokawa, A. Asai, M. Arisawa and S. Shuto, Chem. Commun., 2014, 50, 2445–2447 RSC.
- S. Kawamura, Y. Unno, A. Asai, M. Arisawa and S. Shuto, Bioorg. Med. Chem., 2014, 22, 3091–3095 CrossRef CAS.
- H. Nakamura, M. Watanabe, H. S. Ban, W. Nabeyama and A. Asai, Bioorg. Med. Chem. Lett., 2009, 19, 3220–3224 CrossRef CAS.
- V. S. Korotkov, A. Ludwig, O. V. Larionov, A. V. Lygin, M. Groll and A. De Meijere, Org. Biomol. Chem., 2011, 9, 7791–7798 RSC.
- A. De Meijere, V. S. Korotkov, A. V. Lygin, O. V. Larionov, V. V. Sokolov, T. Graef and M. Es-Sayed, Org. Biomol. Chem., 2012, 10, 6363–6374 RSC.
- M. Groll, V. S. Korotkov, E. M. Huber, A. De Meijere and A. Ludwig, Angew. Chem., Int. Ed., 2015, 54, 7810–7814 CrossRef CAS.
- K. A. Gill, F. Berrué, J. C. Arens, G. Carr and R. G. Kerr, J. Nat. Prod., 2015, 78, 822–826 CrossRef CAS.
- R. Tello-Aburto, L. P. Hallada, D. Niroula and S. Rogelj, Org. Biomol. Chem., 2015, 13, 10127–10130 RSC.
- F. Wolf, J. S. Bauer, T. M. Bendel, A. Kulik, J. Kalinowski, H. Gross and L. Kaysser, Angew. Chem., Int. Ed., 2017, 56, 6665–6668 CrossRef CAS.
- D. Niroula, L. P. Hallada, C. Le Chapelain, S. K. Ganegamage, D. Dotson, S. Rogelj, M. Groll and R. Tello-Aburto, Eur. J. Med. Chem., 2018, 157, 962–977 CrossRef CAS.
- K. Sugawara, M. Hatori, Y. Nishiyama, K. Tomita, H. Kamei, M. Konishi and T. Oki, J. Antibiot., 1990, 43, 8–18 CrossRef CAS.
- T. Oikawa, M. Hasegawa, M. Shimamura, H. Ashino, S.-i. Murota and I. Morita, Biochem. Biophys. Res. Commun., 1991, 181, 1070–1076 CrossRef CAS.
- L. Meng, B. H. B. Kwok, N. Sin and C. M. Crews, Cancer Res., 1999, 59, 2798–2801 CAS.
- B. K. Kyung, J. Myung, N. Sin and C. M. Crews, Bioorg. Med. Chem. Lett., 1999, 9, 3335–3340 CrossRef.
- M. Hanada, K. Sugawara, K. Kaneta, S. Toda, Y. Nishiyama, K. Tomita, H. Yamamoto, M. Konishi and T. Oki, J. Antibiot., 1992, 45, 1746–1752 CrossRef CAS.
- N. Sin, B. K. Kyung, M. Elofsson, L. Meng, H. Auth, B. H. B. Kwok and C. M. Crews, Bioorg. Med. Chem. Lett., 1999, 9, 2283–2288 CrossRef CAS.
- A. Spaltenstein, J. J. Leban, J. J. Huang, K. R. Reinhardt, O. H. Viveros, J. Sigafoos and R. Crouch, Tetrahedron Lett., 1996, 37, 1343–1346 CrossRef CAS.
- M. Groll, K. B. Kim, N. Kairies, R. Huber and C. M. Crews, J. Am. Chem. Soc., 2000, 122, 1237–1238 CrossRef CAS.
- J. Schrader, F. Henneberg, R. A. Mata, K. Tittmann, T. R. Schneider, H. Stark, G. Bourenkov and A. Chari, Science, 2016, 353, 1–6 CrossRef.
- M. Elofsson, U. Splittgerber, J. Myung, R. Mohan and C. M. Crews, Chem. Biol., 1999, 6, 811–822 CrossRef CAS.
- S. D. Demo, C. J. Kirk, M. A. Aujay, T. J. Buchholz, M. Dajee, M. N. Ho, J. Jiang, G. J. Laidig, E. R. Lewis, F. Parlati, K. D. Shenk, M. S. Smyth, C. M. Sun, M. K. Vallone, T. M. Woo, C. J. Molineaux and M. K. Bennett, Cancer Res., 2007, 67, 6383–6391 CrossRef CAS.
- P. L. McCormack, Drugs, 2012, 72, 2023–2032 CrossRef CAS.
- H. J. Zhou, M. A. Aujay, M. K. Bennett, M. Dajee, S. D. Demo, Y. Fang, M. N. Ho, J. Jiang, C. J. Kirk, G. J. Laidig, E. R. Lewis, Y. Lu, T. Muchamuel, F. Parlati, E. Ring, K. D. Shenk, J. Shields, P. J. Shwonek, T. Stanton, C. M. Sun, C. Sylvain, T. M. Woo and J. Yang, J. Med. Chem., 2009, 52, 3028–3038 CrossRef CAS.
- M. J. Lee, D. Bhattarai, J. Yoo, Z. Miller, J. E. Park, S. Lee, W. Lee, J. J. Driscoll and K. B. Kim, J. Med. Chem., 2019, 62, 4444–4455 CrossRef CAS.
- M. Britton, M. M. Lucas, S. L. Downey, M. Screen, A. A. Pletnev, M. Verdoes, R. A. Tokhunts, O. Amir, A. L. Goddard, P. M. Pelphrey, D. L. Wright, H. S. Overkleeft and A. F. Kisselev, Chem. Biol., 2009, 16, 1278–1289 CrossRef CAS.
- G. De Bruin, E. M. Huber, B. T. Xin, E. J. Van Rooden, K. Al-Ayed, K. B. Kim, A. F. Kisselev, C. Driessen, M. Van Der Stelt, G. A. Van Der Marel, M. Groll and H. S. Overkleeft, J. Med. Chem., 2014, 57, 6197–6209 CrossRef CAS.
- G. DeBruin, B. T. Xin, M. Kraus, M. Van Der Stelt, G. A. Van Der Marel, A. F. Kisselev, C. Driessen, B. I. Florea and H. S. Overkleeft, Angew. Chem., Int. Ed., 2016, 55, 4199–4203 CrossRef CAS.
- B. T. Xin, E. M. Huber, G. De Bruin, W. Heinemeyer, E. Maurits, C. Espinal, Y. Du, M. Janssens, E. S. Weyburne, A. F. Kisselev, B. I. Florea, C. Driessen, G. A. Van Der Marel, M. Groll and H. S. Overkleeft, J. Med. Chem., 2019, 62, 1626–1642 CrossRef CAS.
- T. Muchamuel, M. Basler, M. A. Aujay, E. Suzuki, K. W. Kalim, C. Lauer, C. Sylvain, E. R. Ring, J. Shields, J. Jiang, P. Shwonek, F. Parlati, S. D. Demo, M. K. Bennett, C. J. Kirk and M. Groettrup, Nat. Med., 2009, 15, 781–787 CrossRef CAS.
- A. R. Pereira, A. J. Kale, A. T. Fenley, T. Byrum, H. M. Debonsi, M. K. Gilson, F. A. Valeriote, B. S. Moore and W. H. Gerwick, ChemBioChem, 2012, 13, 810–817 CrossRef CAS.
- D. B. B. Trivella, A. R. Pereira, M. L. Stein, Y. Kasai, T. Byrum, F. A. Valeriote, D. J. Tantillo, M. Groll, W. H. Gerwick and B. S. Moore, Chem. Biol., 2014, 21, 782–791 CrossRef CAS.
- J. Almaliti, B. Miller, H. Pietraszkiewicz, E. Glukhov, C. B. Naman, T. Kline, J. Hanson, X. Li, S. Zhou, F. A. Valeriote and W. H. Gerwick, Eur. J. Med. Chem., 2019, 161, 416–432 CrossRef CAS.
- Y. Koguchi, M. Nishio, S. I. Suzuki, K. Takahashi, T. Ohnuki and S. Komatsubara, J. Antibiot., 1999, 52, 1069–1076 CrossRef CAS.
- Y. Koguchi, J. Kohno, S. I. Suzuki, M. Nishio, K. Takahashi, T. Ohnuki and S. Komatsubara, J. Antibiot., 1999, 53, 63–65 CrossRef.
- Y. Koguchi, M. Nishio, S.-I. Suzuki, K. Takahashi and T. Ohnuki, J. Antibiot., 2000, 53, 967–972 CrossRef CAS.
- L. Keller, A. Plaza, C. Dubiella, M. Groll, M. Kaiser and R. Müller, J. Am. Chem. Soc., 2015, 137, 8121–8130 CrossRef CAS.
- J. G. Owen, Z. Charlop-Powers, A. G. Smith, M. A. Ternei, P. Y. Calle, B. V. B. Reddy, D. Montiel and S. F. Brady, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 4221–4226 CrossRef CAS.
- S. Tsukamoto, M. Tatsuno, R. W. M. Van Soest, H. Yokosawa and T. Ohta, J. Nat. Prod., 2003, 66, 1181–1185 CrossRef CAS.
- L. E. Mohamed, H. Gross, A. Pontius, S. Kehraus, A. Krick, G. Kelter, A. Maier, H. H. Fiebig and G. M. König, Org. Lett., 2009, 11, 5014–5017 CrossRef.
- I. E. Mohamed, S. Kehraus, A. Krick, G. M. König, G. Kelter, A. Maier, H. H. Fiebig, M. Kalesse, N. P. Malek and H. Gross, J. Nat. Prod., 2010, 73, 2053–2056 CrossRef CAS.
- A. Noda, E. Sakai, H. Kato, F. Losung, R. E. P. Mangindaan, N. J. De Voogd, H. Yokosawa and S. Tsukamoto, Bioorg. Med. Chem. Lett., 2015, 25, 2650–2653 CrossRef CAS.
- A. Randazzo, C. Debitus, L. Minale, P. G. Pastor, M. J. Alcaraz, M. Payá and L. Gomez-Paloma, J. Nat. Prod., 1998, 61, 571–575 CrossRef CAS.
- L. Margarucci, M. C. Monti, A. Tosco, R. Riccio and A. Casapullo, Angew. Chem., Int. Ed., 2010, 49, 3960–3963 CrossRef CAS.
- L. Margarucci, A. Tosco, R. De Simone, R. Riccio, M. C. Monti and A. Casapullo, ChemBioChem, 2012, 13, 982–986 CrossRef CAS.
- L. M. West and D. J. Faulkner, J. Nat. Prod., 2008, 71, 269–271 CrossRef CAS.
- H. Yang, D. Chen, C. C. Qiuzhi, X. Yuan and Q. P. Dou, Cancer Res., 2006, 66, 4758–4765 CrossRef CAS.
- R. Kannaiyan, M. K. Shanmugam and G. Sethi, Cancer Lett., 2011, 303, 9–20 CrossRef CAS.
- Y. l. Zhong, G. j. Xu, S. Huang, L. Zhao, Y. Zeng, X. f. Xiao, J. l. An, J. Liu and T. Yang, Eur. J. Pharmacol., 2019, 853, 184–192 CrossRef CAS.
- S. M. Kupchan, W. A. Court, R. G. Dailey, C. J. Gilmore and R. F. Bryan, J. Am. Chem. Soc., 1972, 94, 7194–7195 CrossRef CAS.
- L. Lu, J. Kanwar, S. Schmitt, Q. C. Cui, C. Zhang, C. Zhao and Q. P. Dou, Anticancer Res., 2011, 31, 1–10 CAS.
- R. E. Tiedemann, J. Schmidt, J. J. Keats, C. X. Shi, X. Z. Yuan, S. E. Palmer, X. Mao, A. D. Schimmer and A. K. Stewart, Blood, 2009, 113, 4027–4037 CrossRef CAS.
- L. C. Mishra, B. B. Singh and S. Dagenais, Altern. Med. Rev., 2000, 5, 334–346 CAS.
- H. Yang, G. Shi and Q. P. Dou, Mol. Pharmacol., 2007, 71, 426–437 CrossRef CAS.
- S. Tsukamoto, R. Yamanokuchi, M. Yoshitomi, K. Sato, T. Ikeda, H. Rotinsulu, R. E. P. Mangindaan, N. J. de Voogd, R. W. M. van Soest and H. Yokosawa, Bioorg. Med. Chem. Lett., 2010, 20, 3341–3343 CrossRef CAS.
- A. Hirose, H. Maeda, A. Tonouchi, T. Nehira and M. Hashimoto, Tetrahedron, 2014, 70, 1458–1463 CrossRef CAS.
- S. Uesugi, Y. Honmura, M. Nishiyama, K. Kusakabe, A. Tonouchi, T. Yamashita, M. Hashimoto and K. i. Kimura, Bioorg. Med. Chem., 2019, 27, 115161 CrossRef CAS.
- Y. Nagasawa, R. Ueoka, R. Yamanokuchi, N. Horiuchi, T. Ikeda, H. Rotinsulu, R. E. P. Mangindaan, K. Ukai, H. Kobayashi, M. Namikoshi, H. Hirota, H. Yokosawa and S. Tsukamoto, Chem. Pharm. Bull., 2011, 59, 287–290 CrossRef CAS.
- A. H. El-Desoky, H. Kato, K. Eguchi, T. Kawabata, Y. Fujiwara, F. Losung, R. E. P. Mangindaan, N. J. De Voogd, M. Takeya, H. Yokosawa and S. Tsukamoto, J. Nat. Prod., 2014, 77, 1536–1540 CrossRef CAS.
- A. Furusato, H. Kato, T. Nehira, K. Eguchi, T. Kawabata, Y. Fujiwara, F. Losung, R. E. P. Mangindaan, N. J. De Voogd, M. Takeya, H. Yokosawa and S. Tsukamoto, Org. Lett., 2014, 16, 3888–3891 CrossRef CAS.
- H. Kato, A. H. El-Desoky, Y. Takeishi, T. Nehira, E. D. Angkouw, R. E. P. Mangindaan, N. J. de Voogd and S. Tsukamoto, Bioorg. Med. Chem. Lett., 2019, 29, 8–10 CrossRef CAS.
- T. H. Pham, A. Hovhannisyan, D. Bouvier, L. Tian, M. Reboud-Ravaux, G. Melikyan and M. Bouvier-Durand, Bioorg. Med. Chem. Lett., 2012, 22, 3822–3827 CrossRef CAS.
- A. Hovhannisyan, T. H. Pham, D. Bouvier, L. Qin, G. Melikyan, M. Reboud-Ravaux and M. Bouvier-Durand, Bioorg. Med. Chem. Lett., 2013, 23, 2696–2703 CrossRef CAS.
- A. Hovhannisyan, T. H. Pham, D. Bouvier, A. Piroyan, L. Dufau, L. Qin, Y. Cheng, G. Melikyan, M. Reboud-Ravaux and M. Bouvier-Durand, Bioorg. Med. Chem. Lett., 2014, 24, 1571–1580 CAS.
- T. Lindel, Alkaloids: Chem. Biol., 2017, 77, 117–219 CAS.
- R. B. Kinnel, H. P. Gehrken and P. J. Scheuer, J. Am. Chem. Soc., 1993, 115, 3376–3377 CAS.
- I. B. Seiple, S. Su, I. S. Young, C. A. Lewis, J. Yamaguchi and P. S. Baran, Angew. Chem., Int. Ed., 2010, 49, 1095–1098 CrossRef CAS.
- I. B. Seiple, S. Su, I. S. Young, A. Nakamura, J. Yamaguchi, L. Jørgensen, R. A. Rodriguez, D. P. Ömalley, T. Gaich, M. Köck and P. S. Baran, J. Am. Chem. Soc., 2011, 133, 14710–14726 CAS.
- T. A. Lansdell, N. M. Hewlett, A. P. Skoumbourdis, M. D. Fodor, I. B. Seiple, S. Su, P. S. Baran, K. S. Feldman and J. J. Tepe, J. Nat. Prod., 2012, 75, 980–985 CAS.
- P. Beck, T. A. Lansdell, N. M. Hewlett, J. J. Tepe and M. Groll, Angew. Chem., Int. Ed., 2015, 54, 2830–2833 CAS.
- N. M. Hewlett and J. J. Tepe, Org. Lett., 2011, 13, 4550–4553 CrossRef CAS.
- R. T. M. P. De Souza, V. F. Freire, J. R. Gubiani, R. O. Ferreira, D. B. B. Trivella, F. C. Moraes, W. C. Paradas, L. T. Salgado, R. C. Pereira, G. M. Amado Filho, A. G. Ferreira, D. E. Williams, R. J. Andersen, T. F. Molinski and R. G. S. Berlinck, J. Nat. Prod., 2018, 81, 2296–2300 CAS.
- S. Nam, D. M. Smith and Q. P. Dou, J. Biol. Chem., 2001, 276, 13322–13330 CrossRef CAS.
- D. M. Smith, Z. Wang, A. Kazi, L. H. Li, T. H. Chan and Q. P. Dou, Mol. Med., 2002, 8, 382–392 CAS.
- D. M. Smith, K. G. Daniel, Z. Wang, W. C. Guida, T. H. Chan and Q. P. Dou, Proteins: Struct., Funct., Genet., 2004, 54, 58–70 CrossRef CAS.
- W. H. Lam, A. Kazi, D. J. Kuhn, L. M. C. Chow, A. S. C. Chan, Q. Ping Dou and T. H. Chan, Bioorg. Med. Chem., 2004, 12, 5587–5593 CrossRef CAS.
- K. R. Landis-Piwowar, C. Huo, D. Chen, V. Milacic, G. Shi, H. C. Tak and Q. P. Dou, Cancer Res., 2007, 67, 4303–4310 CrossRef CAS.
- H. Yang, D. K. Sun, D. Chen, Q. C. Cui, Y. Y. Gu, T. Jiang, W. Chen, S. B. Wan and Q. P. Dou, Cancer Lett., 2010, 292, 48–53 CrossRef CAS.
- A. Kazi, K. G. Daniel, D. M. Smith, N. B. Kumar and Q. P. Dou, Biochem. Pharmacol., 2003, 66, 965–976 CrossRef CAS.
- S. H. Shim, Phytother. Res., 2011, 25, 615–618 CrossRef CAS.
- D. Chen, K. G. Daniel, M. S. Chen, D. J. Kuhn, K. R. Landis-Piwowar and Q. P. Dou, Biochem. Pharmacol., 2005, 69, 1421–1432 CAS.
- Y. X. Wu and X. Fang, Planta Med., 2010, 76, 128–132 CAS.
- L. Pan, S. Matthew, D. D. Lantvit, X. Zhang, T. N. Ninh, H. Chai, E. J. Carcache De Blanco, D. D. Soejarto, S. M. Swanson and A. D. Kinghorn, J. Nat. Prod., 2011, 74, 2193–2199 CrossRef CAS.
- Y. X. Wu, E. Sato, W. Kimura and N. Miura, Phytother. Res., 2013, 27, 1362–1367 CrossRef CAS.
- Y. Ren, D. D. Lantvit, E. J. C. De Blanco, L. B. S. Kardono, S. Riswan, H. Chai, C. E. Cottrell, N. R. Farnsworth, S. M. Swanson, Y. Ding, X. C. Li, J. P. J. Marais, D. Ferreira and A. D. Kinghorn, Tetrahedron, 2010, 66, 5311–5320 CAS.
- H. Huang, N. Liu, K. Zhao, C. Zhu, X. Lu, S. Li, W. Lian, P. Zhou, X. Dong, C. Zhao, H. Guo, C. Zhang, C. Yang, G. Wen, L. Lu, X. Li, L. Guan, C. Liu, X. Wang, Q. P. Dou and J. Liu, Front. Biosci., 2011, 3, 1315–1325 Search PubMed.
- N. R. Jana, P. Dikshit, A. Goswami and N. Nukina, J. Biol. Chem., 2004, 279, 11680–11685 CrossRef CAS.
- V. Milacic, S. Banerjee, K. R. Landis-Piwowar, F. H. Sarkar, A. P. N. Majumdar and Q. P. Dou, Cancer Res., 2008, 68, 7283–7292 CrossRef CAS.
- W. Zhang, H. Bai, L. Han, H. Zhang, B. Xu, J. Cui, X. Wang, Z. Ge and R. Li, Bioorg. Med. Chem. Lett., 2018, 28, 2459–2464 CrossRef CAS.
- Y. He, J. Huang, P. Wang, X. Shen, S. Li, L. Yang, W. Liu, A. Suksamrarn, G. Zhang and F. Wang, Oncotarget, 2016, 7, 4664–4679 CrossRef.
- H. Yang, P. Zhou, H. Huang, D. Chen, N. Ma, Q. C. Cui, S. Shen, W. Dong, X. Zhang, W. Lian, X. Wang, Q. P. Dou and J. Liu, Int. J. Cancer, 2009, 124, 2450–2459 CrossRef CAS.
- N. Wada, Y. Kawano, S. Fujiwara, Y. Kikukawa, Y. Okuno, M. Tasaki, M. Ueda, Y. Ando, K. Yoshinaga, M. Ri, S. Iida, T. Nakashima, Y. Shiotsu, H. Mitsuya and H. Hata, Int. J. Oncol., 2015, 46, 963–972 CrossRef CAS.
- P. Beck, W. Heinemeyer, A. L. Späth, Y. Elnakady, R. Müller and M. Groll, J. Mol. Biol., 2014, 426, 3108–3117 CrossRef CAS.
- S. Tsukamoto, K. Koimaru and T. Ohta, Mar. Drugs, 2005, 3, 29–35 CrossRef CAS.
- G. E. Chlipala, M. Sturdy, A. Krunic, D. D. Lantvit, Q. Shen, K. Porter, S. M. Swanson and J. Orjala, J. Nat. Prod., 2010, 73, 1529–1537 CrossRef CAS.
- P. A. Osmulski and M. Gaczynska, Mol. Pharmacol., 2013, 84, 104–113 CrossRef CAS.
- M. B. Giletto, P. A. Osmulski, C. L. Jones, M. E. Gaczynska and J. J. Tepe, Org. Biomol. Chem., 2019, 17, 2734–2746 RSC.
| This journal is © The Royal Society of Chemistry 2020 |