
A quantum mechanical study of the mechanism and stereoselectivity of the N-heterocyclic carbene catalyzed [4 + 2] annulation reaction of enals with azodicarboxylates†
Yang
Wang
,
Linjie
Zheng
,
Donghui
Wei
* and
Mingsheng
Tang
*
The College of Chemistry and Molecular Engineering, Center of Computational Chemistry, Zhengzhou University, Zhengzhou, Henan Province 450001, P.R. China. E-mail: donghuiwei@zzu.edu.cn; mstang@zzu.edu.cn
First published on 6th May 2015
Abstract
A systematic theoretical study has been carried out to understand the mechanism and stereoselectivity of [4 + 2] annulation reaction between γ-oxidized enals and azodicarboxylates catalyzed by the N-heterocyclic carbene (NHC). The calculated results reveal that the catalytic cycle can be characterized by three stages (Stages 1, 2, and 3). Stage 1 is the nucleophilic addition of the NHC catalyst to enals upon the intramolecular proton transfer to generate the Breslow intermediate. In this stage, apart from the direct proton transfer mechanism, the H2O (H2O and 2H2O cluster) and bicarbonate anion (HCO3−) mediated proton transfer mechanisms are also investigated; the free energy barrier for the crucial proton transfer steps in Stage 1 is found to be significantly lower by explicit inclusion of the bicarbonate anion (HCO3−). For Stage 2, the removal of the leaving group occurs, followed by C–C bond rotation for the formation of cis-dienolate. Stage 3 is the endo/exo [4 + 2] cycloaddition and dissociation of the catalyst from the final products. The formal [4 + 2] cycloaddition step is calculated to be the enantioselectivity determining step, and the R-configured PR is the predominant product according to the computations, which is in good agreement with the experimental observations. Moreover, the stereoselectivity associated with the chiral carbon center is attributed to the CH–π interaction between Cα−H and the mesityl group of NHC and the variation in the distortion of the dienolate. The mechanistic insights obtained in the present study should be valuable for the other NHC-catalyzed reactions.
1. Introduction
N-Heterocyclic carbenes (NHC) have been widely applied in the form of ligands,1 and Lewis base catalysts.2 As important organocatalysts, NHCs have been successfully used in carbon–carbon and carbon–heteroatom bond formation reactions including cross-benzoin, Stetter, homoenolate, annulation, and cycloaddition reactions.3 In the past few years, Lewis base NHCs have been found to be powerful organocatalysts in the stereoselective cycloaddition reactions of ketenes, including the [2 + 2],4 [2 + 2 + 2],4d,5 and [4 + 2]6 cycloaddition reactions. However, for the annulation reactions catalyzed by NHC, the enals are frequently used as one of the reactants. Due to the special reactivity of the reactants (i.e. ketenes and enals), their NHC-catalyzed cycloaddition/annulation reactions can provide facile and also effective access to a variety of ring skeletons, especially for construction of N/O-containing heterocycles.In the NHC catalyzed annulation reaction, the enals can react with a variety of electrophilic coupling partners, such as alkenes, imines, and ketones. The addition of the NHC catalyst to the enals can lead to different reactive intermediates bearing more than one reactive carbon center of the enals, such as the homoenolate intermediate (β-carbon),7 enolate intermediate (α-carbon),8 and acyl anion equivalent intermediate (carbonyl carbon),9 which allow the inversion of the normal reactivity (umpolung) through formation of Breslow intermediates and serve as the prenucleophiles. In these reactions, the enals can function as two-, three-, and four-carbon synthons undergoing [2 + 4],8e,10 [3 + n] (n = 2, 3, 4),3k,11 and [4 + n] (n = 2, 3)12 annulation reactions. In 2004, Glorius's group7a and Bode's group7b concurrently reported the NHC-catalyzed annulation reaction of enals with aldehydes to afford the γ-lactones, in which the enal β-carbon served as the reactive nucleophilic carbon. Then, the same kind of reaction had been further extensively explored by many other groups. The reactivity of the enal α-carbon reaction has also been well studied by Bode,8a Glorius,8b Scheidt,8c Nair,8d and Chi.8e
In contrast to the α- and β-carbons of enals, the activated γ-carbon of enals working as the four-carbon synthon has been much less studied in the past few years. There are only a few successful examples of NHC-mediated γ-functionalization of enals by activating the enal γ-carbon as nucleophiles.13 In 2012, Chi and coworkers reported the pioneering work on the oxidative NHC-catalyzed [4 + 2] annulation of enals with trifluoromethyl ketones by γ addition;13a then they reported the first NHC-catalyzed [3 + 4] annulation reaction13d of enals with azomethine imines by activating the γ-carbon of enals as the 1,4-dipolarophile. Ye et al. reported the NHC-catalyzed [4 + 2] annulation of γ-oxidized enals with azodicarboxylates in good yield and with excellent enantioselectivity.13b
With the development of NHC-catalyzed annulation reactions of enals in experiment, the theoretical investigations on the mechanisms have also been reported in the literature.14 For example, Bode and coworkers computationally studied the NHC-catalyzed [4 + 2] cycloaddition reaction of α,β-unsaturated aldehydes (C2-synthon) with enones; they identified that the oxyanion-steering mechanism and CH–π interaction are the two crucial interactions for the high selectivity.14c Sunoj et al. performed a theoretical investigation on the mechanism and stereoselectivity in a chiral N-heterocyclic carbene-catalyzed cycloannulation reaction of the homoenolate derived from butenal with pentenone.14d More recently, we have performed the theoretical investigation on the mechanism of the NHC-catalyzed [4 + 2] annulation reaction of enals (C2-synthon) and chalcones, and the computational results show that the acetic acid can assist the proton transfer process.14e To the best of our knowledge, the computational investigation on the mechanism and enantioselectivity of NHC-catalyzed [4 + 2] annulation of enals (C4-synthon) by γ addition has remained hitherto unexplored.
It is noteworthy that the mechanisms and stereoselectivities of NHC-catalyzed [2 + 2],15 [4 + 2],16 [2 + 2 + 2]17 cycloaddition reactions of ketenes have been studied by our group using DFT methods. Actually, the reaction mechanism might be diverse for different NHC catalytic cycloadditions of ketenes; these cycloaddition reactions do not always initiate by the reaction of NHC with ketenes.16 As is known to all, there exists the proton transfer process involved in the NHC-catalyzed annulation reaction of enals, which is remarkably different from the cycloaddition reaction, and this would make the reaction mechanism more complicated. Correspondingly, there should also be multiple mechanisms for NHC-catalyzed annulation reactions of enals, which is because the catalysts, reactants, and additives will influence the proton transfer process involved in these kinds of reactions. Thus, the theoretical investigation is necessary for these special organocatalytic reactions.
In this present study, we aim to disclose the mechanism and enantioselectivity of NHC-catalyzed [4 + 2] annulation reaction of γ-oxidized enals and azodicarboxylates reported by Ye and co-workers (Scheme 1).13b On the basis of the presumptive mechanism proposed by Ye, we suggested the possible mechanism of the reaction of enals with azodicarboxylates catalyzed by NHC (shown in Scheme 2): initially, the NHC nucleophile attacks on the enal to form zwitterionic intermediate I. Subsequently, the intermediate I transforms to the Breslow intermediate IIvia the proton transfer process. Then the removal of the leaving group (LG) generates the trans-dienolate III by abstracting the hydride from the carbonyl oxygen, which affords vinyl enolates as the reactive 1,4-dipolarophiles. The fourth step is the [4 + 2] cycloaddition of III with azodicarboxylates for the formation of the adduct IV, and finally the dissociation of the desired products PR&PS and regeneration of catalyst Cat.
 | ||
| Scheme 1 The NHC-catalyzed [4 + 2] annulation reaction of enals with azodicarboxylates. | ||
 | ||
| Scheme 2 The proposed catalytic cycle for the [4 + 2] annulation reaction. | ||
Nevertheless, there are still some issues that need to be settled as shown in Scheme 2: In the second step, the direct 1,2-proton transfer process would cost quite a high energy barrier due to the large strain in the three-membered ring transition state. Moreover, the reaction proceeds without a protic additive in the reaction system, so how does the 1,2-proton transfer happen? As the design of a new organocatalyst relies on a detailed understanding of the underlying factors that govern the enantioselectivity of these kinds of reactions, so what are the main factors that control the enantioselectivity of this reaction? With these questions as motivation, the present work will pursue a theoretical investigation on the title reaction to not only obtain a preliminary picture from the γ-oxidized enal [4 + 2] annulation reaction, but also explore the factors that control the stereochemistry of this reaction. And we believe that the mechanistic information should be important for understanding the NHC-catalyzed [4 + 2] annulation reactions and providing novel insights into recognizing this kind of reaction in detail.
For the sake of convenience, the [4 + 2] annulation reaction between the γ-oxidized enals (R1, LG![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) OCO2Me, Scheme 2) and the di-tert-butyl azodicarboxylate (R2, Scheme 2) catalyzed by NHC (Cat, Scheme 2) has been chosen as the object of investigation. In the present study, we will give the computational results for the possible reaction mechanisms to illustrate the theoretical methodology for this issue at the molecular level using the density functional theory (DFT), which has been widely used in the study of organic,18 biological reaction mechanisms,19 and others.20
OCO2Me, Scheme 2) and the di-tert-butyl azodicarboxylate (R2, Scheme 2) catalyzed by NHC (Cat, Scheme 2) has been chosen as the object of investigation. In the present study, we will give the computational results for the possible reaction mechanisms to illustrate the theoretical methodology for this issue at the molecular level using the density functional theory (DFT), which has been widely used in the study of organic,18 biological reaction mechanisms,19 and others.20
2. Computational details
Quantum mechanical calculations reported herein were carried out by using density functional theory with the Gaussian 09 suite of programs.21 The solution-phase geometry optimization of all species is performed with the recently developed M06-2X22 density functional along with the 6-31G(d, p) basis set in THF solvent using the integral equation formalism polarizable continuum model (IEF-PCM).23 The harmonic vibrational frequency calculations were performed at the same level of theory as that used for geometry optimizations to provide thermal corrections of Gibbs free energies and make sure that the local minima had no imaginary frequencies, while the saddle points had only one imaginary frequency. Intrinsic reaction coordinates (IRCs)24 were calculated to confirm that the transition state structure connected the correct reactant and product on the potential energy surface, and the natural bond orbital (NBO)25 analysis was employed to assign the atomic charges. The three dimensional structures had been represented in the figures by using CYLView software.26We choose to discuss this theoretical study based on the solution-phase Gibbs free energies calculated by the M06-2X/6-31G(d, p)/IEF-PCM(THF) method rather than Born–Oppenheimer energies, which are the electronic (including nuclear-repulsion) energies plus zero-point vibrational energies (ZPVEs).
3. Results and discussion
As shown in Schemes 3 and 4, the suggested mechanism for each elementary step of the annulation reaction between R1 and R2 catalyzed by Cat includes three stages, i.e. the formation of the Breslow intermediate (Stage 1, Scheme 3), formation of the cis-dienolate (Stage 2, Scheme 4), and formal [4 + 2] cycloaddition and regeneration of the catalyst (Stage 3, Scheme 4). In the following parts of this section, we will give detailed discussions step by step. | ||
| Scheme 3 The possible pathways of Stage 1. | ||
 | ||
| Scheme 4 The possible pathways of Stages 2 and 3. | ||
3.1 Stage 1: formation of the Breslow intermediate
As shown in Scheme 3, there are two steps involved in Stage 1: (1) nucleophilic attack on the γ-oxidized enal R1 by the catalyst Cat, (2) proton transfer of the formed intermediate to give the Breslow intermediate Re/Si-M2. Fig. 1 and 2 depict the Gibbs free energy profile and the optimized structures involved in Stage 1, respectively.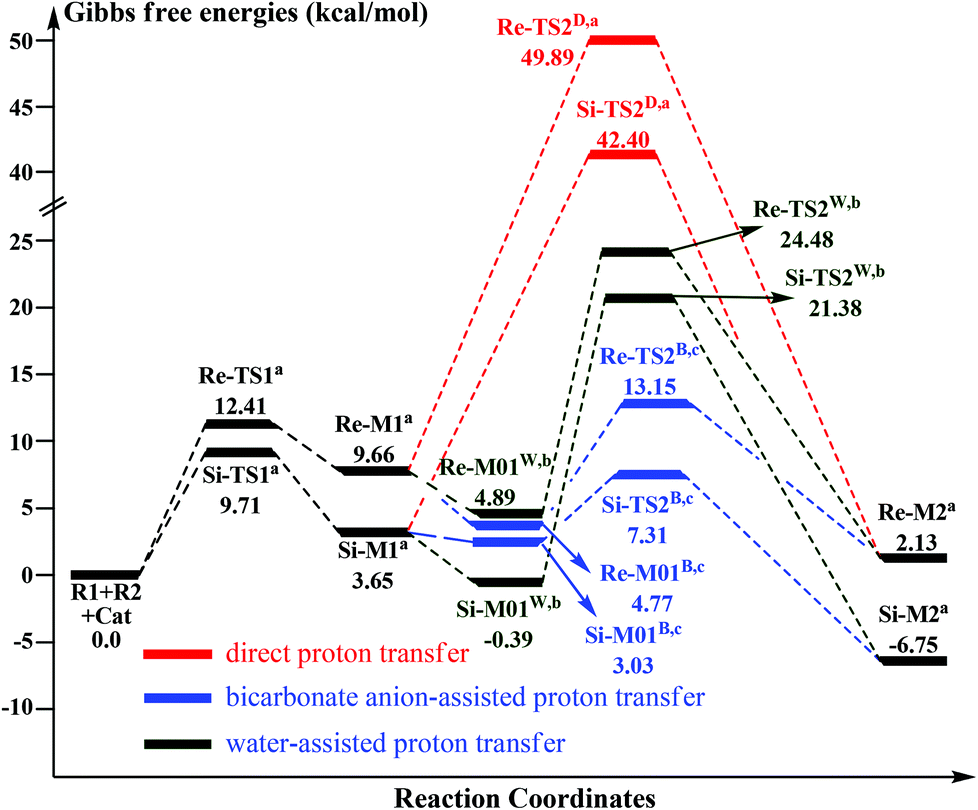 | ||
| Fig. 1 The Gibbs free energy profiles of Stage 1 (aadding the free energy of R2, badding the free energies of H2O and R2, cadding the free energies of the bicarbonate anion and R2.) | ||
 | ||
| Fig. 2 The optimized structures and geometry parameters of the intermediates and transition states involved in Stage 1 (distance in Å and most of the hydrogen atoms are omitted for the sake of clarity). | ||
(i) Direct and HCO3−-assisted proton transfer processes: The direct intramolecular proton transfer process via the transition state Re/Si-TS2D to form the stable Breslow intermediate Re/Si-M2 with the free energy of 2.13/−6.75 kcal mol−1 encounters a significantly higher free energy barrier (49.89/42.40 kcal mol−1, Fig. 1), indicating that the direct proton transfer process is impossible to occur under the experimental conditions and such a possibility is not likely in a non-polar aprotic medium such as THF. Notably, it has been reported that the generation of the Breslow intermediate could involve significant barriers unless a base/acid-assisted proton transfer mechanism is invoked. In view of this, the previously performed HCO3−-assisted proton transfer process, as opposed to a conventional pathway, has been taken into consideration. As illustrated in Scheme 3, the reaction precursor Re/Si-M01B is formed by weak interactions between Re/Si-M1 and HCO3−. Geometrical and structural parameters of the reaction precursor Re/Si-M01B depicted in Fig. 2 show that the intermolecular hydrogen bond makes the zwitterionic Re/Si-M01B more stable. Then the proton HE transfers from CE1 to OB atoms, coupled with the proton HB transferring to the OE atom via the seven-membered ring transition state Re/Si-TS2B. The distances of CE1–HE, HE–OB, OB′–HB, and HB–OE in the transition state Re/Si-TS2B are 1.32/1.29, 1.30/1.36, 1.44/1.11, and 1.05/1.32 Å, respectively, which reveals that the HCO3−-assisted proton transfer step is a concerted but asynchronous process; the formation of the HB–OE bond (1.05/1.32 Å) is a little advanced than the formation of the HE–OB bond (1.30/1.36 Å) in the transition state Re/Si-TS2B. The distance between OE and HE(B) atoms in the Breslow intermediate Re/Si-M2 is 0.97/0.97 Å, demonstrating the complete formation of the OE–HE(B) bond. The energy barrier of this step is 13.15/7.31 kcal mol−1 (Fig. 1), which demonstrates that the HCO3−-assisted proton transfer process occurs more easily than the direct proton transfer process under the experimental conditions. Moreover, we have tried, but failed to locate the HCO3−-assisted five-membered (CE1–HE–OB–HB–OE) ring transition state involved in the proton transfer process, which is easily re-optimized to the seven-membered one.
(ii) The H2O-assisted proton transfer process: It is important at this juncture to infer that the catalytic reaction was carried out without N2/Ar protection at room temperature. This phenomenon shows that there should be traces of water in the solvent, which will mediate the proton transfer and make the H2O-assisted proton transfer pathway feasible. Many other computational studies also demonstrated the similar important role of water or water clusters (2H2O cluster) in the catalytic reactions which involve proton transfer without any protic mediator.27 Prior to generating Re/Si-M2, Re/Si-M1 and H2O first form a complex (Re/Si-M01W) through electrostatic attraction between the two components (the 2H2O cluster assisted proton transfer process has also been considered and discussed in the ESI†). The complex is enthalpically more stable (enthalpy energies are provided in the ESI†), but less stable in free energy than R1 + R2 + Cat, which implies that the electrostatic attraction can contribute to pulling the two components together. After passing through a barrier of 24.48/21.38 kcal mol−1 (Re/Si-TS2W), the Breslow intermediate Re/Si-M2 is formed via a five-membered ring transition state (Re/Si-TS2W). The gradually changed OE–HW, CE1–HE, and OW–HE distances for Re/Si-M01W and Re/Si-TS2W (Fig. 2) illustrate the proton transfer process.
Taken together, three possible pathways for the proton transfer process to afford Re/Si-M2 have been suggested and studied. Based on the discussions above, one can conclude that the HCO3−-assisted proton transfer for the formation of Re/Si-M2via the seven-membered ring transition state Re/Si-TS2B (13.15/7.31 kcal mol−1) is the most energetically feasible than others. In addition, we have failed to locate the transition state for the bimolecular proton transfer process between two Re/Si-M1 molecules. This might be related to the high steric hindrance between the two structures, which makes it difficult by pulling the two molecules of Re/Si-M1 together in a suitable orientation for the proton transfer.
3.2 Stage 2: formation of the cis-dienolate M4endo/exo
Scheme 4 presents the detailed mechanism of the remaining stages (Stages 2 and 3) of the catalytic reaction. There are also two steps involved in Stage 2: the removal of the leaving group (LG) and the rotation of the CE2–CE3 single bond. We term the endo when the dienolate points on the same side of the indane, whereas the dienolate pointing on the opposite side of the indane is named exo. | ||
| Fig. 3 The optimized structures and geometry parameters of the intermediates and transition states involved in Stage 2 (distance in Å, dihedral in °, and most of the hydrogen atoms are omitted for the sake of clarity). | ||
 | ||
| Fig. 4 The Gibbs free energy profiles of Stage 2 and Stage 3 (aadding the free energy of R2. dadding the free energy of LGH and R2. eadding the free energy of LGH. fadding the free energy of Cat and LGH.) | ||
C bonds (CE1CE2 and CE3CE4) to the right relative conformation, which is necessary for the following [4 + 2] cycloaddition process with R2.
3.3 Stage 3: formal [4 + 2] cycloaddition and regeneration of the catalyst
The following process of the catalytic reaction is the [4 + 2] cycloaddition reaction. As shown in Scheme 4, Stage 3 includes two processes: (1) the formal [4 + 2] cycloaddition of cis-dienolate with R2, (2) dissociation of the product with the catalyst and regeneration of the catalyst. The optimized structures involved in Stage 3 are given in Fig. 5. | ||
| Fig. 5 The optimized structures and geometry parameters of the intermediates and transition states involved in Stage 3 (distance in Å and most of the hydrogen atoms are omitted for the sake of clarity). | ||
| Configuration of M4 | Addition face of M4 | Chirality of the CE4 atom |
|---|---|---|
| endo | Re | R |
| endo | Si | S |
| exo | Re | R |
| exo | Si | S |
The geometrical parameters depicted in Fig. 5 show that the formation of the CE4–NA2 bond is preferred over the formation of the CE1–NA1 bond. This phenomenon indicates that the two bonds (i.e. CE1–NA1 and CE4–NA2) are formed by a concerted but highly asynchronous manner. The free energy profile mapped in Fig. 4 reveals that M5Rexo is located 7.75/8.14/12.98 kcal mol−1 lower than M5Rendo/M5Sexo/M5Sendo separately, which implies that M5Rexo will be the significantly dominant isomer from the aspect of thermodynamics. In addition, the energy barriers of the cycloaddition step are 19.62 (TS5Rendo) and 7.07 (TS5Sendo) kcal mol−1 with respect to M4endo for endo addition, whereas those for exo addition are 2.47 (TS5Rexo) and 14.67 (TS5Sendo) kcal mol−1 with respect to M4exo, respectively. Obviously, the endo addition for the formation of M5Rendo and exo addition for the formation of M5Sexo are unfavorable compared to the others, thus in the following parts, we think it is unnecessary to discuss these two possible reaction patterns. The formation of M5Rexo costs the lowest energy barrier and the energy barrier of TS5Rexo is 4.6 kcal mol−1 lower than that of TS5Sendo, which indicates that the formation of M5Rexo is more energy favorable and supports the reported preference to form the R-configuration of the product.
3.4 Origin of the enantioselectivity
In Fig. 6, only the most energy favorable pathways involved in the three states are shown. As discussed above, Stages 1 and 3 both contain more than one step, thus we only provided the energy profiles of pathways with the lowest energy barriers in Fig. 6. For Stage 1, the HCO3−-assisted proton transfer mechanism is the most favorable pathway associated with the energy barrier of 13.15/7.31 kcal mol−1 (Re/Si-TS2B). In Stage 3, the exo addition of R2 to M4Rexo and endo addition of R2 to M4Sendo are energy favorable. Moreover, as can be seen in Fig. 6, the reaction pathway associated with the formation of product PR is the main reaction pathway, and the rate-determining step of the main reaction pathway is identified to be the C–C single bond rotation step with the free energy of 10.57 kcal mol−1 (TS4exo). Furthermore, the chirality center (CE4 atom) is emerged in the formal [4 + 2] cycloaddition step of Stage 3, so we think that this step is the enantioselectivity determining step (R-configuration is predominant). | ||
| Fig. 6 The entire energy profiles of the NHC-catalyzed [4 + 2] annulation reaction. | ||
To explore the reason for the enantioselectivity, we performed the distortion-interaction analysis.28 The relative energies of the transition states TS5Rexo and TS5Sendo parallel the stabilities of the corresponding product–NHC complexes (M5Rexovs.M5Sendo). As both transition states are significantly affected by charge delocalization and steric configuration, we first employed the diastereomeric product complexes M5Rexo and M5Sendo to elucidate the origin of the selectivity. For both structures, we analyzed the contributions of distortion, assuming that the difference in transition state energies will be of similar origin. Here the activation energy is divided into two components, the distortion energy (Edist) and interaction energy (Eint).28 The distortion energy involves geometric and electronic changes to deform the reactants into their transition state geometry, which contains bond stretching, angle decrease or increase, dihedral change, and so on. The interaction energy includes exchange-repulsive and stabilizing electrostatic, polarization, and orbital effects in the transition state structure.
From the results in Table 2, one can conclude that the M4Rexo in M5Rexo is slightly more distorted [ΔΔEdist(M4exo)] from its equilibrium geometry (M4Rexo) than M4Sendo in M5Rendo. This is largely counterbalanced by the distortion of the R2 [ΔΔEdist(R2)], which is more distorted in M5Sendo. And the larger distortion of the R2 in M5Sendo should be the main reason that the free energy of M5Rendo is much lower than that of M5Sendo.
| ΔΔG | ΔΔEdist (M4endo/exo) | ΔΔEdist (R2) | |
|---|---|---|---|
| M5S endo | +12.98 | 0.0 | +27.54 |
| M5R exo | 0.0 | +4.96 | 0.0 |
| TS5S endo | +1.03 | +5.76 | 0.0 |
| TS5R exo | 0.0 | 0.0 | +1.47 |
The analysis of the stereocontrolling TSs is also carried out to figure out the factors that control the stereoselectivity. The distortion results demonstrate that the M4Sendo in TS5Sendo is more distorted than M4Rexo in TS5exo, whereas the R2 is slightly more distorted in TS5Rexo than that in TS5Sendo. As could be anticipated, the M4Rexo benefits from the lower ΔEdist value and the distortion of R2 is offset by large ΔEdist penalties of M4Sendo. Moreover, the transition state with CE1 of the dienolate pointing toward the mesitylene (TS5Rexo) is lower in energy than when it is toward the indane due to a CH–π interaction between CH of the dienolate and the aromatic ring. Specifically, the inner hydrogen on the dienolate carbon is just 2.55 Å from CE1 to mesitylene (Fig. 5) and the distance between C–H of the dienolate and the center of the aromatic ring is ∼2.90 Å, well within the combined van der Waals distance of 2.90 Å. Such CH–π interactions have been observed in the transition states of Diels–Alder reactions, sulfide oxidations, and hydride reductions.29 On the whole, the less distortion and the existence of CH–π interactions make the lower energy barrier of TS5Rexo and the formation of the R-configuration isomer preferred kinetically.
The computed energy difference between the diastereomeric TS5exo and TS5endo is 4.6 kcal mol−1, which corresponds to an enantiomeric excess of >99% in favor of the R isomer. This prediction is in good accordance with the experimentally observed ee of 99%.
4. Conclusion
In this present study, we have analyzed the [4 + 2] annulation reaction between γ-oxidized enals (R1) and di-tert-butyl azodicarboxylate (R2) catalyzed by N-heterocyclic carbene (NHC) using density functional theory. On the basis of our calculations, the reaction is demonstrated to occur through three elementary stages, and for each stage, more than one possible pathway that involved different participation molecules has been investigated. The calculated results reveal that the most favorable pathway contains six elementary steps: the NHC catalyst first reacts with R1 to initiate the reaction, and then the Breslow intermediate is formed by a HCO3−-assisted proton process. Subsequently, the removal of the leaving group occurred together with abstraction of the hydrogen from the OE atom and the next step is the isomerization process to give the cis-dienolate. The fifth step is the endo/exo [4 + 2] addition reaction of R2 to cis-dienolate, and in the final step the NHC catalyst is regenerated and the [4 + 2] cycloaddition products PR&PS are released. The enantioselectivity associated with the chiral carbon center (CE4 atom) turns out to be determined by the Re or Si face addition of R2 with M4endo/exo. All the calculations are consistent with the experimental results.Moreover, the distortion scale of M5 (M5Rexo and M5Sendo) and TS5 (TS5Rexo and TS5Sendo) as well as the CH–π interactions are the key factors that control the stereoselectivity. The use of the bicarbonate anion as the protic medium to assist the proton transfer will provide a new clue for this kind of reaction without any other protic additive.
Acknowledgements
The work described in this paper was supported by the National Natural Science Foundation of China (no. 21303167), China Postdoctoral Science Foundation (no. 2013M530340) and Excellent Doctoral Dissertation Engagement Fund of Zhengzhou University in 2014.Notes and references
- (a) F. Glorius, N-heterocyclic carbene in catalysis – an introduction, Springer-Verlag, Berlin, Heidelberg, 2006, vol. 21, p. 1 Search PubMed; (b) S. P. Nolan, Acc. Chem. Res., 2010, 44, 91 CrossRef PubMed; (c) D. Zhang and G. F. Zi, Chem. Soc. Rev., 2015, 44, 1898 RSC; (d) C. Chen, M. H. Kim and S. H. Hong, Org. Chem. Front., 2015, 2, 241 RSC.
- (a) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606 CrossRef CAS PubMed; (b) F. Glorius and K. Hirano, Nucleophilic carbenes as organocatalysts, Springer-Verlag, Berlin, Heidelberg, 2008, vol. 2, p. 159 Search PubMed; (c) J. L. Moore and T. Rovis, Top. Curr. Chem., 2010, 291, 77 CAS; (d) D. Enders and T. Balensiefer, Acc. Chem. Res., 2004, 37, 534 CrossRef CAS PubMed; (e) V. Nair, S. Vellalath and B. P. Babu, Chem. Soc. Rev., 2008, 37, 2691 RSC; (f) M. Fèvre, J. Pinaud, Y. Gnanou, J. Vignolle and D. Taton, Chem. Soc. Rev., 2013, 42, 2142 RSC.
- (a) J. Kaeobamrung, M. C. Kozlowski and J. W. Bode, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20661 CrossRef CAS PubMed; (b) S. J. Ryan, L. Candish and D. W. Lupton, J. Am. Chem. Soc., 2011, 133, 4694 CrossRef CAS PubMed; (c) T. Y. Jian, L. He, C. Tang and S. Ye, Angew. Chem., Int. Ed., 2011, 50, 9104 CrossRef CAS PubMed; (d) E. M. Phillips, M. Wadamoto, A. Chan and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 3107 CrossRef CAS PubMed; (e) D. Enders, A. Grossmann, J. Fronert and G. Raabe, Chem. Commun., 2010, 46, 6282 RSC; (f) T. Ema, Y. Oue, K. Akihara, Y. Miyazaki and T. Sakai, Org. Lett., 2009, 11, 4866 CrossRef CAS PubMed; (g) D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2011, 133, 1040 CrossRef PubMed; (h) T. Jousseaume, N. E. Wurz and F. Glorious, Angew. Chem., Int. Ed., 2011, 50, 1410 CrossRef CAS PubMed; (i) Q. Kang and Y. Zhang, Org. Biomol. Chem., 2011, 9, 6715 RSC; (j) A. Lee and K. A. Scheidt, Chem. Commun., 2015, 51, 3407 RSC; (k) H. Lu, J. Y. Liu, C. G. Li, J. B. Lin, Y. M. Liang and P. F. Xu, Chem. Commun., 2015, 51, 4473 RSC; (l) X. K. Chen, X. Q. Fang and Y. R. Chi, Chem. Sci., 2013, 4, 2613 RSC.
- (a) X. N. Wang, P. L. Shao, H. Lv and S. Ye, Org. Lett., 2009, 11, 4029 CrossRef CAS PubMed; (b) X. L. Huang, X. Y. Chen and S. Ye, J. Org. Chem., 2009, 74, 7585 CrossRef CAS PubMed; (c) J. Douglas, J. E. Taylor, G. Churchill, A. M. X. Slawin and A. D. Smith, J. Org. Chem., 2013, 78, 3925 CrossRef CAS PubMed; (d) X. N. Wang, L. T. Shen and S. Ye, Org. Lett., 2011, 13, 6382 CrossRef CAS PubMed; (e) T. Wang, X. L. Huang and S. Ye, Org. Biomol. Chem., 2010, 8, 5007 RSC; (f) H. M. Zhang, Z. H. Gao and S. Ye, Org. Lett., 2014, 16, 3079 CrossRef CAS PubMed.
- X. N. Wang, L. T. Shen and S. Ye, Chem. Commun., 2011, 47, 8388 RSC.
- (a) S. M. Leckie, B. Brown, D. Pryde, T. Lebl, A. M. Z. Slawin and A. D. Smith, Org. Biomol. Chem., 2013, 11, 3230 RSC; (b) T. Y. Jian, X. Y. Chen, L. H. Sun and S. Ye, Org. Biomol. Chem., 2013, 11, 158 RSC; (c) T. Y. Jian, P. L. Shao and S. Ye, Chem. Commun., 2011, 47, 2381 RSC.
- (a) C. Burstein and F. Glorius, Angew. Chem., Int. Ed., 2004, 43, 6205 CrossRef CAS PubMed; (b) S. S. Sohn, E. L. Rosen and J. W. Bode, J. Am. Chem. Soc., 2004, 126, 14370 CrossRef CAS PubMed; (c) V. Nair, S. Vellalath, M. Poonoth and E. Suresh, J. Am. Chem. Soc., 2006, 128, 8736 CrossRef CAS PubMed; (d) D. E. A. Raup, B. Cardinal-David, D. Holte and K. A. Scheidt, Nat. Chem., 2010, 2, 766 CrossRef CAS PubMed; (e) X. D. Zhao, D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2011, 133, 12466 CrossRef CAS PubMed; (f) H. Lv, W. Q. Jia, L. H. Sun and S. Ye, Angew. Chem., Int. Ed., 2013, 52, 8607 CrossRef CAS PubMed.
- (a) M. He, J. R. Struble and J. W. Bode, J. Am. Chem. Soc., 2006, 128, 8418 CrossRef CAS PubMed; (b) C. Burstein, S. Tschan, X. L. Xie and F. Glorius, Synthesis, 2006, 2418 CAS; (c) M. Wadamoto, E. M. Phillips, T. E. Reynolds and K. A. Scheidt, J. Am. Chem. Soc., 2007, 129, 10098 CrossRef CAS PubMed; (d) V. Nair, R. R. Paul, K. C. S. Lakshmi, R. S. Menon, A. Jose and C. R. Sinu, Tetrahedron Lett., 2011, 52, 5992 CrossRef CAS PubMed; (e) X. Q. Fang, X. K. Chen and Y. R. Chi, Org. Lett., 2011, 13, 4708 CrossRef CAS PubMed.
- (a) D. A. DiRocco and T. Rovis, J. Am. Chem. Soc., 2011, 133, 10402 CrossRef CAS PubMed; (b) X. Fang, X. Chen, H. Lv and Y. R. Chi, Angew. Chem., Int. Ed., 2011, 50, 11782 CrossRef CAS PubMed; (c) L. H. Sun, Z. Q. Liang, W. Q. Jia and S. Ye, Angew. Chem., Int. Ed., 2013, 52, 5803 CrossRef CAS PubMed.
- L. M. Fang, F. Wang, P. J. Chua, Y. B. Lv, L. J. Zhong and G. F. Zhong, Org. Lett., 2012, 14, 2894 CrossRef PubMed.
- (a) S. H. Hu, B. Y. Wang, Y. Zhang, W. F. Tang, M. Y. Fang, T. Lu and D. Du, Org. Biomol. Chem., 2015, 13, 4661 RSC; (b) S. R. Yetra, S. Mondal, E. Suresh and A. T. Biju, Org. Lett., 2015, 17, 1417 CrossRef CAS PubMed; (c) J. D. Tessier, E. A. O'Bryan, T. B. H. Schroeder, D. T. Cohen and K. A. Scheidt, Angew. Chem., Int. Ed., 2012, 124, 5047 CrossRef PubMed; (d) Z. Q. Liang, Z. H. Guo, W. Q. Jia and S. Ye, Chem. – Eur. J., 2015, 21, 1868 CrossRef CAS PubMed; (e) C. Guo, M. Schedler, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 10232 CrossRef CAS PubMed; (f) A. G. Kravina, J. Mahatthananchai and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 9433 CrossRef CAS PubMed; (g) Y. Zhang, Y. Y. Lu, W. F. Tang, T. Lu and D. Du, Org. Biomol. Chem., 2014, 12, 3009 RSC.
- (a) J. Izquierdo, A. Orue and K. A. Scheidt, J. Am. Chem. Soc., 2013, 135, 10634 CrossRef CAS PubMed; (b) Y. W. Xie, Y. L. Que, T. J. Li, L. Zhu, C. X. Yu and C. S. Yao, Org. Biomol. Chem., 2015, 13, 1829 RSC; (c) H. Lv, W. Q. Jia, L. H. Sun and S. Ye, Angew. Chem., Int. Ed., 2013, 125, 8769 CrossRef PubMed.
- (a) J. M. Mo, X. K. Chen and Y. R. Chi, J. Am. Chem. Soc., 2012, 134, 8810 CrossRef CAS PubMed; (b) X. Y. Chen, F. Xia, J. T. Cheng and S. Ye, Angew. Chem., Int. Ed., 2013, 52, 10644 CrossRef CAS PubMed; (c) R. Liu, C. X. Yu, Z. X. Xiao, T. J. Li, X. S. Wang, Y. W. Xie and C. S. Yao, Org. Biomol. Chem., 2014, 12, 1885 RSC; (d) M. Wang, Z. J. Huang, J. F. Xu and Y. R. Chi, J. Am. Chem. Soc., 2014, 136, 1214 CrossRef CAS PubMed.
- (a) J. Mahatthananchai and J. W. Bode, Acc. Chem. Res., 2013, 47, 696 CrossRef PubMed; (b) R. C. Johnston, D. T. Cohen, C. C. Eichman, K. A. Scheidt and P. H. Y. Cheong, Chem. Sci., 2014, 5, 1974 RSC; (c) S. E. Allen, J. Mahatthananachai, J. W. Bode and M. C. Kozlowski, J. Am. Chem. Soc., 2012, 134, 12098 CrossRef CAS PubMed; (d) P. Verma, P. A. Patni and R. B. Sunoj, J. Org. Chem., 2011, 76, 5606 CrossRef CAS PubMed; (e) Z. Y. Li, D. H. Wei, Y. Wang, Y. Y. Zhu and M. S. Tang, J. Org. Chem., 2014, 79, 3069 CrossRef CAS PubMed.
- (a) D. H. Wei, Y. Y. Zhu, C. Zhang, D. Z. Sun, W. J. Zhang and M. S. Tang, J. Mol. Catal. A: Chem., 2011, 334, 108 CrossRef CAS PubMed; (b) M. M. Zhang, D. H. Wei, Y. Wang, S. J. Li, J. F. Liu, Y. Y. Zhu and M. S. Tang, Org. Biomol. Chem., 2014, 12, 6374 RSC.
- W. J. Zhang, Y. Y. Zhu, D. H. Wei, Y. X. Li and M. S. Tang, J. Org. Chem., 2012, 77, 10729 CrossRef CAS PubMed.
- W. J. Zhang, D. H. Wei and M. S. Tang, J. Org. Chem., 2013, 78, 11849 CrossRef CAS PubMed.
- (a) T. Liu, S. M. Han, L. L. Han, L. Wang, X. Y. Cui, C. Y. Du and S. W. Bi, Org. Biomol. Chem., 2015, 13, 3654 RSC; (b) Q. Zhang, H. Z. Yu and Y. Fu, Org. Chem. Front., 2014, 1, 614 RSC; (c) Y. Qiao and K. L. Han, Org. Biomol. Chem., 2012, 10, 7689 RSC; (d) Y. Wang, D. H. Wei, Z. Y. Li, Y. Y. Zhu and M. S. Tang, J. Phys. Chem. A, 2014, 118, 4288 CrossRef CAS PubMed; (e) F. Godin, M. Duplessis, C. Buonomano, T. Trinh, K. Houde, D. Chapdelaine, J. Rodrigue, A. B. Outros and Y. Guindon, Org. Chem. Front., 2014, 1, 974 RSC; (f) Y. M. Chen, G. A. Chass and D. C. Fang, Phys. Chem. Chem. Phys., 2014, 16, 1078 RSC; (g) Y. Li and D. C. Fang, Phys. Chem. Chem. Phys., 2014, 16, 15224 RSC; (h) Y. Wang, D. H. Wei, W. J. Zhang, Y. Y. Wang, Y. Y. Zhu, Y. Jia and M. S. Tang, Org. Biomol. Chem., 2014, 12, 7503 RSC.
- (a) Y. Qiao, K. L. Han and C. G. Zhan, Org. Biomol. Chem., 2014, 12, 2214 RSC; (b) D. H. Wei, B. L. Lei, M. S. Tang and C. G. Zhan, J. Am. Chem. Soc., 2012, 134, 10436 CrossRef CAS PubMed; (c) D. M. Li, Y. Wang and K. L. Han, Coord. Chem. Rev., 2012, 256, 1137 CrossRef CAS PubMed; (d) D. M. Li, X. Q. Huang and K. L. Han, J. Am. Chem. Soc., 2011, 133, 7416 CrossRef CAS PubMed.
- (a) Y. Ooyama, K. Uenaka and J. Ohshita, Org. Chem. Front., 2015, 2, 552 RSC; (b) Y. Li and Z. Y. Lin, Org. Chem. Front., 2014, 1, 1188 RSC; (c) A. N. Hancock, Y. Kavanagh and C. H. Schiesser, Org. Chem. Front., 2014, 1, 645 RSC; (d) D. Leboeuf, M. Gaydou, Y. H. Wang and A. M. Echavarren, Org. Chem. Front., 2014, 1, 759 RSC; (e) M. Quan, G. Q. Yang, F. Xie, L. Gridney and W. Zhang, Org. Chem. Front., 2015, 2, 398 RSC; (f) L. Zhang and D. C. Fang, J. Org. Chem., 2013, 78, 2405 CrossRef CAS PubMed; (g) J. Y. Tao, D. C. Fang and G. A. Chass, Phys. Chem. Chem. Phys., 2012, 14, 6937 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, M. I. J. Hasegawa, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision C.01), Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- (a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS; (b) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157 CrossRef CAS PubMed; (c) W. J. Zhang, D. G. Truhlar and M. S. Tang, J. Chem. Theory Comput., 2013, 9, 3965 CrossRef CAS.
- (a) B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 106, 5151 CrossRef CAS PubMed; (b) V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995 CrossRef CAS.
- (a) C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154 CrossRef CAS PubMed; (b) C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523 CrossRef CAS.
- (a) A. E. Reed and F. Weinhold, J. Chem. Phys., 1983, 78, 4066 CrossRef CAS PubMed; (b) J. P. Foster and F. Weinhold, J. Am. Chem. Soc., 1980, 102, 7211 CrossRef CAS; (c) E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO Version 3.1 Search PubMed.
- C. Y. Legault, CYLview, 1.0b, Université de Sherbrooke, 2009 (http://www.cylview.org) Search PubMed.
- (a) L. L. Zhao, M. W. Wen and Z. X. Wang, Eur. J. Org. Chem., 2012, 3531 Search PubMed; (b) E. Mercier, B. Fonovic, C. Henry, O. Kwon and T. Dudding, Tetrahedron Lett., 2007, 48, 3617 CrossRef CAS PubMed; (c) Y. Liang, S. Liu, Y. Z. Xia, Y. H. Li and Z. X. Yu, Chem. – Eur. J., 2008, 14, 4361 CrossRef CAS PubMed; (d) Y. Xia, Y. Liang, Y. Chen, M. Wang, L. Jiao, F. Huang, S. Liu, Y. Li and Z. X. Yu, J. Am. Chem. Soc., 2007, 129, 3470 CrossRef CAS PubMed; (e) Y. Liang, S. Liu and Z. X. Yu, Synlett, 2009, 905 CAS; (f) Y. Z. Xia, A. S. Dudnik, Y. H. Li and V. Gevorgyan, Org. Lett., 2010, 12, 5538 CrossRef CAS PubMed; (g) Y. Z. Xia and G. P. Huang, J. Org. Chem., 2010, 75, 7842 CrossRef CAS PubMed; (h) A. S. Dudnik, Y. Z. Xia, Y. H. Li and V. Gevorgyan, J. Am. Chem. Soc., 2010, 132, 7645 CrossRef CAS PubMed.
- C. Y. Legault, Y. Garcia, C. A. Merlic and K. N. Houk, J. Am. Chem. Soc., 2007, 129, 12664 CrossRef CAS PubMed.
- (a) R. Gordillo and K. N. Houk, J. Am. Chem. Soc., 2006, 128, 3543 CrossRef CAS PubMed; (b) C. D. Anderson, T. Dudding, R. Gordillo and K. N. Houk, Org. Lett., 2008, 10, 2749 CrossRef CAS PubMed; (c) M. A. M. Capozzi, C. Centrone, G. Fracchiolla, F. Naso and C. Cardellicchio, Eur. J. Org. Chem., 2011, 4327 CrossRef CAS PubMed; (d) O. Gutierrez, R. G. Iafe and K. N. Houk, Org. Lett., 2009, 11, 4298 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00121h |
| This journal is © the Partner Organisations 2015 |