
Recent advances in photoinduced trifluoromethylation and difluoroalkylation
Xiaolin
Pan
a,
Hongguang
Xia
*b and
Jie
Wu
*ac
aDepartment of Chemistry, Fudan University, 220 Handan Road, Shanghai 200433, China. E-mail: jie_wu@fudan.edu.cn
bDepartment of Biochemistry and Molecular Biology, Zhejiang University School of Medicine, Hangzhou 310058, China. E-mail: hongguangxia@zju.edu.cn
cState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Linglin Road, Shanghai 200032, China
First published on 20th June 2016
Abstract
Recent advances in photoinduced trifluoromethylation and difluoroalkylation under photocatalysis are summarized. Most of the photoredox reactions proceed efficiently under mild conditions with simple operation. Various fluorinated reagents are developed and applied in different transformations. Usually, the photocatalyst in a photocatalytic cycle is initially stimulated to the excited state in the presence of visible light, which then provides an electron to the fluorinated reagent to release a CF3 radical or CF2 radical. The subsequent radical addition to an unsaturated bond with further transformations would produce the corresponding fluorinated products. Alkenes, alkynes, isocyanides, and arenes are frequently employed as substrates for access to fluorochemicals. The advantages of trifluoromethylation and difluoroalkylation under photocatalysis including mild reaction conditions, good functional group tolerance, and experimental ease will make the approaches attractive for further applications.
 Xiaolin Pan | Xiaolin Pan was born in Liaoning (China) in 1989. She received her BS (2011) from the Department of Chemistry at Fudan University. In the year 2011, she continued her study as a Ph.D. candidate at Fudan University, under the guidance of Professor Jie Wu. Her current research interest mainly focuses on the development methods for the synthesis of natural product-like compounds via transition metal catalyzed tandem reactions. |
 Hongguang Xia | Hongguang Xia received his B.Sc. in chemistry from Fudan University in 2006 and he received his Ph.D. degree from the Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences under the supervision of Prof. Junying Yuan (2006–2011). He worked as an Assistant Researcher at the Shanghai Institute of Organic Chemistry (2011–2012). After conducting research as a postdoctoral fellow at Harvard Medical School (2012–2015), he became a Research Fellow in the Department of Biochemistry and Molecular Biology, Zhejiang University School of Medicine. |
 Jie Wu | Jie Wu received his B.Sc. in chemistry from Jiangxi Normal University in 1995 and he pursued his Ph.D. studies at the Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences under the supervision of Prof. Xue-Long Hou (1995–2000). After conducting research as a postdoctoral fellow at Harvard University (2000–2001), he worked as a visiting scientist at the Aaron Diamond AIDS Research Center (2001–2002) and a staff scientist in VivoQuest, Inc. (2002–2004). In 2004, he moved to the Department of Chemistry at Fudan University and held the Professor rank two years later. He received the Thieme Chemistry Journals Award in 2010 and Dow Innovation Challenge Award in 2013. He serves as a member of the Editorial Advisory Board of ACS Combinatorial Science. |
1. Introduction
Molecules bearing fluoro groups are always in great demand due to the wide existence of fluorinated molecules in pharmaceuticals and materials.1 It is known that the incorporation of fluorine atoms into molecules would often significantly improve the medicinal properties of drugs or leading compounds. Therefore, continuous efforts have been made for the construction of fluorinated skeletons and a number of methodologies have been developed in recent years.2 In particular, generation of compounds with CF3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 3 or CF2R4 groups attracts much more attention, with an expectation to improve the physical properties and bioactivities of organic compounds. In spite of various protocols for fluoroalkylation, radical processes5 still made great contributions for access to fluoro-containing molecules with the advantages of high reactivity. Among the routes to afford CF3 and CF2R radicals, a photoredox strategy6 as an efficient approach displays its unique advantages in terms of environment, energy, health and safety. It is notable that the transformations would easily proceed under mild reaction conditions with experimental ease. Among the reports of fluoroalkylation, the groups of MacMillan and Cho have described plenty of pioneer studies. For example, MacMillan and co-workers5g disclosed a facile trifluoromethylation of electron-rich arenes and heterocycles in 2011. During the reaction process, CF3SO2Cl was used as the CF3 source and was activated to release a CF3 radical in the presence of Ru(phen)32+. It was notable that some biologically active trifluoromethylated molecules could be synthesized easily from commercially available photocatalysts and in the presence of a household light bulb by using this strategy. Later, similar transformations were reported by Cho's group.5j With different photocatalysts [Ru(bpy)3Cl2] and CF3 sources (CF3I), Cho and co-workers successfully developed the photoredox trifluoromethylation of heterocycles. MacMillan's group disclosed the asymmetric synthesis of trifluoromethylated aldehydes with different CF3 sources.5k,l Amine catalysts were employed to provide high enantioselectivities through the mechanism of enamine, and a variety of aldehydes showed good performance to accomplish α-trifluoromethylation. Later, the same group5m used a strategy for transforming carbonyl compounds to enolsilanes, providing α-trifluoromethylated ketones, amides and esters in the absence of photocatalysts. Additionally, a one pot procedure for the generation of enolsilanes in situ and trifluoromethylation was accomplished, which was the first example to realize α-trifluoromethylation of carbonyl compounds (except aldehydes) via a photoredox approach. Besides these examples, fluoroalkylation via photoredox catalysis has made rapid progress with different fluoroalkylation reagents in the past few years, and the prospects of utilizing photochemistry into organic fluoroalkylation are still positive.
3 or CF2R4 groups attracts much more attention, with an expectation to improve the physical properties and bioactivities of organic compounds. In spite of various protocols for fluoroalkylation, radical processes5 still made great contributions for access to fluoro-containing molecules with the advantages of high reactivity. Among the routes to afford CF3 and CF2R radicals, a photoredox strategy6 as an efficient approach displays its unique advantages in terms of environment, energy, health and safety. It is notable that the transformations would easily proceed under mild reaction conditions with experimental ease. Among the reports of fluoroalkylation, the groups of MacMillan and Cho have described plenty of pioneer studies. For example, MacMillan and co-workers5g disclosed a facile trifluoromethylation of electron-rich arenes and heterocycles in 2011. During the reaction process, CF3SO2Cl was used as the CF3 source and was activated to release a CF3 radical in the presence of Ru(phen)32+. It was notable that some biologically active trifluoromethylated molecules could be synthesized easily from commercially available photocatalysts and in the presence of a household light bulb by using this strategy. Later, similar transformations were reported by Cho's group.5j With different photocatalysts [Ru(bpy)3Cl2] and CF3 sources (CF3I), Cho and co-workers successfully developed the photoredox trifluoromethylation of heterocycles. MacMillan's group disclosed the asymmetric synthesis of trifluoromethylated aldehydes with different CF3 sources.5k,l Amine catalysts were employed to provide high enantioselectivities through the mechanism of enamine, and a variety of aldehydes showed good performance to accomplish α-trifluoromethylation. Later, the same group5m used a strategy for transforming carbonyl compounds to enolsilanes, providing α-trifluoromethylated ketones, amides and esters in the absence of photocatalysts. Additionally, a one pot procedure for the generation of enolsilanes in situ and trifluoromethylation was accomplished, which was the first example to realize α-trifluoromethylation of carbonyl compounds (except aldehydes) via a photoredox approach. Besides these examples, fluoroalkylation via photoredox catalysis has made rapid progress with different fluoroalkylation reagents in the past few years, and the prospects of utilizing photochemistry into organic fluoroalkylation are still positive.
Usually in the fluoroalkylation reactions, photocatalysts were essential in the presence of visible light. Transition metals, especially [Ru] catalysts and [Ir] catalysts, were most widely employed in trifluoromethylation and difluoromethylation. Moreover, some organic molecules were used as the medium in the process of photocatalysis. The photocatalytic cycle proceeds well through single electron transfer (SET). Fig. 1 displays the reagents of CF3 and CF2 sources in the photochemical reactions, which are classified in this review to demonstrate the fluoroalkylation.
 | ||
| Fig. 1 Reagents of CF3 and CF2 sources. | ||
In some photoredox cycles, a photocatalyst in a photocatalytic cycle is initially stimulated to the excited state in the presence of visible light, which then provides an electron to the fluorinated reagent (Fig. 1) to release a CF3 radical or CF2 radical. It is notable that these fluorine precursors would release electrophilic CF3 and CF2R radicals by a photoredox process. The subsequent radical addition to an unsaturated bond with further transformation would produce the corresponding fluorinated products. Alkenes, alkynes, isocyanides, and arenes were frequently selected for the radical addition to provide access to fluorochemicals (Scheme 1). Meanwhile, fluoroalkyl radicals sometimes were generated through a reductive quenching cycle.
 | ||
| Scheme 1 Photoredox procedure of trifluoromethylation and difluoromethylation. | ||
2. Photoinduced trifluoromethylation and difluoroalkylation
2.1 Trifluoromethylation with Togni's reagent
As one of the most common trifluoromethylation reagents, Togni's reagent 1a is widely used in fluoroalkylation initiated by photocatalysts in the presence of light. During the conversion, the excited photocatalyst would facilitate the C–I bond cleavage to afford a trifluoromethyl radical through single electron transfer (SET). The following radical addition of the trifluoromethyl radical to an alkene, alkyne, isocyanide, or arene would lead to a new radical intermediate, which would then undergo other transformations such as oxidation/nucleophilic attack/cyclization/deprotonation to provide the trifluoromethylated products. In previous reports,5l,7 [Ru] and [Ir] could both act as photocatalysts and motivate Togni's reagents to release ˙CF3 followed by radical additions. Through this strategy, trifluoromethylated alkenes, ketones and lactones were synthesized under mild conditions.In 2014, Masson and co-workers8 disclosed a three-component reaction of difunctionalization of olefins with nucleophilic reagents and Togni's reagent 1a (Scheme 2). The alkenes containing carbamate groups were employed as substrates. As shown in Scheme 2, Ru(bpy)3(PF6)2 was proved to be an effective photocatalyst and could be stimulated in the presence of blue LEDs. The motivated Ru(bpy)3(PF6)2 promoted the cleavage of the C–I bond to release a trifluoromethyl radical via a SET process. The following radical addition to alkene with high regioselectivity could form a new radical, which would undergo oxidation to provide an N-acyliminium cation. The N-acyliminium cation could be easily trapped by nucleophilic reagents including NaN3, KCN, and ROH, leading to a series of trifluoromethylated compounds. Several examples are also presented in Scheme 2, which demonstrated its generality in the transformation.
 | ||
| Scheme 2 Trifluoromethylation of enecarbamates with Togni's reagent 1a catalyzed by Ru(bpy)3(PF6)2. | ||
Among the functionalization of alkenes, organic molecules could act as photosensitizers as well instead of transition metal photocatalysts to initiate the cleavage of O–I bonds giving rise to a trifluoromethyl radical. Scaiano and co-workers9 reported that methylene blue (MB) could be utilized as the photocatalyst in the trifluoromethylation of alkenes, alkynes and heterocycles (Scheme 3). The transformation proceeded smoothly with high efficiency and the reaction pathway was similar to the reactions catalyzed by transition metals.
 | ||
| Scheme 3 Methylene blue (MB) catalyzed trifluoromethylation of alkenes, alkynes and heterocycles. | ||
Some arenes could also react with Togni's reagent 1a to provide trifluoromethylated products. Ma and co-workers10 described that C–H bond cleavage of free anilines would occur to gain access to 2-(trifluoromethyl)anilines (Scheme 4). It was found that Togni's reagent 1a was the best choice as the CF3 precursor during the studies. From the examples listed in Scheme 4, it was clear that the para-position of aniline needed to be protected with substituents to obtain ortho-substituted products. The broad reaction scope was demonstrated, and different functional groups were all tolerated under the conditions. During the transformations, the process of releasing a trifluoromethyl radical and subsequent radical addition was similar to the above reactions. In the catalytic cycle, Ir(IV) acted as an oxidant to convert the radical to a cation, and the following deprotonation generated the trifluoromethyl-substituted anilines.
 | ||
| Scheme 4 Trifluoromethylation of anilines catalyzed by Ir(ppy)3 in the presence of Togni's reagent 1a. | ||
Besides simple functionalization of alkenes, addition to unsaturated bonds followed by annulation involving C–H bond cleavage was reported as well. By using this method, some heterocycles bearing a CF3 group with potential bioactivities could be synthesized in one step with high efficiency.11 Zhu and co-workers11e described some examples, showing that alkenes could be trifluoromethylated with Togni's reagent 1a in the presence of Ru(phen)3Cl2 and the following cyclization would provide CF3-containing compounds bearing a quaternary carbon center (Scheme 5). Some sensitively functional groups such as iodo and hydroxyl groups were compatible in this conversion. Additionally, substrates bearing heterocycles also performed well under the standard conditions. Similar to other trifluoromethylation reactions induced by visible light, the trifluoromethyl radical was produced via a SET process in the presence of light and Ru(phen)3Cl2. The following addition to the double bond of N-aryl acrylamide produced a radical intermediate. Subsequent radical C–H bond functionalization, oxidation, and deprotonation afforded the trifluoromethylated oxindoles. The detection of 2-iodobenzoic acid and the requirement of continuous irradiation of light both supported the proposed mechanism. Later, Wu and co-workers11g developed a catalyst-free radical cyclization of N-arylacrylamides with Togni's reagent enabled by photoenergy. The fluorinated 3,3-disubstituted 2-oxindoles were generated efficiently under ultraviolet irradiation without any metals or photo-redox catalysts. Good functional group tolerance was demonstrated.
 | ||
| Scheme 5 Photo-induced cyclization of N-arylacrylamides with Togni's reagent 1a. | ||
In another example of trifluoromethylation with C–H functionalization reported by Xia and co-workers,12 the transformation went through different pathways according to the different substituents attached to the aromatic ring (Scheme 6). Benzo-heterocycles were generated in most cases. However, the reactions of substrates with an alkoxy group attached onto the para-position of the aromatic ring preferred to form five-membered spirocycles. Similarly, a trifluoromethyl radical was produced from Togni's reagent 1a in the presence of the excited [fac-IrIII(ppy)3]*, which then underwent the radical addition of alkenes. For substrates with substituents except alkoxy groups, 6-endo-cyclization occurred to provide a six-membered benzo-heterocycle radical, while a five-membered spirocyclic radical was formed for substrates with alkoxy groups via 5-exo-cyclization. The following oxidation of the radical to the cation and subsequent deprotonation generated the corresponding products.
 | ||
| Scheme 6 Photo-induced Ir-catalyzed cyclization of N-arylacrylamides with Togni's reagent 1a. | ||
Synthesis of ketones could be achieved via trifluoromethylation of alkenes. In this process, DMSO was employed in the reaction system, which acted not only as a solvent but also as an oxidant to provide the corresponding ketones. Thus, trifluoromethylated ketones could be obtained efficiently under mild reaction conditions. Nearly at the same time, the groups of Akita13 and Guo14 disclosed two similar trifluoromethylations of alkenes with Togni's reagent 1a in the presence of photocatalysts (Scheme 7). Akita13 and co-workers utilized fac-Ir(ppy)3 as the photocatalyst, while Guo's group14 employed Ru(bpy)3(PF6)2 in the reactions. Meanwhile, DMAP and MgSO4 were found to be essential in Guo's report. The photoredox cycle of releasing a trifluoromethyl radical, radical addition to alkene, and oxidation provided the cation intermediate, similar to the above reports. Followed by the nucleophilic attack of DMSO to the cation afforded the trifluoromethylated ketones.
 | ||
| Scheme 7 Photo-induced synthesis of trifluoromethylated ketones. | ||
Trifluoromethyl-containing alkenes could be generated by trifluoromethylation of alkynes, and they also could be obtained via reactions between alkenes and CF3 precursors. For alkenes with leaving groups attached to the double bond, the double bond could be reserved after trifluoromethylation since an elimination process was involved. Akita and co-workers15 described the synthesis of trifluoromethylated alkenes through a reaction of potassium vinyltrifluoroborates with Togni's reagent 1a (Scheme 8). The broad scope of the reactions was demonstrated. For example, the pyridinyl-substituted product could be afforded in 66% yield. A possible mechanism was proposed, which suggested that [Ru(bpy)3](PF6)2 as a photocatalyst could be motivated to release a CF3 radical via a SET process. After addition, oxidation, and elimination, the trifluoromethyl-containing alkenes were produced. Interestingly, despite the existence of MeOH, the elimination of BXn occurred instead of the nucleophilic attack of MeOH to the cation intermediate.
 | ||
| Scheme 8 Synthesis of trifluoromethylated alkenes through a reaction of potassium vinyltrifluoroborates with Togni's reagent 1a. | ||
Later, Zhu and co-workers16 disclosed that trifluoromethylated alkenes could be synthesized as well from the reaction of alkenyl carboxylic acid with Togni's reagent 1a in the presence of Ir(ppy)3 as the photocatalyst (Scheme 9). The examples showed that some sensitive substituents and heterocycles were all compatible under the conditions. Although the conversion seemed to be similar with Akita's work,15 the mechanism indeed proceeded in another way. The failure to obtain the trifluoromethylated TEMPO indicated that the trifluoromethyl radical was not an intermediate in this reaction. Thus, the photocatalyst only reduced Togni's reagent 1a to form a radical anion, which would go through the cleavage of the O–I bond to form an iodine radical. This radical would then undergo addition to α,β-unsaturated carboxylic acids. Subsequent oxidation, decarboxylation and reductive elimination would generate the trifluoromethylated alkenes.
 | ||
| Scheme 9 Synthesis of trifluoromethylated alkenes through a reaction of α,β-unsaturated carboxylic acids with Togni's reagent 1a. | ||
For some substrates bearing allyl moieties, trifluoromethylated allylic compounds could be obtained under the photoinduced conditions. Elimination was still a key step to form the double bond of the final product. In recent examples, the trifluoromethyl radical released from Togni's reagent 1a was added to the double bond of alkenes. Followed by elimination, the expected products were provided. Gouverneur and co-workers17 reported the trifluoromethylation of allylsilanes, leading to trifluoromethylated allylic molecules (Scheme 10). It was found that Ru(bpy)3Cl2·6H2O was the photocatalyst of choice, which would promote the formation of a trifluoromethyl radical and the oxidation of allylsilanes. Three plausible reaction routes were proposed in the meantime. It was reasonable that a trifluoromethyl radical generated in the presence of a photocatalyst would lead to the addition of allylsilanes. After oxidation and subsequent elimination of TMSOMe, the trifluoromethylated allylic compounds would be afforded.
 | ||
| Scheme 10 Generation of trifluoromethylated allylic compounds through a reaction of allylsilanes with Togni's reagent 1a. | ||
In the absence of a leaving group, β-H elimination would occur to provide the trifluoromethylated allylic compounds. As reported by Vincent and co-workers,18 CuII-DMEDA (DMEDA = N,N′-dimethylethylenediamine) could serve as a photocatalyst to promote the release of a trifluoromethyl radical via a SET process (Scheme 11). Various sensitive functional groups such as halide and hydroxyl groups were all compatible during the transformations. O-containing heterocycles could also be synthesized through this approach. The photogenerated Cu(I) species was the actual photocatalyst with high reactivity. After radical addition and oxidation, the subsequent β-H elimination gave the desired products.
 | ||
| Scheme 11 Copper-catalyzed synthesis of trifluoromethylated allylic compounds. | ||
2.2 Trifluoromethylation with Umemoto's reagent
Umemoto's reagents are another kind of CF3 source, which have been used widely in trifluoromethylation under photocatalysis. In the presence of a photocatalyst, the C–S bond cleavage in Umemoto's reagents could easily occur to release a trifluoromethyl radical. Some conversions are similar to those employing Togni's reagent. Among the substrates used to react with Umemoto's reagents, alkenes are the most widespread, and various trifluoromethylated compounds could be generated. Akita and coworkers19 developed the oxy-trifluoromethylation of alkenes in the presence of [Ir] and successfully synthesized various ethers, which could go through elimination to afford trifluoromethylative alkenes.Gouverneur and co-workers20 reported an example of hydrotrifluoromethylation of unactivated alkenes with Umemoto's reagent 1b catalyzed by Ru(bpy)3Cl2 (Scheme 12). It was noteworthy that cholecalciferol from vitamin D3 could be synthesized under mild conditions by using this approach. Although heterocyclic substrates reacted with Umemoto's reagent 1b, the yield was fairly low. The mechanism was proposed, which showed that the trifluoromethyl radical was formed via a SET process involving C–S bond cleavage and its subsequent radical addition to alkenes. In the reaction, methanol served as the solvent as well as the H atom donor in the meantime.
 | ||
| Scheme 12 Hydrotrifluoromethylation of unactivated alkenes with Umemoto's reagent 1b catalyzed by Ru(bpy)3Cl2. | ||
Difunctionalization of alkenes involving trifluoromethylation with Umemoto's reagent 1c has also been developed in recent years. Generally, the photocatalyst was stimulated by visible light to promote the release of a CF3 radical from Umemoto's reagent 1cvia a SET process. The following radical addition to the double bond and oxidation of the radical intermediate afforded a cation, which then reacted with a nucleophile leading to the corresponding products. Akita and co-workers21 disclosed that MeCN and H2O could act as the solvent as well as the nucleophile in the reaction of alkenes with Umemoto's reagent 1c (Scheme 13). In particular, aminotrifluoromethylation of steroids and amino acids was realized in this protocol. A possible reaction pathway revealed that the nucleophilic attack from MeCN to the trifluoromethylated cation underwent a Ritter-type amination. Followed by hydrolyzation, the desired amides were afforded.
 | ||
| Scheme 13 Aminotrifluoromethylation of alkenes with Umemoto's reagent 1c. | ||
Masson and co-workers22 also developed two examples of difunctionalization of alkenes with Umemoto's reagent 1c (Scheme 14). Electron-rich arenes, TMSN3, and amines were utilized as the nucleophiles to react with the cation oxidized from the trifluoromethylated radical. During the reaction process, Ru(bpy)3(PF6)2 served as the photocatalyst to release the CF3 radical from Umemoto's reagent 1c in the presence of visible light.
 | ||
| Scheme 14 Two examples of difunctionalization of alkenes with Umemoto's reagent 1c. | ||
Intramolecular heteroatoms could serve as nucleophiles to trap the cations generated in situ. Generally, alkenes bearing amide, carboxyl, and hydrazone groups were used in such reactions to construct trifluoromethylated heterocycles. The possible reaction pathways were similar to the difunctionalization of alkenes as mentioned above. It was found that in these transformations, Ru(bpy)3(PF6)2 was the choice to release the CF3 radical from Umemoto's reagent 1c in the presence of visible light. Moreover, the base was essential, which depended on the substrates employed.
There were two similar examples by using N-allylamides as the oxygen nucleophiles, as reported by the groups of Xiao23 and Akita.24 In these two reports, the exo-type of products were generated. Xiao and co-workers23 described the synthesis of oxazolines/benzoxazines under mild conditions (Scheme 15). The trifluoromethylated radical and cation were produced in a typical pathway. The oxygen of amide then underwent intramolecular nucleophilic attack to afford the oxazolines. Additionally, Akita and co-workers24 showed that the synthesis of trifluoromethylated heterocycles could be realized at an extremely low temperature with 2,6-lutidine as the base (Scheme 16). The exploration of the reaction scope showed that heterocycles were compatible under the reaction conditions, and steric hindrance was not a barrier in the conversion.
 | ||
| Scheme 15 Synthesis of oxazolines by using Umemoto's reagent 1c. | ||
 | ||
| Scheme 16 Preparation of spiro-heterocycles by using Umemoto's reagent. | ||
Akita and co-workers25 also demonstrated that a carboxylic group could be utilized as an oxygen nucleophile (Scheme 17). Under similar reaction conditions, the trifluoromethylated lactones could be obtained. The reaction of internal alkenoic acids with Umemoto's reagent 1c gave the endo-products, while the reaction of terminal alkenoic acids with Umemoto's reagent underwent exo-cyclization to provide the corresponding exo-products.
 | ||
| Scheme 17 Generation of trifluoromethylated lactones via reactions of alkenoic acids with Umemoto's reagent 1c. | ||
Xiao and co-workers26 further reported that oximes could serve as the oxygen nucleophiles in the reactions of alkenes with Umemoto's reagent 1c (Scheme 18). Additionally, hydrazones could accomplish the similar trifluoromethylated annulation under the same reaction conditions. Thus, β,γ-unsaturated hydrazones/oximes were employed as substrates and only exo-products were generated due to the use of terminal alkenes.
 | ||
| Scheme 18 Reactions of β,γ-unsaturated hydrazones/oximes with Umemoto's reagent 1c. | ||
Among the reactions of Umemoto's reagents and alkenes, continuous efforts have been made to the synthesis of trifluoromethylated alkenes. Usually, the trifluoromethyl radical obtained from Umemoto's reagent in the presence of a photocatalyst would react with alkenes to provide the carbon radical intermediates. These radical intermediates were soon oxidized into cations, which then underwent deprotonation to afford the corresponding trifluoromethylated alkenes.
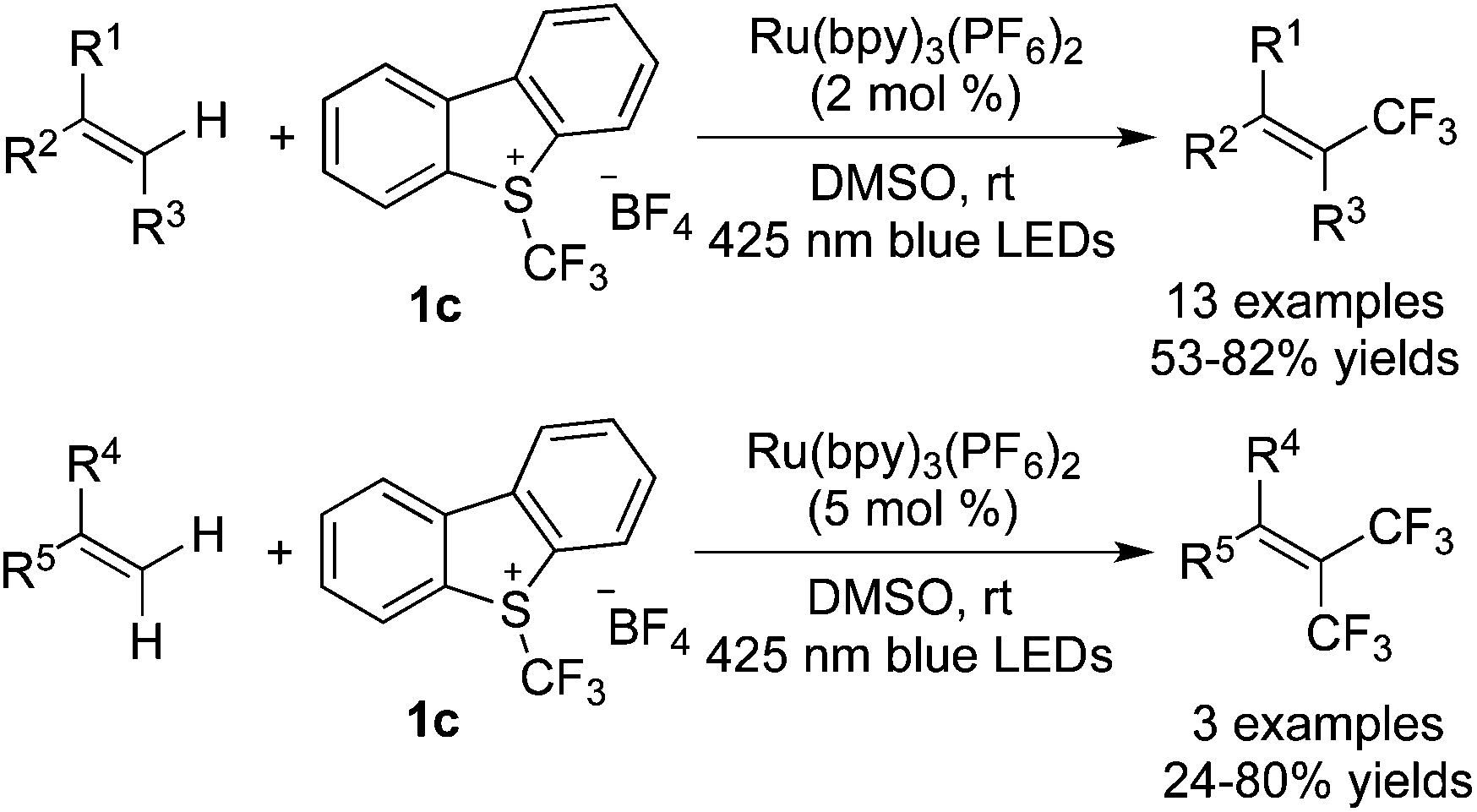
Qing and co-workers27 reported that in the presence of different photocatalysts, the reactions of aryl alkenes afforded trifluoromethylated alkenes with different configurations. In the presence of Ru(bpy)3Cl2·H2O, E-trifluoromethylated alkenes were formed with Togni's reagent 1d (Scheme 19). Interestingly, Z-trifluoromethylated alkenes were obtained in the presence of Ir(ppy)3 when Umemoto's reagent 1c was employed in the above transformation (Scheme 20). Since aryl groups could make radicals more stable, electron-efficient styrenes with N/O-groups attached to aromatic rings were selected as substrates to control the regioselectivity. It was found that the utilization of Ru(bpy)3Cl2·H2O and compound 1d would generate single E-products with high efficiency. However, minor E-products with the majority of Z-products were obtained when Ir(ppy)3 and Umemoto's reagent 1c were employed. Configurations were determined by using different photocatalysts and it might be due to the higher triplet energy of Ir(ppy)3 compared with Ru(bpy)3Cl2·H2O. The triplet excited state of  transformed the ground singlet state of the E-trifluoromethylated alkene to its lowest-energy triplet state, which was easily attenuated to provide a mixture of isomers through isomerization of the E-alkenes.
transformed the ground singlet state of the E-trifluoromethylated alkene to its lowest-energy triplet state, which was easily attenuated to provide a mixture of isomers through isomerization of the E-alkenes.
 | ||
| Scheme 19 Generation of trifluoromethylated alkenes. | ||
 | ||
| Scheme 20 Mono-trifluoromethylation and di-trifluoromethylation of alkenes with Umemoto's reagent 1c. | ||
Akita and co-workers reported the mono-trifluoromethylation and di-trifluoromethylation of alkenes in the presence of [Ru(bpy)3](PF6)2 with Umemoto's reagent 1c (Scheme 20).28 Mono-trifluoromethylated alkenes could be obtained with a slight excess of a CF3 precursor, while di-trifluoromethylation took place with the addition of over 2.0 equivalents of Umemoto's reagent 1c. This work showed that the sp2 C–H bond could undergo cleavage to gain access to mono/di-trifluoromethylated alkenes under nearly the same reaction conditions.
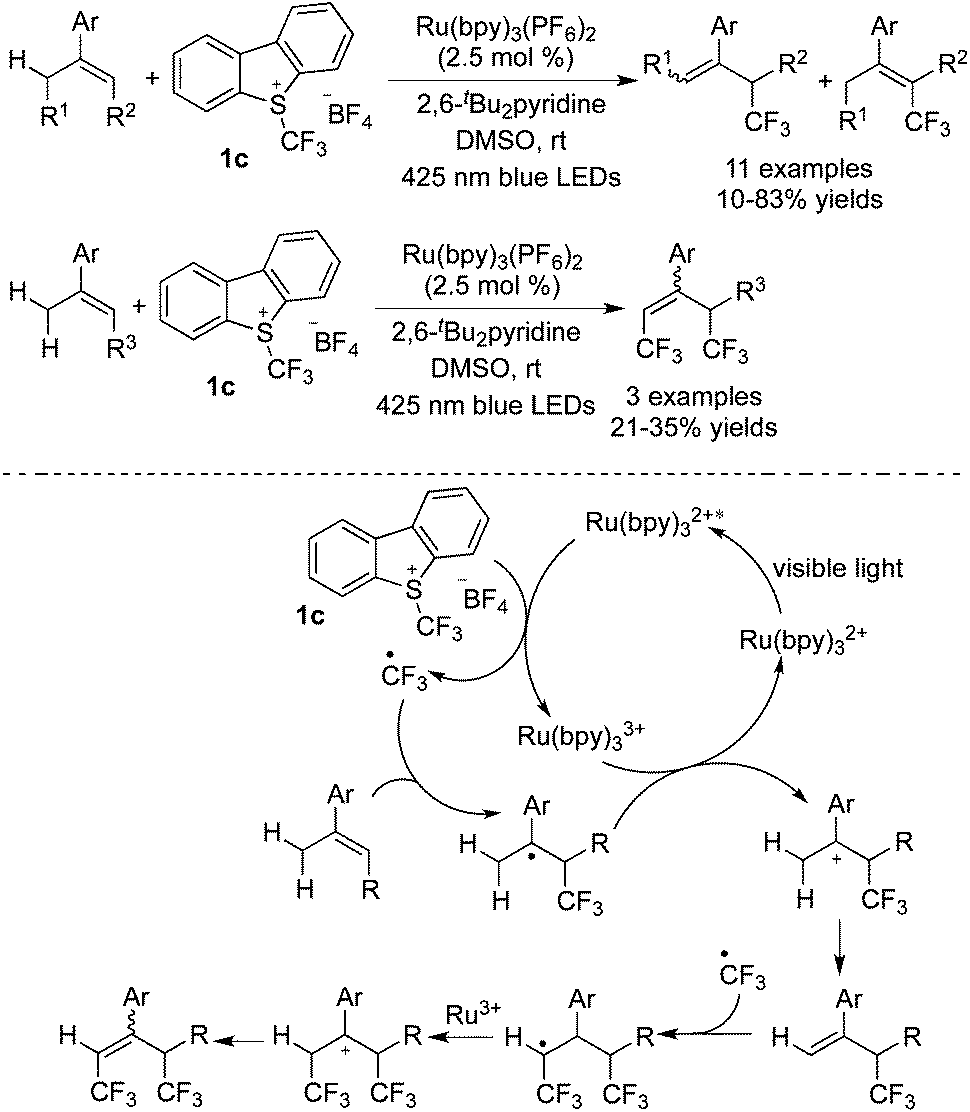
Later, the same group29 developed the mono/di-trifluoromethylation of the allyl C–H bond catalyzed by [Ru(bpy)3](PF6)2 (Scheme 21). For the mechanism displayed in Scheme 21, the first cycle of mono-trifluoromethylation was similar to those conversions as mentioned above. The formation of the mono-trifluoromethylated product provided the possibility of another radical addition of trifluoromethyl. Followed by oxidation and deprotonation, the di-trifluoromethylated products were produced.
 | ||
| Scheme 21 Mono/di-trifluoromethylation of the allyl C–H bond catalyzed by [Ru(bpy)3](PF6)2. | ||
As to the alkenes attached with a hydroxyl group, trifluoromethylation occurred, accompanied by rearrangement to afford ketones. Linear alcohols and cyclic alcohols both underwent rearrangement while the rearrangement of cyclic alcohols resulting in ring-expansion was more favored. Glorius and co-workers30 employed cycloalkanol-substituted styrene derivatives as the substrates to undergo trifluoromethylation with Umemoto's reagent 1b in the presence of [Ru(bpy)3](PF6)2 (Scheme 22). The scope of the reactions revealed that heterocycles and spiro-cycles were all compatible in the reactions. The proposed mechanism indicated the formation of ketones via ring-expansion. TMSOTf played an important role during the process of nucleophilic substitution. 1,2-Alkyl migration caused by ring-expansion resulted in the formation of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond with the release of the silyl group, thus affording trifluoromethylated ketones.
O bond with the release of the silyl group, thus affording trifluoromethylated ketones.
 | ||
| Scheme 22 Photo-induced synthesis of trifluoromethylated ketones. | ||
A similar result for the generation of trifluoromethylated ketones using Umemoto's reagent 1c as the CF3 source was also reported by Kim and co-workers (Scheme 23).31 The reaction was catalyzed by Ru(phen)3Cl2. Except endocyclic alkenes, terminal alkenes were also employed as the substrates. Despite the absence of TMSOTf as a protecting reagent, the transformation proceeded well under the conditions. The plausible mechanism was extremely similar to that proposed by Glorius and coworkers.30 Promoted by OH, 1,2-carbon migration would take place to provide the desired trifluoromethylated ketones.
 | ||
| Scheme 23 Synthesis of trifluoromethylated ketones catalyzed by Ru(phen)3Cl2. | ||
Isocyanides could be used as a partner with Umemoto's reagent as well for the trifluoromethylation reactions. Such a transformation was usually followed by cyclization to form trifluoromethylated isoquinolines. Yu and co-workers32 developed the reactions between isocyanides and Umemoto's reagent 1c in the presence of [Ir] (Scheme 24). Substrates bearing heterocycles could also perform well under the reaction conditions. A plausible reaction pathway was proposed, which revealed that isocyanide would accept the addition of a CF3 radical. Subsequent intramolecular annulation, oxidation and deprotonation afforded the trifluoromethylated quinolines.
 | ||
| Scheme 24 Generation of trifluoromethylated quinolones via a reaction of isocyanide with Umemoto's reagent 1c. | ||
2.3 Trifluoromethylation of alkenes and arenes with CF3SO2X (X = Cl, Na)
CF3SO2X (X = Cl, Na) is a useful trifluoromethyl precursor to produce a trifluoromethyl radical with the release of SO2via a SET process initiated by photocatalysts. CF3SO2Cl would undergo a reductive process to release a trifluoromethyl radical while CF3SO2Na's active mode might be an oxidative process.6b Some pioneer studies2i,5j,33 have revealed that the reactions between enamines, alkenes or arenes and CF3SO2X could successfully accomplish trifluoromethylation of these substrates. For CF3SO2Cl, after releasing a trifluoromethyl radical, a chloride anion was produced at the same time and acted as a nucleophile. It is known that addition of a trifluoromethyl radical to an alkene and oxidation of the radical intermediate would afford a cation. Consequently, the nucleophilic attack of the chloride anion to the cation intermediate resulted in the difunctionalized trifluoromethylated products.Han and co-workers34 reported the trifluoromethylation of alkenes catalyzed by Ru(phen)3Cl2 (Scheme 25). Good functional group tolerance was observed. For instance, heterocycles, iodine, amide, and formyl groups were all compatible under the reaction conditions. As mentioned above, a trifluoromethyl radical along with a chloride anion was released from CF3SO2Cl via a SET process. The chloride anion acted as a nucleophile to trap the cation, leading to the final products.
 | ||
| Scheme 25 Trifluoromethylation of alkenes with CF3SO2Cl catalyzed by Ru(phen)3Cl2. | ||
Dolbier Jr. and co-workers35 described a similar result by employing the same fluoroalkylation reagent and base, catalyzed by Cu(dap)2Cl. Alkenes attached to electron-withdrawing groups were used as the substrates in these transformations (Scheme 26). The reaction scope was extended to other fluoroalkylsulfonyl chlorides, giving rise to a range of fluorine-containing molecules.
 | ||
| Scheme 26 Trifluoromethylation of alkenes with CF3SO2Cl catalyzed by Cu(dap)2Cl. | ||
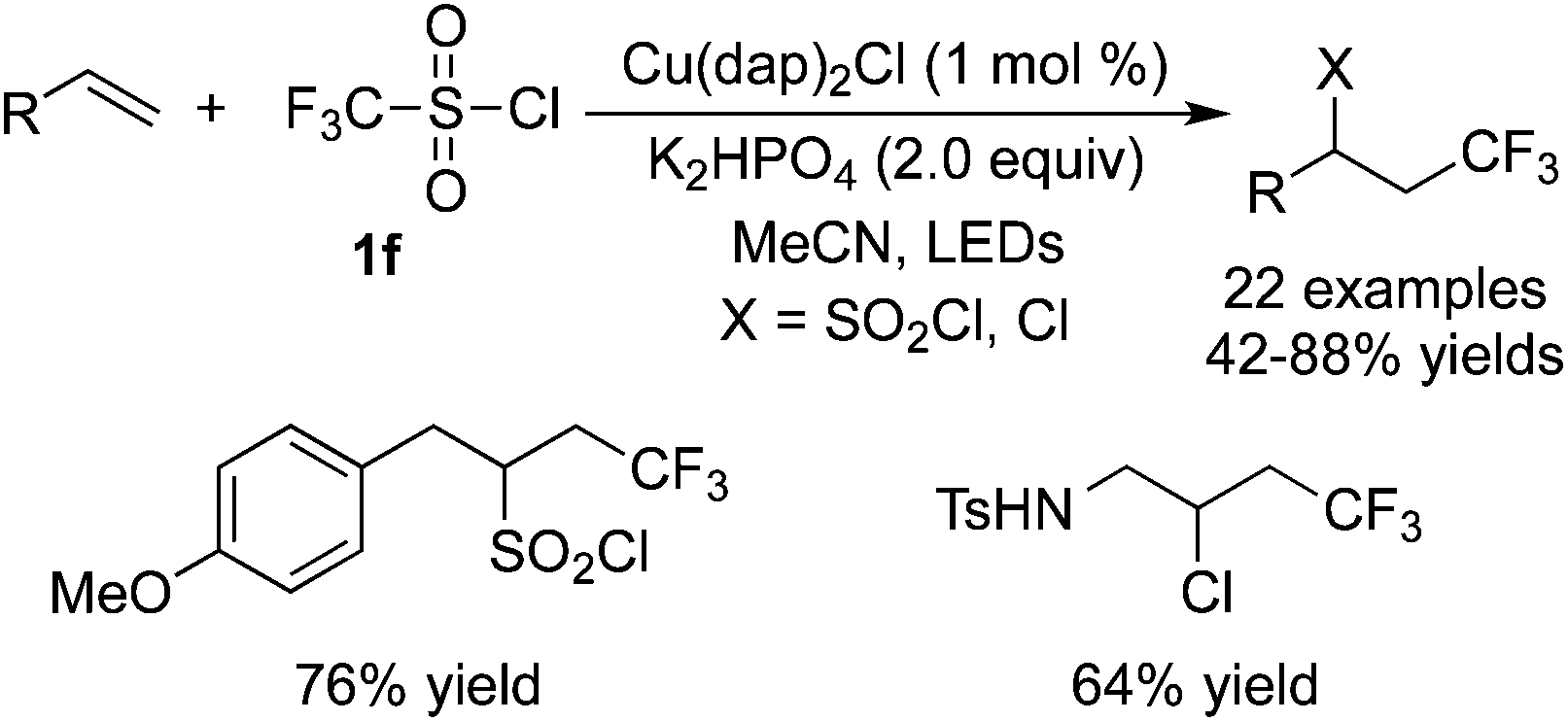
Reactions between CF3SO2Cl and unactivated alkenes were developed by Reiser and co-workers36 (Scheme 27). Either trifluoromethylchlorosulfonylation or chlorotrifluoromethylation of alkenes would occur in the presence of [Cu(dap)2]Cl according to different substrates. It was interesting that chlorotrifluoromethylated products were obtained when substrates with potential coordinating atoms, such as heteroatoms, were used in the reactions. However, substrates without heteroatoms led to trifluoromethylchlorosulfonylation in the presence of [Cu(dap)2]Cl. Furthermore, in the presence of other photocatalysts, such as Ru(bpy)3Cl2, chlorotrifluoromethylated products were generated as the major products while only [Cu(dap)2]Cl favoured trifluoromethylchlorosulfonylation with certain substrates.
 | ||
| Scheme 27 Reactions between CF3SO2Cl and unactivated alkenes. | ||
Hydrotrifluoromethylation of alkenes with CF3SO2Na in the presence of an Ir catalyst has been reported by Li and co-workers recently (Scheme 28).37 Unactivated terminal olefins were used as the substrates, and methanol served not only as a solvent but also a H donor instead of a nucleophile.
 | ||
| Scheme 28 Hydrotrifluoromethylation of unactivated alkenes. | ||
Trifluoromethylation of alkenes with CF3SO2Cl involving intramolecular annulation was also developed. Similar to the process by employing Togni's reagents and Umemoto's reagents, addition of a trifluoromethyl radical and subsequent cyclization produced trifluoromethylated heterocycles in one pot with high efficiency under mild conditions. Dolbier and co-workers38 found that α,β-unsaturated amides were good partners to accept the radical addition of fluoro-containing radicals in the presence of photocatalysts (Scheme 29). Several fluoroalkylsulfonyl chlorides were examined and it was found that different fluoro sources needed different photocatalysts. When Rf (CF3/C4H9/CF2CO2Me) was employed, Ru(phen)3Cl2 was proved to be the best choice. While Rf′ (CF2H, CFH2, CH2CF3) was used as a replacement, fac-Ir(ppy)3 provided better results instead. The transformation proceeded smoothly with high efficiency to produce the exo-trifluoromethylated molecules.
 | ||
| Scheme 29 Photo-induced cyclization of N-arylacrylamides with RfSO2Cl. | ||
Trifluoromethylation of substrates with a sulfonyl group attached to amides by using CF3SO2Cl as the CF3 source was further described by Xia and co-workers (Scheme 30).39 In this transformation, Ru(bpy)3Cl2 was used as the photocatalyst to promote the release of a CF3 radical. Desulfonylation was the key step to produce an amide radical. Two pathways of forming different products are presented in Scheme 30. A direct hydrogen abstraction occurred when the substituent attached onto the nitrogen atom was an aryl group. Relatively, the amide radical attacked the aromatic ring when the substituent attached onto the nitrogen atom was an alkyl group. The subsequent oxidation and deprotonation led to trifluoromethylated oxindoles.
 | ||
| Scheme 30 Trifluoromethylation of substrates with a sulfonyl group attached to amides by using CF3SO2Cl as the CF3 source. | ||
Enol acetates were selected as the substrates for the synthesis of trifluoromethylated ketones, as reported by Yu and co-workers (Scheme 31).40 It was discovered that Ir(ppy)2(dtbbpy)PF6 showed good performance to promote the formation of a trifluoromethyl radical from CF3SO2Cl. As shown in Scheme 30, addition of the trifluoromethyl radical to the double bond provided a carbon radical intermediate, which was then oxidized by Ir(IV) to produce a cation. The subsequent loss of the acetyl cation completed the conversion to form the desired ketones.
 | ||
| Scheme 31 Synthesis of trifluoromethylated ketones from enol acetates. | ||
CF3SO2X could also be used as the CF3 source to react with arenes. Itoha and co-workers41 carried out the fluoroalkylation of electron-rich arenes (attached to the OMe group) with RfSO2Na catalysed by ANQ-2-COOH (Scheme 32). ANQ-2-COOH acted as an electron transfer mediator to promote the release of an electron deficient Rf radical. Electron-rich arenes and heteroarenes were good choices to afford the corresponding products. It was postulated that the electron deficient CF3 radical was preferred to be added to the electron-rich positions.
 | ||
| Scheme 32 Fluoroalkylation of electron-rich arenes with RfSO2Na catalyzed by ANQ-2-COOH. | ||
Li and co-workers42 utilized similar arenes to react with CF3SO2Na, and developed a novel and facile route to generate trifluoromethylated arenes via a photoredox strategy (Scheme 33). Electron-rich arenes and heterocycles were compatible under the reaction conditions. It was notable that in the presence of UV irradiation and acetone, this transformation proceeded smoothly in the absence of transition metals and oxidants. Additionally, trifluoromethylation occurred in the presence of visible light when a mixture of ethyl acetate and diacetyl was employed. The mechanism was different in terms of the release of a trifluoromethyl radical. Acetone was stimulated by light irradiation and could react with CF3SO2Na to form ˙CF3SO2. The subsequent SO2 extrusion occurred to release ˙CF3. Followed by radical addition, oxidation and deprotonation, the trifluoromethylated arenes and heterocycles were produced.
 | ||
| Scheme 33 Synthesis of trifluoromethylated arenes and heterocycles under metal- and oxidant-free conditions. | ||
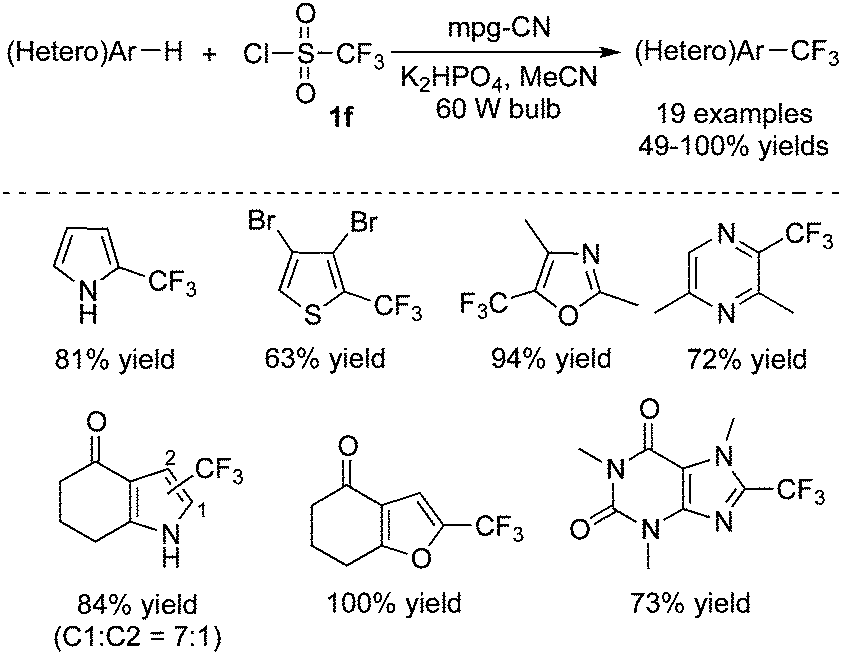
For the synthesis of trifluoromethylated heterocycles, Blechert and co-workers43 used a special recyclable graphitic carbon nitride polymer as the photocatalyst to realize the trifluoromethylation of heterocycles (Scheme 34). Mpg-CN (recyclable mesoporous graphitic carbon nitride) showed good performance to activate CF3SO2Cl, as a result of its appropriate electrochemical behavior. From the examples shown in Scheme 34, it was found that a variety of heterocycles were compatible under the reaction conditions, including N-heterocycles, O-heterocycles, S-heterocycles. Subsequently, Rueping and co-workers developed a photo-induced hydrofluoromethylation of alkenes with CF3SO2Na in a bath under a continuous flow.44 During the transformation, 4,4′-dimethoxybenzophenone was used as the photocatalyst, which was stimulated to promote the release of a trifluoromethyl radical via a SET process.
 | ||
| Scheme 34 Synthesis of trifluoromethylated heterocycles catalyzed by Mpg-CN. | ||
2.4 Trifluoromethylation of alkenes/alkynes/thiophenols with CF3I and its derivatives
As simple trifluoromethylation reagents, CF3I and its derivatives performed well via the cleavage of the C–I bond to provide a trifluoromethyl radical in the presence of a photosensitizer. Subsequent radical addition to unsaturated bonds led to a radical intermediate, which was trapped to form trifluoromethylated molecules. CF3I has been used to synthesize different trifluoromethylated compounds, such as ketones, arenes, alkenes, aza-cyclopropanes and so on. In these transformations,3g,45 either transition metals or organic molecules served as photocatalysts to release the trifluoromethyl radical.Zhao and co-workers46 reported the difunctionalization of styrenes with CF3CH2I in the presence of fac-Ir3+(ppy)3 and oxygen (Scheme 35). Linear alcohols with a CF3 substituent were synthesized under mild reaction conditions. A possible reaction route was proposed. CF3CH2I 1i went through a SET process by the excited fac-Ir(III)(ppy)3. The subsequent deiodination afforded ˙a CH2CF3 radical, which underwent stereoselective addition to the double bond to provide a benzyl radical. This benzyl radical was captured by using O2 rather than water to produce the hydroxyl group, as proved by isotope labeling experiments. It was noteworthy that water could promote the reaction and no desired products were obtained in the absence of water.
 | ||
| Scheme 35 Synthesis of trifluoromethylated alcohols. | ||
With regard to alkynes, the triple bond could easily accept the addition of the CF3 radical to form alkenyl radical intermediates, which underwent subsequent transformation. Cho and co-workers47 disclosed that the multiple products of trifluoromethylated alkenyl iodides, alkenes, and alkynes could be synthesized by using the same substrates (Scheme 36). Terminal alkynes were selected as substrates and CF3I was employed as the CF3 source. The reactions proceeded in different pathways after the addition of the CF3 radical. The alkenyl radical intermediate underwent iodide abstraction from CF3I to provide the corresponding alkenyl iodides. These compounds further proceeded through de-iodination in the presence of a photocatalyst and hydrogen abstraction to give trifluoromethylated alkenes. The trifluoromethylated alkynes were formed by elimination of the hydrogen radical. The detection of trifluoromethylated alkenyl iodides in the reactions and additional experiments of converting alkenyl iodides to trifluoromethylated alkenes under standard reaction conditions both confirmed the reasonability of the proposed pathways.
 | ||
| Scheme 36 Generation of trifluoromethylated alkenyl iodides, alkenes, and alkynes. | ||
It was disclosed by Cho48 that trifluoroethylation of terminal alkynes was realized to synthesize allylic CF3-compounds with CF3CH2I in the presence of visible light (Scheme 37). Iodotrifluoroethylated alkenes and hydrotrifluoroethylated alkenes were obtained under different reaction conditions. The reaction mechanism was very similar to the reactions between CF3I and terminal alkynes with iodotrifluoroethylated alkenes as the key intermediates.
 | ||
| Scheme 37 Generation of trifluoroethylated alkenyl iodides and alkenes. | ||
Cho and co-workers49 also discovered that propargylic alcohols performed well in trifluoromethylation to provide fluorinated ketones, due to the presence of a hydroxyl group in the substrates (Scheme 38). Based on the report as mentioned above,42 it was reasonably proposed that a trifluoromethyl radical was generated in the presence of Ru(bpy)3Cl2 which went through radical addition to form a vinyl radical. Hydrogen abstraction from the amminium radical cation provided allylic alcohol as the key intermediate, which was detected by NMR spectroscopy as well as by GC analysis. Further formation of an allylic radical, isomerization, and hydrogen abstraction produced the corresponding trifluoromethylated ketones.
 | ||
| Scheme 38 Synthesis of fluorinated ketones via trifluoromethylation of propargylic alcohols. | ||
Synthesis of SCF3-containing compounds by using CF3I via trifluoromethylation was also realized. Noël and co-workers50 utilized Ru(bpy)3Cl2 as the photocatalyst in the reactions of thiophenols with CF3I (Scheme 39). Thiophenols, heteroaryl thiols, and aliphatic thiols were all good reactants under the reaction conditions. The generation of a CF3 radical was similar to others as described previously via a SET process. This electrophilic CF3 radical reacted with thiols to form the S–CF3 bond.
 | ||
| Scheme 39 Synthesis of SCF3-containing compounds by using CF3I via trifluoromethylation. | ||
2.5 Trifluoromethylation with other CF3 sources
Besides the common CF3 sources as listed above, other CF3 reagents have also been employed in trifluoromethylation reactions. Guo and co-workers51 focused on the use of TMSCF3 in the trifluoromethylation of benzaldehydes (Scheme 40). In the presence of O2, trifluoromethylated alcohols were obtained with high efficiency. It was demonstrated that benzaldehydes were transformed into triplet intermediates in the presence of light via inter system crossing. In the meantime, homolysis of the C–Si bond of TMSCF3 promoted by light and O2 generated the CF3 radical. Further transformations resulted in the trifluoromethylated alcohols. | ||
| Scheme 40 Trifluoromethylation of benzaldehydes using TMSCF3. | ||
As a shelf-stable and easy-to-use electrophilic CF3 reagent, Yagupol'skii–Umemoto reagent 1e was employed by Akita and co-workers52 to achieve the difunctionalization of alkynes (Scheme 41). The scopes of the reaction showed that the ester group and cycloparaffins were compatible and only E-products were generated with high stereoselectivity. An explanation for the configuration was proposed. The generation of a vinyl radical was similar to other reports. Oxidation of the vinyl radical to a cation and subsequent nucleophilic attack of the triflate anion via anti-addition relative to the electronegative CF3 group occurred to provide the final products.
 | ||
| Scheme 41 Trifluoromethylation of alkynes. | ||
2.6 Difluoroalkylation with BrCF2FG
Among the difluoroalkylation reagents, much attention has been paid to two kinds of reagents classified as HCF2LG (LG = leaving group) and XCF2FG (X = Br, I; FG = functional group). The rare previous reports53 mainly focused on the difluoromethylation of heterocycles. These two CF2 precursors underwent different bond cleavages to provide the corresponding RCF2 radicals. For XCF2FG (X = Br, I), C–Br/C–I bond cleavage was favored and the functional groups were reserved to generate difluoroalkylated products bearing a CF2FG moiety. On the contrary, as a result of removal of the leaving group, HCF2 was attached to difluoromethylated products. Additionally, CF2FG could be transformed to HCF2 under certain reaction conditions.Cho and co-workers54 developed the difluoroalkylation reactions by employing BrCF2CO2Et as the CF2 source and fac-[Ir(ppy)3] as the photocatalyst. Hydrodifluoroalkylated products and alkenyl-difluoroalkylated products were obtained in the presence of different bases and solvents (Scheme 42). Under the irradiation of visible light, fac-[Ir(ppy)3] was stimulated and promoted the cleavage of C–Br to form a difluoroalkyl radical. As a consequence of radical addition to the double bond, the carbon radical intermediates were generated and subsequently underwent H abstraction from DBU to afford the hydrodifluoroalkylated products. Meanwhile, the abstraction of bromide from BrCF2CO2Et occurred in another route. Subsequent elimination of HBr produced alkenyl-difluoroalkylated products.
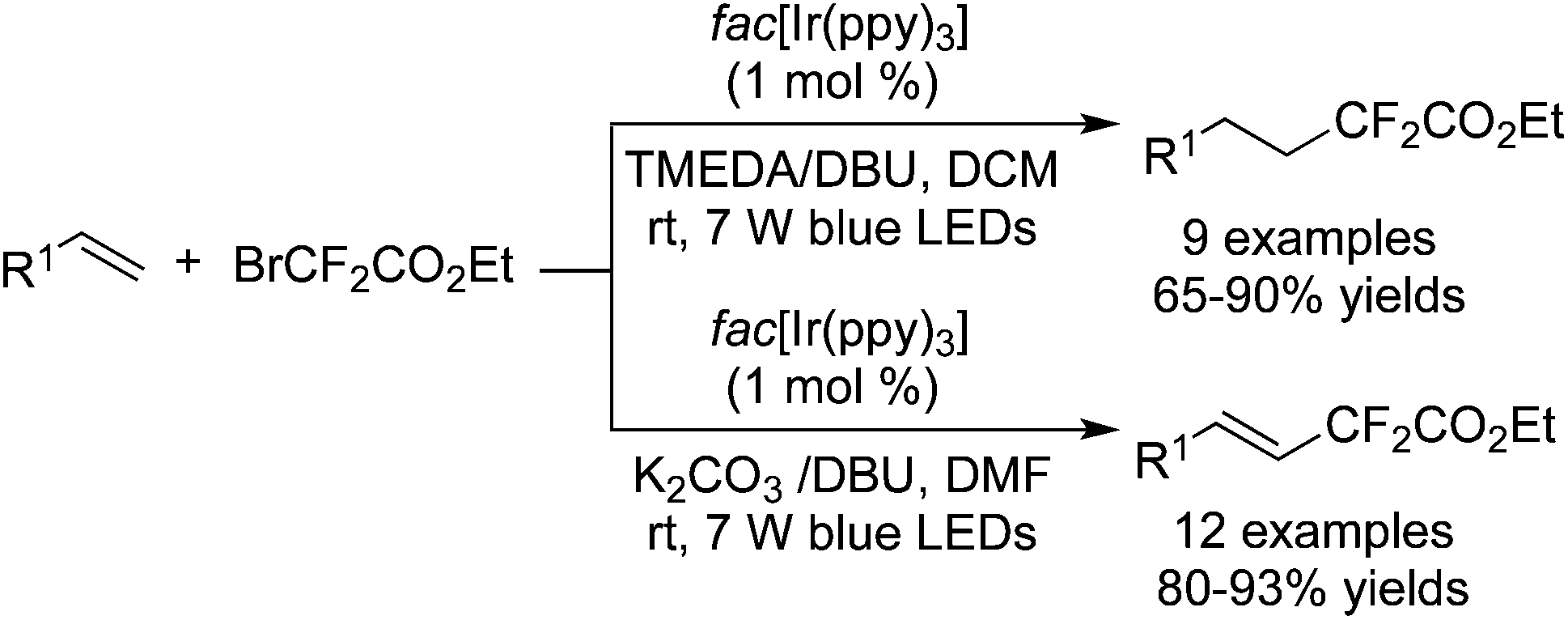 | ||
| Scheme 42 Difluoroalkylation reactions with BrCF2CO2Et catalyzed by fac-[Ir(ppy)3]. | ||
Zhu and co-workers55 disclosed the carbodifluoroalkylation of allyl alcohols in the presence of fac-[Ir(ppy)3] (Scheme 43). In this transformation, aryl migration via the spiro[2.5]octadienyl radical was involved to provide the carbodifluoroalkylated ketones. It was found that BrCF2COR (R = OEt/morpholine) was compatible under the reaction conditions. The reaction process for the generation of a carbon radical intermediate was similar to other difluoroalkylation reactions. This carbon radical intermediate went through intramolecular cyclization. Followed by 1,2-aryl migration, oxidation and deprotonation, the corresponding carbodifluoroalkylated ketones were provided.
 | ||
| Scheme 43 Carbodifluoroalkylation of allyl alcohols catalyzed by fac-[Ir(ppy)3]. | ||
Similar to trifluoromethylation, when α,β-unsaturated amides were employed as substrates in the difluoroalkylation, intramolecular annulation took place to afford fluorinated heterocycles.
Zhu and co-workers56 reported the aryldifluoroacetylation of cinnamamides in the presence of fac-Ir(ppy)3 (Scheme 44). It was found that two patterns of intramolecular annulation were produced according to the different substituents attached to the aromatic ring. The six-membered aryldifluoroacetylated benzocycles were obtained for the substrates with electron-withdrawing groups or no substituents attached to the aromatic rings. When OMe or OH was attached onto the para position of the aromatic rings, the unexpected five-membered azaspiro quinones were generated under different reaction conditions. This work was similar to Xia's report for trifluoromethylation,12 and the reaction pathways for the outcome were almost the same.
 | ||
| Scheme 44 Aryldifluoroacetylation of cinnamamides catalyzed by fac-Ir(ppy)3. | ||
Difluoroalkylation of alkynes and isocyanides involving intramolecular annulation was developed as well. For example, 3-difluoroacetylated coumarins could be obtained when alkynoates were utilized as the substrates in the reaction with BrCF2CO2Et catalyzed by [fac-Ir(ppy)3] (Scheme 45).57
 | ||
| Scheme 45 Generation of 3-difluoroacetylated coumarins. | ||
Yu and co-workers58 reported the reactions between isocyanides and BrCF2CO2Et by using [fac-Ir(ppy)3] as the photocatalyst (Scheme 46). Difluoromethylated phenanthridine derivatives were synthesized via a stepwise procedure. Moreover, several substrates were selected to carry out stepwise experiments and one-pot experiments at the same time. The reaction route was consistent with the difluoroalkylation of alkenes, which proceeded through radical addition followed by intramolecular 6-endo-cyclization.
 | ||
| Scheme 46 Reactions between isocyanides and BrCF2CO2Et by using [fac-Ir(ppy)3] as the photocatalyst. | ||
Cho and co-workers59 found that electron-rich arenes and some heteroaromatics could be used as the substrates to undergo difluoroalkylation with BrCF2CO2Et in the presence of [fac-Ir(ppy)3] as well (Scheme 47). Compared with the electron-rich arenes, heterocycles showed higher reactivity with a lower photocatalyst loading and lower amount of BrCF2CO2Et. Moreover, the α-position of the heterocycles was the first to accept the addition of the CF2 radical, which afforded the major products of the α-substituted heterocycles.
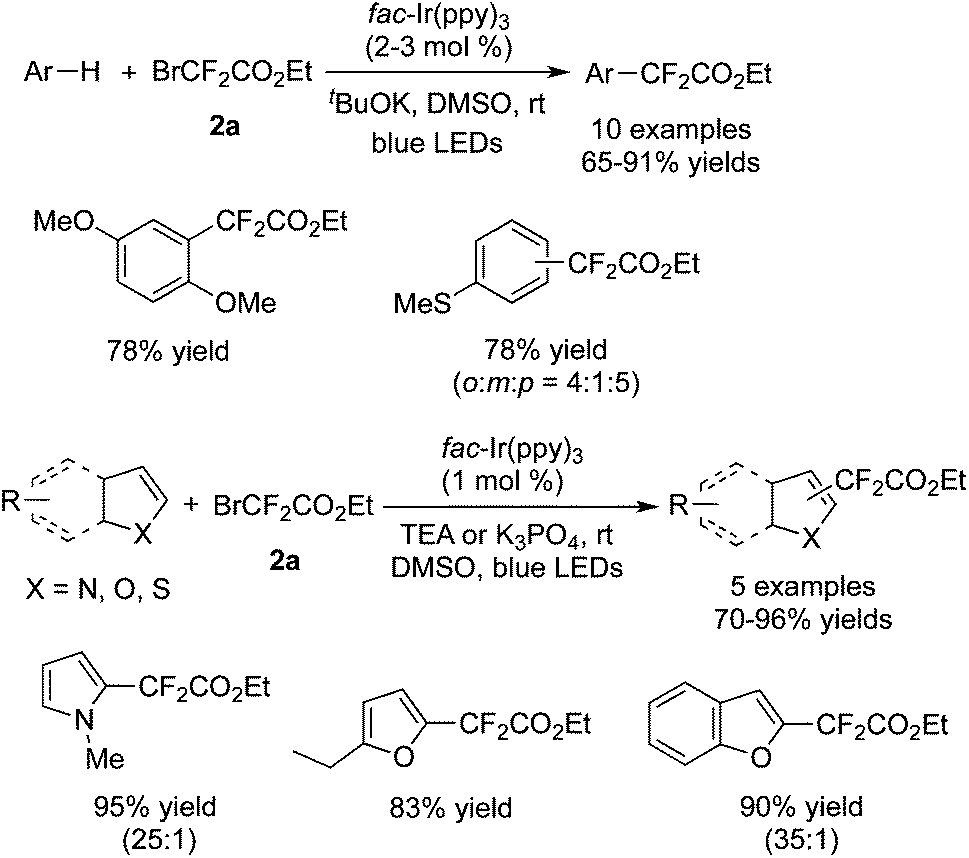 | ||
| Scheme 47 Difluoroalkylation of electron-rich arenes and some heteroaromatics. | ||
Difluoroalkylation of hydrazones involving C–H bond cleavage via an aminyl radical/polar mechanism was reported by Zhu and co-workers (Scheme 48).60 A range of functional groups attached to hydrazones was tolerated under the reaction conditions. Two hypotheses were proposed, which would probably undergo an aminyl radical/polar mechanism and a carbon radical/polar mechanism. The latter could be excluded through computational Gibbs free-energy profiles. For the plausible reaction pathway, it was described that the addition of the CF2 radical to the CN bond produced a nitrogen radical intermediate. The abstraction of hydrogen was blocked due to the stability of the intermediate through a possible three-electron π-bonding interaction. An aminyl radical/polar crossover step occurred. Followed by deprotonation, the corresponding products were produced.
 | ||
| Scheme 48 Difluoroalkylation of hydrazones. | ||
Other difluoroalkylated reagents were also developed. Liu and co-workers61 described the difluoroacetamidation of inactive aromatic compounds and heterocycles with bromodifluoroacetamides in the presence of [fac-Ir(ppy)3]. Arenes with electron-donating groups or electron-withdrawing groups both generated the desired products with high efficiency. Additionally, various bromodifluoroacetamides were employed in the reactions to yield the products including heterocyclic, cyclic and linear amides.
Fluorinated polycyclic lactone scaffolds could be constructed through the difluoroalkylation reaction of active alkenes (Scheme 49).62 In this report, Ir(ppy)3 acted as the photocatalyst and α-bromo-α,α-difluoroacetophenones were used as the CF2 precursors. It was notable that asymmetric synthesis of difluoroalkylated lactones was realized.
 | ||
| Scheme 49 Difluoroacetamidation of inactive aromatic compounds and heterocycles with bromodifluoroacetamides in the presence of [fac-Ir(ppy)3]. | ||
Cai and coworkers63 employed α-aryl-β, β-difluoroenol silyl as the CF2 source to react with arene diazonium tetrafluoroborates in the presence of a small amount of Ru(bpy3)Cl2·6H2O (Scheme 50). It was proposed that aryl radicals were formed to undergo radical addition to carbonyl groups, which could provide new radicals. These new radicals were then oxidized by arene diazonium tetrafluoroborate to cations. An elimination reaction finally occurred to afford ketones which could be converted to difluoromethylarenes under basic conditions.
 | ||
| Scheme 50 Difluoromethylation of arene diazonium tetrafluoroborates. | ||
Since phosphorus-containing compounds show good performance in pharmaceuticals and pesticides, continuous efforts have been made for the synthesis of these compounds. BrCF2PO(OEt)2 was an important CF2 source used in the difluoroalkylation of isoarylcyanides, arenes and heterocycles with the introduction of fluorine and phosphorus in one step.
There were two similar examples nearly reported at the same time by the groups of Wang64 and Liu65 to demonstrate the reactions between BrCF2PO(OEt)2 and isocyanides (Scheme 51). Reactions of substrates with strong electron-withdrawing groups worked well in this transformation. A plausible pathway is proposed in Scheme 48. In the presence of [fac-Ir(ppy)3], addition of the CF2 radical to isocyanide afforded a radical intermediate. Subsequent intramolecular annulation, oxidation and deprotonation produced the corresponding N-heterocycles.
 | ||
| Scheme 51 Difluoroalkylation reaction of BrCF2PO(OEt)2 and isocyanide. | ||
The difluoromethylenephosphonation of arenes and heterocycles with BrCF2PO(OEt)2 in the presence of [fac-Ir(ppy)3] was also described (Scheme 52).66 Reactions of electron-rich and electron-deficient arenes both proceeded smoothly to provide the corresponding products. Different heterocycles were all compatible under the conditions. Moreover, the α-position of the heterocycles was preferred to be decorated with the CF2 group.
 | ||
| Scheme 52 Difluoromethylenephosphonation of arenes and heterocycles with BrCF2PO(OEt)2. | ||
Wang and co-workers67 developed the difluorosulfuration of heterocycles with an iodo-substituted CF2 reagent: PhSO2CF2I in the presence of [Ru(bpy)3]Cl2·6H2O (Scheme 53). In this process, electrophilic radical addition to the heterocycles occurred at the most electron-rich position. Furthermore, desulfonylation of the products provided difluoromethylheterocycles with high efficiency with Mg as the reductant. Additionally, one pot of this transformation from electron-rich heterocycles to difluoromethylheterocycles was feasible with the removal of solvent.
 | ||
| Scheme 53 Difluorosulfuration of heterocycles with PhSO2CF2I in the presence of [Ru(bpy)3]Cl2·6H2O. | ||
2.7 Direct difluoromethylation with HCF2LG
In the past few years, introduction of a CF2H moiety into small molecules under photocatalysis was also developed. Compared with the difluoromethylated approaches for the incorporation of a CF2FG group, the direct introduction of CF2H without the removal of the functional groups after difluoroalkylation was attractive. Among the reagents utilized, HCF2SO2Cl was used widely in direct difluoromethylation.Dolbier and co-workers68 reported the hydrodifluoromethylation of electron-deficient alkenes with HCF2SO2Cl as the CF2 precursor and (TMS)3SiH as the H source (Scheme 54). Reactions of both linear alkenes and exocyclic alkenes proceeded well under the conditions. Various electron-withdrawing groups were compatible in this conversion. It was established that a HCF2 radical was released from HCF2SO2Cl by the excited [fac-Ir(ppy)3]. The subsequent radical addition to electron-deficient alkenes led to an electrophilic radical intermediate. Followed by H abstraction from (TMS)3SiH, the final products were obtained.
 | ||
| Scheme 54 Hydrodifluoromethylation of electron-deficient alkenes with HCF2SO2Cl and (TMS)3SiH. | ||
They further described the difluoromethylation of inactive alkenes in the presence of Cu(dap)2Cl (Scheme 55).69 N/O-Heterocycles could be synthesized by using this strategy. In the transformation, Ag2CO3 was essential to promote the release of the CF2H radical, which was generated via single electron reduction of HCF2SO2Cl by Cu(dap)2Cl. After the radical addition to the double bond, a cation was afforded via oxidation, which initiated the intramolecular nucleophilic attack of nitrogen leading to the difluoromethylated heterocycles.
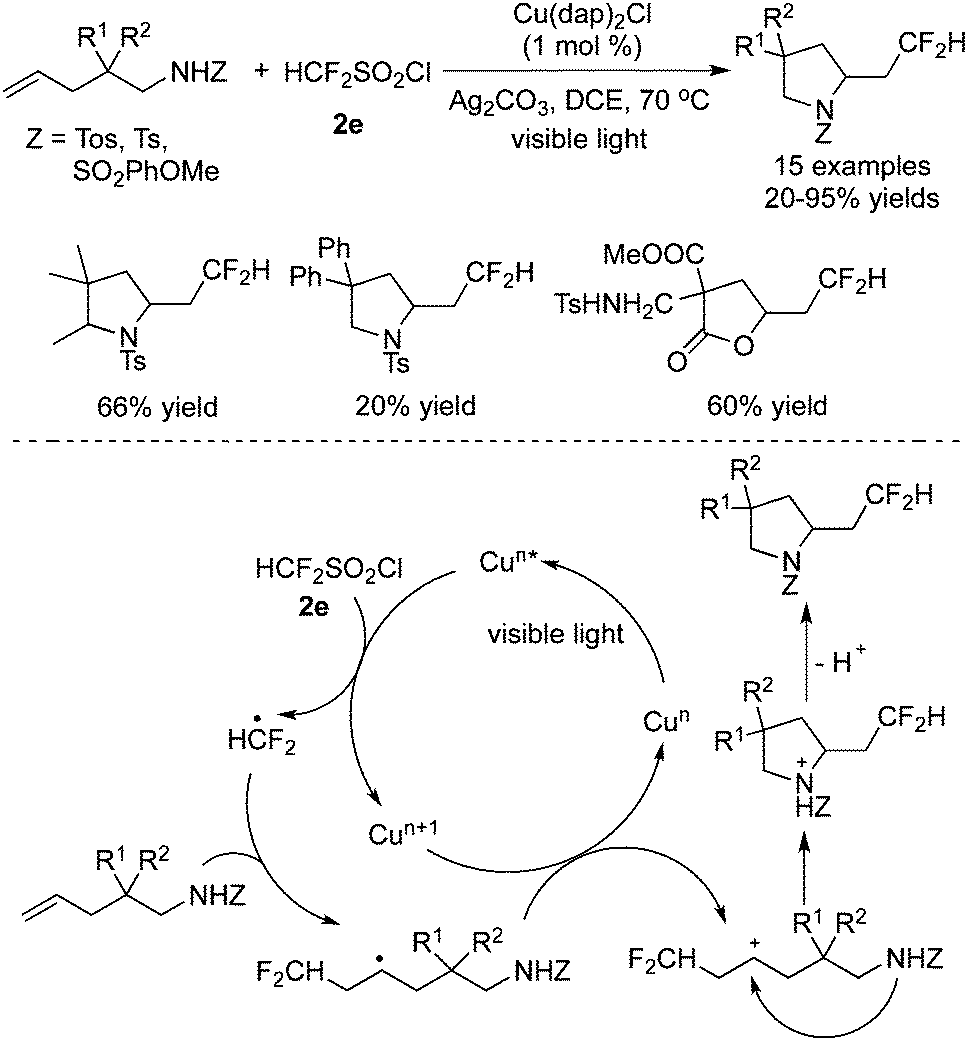 | ||
| Scheme 55 Difluoromethylation of inactive alkenes in the presence of Cu(dap)2Cl. | ||
By employing HCF2SO2Cl as the CF2 source, the direct difluoromethylation of isocyanides in the presence of fac-Ir(ppy)3 without the removal of functional groups was successfully achieved (Scheme 56).70 The scope of this reaction was demonstrated, and the electron-donating groups, electron-withdrawing groups, and heterocyclic groups were all compatible under the reaction conditions.
 | ||
| Scheme 56 Direct difluoromethylation of isocyanides in the presence of fac-Ir(ppy)3. | ||
The direct difluoromethylation of isocyanides with a new CF2 precursor 2h in the presence of [Ru(bpy)3]Cl2·6H2O was developed by Hu and co-workers (Scheme 57).71 Heterocyclic groups and SO2Me were tolerated under the conditions. This method also provided the application of a variety of mono- and trifluoromethyl heteroarylsulfones as the radical fluoroalkylation sources. During the reaction process, sodium carbonate was essential to serve as the base for deprotonation. And carbonate might be of great importance as the electron donor to reduce *[Ru]2+ to [Ru]+, which would indeed initiate the catalyst to release a fluorinated radical.
 | ||
| Scheme 57 Direct difluoromethylation of isocyanides catalyzed by [Ru(bpy)3]Cl2·6H2O. | ||
N-Tosyl-S-difluoromethyl-S-phenylsulfoximine was utilized in direct CF2H of alkenes as well. Akita and co-workers72 developed the difunctionalization of alkenes with N-tosyl-S-difluoromethyl-S-phenylsulfoximine and H2O in the presence of fac-[Ir(ppy)3] (Scheme 58). A range of difluoromethylated alcohols were synthesized. As a shelf-stable CF2 precursor, N-tosyl-S-difluoromethyl-S-phenylsulfoximine was employed to produce a sulfur radical anion via a SET process in the presence of a photocatalyst. Subsequent protonation led to the cleavage of the S–C bond to provide a CF2H radical. Followed by radical addition to the double bond, oxidation and nucleophilic attack by water, the difluoromethylated alcohols were formed.
 | ||
| Scheme 58 Synthesis of difluoromethylated alcohols through difunctionalization of alkenes with N-tosyl-S-difluoromethyl-S-phenylsulfoximine and H2O. | ||
A phosphorus-containing CF2 reagent 2f ([Ph3PCF2Br]+Br−) was developed by Qing and co-workers73 in the hydrodifluoromethylation of inactive alkenes (Scheme 59). A range of substrates were good reactants, providing the corresponding products in moderate to good yields. During the reaction process, tetrahydrofuran acted as the solvent as well as the H source.
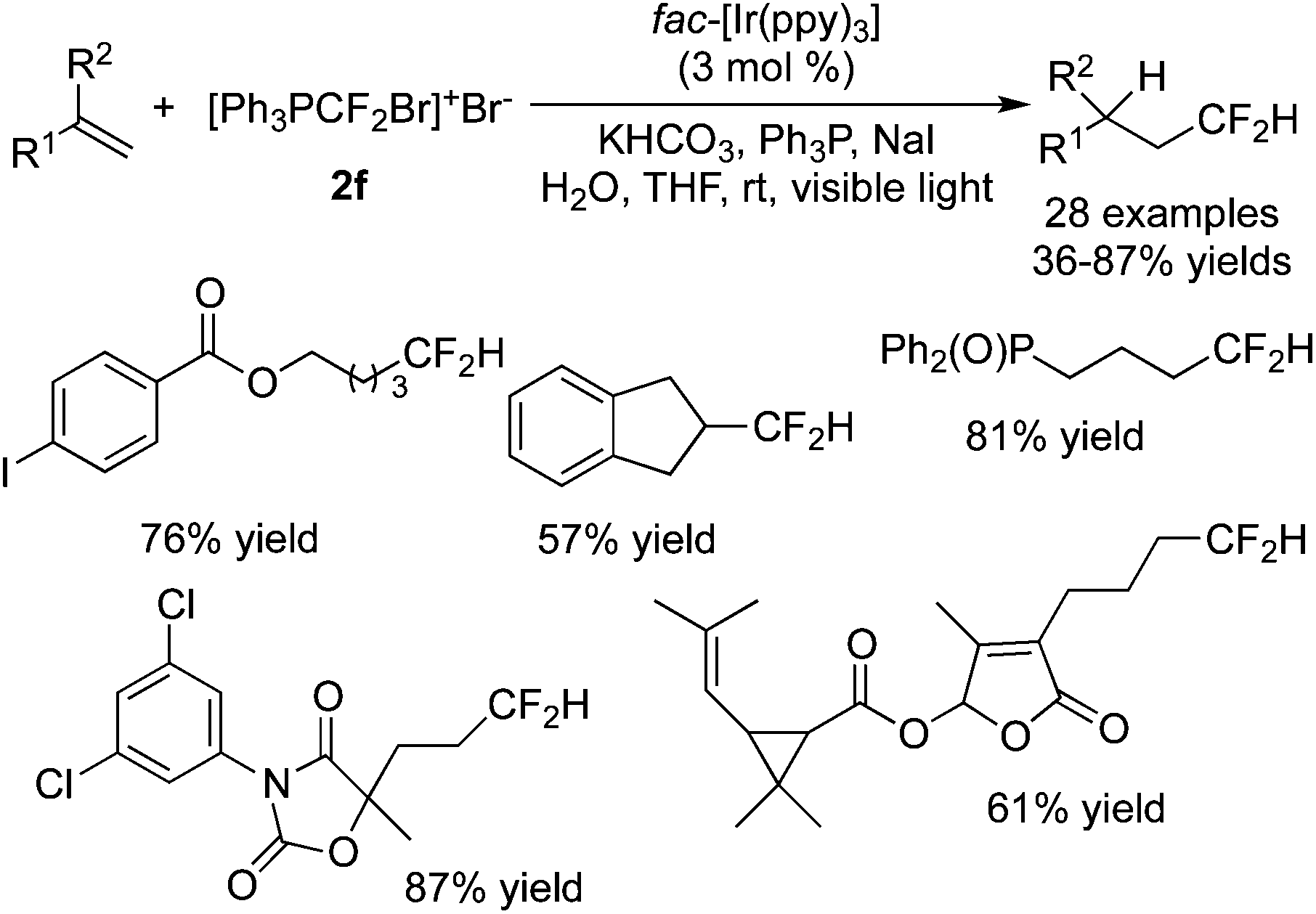 | ||
| Scheme 59 Hydrodifluoromethylation of inactive alkenes. | ||
Recently, Qing and co-workers74 discovered that (bifluoromethyl)triphenylphosphonium bromide 2f could be utilized as the CF2 source to achieve bromodifluoromethylation of inactive alkenes in the presence of Ir(ppy)3 (Scheme 60). One pot transformation through bromodifluoromethylation and elimination led to difluoromethylated alkenes. Copper(II) bromide was added to control the selective generation of bromodifluoromethylated products. Radical addition of ˙CF2H to alkenes provided alkyl radical intermediates. Oxidation by Ir(IV) and the presence of CuBr2 could both afford cations. Followed by nucleophilic attack of the bromide, the corresponding bromodifluoromethylated products were generated. It was also possible to obtain alkylcopper species as key intermediates, which were then transformed to the final products.
 | ||
| Scheme 60 Bromodifluoromethylation of inactive alkenes in the presence of Ir(ppy)3. | ||
3. Conclusions
In conclusion, recent advances in trifluoromethylation and difluoroalkylation induced by photocatalysts are summarized. Most of the photoredox reactions proceed efficiently under mild conditions with simple operation. Various fluorinated reagents are developed and applied in different transformations. Usually, the photocatalyst in a photocatalytic cycle is initially stimulated to the excited state in the presence of visible light, which then provides an electron to the fluorinated reagent to release a CF3 radical or CF2 radical. The subsequent radical addition to unsaturated bonds with further transformation would produce the corresponding fluorinated products. Alkenes, alkynes, isocyanides, and arenes are frequently employed as substrates for access to fluorochemicals. The advantages of trifluoromethylation and difluoroalkylation under photocatalysis including mild reaction conditions, good functional group tolerance, and experimental ease will make the approaches attractive for further applications.Acknowledgements
Financial support from the National Natural Science Foundation of China (no. 21372046 and 21532001) is gratefully acknowledged.Notes and references
- For recent reviews, see:
(a) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2013, 114, 2432 CrossRef PubMed
; (b) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS PubMed
- For selected recent examples, see:
(a) C. Ni, M. Hu and J. Hu, Chem. Rev., 2015, 115, 765 CrossRef CAS PubMed
-
(a) T. Besset, T. Poisson and X. Pannecoucke, Chem. – Eur. J., 2014, 20, 16830 CrossRef CAS PubMed
-
(a) V. V. Levin, A. L. Trifonov, A. A. Zemtsov, M. I. Struchkova, D. E. Arkhipov and A. D. Dilman, Org. Lett., 2014, 16, 6256 CrossRef CAS PubMed
-
(a) X. Liu, F. Xiong, X. Huang, L. Xu, P. Li and X. Wu, Angew. Chem., Int. Ed., 2013, 52, 6962 CrossRef CAS PubMed
- For recent reviews, see:
(a) M. Akita and T. Koike, C. R. Chim., 2015, 18, 742 CrossRef CAS
-
(a)
A. F. Collings and C. Critchley, Artificial photosynthesis, from basic Biology to industrial application, Wiley-VCH, Weinheim, Germany, 2005 CrossRef
- A. Carboni, G. Dagousset, E. Magnier and G. Masson, Org. Lett., 2014, 16, 1240 CrossRef CAS PubMed
- S. P. Pitre, C. D. McTiernan, H. Ismaili and J. C. Scaiano, ACS Catal., 2014, 4, 2530 CrossRef CAS
- J. Xie, X. Yuan, A. Abdukader, C. Zhu and J. Ma, Org. Lett., 2014, 16, 1768 CrossRef CAS PubMed
- Recent reviews and selected examples for the synthesis of 3,3-disubstituted 2-oxindoles via the radical addition reaction involving N-arylacrylamides, see:
(a) J.-R. Chen, X.-Y. Yu and W.-J. Xiao, Synthesis, 2015, 604 Search PubMed
- F. Gao, C. Yang, G. Gao, L. Zheng and W. Xia, Org. Lett., 2015, 17, 3478 CrossRef CAS PubMed
- R. Tomita, Y. Yasu, T. Koike and M. Akita, Angew. Chem., Int. Ed., 2014, 53, 7144 CrossRef CAS PubMed
- L. Li, Q. Chen and Y. Guo, J. Fluorine Chem., 2014, 167, 79 CrossRef CAS
- Y. Yasu, T. Koike and M. Akita, Chem. Commun., 2013, 49, 2037 RSC
- P. Xu, A. Abdukader, K. Hu, Y. Cheng and C. Zhu, Chem. Commun., 2014, 50, 2308 RSC
- S. Mizuta, K. M. Engle, S. Verhoog, O. Galicia-López, M. O'Duill, M. Médebielle, K. Wheelhouse, G. Rassias, A. L. Thompson and V. Gouverneur, Org. Lett., 2013, 15, 1250 CrossRef CAS PubMed
- R. Beniazza, F. Molton, C. Duboc, A. Tron, N. D. McClenaghan, D. Lastécouèresa and J.-M. Vincent, Chem. Commun., 2015, 51, 9571 RSC
- T. Koike, Y. Yasu and M. Akita, Angew. Chem., Int. Ed., 2012, 51, 9567 CrossRef PubMed
- S. Mizuta, S. Verhoog, K. M. Engle, T. Khotavivattana, M. O'Duill, K. Wheelhouse, G. Rassias, M. Medebielle and V. Gouverneur, J. Am. Chem. Soc., 2013, 135, 2505 CrossRef CAS PubMed
- Y. Yasu, T. Koike and M. Akita, Org. Lett., 2013, 15, 2136 CrossRef CAS PubMed
- A. Carboni, G. Dagousset, E. Magnier and G. Masson, Chem. Commun., 2014, 50, 14197 RSC
- Q. Deng, J. Chen, Q. Wei, Q. Zhao, L. Lu and W. Xiao, Chem. Commun., 2015, 51, 3537 RSC
- N. Noto, K. Miyazawa, T. Koike and M. Akita, Org. Lett., 2015, 17, 3710 CrossRef CAS PubMed
- Y. Yasu, Y. Arai, R. Tomita, T. Koike and M. Akita, Org. Lett., 2014, 16, 780 CrossRef CAS PubMed
- Q. Wei, J. Chen, X. Hu, X. Yang, B. Lu and W. Xiao, Org. Lett., 2015, 17, 4464 CrossRef CAS PubMed
- Q. Lin, X. Xu and F. Qing, J. Org. Chem., 2014, 79, 10434 CrossRef CAS PubMed
- R. Tomita, Y. Yasu, T. Koike and M. Akita, Beilstein J. Org. Chem., 2014, 10, 1099 CrossRef PubMed
- M. Asano, R. Tomita, T. Koike and M. Akita, J. Fluorine Chem., 2015, 185, 83 CrossRef
- A. Sahoo, J. Li and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 11577 CrossRef PubMed
- S. B. Woo and D. Y. Kim, J. Fluorine Chem., 2015, 178, 214 CrossRef CAS
- Y. Cheng, X. Yuan, H. Jiang, R. Wang, J. Ma, Y. Zhang and S. Yu, Adv. Synth. Catal., 2014, 356, 2859 CrossRef CAS
- H. Jiang, C. Huang, J. Guo, C. Zeng, Y. Zhang and S. Yu, Chem. – Eur. J., 2012, 18, 15158 CrossRef CAS PubMed
- S. H. Oh, Y. R. Malpani, N. Ha, Y. Jung and S. B. Han, Org. Lett., 2014, 16, 1310 CrossRef CAS PubMed
- X. Tang and W. R. Dolbier Jr., Angew. Chem., Int. Ed., 2015, 54, 4246 CrossRef CAS PubMed
- D. B. Bagal, G. Kachkovskyi, M. Knorn, T. Rawner, B. M. Bhanage and O. Reiser, Angew. Chem., Int. Ed., 2015, 54, 6999 CrossRef CAS PubMed
- L. Zhu, L. Wang, B. Li, B. Fu, C. Zhang and W. Li, Chem. Commun., 2016, 52, 6371 RSC
- X. Tang, C. S. Thomoson and W. R. Dolbier Jr., Org. Lett., 2014, 16, 4594 CrossRef CAS PubMed
- L. Zheng, C. Yang, Z. Xu, F. Gao and W. Xia, J. Org. Chem., 2015, 80, 5730 CrossRef CAS PubMed
- H. Jiang, Y. Cheng, Y. Zhang and S. Yu, Eur. J. Org. Chem., 2013, 5485 CrossRef CAS
- L. Cui, Y. Matusaki, N. Tada, T. Miura, B. Uno and A. Itoha, Adv. Synth. Catal., 2013, 355, 2203 CrossRef CAS
- L. Li, X. Mu, W. Liu, Y. Wang, Z. Mi and C. Li, J. Am. Chem. Soc. 20161385809 Search PubMed.
- M. Baar and S. Blechert, Chem. – Eur. J., 2015, 21, 526 CrossRef CAS PubMed
- Q. Lefebvre, N. Hoffmann and M. Rueping, Chem. Commun., 2016, 52, 2493 RSC
-
(a) Y. Ye and M. S. Sanford, J. Am. Chem. Soc., 2012, 134, 9034 CrossRef CAS PubMed
- L. Li, M. Huang, C. Liu, J. Xiao, Q. Chen, Y. Guo and Z. Zhao, Org. Lett., 2015, 17, 4714 CrossRef CAS PubMed
- N. Iqbal, J. Jung, S. Park and E. J. Cho, Angew. Chem., Int. Ed., 2014, 53, 539 CrossRef CAS PubMed
- G. Roh, N. Iqbal and E. J. Cho, Chin. J. Chem. 201634459 Search PubMed.
- S. Park, J. M. Joo and E. J. Cho, Eur. J. Org. Chem., 2015, 4093 CrossRef CAS
- N. J. W. Straathof, B. J. P. Tegelbeckers, V. Hessel, X. Wang and T. Noël, Chem. Sci., 2014, 5, 4768 RSC
- J. Sun, X. Peng and H. Guo, Tetrahedron Lett., 2015, 56, 797 CrossRef CAS
- R. Tomita, T. Koike and M. Akita, Angew. Chem., Int. Ed., 2015, 54, 12923 CrossRef CAS PubMed
-
(a) S. Murakami, H. Ishii, T. Tajima and T. Fuchigami, Tetrahedron, 2006, 62, 3761 CrossRef CAS
- B. Yu, N. Iqbal, S. Park and E. J. Cho, Chem. Commun., 2014, 50, 12884 RSC
- P. Xu, K. Hu, Z. Gu, Y. Cheng and C. Zhu, Chem. Commun., 2015, 51, 7222 RSC
- Z. Gu, H. Zhang, P. Xu, Y. Cheng and C. Zhu, Adv. Synth. Catal., 2015, 357, 3057 CrossRef CAS
- W. Fu, M. Zhu, G. Zou, C. Xu, Z. Wang and B. Ji, J. Org. Chem., 2015, 80, 4766 CrossRef CAS PubMed
- X. Sun and S. Yu, Org. Lett., 2014, 16, 2938 CrossRef CAS PubMed
- J. Jung, E. Kim, Y. You and E. J. Cho, Adv. Synth. Catal., 2014, 356, 2741 CrossRef CAS
- P. Xu, G. Wang, Y. Zhu, W. Li, Y. Cheng, S. Li and C. Zhu, Angew. Chem., Int. Ed., 2016, 55, 2939 CrossRef CAS PubMed
- L. Wang, X. Wei, W. Jia, J. Zhong, L. Wu and Q. Liu, Org. Lett., 2014, 16, 5842 CrossRef CAS PubMed
- C. Qu, P. Xu, W. Ma, Y. Cheng and C. Zhu, Chem. Commun., 2015, 51, 13508 RSC
- Y. Wu, G. Lu, B. Zhou, M. Bu, L. Wan and C. Cai, Chem. Commun. 2016525965 Search PubMed.
- M. Zhu, W. Fu, G. Zou, C. Xu and Z. Wang, J. Fluorine Chem., 2015, 180, 1 CrossRef CAS
- S. Wang, W. Jia, L. Wang and Q. Liu, Eur. J. Org. Chem., 2015, 6817 CrossRef CAS
- L. Wang, X. Wei, W. Lei, H. Chen, L. Wu and Q. Liu, Chem. Commun., 2014, 50, 15916 RSC
- Y. Su, Y. Hou, F. Yin, Y. Xu, Y. Li, X. Zheng and X. Wang, Org. Lett., 2014, 16, 2958 CrossRef CAS PubMed
- X. Tang, Z. Zhang and W. R. Dolbier Jr., Chem. – Eur. J., 2015, 21, 18961 CrossRef CAS PubMed
- Z. Zhang, X. Tang, C. S. Thomoson and W. R. Dolbier Jr., Org. Lett., 2015, 17, 3528 CrossRef CAS PubMed
- Z. Zhang, X. Tang and W. R. Dolbier Jr., Org. Lett., 2015, 17, 4401 CrossRef CAS PubMed
- J. Rong, L. Deng, P. Tan, C. Ni, Y. Gu and J. Hu, Angew. Chem., Int. Ed., 2016, 55, 2743 CrossRef CAS PubMed
- Y. Arai, R. Tomita, G. Ando, T. Koike and M. Akita, Chem. – Eur. J., 2016, 22, 1262 CrossRef CAS PubMed
- Q. Lin, X. Xu, K. Zhang and F. Qing, Angew. Chem., Int. Ed., 2016, 55, 1479 CrossRef CAS PubMed
- Q. Lin, Y. Ran, X. Xu and F. Qing, Org. Lett., 2016, 18, 2419 CrossRef CAS PubMed
| This journal is © the Partner Organisations 2016 |