
Catalytic wet air oxidation of aqueous solution of phenol in a fixed bed reactor over Ru catalysts supported on ceria promoted MCM-41
Dilip Kumar Mondala,
Chandona Mondala and
Shyamal Roy*ab
aDepartment of Chemical Engineering, Jadavpur University, Kolkata-700032, West Bengal, India. E-mail: shyamal.roy@rutgers.edu
bChemical and Biochemical Engineering Department, Rutgers, The State University of New Jersey, Piscataway, NJ 08854, USA
First published on 14th November 2016
Abstract
Catalytic wet air oxidation (CWAO) of phenol was carried out in a fixed bed reactor over a noble metal (Ru) supported on silica MCM-41 material. The addition of small amounts of ceria as a promoter to the MCM-41 material enhanced both the activity and selectivity of Ru in this reaction, with significant conversions of about 99% obtained at 413 K and 18 bar oxygen pressure. The oxidation of phenol required moderate temperature and oxygen pressure, and the oxidizing capacity of the catalyst required adjustment. The activation energy of the oxidation reaction was found to be 33 kJ mol−1 at 413 K using the ceria promoted Ru/MCM-41 catalyst. The supports and catalysts were characterized using the Brunauer–Emmett–Teller (BET) surface area, temperature programmed reduction (TPR), CO chemisorption, temperature programmed desorption (TPD), X-ray diffraction (XRD), scanning electron microscopy (SEM), energy dispersive X-ray analysis (EDX), transmission electron microscopy (TEM), and inductively coupled plasma atomic emission spectroscopy (ICP-AES).
1. Introduction
Catalytic wet air oxidation (CWAO) has recently emerged as one of the wastewater treatment technologies and has proven to have promising effects on the oxidation of highly toxic and non-biodegradable industrial effluent.1 The reaction is carried out under oxygen pressure (5–200 bar) at elevated temperatures (125–320 °C) in the presence of a heterogeneous catalyst (oxides, supported noble metals).2 Phenolic pollutants are mainly distributed in the terrestrial and aquatic environments due to the widespread use of pesticides, pharmaceutical products and chemical raw materials, which are synthesized and manufactured in large amounts using phenolic compounds.3 Because of their toxicity, phenolic pollutants can be treated by microorganisms at low concentrations.4 The inability of conventional methods to effectively remove toxic organic pollutants has made it evident that new, compact and more efficient processes are required.5 Several treatment technologies are available that can be used depending on the nature and volume of the effluent. The treatment of wastewater, containing high concentrations of phenolic compounds, mainly using wet air oxidation (WAO),6 chemical oxidation,7 sonolysis technology8 and photocatalysis techniques has been reported.9In recent years, microwave technology has attracted the attention of researchers due to its property of molecular level heating, which leads to homogeneous and quick thermal reactions.10 However, it suffers from some disadvantages, including harsh reaction conditions (high temperature and high pressure), high capital costs, high noise and low quantum efficiency, which need to be solved and improved with their further development.11
Although incineration is the most efficient and useful method, because of the high energetic costs involved and above all, the release of noxious compounds (oxides of sulfur and nitrogen, furan) into the atmosphere, incineration is suitable only as an end-of-pipe treatment or when the chemical oxygen demand (COD) of the effluents is higher than 300 g L−1.12 Biological treatment is a low cost and simple method; however, the toxicity of the effluent makes this treatment ineffective for organic concentrations above 70–200 mg L−1. Potential technologies for COD removal between these two extremes include physicochemical treatment methods, air based oxidation, and chemical oxidation. In terms of energy efficiency, wet air oxidation (WAO) and catalytic wet air oxidation (CWAO) are suitable for the treatment of wastewater with COD loads below or within the given limits.13 Herbicide removal,14 oxidative treatment of pulp and paper mill effluents,15 pretreatment of water from alkaloid plants (typically high strength industrial wastewater with COD around 27 g L−1),16 treatment of printing and dyeing wastewater from the textile industry,17 and treatment of wastewater from H-acid manufacturing process18 are recent examples of the applications of this technology.
In wet air or thermal liquid phase oxidation (WAO) processes, the generation of active oxygen species, such as hydroxyl radicals, takes place at high temperature and pressure and is known to have a great potential for treating effluents containing high organic matter content (chemical oxygen demand (COD) 10–100 g L−1) or toxic contaminants, for which direct biological purification is unfeasible.19
The WAO process has well known capacities for breaking down biologically refractory compounds to simpler and easily treated materials before their release into the environment. This aqueous phase oxidation process takes place under high reaction temperatures (473–593 K) and pressures (20–200 bar) by means of hydroxyl radicals.20 In the WAO processes, the organic contaminants dissolved in water are either partially degraded to biodegradable intermediates by an oxidizing agent or mineralized to non-toxic inorganic compounds, such as CO2, H2O, and inorganic salts, which remain in the aqueous phase. Sulphur is converted to sulphate, halogens to halides, and phosphorus to phosphates. Organic nitrogen may produce ammonia, nitrate, and nitrogen. As compared to the other thermal processes, WAO does not produce NOx, SOx, HCl, dioxins, furans, fly ash etc.21
One of the main drawbacks of the WAO process is its inability to achieve the complete mineralization of organics since some low molecular weight oxygenated compounds (especially acetic and propionic acids, as well as methanol, ethanol, and acetaldehyde), originally present in the wastewater or accumulated in the liquid phase during the oxidation process, are resistant to their further transformation into carbon dioxide.22 Comparing with conventional wet air oxidation, catalytic wet air oxidation (CWAO) has lower energy requirements. Due to the presence of a catalyst, much higher oxidation rates are achieved, and consequently, one can use less severe reaction conditions to reduce the chemical oxygen demand to the same degree as in the case of the non-catalytic process. In the CWAO process, organics are oxidized to non-toxic inorganic compounds, such as CO2, H2O, and heteroatom dissolved ions, at much lower temperatures and pressures than in the non-catalyzed thermal processes.23
By using a suitable catalyst, the operating conditions needed for the WAO process can be lowered to more amenable values (363–493 K, 1–50 bar) with consequent economic advantages and without any loss in the degradation efficiency.24 Although, homogeneous catalysts are very efficient for this process,25 their use require an additional separation step for the removal of toxic ions from the solution. Since heterogeneous catalysts can be easily removed, their development and optimization has been the subject of several studies in recent decades.26 Efficient approaches towards improving the performance of the catalysts used in such processes include changing the active phase (carbide, nitride, phosphide, etc.), using a different catalyst preparation method (co-precipitation, chemical vapor deposition, etc.) or applying new types of catalyst supports (CNTs and mesoporous silicas such as MCM-41).27 These ordered mesoporous materials are considered ideal hosts for nanoparticles due to their large surface area and well defined pore structure.28 Most importantly, their pore sizes can be tuned within the nanometer range, and the pore surface can be functionalized with various metal oxides and organic groups. Among these materials, silica MCM-41 has been extensively studied due to its 2D structure with interconnected pores.29 MCM-41 is a mesoporous silica molecular sieve with high surface area, tunable uniform hexagonal channels ranging from 5 to 30 nm, and thick framework walls (3–6 nm), which attribute for it to be thermally and hydrothermally robust.30 These attractive textural properties of silica MCM-41 makes it a potential alternative to the commonly used γ-Al2O3 support due to the fact that its pore size can be manipulated via controlling the synthesis parameters, while maintaining a relatively high surface area.31
This study was mainly aimed at determining the initial activities and kinetic parameters considering the total oxidation of the aqueous solutions of phenol on the silica MCM-41 supported ruthenium catalysts into carbon dioxide under moderate temperature and pressure conditions using oxygen as an oxidizing agent. Phenol is a low molecular weight organic compound that is resistant to oxidation. It is a good model system because it occurs in most of the degradation pathways for more complex organic compounds. Several experimental techniques, such as BET surface area, temperature-programmed reduction (TPR), CO chemisorption, temperature-programmed desorption (TPD), X-ray diffraction (XRD), scanning electron microscopy (SEM), energy dispersive X-ray analysis (EDX), inductively coupled plasma-atomic emission spectroscopy (ICP-AES), and transmission electron microscopy (TEM), were used to determine the suitability of the ceria promoted silica MCM-41 solid supported metal catalysts for the catalytic wet air oxidation of phenol.
A kinetic analysis describing the observed activation phenomena in the ceria promoted Ru/MCM-41 and Ru/MCM-41 catalysts was investigated using the experimental data obtained in the CWAO of the studied systems. To our knowledge, there are no previous reports on the treatments using the CWAO of phenol under similar conditions. Although silica MCM-41 supported ruthenium group metals are much more resistant to leaching in acidic solutions than the catalysts based on carbon supports, they have been rarely employed in CWAO.
2. Experimental
2.1. Catalytic performance studies
The reaction was carried out in a continuous upflow catalytic fixed bed reactor having an ability to operate under up to 50 bar pressure and at 773 K temperature and equipped with a high pressure piston pump (Fig. 1). The catalyst extrudates were placed at the centre of the reactor throughout the kinetic study. When the amount of the catalyst was 6 g, the exact length of the catalytic bed was 10 cm. The remaining space of the catalyst bed on the top and the bottom of the reactor was filled with glass beads to avoid the entrance and exit effects. The catalyst was reduced in situ by hydrogen at 453 K for 5 h and the reactor was brought to the desired temperature before the reactant was introduced. Phenol oxidation was carried out without the catalyst and with the support without metal under the following conditions: different temperatures (313–423 K), pressures (1–18 bar), feed weight hour space velocity (WHSV) (1–10 h−1), oxygen to phenol molar ratio (1–14), metal loading (0.2–0.7 wt% of Ru) and ceria loading (1–18 wt%) in the Ru/MCM-41 catalyst. The progress of the reaction was monitored by withdrawing the liquid samples from the sample collecting vessel at regular time intervals and analyzed using gas chromatography (Agilent Technologies 6890 N, USA). The gas phase was analyzed by a GC equipped with a katharometer. A Porapak Q packed column (1/4 in, 0.5 m) was used to analyze CO2 using hydrogen as the carrier gas. Carboxylic acids were separated on an organic acid analysis column (Aminex HPX87H (3007.8 mm)). The mobile phase was a 0.004 M H2SO4 solution (0.8 mL min−1). A diode array UV/visible detector (Thermo Finnigan UV6000LP) coupled with a refractive index detector (RI 150, Thermo Finnigan) was used for the quantification. The column temperature was stabilized at 30 °C. To complete the carbon balance, TOC was measured with an OI Analytical (1020A) TOC analyzer, which employs high temperature combustion oxidation coupled with non-dispersive infrared detection technology. The retention times for the different compounds were determined by injecting pure compounds under the identical GC conditions. The activity was described in terms of the oxidation of phenol using a specific reaction rate according to eqn (1)
 | (1) |
 | ||
| Fig. 1 Schematic of the experimental set-up for the oxidation of phenol. | ||
The oxidation of phenol was observed to occur readily within the studied temperature range and the conversion of phenol to CO2 and H2O was found to be 99% at 413 K. The reactor exhibited no temperature or pressure gradients across the catalyst bed and there were no diffusional limitations for the transport of reactant and product molecules.
2.2. Material balance
The products of the oxidation of phenol were CO2 and H2O, and these can lead to the formation of other carboxylic acids as intermediates. The phenol samples were analysed using high performance liquid chromatography (HPLC) (column: Nova Pak C18 (Waters)), with 1 mL min−1 flow rate of the mobile phase (MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O:H3PO4 = 40:60:0.5 volume%) analyzer, for the possible presence of organic compounds other than phenol. Fig. 2 shows only one peak, indicating the absence of other organic compounds.
:H2O:H3PO4 = 40:60:0.5 volume%) analyzer, for the possible presence of organic compounds other than phenol. Fig. 2 shows only one peak, indicating the absence of other organic compounds.
 | ||
| Fig. 2 HPLC analysis of the sample after the reaction run. | ||
The material balance for phenol was checked by measuring the total organic carbon (TOC) in the samples using a TOC analyzer (Analytik Jena, multi N/C 2100), the operation of which is based on the combustion/non-dispersive infrared gas analysis. The total organic carbon values for the fresh phenol solution and for that obtained after the oxidation were determined for the experiments performed using the Ru/CeO2-MCM-41 catalyst. A close agreement between the degradation values of phenol determined by the GC method and those determined by the TOC analyzer was noticed.
2.3. Catalyst preparation
:1H2SO4 (diluted 1:1 v/v). A gel was obtained at this stage and was stirred for further 30 min. The polypropylene container was now closed and allowed to age at room temperature for 24 h without stirring. The resultant gel was filtered, washed with DDW to remove the adhering ions, and dried at 120 °C followed by calcination at 450 °C for 6 h at a heating rate of 2 °C min−1. The final material obtained was used for all the further studies.2.4. Characterization of the catalysts
As shown in Fig. 3, all the samples exhibited type IV isotherms with H1 hysteresis loops in the relative pressure (P/P0) range between 0.6 and 0.8, which is characteristic of highly ordered mesoporous materials. This revealed that the ordered mesoporous structure was retained after the loading of Ru. The corresponding pore size distributions calculated by the Barrett–Joyner–Halenda (BJH) method were around 3.5 nm (Fig. 4).
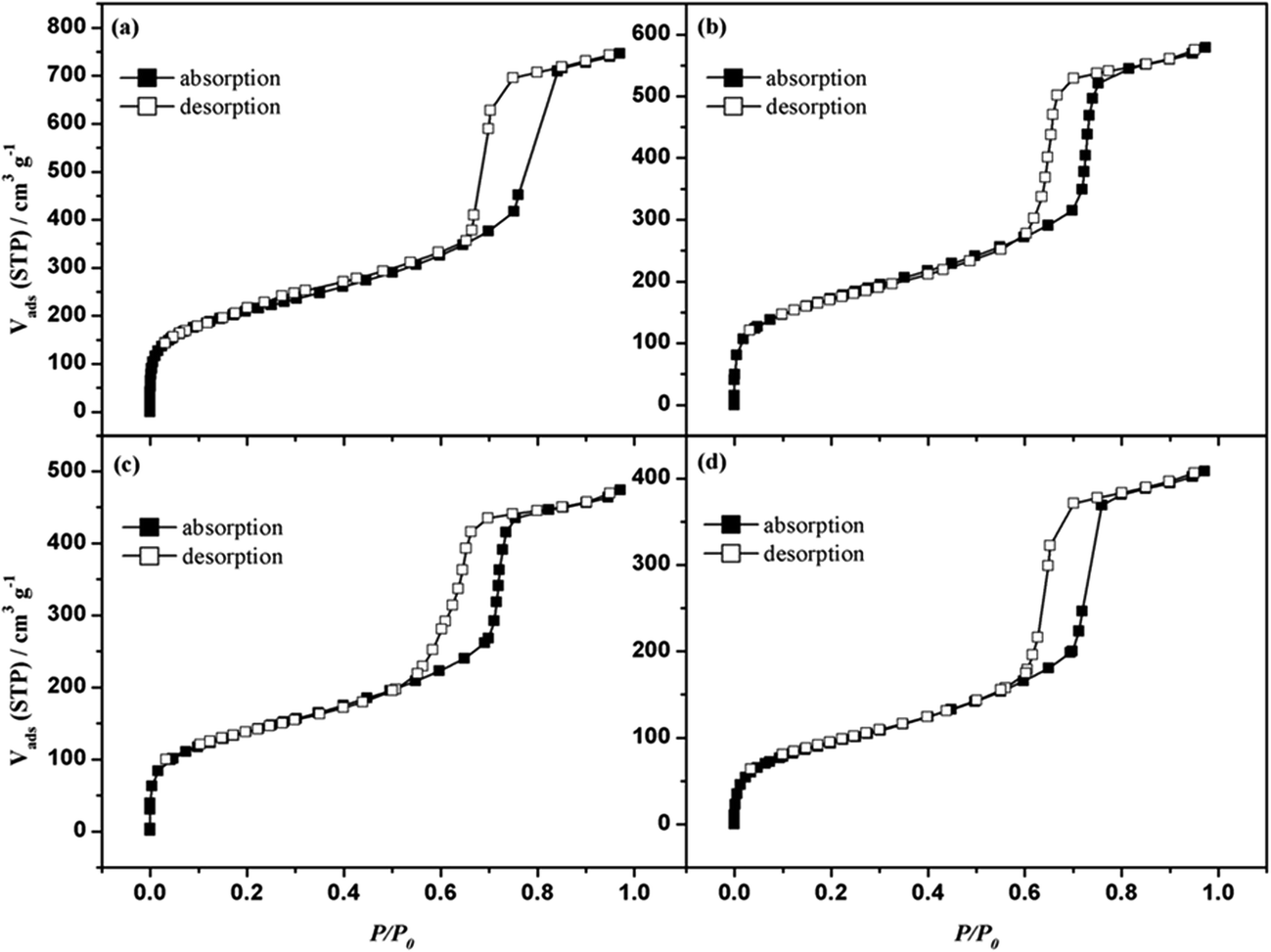 | ||
| Fig. 3 Nitrogen adsorption–desorption isotherms of the (a) MCM-41, (b) CeO2(14 wt%)-MCM-41, (c) Ru(0.5 wt%)/CeO2(14 wt%)-MCM-41cal, and (d) Ru(0.5 wt%)/MCM-41cal. | ||
 | ||
| Fig. 4 TPR profiles of Ru/CeO2-MCM-41 catalyst. | ||
The MCM-41 support displayed the highest specific area value in comparison to the other samples (650 m2 g−1) (Table 1) and the orders of the specific area, average pore diameter and average pore volume are given in Table 2.
| Catalyst/support | BET surface area (m2 g−1) | Total pore vol (cm3 g−1) | Pore diameter (nm) | Metal dispersion (%) | Metal particle size (nm) | Acidity using NH3 (μmol g−1) | Ru (wt%) | Rate [g mol (gcat min)−1] | Activation energy (kJ mol−1) |
|---|---|---|---|---|---|---|---|---|---|
| a Chemical compositions of the catalysts (Ru) were determined by inductively coupled plasma-atomic emission spectroscopy (ICP-AES).b BET specific area, pore volume, and average pore diameter as measured by N2 adsorption–desorption isotherms at 77 K.c Average metal particle size of the reduced samples at 453 K as measured by CO chemisorptions.d Metal dispersion using CO chemisorptions.e Acidity of the reduced samples and bare carriers as measured by TPD of adsorbed NH3. | |||||||||
| MCM-41 | 650 | 1.188 | 2.5–4.6 | 135.70 | |||||
| CeO2(14 wt%)-MCM-41 | 620 | 0.78 | 2.5–4.7 | 305.58 | — | ||||
| Ru(0.5 wt%)/MCM-41cal | 550 | 0.745 | 2.5–4.3 | 80 | 1.2 | 320.55 | 0.45 | 1.3 × 10−4 | 39 |
| Ru(0.5 wt%)/CeO2(10 wt%)-MCM-41cal | 570 | 0.751 | 2.55–4.61 | 88 | 1.5 | 0.458 | |||
| Ru(0.5 wt%)/CeO2(12 wt%)-MCM-41cal | 575 | 0.753 | 2.55–4.62 | 91 | 1.4 | 0.46 | |||
| Ru(0.5 wt%)/CeO2(14 wt%)-MCM-41cal | 580 | 0.756 | 2.55–4.63 | 93 | 1.3 | 335.76 | 0.47 | 1.82 × 10−4 | 33 |
| Ru(0.5 wt%)/CeO2(16 wt%)-MCM-41cal | 574 | 0.752 | 2.55–4.60 | 89 | 1.2 | 0.456 | |||
| Ru(0.5 wt%)/CeO2(18 wt%)-MCM-41cal | 569 | 0.75 | 2.55–4.59 | 87 | 1.1 | 0.454 | |||
| Ru(0.5 wt%)/MCM-41spent | 430 | 0.642 | 2.5–3.9 | 1.6 | |||||
| Ru(0.5 wt%)/CeO2(14 wt%)-MCM-41spent | 565 | 0.722 | 2.5–4.58 | 1.4 | |||||
| Specific area | Pore diameter | Pore volume |
|---|---|---|
| MCM-41 > CeO2(14 wt%)-MCM-41 > Ru(0.5 wt%)/CeO2(14 wt%)-MCM-41cal > Ru(0.5 wt%)/CeO2(12 wt%)-MCM-41cal > Ru(0.5 wt%)/CeO2(16 wt%)-MCM-41cal > Ru(0.5 wt%)/CeO2(10 wt%)-MCM-41cal > Ru(0.5 wt%)/CeO2(18 wt%)-MCM-41cal > Ru(0.5 wt%)/CeO2(14 wt%)-MCM-41spent > Ru(0.5 wt%)/MCM-41cal > Ru(0.5 wt%)/MCM-41spent | CeO2(14 wt%)-MCM-4 > Ru(0.5 wt%)/CeO2(14 wt%)-MCM-41cal > Ru(0.5 wt%)/CeO2(12 wt%)-MCM-41cal > Ru(0.5 wt%)/CeO2(10 wt%)-MCM-41cal > Ru(0.5 wt%)/CeO2(16 wt%)-MCM-41cal > Ru(0.5 wt%)/CeO2(18 wt%)-MCM-41cal > MCM-41 > Ru(0.5 wt%)/CeO2(14 wt%)-MCM-41spent > Ru(0.5 wt%)/MCM-41cal > Ru(0.5 wt%)/MCM-41spent | MCM-41 > CeO2(14 wt%)-MCM-41 > Ru(0.5 wt%)/CeO2(14 wt%)-MCM-41cal > Ru(0.5 wt%)/CeO2(12 wt%)-MCM-41cal > Ru(0.5 wt%)/CeO2(16 wt%)-MCM-41cal > Ru(0.5 wt%)/CeO2(10 wt%)-MCM-41cal > Ru(0.5 wt%)/CeO2(18 wt%)-MCM-41cal > Ru(0.5 wt%)/MCM-41cal > Ru(0.5 wt%)/CeO2(14 wt%)-MCM-41spent > Ru(0.5 wt%)/MCM-41spent |
The specific SBET values of the supports decreased upon the incorporation of the Ru components (Table 1). The highest specific area value was observed in the calcined Ru/CeO2-MCM-41 in comparison to the calcined Ru/MCM-41 catalyst. The average pore diameter in the calcined Ru(0.5 wt%)/CeO2(14 wt%)-MCM-41 and Ru(0.5 wt%)/MCM-41 samples decreased slightly with the deposition of the active components, ca. 0.01 and 0.15 nm, respectively. However, the Ru(0.5 wt%)/CeO2(14 wt%)-MCM-41 sample almost preserved the pore diameter in comparison to the homologous CeO2(14 wt%)-MCM-41 support. Once again, the lower pore volume was observed in the Ru(0.5 wt%)/MCM-41cal and Ru(0.5 wt%)/MCM-41spent samples. Moreover, it seems that the type of the mesostructured material had an important role in the structural parameters of the supported catalysts.
The Ru(0.5 wt%)/MCM-41spent catalyst exhibited the higher decrease in the specific area of the sample, suggesting that under the conditions of oxidation (employed in this study), larger carbon and Ru particles formed, which blocked the pores. This is in agreement with the pore diameter values. The pore diameter decreased ca. 0.35 nm in the Ru(0.5 wt%)/MCM-41spent sample, whereas it decreased ca. 0.06 nm in the Ru(0.5 wt%)/CeO2(14 wt%)-MCM-41spent sample, almost remaining the same. When comparing the structural parameters of the calcined catalysts with those of the spent catalyst in the oxidation of phenol, it can be noted that the Ru(0.5 wt%)/CeO2(14 wt%)-MCM-41spent catalysts largely preserved the structural parameters of the freshly calcined state. However, a significant decrease in the structural parameters of Ru(0.5 wt%)/MCM-41spent catalyst was observed. The origin of this behavior was not very clear; however, it proved that the Ru(0.5 wt%)/MCM-41 catalyst underwent major changes in its structural parameters during the reaction and this should be reflected in their catalytic performance.
After the TPR experiment, the CO chemisorption was determined at 50 °C in a 20 mL min−1 stream of He using a pulsed chemisorption technique, in which 0.1 mL CO/He (CO: 5 vol%) pulses of each gas were utilized. The metal particle size was estimated assuming a CO:Ru = 1:1 surface stoichiometry.
 | ||
| Fig. 5 (a) Small angle XRD profiles of MCM-41, CeO2-MCM-41, Ru/MCM-41, and Ru/CeO2-MCM-41 catalysts. (b) Broad angle XRD profiles of MCM-41, CeO2-MCM-41, Ru/MCM-41, and Ru/CeO2-MCM-41 catalysts. | ||
 | ||
| Fig. 6 (a) SEM image of the Ru/MCM-41 catalyst. (b) SEM image of Ru/CeO2-MCM-41 catalyst. | ||
| Catalyst | Ru (wt%) | Si (wt%) | O2 (wt%) | Ce (wt%) |
|---|---|---|---|---|
| MCM-41 | — | 33 ± 1 | 65.8 ± 0.5 | — |
| CeO2-MCM-41 | — | 32 ± 1 | 64.0 ± 0.5 | 2 ± 0.5 |
| Ru/MCM-41 | 0.38 ± 0.12 | 33 ± 1 | 65 ± 0.5 | |
| Ru/CeO2-MCM-41 | 0.4 ± 0.15 | 32 ± 1 | 63.45 ± 1 | 1.5 ± 0.5 |
| Catalyst | Ru metal loading (wt%) | Ru metal content before reaction (wt%) | Ru metal content after reaction (wt%) |
|---|---|---|---|
| Ru/MCM-41 | 0.5 | 0.4 ± 0.022 | 0.4 ± 0.016 |
| Ru/CeO2-MCM-41 | 0.5 | 0.46 ± 0.026 | 0.46 ± 0.023 |
The TEM images of the calcined Ru(0.5 wt%)/MCM-41 and Ru(0.5 wt%)/CeO2(14 wt%)-MCM-41 catalysts are shown in Fig. 7a and b, respectively. The TEM technique showed a well-ordered cylindrical channel-like pore morphology with a 2D hexagonal mesostructure. The TEM images provide a pictorial evidence of an ordered mesostructured MCM-41 silicate material. These images confirmed that the mesoporous and well-ordered structure of MCM-41 was preserved even after loading the metals and that the metals were well-dispersed in the pore channels since no patches of the metal aggregates or agglomerates were found in the samples. The average distance between the two consecutive centers of the hexagonal pores, estimated from the TEM images, was in the range of 2.5–4.9 nm, which is consistent with the N2 physisorption results.
 | ||
| Fig. 7 (a) TEM image of Ru(0.5 wt%)/MCM-41 catalyst (a) ×20k magnification. (b) TEM image of Ru(0.5 wt%)/CeO2(14 wt%)-MCM-41 catalyst (b) ×20k magnification. | ||
3. Results and discussion
Phenol oxidation was first investigated with the supports (MCM-41 and CeO2-MCM-41) without metal and it was found that at 413 K and 18 bar pressure, the phenol conversions were 9% and 12%, respectively. For the same standard conditions but in the presence of the Ru/MCM-41 catalyst with a ruthenium metal loading of 0.5 wt%, the conversion was about 80%, which increased to 99% in the presence of the ceria promoted Ru(0.5 wt%)/CeO2(10 wt%)-MCM-41 catalyst. The conversion curves followed the behavior described in Fig. 8a. However, since there is no pure phenol solution in industrial effluents, the activity data of the Ru(0.5 wt%)/CeO2(10 wt%)-MCM-41 catalyst was tested for the treatment of a simulated phenol containing solution with toxic bisphenol,27 oxalic acid,2 and acetic acid.29 The results, as shown in Fig. 8c, indicated that the conversion of phenol was 99%, bisphenol 42%, oxalic acid 17.5% and acetic acid 28%, as the catalyst was very specific to the reactants. Since 99% phenol conversion was achieved with the Ru(0.5 wt%)/CeO2(10 wt%)-MCM-41 catalyst under the applied conditions, further study was carried out with phenol as the model compound. | ||
| Fig. 8 (a) Effect of temperature on phenol conversion. Reaction conditions: pressure: 18 bar, WHSV: 1 h−1, oxygen/phenol mole ratio: 13. (b) Arrhenius plot of ln(k) as a function of 1/T for Ru(0.5 wt%)/MCM-41 and Ru(0.5 wt%)/CeO2(14 wt%)-MCM-41 catalysts. (c) Activity of the Ru/CeO2-MCM-41 catalyst using aqueous solution of phenol, bisphenol, oxalic acid, and acetic acid. Temperature: 413 K, pressure: 18 bar, WHSV: 1 h−1, oxygen to phenol mole ratio: 13. | ||
3.1. Effect of the reaction temperature on the conversion of phenol
The effect of temperature has been studied by varying the temperature from 313 to 423 K. The other process variables, such as pressure, WHSV, and oxygen/phenol molar ratio, were kept constant at 18 bar, 1.0 h−1 and 13, respectively, during the experiments. The results shown in Fig. 8a indicate that an increase in temperature favored the percentage of phenol conversion as expected. This is due to the fact that the rate constant increases with an increase in the reaction temperature, leading to the enhancement of phenol degradation since this process is considered to be kinetically controlled.However, the rate of increase in the oxidation was slightly slow at higher temperatures as compared to that at lower temperatures. Thus, it can be observed from Fig. 8a that a 10 K rise in temperature, from 413 to 423 K only, results in about a 1.5% change in the phenol conversion. Hence, operating the oxidation process at about 413 K, a maximum phenol removal of about 99% can be achieved using the optimum Ru/CeO2-MCM-41 catalyst. The experimental runs were repeated at 413 K and the results were compared with those of the previous runs to ascertain the reproducibility of the data obtained. The results of the error analyses indicate that the data are quite reproducible with only a narrow margin of error of 1.2%.
The reaction rate constants were calculated from the experimental data and the results were plotted as ln(k) vs. 1/T, as shown in Fig. 8b, for the Ru(0.5 wt%)/MCM-41 and Ru(0.5 wt%)/CeO2(14 wt%)-MCM-41 catalysts. The activation energy of the reaction using the Ru(0.5 wt%)/MCM-41 catalyst was found to be 39 kJ mol−1, whereas the activation energy using the ceria promoted Ru(0.5 wt%)/CeO2(14 wt%)-MCM-41 catalyst was found to be 33 kJ mol−1 (Fig. 8b). The value of the activation energy for the ceria promoted Ru(0.5 wt%)/CeO2(14 wt%)-MCM-41 catalyst was lower than that for the Ru(0.5 wt%)/MCM-41 catalyst, indicating the higher activity of the ceria promoted catalyst. This is due to the fact that in the present study the oxidation was carried out using a catalyst that facilitates a chemical reaction and proceeds by a different pathway with a lower energy barrier. The activity was described in terms of the oxidation of phenol using a specific reaction rate at 413 K, 18 bar pressure, and 3 h on stream according to eqn (1) and is given in Table 1. It was observed that the reaction rate for phenol oxidation was higher on the Ru(0.5 wt%)/CeO2(14 wt%)-MCM-41 catalyst than that on the Ru(0.5 wt%)/MCM-41 catalyst. The hexagonal nature of the mesoporous materials and their thermal and hydrothermal stability increased when the transition metals were anchored into the framework of the mesoporous silicate.33,34 Substitution of foreign ions (Ce3+, Al3+, Zr4+ etc.) into the silicate framework is an efficient route to enhance the acidity and stability of mesoporous silica (Table 1).35,36 It can be further noticed from Table 1 that the addition of Ce on the MCM-41 support increased the metal dispersion and acidity. Some authors have reported that Al−, Ti− or Zr− containing SBA-15 provides a better dispersion of the metal species when compared to the pure SBA-15 and alumina supported catalysts.37,38
The phenol oxidation reaction was found to be first order (n = 1) with respect to phenol and Thiele modulus, and the Weisz–Prater criterion was applied to determine the rate limiting step.29 When the value of Thiele modulus is small, the surface reaction is usually rate limiting. In the present study, the value of the Thiele modulus was found to be 0.79, indicating that the surface reaction was the rate limiting step. The Weisz–Prater criterion was applied to determine if the internal diffusion was limiting the reaction. The value of Weisz–Prater criterion was found to be 1.95 × 10−5 (˂1), which indicated that there were no diffusion limitations and consequently, no concentration gradient existed within the catalyst pores.
3.2. Effect of the oxygen to phenol molar ratio on the conversion of phenol
The choice of an appropriate ratio of oxygen to phenol is very crucial for the catalytic wet air oxidation. Generally, an increase in the oxygen partial pressure increases the rate of oxidation, which in turn increases the rate of the removal of phenolic compounds. The use of a higher oxygen pressure also enriches the catalyst and reduces deactivation. However, a very high value of the oxygen to phenol molar ratio may increase the cost of the process. Thus, an optimum value of the oxygen to phenol molar ratio is always desired. In the present study, the effect of the oxygen to phenol molar ratio on the oxidation of phenol was determined by varying it from 1 to 14, maintaining the temperature, pressure and WHSV at 413 K, 18 bar, and 1.0 h−1, respectively.It can be observed from Fig. 9 that the removal of phenolic compounds significantly increased as the oxygen to phenol molar ratio was increased up to 13. However, the effect began to level off beyond this threshold, rendering further increments economically non-beneficial to the process. This observation was explained by Bej et al.39 and was attributed to the pseudo first order dependence of the rate of oxidation reaction on the oxygen partial pressure at its high value corresponding to an oxygen to phenol molar ratio of 13. Thus, the present study indicates that an oxygen/phenol ratio of about 13 should be maintained for achieving maximum phenol conversion.
 | ||
| Fig. 9 Effect of oxygen to phenol molar ratio on the phenol conversion. Reaction conditions: temperature: 413 K, pressure: 18 bar, WHSV: 1 h−1, catalyst used: Ru(0.5 wt%)/CeO2(14 wt%)-MCM-41. | ||
3.3. Effect of the oxygen partial pressure on the conversion of phenol
One crucial process parameter that directly affects the oxidation conversions is the oxygen partial pressure. For instance, in the CWAO of phenol, it was observed that below a certain oxygen partial pressure it becomes impossible to increase the conversion of phenol even if the operating temperature is increased. As a result, the effect of pressure on the catalytic wet air oxidation of phenol was studied at a temperature, WHSV, and oxygen to phenol molar ratio of 413 K, 1 h−1, and 13, respectively, and is represented in Fig. 10. | ||
| Fig. 10 Effect of pressure on the phenol conversion. Reaction conditions: temperature: 413 K, WHSV: 1 h−1, oxygen to phenol molar ratio: 13, catalyst used: Ru(0.5 wt%)/CeO2(14 wt%)-MCM-41. | ||
As the partial pressure of oxygen increased, the concentration of oxygen in the solution also increased, according to Henry's law, and consequently, the reaction rate was enhanced. As was expected, increase in the partial pressure of oxygen resulted in an increase in the extent of phenol conversion by increasing the catalyst's activity.
It is known that the major role of the catalyst is to provide the required reaction interface for the reactants, thereby promoting the interactions between the effluent constituents (i.e. phenol, carboxylic acid etc.) and oxygen. However, it is noteworthy that excessively high oxygen pressures may only serve to saturate the catalyst and any further increase in the oxygen partial pressure tended to affect the phenol conversions by only a slight margin. For the optimized Ru/CeO2-MCM-41 catalyst study, one can observe from Fig. 10 that an increase in the oxygen pressure from 14 to 18 bar only resulted in a less than 2.5% conversion during the oxidation process.
3.4. Effect of the feed weight hourly space velocity on the conversion of phenol
The effect of the feed weight hourly space velocity (WHSV) on the conversion of phenol was studied by varying the WHSV from 1 to 10 h−1 at a constant temperature of 413 K, pressure of 18 bar, and an oxygen/phenol molar ratio of 13. The experimental results are depicted in Fig. 11 and suggest that the phenol conversion was found to decrease with an increase in the feed WHSV of phenol that can be explained by two different ways. With an increase in the liquid feed rate, the wetted fractions of the catalyst as well as the gas liquid and liquid solid mass transfer coefficients also increase. At a lower liquid feed rate, the catalyst particles are partially wetted and under these conditions, the conversion increases due to a direct transfer of the gas phase reactant to the catalyst surface, already internally wetted due to the capillary forces. Therefore, with an increase in the liquid feed rate, an increase in the wetted fraction was expected to retard the conversion, whereas an increase in the external mass transfer coefficients will enhance the conversion.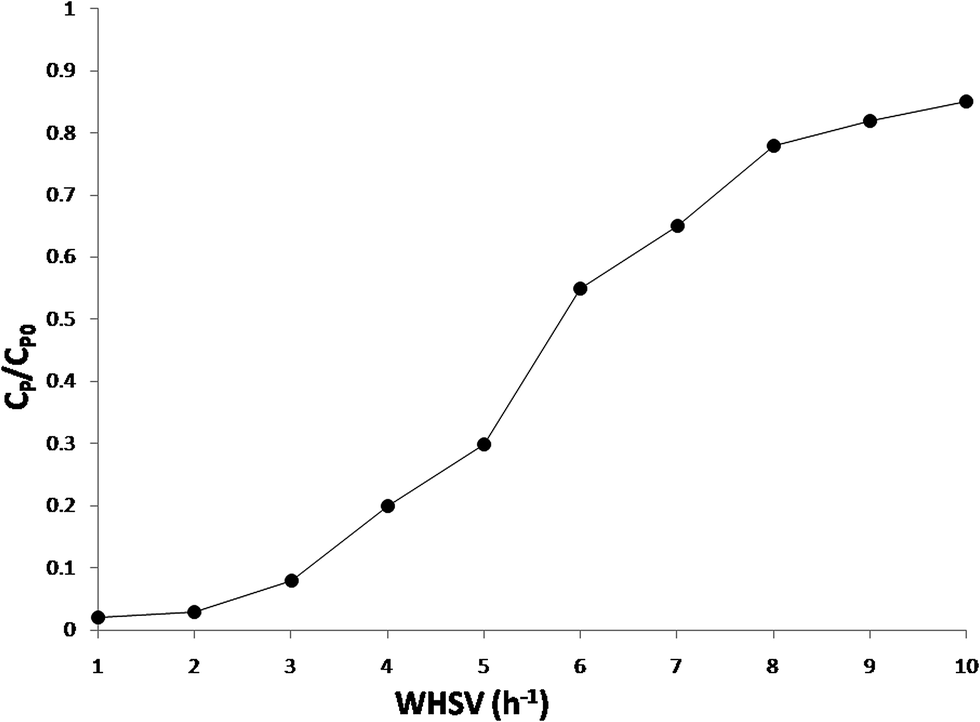 | ||
| Fig. 11 Effect of weight hourly space velocity on the phenol conversion. Reaction conditions: temperature: 413 K, pressure: 18 bar, oxygen to phenol molar ratio: 13, catalyst used: Ru(0.5 wt%)/CeO2(14 wt%)-MCM-41. | ||
Another possible reason for the observed decrease in the conversion was due to the shorter residence time of the reactants under a high feed WHSV. However, the former explanation was applicable to the large catalyst bed but in this study a low amount of the catalyst (around 6 g, smaller catalyst bed) was used, and hence the latter explanation is more appropriate for this study. It could be concluded from the WHSV studies that under these conditions about 99% phenol conversion can be achieved over the optimum Ru/CeO2-MCM-41 catalyst.
3.5. Effect of Ru loading (wt%) on the conversion of phenol
Experiments were performed by varying the ruthenium content within the catalyst support by increasing the concentration of the ruthenium acetylacetonate. A series of experiments were performed using different Ru loadings from 0.2 to 0.7 wt% and the results are shown in Fig. 12. It can be noticed that there is an increase in the phenol degradation with an increase in the loading of Ru from 0.2 to 0.5 wt%. It can be observed that the maximum conversion (about 99%) was obtained at a 0.5 wt% loading of Ru metal and beyond 0.5 wt% loading of Ru, there was hardly any increase in the phenol degradation. This may be due to the fact that enough active catalyst sites were available at a 0.5 wt% catalyst loading for the reaction to occur, and a decrease in the active catalyst sites due to a higher catalyst loading will not enhance the reaction. The CO chemisorption tests showed that maximum amount of metal (80% of 0.5 wt% Ru) was available for reaction in the case of 0.5 wt% Ru loading (Table 5). Beyond the 0.5 wt% metal loading, the metal available for the reaction decreased due to either the blocking of the pores of the catalyst support by the metal precursor or the agglomeration of the metal in the case of higher loading of metal.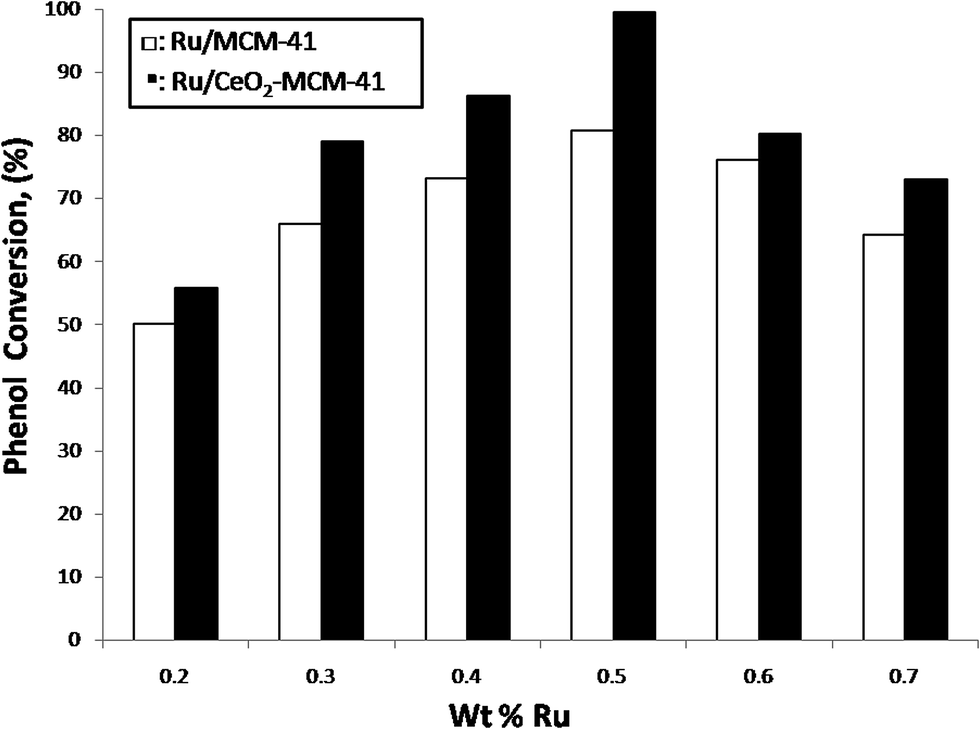 | ||
| Fig. 12 Effect of Ru loading (wt%) on the phenol conversion. Reaction conditions: temperature: 413 K, pressure: 18 bar, WHSV: 1 h−1, oxygen to phenol molar ratio: 13. | ||
| Ru metal loading (wt%) | Ru metal dispersion (%) |
|---|---|
| 0.2 | 85 |
| 0.3 | 82.5 |
| 0.4 | 81 |
| 0.5 | 80 |
| 0.6 | 64 |
| 0.7 | 51 |
3.6. Effect of the ceria loading (wt%) in the Ru/MCM-41 catalyst on the conversion of phenol
Experiments were performed using different ceria amounts (1–18 wt%) with the 0.5 wt% Ru loading to determine the optimum amount of ceria for the degradation of phenol and the results are shown in Fig. 13. The degradation of phenol was found to increase with an increase in ceria loading from 1 wt% to 14 wt%. The catalyst with 14 wt% ceria displayed the best result, with 99% conversion at 413 K. A further increase in ceria loading beyond 14 wt% led to a decrease in the degradation of phenol. This may be due to the reduction in the number of active catalyst sites.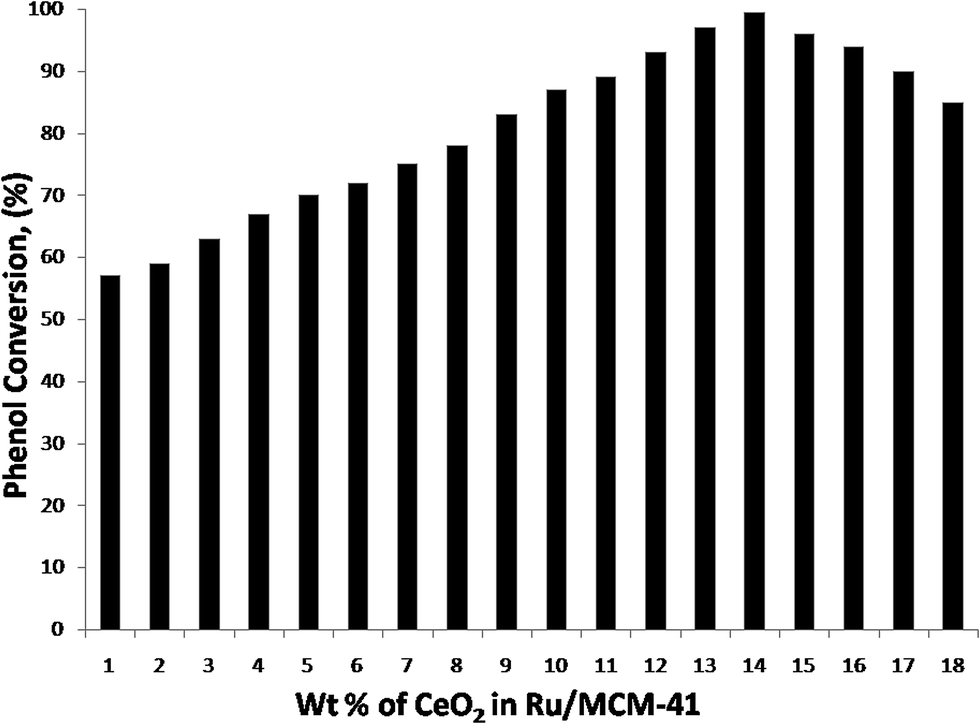 | ||
| Fig. 13 Effect of ceria loading on the phenol degradation. Reaction conditions: temperature: 413 K, pressure: 18 bar, WHSV: 1 h−1, oxygen to phenol molar ratio: 13. | ||
The CO chemisorption results reported in Table 6 show that the dispersion of Ru was at a maximum of about 93% with a ceria loading of 14 wt% because ceria promoted the dispersion of Ru and that strong Ru ceria interactions may have a profound effect on the oxygen storage capacity.40 Kim and Ihm41 reported that Ru was largely dispersed at a lower ceria loading (under 14 wt%) and Ru–Ce-support interactions may exist. The ruthenium crystallite size was calculated from Fig. 5b using the half width of the Ru peak in the XRD curve of the catalyst using the Scherrer equation (Table 6) and error in the Ru particle size was found to be ±2%. From Table 6, we can see that the Ru crystallite size on Ru/CeO2-MCM-41 was smaller than that on Ru/MCM-41, which indicates that ceria decreased the Ru crystallite size and inhibited Ru crystallite growth during the reaction. The strong interactions between Ru and ceria may result in a larger extent of boundary area between cerium oxide and ruthenium, and the active sites involve both Ru and ceria. It was found that during the CWAO process, some of the cerium was in the state of Ce(III).42 The lower valance state of cerium may dissociatively adsorb oxygen or water, and the resulting ad-species-O may be transferred to adjacent ruthenium and reacted with the surface carbon species to produce CO2 and H2O. Zhuang43 reported that cerium oxide itself oxidizes hydrocarbon and although its activity is several orders lower than that of the other transition metal oxides, when noble metals coexist with cerium oxide, the reaction will be greatly accelerated. Therefore, the activity of the Ru/CeO2-MCM-41 catalyst increased. When the amount of ceria was increased beyond 14 wt% on the surface of the catalyst, Ce(III) ion, which is rich in electrons, interacted with Ru atom, making Ru atom more electron enriched; therefore, the degradation activity of Ru for phenol was decreased.
| a Ruthenium crystallite size as calculated by the Scherrer equation. | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ceria loading (wt%) | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 |
| Ru loading (0.5 wt%) | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| Ru metal dispersion (%) | 80 | 80.5 | 81 | 82 | 83 | 84 | 84.5 | 85 | 86 | 87 | 88 | 89 | 91 | 92 | 93 | 91 | 89 | 88 | 87 |
| Particle size before the reaction (Å) | 20.8 | 19.6 | 19 | 18.5 | 18 | 17.5 | 17 | 16.4 | 16 | 15.5 | 15 | 14.6 | 14 | 13.5 | 13 | 12.5 | 12 | 11.5 | 11 |
| Particle size after the reaction (Å) | 25.7 | 25 | 24.6 | 24 | 23.5 | 23 | 22.5 | 22 | 21.4 | 21 | 20.5 | 20 | 19.4 | 19 | 18.5 | 18 | 17.5 | 17 | 16.6 |
The conversions obtained with the Ru(0.5 wt%)/CeO2(14 wt%)-MCM-41 and Ru(0.5 wt%)/MCM-41 catalysts were far higher than those obtained with the bare support, confirming the catalytic effect. By determining the metallic contents of the catalysts before and after the reaction, it can be concluded that almost no metal leaching occurred with these catalytic systems (Table 4). Noble metals are thermodynamically stable in their zero-valent state under a very broad range of conditions and specifically, under the conditions employed in this work, thus leading to the absence of leaching.
3.7. Catalyst stability study
The phenol oxidation experiments were performed for a period of 15 days to study the activity of the catalyst (Fig. 14). It was noticed that the phenol oxidation was stable during the reaction time and the catalyst did not show deactivation effects. EDX analysis of the spent catalysts was performed and the images showed no carbon peak in both the spent Ru(0.5 wt%)/CeO2(14 wt%)-MCM-41 and Ru(0.5 wt%)/MCM-41 catalysts and showed only silica (Si: 50.91 wt%), ruthenium (Ru: 0.27 wt%), cerium (Ce: 2.52 wt%) and oxygen (O: 46.30 wt%) in the spent catalysts. Thermogravimetric analysis (TGA) of the catalysts was also carried out and a significant weight loss during the initial heating up period in the temperature range 100–300 °C was observed. This weight loss was mostly due to the removal of moisture from the catalyst surface and that retained inside the pores, which suggested that a higher temperature is required to remove the excess moisture. Further increase in temperature did not lead to any significant change in the weight of the catalysts. Deactivation of the spent Ru(0.5 wt%)/CeO2(14 wt%)-MCM-41 and Ru(0.5 wt%)/MCM-41 catalysts due to metal sintering was investigated using the BET surface area analyzer. There was no metal sintering observed in the spent catalysts as the BET surface area closely agreed with the BET surface area of the fresh catalysts. | ||
| Fig. 14 Catalyst stability plot for 15 days reaction duration using Ru(0.5 wt%)/CeO2(14 wt%)-MCM-41 catalyst. Temperature: 413 K, pressure: 18 bar, WHSV: 1 h−1, oxygen to phenol molar ratio: 13. | ||
4. Conclusions
CWAO experiments were conducted in a continuous upflow fixed bed reactor at temperatures up to 423 K and under an oxygen partial pressure of 18 bar. Among all the catalysts studied, the presence of the Ru(0.5 wt%)/CeO2(14 wt%)-MCM-41 catalyst exhibited the most efficient destruction of phenol. Moreover, under the reaction conditions (413 K and ruthenium loading of 0.5 wt%) 99% phenol removal was achieved, and the phenol conversion rate increased upon increasing the reaction temperature and partial pressure of oxygen. The type of mesostructured material used in this study has an important effect on the dispersion of Ru metal and the highest dispersion was observed for the catalysts prepared using the ceria promoted MCM-41 materials, and their samples had the higher catalytic performances in the oxidation of phenol under the conditions employed in this study. The higher catalytic activity in this sample was related to the dispersion of Ru precursor and the acidity of the sample.The dispersion of Ru was at its maximum (93%) with a ceria loading of 14 wt% because ceria promoted the dispersion of Ru and that strong Ru ceria interactions may have a profound effect on the oxygen storage capacity. Moreover, the Ru crystallite size on Ru/CeO2-MCM-41 was smaller than that found on Ru/MCM-41, which indicates that ceria can decrease the Ru crystallite size and inhibit Ru crystallite growth during the reaction.
Within 15 days of reaction, the Ru/CeO2-MCM-41 catalyst displayed a good stability in the fixed bed reactor with an upflow of phenol. Since no catalyst leaching occurred and significant phenol conversions were obtained, the ceria promoted Ru/CeO2-MCM-41 catalysts can be regarded as the promising heterogeneous catalysts for the degradation of hazardous phenolic compounds, without concern over environmental risk due to active metal leaching from the reactor system and high initial investments.
Acknowledgements
The authors gratefully acknowledge the financial support provided by the Chemical Engineering Department, Jadavpur University, Kolkata, India. The authors also acknowledge Prof. A. K. Biswas (Former Professor and Head, Chemical Engineering Department, Indian Institute of Technology, Kharagpur, India) for their support.Notes and references
- A. E. Monterosa, G. Lafayea, A. Cervantesc, G. D. Angelc, J. Barbier and G. Torresb, Catal. Today, 2015, 58, 564–569 CrossRef.
- S. Roy and A. K. Saroha, RSC Adv., 2014, 4, 56838–56847 RSC.
- S. V. Mishra, V. M. Mahajani and J. B. Joshi, Ind. Eng. Chem. Res., 1995, 34, 2–48 CrossRef.
- F. Luck, Catal. Today, 1999, 53, 81–91 CrossRef CAS.
- J. C. Spain, P. A. van Veld, C. A. Monti, P. H. Pritchard and C. R. Cripe, Appl. Environ. Microbiol., 1984, 48, 944–950 CAS.
- S. Roy, M. Vashishtha and A. K. Saroha, J. Eng. Sci. Technol. Rev., 2010, 3, 95–107 CAS.
- S. Roy and A. K. Saroha, Indian Chem. Eng., 2011, 53, 136–151 CrossRef CAS.
- K. R. Rogers and J. M. van Emon, Immunoassay for p-nitrophenol in urine, EPA Report no. EPA/600/A-93/074, USA, 1993 Search PubMed.
- S. Tanjore and T. Viraraghavan, Int. J. Environ. Stud., 1994, 45, 155–164 CrossRef CAS.
- J. C. Spain and D. T. Gbison, Appl. Environ. Microbiol., 1991, 57, 812–819 CAS.
- G. Deiber, J. N. Foussard and H. Debellefontaine, Environ. Pollut., 1997, 96, 311–319 CrossRef CAS PubMed.
- J. Kiwi, C. Pulgarin and P. Peringer, Appl. Catal., B, 1994, 3, 335–350 CrossRef CAS.
- M. R. Hoffmann, I. Hua and R. Hochemer, Ultrason. Sonochem., 1996, 3, S163–S172 CrossRef CAS.
- M. S. Dieckmann and K. A. Gray, Water Res., 1996, 30, 1169–1183 CrossRef CAS.
- J. A. Menéndez, M. Inguanzo and J. J. Pis, Water Res., 2002, 36, 3261–3264 CrossRef.
- H. Debellefontaine and J. N. Foussard, Waste Manage., 2000, 20, 15–25 CrossRef CAS.
- C. C. Manole, J. L. Carine, W. Anne-Marie and D. Henri, Ind. Eng. Chem. Res., 2007, 46, 8388–8396 CrossRef.
- O. Gimeno, P. Plucinski, S. T. Kolaczkowski, F. J. Rivas and P. M. Alvarez, Ind. Eng. Chem. Res., 2003, 42, 1076–1086 CrossRef CAS.
- D. B. Akolekar, S. K. Bhargava, I. Shirgoankar and J. Prasad, Appl. Catal., A, 2002, 236, 255–262 CrossRef CAS.
- Y. Kacar, E. Alpay and V. K. Ceylan, Water Res., 2003, 37, 1170–1176 CrossRef CAS PubMed.
- X. J. Hu, L. C. Lei, G. H. Chen and P. L. Yue, Water Res., 2001, 35, 2078–2080 CrossRef CAS PubMed.
- W. P. Zhu, Y. J. Bin, Z. H. Li, Z. P. Jiang and T. Yin, Water Res., 2002, 36, 1947–1954 CrossRef CAS PubMed.
- A. Pintar, Catal. Today, 2003, 77, 451–462 CrossRef CAS.
- S. K. Bhargava, J. Tardio, J. Prasad, K. Folger, D. B. Akolekar and S. C. Grocott, Ind. Eng. Chem. Res., 2006, 45, 1221–1258 CrossRef CAS.
- U. S. Kulkarni and S. G. Dixit, Ind. Eng. Chem. Res., 1991, 30, 1916–1920 CrossRef CAS.
- S. Hosokawa, H. Kanai, K. Utani, Y. Taniguchi, Y. Saito and S. Imamura, Appl. Catal., B, 2003, 45, 181–187 CrossRef CAS.
- B. Erjavec, R. Kaplan, P. Djinovic and A. Pintar, Appl. Catal., B, 2013, 132, 342–352 CrossRef.
- P. Gallezot, S. Chaumet, A. Perrard and P. Isnard, J. Catal., 1997, 168, 104–109 CrossRef CAS.
- S. Roy and S. Datta, Ind. Eng. Chem. Res., 2013, 52, 17360–17368 CrossRef CAS.
- S. Roy, B. L. Newalkar and S. Datta, Can. J. Chem. Eng., 2014, 92, 1034–1040 CrossRef CAS.
- R. Ryoo, C. H. Ko, M. Kruk, V. Antochshuk and M. Jaroniec, J. Phys. Chem. B, 2000, 104, 11465–11471 CrossRef CAS.
- D. Zhao, Q. Huo, J. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS.
- Y. Cao, J. C. Hu, P. Yang, W. L. Dai and K. N. Fan, Chem. Commun., 2003, 908–909 RSC.
- T. A. Zepeda, B. Pawelec, J. L. G. Fierro and T. Halachev, J. Catal., 2006, 242, 254–269 CrossRef CAS.
- N. Kostova, A. Spojakina, K. Jiratova, O. Solcova, L. Dimitrov and L. Petrov, Catal. Today, 2001, 65, 217–223 CrossRef CAS.
- T. Zepeda, B. Pawelec, J. L. G. Fierro, A. Olivas, S. Fuenres and T. Halachev, Microporous Mesoporous Mater., 2008, 111, 157–170 CrossRef CAS.
- O. Y. Gutierrez, D. Valencia, G. A. Fuentes and T. Klimova, J. Catal., 2007, 249, 140–153 CrossRef CAS.
- O. Y. Gutierrez, G. A. Fuentes, C. Salcedo and T. Klimova, Catal. Today, 2006, 116, 485–497 CrossRef CAS.
- S. K. Bej, A. K. Dalai and J. Adjaye, Pet. Sci. Technol., 2002, 20, 867–877 CrossRef CAS.
- A. Trovarelli, Catal. Rev.: Sci. Eng., 1996, 38, 439–520 CAS.
- S. Kim and S. Ihm, Ind. Eng. Chem. Res., 2002, 41, 967–1972 Search PubMed.
- O. Gu, W. Chu, Z. L. Zhen Gao, Z. L. Yu and S. Y. Yuan, Stud. Surf. Sci. Catal., 1998, 119, 855–860 CrossRef.
- Q. Zhuang, Y. Y. Qin and L. Chang, Appl. Catal., 1991, 70, 1–8 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |