
Rhodium/copper-cocatalyzed annulation of benzylamines with diazo compounds: access to fused isoquinolines†
Qiang
Wang
ab and
Xingwei
Li
*a
aDalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: xwli@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 26th July 2016
Abstract
Benzylamine has been applied as an arene source in C–H activation and coupling with different types of diazo compounds, leading to the synthesis of fused isoquinolines. This occurs via a mild synergistic rhodium- and copper-catalyzed process. Moreover, ecofriendly O2 has been used as a terminal oxidant with high efficiency.
Isoquinolines are a ubiquitous structural motif in numerous pharmaceuticals and biologically active compounds.1 Recently, substantial progress has been made in the Rh(III)- and Ru(II)-catalyzed C–H bond functionalization of aromatic imines or oximes, which has streamlined access to synthetically important isoquinolines in an atom- and step-economical fashion without pre-activation of the substrates.2 In this process, the lone pair of the nitrogen offers sufficient chelation-assistance which leads to metalation of an ortho-C–H bond. Nevertheless, these reports are limited by the demonstrated scope of imine or oxime substrates, which are simple and relatively stable aromatic ketimines or ketoximes. Thus, it is difficult to develop practical methods to construct 1-unsubstituted isoquinolines, in spite of the vast prevalence of these scaffolds in pharmaceutical molecules and natural products (Fig. 1).3 One of the reasons is that aldimines are prone to hydrolysis under the catalytic conditions.4 To steer around this intrinsic obstacle, Miura and co-workers developed a practical method in which readily available benzylamines are transferred to related aldimines in situ which then coupled with alkynes to form isoquinolines via a Rh/Cu-catalyzed oxidative cyclization (Scheme 1).5
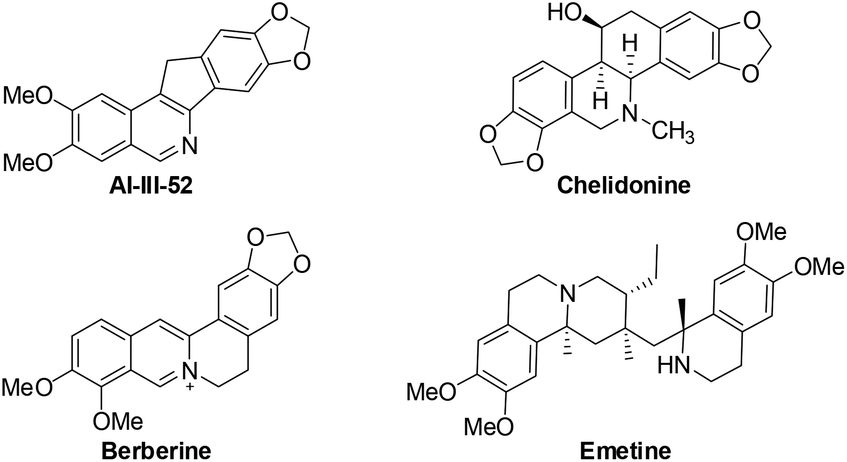 | ||
| Fig. 1 Bioactive compounds containing 1-unsubstituted isoquinoline moieties. | ||
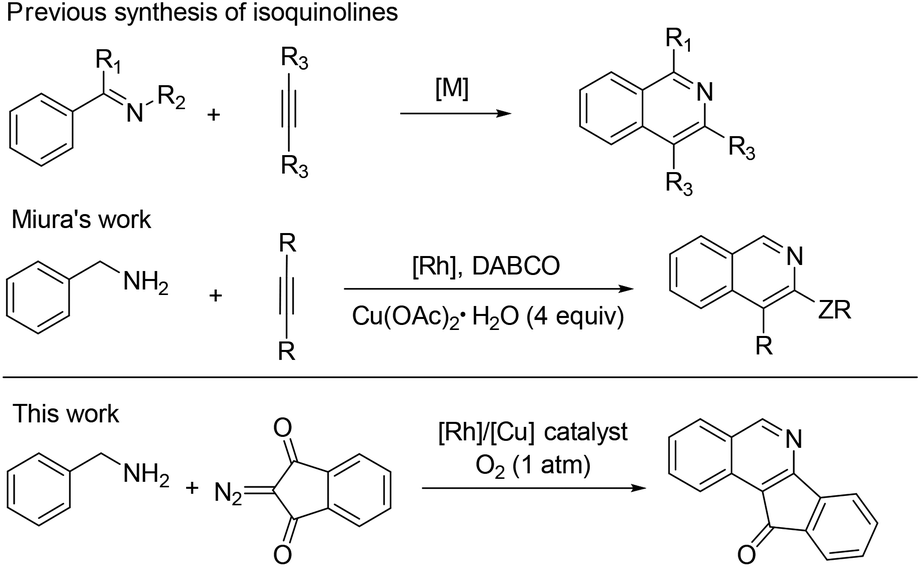 | ||
| Scheme 1 Strategies for the synthesis of isoquinolines. | ||
The use of diazo compounds for coupling with arenes via transition-metal-catalyzed C–H activation has recently received much attention. The pioneering work by Yu indicated that an oxime group could induce ortho-C–H activation and coupling with diazomalonates in the presence of a Rh(III) catalyst.6 Afterwards many groups further took advantage of the versatile reactivity of diazo compounds to assemble various nitrogen-containing heterocycles.7 Inspired by these results, we envisioned the feasibility of C–H activation and annulation of benzylamines with diazocarbonyl compounds under rhodium catalysis.
In Miura's work, the benzylic amine was oxidized to an imine by using a stoichiometric amount of Cu(OAc)2·H2O under rather harsh conditions. Given the importance of 1-unsubstituted and fused isoquinolines and the limitations of previous methods, we aim to adopt a clean oxidation of primary amines to imines and directly use the imine as an intermediate for further C–H functionalization under mild conditions. The oxidative transformation of amines using a green oxidant such as oxygen attracted our attention.2s,5 We now report an efficient aerobic synthesis of isoquinolines starting from readily available benzylamines and diazo compounds via synergistic rhodium/copper catalysis. More importantly, access to such fused isoquinolines through traditional methods is a great challenge.8
We selected the coupling of benzylamine 1a with 2-diazo-1H-indene-1,3(2H)-dione 2a as the model reaction (Table 1). Initially, the reaction of 1a and 2a catalyzed by [Cp*RhCl2]2/AgSbF6 in the presence of a stoichiometric amount of Cu(OAc)2 afforded isoquinoline 3a in 30% yield (entry 1). The reaction proceeded giving a similar yield when conducted under an O2 atmosphere with 20 mol% of Cu(OAc)2 (entry 2). A further increase of the reaction temperature to 80 °C gave rise to an improved yield of 75% (entry 3). However, a further increase of the temperature led to a lower yield (entry 4). Switching to other solvents such as THF and TFE gave no desired reaction (entries 5 and 6). Employment of other copper salts such as CuCl2 and CuSO4 also resulted in poor efficiency (entries 7 and 8). Further improvement was attained when Zn(OTf)2 (20 mol%) was introduced as an additive (entry 9). Control experiments confirmed that essentially no desired product was detected when either the Rh catalyst or the Cu additive was omitted. Moreover, air was not an efficient oxidant for this transformation (entry 14). Consequently, the following reaction conditions have been identified: [Cp*RhCl2]2 (4 mol%), AgSbF6 (16 mol%), Cu(OAc)2 (20 mol%) and Zn(OTf)2 (20 mol%) in DCE at 80 °C under O2 (1 atm).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | [Cu] | Additive | Solvent | Atmosphere | T (°C) | 3a (%) |
| a Reaction conditions: 1a (0.10 mmol) and diazo compound 2a (0.11 mmol) with [Cp*RhCl2]2 (4 mol%), AgSbF6 (16 mol%), [Cu] (20 mol%) and additive (20 mol%), DCE (3 mL), 80 °C, 12 h, yield was isolated. b 2 equiv. of Cu(OAc)2 were added. c In the absence of Rh(III). | ||||||
| 1 | Cu(OAc)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
— | DCE | N2 | 60 | 30 |
| 2 | Cu(OAc)2 | — | DCE | O2 | 60 | 33 |
| 3 | Cu(OAc)2 | — | DCE | O2 | 80 | 75 |
| 4 | Cu(OAc)2 | — | DCE | O2 | 100 | 66 |
| 5 | Cu(OAc)2 | — | TFE | O2 | 80 | — |
| 6 | Cu(OAc)2 | — | THF | O2 | 80 | — |
| 7 | CuCl2 | — | DCE | O2 | 80 | 16 |
| 8 | CuSO4 | — | DCE | O2 | 80 | 21 |
| 9 | Cu(OAc) 2 | Zn(OTf) 2 (20 mol%) | DCE | O 2 | 80 | 93 |
| 10c | Cu(OAc)2 | Zn(OTf)2 (20 mol%) | DCE | O2 | 80 | — |
| 11 | — | Zn(OTf)2 (20 mol%) | DCE | O2 | 80 | — |
| 12 | — | Fe(acac)2 (20 mol%) | DCE | O2 | 80 | — |
| 13 | — | MnO2 (20 mol%) | DCE | O2 | 80 | — |
| 14 | Cu(OAc)2 | Zn(OTf)2 (20 mol%) | DCE | Air | 80 | 57 |
With the optimal conditions in hand, we scrutinized the scope of the aryl amines in the coupling with 2-diazo-1H-indene-1,3(2H)-dione (2a). Amines bearing electron-donating and -withdrawing groups in the aryl ring were well-tolerated, and the corresponding products were isolated in good to excellent yields (Scheme 2). The same observation applied to benzylamines bearing different ortho substituents (3n–q). Completely regioselective annulation occurred at the less hindered position for most meta-substituted substrates (3k–l), except that the meta-fluoro-substituted benzylamine 1m showed a considerable secondary directing group effect,9 thus leading to C–H functionalization at the sterically more hindered site (3m). Moreover, α-methyl and α-phenyl benzylamines also exhibited good reactivity (3r and 3s), where no significant effect of the steric hindrance of the α-substituent was observed (Table 2).
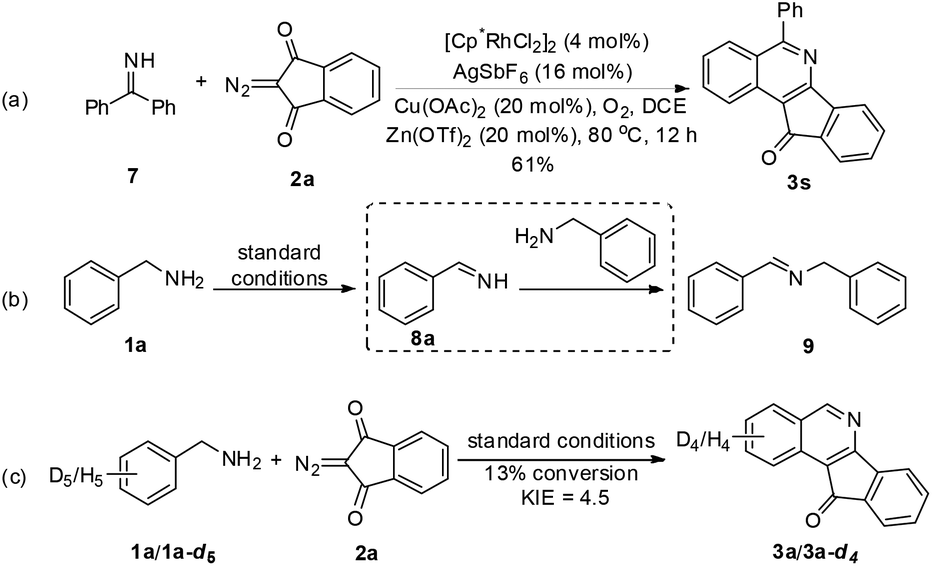 | ||
| Scheme 2 Mechanistic studies. | ||
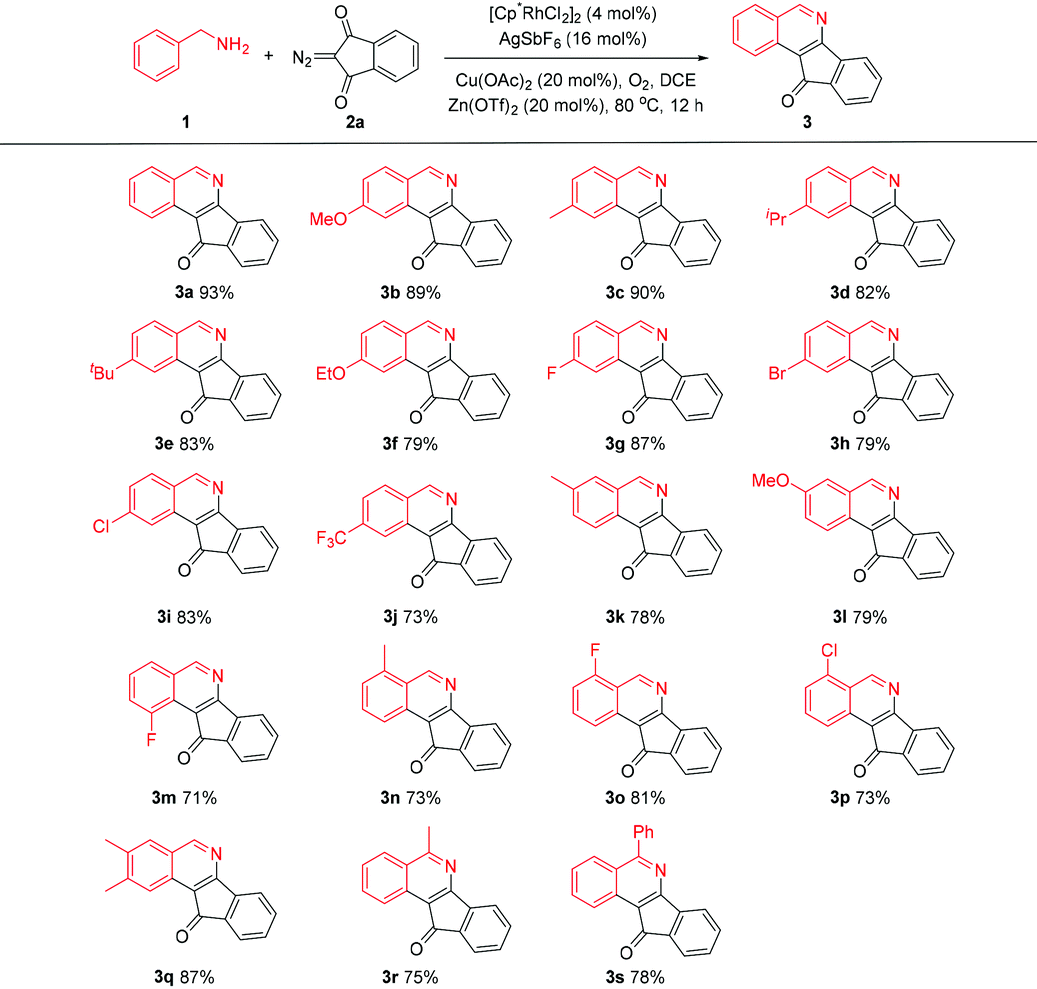
We next explored the applicability of other diazo compounds under the standard conditions. A variety of α-diazo esters reacted smoothly with the benzylamines to afford the corresponding isoquinolines in moderate to good yields (Table 3).
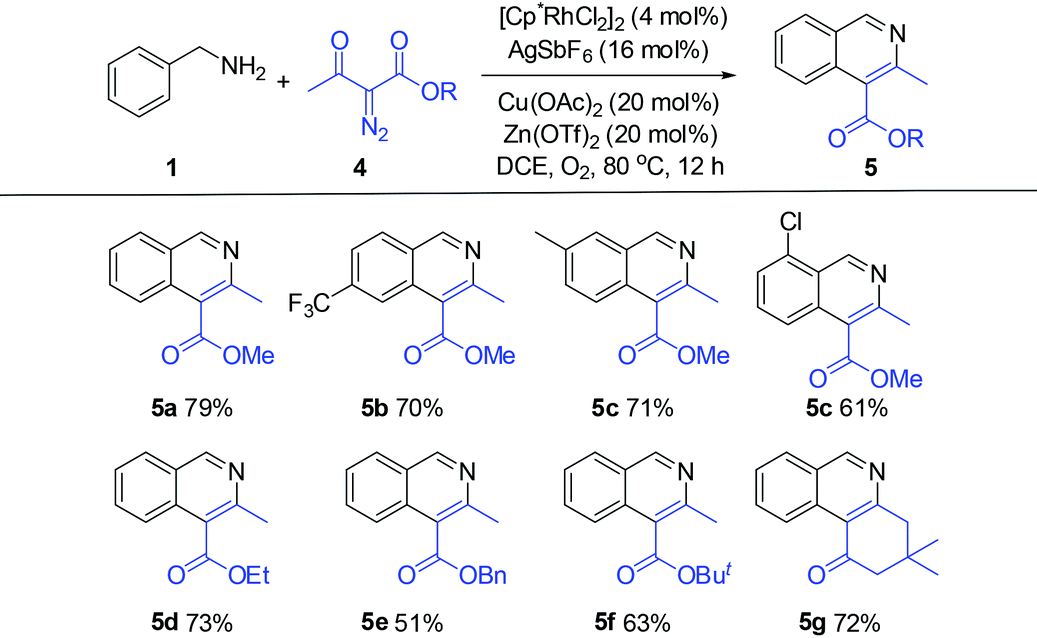
The obtained products could be readily derivatized. The carbonyl group was reduced to a methylene group via a Wolff–Kishner reduction (eqn (1)),10 and the final product 6 has the same scaffold with the TOP1 inhibitor AI-III-52.3a Moreover, the reaction can be successfully scaled up to 2 mmol without much loss of yield even with a reduced loading of the catalyst and the additive (eqn (2)).
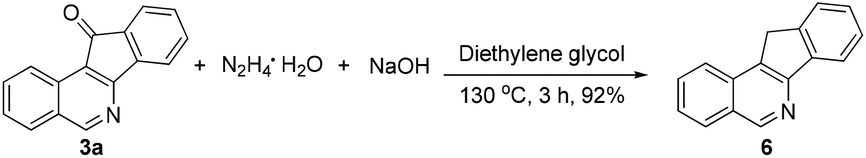 | (1) |
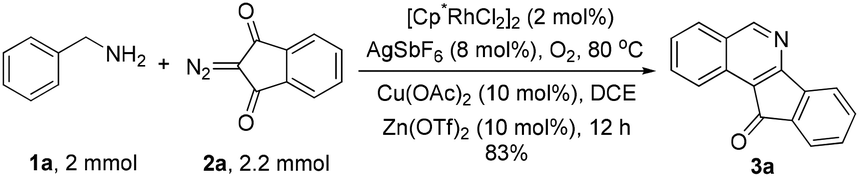 | (2) |
To gain mechanistic insight into the reaction, benzophenone imine (7) was treated with 2a under the standard reaction conditions, and the same annulation product 3s was isolated in 61% yield (Scheme 2). Additionally, benzylamine 1a was transformed to imine 9 (determined by GC-MS) in the absence of a coupling partner under the standard conditions, in which an unstable imine 8a is proposed to undergo condensation with another molecule of 1a,11 which was consistent with Miura's work.5 These results indicate that amine oxidation likely took place prior to the C–H bond activation. Subsequently, the intermolecular isotope effect (kH/kD = 4.5) indicated that C–H bond cleavage was possibly involved in the turnover-limiting step of this transformation.
On the basis of above experimental results and the literature reports,12 a plausible mechanism has been proposed (Scheme 3). Cyclometalation of aldimine 8a generated in situ gives a rhodacyclic intermediate IIvia a proposed concerted metalation/deprotonation (CMD) process.13 Interactions with an incoming diazo substrate then occur to form a rhodium–carbene intermediate III by dediazonization. Subsequently, the intermediate III underwent migratory insertion to afford a six-membered rhodacyclic intermediate IV. Proto-demetalation of IV gave an alkylated imine intermediate V and regenerated the active species I. Finally, nucleophilic cyclization–condensation of V produces the final product 3a.
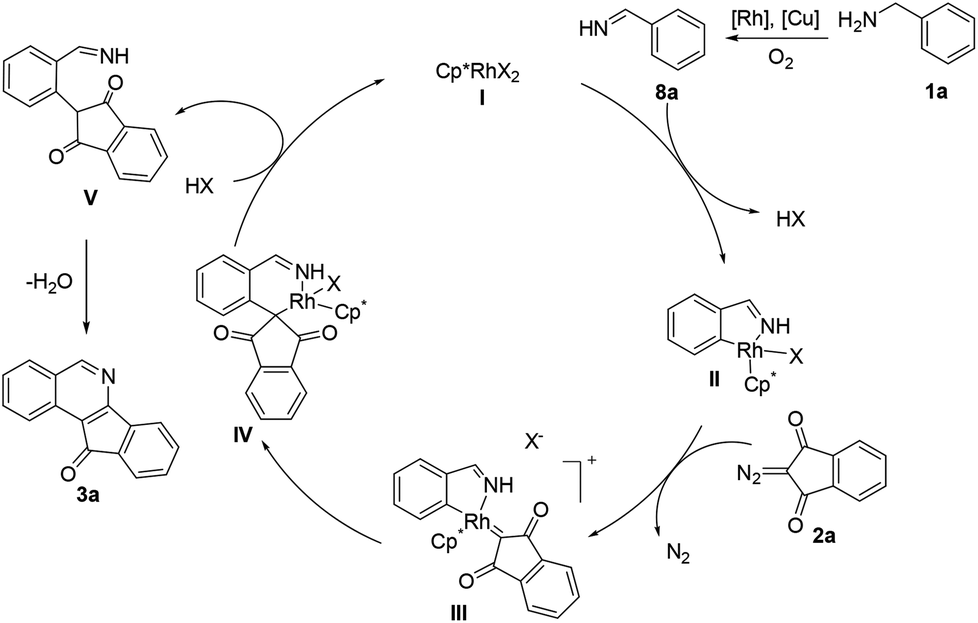 | ||
| Scheme 3 Proposed mechanism. | ||
Conclusions
In conclusion, we have developed a novel system of Rh/Cu co-catalyzed synthesis of condensed isoquinolines in which readily accessible benzylamines serve as efficient building blocks via a dehydrogenation process and the ecologically friendly O2 can be used as the terminal oxidant. Further synthetic applications to bioactive molecules are underway in our laboratory.Acknowledgements
Financial support from the NSFC (no. 21525208 and 21272231) is gratefully acknowledged.Notes and references
- (a) J. D. Scott and R. M. Williams, Chem. Rev., 2002, 102, 1669 CrossRef CAS PubMed; (b) K. W. Bentley, Nat. Prod. Rep., 2006, 23, 444 RSC; (c) J. Ziegler and P. J. Facchini, Annu. Rev. Plant Biol., 2008, 59, 735 CrossRef CAS PubMed; (d) K. Bhadra and G. S. Kumar, Med. Res. Rev., 2011, 31, 821 CrossRef CAS PubMed; (e) R. Alajarn and C. Burgos, in Modern Heterocyclic Chemistry, ed. J. Alvarez-Builla, J. J. Vaquero and J. Barluenga, Wiley-VCH, Weinheim, 2011, vol. 3, pp. 1527–1629 Search PubMed; (f) N. L. Subasinghe, J. Lanter, T. Markotan, E. Opas, S. McKenney, C. Crysler, C. Hou, J. O'Neill, D. Johnson and Z. Sui, Bioorg. Med. Chem. Lett., 2013, 23, 1063 CrossRef CAS PubMed.
- (a) T. Fukutani, N. Umeda, K. Hirano, T. Satoh and M. Miura, Chem. Commun., 2009, 34, 5141 RSC; (b) N. Guimond and K. Fagnou, J. Am. Chem. Soc., 2009, 131, 12050 CrossRef CAS PubMed; (c) P. C. Too, Y.-F. Wang and S. Chiba, Org. Lett., 2010, 12, 5688 CrossRef CAS PubMed; (d) X. Zhang, D. Chen, M. Zhao, J. Zhao, A. Jia and X. Li, Adv. Synth. Catal., 2011, 353, 719 CrossRef CAS; (e) T. K. Hyster and T. Rovis, Chem. Commun., 2011, 47, 11846 RSC; (f) P. C. Too, S. H. Chua, S. H. Wong and S. Chiba, J. Org. Chem., 2011, 76, 6159 CrossRef CAS PubMed; (g) X. Wei, M. Zhao, Z. Du and X. Li, Org. Lett., 2011, 13, 4636 CrossRef CAS PubMed; (h) L. Zheng, J. Ju, Y. Bin and R. Hua, J. Org. Chem., 2012, 77, 5794 CrossRef CAS PubMed; (i) D.-S. Kim, J.-W. Park and C.-H. Jun, Adv. Synth. Catal., 2013, 355, 2667 CrossRef CAS; (j) S.-C. Chuang, P. Gandeepan and C.-H. Cheng, Org. Lett., 2013, 15, 5750 CrossRef CAS PubMed; (k) W. Liu, X. Hong and B. Xu, Synthesis, 2013, 2137 CAS; (l) Z. Shi, D. C. Koester, M. Boultadakis-Arapinis and F. Glorius, J. Am. Chem. Soc., 2013, 135, 12204 CrossRef CAS PubMed; (m) D. Zhao, F. Lied and F. Glorius, Chem. Sci., 2014, 5, 2869 RSC; (n) W. Han, G. Zhang, G. Li and H. Huang, Org. Lett., 2014, 16, 3532 CrossRef CAS PubMed; (o) Q. Wang, Y. Li, Z. Qi, F. Xie, Y. Lan and X. Li, ACS Catal., 2016, 6, 1971 CrossRef CAS; (p) X. Wang, A. Lerchen and F. Glorius, Org. Lett., 2016, 18, 2090 CrossRef CAS PubMed; (q) J. H. Kim, S. Greßies and F. Glorius, Angew. Chem., Int. Ed., 2016, 55, 5577 CrossRef CAS PubMed; (r) Q. Wang and X. Li, Org. Lett., 2016, 18, 2102 CrossRef CAS PubMed; (s) X. Wang and N. Jiao, Org. Lett., 2016, 18, 2150 CrossRef CAS PubMed; (t) H. Wang, L. Li, S. Yu, Y. Li and X. Li, Org. Lett., 2016, 18, 2914 CrossRef CAS PubMed. For related reviews, see: (u) J. WencelDelord, T. Droge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC; (v) L. Ackermann, Acc. Chem. Res., 2014, 47, 281 CrossRef CAS PubMed; (w) G. Song and X. Li, Acc. Chem. Res., 2015, 48, 1007 CrossRef CAS PubMed.
- (a) Y. Song, Z. Shao, T. S. Dexheimer, E. S. Scher, Y. Pommier and M. Cushman, J. Med. Chem., 2010, 53, 1979 CrossRef CAS PubMed; (b) A. Panzer, A. M. Joubert, P. C. Bianchi, E. Hamel and J. C. Seegers, Eur. J. Cell Biol., 2001, 80, 111 CrossRef CAS PubMed; (c) A. Nasreen, F. Akhtar, M. S. Shekhani, J. Clardy, M. Parvez and M. I. Choudhary, J. Nat. Prod., 1997, 60, 472 CrossRef PubMed; (d) W. Wu, H.-R. Zhou, S. J. Bursian, J. E. Link and J. J. Pestka, Arch. Toxicol., 2016, 90, 997 CrossRef CAS PubMed.
- (a) J. J. Cornejo, K. D. Larson and G. D. Mendenhall, J. Org. Chem., 1985, 50, 5382 CrossRef CAS; (b) D. R. Boyd, P. B. Coulter, R. Hamilton, N. T. Thompson, N. D. Sharma and M. E. Stubbs, J. Chem. Soc., Perkin Trans. 1, 1985, 2123 RSC.
- K. Morimoto, K. Hirano, T. Satoh and M. Miura, Chem. Lett., 2011, 40, 600 CrossRef CAS.
- W. Chan, S. Lo, Z. Zhou and W. Yu, J. Am. Chem. Soc., 2012, 134, 13565 CrossRef CAS PubMed.
- (a) T. K. Hyster, K. E. Ruhl and T. Rovis, J. Am. Chem. Soc., 2013, 135, 5364 CrossRef CAS PubMed; (b) B. Ye and N. Cramer, Angew. Chem., Int. Ed., 2014, 53, 7896 CrossRef CAS PubMed; (c) J. Li, M. Tang, L. Zang, X. Zhang, Z. Zhang and L. Ackermann, Org. Lett., 2016, 18, 2742 CrossRef CAS PubMed.
- (a) M. A. Campo and R. C. Larock, Org. Lett., 2000, 2, 3675 CrossRef CAS PubMed; (b) M. A. Campo and R. C. Larock, J. Org. Chem., 2002, 67, 5616 CrossRef CAS PubMed.
- D. Balcells, E. Clot and O. Eisenstein, Chem. Rev., 2010, 110, 749 CrossRef CAS PubMed.
- N. Kishner, J. Russ. Phys. – Chem. Soc., 1911, 43, 582 Search PubMed.
- X. Lang, H. Ji, C. Chen, W. Ma and J. Zhao, Angew. Chem., Int. Ed., 2011, 50, 3934 CrossRef CAS PubMed.
- (a) S. Sharma, S. H. Han, S. Han, W. Ji, J. Oh, S.-Y. Lee, J. S. Oh, Y. H. Jung and I. S. Kim, Org. Lett., 2015, 17, 2852 CrossRef CAS PubMed; (b) S. H. Park, J. Kwak, K. Shin, J. Ryu, Y. Park and S. Chang, J. Am. Chem. Soc., 2014, 136, 2492 CrossRef CAS PubMed; (c) D. Yu, M. Suri and F. Glorius, J. Am. Chem. Soc., 2013, 135, 8802 CrossRef CAS PubMed.
- (a) D. L. Davies, S. M. A. Donald, O. Al-Duaij, S. A. Macgregor and M. Pölleth, J. Am. Chem. Soc., 2006, 128, 4210 CrossRef CAS PubMed; (b) D. Lapointe and K. Fagnou, Chem. Lett., 2010, 39, 1118 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data for new compounds. See DOI: 10.1039/c6qo00287k |
| This journal is © the Partner Organisations 2016 |