
DNA and BSA binding, anticancer and antimicrobial properties of Co(II), Co(II/III), Cu(II) and Ag(I) complexes of arylhydrazones of barbituric acid†
Jessica Palmucciab,
Kamran T. Mahmudov*ac,
M. Fátima C. Guedes da Silva*a,
Fabio Marchettib,
Claudio Pettinarid,
Dezemona Petrellie,
Luca A. Vitalif,
Luana Quassintif,
Massimo Bramuccif,
Giulio Lupidi*f and
Armando J. L. Pombeiro*a
aCentro de Química Estrutural, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001 Lisbon, Portugal. E-mail: kamran_chem@mail.ru; kamran_chem@yahoo.com; fatima.guedes@tecnico.ulisboa.pt; pombeiro@tecnico.ulisboa.pt
bSchool of Science and Technology, University of Camerino, Chemistry Section, via S. Agostino 1, 62032 Camerino, Italy
cDepartment of Chemistry, Baku State University, Z. Xalilov Str. 23, Az 1148 Baku, Azerbaijan
dSchool of Pharmacy, University of Camerino, Chemistry Section, via S. Agostino 1, 62032 Camerino, Italy
eSchool of Biosciences and Veterinary Medicine, University of Camerino, Piazza dei Costanti 4, 62032 Camerino, Italy
fSchool of Pharmacy, University of Camerino, Biological Section, via Gentile III da Varano, 62032 Camerino, Italy. E-mail: giulio.lupidi@unicam.it
First published on 18th December 2015
Abstract
Two new cocrystalline compounds, (Hen)(H2L2)·2/3H2O (2) and (Him)(H3L3)·2H2O (8), were prepared by the reaction of 5-(2-(4-chlorophenyl)hydrazono)pyrimidine-2,4,6(1H,3H,5H)-trione (H3L2) and the sodium salt of 2-(2-(2,4,6-trioxotetrahydropyrimidin-5(2H)-ylidene)hydrazinyl)benzenesulfonic acid (H4L3), [Na(H3L3)(μ-H2O)(H2O)2]2 (1) with protonated ethylenediamine (Hen) and imidazole (Him), respectively. By using 5-(2-(2-hydroxyphenyl)hydrazono)pyrimidine-2,4,6(1H,3H,5H)-trione (H4L1) and 1, several known CuII, CoII, CoII/III and new AgI complexes, [Cu(H2L1)(H2O)(im)]·3H2O (3), [Co(H2O)6][Co(H2L1)2]2·8H2O (4), [Co(H2L3)(im)3] (5), [Cu(H2L3)(im)2]·H2O (6), [Ag(H2O)(μ-H3L3)]n (7) and [Co(H2O)6][H3L3]2·8H2O (9), were also prepared in order to study their DNA and BSA binding, anticancer and antimicrobial properties. The complexes are able to interact with DNA and BSA with high binding constant values. In particular, complex 4 strongly intercalates DNA and binds BSA. The antimicrobial activity of all compounds, determined against a panel of reference bacterial and fungal strains, indicates that only 2 and 7 possess antimicrobial activity. The same compounds 2 and 7 show a pronounced antiproliferative activity against the human tumor cell lines A375, MDA-MB 231 and HCT116.
Introduction
Although barbituric acid is pharmacologically inactive, its derivatives possess very interesting pharmaceutical properties and can lead to drugs that act as central nervous system depressants, as sedatives (in small doses)/hypnotic (in larger doses) pharmaceuticals, anticonvulsants and anesthetics.1–5 Currently, therapeutic applications of barbiturates are expanding in connection with their antitumor, anti-invasive and anti-angiogenic effects.2–5 The pharmaceutical properties of barbiturates mainly depend on the side groups attached to the C5 atom of the pyrimidine ring (Scheme 1a).1–5 Several methods for the modification/functionalization of barbituric acid have been reported,6–11 and one of the most common and widely used concerns the Japp–Klingemann reaction, in which barbituric acid reacts with diazonium salts leading to arylhydrazones of barbituric acid (AHBAs) (Scheme 1b) in a basic medium.6,8 However, to our knowledge, pharmaceutical properties of AHBAs have not yet been studied, what is explored in this work, including their cocrystals and metal complexes. | ||
| Scheme 1 Molecular structure of barbituric acid (a) and AHBAs (b). | ||
Poor aqueous solubility is one of the most serious formulation challenges facing the pharmaceutical industry today. Approximately 40% of the drugs discovered through combinatorial chemistry and high throughput screening have an aqueous solubility of less than 10 μM.12–14 Limited aqueous solubility of drug molecules can lead to poor bioavailability and constitutes a significant risk factor in low oral absorption.15 A number of strategies have been employed for increasing the dissolution rate of poorly aqueous soluble pharmaceuticals, specifically in solid dosage formulations, the most applied technique for dissolution enhancement being cocrystal/salt formation.16,17 As far as we know, before the current work, cocrystals of AHBAs had not yet been isolated and structurally characterized, and their antimicrobial and antitumor activity and DNA/protein binding studies had not been performed.
Thus, in this work we focused on the following aims: (i) synthesis of new AHBA ligands and their cocrystals with ethylenediamine (en) and imidazole (im); (ii) preparation of new AgI and known water soluble CoII, CoII/III and CuII–AHBA complexes (Scheme 2);18 (iii) evaluation of the antimicrobial and antiproliferative activities all AHBA ligands and their cocrystals and CoII, CoII/III, CuII and AgI complexes; (iv) investigation of the binding mode of AHBAs and their cocrystals and complexes to DNA and BSA albumin, that should play a role in the pharmacokinetics and pharmacodynamics of such potential drugs.
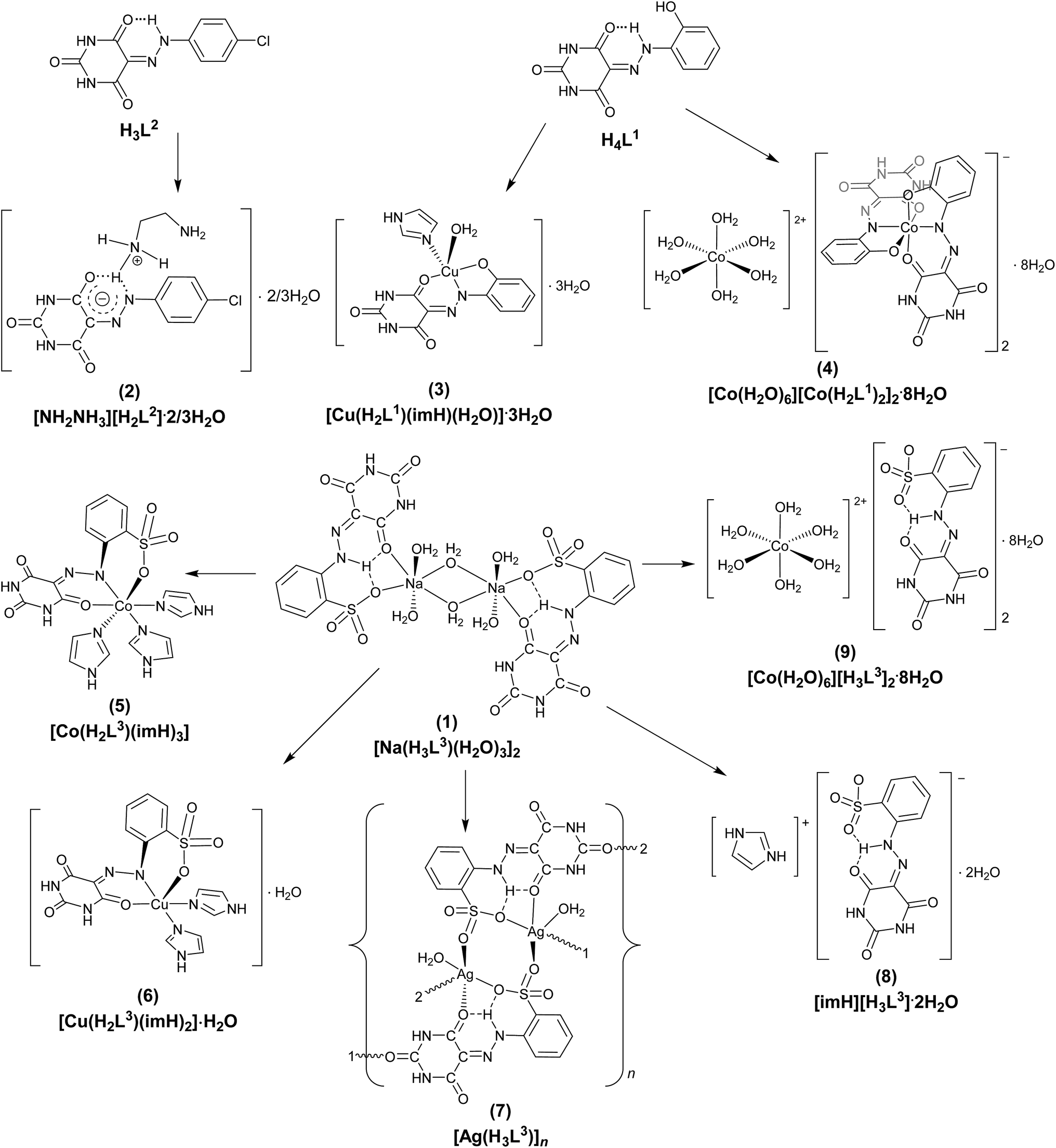 | ||
| Scheme 2 Schematic representations of H4L1, H3L2 and 1–9. | ||
Results and discussion
Synthesis and characterization of AHBA ligands their cocrystals and CoII/III, CuII and AgI complexes
Water soluble 5-(2-(2-hydroxyphenyl)hydrazono)pyrimidine-2,4,6(1H,3H,5H)-trione (H4L1), sodium salt of 2-(2-(2,4,6-trioxotetrahydropyrimidin-5(2H)-ylidene)hydrazinyl)benzene-sulfonic acid (H4L3), [Na(H3L3)(μ-H2O)(H2O)2]2 (1), [Cu(H2L1)(H2O)(im)]·3H2O (3), [Co(H2O)6][Co(H2L1)2]2·8H2O (4), [Co(H2L3)(im)3] (5), [Cu(H2L3)(im)2]·H2O (6) and [Co(H2O)6][H3L3]2·8H2O (9) (Scheme 2) were reported earlier by us,18 and hence will not be discussed.5-(2-(4-Chlorophenyl)hydrazono)pyrimidine-2,4,6(1H,3H,5H)-trione (H3L2) (Scheme 2) was prepared by a modified known18–22 aqueous diazotization of 4-chloroaniline and subsequent coupling with barbituric acid in basic medium. H3L2 is highly soluble in DMSO, DMF, water, methanol, acetone and acetonitrile. The 1H and 13C-NMR spectra of H3L2 support the hydrazone form and ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–N
N–N![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) – signal is observed at δ 14.03 in the 1H-NMR spectrum, while the 13C-NMR spectrum shows two separate singlets for the CO and CO⋯H of pyrimidine moiety at δ 159.76 and 161.92, respectively (Fig. 1S and 2S†). The IR spectrum of H3L2 shows ν(NH) vibrations at 3260, 3085 and 2813 cm−1, while ν(CO) at 1753 and 1653, ν(CO⋯H) at 1588 and ν(CN) at 1513 cm−1 (Fig. 3S†), supporting the existence of the H-bonded hydrazone structure in the solid state.
– signal is observed at δ 14.03 in the 1H-NMR spectrum, while the 13C-NMR spectrum shows two separate singlets for the CO and CO⋯H of pyrimidine moiety at δ 159.76 and 161.92, respectively (Fig. 1S and 2S†). The IR spectrum of H3L2 shows ν(NH) vibrations at 3260, 3085 and 2813 cm−1, while ν(CO) at 1753 and 1653, ν(CO⋯H) at 1588 and ν(CN) at 1513 cm−1 (Fig. 3S†), supporting the existence of the H-bonded hydrazone structure in the solid state.
Cocrystals of (Hen)(H2L2)·2/3H2O (2) were obtained from reaction of H3L2 with ethylenediamine (en) in methanol (Schemes 2 and 1S†). The IR spectrum of 2 displays ν(CO) at 1693 cm−1, value that is significantly shifted in relation to that (1753 cm−1) of the free ligand (Fig. 4S†). The 1H-NMR spectrum of 2 in DMSO-d6 solution at room temperature confirms the disappearance of the hydrazone proton (Fig. 5S and 6S†). The ESI mass spectrum of 2 dissolved in methanol shows the parent peak at m/z = 327.2 [C12H15ClN6O3 + H]+. Elemental analyses are consistent with the proposed formulation, which is also supported by X-ray crystallography (see below).
Another cocrystalinde compound, [(Him)(H3L3)]·2H2O (8), was isolated by treatment of 1 with imidazole (im) in the presence of HNO3 in methanol (Scheme 2). Whereas, the first AgI complex with an AHBA ligand, [Ag(H2O)(μ-H3L3)]n (7), is obtained in acidic medium (with addition of HNO3) from reaction of 1 with AgNO3 in acetone–water mixture (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) at room temperature (Scheme 2). Both compounds, 7 and 8, were characterized by elemental analysis, 1H and 13C NMR spectroscopies (Fig. 7S–12S†), ESI-MS, IR spectroscopy and single crystal X-ray diffraction. In the IR spectra of 7 and 8, the ν(CO) signal appears at 1689 and 1698 cm−1 (Fig. 13S and 14S†), respectively, values that are significantly shifted in relation to the corresponding signal of 1 (1677 cm−1, Fig. 15S†). The ESI mass spectra of 7 and 8 dissolved in CH3OH show the peaks at m/z = 839.1 [2{[Ag(H2O)(μ-H3L3)]n} − 2H2O + H]+ and 312.1 [H3L3 + H]+. In the 1H NMR spectra of 7 and 8, the hydrazone protons are observed at 14.78 and 14.79 ppm, respectively (Fig. 7S and 9S†). Elemental analyses are consistent with the proposed formulations which are also proved by X-ray analysis (see below). Both compounds are soluble in polar solvents such as water, methanol, DMF and DMSO.
:1, v/v) at room temperature (Scheme 2). Both compounds, 7 and 8, were characterized by elemental analysis, 1H and 13C NMR spectroscopies (Fig. 7S–12S†), ESI-MS, IR spectroscopy and single crystal X-ray diffraction. In the IR spectra of 7 and 8, the ν(CO) signal appears at 1689 and 1698 cm−1 (Fig. 13S and 14S†), respectively, values that are significantly shifted in relation to the corresponding signal of 1 (1677 cm−1, Fig. 15S†). The ESI mass spectra of 7 and 8 dissolved in CH3OH show the peaks at m/z = 839.1 [2{[Ag(H2O)(μ-H3L3)]n} − 2H2O + H]+ and 312.1 [H3L3 + H]+. In the 1H NMR spectra of 7 and 8, the hydrazone protons are observed at 14.78 and 14.79 ppm, respectively (Fig. 7S and 9S†). Elemental analyses are consistent with the proposed formulations which are also proved by X-ray analysis (see below). Both compounds are soluble in polar solvents such as water, methanol, DMF and DMSO.
X-ray diffraction analyses
Crystals of 2 and 8 suitable for X-ray diffraction analysis were obtained upon crystallization from methanol or water–acetone mixtures (1:4, v/v) (7). Representative plots of 2, 7 and 8 are depicted in Fig. 1. Crystallographic data and refinement parameters are given in Table 1S,† selected bond distances and angles in Table 2S.† H-contacts for 2, 7 and 8 are outlined in Table 3S† and depicted in Fig. 16S–19S†.
 | ||
| Fig. 1 X-ray molecular structures of 2, 7 and 8 with atom numbering schemes. In 2, only one hydrazine anion and one ethylenediammonium cation is shown. | ||
In 2 and 8 the (H2L2)− or (H3L3)− species cocrystallized with monoprotonated ethylene diamine (2) or imidazolium (8) cations, respectively, apart from two water molecules, and can be roughly planar (in 8) or present a considerable twisting (in 2) as measured by the angles between the least square plane of the heteronuclear ring (plane H) and the least square plane of the phenyl ring (plane P; Table 3S†).
Compound 2 crystallizes in the monoclinic space group P21/c with three monoprotonated ethylene diamines, three (H2L2)− anions and two water molecules. Due to deprotonation of hydrazone unit of H3L2 by en, its “resonance assisted hydrogen bond” (RAHB)7 system is converted into “charge assisted hydrogen bond” (CAHB)7 (Scheme 1S†), with medium-to-weak intermolecular hydrogen-bond contacts involving the (H2L2)− monoanion varying in the (donor⋯acceptor) range distances of 2.785(8)–3.296(8) Å (Table 3S†). Together with keto-O atoms, every hydrazo/azo group of (H2L2)− in 2 acts as acceptor from two Hen+ or from one of these cations and a molecule of water, to form two (or even three) bifurcated hydrogen bonds (Fig. 16S and Table 3S†). The nitrogen and oxygen atoms of pyrimidine trione moieties of (H2L2)− are further linked via intermolecular H-contacts giving rise to an intricate 3D network (Fig. 17S†).
The asymmetric unit of 7 contains one AgI cation, a (H3L3)− anion and a coordinated water molecule. The structure is a 1D polymer that runs along the crystallographic b direction with the metal cation adopting a distorted square pyramidal geometry (τ5 = 0.35)23 filled by the oxygen atom from water, two Osulfonyl-atoms and two Oketone-atoms from three adjacent symmetry generated hydrazone moieties (Fig. 18S†). An interesting feature of polymer 7 arises from the μ-OOSO bridging mode of the sulfonyl groups of the organic ligands which results in the generation of {Ag2S2O4} dimetallic cores (Fig. 19S†). These cores are then doubly bound by two Oketone-atoms from the pyrimidine trione moieties thus giving rise to an infinite 1D double chain. Therefore, this double chain comprises additional cavities formed by repeating 12-membered dimetallic –{AgOketoneCNCOketone}2–, and 10-membered monometallic –{AgOsulfonylSC2N2C2Oketone}– rings. In 7 the (H3L3)− anion preserves the strong RAHB linking the NH-moiety of the hydrazone unit (N1–H1N) in a three-centered interaction with coordinated Oketone- and Osulfonyl-atoms. The non-coordinated Osulfonyl-atom (O12) is involved in an intra-chain H-bond contact acting as acceptor towards HN(3) (donor site) of pyrimidine moiety.
The structure of 8 contains three-centred RAHB/CAHB interactions7 in the anionic hydrazone units involving the Oketone- and Osulfonyl-atoms, which relates to the synergistic mutual reinforcement of intramolecular hydrogen bonding due to π-electron delocalization. The typical double bond lengths of Cpyrimidine–Nhydrazone in the structure of 8 [1.326(4) Å] and the formation of intramolecular N–H⋯O RAHB contacts indicate that hydrazone is the preferred tautomeric form in these cases. The structure of 8 contains trapped water molecules, which interact among themselves and with the imidazolium and pyrimidine trione moiety via intermolecular hydrogen bonds. Such interactions, together with others involving the O- and N-atoms of pyrimidine trione rings (Table 3S and Fig. 20S†), give rise to intricate 3D supramolecular frameworks.
O, while the bands at 388–414 nm are mainly of n–π* type in NN.24–26 Similarly the sodium complex 1 shows analogous features, even if the band at the lower wavelength seems splitted into two very close absorptions.With respect to H4L1, in the spectra of its complexes 3 and 4, containing Cu(II) and Co(II/III) respectively, a new broad and very intense absorption band appears in the 420–480 nm range, which suggests n–π* and π–π* electronic transitions in the obtained compounds. Thus, new band can be attributed to a deprotonated form of the ligand moiety arising upon coordination to a metal centre and can be assigned to electronic transitions to bring about the conjugation between the metallocycles and the aromatic ring system.
The spectra of metal the complexes 5–9 are essentially similar to that of the ligand in the sodium salt 1, apart for the Co(II) and Cu(II) complexes 5 and 6, respectively, where a broadening of the band at lower wavelength is observed, due to an additional band partially obscured by the formed one. No d–d or CT transitions were observed in any of the CoII and CuII complexes and this may be due to the weakness of the bands at such low concentrations of the solutions (ca. 10−4 M) or because they are obscured by the intense bands.
 | ||
| Fig. 2 Absorption spectral traces of complex 4 buffer (10 mM, pH 7.2) after gradual addition of calf thymus DNA (from a = 0 to h = 24 μM). Inset shows the plot of [DNA]/|εa − εf| vs. [DNA] (eqn (1) in ESI†). | ||
As reported in Fig. 2, fixed amounts of complex 4 were titrated with increasing amount of ct-DNA (0–26 μM), resulting in an overall hyperchromism at the intraligand absorption bands in the 240–280 nm range of the complex. In general, the “hyperchromic effect” is related to corresponding changes of DNA on its conformation and structure, after coordination with metal complexes.29 Hypochromism results from contraction of DNA in the helix axis, while hyperchromism arises from the damage of DNA double helix structure.30,31 An “hyperchromic effect” has been observed for all the tested compounds, while a moderate red shift of 1–4 nm was observed only for complexes 5 and 7. The hyperchromic effect with red shift seems to suggest that complexes 5 and 7 bind to DNA by external contact possibly via electrostatic binding, as previously observed for other metal complexes.32
For complexes 1, 3, 4, 6, 8 and 9, or proligands H4L1 and H3L2, no shift of the absorption maximum was observed and this behaviour can be probably related to a different DNA binding propensity. To further evaluate the binding affinity of complexes and cocrystals with ct-DNA, the intrinsic binding constant has also been determined by using eqn (1) (ESI†), detecting the changes of absorbance for of different concentrations of ct-DNA. The values of the binding constant Kb, obtained from for compounds 1–9 are reported in Table 1 and can be resumed according to the following trend: 4 ≫ 2 > 7 > 3 ≈ H4L1 ≈ 8 ≈ 5 ≈ 9 > 6 ≈ H3L1 ≈ 1.
| Compound | BSA binding | BSA binding | ct-DNA binding | EB–ct-DNA binding | EB–ct-DNA binding | DAPI–ct-DNA binding |
|---|---|---|---|---|---|---|
| KSV × 104 M−1 | Kq × 1013 M−1 s−1 | Kb × 103 M−1 | KSV × 103 M−1 | Kapp × 105 M−1 | KSV × 103 M−1 | |
| 1 | 8.80(±0.36) | 0.88(±0.03) | 0.75(±0.24) | ND | ND | 18.65(±0.20) |
| 2 | 16.42(±0.40) | 1.64(±0.04) | 13.6(±0.27) | 4.95(±0.24) | 2.47 | 13.61(±0.78) |
| 3 | 10.48(±0.22) | 1.04(±0.02) | 1.76(±0.34) | 11.73(±0.12) | 5.86 | 9.45(±0.36) |
| 4 | 96.82(±0.40) | 9.68(±0.04) | 54.9(±0.10) | 28.54(±0.05) | 14.27 | 70.05(±2.51) |
| 5 | 26.31(±0.12) | 2.63(±0.02) | 1.14(±0.36) | 21.66(±0.10) | 10.83 | 13.47(±0.10) |
| 6 | 29.64(±0.85) | 2.96(±0.08) | 0.85(±0.21) | 5.42(±0.30) | 2.71 | 13.38(±0.21) |
| 7 | 17.28(±0.80) | 1.72(±0.08) | 3.33(±0.60) | 0.53(±0.02) | 0.26 | 8.55(±1.20) |
| 8 | 25.55(±0.30) | 2.55(±0.03) | 1.17(±0.44) | 1.56(±0.25) | 0.78 | 28.60(±0.06) |
| 9 | 16.50(±0.90) | 1.65(±0.09) | 1.12(±0.35) | 21.84(±0.07) | 1.09 | 21.90(±0.60) |
| H3L2 | 18.24(±1.76) | 1.82(±0.17) | 0.78(±0.21) | 0.88(±0.06) | 0.44 | 1.39(±0.21) |
| H4L1 | 16.46(±1.16) | 1.64(±0.11) | 1.36(±0.40) | 8.93(±0.61) | 4.46 | 27.33(±0.52) |
Fig. 3 reports the emission spectra of the EB–ct-DNA in the presence of increasing amounts of complex 4. The addition of 4 (different concentrations) to the EB-bound ct-DNA solution caused a reduction in emission intensity, indicating that complex 4 substitutes ct-DNA-bound EB and binds to ct-DNA with different affinity. In order to understand quantitatively the magnitude of the binding strength of complexes 1–9 and proligands H4L1 and H3L2 with ct-DNA, the quenching ability has been then analyzed by the Stern–Volmer equation and KSV values are shown in Table 1. From data on Table 1 a low correlation between Kb and KSV values was observed, probably due to the different sensitivity of the two techniques used. Additionally we have evaluated the binding constant Kapp (Table 1), and the values obtained for compounds 3, 4, 5, 9 and the proligand H4L1 suggest that they intercalated strongly to DNA. A similar mechanism is hypothesized for the other compounds and the free proligand that bind to DNA with lower affinity.
 | ||
| Fig. 3 Emission spectra of EB bound to ct-DNA in the presence of complex 4 with increasing concentration (from a = 0 to I = 39 μM); ([EB] = 5 μM, [ct-DNA] = 50 μM). Inset: Stern–Volmer plot of F0/F vs. [complex 4] for the titration of EB–ct-DNA system with complex 4. | ||
Fig. 23S (ESI†) reports the emission quenching spectra of EB–ct-DNA solutions in the presence of all complexes and proligands. Data from Table 1 suggest a stronger interaction of complexes 3, 4, 5 and 9 with EB–ct-DNA (4 > 5 ≈ 9 > 3 > H4L1), than the remaining compounds, showing a lower affinity for the EB–ct-DNA complex in the following order: 6 ≈ 2 > 8 > H3L2 > 7. Moreover, the high Kapp values obtained (about 105 M−1) suggest that our compounds intercalate strongly to DNA with similar mechanism, i.e. through the replacement of EB from DNA, but with different affinity.
As shown in Fig. 4, the addition of 4, with increasing concentration, to the DAPI–ct-DNA complex reduces its fluorescence intensity, thereby clearly suggesting that complex 4 can display DAPI to a considerable extent and can be able to bind to the minor groove of DNA. Results for all complexes and proligands are reported in Fig. 24S (ESI†). Of particular interest is the behaviour of complex 7 and the cocrystalline 2: in fact they do not decrease the fluorescence of DAPI–ct-DNA but produce an hyperchromic effect with an increase of fluorescence and a blue shift of the emission maximum. Compounds 2 and 7 interact with the system DAPI–ct-DNA with different affinity, but the presence of isosbestic points indicates an equilibrium state between the complexes and DAPI–ct-DNA (Fig. 24S†). The significant hyperchromic effects, as well as the moderate blue shift of the emission maximum, support the existence of a strong interaction between compounds 2 and 7 and the whole system DAPI–ct-DNA, without expulsion of DAPI molecules from the minor grove of DNA. A possible interaction between the metal compounds and the DAPI–ct-DNA system can be through external electrostatic interaction between the metal compounds and the DAPI–ct-DNA system. A similar situation has been recently found in the case of water-soluble tris(pyrazolyl) methanesulfonate silver(I) derivatives of N-methyl-1,3,5-triaza-7-hosphaadamantane salt.37
 | ||
| Fig. 4 Emission fluorescence spectra of DAPI–ct-DNA complex upon excitation at 338 nm in the presence of 4 with increasing concentration (from a = 0 to I = 36 μM). Experiments were performed in 10 mM phosphate buffer, pH 7.2. Inset: Stern–Volmer plot of F0/F vs. [complex 4] for the titration of DAPI–ct-DNA complex with complex 4. | ||
The Stern–Volmer quenching constants for the interaction of complexes and proligands with DAPI–ct-DNA were evaluated and reported in Table 1. The values of KSV show also in this case that complex 4 has the highest affinity towards DNA. The affinity of the tested compounds follow the order 4 ≫ 8 ≈ H4L1 > 9 > 1 while a lower activity was observed for the remaining species (2 ≈ 5 ≈ 6 > 3 ≈ 7 ≈ 9 > H3L2). It's worth to note that, after 4, the co-crystal 8 seems very able to display efficiently DAPI from the minor grove of DNA helix, in accordance with previous findings on several imidazolium salts.38
From all data of Table 1 we can conclude that complexes 3, 4, 5, 6, 7 and 8 display a very high affinity for DNA, but probably with different types of association, based on the value of their association constants. Interestingly, the Co(II) complex 4 was that with the highest activity which preferentially seems to bind to minor groove of DNA.
The highest affinity of the ionic complex 4 for both EB–ct-DNA and DAPI–ct-DNA, with respect to all other compounds, is likely related to its bulkiness, being composed by a cationic [Co(OH2)6]2+ unit and two anionic [Co(H2L1)2]− counterparts. Such very big system displays the highest ability, among the others, to replace EB from the major grove of DNA and also DAPI from the minor grove of DNA. Moreover, also the binding constant Kb of 4 is the highest among the others, and ca. 50 times higher than that exhibited by compound 9, containing the same cationic unit [Co(OH2)6]2+, but only an anionic (H3L3)− ligand. This means that there is a contribution of the anionic [Co(H2L1)2]− unit of compound 4 in its ability to bind DNA and to replace EB and DAPI from major and minor groves, respectively. Such contribution can be tentatively explained if we consider the possibility of ionic pairs formation such as {[Co(OH2)6][Co(H2L1)2]}+ for compound 4 and {[Co(OH2)6](H3L3)}+ for compound 9, which may behave as single positive systems able to interact with the negative backbone of phosphate groups in DNA through electrostatic interactions and then intercalation. Differently to the simple anion (H3L3)− in compound 9, the anionic unit [Co(H2L1)2]− in compound 4 contains a d6 low spin Co(III) center, which is able to strongly attract and concentrate the whole negative change on it, by depleting the electronic cloud from the periphery of the complex. Additionally, DNA double helix possesses many hydrogen-bonding sites positioned on the edges of the DNA bases, so that it's quite possible that the (H2L1)− functionalities could participate in H-bonding with the DNA base pairs and intercalate the DNA helix,39 even if the (H2L1)− ligands are part of an anionic complex.
 | ||
| Fig. 5 Emission spectra of BSA (15 × 10−6 M) in 20 mM phosphate buffer (pH = 7.4) and 25 °C. Excitation carried out at 285 nm. The arrow (with its direction) shows that the increasing concentration of complex 4 (from a = 0 to m = 30 μM) is accompanied by a quenching of BSA fluorescence intensity. Inset, Stern–Volmer plots of F0/F vs. [complex 4] for the titration of BSA with complex 4. | ||
The Stern–Volmer constants of binding to BSA for all complexes and proligands are reported in Table 1. Complex 4 is also in this case the most active complex towards the binding to BSA, with a Stern–Volmer constant about 3 times higher than those of the other complexes.
Also in this case there is a different ability in binding BSA by the ionic compounds 4 and 9 containing the same cationic unit [Co(OH2)6]2+ but two different anionic counterparts, [Co(H2L1)2]− and [(H2L3)]−. So that, even if we can imagine that the primary interaction with BSA protein chain could be with the cationic unit [Co(OH2)6]2+, possibly through replacement of some water molecule from a labile d7 Co(II) centre, we cannot exclude some steric contribution from the anionic [Co(H2L1)2]− unit. Beside this, compounds 5, 6 and 8 show essentially the same activity towards the BSA (Table 1), likely due to the hydrophobicity of substituents in the ligand and not to the type of metal in the complex. Whereas, compounds 2, 7 and 9 bind to BSA with moderate affinity, similar to that of proligands H4L1 and H3L2.
It is well known that there are two quenching mechanisms involved in quenching process, which are usually classified as dynamic quenching and static quenching. Dynamic quenching refers to a process where the fluorophore and the quencher come into contact during the lifetime of the excited state, whereas static quenching refers to fluorophore–quencher complex formation.45 According to the literature, for dynamic quenching the maximum scatter collision quenching constant of various quenchers with the biopolymer is 2.0 × 1010 M−1 s−146 and the fluorescence lifetime of the biopolymer is 10−8 s.47
Table 1 shows the values of KSV and Kq = KSV/τ0. The obtained values of Kq were larger than the limiting diffusion rate constant of the biomolecule (2.0 × 1010 M−1 s−1), which indicates that the fluorescence quenching is caused by a specific interaction between BSA and the different compounds tested. Therefore, the quenching mechanism mainly arises from the formation of BSA-complex rather than dynamic quenching. In conclusion, the static quenching is dominant in these system.48
Biological activity of proligands H4L1, H3L2, cocrystals 2, 8 and metal complexes 1, 3–7, 9
| Cell line (IC50 μM) | |||
|---|---|---|---|
| A375b | MDA-MB 231c | HCT116d | |
| a The IC50 value is the concentration of compound that affords a 50% reduction in cell growth (after 72 h of incubation). C.I. = confidence interval.b Human malignant melanoma cell line.c Human breast adenocarcinoma cell line.d Human colon carcinoma cell line.e The two-tailed P value is <0.0001, considered extremely significant (compound 7 vs. AgNO3).f The two-tailed P value is <0.0001, considered extremely significant (compound 7 vs. cisplatin).g The two-tailed P value is 0.0002, considered extremely significant (compound 7 vs. cisplatin). | |||
| H4L1 | 79.3 | >100 | >100 |
| 95% C.I. | 68.6–82.4 | ||
| H3L2 | >100 | >100 | >100 |
| 95% C.I. | |||
| 1 | >100 | >100 | >100 |
| 95% C.I. | |||
| 2 | 17.43 | 24.81 | 17.28 |
| 95% C.I. | 16.60–18.31 | 23.10–26.66 | 16.74–17.83 |
| 3 | >100 | >100 | >100 |
| 95% C.I. | |||
| 4 | >100 | >100 | >100 |
| 95% C.I. | |||
| 5 | >100 | >100 | >100 |
| 95% C.I. | |||
| 6 | >100 | >100 | >100 |
| 95% C.I. | |||
| 7 | 0.97e,g | 1.78e,f | 1.14e,f |
| 95% C.I. | 0.93–1.00 | 1.65–1.91 | 1.04–1.17 |
| 8 | >100 | >100 | >100 |
| 95% C.I. | |||
| 9 | >100 | >100 | >100 |
| 95% C.I. | |||
| AgNO3 | 2.73 | 11.20 | 6.01 |
| 95% C.I. | 2.37–2.92 | 9.90–12.67 | 5.66–6.38 |
| Cisplatin | 0.59 | 7.12 | 4.92 |
| 95% C.I. | 0.51–0.71 | 5.99–7.62 | 4.51–5.50 |
| Compound | S. aureus ATCC 29213 | E. faecalis ATCC 29212 | E. coli ATCC 25922 | P. aeruginosa ATCC 27853 | C. albicans ATCC 24433 |
|---|---|---|---|---|---|
| H4L1 | 6.0 ± 0 | 6.0 ± 0 | 6.0 ± 0 | 6.0 ± 0 | 6.0 ± 0 |
| H3L2 | 6.0 ± 0 | 6.0 ± 0 | 6.0 ± 0 | 6.0 ± 0 | 6.0 ± 0 |
| 1 | 6.0 ± 0 | 6.0 ± 0 | 6.0 ± 0 | 6.0 ± 0 | 6.0 ± 0 |
| 2 | 8.7 ± 0.2 | 6.5 ± 0.5 | 10.2 ± 0.2 | 11.0 ± 0.5 | 11.0 ± 1.0 |
| 3 | 6.0 ± 0 | 6.0 ± 0 | 6.0 ± 0 | 6.0 ± 0 | 6.0 ± 0 |
| 4 | 6.0 ± 0 | 6.0 ± 0 | 6.0 ± 0 | 6.0 ± 0 | 6.0 ± 0 |
| 5 | 6.0 ± 0 | 6.0 ± 0 | 6.0 ± 0 | 6.0 ± 0 | 6.0 ± 0 |
| 6 | 6.0 ± 0 | 6.0 ± 0 | 6.0 ± 0 | 6.0 ± 0 | 6.0 ± 0 |
| 7 | 10.5 ± 0.5 | 9.5 ± 0.5 | 11.0 ± 1.0 | 12.5 ± 0.5 | 15.5 ± 0.5 |
| 8 | 6.0 ± 0 | 6.0 ± 0 | 6.0 ± 0 | 6.0 ± 0 | 6.0 ± 0 |
| 9 | 6.0 ± 0 | 6.0 ± 0 | 6.0 ± 0 | 6.0 ± 0 | 6.0 ± 0 |
| Compound | S. aureus ATCC 29213 | E. faecalis ATCC 29212 | E. coli ATCC 25922 | P. aeruginosa ATCC 27853 | C. albicans ATCC 24433 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC | MIC | MBC | |
| 2 | 64–128 | 256 | 128 | 256 | 128 | 128 | 64 | 64 | 32 | 32 |
| 7 | 32 | 64 | 64 | 64 | 16 | 16 | 8 | 16 | 4 | 4 |
One of them (2) is the cocrystal of H3L2 with ethylenediamine (en), the second one (7) is the complex of 1 with silver. MIC values range from 32 to 128 for the complex 2 and from 4 to 64 μM in the case of complex 7. Both active complexes possess a bactericidal activity. Proligands H4L1 and H3L2 and complexes 2–6, 8 and 9 do not possess antimicrobial activity against the tested strains.
Conclusions
In the present work, for the new series of CoII, CoII,III, CuII and AgI complexes of AHBA and cocrystalline species with ethylenediamine and imidazole, we evaluated different biological properties such as anticancer and antimicrobial activity and DNA and BSA binding abilities. Among all the compounds screened, only the AgI complex (7) showed remarkable cytotoxic and antibacterial activity (higher of that of AgNO3) while a lower activity was found for the cocrystalline (2), and no activity was revealed by the other tested derivatives. They are both different to all other proligands and metal complexes, in fact they seem to afford external electrostatic interactions with the DAPI–ct-DNA system without a displacement of the dye. Although the CoII/III complex 4 was able to bind strongly with DNA, it seems however to be not active towards the growth of tumor cells and bacteria.Experimental
Materials and instrumentation
The 1H and 13C NMR spectra were recorded at room temperature on a Bruker Avance II + 300 (UltraShield™ Magnet) spectrometer operating at 300.130 and 75.468 MHz for proton and carbon-13, respectively. The chemical shifts are reported in ppm using tetramethylsilane as the internal reference. The infrared spectra (4000–400 cm−1) were recorded on a BIO-RAD FTS 3000MX instrument in KBr pellets. Carbon, hydrogen, and nitrogen elemental analyses were carried out by the Microanalytical Service of the Instituto Superior Técnico. Electrospray mass spectra (ESI-MS) were run with an ion-trap instrument (Varian 500-MS LC Ion Trap Mass Spectrometer) equipped with an electrospray ion source. For electrospray ionization, the drying gas and flow rate were optimized according to the particular sample with 35 p.s.i. nebulizer pressure. Scanning was performed from m/z 100 to 1200 in methanol solution. The compounds were observed in the negative or positive mode (capillary voltage = 80–105 V).Synthesis of H3L2
O), 1588 ν(CO⋯H), 1513 ν(CN) cm−1. 1H NMR (300.130 MHz) in DMSO-d6, internal TMS, δ (ppm): 7.48–7.63 (4H, Ar-H), 11.31 (s, 1H, N–H), 11.52 (s, 1H, N–H), 14.03 (s, 1H, N–H). 13C{1H} NMR (75.468 MHz, DMSO-d6). δ: 118.11 (Ar-Cl), 118.30 and 129.54 (4Ar-H), 129.70 (CN), 140.46 (Ar-NHN), 149.80 and 159.76 (CO), 161.92 (CO⋯H).2: yield, 284 mg, 84% (based on H3L2). It is soluble in water, methanol, DMF, DMSO and only slightly in acetonitrile, acetone and chloroform. Anal. calcd for C36H49Cl3N18O11 (Mr = 1016.25): C, 42.55; H, 4.86; N, 24.81; found: C, 42.27; H, 4.94; N, 24.88%. ESI-MS: m/z: 327.2 [C12H15ClN6O3 + H]+. IR (KBr): 3446 and 3355 ν(OH), 3157 and 3062 ν(NH), 1693 and 1655 ν(CO), 1579 ν(CN) cm−1. 1H NMR (300.130 MHz) in DMSO-d6, internal TMS, δ (ppm): 2.66 (4H, CH2), 4.68 (6H, NH3+), 7.38–7.49 (4H, Ar-H). 13C{1H} NMR (75.468 MHz, DMSO-d6). δ: 41.50 (2CH2), 116.75 (Ar-Cl) and (Ar-NHN), 120.13 (2Ar-H), 128.80 (2Ar-H), 129.10 (CN), 149.06, 151.85 and 161.13 (CO).
:4, v/v), then 200 μL of HNO3 (65% w/w) were added at room temperature. The addition of 338 mg (2 mmol) of AgNO3 together with 10 mL of distilled water resulted in a clear solution. The resulting solution was allowed to stand in darkness room at room temperature and yellow crystals of 7 were obtained after 1 day.7: yield, 236 mg, 54% (based on Ag). It is soluble in water, DMSO, methanol and DMF. Anal. calcd for C10H9AgN4O7S (Mr = 437.13): C, 27.48; H, 2.08; N, 12.82%. Found: C, 27.51; H, 2.09; N, 13.19%. IR (KBr): 3480 ν(OH), 3211 and 3067 ν(NH), 1736, 1689 and 1663 ν(CO), 1579 ν(CN) cm−1. ESI-MS: m/z: 839.1 [2Mr − 2H2O + H]+. 1H NMR (300.130 MHz) in DMSO-d6, internal TMS, δ (ppm): 7.20–7.77 (4H, Ar-H), 11.21 (s, 1H, N–H), 11.33 (s, 1H, N–H), 14.78 (s, 1H, N–H). 13C{1H} NMR (75.468 MHz, DMSO-d6). δ: 116.14, 118.16, 124.87 and 127.50 (Ar-H), 130.31 (Ar-NHN), 135.96 (CN), 138.16 (Ar-SO3Ag), 150.00 and 160.22 (CO), 160.62 (CO⋯H).
8: yield, 254 mg, 61% (based on im). It is soluble in water, methanol, DMF, DMSO and only slightly in acetonitrile, acetone and chloroform. Anal. calcd for C13H16N6O8S (Mr = 416.37): C, 37.50; H, 3.87; N, 20.18%. Found: C, 37.26; H, 3.62; N, 20.20%. IR (KBr): 3446 ν(OH), 3202, 3063 and 2862 ν(NH), 1739, 1698 and 1675 ν(CO), 1581 ν(CN) cm−1. ESI-MS: m/z: 312.1 [H3L3 + H]+. 1H NMR (300.130 MHz) in DMSO-d6, internal TMS, δ (ppm): 7.19–7.77 (4H, Ar-H and 2H, C–H im), 9.08 (s, 1H, C–H im), 11.20 (s, 1H, N–H), 11.33 (s, 1H, N–H), 14.21 (s, 1H, N–H im), 14.79 (s, 1H, N–H). 13C{1H} NMR (75.468 MHz, DMSO-d6). δ: 116.14, 118.16, 124.87 and 127.50 (Ar-H), 130.31 (Ar-NHN), 135.96 (CN), 138.16 (Ar-SO3−), 150.00 and 160.22 (CO), 160.62 (CO⋯H).
X-ray structure determinations
X-ray quality single crystals of the compounds were mounted in a nylon loop and measured at 150 K (7 and 8) or at room temperature (2). Intensity data were collected using a Bruker AXS-KAPPA APEX II diffractometer or a Bruker APEX-II PHOTON 100 diffractometer with graphite monochromated Mo-Kα (λ 0.71069) radiation. Data were collected using phi and omega scans of 0.5° per frame and a full sphere of data was obtained. Cell parameters were retrieved using Bruker SMART41 software and refined using Bruker SAINT49 on all the observed reflections. Absorption corrections were applied using SADABS.49 Structures were solved by direct methods by using the SHELXS package50 and refined with SHELXL-2014.50 Calculations were performed using the WinGX System-Version 1.80.03.51 The hydrogen atoms of water, hydrazine or ammonium groups were found in the difference Fourier map and the isotropic thermal parameters were set at 1.5 times the average thermal parameters of the belonging oxygen or nitrogen atoms, frequently with their distances restrained by using the DFIX and DANG commands. Coordinates of hydrogen atoms bonded to carbon atoms were included in the refinement using the riding-model approximation with the Uiso(H) defined as 1.2Ueq of the parent aromatic or methylene atoms. Least square refinements with anisotropic thermal motion parameters for all the non-hydrogen atoms and isotropic for the remaining atoms were employed. CCDC 1055318, 1055323 and 1055324 contain the supplementary crystallographic data for this paper.†| [DNA]/|εa − εf| = [DNA]/|εb − εf| + 1/Kb|εb − εf| | (1) |
| Fc = Fm × e(A1+A2)/2 | (2) |
| F0/Fc = 1 + kQτ0[C] = 1 + KSV[C] | (3) |
| I0/I = 1 + KSV[compound] | (4) |
The binding constant Kapp was obtained by the eqn (5):
| KEB[EB] = Kapp[compound] | (5) |
Acknowledgements
This work has been partially supported by the Foundation for Science and Technology (FCT), Portugal (UID/QUI/00100/2013 project). M. F. C. G. S. and A. J. L. P. acknowledge the Russian Science Foundation for support in the structural analysis and help in organizing the International Laboratory (grant 14-43-00017). The authors acknowledge the Portuguese NMR Network (IST-UTL Centre) for access to the NMR facility, and the IST Node of the Portuguese Network of mass-spectrometry (Dr Conceição Oliveira) for the ESI-MS measurements. The authors also acknowledge the financial support by the University of Camerino (Fondo di Ateneo per la Ricerca 2011–2012) and Nuova Simonelli Company.References
- C. J. Suckling, in Trends in Medicinal Chemistry, ed. H. van der Goot, G. Domany, L. Pallos and H. Timmerman, Elsevier, Amsterdam, 1989, p. 805 Search PubMed.
- E. Maquoi, N. E. Sounni, L. Devy, F. Olivier, F. Frankenne, H.-W. Krell, F. Grams, J.-M. Foidart and A. Noel, Clin. Cancer Res., 2004, 10, 4038–4047 CrossRef CAS PubMed.
- H.-J. Breyholz, S. Wagner, A. Faust, B. Riemann, C. Hoeltke, S. Hermann, O. Schober, M. Schafersa and K. Kopka, ChemMedChem, 2010, 5, 777–789 CrossRef CAS PubMed.
- P. Bartsch, D. Cataldo, R. Endele, B. Evrard, J.-M. Foidart, H.-W. Krell and G. Zimmermann, Int. Appl. No. PCT EP, 003348, 2005.
- F. Grams, H. Brandstetter, S. D'Alo, D. Geppert, H.-W. Krell, H. Leinert, V. Livi, E. Menta, A. Oliva and G. Zimmermann, Biol. Chem., 2001, 382, 1277–1285 CrossRef CAS PubMed.
- K. T. Mahmudov, M. N. Kopylovich, A. M. Maharramov, M. M. Kurbanova, A. V. Gurbanov and A. J. L. Pombeiro, Coord. Chem. Rev., 2014, 265, 1–37 CrossRef CAS.
- K. T. Mahmudov, M. N. Kopylovich and A. J. L. Pombeiro, Coord. Chem. Rev., 2013, 257, 1244–1281 CrossRef CAS.
- K. T. Mahmudov, M. F. C. Guedes da Silva, M. Glucini, M. Renzi, K. C. P. Gabriel, M. N. Kopylovich, M. Sutradhar, F. Marchetti, C. Pettinari, S. Zamponi and A. J. L. Pombeiro, Inorg. Chem. Commun., 2012, 22, 187–189 CrossRef CAS.
- C. Saravanan, S. Easwaramoorthi and L. Wang, Dalton Trans., 2014, 43, 5151–5157 RSC.
- M. Klikar, F. Bureš, O. Pytela, T. Mikysek, Z. Padělková, A. Barsella, K. Dorkenoo and S. Achelle, New J. Chem., 2013, 37, 4230–4240 RSC.
- M. Szostak, B. Sautier and D. J. Procter, Chem. Commun., 2014, 50, 2518–2521 RSC.
- H. D. Williams, N. L. Trevaskis, S. A. Charman, R. M. Shanker, W. N. Charman, C. W. Pouton and C. J. H. Porte, Pharmacol. Rev., 2013, 65, 315–499 CrossRef PubMed.
- C. A. Lipinski, Curr. Drug Discovery Technol., 2001, 4, 17–19 Search PubMed.
- C. A. Lipinski, Am. Pharm. Rev., 2002, 5, 82–85 Search PubMed.
- E. M. Merisko-Liversidge and G. G. Liversidge, Toxicol. Pathol., 2008, 36, 43–48 CrossRef CAS PubMed.
- L. S. Reddy, N. J. Babu and A. Nangia, Chem. Commun., 2006, 1369–1371 RSC.
- R. V. Prasad, M. G. Rakesh, R. M. Jyotsna, S. T. Mangesh, P. S. Anita and P. K. Mayur, Int. J. Pharm., Chem. Biol. Sci., 2012, 1, 725–736 Search PubMed.
- J. Palmucci, K. T. Mahmudov, M. F. C. Guedes da Silva, L. M. D. R. S. Martins, F. Marchetti, C. Pettinari and A. J. L. Pombeiro, RCS Adv., 2015, 5, 84142–84152 CAS.
- A. M. Maharramov, R. A. Aliyeva, I. A. Aliyev, F. G. Pashaev, A. G. Gasanov, S. I. Azimova, R. K. Askerov, A. V. Kurbanov and K. T. Mahmudov, Dyes Pigm., 2010, 85, 1–6 CrossRef CAS.
- K. T. Mahmudov, A. M. Maharramov, R. A. Aliyeva, I. A. Aliyev, R. K. Askerov, R. Batmaz, M. N. Kopylovich and A. J. L. Pombeiro, J. Photochem. Photobiol., A, 2011, 219, 159–165 CrossRef CAS.
- S. R. Gadjieva, T. M. Mursalov, K. T. Mahmudov and F. M. Chyragov, Russ. J. Coord. Chem., 2006, 32, 304–308 CrossRef.
- K. T. Mahmudov, R. A. Aliyeva, S. R. Gadjieva and F. M. Chyragov, J. Anal. Chem., 2008, 63, 435–438 CrossRef.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- Y. Gülseven, E. Taşal, İ. Sıdır, T. Güngör, H. Berber and C. Öğretir, Int. J. Hydrogen Energy, 2009, 34, 5255–5259 CrossRef.
- A. Airinei, E. Rusu and D. Dorohoi, Spectrosc. Lett., 2001, 34, 65–74 CrossRef CAS.
- İ. Sıdır, E. Taşal, Y. Gülseven, T. Güngör, H. Berber and C. Öğretir, Int. J. Hydrogen Energy, 2009, 34, 5267–5273 CrossRef.
- R. S. Kumar, K. Sasikala and S. Arunachalam, J. Inorg. Biochem., 2008, 102, 234–241 CrossRef CAS PubMed.
- Z.-C. Liu, B.-D. Wang, B. Li, Q. Wang, Z.-Y. Yang, T.-R. Li and Y. Li, Eur. J. Med. Chem., 2010, 45, 5353–5361 CrossRef CAS PubMed.
- K. Akdi, R. A. Vilaplana, S. Kamah and F. González-Vílchez, J. Inorg. Biochem., 2005, 99, 1360–1368 CrossRef CAS PubMed.
- Q. Li, P. Yang, H. Wang and M. Guo, J. Inorg. Biochem., 1996, 64, 181–195 CrossRef CAS PubMed.
- S. Shi, J. Liu, J. Li, K.-C. Zheng, X.-M. Huang, C.-P. Tan, L.-M. Chen and L.-N. Ji, J. Inorg. Biochem., 2006, 100, 385–395 CrossRef CAS PubMed.
- (a) S. Tabassum, M. Afzal and F. Arjmand, J. Photochem. Photobiol., B, 2012, 115, 63–72 CrossRef CAS PubMed; (b) S. Tabassum, M. Zaki, M. Afzal and F. Arjmand, Eur. J. Med. Chem., 2014, 74, 509–523 CrossRef CAS PubMed.
- F. J. Meyer-Almes and D. Porschke, Biochemistry, 1993, 32, 4246–4253 CrossRef CAS PubMed.
- J. B. LePecq and C. Paoletti, J. Mol. Biol., 1967, 27, 87–106 CrossRef CAS PubMed.
- R. F. Pasternack, M. Cacca, B. Keogh, T. A. Stephenson, A. P. Williams and F. J. Gibbs, J. Am. Chem. Soc., 1991, 113, 6835–6840 CrossRef CAS.
- E. N. Zaitsev and S. C. Kowalczykowski, Nucleic Acids Res., 1998, 26, 650–654 CrossRef CAS PubMed.
- P. Smoleński, C. Pettinari, F. Marchetti, M. F. C. Guedes da Silva, G. Lupidi, G. V. Badillo Pazmay, D. Petrelli, L. A. Vitali and A. J. L. Pombeiro, Inorg. Chem., 2015, 54, 434–440 CrossRef PubMed.
- A. Pabbathi and A. Samanta, J. Phys. Chem. B, 2015, 119, 11099–11105 CrossRef CAS PubMed.
- M. Shamsi, S. Yadav and F. Arjmand, J. Photochem. Photobiol., B, 2014, 136, 1–11 CrossRef CAS PubMed.
- J. Lu, A. J. Stewart, P. J. Sadler, T. J. Pinheiro and C. A. Blindauer, Biochem. Soc. Trans., 2008, 36, 1317–1321 CrossRef CAS PubMed.
- A. M. Merlot, D. S. Kalinowski and D. R. Richardson, Front. Physiol., 2014, 12, 1–7 Search PubMed , ID 299.
- O. K. Abou-Zied and O. I. K. Al-Shihi, J. Am. Chem. Soc., 2008, 130, 10793–10801 CrossRef CAS PubMed.
- D. Senthil Raja, N. S. P. Bhuvanesh and K. Natarajan, Inorg. Chem., 2011, 50, 12852–12866 CrossRef PubMed.
- M. Tan, W. Liang, X. Luo and Y. Y. Gu, J. Chem., 2013, 1–6 CrossRef , ID 308054.
- J. Jayabharathi, K. Jayamoorthy, V. Thanikachalam and R. Sathishkumar, Spectrochim. Acta, Part A, 2013, 108, 146–150 CrossRef CAS PubMed.
- (a) X. Zhang, L. Li, Z. Xu, Z. Liang, J. Su, J. Huang and B. Li, PLoS One, 2013, 8, e59106 CAS; (b) H. Cao, D. Wu, H. Wang and M. Xu, Spectrochim. Acta, Part A, 2009, 73, 972–975 CrossRef PubMed.
- I. Timtcheva, V. Maximova, T. Deligeorgiev, N. Gadjev, K. H. Drexhage and I. Petkova, J. Photochem. Photobiol., B, 2000, 58, 130–135 CrossRef CAS.
- W. R. Ware, J. Phys. Chem., 1962, 66, 455–458 CrossRef CAS.
- Bruker, APEX2 & SAINT, AXS Inc., Madison, WI, 2004 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- S. Tabassum, S. Amir, F. Arjmand, C. Pettinari, F. Marchetti, N. Masciocchi, G. Lupidi and R. Pettinari, Eur. J. Med. Chem., 2013, 60, 216–232 CrossRef CAS PubMed.
- A. Wolfe, G. H. Shimer and T. Meehan, Biochemistry, 1987, 26, 6392–6396 CrossRef CAS PubMed.
- A. M. Pyle, J. P. Rehmann, R. Meshoyrer, C. V. Kumar, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1989, 111, 3051–3058 CrossRef CAS.
- O. Stern and M. Volmer, Z. Med. Phys., 1919, 20, 183–188 CAS.
- M. Lee, A. L. Rhodes, M. D. Wyatt, S. Forrow and J. A. Hartley, Biochemistry, 1993, 32, 4237–4245 CrossRef CAS PubMed.
- N. Shahabadi, M. Maghsudi, M. Mahdavi and M. Pourfoulad, DNA Cell Biol., 2012, 31, 122–127 CrossRef CAS PubMed.
- L. Quassinti, F. Maggi, L. Barboni, M. Ricciutelli, M. Cortese, F. Papa, C. Garulli, C. Kalogris, S. Vittori and M. Bramucci, Fitoterapia, 2014, 97, 133–141 CrossRef CAS PubMed.
- CLSI, Performance standards for antimicrobial susceptibility testing, 19th informational supplement M100-S19, Clinical and Laboratory Standards Institute, Wayne, PA, 2009, p. 20 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystal data, experimental parameters and selected details of the refinement calculations (for 2 and 8), electronic absorption and emission fluorescence spectra of compounds 1–9. CCDC 1055318, 1055323 and 1055324 contain the supplementary crystallographic data for compounds 2, 7 and 8, respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra20157h |
| This journal is © The Royal Society of Chemistry 2016 |