
Microgel-engineered temperature-responsive microcapsules at liquid interfaces for sequential biocatalytic reactions†
Divya
Gaur
a,
Wasia
Khanam
b,
Nidhi C.
Dubey
b and
Bijay P.
Tripathi
 *a
*a
aFunctional Materials & Membranes Laboratory, Department of Materials Science & Engineering, Indian Institute of Technology Delhi, New Delhi, 110016, India. E-mail: bptripathi@mse.iitd.ac.in; drbptripathi@gmail.com
bDepartment of Molecular Medicine, Jamia Hamdard, New Delhi 110062, India
First published on 24th April 2025
Abstract
Microgels, with their adjustable deformability and responsiveness, offer a promising alternative to prepare microcompartments via template-directed assembly onto emulsion droplets. Herein, we introduce a robust and reproducible strategy to synthesize responsive microgel poly(N-isopropylacrylamide-co-serine) (PNSER)-stabilized Pickering emulsions (both water-in-oil (W/O) and oil-in-water (O/W)) to design tunable vesicles (called microgelsomes) that could encapsulate hydrophobic and hydrophilic molecules. Unlike previous approaches that typically support only one emulsion type, our system uniquely accommodates both, offering a versatile platform for encapsulating a broad spectrum of active biomolecules within a single framework. The system was validated by applying it to two catalytic systems: antioxidation activity of lipophilic vitamin E and a bienzymatic cascade reaction involving laccase and transaminase to catalyze the stereoselective amination of racemic alcohols, yielding chiral amines, important for the synthesis of pharmaceutical drugs. We addressed the challenge of using two different pH requirements for the enzymes by conducting the cascade in a one-pot reaction at a single pH, thereby enhancing the efficiency and simplicity of the process. Impressively, the one-pot bienzymatic cascade produced 95–98% conversion, 2-fold higher than that of their free counterpart (43–50%) and comparable to that of the two-step sequential reaction performed at their respective optimal pH values.
Green foundation1. This work introduces a synthetic strategy to fabricate biocatalytic microcompartments (microgelsomes) with tunable permeability, by templating microgels on both water-in-oil and oil-in-water emulsion droplets. Serine-based microgelsomes allow encapsulation of biomolecules varying from lipophilic vitamin E to hydrophilic enzymes. The microgelsomes efficiently performed a two-step and one-pot bienzymatic reaction converting phenols to ketones and enantioselective amines.2. The main drawback of coupling laccase and transaminase is their different operational pH values, which is overcome by co-encapsulation of enzymes in microgelsomes, allowing a one-pot cascade reaction with similar activity to two-step bienzymatic reactions operated at their optimal pH. This strategy reduces operational steps, improving the process ‘pot-economy’. 3. The microgelsomes could also be used for encapsulation of enzymes for multi-step cascade reactions, as a step towards cellular mimics. The synthesized microgels can be investigated for the preparation of compartments using a water-in-water emulsion droplet as a template. |
1. Introduction
Cell-free synthetic biology has emerged as a more sustainable and greener alternative to conventional industrial catalytic processes and includes reconstitution of cellular components in a ‘minimalist’ format to efficiently perform chemical reactions.1–5 While enzymatic cascades have seen remarkable developments, enzyme encapsulation is considered one of the key strategies for optimizing their performance, especially in industrial contexts.6–10 This technique not only simplifies downstream processing but also boosts enzyme operational stability, facilitates easier recovery, and enables reuse. The encapsulation of complex biochemical pathways within the selectively permeable cell-like membrane preserves them against potential damage, allowing transfer of small molecules and regulation by external stimuli from neighboring cells, often through molecular diffusion.11 This has inspired the development of various bottom-up strategies to construct biomimetic compartments with vesicular architectures like liposomes,12 polymersomes,13 microfluidic channels,14–16 and water–oil emulsion droplets.17–19 These scaffolds have been extensively designed as biomimetic reactors and enclose minimal components for simple to multistep enzymatic pathways (enzymes, cofactors, substrates, and genetic circuits), exhibiting observably enhanced reaction rates.20,21Despite notable advancements, most of these biomimetic scaffolds require complex preparation techniques or exhibit an incompatible interior solvent phase, which limits the control of the internal microenvironment and exhibits low encapsulation efficiency.22,23 Consequently, the ability to encapsulate enzymes without significantly compromising their activity or selectivity, maintaining efficient mass transport, and preserving the stability of enzymes and the scaffold is a highly sought-after characteristic for biocatalysts in this field. In this context, Pickering emulsions, stabilized by a wide range of colloidal particles, offer a promising platform for easy and efficient compartmentalization of molecules within a colloid particle-constituted membrane (colloidosomes).24,25 Owing to their chemical versatility, surface tunability, and microporous structures,26–28 these amphiphilic colloids have been employed to design hierarchical scaffolds with biomimetic functions (encapsulation, catalysis, growth, and chemical communication).29 This attribute is crucial in the domains of biomedicine and catalysis, especially biphasic catalysis involving bioactive encapsulants. However, the stability of colloidosomes stabilized via non-covalent interactions is not sufficient to maintain their framework upon complete phase transfer.24 Hence, both the development of new nanoparticles and the design of compartments with responsive and tunable membrane permeability, capable of encapsulating active biomacromolecules, are highly desired.
The development of microgel-derived colloidosomes offers a promising solution for biomimetic scaffolds. Featuring a loosely crosslinked polymeric network, microgels are soft, hydrated, and deformable structures with tunable porosity and tailored functionality, and are known to create a favorable reaction medium for the biomolecules.30 Microgels have been extensively used as excellent enzyme carriers, offering high enzyme stability and loading efficiency.30 While rigid colloids and surfactants require external energy to overcome their energy barrier for spontaneous adsorption at liquid–liquid interfaces, microgels, positioned between these two material types, display inherent and specific interfacial properties,31 leading to their spontaneous and inhomogeneous deformation at the oil–water interface.32,33 Additionally, these microgel-laden microdroplets exhibit greater stability against coalescence than conventional surfactant-stabilized emulsions and thus can be leveraged as robust compartments for housing bioactives.34 Different physicochemical properties of microgels have been investigated to understand the adsorption behavior of microgels at liquid interfaces.34–38 Interestingly, exploiting these microgel-stabilized emulsion droplets as templates, microgelsomes can be constructed with a selectively permeable and stimuli-responsive membrane and thus exhibit a vesicular architecture.39–43 These characteristic features allow for the encapsulation and programmed transport of specific molecules, offering a rational design of biomimetic protocells.
Expanding the niche of microgelsomes as biomimetic microreactors, herein, we developed a ‘cell-like’ system that could exhibit both aqueous and non-aqueous cores. In contrast to existing approaches, which generally stabilize only one type of emulsion,42–45 our system is unique in its ability to stabilize both O/W and W/O emulsion droplets, offering a versatile platform for encapsulating a broad spectrum of active biomolecules (hydrophilic and lipophilic) within a single framework. The system's efficiency was demonstrated through its application in two distinct reactions catalysed by lipophilic vitamin E and hydrophilic enzymes: S-transaminase (STH) and laccase compartmentalized within the oil-filled and water-filled microgelsomes, respectively. STH is known for its stereoselective conversion of prochiral ketones to chiral amines, important precursors in agrochemical and pharmaceutical synthesis. However, the broader application of STH in organic syntheses, while also being promising, has recently gained momentum due to earlier challenges such as product inhibition and the limited substrate scope.46–49 Selective alcohol amination to chiral amines has been largely exploited via well-designed enzymatic cascades, majorly by coupling an alcohol dehydrogenase (ADH) with STH or amine dehydrogenases (AmDHs).50,51 However, the high specificity of ADHs necessitates the use of both stereoselective forms to catalyze the conversion of racemic alcohols to ketones.52
Combining STH with laccase provides an alternative cascade, catalyzing the non-selective oxidation of alcohols to ketones, followed by their stereoselective amination by STH.46,48,52 However, a primary challenge in effectively operating this cascade is the alignment of their optimal conditions for pH, co-solvents, and the use of isopropyl amines.53,54 Furthermore, their individual catalytic rates and robustness must be balanced, and any potential inhibitory effects between them must be eliminated. Although effective, the previously mentioned strategies employed STH and laccase in a one-pot, two-step cascade where the reaction pH was adjusted over time to activate each enzyme sequentially.55–57
2. Results and discussion
2.1. Synthesis and characterization of the Ser-Ac monomer and the PNSER microgel
To stabilize both O/W and W/O emulsions, anionic core–shell PNSER microgels were prepared via free-radical precipitation copolymerization of NIPAM and N,N′-methylene bisacrylamide (BIS) generating PNIPAM particles which were subsequently used as core particles for further polymerization with O-acryloyl L-serine (Ser-Ac) (Fig. 1). To design microgels for the preparation of responsive, self-assembled compartments, it is important to incorporate easily reactive, charged functional groups to ensure facile interlinking of microgels at the interface that allows formation of stable compartments upon complete phase transfer to the aqueous medium. Polymerization with O-acryloyl modified amino-acids generates free amine and carboxylic groups on the polymer skeleton that render surface charge to the microgels and allow interlinking of microgels at the liquid interfaces post-microgel assembly. O-acryloylation of L-serine was achieved by chelating amine and carboxylic acid moieties via a 5-membered ring copper complex, followed by a reaction with acryloyl chloride. The copper complexation was removed by washing with 8-hydroxyquinoline, producing an ampholytic monomer, Ser-Ac (Fig. S1, ESI†). The chemical structure of the monomer was confirmed using 1H NMR and FTIR spectroscopy (Fig. S2, ESI†).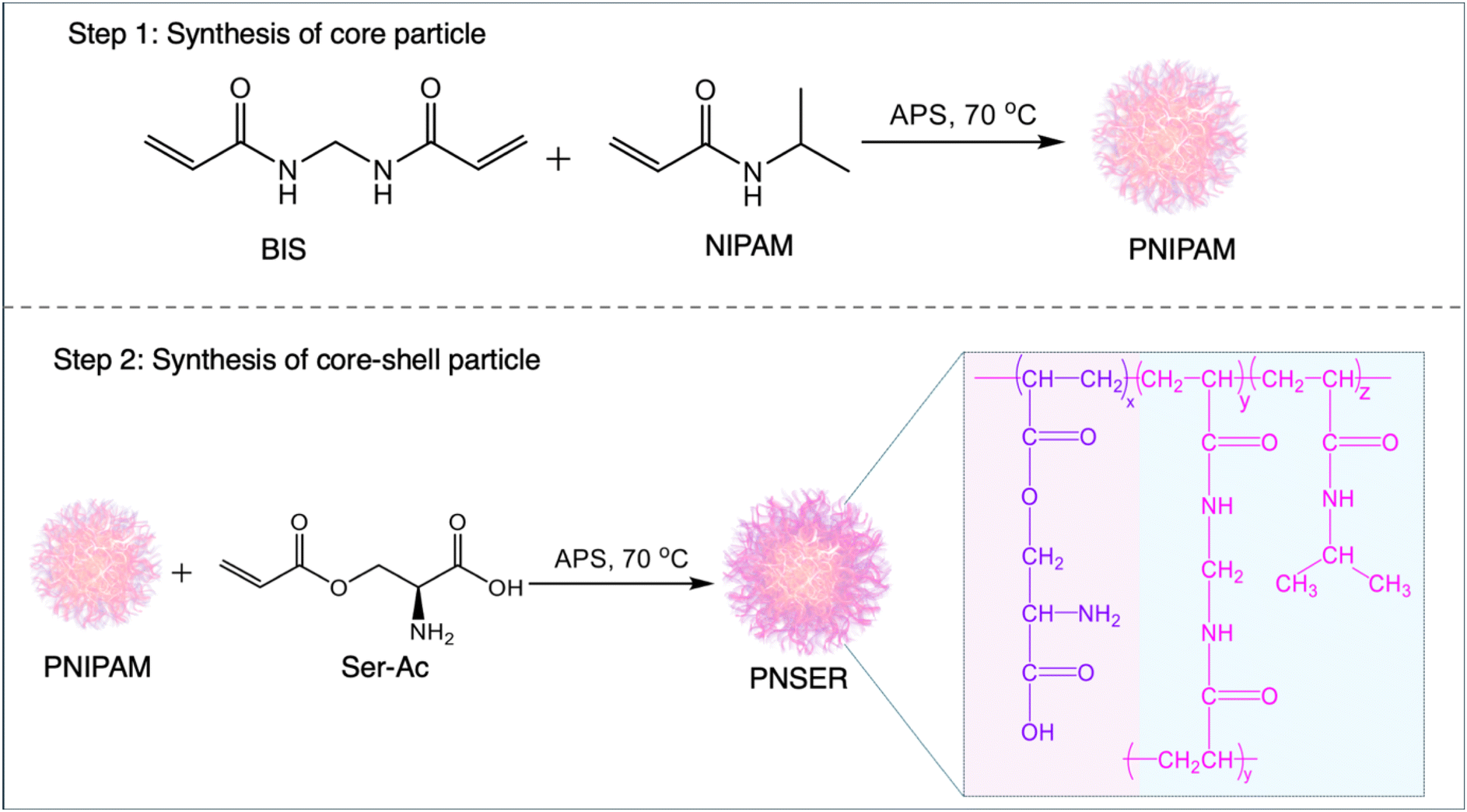 | ||
| Fig. 1 Stepwise representation of the synthesis of the PNSER microgel. Step 1 shows the chemical route to the synthesis of PNIPAM nanoparticles which serve as seed particles for Step 2: polymerization of Ser-Ac on PNIPAM core particles generating core–shell PNSER microgels. | ||
The PNIPAM core particles were functionalized with the Ser-Ac monomer via surfactant-assisted free-radical polymerization to form a stable core–shell microgel structure. The core–shell morphology was designed to provide a high surface density of amine groups on the microgel particles, facilitating their interlinking at liquid–liquid interfaces and promoting the formation of stable microgelsomes. PNIPAM stabilizes the particle structure and imparts temperature-dependent size tunability.58 Its thermoresponsive behavior arises from its chemical structure, which features both hydrophilic amide groups and hydrophobic isopropyl moieties.59 Because of this, PNIPAM shows reversible coil-to-globule transition as the temperature is varied above or below the lower critical solution temperature (LCST) (32 °C). The first step in the synthesis of the PNIPAM core involved polymerization of NIPAM with BIS in the presence of SDS, initiated by APS. In the second step, the preformed core served as a seed for shell formation by incorporating the amphiphilic comonomer Ser-Ac, resulting in amine-decorated core–shell microgels. During core synthesis, a homogeneous nucleation mechanism, coupled with the anionic surface charge density from SDS and sulfate ions from APS, led to the formation of well-defined particles. In the second step of shell synthesis, the presence of APS and Ser-Ac together facilitated the chain transfer mechanism, playing a crucial role in modulating the final size, surface charge, and morphology of the microgel particles.60 While BIS, a bifunctional crosslinker, results in the formation of a dense 3D crosslinked core, the polymerization of Ser-Ac in the second step results in the formation of a negatively charged, core–shell microgel. To obtain stable PNSER microgels, the composition of core particles was optimized by varying the crosslinker (BIS) amount (0.93 mol%, 1.34 mol%, 1.87 mol%) and they were then used as seed particles for the polymerization of Ser-Ac (Table S1, ESI†).
The core–shell morphologies of microgel particles, namely, PNSER_10, PNSER_15, and PNSER_20, were characterized by TEM. As shown in Fig. S5(A–C), ESI,† PNSER_10 with a low crosslinker amount (0.93 mol%) shows less contrast between the core and shell, in comparison with PNSER_15 and PNSER_20 microgel particles. The TEM image of PNSER_20 (Fig. S5(C), ESI†) shows a prominent dense core and a less dense shell around it. Furthermore, the spherical morphology of PNSER microgels in the dried state was confirmed using FE-SEM (Fig. S5(D–F), ESI†) and AFM images (Fig. S5(G–I), ESI†). As revealed by the FE-SEM images, the morphology of PNSER_10 and PNSER_15 particles is not delineated, while PNSER_20 shows distinct core–shell particles with uniform size distribution with an average size of 169.2 ± 0.02 nm (Fig. S5(D–F), ESI†). This can be attributed to the variable amount of crosslinkers used in the synthesis of microgels that reinforce the microgel framework.
Furthermore, the successful polymerization of Ser-Ac on PNIPAM particles was confirmed by FTIR spectroscopy, as depicted in Fig. 2A. In comparison with PNIPAM, the FTIR spectra of PNSER exhibited a broad prominent peak at 3400 cm−1 corresponding to the N–H stretching of the primary amine group and O–H stretching of the carboxylic group present in serine. In addition, the FTIR spectra displayed characteristic peaks at 1645 cm−1 and 1541 cm−1 for amide I and amide II moieties present in the NIPAM backbone. The swellability of PNSER microgel particles with response to temperature was analyzed using dynamic light scattering (DLS). As shown in Fig. 2B, PNSER microgels exhibited a gradual decrease in size with an increase in temperature. Hydrodynamic sizes for PNSER_20, PNSER_15, and PNSER_10 decrease from 530.7 ± 94.1, 851.3 ± 88.8, and 1393.1 ± 96 nm at 24 °C to 208.9 ± 0.1, 213 ± 10.9, and 207.9 ± 6.7 nm at 50 °C, respectively, showing an LCST around 30 °C. Above the LCST, the PNSER microgel starts to collapse, showing an average size of 209.9 ± 2.7 nm in the collapsed state, at 50 °C. The decrease in the LCST can be attributed to the successful polymerization of the hydrophilic monomer Ser-Ac on the PNIPAM core particles.59,61 Additionally, the aqueous dispersion of microgels was thermocycled between temperatures 24 and 50 °C to study the structural stability of microgels in response to temperature. The microgels exhibited temperature-dependent size reversibility, as observed from temperature-recycling data shown in Fig. 2C, indicating the robust microgel structure that could retain the morphology after repeated swelling and collapse with temperature. The microgels show marked variation in the sizes in the dried and hydrated states, as evident from the microscopic and DLS studies, signifying high water absorption capacity and swelling behavior of microgels, a peculiar characteristic of the PNIPAM-derived microgels.
 | ||
| Fig. 2 Characterization of PNSER microgel particles synthesized using different crosslinker (BIS) amounts: 0.93 mol% (PNSER_10), 1.34 mol% (PNSER_15), and 1.87 mol%, (PNSER_20). (A) FTIR spectra of PNIPAM and different PNSER microgels and (B) hydrodynamic size variation in PNSER microgels in response to temperature. (C) Temperature-induced size reversibility of the PNSER_20 microgel dispersed in water, (D) surface charge on microgels dispersed in aqueous solutions with varying pHs (pH 3–12), and (E) interfacial tension exhibited by the aqueous dispersion of PNSER microgels in 2-ethyl 1-hexanol (oil phase). The error bars are calculated from the data of three repeat experiments. | ||
The surface charge on PNSER microgels was studied using zeta potential in the pH range 3–11, and the corresponding data are displayed in Fig. 2D. The microgels were found to be anionic, and the zeta potential was found to be in the range of +0.5 to −2.5 mV between pH 3 and 11 for all the three different microgels. A more negative surface charge was observed on the microgels with increasing pH. This can be attributed to the deprotonation of the carboxylic groups at higher pH values. The isoelectric point for microgels was observed at pH 5.5, close to that of serine,62 thus confirming the successful incorporation of the comonomer, Ser-Ac, in the microgel. The serine-functionalized microgels endowed the microgels with good swellability in both oil (2-ethyl hexanol) and water, resulting in uniform microgel dispersions in both phases. This can be attributed to the surface charge on the microgel created by the amine and carboxylic groups in the serine shell, which enhances the interfacial properties of the PNSER microgels. More importantly, when compared to PNIPAM microgels, PNSER exhibited higher interfacial tension, enabling stabilization of both the O/W and W/O Pickering emulsions. As observed from the interfacial tension (γ) studies shown in Fig. 2E, microgels synthesized using 0.93 mol%, 1.34 mol%, and 1.87 mol% crosslinkers displayed γ values at 4.4, 4.9, and 5.2 mN m−1, respectively. The γ value is observed to increase with the crosslinker amount, showing the effect of core–shell morphology of microgels on their adsorption on liquid interfaces.
2.2. Preparation of microgelsomes using Pickering emulsions
Following the investigation of the physicochemical properties, these serine-functionalized microgels were used to prepare micron-sized vesicular structures called microgelsomes via their template-directed assembly onto emulsion droplets. Unlike conventional Pickering systems that require distinct hydrophilic or hydrophobic particles to stabilize O/W or W/O emulsions, respectively, PNSER microgels can stabilize both emulsion types owing to their intrinsic amphiphilic architecture. Previous studies have achieved such duality through selective surface modification to impart hydrophilic or hydrophobic functionalities;63–65 in contrast, our system inherently enables tunable lumen composition and the co-encapsulation of both hydrophilic and hydrophobic cargoes within a single microgel building block.Fig. 3(A and B) schematically elucidates the assembly of PNSER microgels on W/O and O/W emulsion droplets to form water- and oil-filled microgelsomes, respectively. To prepare microgelsomes with an aqueous interior, an aqueous dispersion of microgels was emulsified with 2-ethyl hexanol at a jw value of 0.3, resulting in the formation of monodisperse, spherical microcapsules with an aqueous interior, dispersed in the oil phase. To enhance the structural integrity of the microgelsomes upon transfer to a complete aqueous phase, the system was subjected to post-assembly treatment with FeCl3, facilitating interfacial crosslinking via the coordination of Fe3+ ions with primary amine groups on the adjacent microgel particles. This metal–ligand complexation effectively reinforces the microgel shell by promoting interparticle bridging at the oil–water interface (Fig. 3A). The interlinking of microgels was performed at pH 5, the pI for PNSER microgels, resulting in the formation of interlinked stable structures.
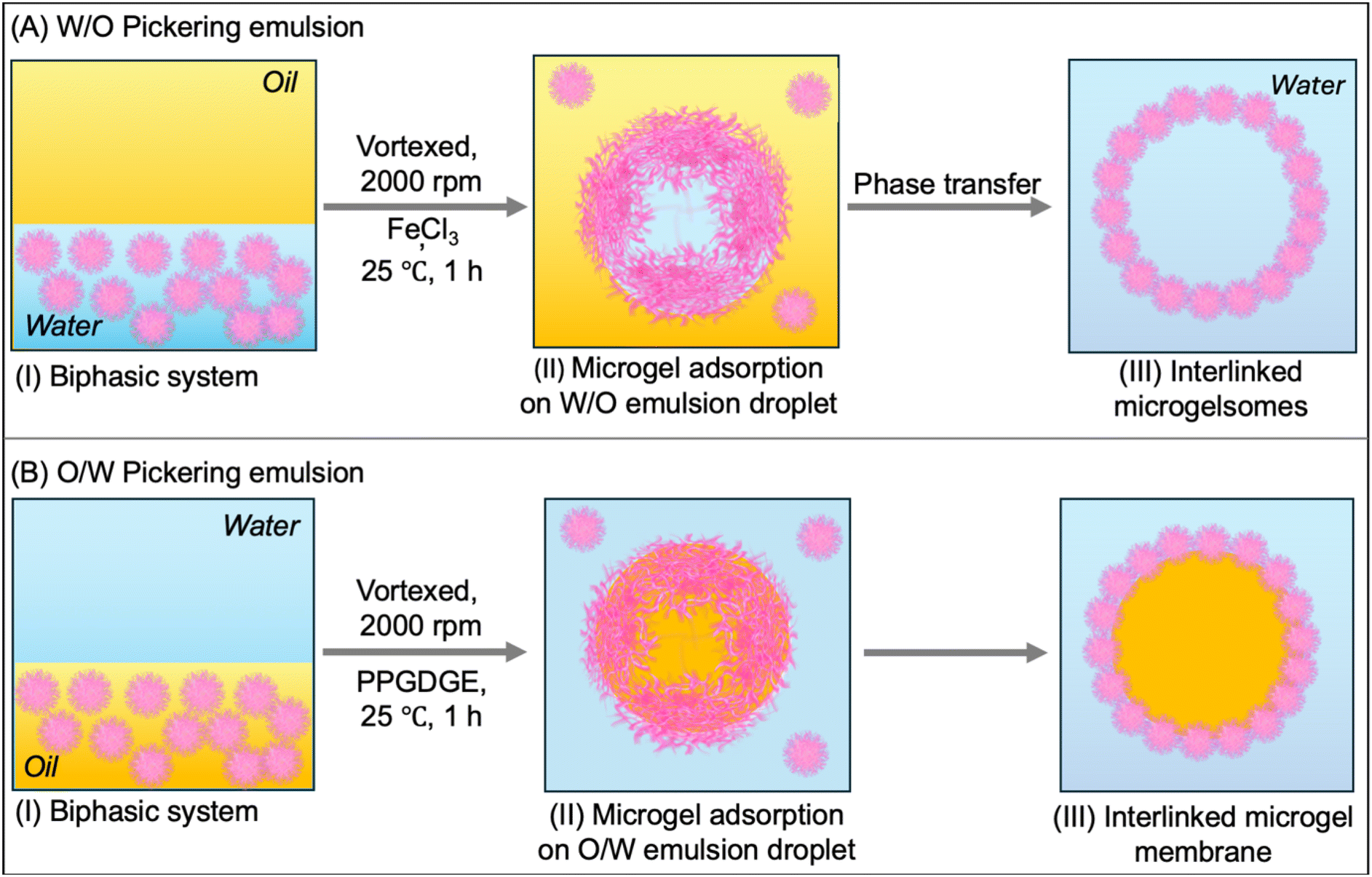 | ||
| Fig. 3 Construction of microgelsomes by assembly of PNSER microgels (A) on W/O and (B) O/W emulsion droplets. Stepwise representation of microgelsome assembly. (I) Biphasic system containing a dispersion of the microgel in the lower phase. (II) The biphasic system was vortexed resulting in the formation of microgel-stabilized emulsion droplets homogeneously dispersed in a continuous phase. (III) The microgel particles were interlinked at the droplet interface by the addition of FeCl3 and PPGDGE as crosslinkers for W/O emulsion and O/W emulsion droplets. The W/O microgelsomes were transferred to a complete aqueous phase in the final step. | ||
The interlinked microgelsomes were separated from the unbound microgel particles by water washing using a separatory funnel. Upon water addition, the excess microgels preferentially partitioned into the aqueous phase, which was carefully decanted. Subsequently, the excess oil phase was injected out, followed by dialysis against an ethanol–water mixture (50%) and then pure water, enabling complete removal of residual oil and successful transfer of the microgelsomes into an all-aqueous environment. The purified microgelsomes were stored at room temperature for further characterization and application.
For the preparation of microgelsomes with an oil-filled lumen, microgel dispersion in 2-ethyl hexanol along with the encapsulants was mixed with water at a jo value of 0.3, resulting in the formation of spherical, oil-filled microgelsomes dispersed in the aqueous phase. Microgels adsorbed at the oil–water interface were covalently crosslinked with neighboring particles using PPGDGE, which undergoes an amine–epoxy reaction with the primary amine groups of PNSER microgels. This crosslinking occurs via a nucleophilic ring-opening of the epoxide groups by amines, as illustrated in Fig. 3B. The morphology of the fabricated microgelsomes was characterized using FE-SEM and optical microscopy. Air-dried samples of the aqueous microgelsome dispersion were imaged using FE-SEM (Fig. 4A), which revealed collapsed vesicular structures with rough surfaces composed of densely packed microgels forming the vesicle membrane. The optical image of the aqueous microgelsome dispersion (Fig. 4B) displays their hydrated, spherical morphology. The adsorption of microgels at the W/O interface was further confirmed using CLSM imaging of FITC-labeled microgel-derived microgelsomes (Section S2.9., ESI†), which revealed a well-defined fluorescent green shell encapsulating a spherical core (Fig. 4C). Additionally, the formation of O/W microgelsomes was validated using Nile red-stained oil, resulting in a red fluorescent core indicative of successful oil encapsulation (Fig. 4D).
 | ||
| Fig. 4 Characterization of microgelsomes prepared using PNSER microgels. (A) FE-SEM image of dried microgelsomes, with the inset image showing their collapsed morphology. (B) Optical image of microgelsomes prepared using a W/O emulsion droplet. CLSM images of interlinked microgelsomes prepared using (C) FITC-labelled PNSER microgels templated on the W/O emulsion droplet, and (D) PNSER microgels templated on the O/W emulsion droplet stained with hydrophobic Nile red (red fluorescent dye). | ||
The microscopic images further confirmed the narrow size distribution of the microcapsules with a mean diameter determined from the statistical analysis of 50 particles at 1.5 ± 1.18 μm (Fig. 4B). Strikingly, the size of the microgelsomes formed using PNSER microgels was observed to depend on the amount of crosslinker used for the microgel synthesis66 (Fig. S7(A–C), ESI).† The average size of the microgelsomes was found to increase with the crosslinker (BIS) amount. The synthesized microgels PNSER_10, PNSER_15, and PNSER_20 formed microgelsomes with sizes 1.05 ± 0.29 μm, 2.07 ± 0.63 μm, and 3.19 ± 0.51μm, respectively. We further investigated the storage stability of the microgelsomes dispersed in the aqueous phase. The photographic and the FE-SEM images demonstrate the structural instability of the microgelsomes below 4 °C and high mechanical compaction, resulting in the disassembly of the microgelsome structure (Fig. S8(A–D), ESI†), whereas, when stored at room temperature, the microgelsomes exhibited explicit stability for over a month, with an intact spherical morphology as depicted in the FE-SEM image (Fig. S8(E), ESI†).
These microscopic observations confirm the stable interlinking of microgels at the droplet interface, resulting in the formation of micrometer-sized, membrane-bound vesicles that maintain structural integrity over extended periods under ambient storage conditions, highlighting the formation of robust microgelsomes with a tunable lumen composition.
2.3. Biomimetic characteristics of microgelsomes
The microgelsomes were engineered to replicate the vesicular architecture, featuring an internal compartment enclosed by a membrane that acts as a selective barrier to the external environment, mimicking the semi-permeable nature of biological cells. This design enables the compartmentalization of functional molecules, while the responsive and tailorable membrane permits controlled diffusion of substrates and products, thus facilitating cell-like in situ reactions within a confined environment. | ||
| Fig. 5 Encapsulation of molecules. Confocal images depicting the compartmentalization of fluorescently labelled encapsulants: (A) FITC-dextran, (B) vitamin E, (C) FITC-STH, (D) RITC-lac, and (E) FITC-STH_RITC-lac in microgelsomes. | ||
 | ||
| Fig. 6 Permeability studies of encapsulants. Release profiles of encapsulated Rh6G (A) at different temperatures (20 °C–55 °C) and (B) at room temperature for 100 min, recorded at 530 nm spectrophotometrically. (C) Release of encapsulated FITC-dextran at temperatures 25 °C–65 °C. Error bars show data for three repeat experiments. | ||
These findings demonstrate that the microgelsome architecture supports efficient encapsulation and retention of high molecular weight molecules, while the semi-permeable membrane facilitates controlled diffusion of low molecular weight species. This combination of molecular confinement and selective transport emphasizes their potential for use as biomimetic reactor systems.
2.3.3.1. Oil-in-water (O/W) emulsion: encapsulation and antioxidant activity of vitamin E
Vitamin E is a lipophilic vitamin known for its antioxidant, neuroprotection, and anti-inflammatory properties. However, its applications are often restricted due to its sensitivity to oxygen, high temperatures, alkaline conditions, and light. In this regard, encapsulation has proved to be an effective route to protect vitamin E for its application in the pharmaceutical and food industries.71 The encapsulation of vitamin E was chosen to demonstrate the compatibility and stability of lipophilic molecules within the oil-filled microgelsomes, showcasing the platform's ability to host hydrophobic species with potential applications in biocatalysis, drug delivery, and active compound stabilization. In this study, vitamin E was encapsulated in the microgelsomes using the O/W Pickering emulsion method using a long-chain triacylglycerol as a carrier oil. For the encapsulation, vitamin E was first homogenized in oil containing uniformly dispersed PNSER microgels, followed by emulsification with water. For the comparison of antioxidant activity, vitamin E dissolved in the oil phase was used as a control. The antioxidant activity of encapsulated vitamin E (En_Vit E) was evaluated and compared with that of its free form (F_Vit E) using the antioxidation of ABTS as a model reaction, shown in Fig. 7A. The reaction involves the vitamin E-catalyzed reduction of blue-colored ABTS˙+ solution to colorless ABTS.72 The reduction of the ABTS˙+ solution to a colorless ABTS solution by En_Vit E is presented in Fig. 7B. Both En_Vit E and F_Vit E showed comparable antioxidant activities, as evident from Fig. 7C, indicating that the stability of vitamin E was preserved after encapsulation, thereby providing an effective compartmentalization method. The slight deviation in the activity of the encapsulated form can be attributed to the slow rate of the diffusion-mediated transfer of the substrate ABTS˙+ across the microgelsome membrane.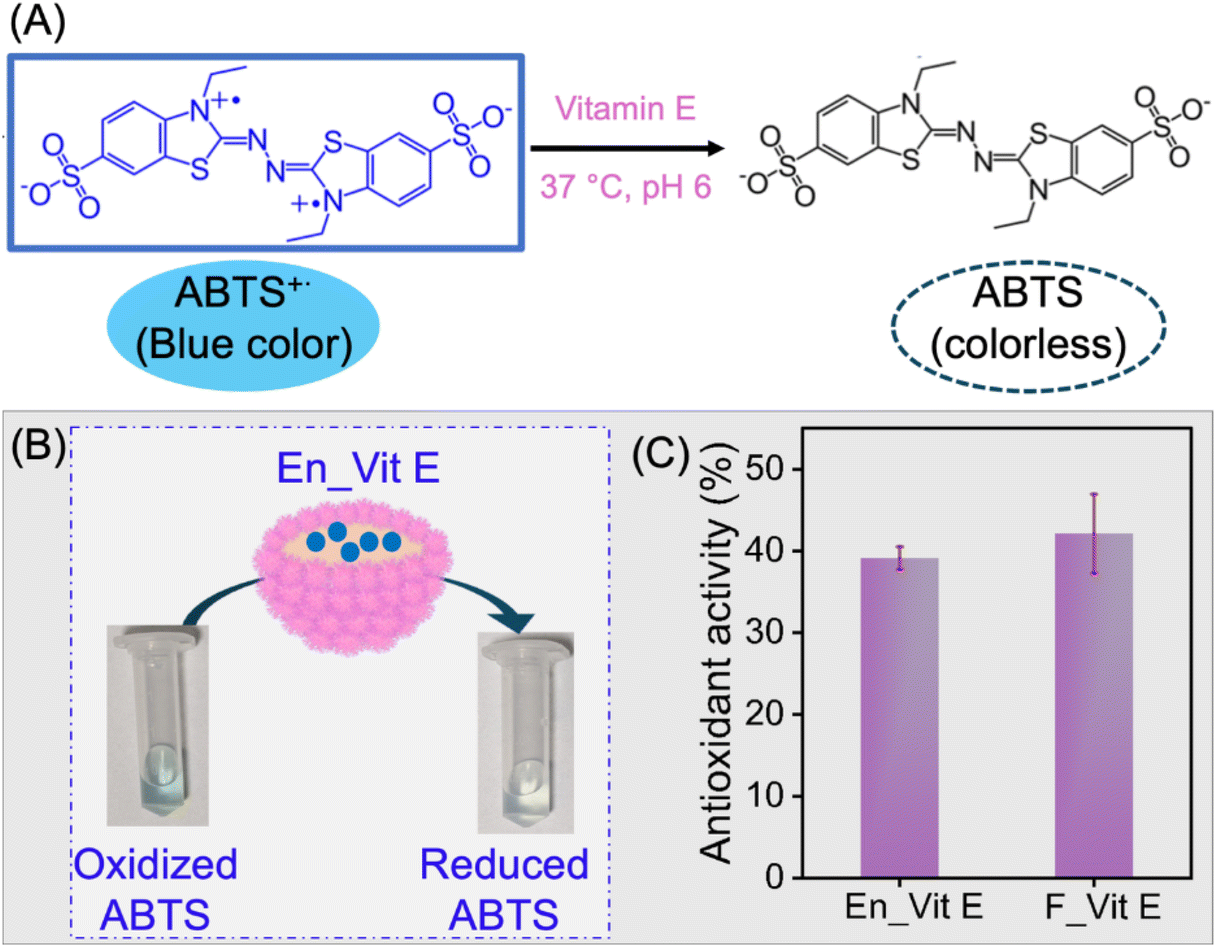 | ||
| Fig. 7 Antioxidation activity of vitamin E. (A) Schematic reaction for the vitamin E mediated reduction of blue-colored ABTS˙+ to colorless ABTS. (B) Experimental image showing the antioxidant activity of encapsulated vitamin E. (C) Antioxidant activity of free (F_Vit E) and encapsulated vitamin E (En_Vit E). Error bars show data for three repeat experiments. | ||
2.3.3.2. Water-in-oil (W/O) emulsion: biosynthesis of chiral amino molecules using the bienzymatic reaction of laccase and S-transaminase
By leveraging the capability of the microgelsomes to encapsulate high molecular weight biomolecules within the selectively permeable, temperature-responsive microgel membrane, these scaffolds were developed into biocatalytic microreactors. These microgel-stabilized W/O droplets have been shown to be more stable against coalescence in comparison with the commonly used surfactant-stabilized emulsions73 and can accordingly be harnessed as robust compartments to host catalytically active microgelsomes. Enzymes laccase and STH were encapsulated within microgelsomes and the catalytic performance of our biomimetic microreactors in the synthesis of chiral amines was examined. The two enzymes operate optimally at different pH levels, making their coupled reaction inherently challenging. A key strategy to overcome this incompatibility has been the use of a one-pot, two-step bienzymatic reaction, in which the components required for the second step are introduced after completion of the first along with a controlled adjustment of the reaction pH to suit the second enzyme's optimal conditions.In an effort to improve the existing strategy, the developed microgelsomes were evaluated for their performance in a one-pot bienzymatic reaction. Their catalytic efficiency was compared with that of the corresponding two-step sequential reaction and the free enzyme counterparts. Accordingly, the two chemical pathways employed are as follows (Fig. 8). The first route involved a two-step sequential reaction (Free lac_S1, Free STH_S2, En lac_S1, and En STH_S2) performed at the optimum pH of enzymes. This chemical route involved the conversion of 1-phenyl ethanol to acetophenone by laccase/TEMPO at pH 5, which was then transferred to reaction media (pH 8) containing alanine and PLP, for second catalysis to S-MBA by STH (Fig. 8(A)). The second route was devised as a greener alternative to this cascade reaction via the co-encapsulation of STH and laccase within the microgelsomes to perform a one-pot bienzymatic reaction (En_LaSh) at a single pH in aqueous media (Fig. 8(B)).
 | ||
| Fig. 8 Bienzymatic reaction route for (A) the two-step sequential reaction and (B) one-pot synthesis of chiral amine; S-MBA from racemic 1-phenyl ethanol. | ||
Remarkably, the activity of the biocatalytic microgelsomes (En lac_S1, En STH_S2, and En LaSh) was observed to be significantly higher, leading to an increased conversion of 95–98%, which is 2 times higher than that of their free forms (Free lac_S1, Free STH_S2, and Free_LaSh), as shown in Fig. 9(A) and Table 1. Importantly, the activity of the encapsulated one-pot catalysis, En LaSh performed at pH 5 was found to be comparable to that of the two-step sequential reaction and 2-fold higher than that of the one-pot cascade using their free forms. The results suggest the favorable microenvironment offered by the microgelsomes in operating the two enzymes at a single pH. The latter strategy of enzyme co-encapsulation within the microgelsome framework featured high reaction efficiency, reducing operational steps and thus improving the process ‘pot-economy’.
 | ||
| Fig. 9 Activity of biocatalytic microgelsomes. (A) Schematics of the synthesis of a chiral amine via a compartmentalized bienzymatic cascade reaction showing the two routes adopted for experimentation: two-step sequential and one-pot reaction. Laccase-mediated conversion of 1-phenyl ethanol to acetophenone, in the presence of the mediator TEMPO, and subsequent catalysis of acetophenone to S-MBA by STH in the presence of the substrate alanine and the cofactor PLP. Relative activity of free and encapsulated enzymes at (B) 30 °C (below the LCST) and (C) 40 °C (above the LCST) illustrating the effect of temperature-mediated membrane-gated enzyme catalysis for a programmable reaction. Error bars show data for three repeat experiments. | ||
| Enzyme | Conversion (%) |
|---|---|
| Free lac_S1 | 54.80 |
| En_lac_S1 | 94.86 |
| Free STH_S2 | 90.68 |
| En_STH_S2 | 97.98 |
| Free_LaSh | 43.34 |
| En_LaSh | 95.53 |
Additionally, the selective permeability of the microgelsomes in response to temperature was exploited to develop a thermally gated microgel membrane, to regulate the accessibility of the substrate molecules from the outer continuous phase to the confined enzymes within the microcompartments, in response to temperature. The catalytic activity of microgelsomes was investigated at 40 °C (above the LCST of PNSER microgels). The bioactivity of the microgelsomes was investigated by monitoring the conversion of 1-phenyl ethanol to acetophenone and S-MBA using HPLC and further confirmed by GC-MS (Fig. S11–S13, ESI†). The enzymatic activity results obtained at 30 °C differed significantly from those observed at 40 °C. As shown in Fig. 9C, no product formation was observed in the case of biocatalytic microgelsomes; meanwhile, almost 100% relative enzymatic activity was observed for the free enzymes. These results can be attributed to the temperature-responsive behavior of PNSER microgels, which causes the microgel membrane to collapse at temperatures above its LCST, preventing the diffusion of substrates into the vesicle and, thus, switching off the catalytic activity of microgelsomes at temperatures above the LCST.
Furthermore, the kinetic profiles of the encapsulated and free enzymes (Fig. S14, ESI†) showed that the encapsulated enzymes exhibited higher Km values (Table 2), En_lac_S1: 24.96 mM and En STH_S2: 0.23 mM, than their free forms, Free lac_S1: 5.8 mM and Free STH_S2: 0.09 mM. The increased Km could be ascribed to the slow rate of diffusion-mediated molecular transport across the membrane. These results clearly show that the encapsulation of enzymes in microgelsomes leads to cell-like biocatalytic reactors, that is, microscopic reaction compartments with enzymes, capable of functioning upon induction.
| Free lac | En_lac | Free STH | En_STH | |
|---|---|---|---|---|
| V max (mM min−1) | 0.008 | 0.016 | 17.99 | 12.59 |
| K m (mM) | 5.8 | 24.96 | 0.093 | 0.227 |
3. Conclusions
In summary, we present a simple and eco-friendly approach to developing versatile microgelsomes for the compartmentalization of different biomolecules varying from lipophilic to hydrophilic in nature. The supramolecular metal–organic coordination complex of Fe3+ and the amine moiety for interlinking adsorbed microgels at the W/O interface was used to facilitate the stability of microgelsomes in the all-aqueous phase. The microgelsomes exhibited dynamic and selective molecular permeability at liquid–liquid phase boundaries, protecting the encapsulated molecules from the outer environment while maintaining close contact with the outer medium. We developed microbioreactors with a tunable lumen and validated their compartmentalization efficiency by the encapsulation of lipophilic vitamin E and hydrophilic enzymes, which could perform catalysis within the gated polymeric compartments. The co-encapsulation of laccase and STH within microgelsomes addresses two significant drawbacks of this bienzymatic cascade: different operational pH and use of organic solvents for the substrate solubility,74 generating industrially significant chiral amines. Our approach represents a robust and environmentally friendly approach to constructing programmable biocatalytic reactors and compartmentalization of multienzymes under mild reaction conditions. To our knowledge, no similar systems have been demonstrated in the literature. We believe that the system presents a significant advancement in biocatalysis and biomolecule encapsulation and can be employed to design other stimuli-responsive microgels and derived microgelsomes to develop bioreactors, biosensors, and programmable drug delivery vehicles.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
DG thanks the Ministry of Education, Government of India for the Prime Minister Research Fellowship. Funding support (file no.: 5/3/8/69/2020-ITR) from the Indian Council of Medical Research, Government of India, is gratefully acknowledged. NCD acknowledges the financial support from the Department of Science and Technology (DST, Gov. of India) under the DST-INSPIRE Faculty program (INSPIRE code: IFA17-LSPA91). The authors would also like to acknowledge the Central Research facility, IIT Delhi.References
- J. U. Bowie, S. Sherkhanov, T. P. Korman, M. A. Valliere, P. H. Opgenorth and H. Liu, Trends Biotechnol., 2020, 38, 766–778 CrossRef CAS PubMed.
- K. S. Rabe, J. Müller, M. Skoupi and C. M. Niemeyer, Angew. Chem., Int. Ed., 2017, 56, 13574–13589 CrossRef CAS PubMed.
- M. W. Ullah, S. Manan, M. Ul-Islam, W. A. Khattak, K. A. Khan, J. Liu, G. Yang and J. Sun, Green Chem., 2023, 25, 4912–4940 RSC.
- M. Teshima, V. P. Willers and V. Sieber, Curr. Opin. Biotechnol., 2023, 79, 102868 CrossRef CAS PubMed.
- S. P. France, L. J. Hepworth, N. J. Turner and S. L. Flitsch, ACS Catal., 2017, 7, 710–724 CrossRef CAS.
- E. L. Bell, W. Finnigan, S. P. France, A. P. Green, M. A. Hayes, L. J. Hepworth, S. L. Lovelock, H. Niikura, S. Osuna, E. Romero, K. S. Ryan, N. J. Turner and S. L. Flitsch, Nat. Rev. Methods Primers, 2021, 1, 46 CrossRef CAS.
- A. Basso and S. Serban, Mol. Catal., 2019, 479, 110607 CrossRef CAS.
- M. A. Huffman, A. Fryszkowska, O. Alvizo, M. Borra-Garske, K. R. Campos, K. A. Canada, P. N. Devine, D. Duan, J. H. Forstater, S. T. Grosser, H. M. Halsey, G. J. Hughes, J. Jo, L. A. Joyce, J. N. Kolev, J. Liang, K. M. Maloney, B. F. Mann, N. M. Marshall, M. McLaughlin, J. C. Moore, G. S. Murphy, C. C. Nawrat, J. Nazor, S. Novick, N. R. Patel, A. Rodriguez-Granillo, S. A. Robaire, E. C. Sherer, M. D. Truppo, A. M. Whittaker, D. Verma, L. Xiao, Y. Xu and H. Yang, Science, 2019, 366, 1255–1259 CrossRef CAS PubMed.
- S. Kumar, M. Karmacharya and Y.-K. Cho, Small, 2023, 19, 2202962 CrossRef CAS PubMed.
- H. Gröger, F. Gallou and B. H. Lipshutz, Chem. Rev., 2023, 123, 5262–5296 CrossRef PubMed.
- V. E. Deneke and S. Di Talia, J. Cell Biol., 2018, 217, 1193–1204 CrossRef CAS PubMed.
- K. P. Adamala, D. A. Martin-Alarcon, K. R. Guthrie-Honea and E. S. Boyden, Nat. Chem., 2017, 9, 431–439 CrossRef CAS PubMed.
- Y. Zhu, S. Cao, M. Huo, J. C. M. van Hest and H. Che, Chem. Sci., 2023, 14, 7411–7437 RSC.
- A. M. Tayar, E. Karzbrun, V. Noireaux and R. H. Bar-Ziv, Nat. Phys., 2015, 11, 1037–1041 Search PubMed.
- E. Karzbrun, A. M. Tayar, V. Noireaux and R. H. Bar-Ziv, Science, 2014, 345, 829–832 CrossRef CAS PubMed.
- A. S. Zadorin, Y. Rondelez, G. Gines, V. Dilhas, G. Urtel, A. Zambrano, J.-C. Galas and A. Estevez-Torres, Nat. Chem., 2017, 9, 990–996 CrossRef CAS PubMed.
- A. J. Genot, A. Baccouche, R. Sieskind, N. Aubert-Kato, N. Bredeche, J. F. Bartolo, V. Taly, T. Fujii and Y. Rondelez, Nat. Chem., 2016, 8, 760–767 CrossRef CAS PubMed.
- A. N. Zaikin and A. M. Zhabotinsky, Nature, 1970, 225, 535–537 CrossRef CAS PubMed.
- M. Weitz, J. Kim, K. Kapsner, E. Winfree, E. Franco and F. C. Simmel, Nat. Chem., 2014, 6, 295–302 CrossRef CAS PubMed.
- D. Gaur, N. C. Dubey and B. P. Tripathi, Adv. Colloid Interface Sci., 2022, 299, 102566 CrossRef CAS PubMed.
- L. Velasco-Garcia and C. Casadevall, Commun. Chem., 2023, 6, 263 CrossRef CAS PubMed.
- N. Gao, T. Tian, J. Cui, W. Zhang, X. Yin, S. Wang, J. Ji and G. Li, Angew. Chem., Int. Ed., 2017, 56, 3880–3885 CrossRef CAS PubMed.
- E. Amstad, ACS Macro Lett., 2017, 6, 841–847 CrossRef CAS.
- J. Pan, J. Chen, X. Wang, Y. Wang and J.-B. Fan, Interdiscip. Med., 2023, 1, e20230014 CrossRef.
- Z. Gao, X. Cui and J. Cui, Supramol. Mater., 2022, 1, 100015 Search PubMed.
- K. Yang, Y. Liu, Y. Liu, Q. Zhang, C. Kong, C. Yi, Z. Zhou, Z. Wang, G. Zhang, Y. Zhang, N. M. Khashab, X. Chen and Z. Nie, J. Am. Chem. Soc., 2018, 140, 4666–4677 CrossRef CAS PubMed.
- L. Wang, Y. Liu, J. He, M. J. Hourwitz, Y. Yang, J. T. Fourkas, X. Han and Z. Nie, Small, 2015, 11, 3762–3767 CrossRef CAS PubMed.
- X. Yu, K. Yue, I.-F. Hsieh, Y. Li, X.-H. Dong, C. Liu, Y. Xin, H.-F. Wang, A.-C. Shi, G. R. Newkome, R.-M. Ho, E.-Q. Chen, W.-B. Zhang and S. Z. D. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10078–10083 CrossRef CAS PubMed.
- M. Wei, Y. Lin and Y. Qiao, Giant, 2023, 13, 100143 CrossRef CAS.
- N. C. Dubey, D. Gaur and B. P. Tripathi, J. Polym. Sci., 2023, 61, 1730–1748 CrossRef CAS.
- F. A. Plamper and W. Richtering, Acc. Chem. Res., 2017, 50, 131–140 CrossRef CAS PubMed.
- B. P. Binks, Curr. Opin. Colloid Interface Sci., 2002, 7, 21–41 CrossRef CAS.
- M. A. Fernandez-Rodriguez, A. Martín-Molina and J. Maldonado-Valderrama, Adv. Colloid Interface Sci., 2021, 288, 102350 CrossRef CAS PubMed.
- S. Stock and R. von Klitzing, Curr. Opin. Colloid Interface Sci., 2022, 58, 101561 CrossRef CAS.
- M. M. Schmidt, S. Bochenek, A. A. Gavrilov, I. I. Potemkin and W. Richtering, Langmuir, 2020, 36, 11079–11093 CrossRef CAS PubMed.
- S. Schmidt, T. Liu, S. Rütten, K.-H. Phan, M. Möller and W. Richtering, Langmuir, 2011, 27, 9801–9806 CrossRef CAS PubMed.
- M. Destribats, M. Wolfs, F. Pinaud, V. Lapeyre, E. Sellier, V. Schmitt and V. Ravaine, Langmuir, 2013, 29, 12367–12374 CrossRef CAS PubMed.
- W. Wang, A. H. Milani, L. Carney, J. Yan, Z. Cui, S. Thaiboonrod and B. R. Saunders, Chem. Commun., 2015, 51, 3854–3857 RSC.
- C. G. Palivan, L. Heuberger, J. Gaitzsch, B. Voit, D. Appelhans, B. Borges Fernandes, G. Battaglia, J. Du, L. Abdelmohsen, J. C. M. van Hest, J. Hu, S. Liu, Z. Zhong, H. Sun, A. Mutschler and S. Lecommandoux, Biomacromolecules, 2024, 25, 5454–5467 CrossRef CAS PubMed.
- R. Toor, A. N. Copstein, C. Trébuchet, B. Goudeau, P. Garrigue, V. Lapeyre, A. Perro and V. Ravaine, J. Colloid Interface Sci., 2023, 630, 66–75 CrossRef CAS PubMed.
- R. Tamate, T. Ueki and R. Yoshida, Angew. Chem., Int. Ed., 2016, 55, 5179–5183 CrossRef CAS PubMed.
- X. Guan, Y. Liu, Z. Wan, Y.-L. S. Tse and T. Ngai, Chem. Sci., 2022, 13, 6205–6216 RSC.
- D. Gaur, N. C. Dubey and B. P. Tripathi, Biomacromolecules, 2024, 25, 1108–1118 CrossRef CAS PubMed.
- L. Lei, Q. Zhang, S. Shi and S. Zhu, Langmuir, 2017, 33, 6108–6115 CrossRef CAS PubMed.
- X. Guan, Y. Liu, L. Li, M.-H. Kwok, M. Ding, H. Jiang and T. Ngai, Adv. Sci., 2025, 12, 2415642 CrossRef CAS PubMed.
- R. C. Simon, N. Richter, E. Busto and W. Kroutil, ACS Catal., 2014, 4, 129–143 CrossRef CAS.
- W. Khanam and N. C. Dubey, Mater. Today Chem., 2022, 24, 100922 CrossRef CAS.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef CAS PubMed.
- D. Ghislieri and N. J. Turner, Top. Catal., 2014, 57, 284–300 CrossRef CAS.
- F.-F. Chen, Y.-Y. Liu, G.-W. Zheng and J.-H. Xu, ChemCatChem, 2015, 7, 3838–3841 CrossRef CAS.
- F. G. Mutti, T. Knaus, N. S. Scrutton, M. Breuer and N. J. Turner, Science, 2015, 349, 1525–1529 CrossRef CAS PubMed.
- L. Martínez-Montero, V. Gotor, V. Gotor-Fernández and I. Lavandera, Green Chem., 2017, 19, 474–480 RSC.
- M. D. Truppo, J. D. Rozzell, J. C. Moore and N. J. Turner, Org. Biomol. Chem., 2009, 7, 395–398 RSC.
- J. T. C. Brown and R. M. Phelan, Org. Process Res. Dev., 2023, 27, 1293–1299 CrossRef CAS.
- S. Mathew, D. Renn and M. Rueping, ACS Catal., 2023, 13, 5584–5598 CrossRef CAS.
- R. S. Correia Cordeiro, N. Ríos-Lombardía, F. Morís, R. Kourist and J. González-Sabín, ChemCatChem, 2019, 11, 1272–1277 CrossRef CAS.
- J. Albarrán-Velo, I. Lavandera and V. Gotor-Fernández, ChemBioChem, 2020, 21, 200–211 CrossRef PubMed.
- B. Sierra-Martin, J. R. Retama, M. Laurenti, A. F. Barbero and E. L. Cabarcos, Adv. Colloid Interface Sci., 2014, 205, 113–123 CrossRef CAS PubMed.
- Z. M. O. Rzaev, S. Dinçer and E. Pişkin, Prog. Polym. Sci., 2007, 32, 534–595 CrossRef CAS.
- N. C. Dubey, B. P. Tripathi, M. Stamm and L. Ionov, Biomacromolecules, 2014, 15, 2776–2783 CrossRef CAS PubMed.
- R. Singh, S. A. Deshmukh, G. Kamath, S. K. R. S. Sankaranarayanan and G. Balasubramanian, Comput. Mater. Sci., 2017, 126, 191–203 CrossRef CAS.
- K. Kobayashi, in Encyclopedia of Astrobiology, ed. M. Gargaud, R. Amils, J. C. Quintanilla, H. J. Cleaves, W. M. Irvine, D. L. Pinti and M. Viso, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 1503–1503 Search PubMed.
- H. Jiang, X. Hu, W. Jiang, X. Guan, Y. Li and T. Ngai, Langmuir, 2022, 38, 12273–12280 CrossRef CAS PubMed.
- M. Lan, Y. Song, S. Ou, J. Zheng, C. Huang, Y. Wang, H. Zhou, W. Hu and F. Liu, Langmuir, 2020, 36, 14991–14998 CrossRef CAS PubMed.
- B. P. Binks and D. Yin, Soft Matter, 2016, 12, 6858–6867 RSC.
- F. Camerin, M.Á Fernández-Rodríguez, L. Rovigatti, M.-N. Antonopoulou, N. Gnan, A. Ninarello, L. Isa and E. Zaccarelli, ACS Nano, 2019, 13, 4548–4559 CrossRef CAS PubMed.
- M. Forlin, R. Lentini and S. S. Mansy, Curr. Opin. Chem. Biol., 2012, 16, 586–592 CrossRef CAS PubMed.
- P. Stano and P. L. Luisi, Curr. Opin. Biotechnol., 2013, 24, 633–638 CrossRef CAS PubMed.
- N. Ichihashi and T. Yomo, Curr. Opin. Chem. Biol., 2014, 22, 12–17 CrossRef CAS PubMed.
- Y. Sato, K. Komiya, I. Kawamata, S. Murata and S.-i. M. Nomura, Chem. Commun., 2019, 55, 9084–9087 RSC.
- A. M. Ribeiro, B. N. Estevinho and F. Rocha, Food Hydrocolloids, 2021, 121, 106998 CrossRef CAS.
- A. Cano, M. Acosta and M. B. Arnao, Redox Rep., 2000, 5, 365–370 CrossRef CAS PubMed.
- R. Aveyard, B. P. Binks and J. H. Clint, Adv. Colloid Interface Sci., 2003, 100–102, 503–546 CrossRef CAS.
- L. Prout, H. C. Hailes and J. M. Ward, RSC Adv., 2024, 14, 4264–4273 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5gc00775e |
| This journal is © The Royal Society of Chemistry 2025 |