
Asymmetric synthesis of γ-aryl-substituted GABA derivatives via a highly diastereoselective Rh-catalyzed boronic acid addition at room temperature†
P. Veeraraghavan
Ramachandran
*,
Wataru
Mitsuhashi
and
Bidyut
Biswas
Herbert C. Brown Center for Borane Research, Department of Chemistry, Purdue University, 560 Oval Dr. West Lafayette, IN 47907, USA. E-mail: chandran@purdue.edu; Fax: +1 765 494 5303; Tel: +1 765 494 0239
First published on 5th June 2015
Abstract
A highly diastereoselective Rh-catalyzed boronic acid addition to enantiopure sulfinylimines providing γ-aryl GABA derivatives has been described. The reaction proceeds in protic solvents at room temperature and the starting material is readily prepared. The novel protocol enables the introduction of a variety of aryl substituents onto an unactivated sulfinylimine under mild conditions.
“GABA (γ-aminobutyric acid) is the main inhibitory neurotransmitter in the adult mammalian central nervous system (CNS).”1 It is critical in balancing neuronal excitation and inhibition, and has been involved in a large number of GABAergic diseases,2 which include neurological disorders such as Huntington's and Parkinson's disease, musculoskeletal and pain disorders, addiction and drug-withdrawal syndromes, epilepsy and seizures, anesthesia, liver diseases and hepatic encephalopathy, cognition, learning and memory disorders, premenstrual and other hormonal disorders, and many other related conditions.3 The therapeutic applications of GABA derivatives are limited by the inability of GABA to cross the blood–brain barrier, thus preventing oral administration. The preparation and pharmacological effect of many structural analogs of GABA for the treatment of CNS disorders has drawn attention of medicinal chemists (Fig. 1).4 Examples of these medicines are the β-(4-chloro)phenyl GABA analog baclofen,5 the β-substituted GABA analog gabapentin (Neurontin®, Pfizer, NY, USA) and pregabalin (Lyrica©, Pfizer) that are effective in the treatment of neuropathic pain via a mechanism which involves binding to the α2δ subunit of voltage-gated Ca2+ channels.6 Also, the γ-vinyl GABA analog vigabatrin (Sabril®, Lundbeck, Copenhagen, Denmark), an effective inhibitor of GABA transaminase, is prescribed for treating epileptic seizures.7
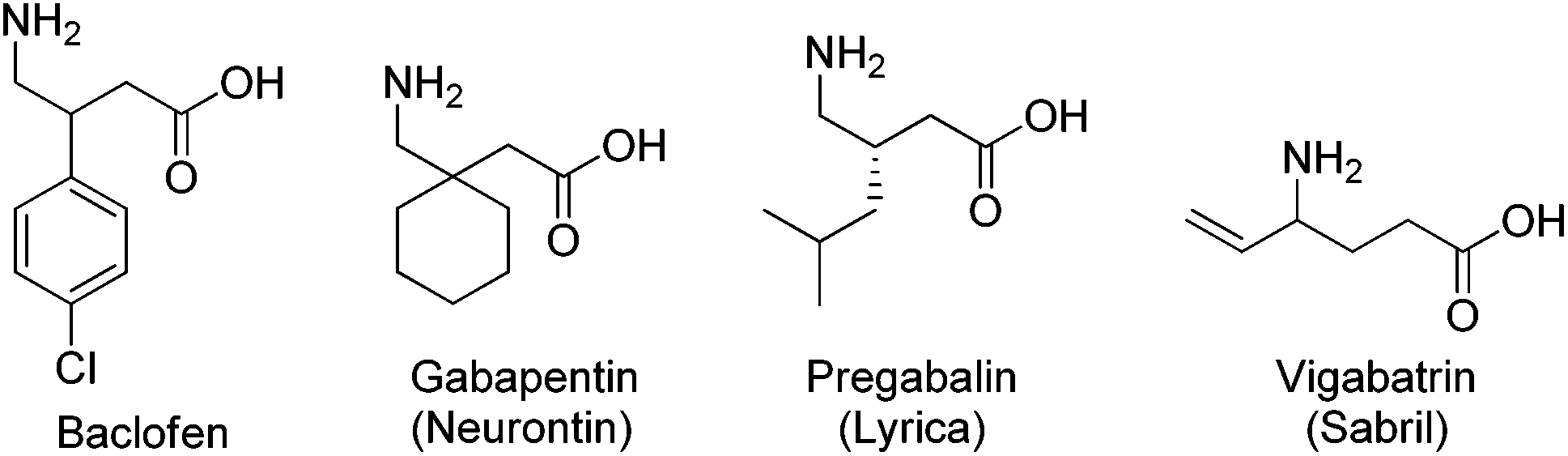 | ||
| Fig. 1 GABAergic drugs. | ||
GABA receptors are categorized into three major classes: GABAA, GABAB, and GABAC. GABAA and GABAC receptors are ligand-gated ion channels, on the other hand GABAB receptors are G protein coupled receptors. GABAC receptors differ considerably from GABAA receptors in terms of its agonist and antagonist structural profiles. Both sub-families of GABA-activated ligand-gated ion channels are targets for drug development.8 The area of GABAergic drugs is growing rapidly in the field of medicinal chemistry and neurodegenerative diseases. Although there are several procedures for the preparation of α- and β-substituted GABA derivatives,9 the corresponding γ-substituted derivatives have not received similar attention.
A number of useful protocols to synthesize chiral α- and β-amino acids have been developed using a t-butanesulfinamide.10 On the basis of the initial report by Miyaura and co-workers on the addition of arylboronic acids to sulfonylimine,11 Ellman and co-workers described the first Rh-catalyzed diastereoselective arylboronic acid addition to sulfinylimines,12 and further expanded the methodology for the synthesis of α-amino acids.13 In addition to the above high temperature protocols, the first boronic acid addition to sulfinylimines at room temperature was reported by Bolshan and Batey.14 Excellent yields have been achieved with activated sulfinylimines and boronic acids. However, the reaction with unactivated aliphatic sulfinylimines has remained a challenge. A diastereoselective synthesis of γ-substituted γ-amino acids by using a sulfinamide via the sequence of sulfinylimine formation and diastereoselective reduction has been reported recently.15 Additionally, the syntheses of α-methylene γ-substituted γ-amino acids have been reported by Lin16 and Yus17 and co-workers.
We have been interested in the synthesis of GABA derivatives, and have reported a protocol involving the asymmetric allylation of N-alumino- or -boryl imines, prepared from commercially available nitriles, followed by hydroboration and oxidation.18 Considering the importance of γ-substituted γ-amino acids,7 it is surprising that a direct synthesis has not been reported. We envisaged that the application of the above Rh-catalyzed addition of arylboronic acids to 4-sulfinylimino butanoate can lead to the preparation of γ-substituted GABA derivatives, thus providing a useful and powerful method to access this important class of compounds. The inherent lower reactivity of the aliphatic sulfinylimine was a concern. Allylation of these types of inactivated sulfinylimines is known.19 However, to the best of our knowledge, Rh-catalyzed addition of boronic acids14 remains a challenge. Herein we describe the successful preparation of the target molecules using the Ellman protocol.
We began the investigation by using a racemic sulfinylimine (rac-1), prepared as described21 (see the ESI†), as the substrate. PhB(OH)2 (2 equiv.) was chosen as the model reagent along with 10 mol% of Rh[(COD)(MeCN)2]BF4 and 2 equiv. of Et3N in dioxane/H2O (v/v = 1/2) at room temperature. The reaction was monitored by TLC and provided rac-2 in 43% isolated yield after purification. We then optimized for catalyst loading, solvent, temperature, additives, and boron source. The most common solvent for the Rh-catalyzed boronic acid addition is dioxane. However, the less harmful22 2-propanol gave an improved yield of rac-2, with decreased catalyst loading (5 mol%) during the phenylboronic acid addition to rac-1. Other alcoholic solvents such as MeOH, nPrOH, nBuOH and tBuOH did not improve the yield further. The equivalencies of phenylboronic acid and triethylamine were also varied, but the yield did not improve further. Seeking more efficient conditions, several phenylboronic acid derivatives were screened to optimize the reaction. As can be seen from Table 1, all of these derivatives, except for the potassium trifluoroborate salt, gave similar yields. However, due to its performance in diastereoselectivity and lower molecular weight, commercially available boronic acids were used for further reactions.
|
|
|||
|---|---|---|---|
| Entry | Boronic acid derivatives | Yielda (%) | drb |
a Isolated yield.
b Determined by comparing 1H NMR of diastereomers prepared according to the literature.20
c The yield of the product (rac-2) was estimated using the ratio obtained by 1H NMR since it was difficult to separate from pinacol.
d The recovered starting material was a mixture with impurities which were difficult to separate.
e The amount of recovery is estimated by 1H NMR of the crude product (rac-1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :rac-2 = 73:27). :rac-2 = 73:27).
|
|||
| 1 | PhB(OH)2 | 75 | 97:3 |
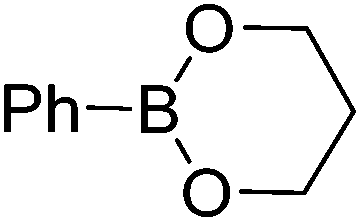
| 2 |

|
71c | 94:6 |
| 3 |
|
71 | 96.5:3.5 |
| 4 | PhBF3K | 23d,e | 92:8 |
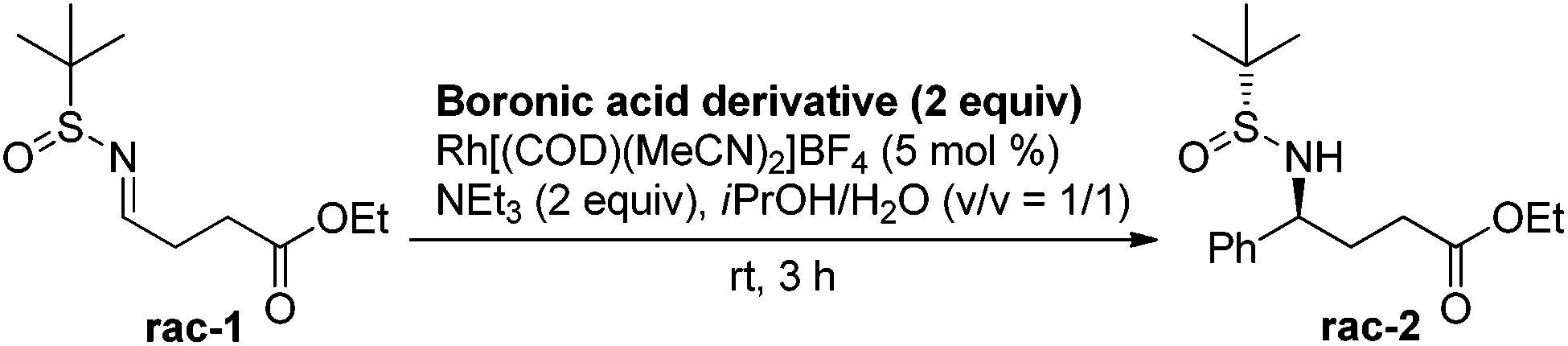
Having standardized the conditions for the preparation of rac-2, we attempted the reaction of phenylboronic acid with an enantiopure (R)-isomer of the sulfinylimine (1). The reaction was complete as before and the product 2a was isolated in 77% yield. The diastereomeric ratio was determined by 1H NMR (and/or 19F NMR) spectroscopy analysis of the NH and/or benzylic protons by comparing with epimers prepared according to the literature.20 We were pleased to observe a diastereomeric ratio of 97.5:2.5. The absolute configuration of the product (2a) was confirmed by the rotation of the lactamized product [(−)-(S)-5-phenylpyrrolidin-2-one] reported in the literature.23 On the basis of analogy with 2a, we believe that we have obtained the (4S)-isomer for all of the GABA derivatives 2b–g described subsequently.
The generality of the Rh-catalyzed addition of arylboronic acids to chiral sulfinylimine (1) was demonstrated with a series of boronic acids under the optimized conditions (Table 2). Good yields were observed with electron neutral and rich arylboronic acids (entries 1–3). Weakly deactivated 4-halogenated arylboronic acids resulted in moderate yields and the starting material was also recovered (entries 4 and 5). A sterically hindered (entry 6) and inductively deactivated boronic acid (entry 7) were less reactive and gave lower yields with the recovery of more starting material (∼50% on the basis of the recovered starting material). Extending the reaction time for those in entries 4–7 did not improve the yield, but rather adversely affected the dr. This was attributed to the electron-withdrawing group at the 4-position affecting the benzylic proton of the product. Overall, the diastereoselectivities are excellent (96:4–97.5:2.5), but the yields are strongly affected by the (sterically or electronically) deactivating functional groups in boronic acids. The optimized reaction time was 3 h since longer reaction time negatively affected the diastereoselectivity. Under this condition, the byproducts which decreased the product yields were mostly the hydrolyzed imine (aldehyde and sulfinamide). We also attempted the reaction of a heteroaromatic boronic acid, 2-thiophenylboronic acid. Unfortunately, the reaction, monitored by TLC, did not proceed.
|
|
|||
|---|---|---|---|
| Entry | Product | Yielda (%) | drb |
| a Isolated yield. b Determined by comparing 1H NMR (NH or benzylic H) (and 19F NMR) of diastereomers prepared according to the literature.20 c The yield on the basis of the recovered starting material. | |||
| 1 |

|
77 | 97.5:2.5 |
| 2 |
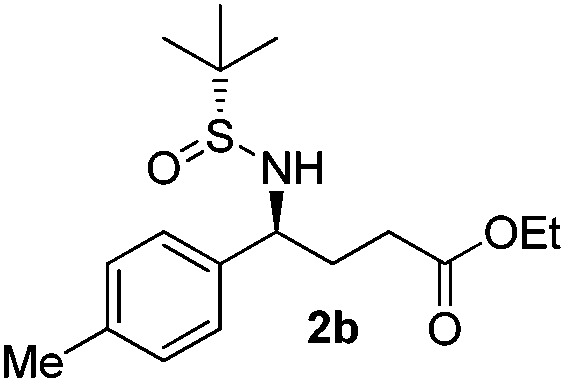
|
81 | 97:3 |
| 3 |
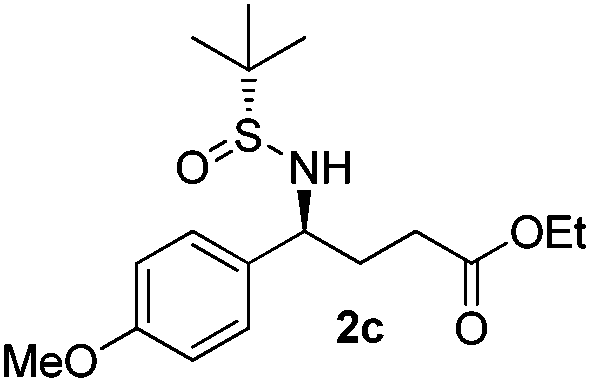
|
64 | 96:4 |
| 4 |
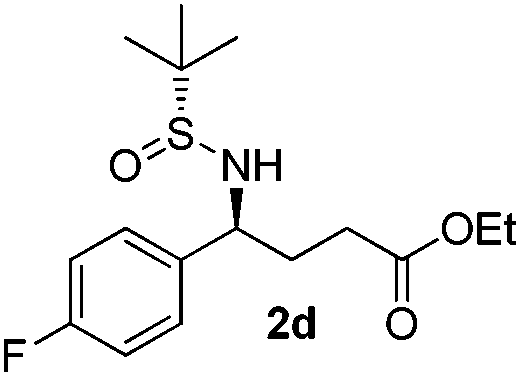
|
49 (63)c | 97:3 |
| 5 |

|
43 (52)c | 97:3 |
| 6 |
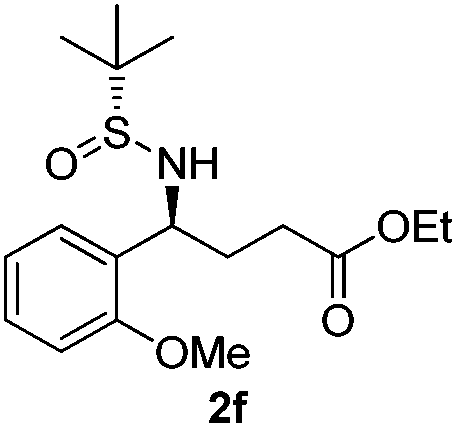
|
28 (52)c | 97:3 |
| 7 |
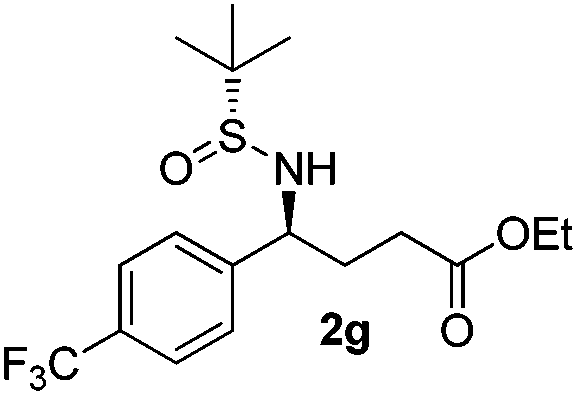
|
21 (54)c | 97:3 |

Conclusions
In conclusion, we have reported a diastereoselective synthesis of GABA derivatives via a Rh-catalyzed boronic acid addition to enantiopure t-butanesulfinyl imine. A variety of arylboronic acids, including those with a sterically hindered and electron-withdrawing substituent, are applicable to the methodology. The simple preparation of the chiral imine, coupled with the facile diastereoselective addition of boronic acids, makes this reaction attractive for the preparation of a variety of γ-aryl γ-amino acids. Further work to improve the yields and include the addition of alkylboronic acids is in progress.Experimental
General
All reactions were performed at room temperature and under a nitrogen atmosphere in either flame or oven-dried glassware unless otherwise noted. All TLC analysis was performed using aluminum-backed thin-layer chromatography plates (Dynamic Absorbent Inc., 200 μm thickness, F-254 Indicator). Flash chromatography was performed using 230–400 mesh, 60 Å pore diameter flash chromatography gel. All chromatography elutions were gradient in nature, eluting first with hexanes, followed by incorporating more polar solvents as appropriate. 1H, 13C, and 19F spectra were recorded at room temperature on a Varian INOVA 300 MHz or Bruker 400 MHz. Chemical shifts (δ values) are reported in parts per million, and are referenced to tetramethylsilane. 19F NMR is referenced internal to the spectrometer. Data are reported as: δ value, multiplicity, and integration (s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, h = hextet, br = broad). 1H NMR (and/or 19F NMR) was used as the measurement of dr by comparing the relative integration values of the benzylic or NH protons. Optical rotations were measured on an Autopol III automatic polarimeter, and are reported against the “Sodium D” line at 25 °C ([α]25D).General procedure for Rh-catalyzed arylboronic acids
To ArB(OH)2 (0.4 mmol) and Rh[(COD)(MeCN)2]BF4 (3.8 mg, 0.01 mmol) in a 1 dram vial, 1 (0.2 mmol) in degassed iPrOH/H2O (1 mL, v/v = 1:1) and triethylamine (56 μL, 0.4 mmol) were added at 0 °C. After stirring for 3 h at rt, the crude product was diluted with water (5 mL) and extracted with EtOAc (3 × 10 mL). The combined organic layers were then washed with brine (15 mL) and filtered through Na2SO4, and the solvent was removed under reduced pressure. The crude product was purified by column chromatography to provide the addition products 2.
(S)-Ethyl 4-[(R)-1,1-dimethylethylsulfinamido]-4-phenylbutanoate (2a)15
77% yield, 97.5:2.5 dr. [α]25D = −121 (c 0.86, CHCl3). 1H NMR (CDCl3) δ 7.38–7.26 (m, 5H), 4.43 (td, J = 6.8, 2.7 Hz, 1H), 4.09 (q, J = 7.1 Hz, 2H), 3.79 (d, J = 2.7 Hz, 1H), 2.39–2.07 (m, 4H), 1.23 (t, J = 7.1 Hz, 3H), 1.19 (s, 9H). 13C NMR (CDCl3) δ 172.9, 141.0, 128.3, 127.5, 127.3, 60.5, 58.7, 55.5, 33.2, 30.7, 22.5, 14.1. Mass (ESI+): calc. for [M + H]+ 312.2, found 312.2; for [M + Na]+ 334.2, found 334.3.
(S)-Ethyl 4-[(R)-1,1-dimethylethylsulfinamido]-4-(4-methylphenyl)butanoate (2b)
81% yield, 97:3 dr. [α]25D = −82.9 (c 2.43, CHCl3). 1H NMR (CDCl3) δ 7.23–7.04 (m, 4H), 4.37 (dt, J = 6.8, 2.7 Hz, 1H), 4.07 (q, J = 7.2 Hz, 2H), 3.75 (d, J = 2.7 Hz, 1H), 2.32 (s, 3H), 2.28–2.00 (m, 4H), 1.22 (d, J = 7.2 Hz, 3H), 1.17 (s, 9H). 13C NMR (CDCl3) δ 172.9, 137.8, 137.2, 129.0, 127.2, 60.5, 58.3, 55.4, 33.2, 30.7, 22.5, 21.1, 14.1. Mass (ESI+): calc. for [M + H]+ 326.2, found 326.1; for [M + Na]+ 348.2, found 348.2.
(S)-Ethyl 4-[(R)-1,1-dimethylethylsulfinamido]-4-(4-methoxyphenyl)-butanoate (2c)
64% yield, 96:4 dr. [α]25D = −78.6 (c 1.66, CHCl3). 1H NMR (CDCl3) δ 7.21 (d, J = 8.7 Hz, 2H), 6.87 (d, J = 8.7 Hz, 2H), 4.46–4.31 (m, 1H), 4.09 (q, J = 7.1 Hz, 2H), 3.80 (s, 3H), 3.73 (d, J = 2.1 Hz, 1H), 2.33–2.02 (m, 4H), 1.23 (t, J = 7.1 Hz, 3H), 1.18 (s, 9H). 13C NMR (CDCl3) δ 173.2, 159.2, 133.0, 128.7, 113.9, 60.6, 58.1, 55.5, 55.2, 33.3, 30.8, 22.6, 14.2. Mass (ESI+): calc. for [M + Na]+ 364.2, found 364.1; for [M + K]+ 380.1, found 380.0.
(S)-Ethyl 4-[(R)-1,1-dimethylethylsulfinamido]-4-(4-fluorophenyl)butanoate (2d)
49% yield (63% on the basis of the recovered starting material), 97:3 dr. [α]25D = −82.7 (c 1.48, CHCl3). 1H NMR (CDCl3) δ 7.34–7.20 (m, 2H), 7.03 (m, 2H), 4.43 (m, 1H), 4.10 (q, J = 7.2 Hz, 2H), 3.81 (s, 1H), 2.40–2.18 (m, 2H), 2.18–2.00 (m, 2H), 1.24 (t, J = 7.2 Hz, 3H), 1.19 (s, 9H). 13C NMR (CDCl3) δ 173.2, 162.3 (d, JC–F = 246.1 Hz), 137.0, 129.2 (d, JC–F = 8.1 Hz), 115.5 (d, J = 21.5 Hz), 60.7, 58.0, 55.6, 33.3, 30.7, 22.6, 14.2. 19F NMR (CDCl3) δ −115.71. Mass (ESI+): calc. for [M + H]+ 330.2, found 330.1; for [M + Na]+ 352.1, found 352.2; [M + K]+ 368.1, found 368.1.
(S)-Ethyl 4-[(R)-1,1-dimethylethylsulfinamido]-4-(4-chlorophenyl)butanoate (2e)
43% yield, (52% on the basis of the recovered starting material), 97:3 dr. [α]25D = −82.4 (c 1.19, CHCl3). 1H NMR (CDCl3) δ 7.32 (d, J = 8.5 Hz, 2H), 7.24 (d, J = 8.5 Hz, 2H), 4.42 (td, J = 6.9, 2.5 Hz, 1H), 4.10 (q, J = 7.2 Hz, 2H), 3.85 (d, J = 2.2 Hz, 1H), 2.39–2.19 (m, 2H), 2.19–2.00 (m, 2H), 1.24 (t, J = 7.2 Hz, 3H), 1.19 (s, 9H). 13C NMR (CDCl3) δ 173.1, 139.8, 133.4, 128.8, 128.7, 60.7, 58.0, 55.5, 33.0, 30.6, 22.5, 14.1. Mass (ESI+): calc. for [M + Na]+ 368.1, found 368.2; [M + K]+ 384.1, found 384.1.
(S)-Ethyl 4-[(R)-1,1-dimethylethylsulfinamido]-4-(2-methoxyphenyl)-butanoate (2f)
28% yield, (52% on the basis of the recovered starting material), 97:3 dr. [α]25D = −56.9 (c 0.93, CHCl3). 1H NMR (CDCl3) δ 7.31–7.20 (m, 2H), 7.00–6.76 (m, 2H), 4.73 (m, 1H), 4.10 (t, J = 7.1 Hz, 2H), 3.83 (s, 3H), 2.50–2.02 (m, 4H), 1.23 (t, J = 7.1 Hz, 3H), 1.17 (s, 9H). 13C NMR (CDCl3) δ 173.3, 156.6, 129.5, 128.3, 127.8, 120.3, 110.5, 60.4, 55.6, 55.2, 55.1, 31.8, 31.0, 22.5, 14.2. Mass (ESI+): calc. for [M + Na]+ 364.2, found 364.3; [M + K]+ 380.1, found 380.1.
(S)-Ethyl 4-[(R)-1,1-dimethylethylsulfinamido]-4-(4-trifluoromethylphenyl)butanoate (2g)
21% yield, (54% on the basis of the recovered starting material), 97:3 dr. [α]25D = −74.2 (c 0.75, CHCl3). 1H NMR (CDCl3) δ 7.61 (d, J = 8.1 Hz, 2H), 7.44 (d, J = 8.1 Hz, 2H), 4.52 (td, J = 6.8, 2.6 Hz, 1H), 4.11 (q, J = 7.2 Hz, 2H), 4.05–3.92 (m, 1H), 2.45–2.23 (m, 2H), 2.13 (m, 2H), 1.24 (t, J = 7.2 Hz, 3H), 1.21 (s, 9H). 13C NMR (CDCl3) δ 173.2, 145.7, 130.1 (q, J = 32.7 Hz), 127.9, 125.6 (d, J = 3.5 Hz), 122.7, 60.9, 58.4, 55.8, 33.1, 30.7, 22.6, 14.2. 19F NMR (CDCl3) δ −64. Mass (ESI+): calc. for [M]+ 381.2, found 381.0; [M + Na]+ 402.1, found 402.3.
Acknowledgements
We thank the Herbert C. Brown Center for Borane Research for financial assistance and Mr. Eric Jones for some preliminary experiments.Notes and references
- M. Farrant and N. Zoltan, Nat. Rev. Neurosci., 2005, 6, 215 CrossRef CAS PubMed.
- For a special focus issue on GABAergic drugs, seeFuture Med. Chem., 2011, 3, 139.
- (a) K. Gajcy, S. Lochynski and T. A. Librowski, Curr. Med. Chem., 2010, 17, 2338 CrossRef CAS; (b) M. Ordóñez and C. Cativiela, Tetrahedron: Asymmetry, 2007, 18, 3 CrossRef PubMed.
- P. Yogeeswari, J. V. Ragahendran and D. Sriram, Recent Pat. CNS Drug Discovery, 2006, 1, 113 CrossRef CAS.
- R. N. Brogden, T. M. Speight and G. S. Avery, Drugs, 1974, 8, 1 CrossRef CAS PubMed.
- B. A. Lauria-Homer and R. B. Phol, Expert Opin. Invest. Drugs, 2003, 12, 663 CrossRef PubMed.
- J. B. Bolton, E. Rimmer, J. Williams and A. Richens, Br. J. Clin. Pharmacol., 1989, 27(suppl. 1), S35 CrossRef PubMed.
- M. Chebib and G. A. R. Johnston, J. Med. Chem., 2000, 43, 1427 CrossRef CAS.
- Y. Zhu, S. Khumsubdee, A. Schaefer and K. Burgess, J. Org. Chem., 2011, 76, 7449 CrossRef CAS PubMed.
- M. T. Robak, M. A. Herbage and J. A. Ellman, Chem. Rev., 2010, 110, 3600 CrossRef CAS PubMed.
- M. Ueda, A. Saito and N. Miyaura, Synlett, 2000, 1637 CAS.
- D. J. Weix, Y. Shi and J. A. Ellman, J. Am. Chem. Soc., 2005, 127, 1092 CrossRef CAS PubMed.
- M. A. Beenen, D. J. Weix and J. A. Ellman, J. Am. Chem. Soc., 2006, 128, 6304 CrossRef CAS PubMed.
- Y. Bolshan and R. A. Batey, Org. Lett., 2005, 7, 1481 CrossRef CAS PubMed.
- L. R. Reddy, K. Prasad and M. Prashad, J. Org. Chem., 2012, 77, 6296 CrossRef PubMed.
- A. Shen, M. Liu, Z.-S. Jia, M.-H. Xu and G.-Q. Lin, Org. Lett., 2010, 12, 5154 CrossRef CAS PubMed.
- H. K. Dema, F. Foubelo and M. Yus, Heterocycles, 2011, 82, 1411 CAS.
- (a) P. V. Ramachandran and T. E. Burghardt, Chem. – Eur. J., 2005, 11, 4387 CrossRef CAS PubMed; (b) P. V. Ramachandran and D. Biswas, Org. Lett., 2007, 9, 3025 CrossRef CAS PubMed.
- O. S. R. Barros, J. A. Sirvent, F. Foubelo and M. Yus, Chem. Commun., 2014, 50, 6898 RSC.
- K. Brak, K. T. Barrett and J. A. Ellman, J. Org. Chem., 2009, 74, 3606 CrossRef CAS PubMed.
- G. Liu, D. A. Cogan, T. D. Owens, T. P. Tang and J. A. Ellman, J. Org. Chem., 1999, 64, 1278 CrossRef CAS.
- Dioxane is a carcinogen while 2-propanol is safe (MSDS, Sigma-Aldrich).
- P. Camps, T. Gómez, D. Muñoz-Torrero, J. Rull, L. Sánchez, F. Boschi, M. Comes-Franchini, A. Ricci, T. Calvet, M. Font-Bardia, E. De Clercq and L. Naesens, J. Org. Chem., 2008, 73, 6657 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of 1 and additional experimental details, 1H and 13C NMR spectra of all products, and 19F NMR spectra of 2d and 2g. See DOI: 10.1039/c5qo00133a |
| This journal is © the Partner Organisations 2015 |