
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic [3 + 2]-annulation via sodium tetraarylborate: a fundamental approach for synthesizing 1,4,2-diazaborole analogs†
Hao-Ni
Qin
a,
Hao-Wen
Jiang
a,
Yi
Zhao
b,
Saira
Qurban
a,
Ke-Chun
Wang
a and
Peng-Fei
Xu
 *a
*a
aState Key Laboratory of Applied Organic Chemistry, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, Gansu, China. E-mail: xupf@lzu.edu.cn
bTecon Pharmaceutical Co., Ltd, No. 109, Jintun Road, Urumqi City 830023, Xinjiang, China
First published on 13th January 2025
Abstract
Substantial advancements have been achieved in the field of photocatalytic borylation utilizing 4c-7e Lewis base-boryl radicals. However, the utilization of 3c-5e neutral boryl radicals for C–B bond formation remains relatively underexplored due to their inherent instability. In this study, we successfully demonstrated the direct construction of C–B bonds using sodium tetraarylborate as a key reagent. This was accomplished by effectively stabilizing diaryl boryl radicals with nitrile compounds, thereby facilitating the synthesis of valuable boron-containing compounds. Overall, our research elucidates the significant role played by sodium tetraarylborate in enabling an efficient and versatile approach for synthesizing of 1,4,2-diazaborole analogs through a photocatalyzed [3 + 2]-annulation reaction. This mild and adaptable methodology expands synthetic strategies for obtaining diverse derivatives of 1,4,2-diazaboroles, with the RCN–BAr2 complex serving as an effective boron–nitrogen synthon that opens up pathways to multiple boron–nitrogen heterocycles. Furthermore, this breakthrough significantly enhances the applicability of sodium tetraarylborate in photoredox catalysis.
Introduction
Boron-containing compounds have become focal points in both scientific research and industrial development due to their unique chemical properties and diverse applications.1–5 Since the late 1990s, small boron-based molecules have garnered significant interest in the fields of chemistry and biomedicine.6–18 Among these, boron–nitrogen heterocycles stand out for their distinctive properties, finding utility in manufacturing, optoelectronics, and cosmetics.19–37 Representative cases are illustrated in Scheme 1A. One notable instance is 1,4,2-diazaborole, a five-membered boron–nitrogen heterocycle with the boron and nitrogen atoms occupying positions 1, 4, and 2 respectively. Structurally resembling 2-imidazoline,38–40 the inclusion of boron imparts unique electronic properties and reactivity to this compound. These attributes enhance its appeal for applications in organic chemistry and materials science while attracting growing interest from researchers.28–37 | ||
| Scheme 1 Research background. | ||
Ongoing research on the analogs of 1,4,2-diazaborole has been primarily focused on its exploration and synthesis optimization, with various boron reagents developed to date (Scheme 1B). As early as 1984, the Sadhu group devised a strategy utilizing α-iodoboronic ester in combination with amidine and n-BuLi to synthesize a single compound 1,4,2-diazaborole, achieving moderate yields.29 Subsequently, the Pawelke group synthesized the key boron reagents known as azoniaboratacyclopropanes which underwent ring-opening and expansion reactions to yield several derivatives of 1,4,2-diazaborole.30 Later on, the Erker group established a mild three-component sequential protocol using their described FmesBH2 along with isocyanides and nitriles. This showcased an efficient synthetic strategy.36 Recently, the Miyata group has developed a mild synthetic strategy using potassium α-aminoborate and azirine, thereby providing opportunities for further exploration.37 Despite recent advancements, there is still a strong demand for a more concise, efficient, and step-economical synthesis of 1,4,2-diazaborole analogs from readily available starting materials. We propose investigating the utilization of an economical and commercially available boron reagent for their direct synthesis.
In the field of photocatalytic borylation, significant advancements have been made in the realm of boryl radical chemistry in recent years.41–68 While Lewis base-boryl radicals, such as N-heterocyclic carbene (NHC) boryl radicals46–54 and amine boryl radicals,55–62 have been extensively studied, our group has also conducted relevant studies in this field.57,59 However, compared to the well-studied 4c-7e boryl radicals, the inherently unstable 3c-5e neutral boryl radicals have received considerably less attention. Amongst the precursors for neutral boryl radical, sodium tetraarylborate has garnered attention since the 1960s due to its affordability and stability.63,64 Nevertheless, its role in photochemistry has primarily focused on biaryl compound synthesis while leaving unexplored potential for generating neutral boryl radicals.65 In 2022, the Xia group achieved a significant breakthrough by employing neutral diaryl boryl radicals for the activation of the C–O bond in benzyl alcohol.66 The Wang group further employed diaryl boryl radicals as precursors for aryl radicals in reactions.67 Subsequently, the Sharma group demonstrated a core strategy by using diphenyl boryl radicals for C–Br bond activation.68 The aforementioned research studies successfully employed neutral boryl radicals derived from sodium tetraarylborate in photoredox catalysis to generate key carbon radicals; however, their application in C–B bond formation for boron-containing compounds remains unexplored (Scheme 1C).
Inspired by groundbreaking previous research, our focus is on the development of a catalytic strategy for synthesizing 1,4,2-diazaborole analogs, a distinct class of five-membered boron–nitrogen heterocycles. The [3 + 2]-annulation reaction serves as an efficient method for rapidly constructing five-membered carbon (hetero) cycles. Therefore, we propose to investigate whether neutral diphenyl boryl radicals can facilitate this reaction with suitable substrates, thereby enabling the rapid synthesis of 1,4,2-diazaborole analogs.
Building on the aforementioned literature, we propose that nitrile compounds, possessing a lone pair of electrons on the nitrogen atom, can function as Lewis bases by coordinating with electron-deficient diphenyl boryl radicals to yield intermediate II, specifically the MeCN–BPh2 complex.34,69 Intermediate II features two reactive sites: an electrophilic cyanide carbon and a nucleophilic boryl radical. This imparts significant potential for it to serve as a [3 atoms]-synthon in [3 + 2]-annulation reactions. Furthermore, we hypothesize that this synthon could interact with imines to construct the C–B bond and facilitate the synthesis of 1,4,2-diazaborole analogs through a [3 + 2]-annulation pathway.
Results and discussion
To validate the feasibility of our hypothesis, imine 1a and sodium tetraphenylborate 2a were selected as model substrates and [Ir(dF(CF3)ppy)2dtbbpy]PF6 was utilized as the photocatalyst under blue light irradiation in acetonitrile, resulting in the successful synthesis of target compound 4aa with a yield of 47%. After extensive optimization, it was discovered that [Ir(dF(CF3)ppy)2dtbbpy]PF6 exhibited the highest efficacy (Table 1: entries 1–5), and using 3.0 equivalents of 2a led to a significantly improved yield (entries 6–8). The optimization process involved fine-tuning the quantities of the photocatalyst, a 4 Å molecular sieve, solvent and reaction time, resulting in an enhanced yield of 78% (entries 9–14) (ESI Tables S2–S4†). Considering the susceptibility of imine 1a to hydrogenation and dimerization, we systematically varied its amount alongside boron source 2a as a reference point (ESI Table S5†). The highest yield of 91% was achieved with an equivalent ratio of 3.5 for compound 1a (entry 15). Subsequently, the conditions outlined in entry 15 were identified as the optimal reaction parameters. Control experiments unequivocally demonstrate that both the photocatalyst and light play indispensable roles in this [3 + 2]-annulation reaction (entries 16–17).|
|
|||||
|---|---|---|---|---|---|
| Entry | PC |
1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2a :2a |
Solvent | Additive | Yielda,b |
| a Isolated yield. b Unless otherwise noted, reaction conditions are as follows: imine (0.05 mmol), NaBPh4 (x mmol), PC (3 mol%), MeCN (0.5 mL), 30 W blue LEDs, 0 °C, 12 h, under an argon atmosphere. c [Ir(dF(CF3)ppy)2dtbbpy]PF6 (4 mol%). d Reaction time extended to 24 h. e Reaction time extended to 30 h. f Reaction time extended to 27 h. | |||||
| 1 | PC1 | 1:2 |
MeCN | — | 47 |
| 2 | PC2 | 1:2 |
MeCN | — | 33 |
| 3 | PC3 | 1:2 |
MeCN | — | 22 |
| 4 | PC4 | 1:2 |
MeCN | — | 0 |
| 5 | PC5 | 1:2 |
MeCN | — | 0 |
| 6 | PC1 | 1:1 |
MeCN | — | 34 |
| 7 | PC1 | 1:3 |
MeCN | — | 49 |
| 8 | PC1 | 1:4 |
MeCN | — | 44 |
| 9 | PC1 | 1:3 |
MeCN | — | 51c |
| 10 | PC1 | 1:3 |
MeCN | 4 Å MS (20 mg) | 55c |
| 11 | PC1 | 1:3 |
MeCN + DCM | 4 Å MS (20 mg) | 60c |
| 12 | PC1 | 1:3 |
MeCN + DCM | 4 Å MS (20 mg) | 73c,d |
| 13 | PC1 | 1:3 |
MeCN + DCM | 4 Å MS (20 mg) | 58c,e |
| 14 | PC1 | 1:3 |
MeCN + DCM | 4 Å MS (20 mg) | 78c,f |
| 15 | PC1 |
3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) .5:1 .5:1
|
MeCN + DCM | 4 Å MS (20 mg) | 91c , |
| 16 | Without PC | Trace | |||
| 17 | Without light | N.R. | |||

After establishing the optimal reaction conditions, we proceeded to expand our investigation in order to assess the substrate scope and generality of the reaction. Initially, a variety of imines derived from aromatic amines were subjected to testing. As indicated in Table 2, the incorporation of electron-donating groups such as methoxy (4aa), tert-butyl (4ab) and phenyl (4ac) onto the aniline aromatic ring consistently resulted in favorable results, thereby demonstrating compatibility within the reaction system. However, it should be noted that introducing an amido group (4ad) led to a modest reduction in yield. Furthermore, both unsubstituted aniline-derived imine (4ae) and 2-naphthylamine-derived imine (4af) exhibited commendable yields. In contrast, halogen-substituted anilines (4ag–4ai) showed a slight decrease in yields. Notably, electron-withdrawing groups such as acetyl (4aj), cyano (4ak), and ester groups (4al) were well tolerated, resulting in a slight reduction in yields. Next, different boron sources were investigated. Substituting the phenyl group in sodium tetraphenylborate with 4-methylphenyl (4ba) and 2-naphthyl (4bb) still afforded the target product, albeit with reduced yield. Subsequently, nitriles were tested. In addition to acetonitrile, cyclopropyl nitrile (4ca), isobutyronitrile (4cb) and 4-methoxybenzenecarbonitrile (4cc) also underwent successful transformation, demonstrating the method's broad applicability. The configuration of compound 4aa was determined by X-ray crystal structure analysis (CCDC 2381718).

The reaction's scope and versatility were evaluated by testing various imines derived from α-ketoesters. As indicated in Table 3, the reaction provided moderate to good yields for imines derived from α-ketoesters, with increased steric hindrance resulting in lower yields (4am–4ar). Subsequently, the influence of electron-donating substituents on the aromatic ring of α-ketoesters, such as methyl (4as), ethyl (4at), tert-butyl (4au), phenyl (4av), and methoxy groups (4aw), was investigated, yielding slightly lower results compared to the model substrate.
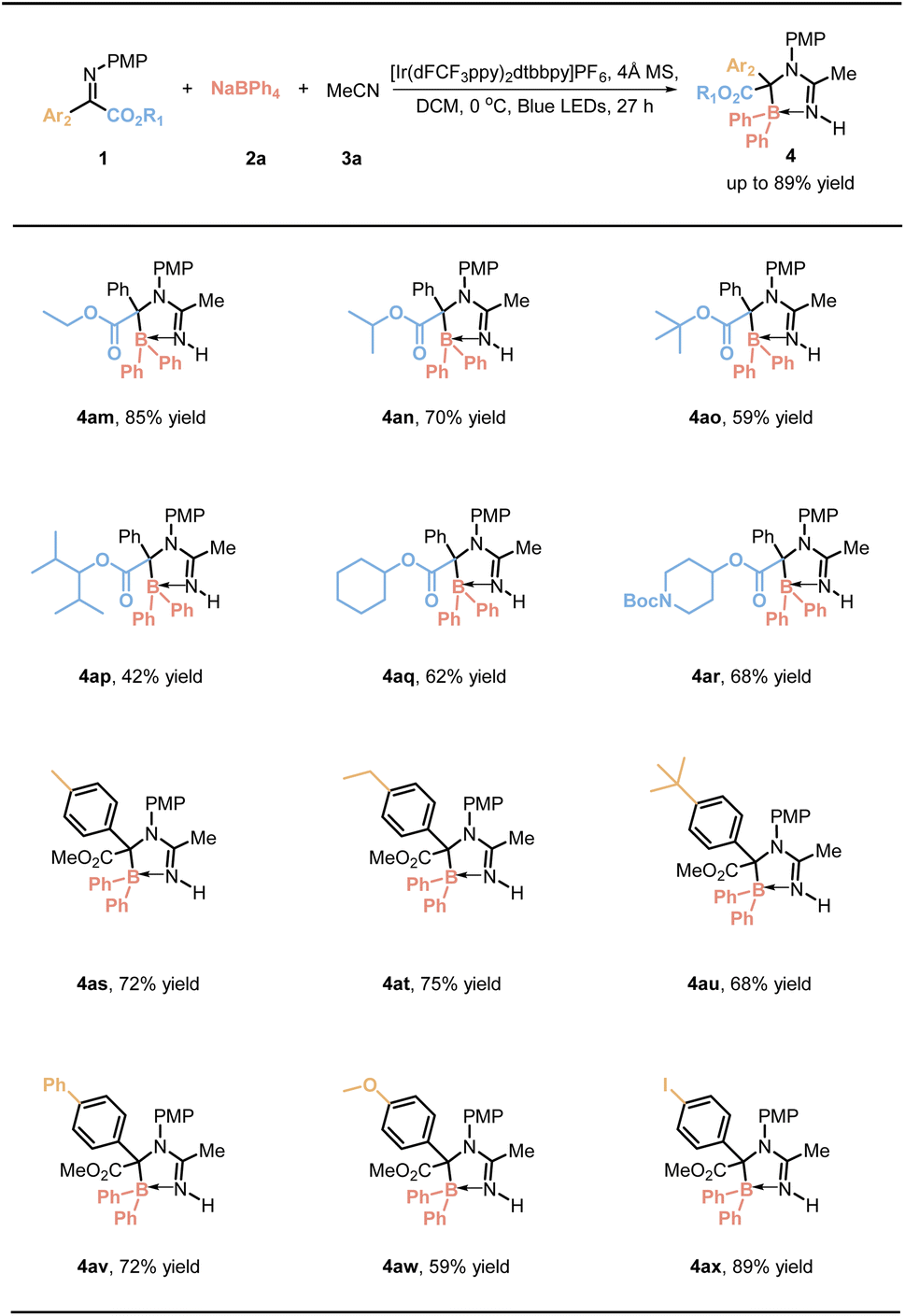
Remarkably, when an iodine substituent was introduced at the para position (4ax), a yield of 89% was achieved, demonstrating the robustness of the reaction pathway against this substitution.
To further broaden the applicability of our methods, sodium tetraphenylborate can also undergo photocatalytic [3 + 2]-annulation reactions with alkenes, leading to the generation of additional classes of boron–nitrogen heterocycles. This shows that introducing both electron-withdrawing group-substituted styrenes (6a–6c) and electron-rich 1,1-diphenylethylene (6d) into the template reaction yields the desired azaborole in moderate to good efficiency (Table 4). To demonstrate the practicality of our method, we conducted a scale-up experiment, successfully synthesizing 1.1 grams of the target product with a yield of 77% (Table 5a). These results highlight the advantages of our strategy for synthesizing diverse boron–nitrogen heterocycles, including mild reaction conditions and broad functional group compatibility.

|

|
We then conducted a series of experiments to elucidate the reaction mechanism. Under standard conditions, the reaction was inhibited by radical scavengers, and the corresponding product formed through radical trapping was identified using high-resolution mass spectrometry (HR-MS) (Table 5b). Control experiments demonstrated that both light irradiation and the presence of a photocatalyst are indispensable for the reaction to occur (Table 1: entries 16–17). A radical clock experiment employing 1-phenylvinylcyclopropane and sodium tetraphenylborate yielded the formation of a cyclopropane ring-opened product, which was identified by HR-MS (Table 5c). These experimental findings provide support for a photoredox radical process. Fluorescence quenching experiments revealed that both imine and sodium tetraphenylborate can quench the excited state of the photocatalyst; however, imine 1a exhibited a more pronounced effect (Table 5d, left). Further light on/off experiments reveal that any chain propagation process was transient and that light played a crucial role in the formation of the product (Table 5d, right). The existence of the radical intermediate II is indirectly confirmed by the hydrogen abstraction reaction (ESI Fig. S3†). Additionally, the dimeric product of the reduced imine, confirmed by HR-MS, provides evidence for the radical coupling pathway leading to the target product (Scheme 2). However, it cannot be completely ruled out that direct attack by the boryl radical on the imine via a single-electron transfer mechanism may occur (ESI Fig. S8†).
 | ||
| Scheme 2 Proposed mechanism. | ||
Based on the mechanistic studies, a plausible reaction mechanism is proposed in Scheme 2. Upon irradiation, an excited IrIII undergoes single-electron transfer (SET) on the imine to deliver the α-amino radical anion I and a resulting IrIV. Subsequently, this IrIV species is reduced back to its ground state by NaBPh4, while simultaneously oxidizing NaBPh4 to produce a diphenyl boryl radical. In the presence of acetonitrile, it is hypothesized that nitrogen initially coordinates with the diphenyl boryl radical, forming intermediate II—indirectly confirmed by HR-MS. Intermediate I and II then engage in a radical–radical cross-coupling reaction to form III. The nitrogen anion in III subsequently undergoes nucleophilic addition with the cyano group, ultimately resulting in the target 1,4,2-diazaborole product.
Conclusions
We present a mild and efficient method for the formation of C–B bonds using sodium tetraarylborate, which stabilizes neutral boryl radicals with nitriles. This methodology enables the synthesis of 1,4,2-diazaborole analogs via photoredox catalyzed [3 + 2]-annulation. This approach offers gentle reaction conditions, facile substrate preparation, broad applicability, and flexible compatibility with various functional groups. Beyond expanding synthetic routes for 1,4,2-diazaborole analogs, this study highlights the versatility of sodium tetraarylborate in photoredox catalysis and its potential in boron–nitrogen heterocycle synthesis. Our ongoing research aims to further explore innovative applications of this compound in boron chemistry.Data availability
All experimental data associated with this work are available in the ESI.†Author contributions
H.-N. Q. planned and conducted most of the experiments; H.-W. J. offered constructive suggestions and revised the manuscript; Y. Z. supervised and supported this project. Saira Qurban and K.-C. W. revised the manuscript; P.-F. X. conceptualised and supervised the project and provided critical feedback during the whole process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the National Natural Science Foundation of China (NSFC, 21871116 and U22A20390), supported by the Xinjiang Uygur Autonomous Region Science and Technology Department's Project [Xincaihang (2023-211)], the Major Research Program of Gansu Province (23JRRA1512), and the “111” Program from the Ministry of Education of the People's Republic of China.Notes and references
- S. J. Baker, C. Z. Ding, T. Akama, Y.-K. Zhang, V. Hernandez and Y. Xia, Future Med. Chem., 2009, 1, 1275–1288 CrossRef CAS.
- R. Thompson, Pure Appl. Chem., 1974, 39, 547–559 CrossRef CAS.
- Y. Guan and Y. Zhang, Chem. Soc. Rev., 2013, 42, 8106 RSC.
- W. L. A. Brooks and B. S. Sumerlin, Chem. Rev., 2016, 116, 1375–1397 CrossRef CAS.
- M. Fan, X. Liang, Q. Li, L. Cui, X. He and X. Zou, Chin. Chem. Lett., 2023, 34, 107275 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- K. S. Yoo, C. H. Yoon and K. W. Jung, J. Am. Chem. Soc., 2006, 128, 16384–16393 CrossRef CAS PubMed.
- C. M. Crudden, B. W. Glasspoole and C. J. Lata, Chem. Commun., 2009, 44, 6704 RSC.
- A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef CAS.
- C. Poon, D. Wu and V. W. Yam, Angew. Chem., Int. Ed., 2016, 55, 3647–3651 CrossRef CAS.
- P. Andrés, G. Ballano, M. I. Calaza and C. Cativiela, Chem. Soc. Rev., 2016, 45, 2291–2307 RSC.
- G. F. S. Fernandes, W. A. Denny and J. L. Dos Santos, Eur. J. Med. Chem., 2019, 179, 791–804 CrossRef CAS PubMed.
- A. Šterman, I. Sosič, S. Gobec and Z. Časar, Org. Chem. Front., 2019, 6, 2991–2998 RSC.
- J. Plescia and N. Moitessier, Eur. J. Med. Chem., 2020, 195, 112270 CrossRef CAS.
- M. A. Dymova, S. Y. Taskaev, V. A. Richter and E. V. Kuligina, Cancer Commun., 2020, 40, 406–421 CrossRef PubMed.
- A. N. Tevyashova and M. V. Chudinov, Russ. Chem. Rev., 2021, 90, 451–487 CrossRef.
- L. Ji and H. Zhou, Tetrahedron Lett., 2021, 82, 153411 CrossRef CAS.
- R. J. Grams, W. L. Santos, I. R. Scorei, A. Abad-García, C. A. Rosenblum, A. Bita, H. Cerecetto, C. Viñas and M. A. Soriano-Ursúa, Chem. Rev., 2024, 124, 2441–2511 CrossRef CAS PubMed.
- R. T. Paine and C. K. Narula, Chem. Rev., 1990, 90, 73–91 CrossRef CAS.
- Q. Liu, M. S. Mudadu, H. Schmider, R. Thummel, Y. Tao and S. Wang, Organometallics, 2002, 21, 4743–4749 CrossRef CAS.
- H. F. Bettinger, T. Kar and E. Sánchez-García, J. Phys. Chem. A, 2009, 113, 3353–3359 CrossRef CAS.
- N. Boens, V. Leen and W. Dehaen, Chem. Soc. Rev., 2012, 41, 1130–1172 RSC.
- J. Lu, S. Ko, N. R. Walters, Y. Kang, F. Sauriol and S. Wang, Angew. Chem., Int. Ed., 2013, 52, 4544–4548 CrossRef CAS PubMed.
- Y. Valadbeigi, New J. Chem., 2018, 42, 18777–18786 RSC.
- J.-M. Yang, F.-K. Guo, Y.-T. Zhao, Q. Zhang, M.-Y. Huang, M.-L. Li, S.-F. Zhu and Q.-L. Zhou, J. Am. Chem. Soc., 2020, 142, 20924–20929 CrossRef CAS.
- J. J. Blackner, O. M. Schneider, W. O. Wong and D. G. Hall, J. Am. Chem. Soc., 2024, 146, 19499–19508 CrossRef CAS PubMed.
- X. Zhang, W. Su, H. Guo, P. Fang, K. Yang and Q. Song, Angew. Chem., Int. Ed., 2024, 63, e202318613 CrossRef CAS PubMed.
- M. Nakahara, Y. Wada, M. Kawano, K. Hanaya, T. Sugai and S. Higashibayashi, Chem. Lett., 2024, 53, upae069 CrossRef.
- D. S. Matteson and K. M. Sadhu, Organometallics, 1984, 3, 614–618 CrossRef CAS.
- A. Ansorge, D. J. Brauer, H. Biirger, T. Hagen and G. Pawelke, J. Org. Chem., 1994, 467, l–11 Search PubMed.
- B. Su, Y. Li, R. Ganguly, J. Lim and R. Kinjo, J. Am. Chem. Soc., 2015, 137, 11274–11277 CrossRef CAS.
- A. R. Cabrera, R. S. Rojas, M. Valderrama, P. Plüss, H. Berke, C. G. Daniliuc, G. Kehr and G. Erker, Dalton Trans., 2015, 44, 19606–19614 RSC.
- B. Su, Y. Li, R. Ganguly and R. Kinjo, Angew. Chem., Int. Ed., 2017, 56, 14572–14576 CrossRef CAS PubMed.
- Y. Katsuma, L. Wu, Z. Lin, S. Akiyama and M. Yamashita, Angew. Chem., Int. Ed., 2019, 58, 317–321 CrossRef CAS PubMed.
- Y. Lebedev, C. Apte, S. Cheng, C. Lavigne, A. Lough, A. Aspuru-Guzik, D. S. Seferos and A. K. Yudin, J. Am. Chem. Soc., 2020, 142, 13544–13549 CrossRef CAS PubMed.
- J. Li, C. G. Daniliuc, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2021, 60, 27053–27061 CrossRef CAS.
- T. Oba, H. Takahashi, M. Roppongi and K. Miyata, Tetrahedron Lett., 2022, 102, 153953 CrossRef CAS.
- V. Sharma, S. Peddibhotla and J. J. Tepe, J. Am. Chem. Soc., 2006, 128, 9137–9143 CrossRef CAS PubMed.
- C. Karmel, C. Z. Rubel, E. V. Kharitonova and J. F. Hartwig, Angew. Chem., Int. Ed., 2020, 59, 6074–6081 CrossRef CAS.
- P. Bousquet, A. Hudson, J. A. García-Sevilla and J.-X. Li, Pharmacol. Rev., 2020, 72, 50–79 CrossRef CAS.
- D. Zhao and Z. Xie, Angew. Chem., Int. Ed., 2016, 55, 3166–3170 CrossRef CAS.
- Y. Su and R. Kinjo, Coord. Chem. Rev., 2017, 352, 346–378 CrossRef CAS.
- M. Chen, J. Xu, D. Zhao, F. Sun, S. Tian, D. Tu, C. Lu and H. Yan, Angew. Chem., Int. Ed., 2022, 61, e202205672 CrossRef CAS.
- S. Li and Z. Xie, J. Am. Chem. Soc., 2022, 144, 7960–7965 CrossRef CAS PubMed.
- H. Ren, P. Zhang, J. Xu, W. Ma, D. Tu, C. Lu and H. Yan, J. Am. Chem. Soc., 2023, 145, 7638–7647 CrossRef CAS PubMed.
- N. Zhou, X. Yuan, Y. Zhao, J. Xie and C. Zhu, Angew. Chem., Int. Ed., 2018, 57, 3990–3994 CrossRef CAS PubMed.
- W. Xu, H. Jiang, J. Leng, H. Ong and J. Wu, Angew. Chem., Int. Ed., 2020, 59, 4009–4016 CrossRef CAS.
- W. Dai, S. J. Geib and D. P. Curran, J. Am. Chem. Soc., 2020, 142, 6261–6267 CrossRef PubMed.
- P. Xia, D. Song, Z. Ye, Y. Hu, J. Xiao, H. Xiang, X. Chen and H. Yang, Angew. Chem., Int. Ed., 2020, 59, 6706–6710 CrossRef CAS.
- J. Qi, F. Zhang, J. Jin, Q. Zhao, B. Li, L. Liu and Y. Wang, Angew. Chem., Int. Ed., 2020, 59, 12876–12884 CrossRef CAS.
- C. Zhu, J. Dong, X. Liu, L. Gao, Y. Zhao, J. Xie, S. Li and C. Zhu, Angew. Chem., Int. Ed., 2020, 59, 12817–12821 CrossRef CAS.
- T. Taniguchi, Chem. Soc. Rev., 2021, 50, 8995–9021 RSC.
- T.-Y. Peng, F.-L. Zhang and Y.-F. Wang, Acc. Chem. Res., 2023, 56, 169–186 CrossRef CAS.
- Y. Xie, R. Zhang, Z. Chen, M. Rong, H. He, S. Ni, X. He, W. Xiao and J. Xuan, Adv. Sci., 2024, 11, 2306728 CrossRef CAS.
- J. H. Kim, T. Constantin, M. Simonetti, J. Llaveria, N. S. Sheikh and D. Leonori, Nature, 2021, 595, 677–683 CrossRef CAS PubMed.
- L. Capaldo, T. Noël and D. Ravelli, Chem Catal., 2022, 2, 957–966 CrossRef CAS.
- H.-W. Jiang, W.-L. Yu, D. Wang and P.-F. Xu, ACS Catal., 2024, 14, 8666–8675 CrossRef CAS.
- W. Li, Z. Huang, D. Zhong and H. Li, Org. Lett., 2024, 26, 2062–2067 CrossRef CAS.
- H.-W. Jiang, H.-N. Qin, A.-L. Wang, R. Zhang and P.-F. Xu, Org. Lett., 2024, 26, 9282–9287 CrossRef CAS PubMed.
- C. S. Buettner, C. Stavagna, M. J. Tilby, B. Górski, J. J. Douglas, N. Yasukawa and D. Leonori, J. Am. Chem. Soc., 2024, 146, 24042–24052 CrossRef CAS.
- Z. Zhang, M. J. Tilby and D. Leonori, Nat. Synth., 2024, 3, 1221–1230 CrossRef.
- K.-R. Li, X.-C. He, J. Gao, Y.-L. Liu, H.-B. Chen, H.-Y. Xiang, K. Chen and H. Yang, J. Org. Chem., 2024, 89, 12658–12667 CrossRef CAS.
- J. L. R. Williams, J. C. Doty, P. J. Grisdale, R. Searle, T. H. Regan, G. P. Happ and D. P. Maier, J. Am. Chem. Soc., 1967, 89, 5153–5157 CrossRef CAS.
- J. C. Doty, P. J. Grisdale, T. R. Evans and J. L. R. Williams, J. Org. Chem., 1971, 32, C35–C37 CrossRef CAS.
- A. Music, A. N. Baumann, F. Boser, N. Müller, F. Matz, T. C. Jagau and D. Didier, Chem.–Eur. J., 2021, 27, 4322–4326 CrossRef CAS PubMed.
- W.-D. Li, Y. Wu, S.-J. Li, Y.-Q. Jiang, Y.-L. Li, Y. Lan and J.-B. Xia, J. Am. Chem. Soc., 2022, 144, 8551–8559 CrossRef CAS.
- F. Yue, H. Ma, P. Ding, H. Song, Y. Liu and Q. Wang, ACS Cent. Sci., 2023, 9, 2268–2276 CrossRef CAS.
- S. Pillitteri, R. Walia, E. V. Van Der Eycken and U. K. Sharma, Chem. Sci., 2024, 15, 8813–8819 RSC.
- M. Frick, E. Kaifer and H. Himmel, Angew. Chem., Int. Ed., 2017, 56, 11645–11648 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2381718. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc08085h |
| This journal is © The Royal Society of Chemistry 2025 |