
Antileishmanial activity of sp2-iminosugar derivatives†
Elena M. Sánchez-Fernández‡
a,
Verónica Gómez-Pérez‡b,
Raquel García-Hernándezb,
José Manuel García Fernándezc,
Gabriela B. Platad,
José M. Padrónd,
Carmen Ortiz Mellet§
*a,
Santiago Castanys§*b and
Francisco Gamarro§*b
aDepartamento de Química Orgánica, Facultad de Química, Universidad de Sevilla, Apartado 553, E-41071, Spain. E-mail: mellet@us.es
bInstituto de Parasitología y Biomedicina “López-Neyra”, IPBLN-CSIC, Parque Tecnológico de Ciencias de la Salud, 18016-Granada, Spain
cInstituto de Investigaciones Químicas (IIQ), CSIC – Universidad de Sevilla, Avda. Américo Vespucio 49, 41092, Sevilla, Spain
dBioLab, Instituto Universitario de Bio-Orgánica “Antonio González”, Centro de Investigaciones Biomédicas de Canarias, Universidad de La Laguna, 38206, La Laguna, Spain
First published on 17th February 2015
Abstract
A series of sp2-iminosugar-type glycomimetics bearing S-linked pseudoglycoside substituents (sulfide, sulfoxide and sulfone derivatives) has been synthesized and evaluated as new potential drugs against the protozoan parasite Leishmania, responsible of leishmaniasis, the second most relevant parasitic disease after malaria. All the prepared compounds share a bicyclic 5N,6O-oxomethylidenenojirimycin glycone-like moiety bearing a substitution pattern of configurational complementarity with the natural α-glucosides and incorporate either an n-octyl or n-dodecyl aglycone-like substituent. Not surprisingly, they behaved as potent to moderate competitive inhibitors of α-glucosidase (inhibition constants, Ki, in the range 1.3 to 447 μM). Evaluation of the antileishmanial activity indicated that the dodecyl pseudoglycosides present a significant antiparasitic activity in intracellular amastigotes of Leishmania donovani, the clinically relevant form of the parasite. The antileishmanial effect seems to be associated with the anticancer and proapoptotic activity of the glycomimetics, but not with the α-glucosidase inhibitory efficiency. The (SS)-configured dodecylsulfoxide derivative 4, exhibiting the most favourable activity/toxicity profile, was further assayed in combination treatment with miltefosine, the first oral antileishmanial drug, using the fixed ratio isobologram method. The interaction between derivative 4 and 0.1, 0.2 and 0.3 μM miltefosine was classified as synergistic, showing combination indices of 0.78, 0.76 and 0.80, respectively. Additionally, a miltefosine resistant Leishmania line and the wild-type strain showed similar susceptibility to derivative 4. The results illustrate the potential of sp2-iminosugar pseudoglycosides as promising prototypes for the development of new therapeutic strategies for leishmaniasis.
Introduction
Leishmaniasis is a broad spectrum disease caused by protozoan parasites of the genus Leishmania, which are transmitted by the bite of infected sandflies. It is one of the world's most neglected diseases affecting 12 million people in 98 countries, with 350 million people considered at risk of infection and 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths per year.1 Leishmania donovani is responsible for visceral leishmaniasis (VL) in the Indian subcontinent and East Africa, a disease that is lethal in the absence of treatment. Current leishmaniasis treatment relies exclusively on chemotherapy, such as pentavalent antimonials, amphotericin B, miltefosine, and paromomycin. These first-line drugs have a limited efficacy due to growing resistance, frequent side effects and the high cost of treatment.2 Therefore, the World Health Organization (WHO) has recommended combination treatment in order to increase the effective life of the available medicines, reducing the treatment duration and cost and the probability of selection of drug-resistant parasites.3 Miltefosine is an alkylphosphocholine originally developed as an anticancer drug that has become the first oral drug to treat leishmaniasis.4 Miltefosine monotherapy regimen is well tolerated, except for mild gastrointestinal side effects, although it is potentially teratogenic. Furthermore, experimental resistance to miltefosine is very easily achieved,5 suggesting the need to introduce new therapeutic strategies to prevent treatment failure. Several miltefosine-containing combined treatments for VL have been conducted with favourable results in India,6 and others are currently being explored in multiple controlled clinical trials in East Africa.7 These results strongly suggest that the success of the on going efforts against leishmaniasis will critically depend on the identification of new active molecules that could broaden the current multidrug formulation options.
000 deaths per year.1 Leishmania donovani is responsible for visceral leishmaniasis (VL) in the Indian subcontinent and East Africa, a disease that is lethal in the absence of treatment. Current leishmaniasis treatment relies exclusively on chemotherapy, such as pentavalent antimonials, amphotericin B, miltefosine, and paromomycin. These first-line drugs have a limited efficacy due to growing resistance, frequent side effects and the high cost of treatment.2 Therefore, the World Health Organization (WHO) has recommended combination treatment in order to increase the effective life of the available medicines, reducing the treatment duration and cost and the probability of selection of drug-resistant parasites.3 Miltefosine is an alkylphosphocholine originally developed as an anticancer drug that has become the first oral drug to treat leishmaniasis.4 Miltefosine monotherapy regimen is well tolerated, except for mild gastrointestinal side effects, although it is potentially teratogenic. Furthermore, experimental resistance to miltefosine is very easily achieved,5 suggesting the need to introduce new therapeutic strategies to prevent treatment failure. Several miltefosine-containing combined treatments for VL have been conducted with favourable results in India,6 and others are currently being explored in multiple controlled clinical trials in East Africa.7 These results strongly suggest that the success of the on going efforts against leishmaniasis will critically depend on the identification of new active molecules that could broaden the current multidrug formulation options.
Iminosugars,8 nitrogen-in-the-ring carbohydrate mimics (glycomimetics), have been proposed as potential candidates for the development of new antiparasite drugs.9 These natural or synthetic polyhydroxylated alkaloids can interact with a range of carbohydrate processing enzymes such as glycosyltransferases, glycosidases and nucleoside-processing enzymes, thereby interfering in many biological processes of medicinal interest.10 Thus, iminosugar derivatives such as 1-deoxynojirimycin (DNJ) or castanospermine (CS) have been described as immunosuppressive,11 antitumor12 and antiviral agents.13 Recently, Ruhela et al.14 have reported a new family of bicyclic iminosugars behaving as inhibitors of elongating α-D-mannosyl phosphate transferase of microsomal membranes of L. donovani, suggesting that these glycomimetics could be also considered as promising antileishmanial drugs. The broad range of potential activities of iminosugars represents however a limitation for their clinical application, emphasizing the need for developing more selective leads.
Replacement of the endocyclic amine-type nitrogen atom characteristic of classical iminosugars by a sp2-hybridized pseudoamide-type nitrogen (guanidine, urea, carbamate, isourea, or their thio-analogues) has been shown to afford a new family of glycomimetics (sp2-iminosugars) with unprecedented abilities to discriminate between different glycosidase isoenzymes.15 Several sp2-iminosugars are currently under investigation as pharmacological chaperones for lysosomal storage disorders, including Gaucher,16 Fabry17 and GM1 gangliosidosis diseases.18 Interestingly, compounds with pseudoglycoside structure19 have been found to exhibit antitumor activity, which was ascribed to their ability to interfere with N-glycoprotein biosynthesis by inhibiting the neutral endoplasmic reticulum α-glucosidase. The S-linked octyl glycoside bicyclic nojirimycin analogue 1 was particularly efficient at this respect, being able to arrest the cell cycle and induce apoptosis in breast cancer cell lines without affecting normal cells.20 Since several compounds with antileishmanial activity have been shown to act through programmed cell death mechanisms,21 assaying the potential of sp2-iminosugar S-pseudoglycosides for the treatment of leishmaniasis seemed intriguing. Following our efforts in this field, here we present the synthesis of new sp2-iminosugar thioglycoside- (2), glycosyl sulfoxide- (3–6) and glycosyl sulfone-type (7 and 8) derivatives (Fig. 1). The compounds were first assayed for their inhibitory activity towards a panel of commercial glycosidases. Next, the antileishmanial activity of derivatives 1–8, was evaluated against promastigotes, axenic and intracellular amastigotes of L. donovani. The known19a N- and C-pseudoglycoside derivatives 9 and 10 were also included in this study to check the effect of the nature of the glycosidic linkage in the biological activity. Cytotoxicity against the human monocytic cell line THP-1 and the fibroblast cell line MRC-5 was also assessed in order to exclude those molecules showing unfavourable toxicological profile for further development.
 | ||
| Fig. 1 Chemical structure of sp2-iminosugars evaluated in this study. | ||
Results and discussion
Synthesis
The octyl- and dodecyl-thioglycoside sp2-iminosugar derivatives 1 and 2 were readily synthesized from the per-O-acetylated bicyclic nojirimycin derivative 11 (ref. 22) by reaction with commercial 1-octanethiol or 1-dodecanethiol in the presence of boron trifluoride etherate (BF3·OEt2)23 at 0 °C and conventional deacetylation of the resulting adducts (Scheme 1; overall yield 85–95%). The stereochemical outcome of this reaction is remarkable, affording in both cases the α-anomer (1R) 12 or 13, respectively, as the major diastereomer (α:β ratio 20:1), in spite of the participating character of the acetyl group vicinal to the pseudoanomeric position. This result underlines the utmost influence of the anomeric effect in the reactivity and stability of sp2-iminosugars, favoring the axial orientation of pseudoanomeric substituents. Compounds 12 and 13 can satisfy this requirement in the 4C1 chair conformation, whereas the β-anomers (1S) 12β or 13β have to adopt a skew-boat conformation at the six-membered ring, a less favourable situation, to fulfill the anomeric effect. Compound 13β was isolated in sufficient amount to allow subsequent conventional deacetylation. The fully unprotected compound 2β adopted instead the 4C1 conformation in water solution, with the S-dodecyl substituent in equatorial orientation, probably due to the expected weakening of the electrostatic contribution to the anomeric effect in polar solvents.
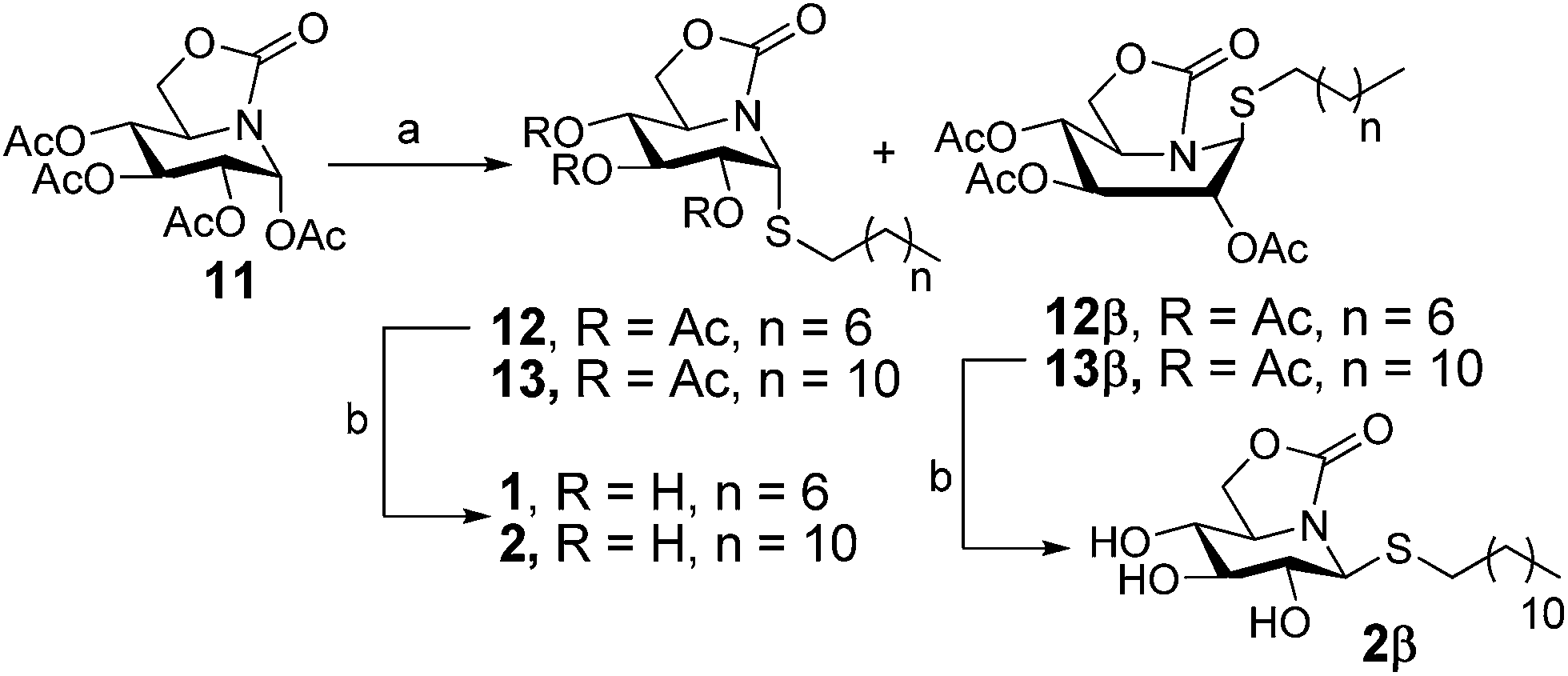 | ||
| Scheme 1 Synthesis of pseudoalkylthioglycoside iminosugar derivatives 1 and 2. Reagents and conditions: (a) RSH, BF3·OEt2, DCM, 0 °C to r.t.; (b) NaOMe (1 M), MeOH, r.t. | ||
Oxidation of the acetylated α-pseudoglycosyl sulfides 12 and 13 by treatment with one equivalent of m-chloroperoxybenzoic acid (mCPBA) at 0 °C for 10 min (ref. 24) afforded the corresponding acetylated glycosyl sulfoxides as 1:1 mixtures of the (SS) and (RS) diastereomers at the chiral S-atom (14 and 15 from 12; 16 and 17 from 13, respectively). The optically pure diastereomers could be separated by column chromatography and further deacetylated to afford the target fully unprotected (SS) and (RS) octyl- (3 and 5) and dodecyl- (4 and 6) sulfoxides. When oxidation of the acetylated sulfides 12 and 13 was effected with an excess of mCPBA, the peracetylated α-configured sulfones (18 and 19) could be isolated in 78–90% yield. Deprotection by conventional deacetylation resulted in the unprotected sulfones 7 and 8 (Scheme 2).
 | ||
| Scheme 2 Synthesis of sulfinyl and sulfonyl iminosugar derivatives 3–6 and 7–8. Reagents and conditions: (a) MCPBA (1 equiv.), DCM, 0 °C, 10 min, 69% (global yield for the octylsulfinyl derivatives), 74% (global yield for the dodecylsulfinyl derivatives); (b) NaOMe (1 M), MeOH, r.t., 15 min, 85–93%; (c) NaOMe (1 M), MeOH, r.t., 30 min, 87–88%. (d) MCPBA (2 equiv.), DCM, 0 °C, 10 min, 78–90%; (e) NaOMe (1 M), MeOH, r.t., 20 min, 83–84%. | ||
It is well known the importance of chirality in drugs, hence the need of determining the stereochemistry of the chiral sulfur in drugs bearing sulfoxides as they may display different chemical and pharmacological behaviour.25 Although no single crystals of the diastereomeric sulfoxides 3 and 4 or 5 and 6 suitable for X-ray diffraction could be obtained, some diagnostic chemical shift differences in the respective 1H NMR spectra allowed the tentative assignment of their absolute configuration. Notably, the H-5 resonance was deshielded by 0.2 ppm in derivatives 3 and 4 as compared with 5 and 6. Literature data on alkyl α-D-glycosyl sulfoxides support then the (SS) assignment for the first ones, where H-5 and the sulfoxide oxygen would be located in close proximity in the more favourable exoanomeric-type conformation, that is, with C-2 in the ring and the exocyclic methylene carbon in anti disposition.26a The lower magnetic nonequivalence (Δδ) of the methylene protons vicinal to the chiral sulfur atom (SOCH2) for 3 and 4 (35 Hz) as compared with 5 and 6 (70 Hz) is also in agreement with the NMR properties reported in the literature for (SS) and (RS) alkyl α-D-glycosyl sulfoxides, respectively.26b
Glycosidase inhibitory activity
Previous studies on sp2-iminosugars pointed to a relationship between their ability to inhibit neutral α-glucosidase and their proapoptotic activity. In order to ascertain if α-glucosidase inhibition can be used as a preliminary criterion to select candidates against leishmaniasis, and also to discard broad range glycosidase inhibitors with potential secondary effects, we first evaluated the inhibitory activity of compounds 1–8 against a panel of commercial glycosidases. The corresponding data are summarized in Table 1. None of them showed inhibition towards α-mannosidase (jack beans), β-mannosidase (Helix pomatia), α-galactosidase (green coffee) or β-galactosidase (E. coli), in agreement with their D-gluco configurational pattern. As expected, the pseudoglycosyl sulfides 1 and 2 exhibited strong inhibitory activity against the neutral α-glucosidase II (yeast), with Ki values in the low μM range (1.3–3.4 μM), regardless of the length of the aliphatic chain. However, their oxidized analogues bearing the sulfinyl chain displayed remarkable differences not only in activity but also in selectivity depending on the configuration of the new stereogenic centre generated after oxidation. Thus, the (RS)-octylsulfoxide 5 (Ki = 14.3 μM) was a 3-fold more potent inhibitor against this enzyme than 3 (Ki = 44 μM), which on the contrary was a stronger inhibitor of β-glucosidase (Ki = 37 and 214 μM for 3 and 5, respectively). Somehow surprisingly, the inhibitory activity dramatically decreased for the dodecyl sulfoxides 4 and 6 (Ki values against α-glucosidase 272 and 447 μM, respectively), whereas both the octyl and dodecyl sulfone derivatives 7 and 8 behaved as strong inhibitors of this enzyme.Antileishmanial activity and cellular toxicity of the novel iminosugar pseudoglycoside inhibitors
Antileishmanial activity of the S-pseudoglycoside sp2-iminosugar compounds 1–8 has been evaluated against promastigotes, axenic amastigotes and intracellular amastigotes of L. donovani, using a MTT-based assay, resazurin or luciferin assay, respectively. The previously reported 1-octylamino (9) and 1-C-octyl (10) derivatives (Fig. 1) were also included in this study. Leishmania has two major life cycle stages: promastigotes, which are easily cultured in suspension, and intracellular amastigotes, which are more difficult to maintain in vitro since they require macrophages as host cells with highly acidic intracellular environment. Considered as an intermediate form, axenic amastigotes are adapted to grow in a medium that mimics the intracellular conditions of macrophages. Cell viability was evaluated by determining the concentration of compound required to inhibit the growth of parasites by 50% (EC50). Neither of the compounds assayed presented significant activity against the extracellular promastigote forms (Table 2); however, compounds with the longer dodecyl aliphatic chain (2, 4, 6, 8) showed moderate antileishmanial activity against axenic and intracellular amastigotes, suggesting that the activity of this set of compounds relies specifically on the clinically relevant form of the parasite (Table 2). Except for sulfide 2, these compounds were more efficient against intracellular amastigotes as compared with axenic amastigotes, which points to some additional host cell-mediated effector mechanisms implied in intracellular parasite killing by the sulfoxide (4 and 6) and the sulfone (8) pseudoglycosides.27 A similar scenario has been described for miltefosine activity,28 highlighting the interest of these sp2-iminosugar derivatives as promising antileishmanial candidates for further drug development.| Compound | Promastigotes L. donovani Dd8 EC50 μM | Axenic amastigotes L. donovani HU3 EC50 μM | Intracellular amastigotes Dd8 EC50 μM, [SI]b | THP-1 EC50 μM | MRC-5 EC50 μM |
|---|---|---|---|---|---|
| a Parasites were grown as described in the Experimental section for 72 h at 28 °C (promastigotes) or 37 °C (axenic and intracellular amastigotes) in the presence of increasing concentrations of compounds. THP-1 and MRC-5 cells were grown as described in the Experimental section for 72 h at 37 °C, in the presence of increasing concentrations of compounds. Cell viability was determined using an MTT-based assay (promastigotes), resazurin assay (axenic amastigotes) or luciferase assay (intracellular amastigotes). Miltefosine was used as the reference antileishmanial agent. Data are means of EC50 ± SD from three independent experiments.b Selectivity indices [SI] were calculated by dividing the EC50 values for MRC-5 cells by that for intracellular amastigotes. | |||||
| 1 | >100 | >100 | 73.31 ± 1.97 [0.79] | 122.33 ± 5.79 | 58.14 ± 12.62 |
| 2 | 87.79 ± 3.47 | 14.52 ± 1.03 | 20.48 ± 3.59 [2.10] | 70.46 ± 3.75 | 43.07 ± 9.83 |
| 3 | >100 | >100 | >100 | 218.74 ± 0.45 | 175.54 ± 14.79 |
| 4 | >100 | 24.38 ± 1.68 | 10.80 ± 0.27 [8.74] | 118.83 ± 3.37 | 94.35 ± 20.93 |
| 5 | >100 | >100 | >100 | 161.79 ± 20.27 | 190.10 ± 0.14 |
| 6 | >100 | 60.52 ± 17.35 | 33.29 ± 7.36 [3.59] | 68.41 ± 6.79 | 119.40 ± 17.91 |
| 7 | >100 | >100 | >100 | 285.26 ± 55.14 | 159.79 ± 11.75 |
| 8 | >100 | 46.64 ± 2.92 | 19.08 ± 0.84 [3.17] | 99.58 ± 12.47 | 60.23 ± 2.86 |
| 9 | >100 | >100 | 47.00 ± 8.80 [3.29] | 208.69 ± 10.32 | 154.52 ± 5.30 |
| 10 | >100 | >100 | >100 | 203.18 ± 23.56 | 54.76 ± 6.24 |
| Miltefosine | 6.60 ± 1.57 | 1.40 ± 0.19 | 0.44 ± 0.08 [130.86] | 26.86 ± 3.08 | 57.58 ± 6.38 |
The critical influence of the length of the aliphatic chain of the sp2-iminosugar pseudoglycosides in the antileishmanial activity is remarkable. Thus, the inhibitory effect on intracellular amastigotes was drastically reduced or abolished on going from the dodecyl (2, 4, 6 or 8) to the octyl pseudoglycoside counterparts (1, 3, 5 or 7, respectively). Considering that these parasite forms reside and multiply inside the infected mammalian host cells, the observed differences may result from a greater cellular permeability for the more lipophilic dodecyl derivatives. In any case, the data discard a direct relationship between the α-glucosidase inhibition potency and the antileishmanial activity. Indeed, a preliminary assay of the anticancer activity of these compounds towards a panel of cancer cell lines likewise indicated a higher antiproliferative activity for the dodecyl versus the octyl pseudoglycosides (data not shown). It seems therefore that anticancer and antileishmanial activity could be actually linked in this family of glycomimetics, but the previously advanced hypothesis of inhibition of α-glucosidase being at the origin of the biological activity must be taken with caution.29
Cytotoxicity of compounds has been tested against the human lung fibroblast cell line MRC-5 and against the human monocytic leukemia cell line THP-1, the host cell used in the assay with intracellular amastigotes. In general, the MRC-5 cells are more susceptible than the THP-1 cells and most compounds show moderate cell toxicity against MRC-5, with EC50 values between 50 and 190 μM. Taking into account the mammalian cytotoxicity, compound 4 was the most promising candidate, with a significant activity against intracellular amastigotes of L. donovani Dd8 (EC50 10.80 ± 0.27 μM) and a relatively low toxicity against THP-1 and MRC-5 cell lines (EC50 118.83 ± 3.37 and 94.35 ± 20.93 μM, respectively; Table 2).
Finally, we evaluated the activity of compound 4 towards intracellular amastigotes of a previously described L. donovani line (M-40R) resistant to miltefosine, the first oral drug against leishmaniasis.30 In spite of being >29-fold more resistant to miltefosine (EC50 > 40 μM) relative to the L. donovani HU3 wild-type strain (EC50 1.36 ± 0.12 μM), the susceptibility of the M-40R line to compound 4 was similar to the wild-type strain, with EC50 values of 20.55 ± 1.78 and 19.19 ± 0.70 μM, respectively.
Drug combination with miltefosine
Taking into consideration the recommendation of WHO with respect to the use of drug combinations in the chemotherapy of leishmaniasis,3 we next investigated the effect of the combination of the most active sp2-iminosugar candidate 4 with miltefosine. For that purpose, intracellular amastigotes of L. donovani were treated with different concentrations of compound 4 (2, 4, 8 and 16 μM) at fixed concentrations of miltefosine (0.1, 0.2 and 0.3 μM), as described in the Experimental section. Dose-response curves showed that the combined treatment of 4 and miltefosine was more effective at inhibiting the parasite growth as compared with compound 4 alone (Fig. 2, Table 3). | ||
| Fig. 2 Effects of combination of compound 4 with miltefosine on L. donovani intracellular amastigotes viability. Intracellular L. donovani amastigotes in infected THP-1 cells were grown and exposed to drug pressure for 72 h at 37 °C in the presence of increasing concentrations of compounds 4 (2, 4, 8 and 16 μM) + miltefosine (0.1, 0.2 and 0.3 μM). Parasite viability was determined using the luciferase assay (as described in the Experimental section). Data are means of EC50 ± SD from three independent experiments. Significant differences were determined using Student's t test (*p < 0.01; **p < 0.001). | ||
| Compound | EC50 μM | |
|---|---|---|
| a Intracellular L. donovani amastigotes in infected THP-1 cells were grown and exposed to drug pressure for 72 h at 37 °C in the presence of increasing concentrations of compounds. Parasite viability was determined using the luciferase assay (as described in the Experimental section). Miltefosine was used as the reference antileishmanial agent. Data are means of EC50 ± SD from three independent experiments. | ||
| 4 | 10.80 ± 0.27 | |
| 4 | +Miltefosine 0.1 μM | 6.44 ± 1.05 |
| 4 | +Miltefosine 0.2 μM | 3.56 ± 0.16 |
| 4 | +Miltefosine 0.3 μM | 1.31 ± 0.19 |
| Miltefosine | 0.44 ± 0.08 | |
The combination of compound 4 with 0.1, 0.2 and 0.3 μM miltefosine induced a decrease of the EC50 values from 10.8 μM to 6.4, 3.6 and 1.3 μM, respectively (Table 3). The effect of combination treatment (synergy, additivity or antagonism) was determined by using the classic isobologram method31 and the combination index (CI) for each 4–miltefosine combination. Isobolographic analysis (Fig. 3) showed that all the interactions of compound 4 with miltefosine were synergistic in all the assayed combinations. CI values of compound 4 combined with 0.1, 0.2 and 0.3 μM miltefosine were 0.78, 0.76 and 0.80 respectively, indicating a moderate synergism.32
 | ||
| Fig. 3 Isobologram analysis for the combinations of compound 4 and miltefosine. The line indicates synergy, additivity or antagonism when the points are located below, on or above the line, respectively. (■) 4 + 0.1 μM miltefosine, (▲) 4 + 0.2 μM miltefosine, (●) 4 + 0.3 μM miltefosine. Data are means ± SD of three independent experiments. | ||
We have further evaluated the toxicity of the different combinations of compound 4 with miltefosine in THP-1 and MRC-5 cells. At the concentrations used, the drug combinations were not cytotoxic (data not shown). Overall, the ensemble of results supports that sp2-iminosugar pseudoglycosides can be considered as promising molecules for the development of new combination therapies against leishmaniasis. The advantage of combination therapy includes an increased effectiveness of the drug, reduced dosage, decreased toxicity and a delay or prevention on the appearance of drug resistance.
Conclusions
In conclusion, the results disclosed in this study provide the first evidence of antileishmanial activity of sp2-iminosugar derivatives. The possibility of using these compounds in combination therapy with miltefosine is particularly interesting in view of the observed synergistic effect. Although much research is still needed to ascertain the exact mechanism of action, the current body of data suggests a relationship between anticancer (proapoptotic) and antileishmanial activity in this family of glycomimetics. No direct relationship between these biological activities and the inhibition of glycosidases has been established so far, however. Indeed, glycomimetics can potentially interact with a range of additional enzymes and receptors, including glycosyltransferases,33 lectins34 and chaperones,35 that can interfere in cell proliferation and cell death. In any case, the antileishmanial activity and the selectivity towards the intracellular form of the parasite is strongly dependent on the nature of the aglycone-type substituent, underlining the importance of developing diversity-oriented synthetic strategies allowing optimization of non-glycone interactions for specific applications.36Experimental
General methods
Reagents and solvents were purchased from commercial sources and used without further purification. Optical rotations were measured with a JASCO P-2000 polarimeter, using a sodium lamp (λ = 589 nm) at 22 °C in 1 cm or 1 dm tubes. NMR experiments were performed at 300 (75.5), 400 (100.6) and 500 (125.7) MHz. 1-D TOCSY as well as 2-D COSY and HMQC experiments were carried out to assist on signal assignment. For ESI mass spectra, 0.1 pm sample concentrations were used, the mobile phase consisting of 50% aq. MeCN at 0.1 mL min−1. Thin-layer chromatography was performed on precoated TLC plates, silica gel 30F-245, with visualization by UV light and by carring with 10% H2SO4 or 0.2% w/v cerium(IV) sulphate-5% ammonium molybdate in 2 M H2SO4 or 0.1% ninhydrin in EtOH. Column chromatography was performed on Chromagel (silice 60 AC.C 70–200 μm). Elemental analyses were performed at the Servicio de Microanálisis del Instituto de Investigaciones Químicas de Sevilla, Spain.For the biological assays, stock solutions of the synthesized compounds in DMSO at 10 mM were prepared. Triton X-100, paraformaldehyde, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), resazurin and phorbol 12-myristate 13-acetate (PMA), were purchased from Sigma-Aldrich (St. Louis, MO). Miltefosine was purchased from Zentaris GmbH (Frankfurt am Main, Germany). DMNPE-luciferin {D-luciferin-1[-(4,5-dimethoxy-2-nitrophenyl)ethyl ester]}, hygromycin B, and 4,6-diamidino-2-phenylindole dihydrochloride (DAPI) were purchased from Invitrogen (Carlsbad, CA). Kit Luciferase Assay System was purchased from Promega. L-Glutamine and penicillin/streptomycin were obtained from Gibco. All chemicals were of the highest quality available. The peracetylated bicyclic nojirimycin derivative (11),22 and the (1R)-1-octylamino- (9) and (1R)-1-octyl-5N,6O-oxomethylidenenojirimycin (10) derivatives were prepared according to previously reported procedures.19a
Leishmania culture conditions
Promastigotes of L. donovani MHOM/IND/80/Dd8, L. donovani MHOM/ET/67/HU3 and the miltefosine-resistant line M-40R30 used in this study were grown at 28 °C in RPMI 1640-modified medium (Invitrogen) supplemented with 20% heat-inactivated fetal bovine serum (iFBS, Invitrogen).37 Additionally, L. donovani MHOM/IND/80/Dd8 with luciferase gene integrated into the parasite genome (L. donovani-LUC) was grown under the same conditions with 100 μg mL−1 of hygromycin B (unpublished results). Axenic L. donovani MHOM/ET/67/HU3 amastigote parasites (provided by Dr L. Maes form LMPH, University of Antwerp, Belgium) were grown in Schneider medium supplemented with 20% iFBS, pH 5.4 at 37 °C and 5% CO2.Susceptibility analysis in Leishmania promastigotes
In drug susceptibility assays, log phase L. donovani promastigotes were incubated at 28 °C for 72 h in the presence of increasing concentrations of compounds. Cell viability was determined by the MTT colorimetric assay, as described previously.38 Miltefosine was used as standard antileishmanial agents.Susceptibility analysis in Leishmania axenic amastigotes
Axenic amastigotes of L. donovani (106 cells per mL) in a 96-well plate were incubated with increasing concentrations of compounds for 72 h at 37 °C, followed by a resazurin-based assay.39 Briefly, 40 μL of resazurin (0.02% in MilliQ water) were added to each well, incubated for 24 h at 37 °C and fluorescence was detected at 550–590 nm.Cell lines culture and determination of cellular toxicity
Human myelomonocytic cell line THP-1 were grown at 37 °C and 5% CO2 in RPMI-1640 supplemented with 10% iFBS, 2 mM glutamate, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin. 3 × 104 cells per well in 96-well plates were differentiated to macrophages with 20 ng mL−1 of PMA treatment for 48 h followed by 24 h of culture in fresh medium.40 MRC-5 cells, a SV-40 transformed human fetal lung fibroblast cell line, were maintained at 37 °C and 5% CO2 in DMEM supplemented with 10% iFBS, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin. Cells were harvested by treatment with 0.05% (w/v) trypsin plus 0.48 mM EDTA for 5 min, diluted to 4 × 104 cells per mL in 96-well plates and incubated at 37 °C and 5% CO2 before toxicity assay.41 Cellular toxicity of all compounds was determined using the colorimetric MTT-based assay after incubation at 37 °C for 72 h in the presence of increasing concentrations of compounds.38 The results are expressed as EC50 values, as the concentration of compound that reduce cell growth by 50% versus untreated control cells.Susceptibility analysis in intracellular Leishmania amastigotes
Macrophage-differentiated-THP-1 cells, which are considered a suitable model for human macrophages, were plated at a density of 3 × 104 or 3 × 105 macrophages per well in 96-well white polystyrene microplates or 24-well tissue culture chamber slides, respectively, and were infected at a macrophage/parasite ratio of 1:10 with L. donovani promastigotes. 24 h after infection at 35 °C and 5% CO2, extracellular parasites were removed by washing with serum-free medium. Infected cell cultures were then incubated at different compound concentrations in RPMI 1640 medium plus 10% iFBS at 37 °C with 5% CO2 for 72 h. To determine the susceptibility of L. donovani-LUC amastigotes, infected macrophages maintained in 96-well plates were lysed and the luminescence intensity was measured as indicative of the intracellular parasite growth, using the Luciferase Assay System Kit (Promega, Madison, Wis.) according to the instructions of the supplier. To determine the susceptibility of L. donovani HU3 amastigotes, infected macrophages maintained in 24-well plates were fixed for 30 min at 4 °C with 2.5% paraformaldehyde phosphate-buffered saline (PBS; 1.2 mM KH2PO4, 8.1 mM Na2HPO4, 130 mM NaCl and 2.6 mM KCl adjusted to pH 7) and permeabilized with 0.1% Triton X-100 in PBS for 30 min. Intracellular parasites and macrophages were detected by nuclear staining with ProLong® Gold antifade reagent plus DAPI (Invitrogen). The percentage of infection and the mean number of amastigotes in the infected macrophages were determined in 200 macrophages per well.
Drug interaction analysis
To analyse the combination of miltefosine with the most active compound (4), intracellular amastigotes of L. donovani were treated with increasing concentrations of the compound at fixed concentrations of miltefosine (0.1, 0.2 and 0.3 μM) and cell viability was evaluated by luciferase assay as described above. A classical isobologram was constructed by plotting two drugs concentrations on the x-axis and y-axis respectively in a two-coordinate plot.42 The line connecting the concentration of both drugs required to produce a certain dose–response (e.g. EC50) is the line of additivity. The concentrations of the two drugs used in combination are place in the same plot, indicating synergy, additivity or antagonism when they are located below, on or above that line, respectively. The combination index (CI) was used to express synergism (CI < 1), additivity (CI = 1) or antagonism (CI > 1) and was calculated according to the classic isobologram equation.30Statistical analysis
All assays were performed in triplicates. Data are presented as the mean ± SD for three independent experiments. Statistical significance was calculated using Student's t-test. Differences were considered significant at a level of p < 0.01.General procedure for the preparation of pseudothioglycoside sp2-iminosugar derivatives
To a stirred solution of 11 (404 mg, 1.08 mmol) in anhydrous CH2Cl2 (20 mL) at 0 °C, BF3·Et2O (0.48 mL, 3.78 mmol, 3.5 equiv.) and the corresponding n-alkylthiol (2.27 mmol, 2.1 equiv.) were dropwise added under N2 atmosphere. The mixture was stirred for 60 min (TLC monitoring), diluted with CH2Cl2 (80 mL) and washed with water (15 mL), aq. NaHCO3 (15 mL) and water (15 mL), dried (Na2SO4) and concentrated to afford the corresponding per-O-acetylated thioglycosides 12/12β and 13/13β (α:β ratio 20:1; H-1 integration). The pure anomers were obtained after separation by column chromatography (1:3 EtOAc–cyclohexane). Conventional de-O-acetylation of the major α-anomers 12 and 13 with NaOMe in MeOH and subsequent column chromatography (20:1 → 9:1 DCM–MeOH) of the crude product afforded the target fully unprotected thioglycosides 1 and 2 in 92% and quantitative yield, respectively.
The octyl pseudothioglycosides 12/12β and 1 exhibited identical physicochemical data to those already reported in a preliminary communication.19a The corresponding data for the α-configured thiododecyl analogues 13 and 2 are listed hereinafter, whereas data for the minor β-diastereomers 13β and 2β are collected in the ESI.†
:1 EOAc–cyclohexane). [α]D +72.5 (c 1.0 in DCM). 1H NMR (500 MHz, CDCl3) δ 5.66 (d, 1H, J1,2 = 5.8 Hz, H-1), 5.41 (t, 1H, J2,3 = J3,4 = 9.8 Hz, H-3), 4.95 (dd, 1H, H-2), 4.92 (t, 1H, J4,5 = 9.5 Hz, H-4), 4.44 (dd, 1H, J6a,6b = 9.0 Hz, J5,6a = 8.5 Hz, H-6a), 4.27 (dd, 1H, J5,6b = 6.6 Hz, H-6b), 4.15 (ddd, 1H, H-5), 2.60 (m, 1H, SCH2), 2.47 (m, 1H, SCH2), 2.08 (s, 3H, MeCO), 2.05 (s, 3H, MeCO), 2.02 (s, 3H, MeCO), 1.63–1.51 (m, 2H, SCH2CH2), 1.36–1.24 (m, 18H, CH2), 0.89 (t, 3H, 3JH,H = 7.0 Hz, CH3). 13C NMR (125.7 MHz, CDCl3) δ 170.1–169.6 (CO ester), 155.5 (CO carbamate), 72.8 (C-4), 70.4 (C-2), 70.0 (C-3), 66.4 (C-6), 57.9 (C-1), 51.4 (C-5), 32.0–22.8 (CH2), 20.7–20.6 (MeCO), 14.2 (CH3). ESIMS: m/z 538.2 [M + Na]+. Anal. calcd for C25H41NO8S: C 58.23, H 8.01, N 2.72, S 6.22. Found: C 58.31, H 8.121, N 2.65, S 6.17.:1 DCM–MeOH). [α]D +94.8 (c 1.0 in MeOH) 1H NMR (500 MHz, CD3OD) δ 5.25 (d, 1H, J1,2 = 5.6 Hz, H-1), 4.55 (t, 1H, J6a,6b = J5,6a = 8.7 Hz, H-6a), 4.27 (dd, 1H, J5,6b = 5.8 Hz, H-6b), 3.92 (ddd, 1H, J4,5 = 9.5 Hz, H-5), 3.65 (dd, 1H, J2,3 = 9.6 Hz, H-2), 3.53 (t, 1H, J3,4 = 9.2 Hz, H-3), 3.32 (dd, 1H, H-4), 2.61–2.56 (m, 1H, SCH2), 2.53–2.48 (m, 1H, SCH2), 1.68–1.54 (m, 2H, SCH2CH2), 1.43–1.39 (m, 2H, CH2), 1.36–1.29 (m, 16H, CH2), 0.90 (t, 3H, 3JH,H = 7.0 Hz, CH3). 13C NMR (125.7 MHz, CD3OD) δ 158.4 (CO), 75.7 (C-4), 75.2 (C-3), 72.5 (C-2), 68.2 (C-6), 62.5 (C-1), 54.5 (C-5), 33.0–23.7 (CH2), 14.4 (CH3). ESIMS: m/z 388.0 [M − H]−. Anal. calcd for C19H35NO5S: C 58.58, H 9.06, N 3.60, S 8.23. Found: C 58.38, H 8.98, N 3.88, S 8.57.General procedure for the preparation of sulfoxide derivatives from sulfide precursors
To a solution of (1R)-1-octyl(dodecyl)thio-5N,6O-oxomethylidenenojirimycin (12 or 13) (0.23 mmol) in DCM (6 mL), MCPBA (70%, 41 mg, 0.23 mmol) was added at 0 °C. The reaction mixture was stirred for 10 min (TLC monitoring), diluted with DCM (50 mL), washed with aqueous NaHCO3 (10 mL), brine (10 mL), dried (MgSO4) and concentrated under reduced pressure. The resulting crude was purified by column chromatography to give the corresponding octyl(dodecyl)sulfoxide.:1 EtOAc–cyclohexane). Yield: 33 mg (30%). Rf 0.55 (3:2 EtOAc–cyclohexane). [α]D +38.6 (c 0.8 in DCM). 1H NMR (500 MHz, CDCl3) δ 5.85 (t, 1H, J2,3 = J3,4 = 10.0 Hz, H-3), 5.25 (dd, 1H, J1,2 = 7.1 Hz, H-2), 4.98 (t, 1H, J4,5 = 9.0 Hz, H-4), 4.91 (d, 1H, H-1), 4.66 (td, 1H, J5,6a = 9.0 Hz, J5,6b = 5.1 Hz, H-5), 4.44 (t, 1H, J6a,6b = 9.0 Hz, H-6a), 4.22 (dd, 1H, H-6b), 2.79 (ddd, 1H, 2JH,H = 13.2 Hz, 3JH,H = 9.0 Hz, 3JH,H = 6.0 Hz, SOCH2), 2.62 (ddd, 1H, SOCH2), 2.07–1.97 (3s, 9H, MeCO), 1.80–1.10 (m, 12H, CH2), 0.81 (t, 3H, 3JH,H = 7.0 Hz, CH3). 13C NMR (125.7 MHz, CDCl3) δ 170.1–169.2 (CO ester), 156.6 (CO carbamate), 71.6 (C-4), 69.6 (C-3), 69.4 (C-2), 66.5 (C-6, C-1), 54.5 (C-5), 49.7 (SOCH2), 31.7–22.5 (CH2), 20.6–20.5 (MeCO), 14.0 (CH3). ESIMS: m/z 498.1 [M + Na]+. Anal. calcd for C21H33NO9S: C 53.04, H 6.99, N 2.95, S 6.74. Found: C 52.78, H 6.67, N 2.63, S 6.53.:1 EtOAc–cyclohexane). Yield: 44 mg (39%). Rf 0.40 (3:2 EtOAc–cyclohexane). [α]D +33.5 (c 1.0 in DCM). 1H NMR (500 MHz, CDCl3) δ 5.50 (dd, 1H, J2,3 = 7.0 Hz, J3,4 = 5.0 Hz, H-3), 5.45 (bt, 1H, H-2), 4.98 (d, 1H, J1,2 = 5.5 Hz, H-1), 4.85 (dd, 1H, J4,5 = 8.3 Hz, H-4), 4.40 (d, 2H, J5,6 = 5.5 Hz, H-6a, H-6b), 3.99 (dt, 1H, H-5), 2.88 (ddd, 1H, 2JH,H = 13.5 Hz, 3JH,H = 9.5 Hz, 3JH,H = 7.0 Hz, SOCH2), 2.75 (ddd, 1H, SOCH2), 2.08–2.00 (3s, 9H, MeCO), 1.90–1.66 (m, 2H, SO2CH2CH2), 1.46–1.15 (m, 10H, CH2), 0.81 (t, 3H, 3JH,H = 7.0 Hz, CH3). 13C NMR (125.7 MHz, CDCl3) δ 169.8–168.5 (CO ester), 156.3 (CO carbamate), 72.2 (C-4), 68.8 (C-3), 67.4 (C-1), 67.3 (C-6), 67.1 (C-2), 54.4 (C-5), 50.5 (SO2CH2), 31.7–22.5 (CH2), 20.5–20.4 (MeCO), 14.0 (CH3). ESIMS: m/z 498.1 [M + Na]+. Anal. calcd for C21H33NO9S: C 53.04, H 6.99, N 2.95, S 6.74. Found: C 53.11, H 7.08, N 2.79, S 6.52.:5 MeOH–EtOAc). [α]D +63.5 (c 1.0 in MeOH). 1H NMR (500 MHz, CD3OD) δ 4.62 (t, 1H, J5,6a = J6a,6b = 9.0 Hz, H-6a), 4.28 (dd, 1H, J5,6b = 6.4 Hz, H-6b), 4.06 (ddd, 1H, J4,5 = 9.5 Hz, H-5), 3.94 (dd, 1H, J2,3 = 9.5 Hz, J1,2 = 6.6 Hz, H-2), 3.82 (t, 1H, J3,4 = 9.5 Hz, H-3), 3.35 (t, 1H, H-4), 2.97 (ddd, 1H, 2JH,H = 13.0 Hz, 3JH,H = 9.0 Hz, 3JH,H = 7.0 Hz, SO2CH2), 2.90 (ddd, 1H, SO2CH2), 1.87–1.72 (m, 2H, SO2CH2CH2), 1.55–1.27 (m, 10H, CH2), 0.91 (t, 3H, 3JH,H = 7.0 Hz, CH3). 13C NMR (125.7 MHz, CD3OD) δ 159.4 (CO), 75.7 (C-3), 74.8 (C-4), 72.5 (C-1), 71.6 (C-2), 68.7 (C-6), 57.5 (C-5), 52.3 (SOCH2), 32.9–23.7 (CH2), 14.4 (CH3). ESIMS: m/z 372.0 [M + Na]+. Anal. calcd for C15H27NO6S C 51.56, H 7.79, N 4.01, S 9.18. Found: C 51.33, H 7.54, N 3.79, S 8.85.:5 MeOH–EtOAc). [α]D +53.6 (c 0.5 in MeOH). 1H NMR (500 MHz, CD3OD) δ 4.89 (d, 1H, J1,2 = 5.9 Hz, H-1), 4.58 (t, 1H, J5,6a = J6a,6b = 9.0 Hz, H-6a), 4.30 (dd, 1H, J5,6b = 5.0 Hz, H-6b), 4.07 (dd, 1H, J2,3 = 9.0 Hz, H-2), 3.94 (t, 1H, J3,4 = 9.0 Hz, H-3), 3.91 (ddd, 1H, J4,5 = 8.0 Hz, H-5), 3.39 (dd, 1H, H-4), 3.14 (ddd, 1H, 2JH,H = 13.2 Hz, 3JH,H = 9.5 Hz, 3JH,H = 7.0 Hz, SOCH2), 3.00 (ddd, 1H, SOCH2), 1.90–1.72 (m, 2H, SO2CH2CH2), 1.58–1.27 (m, 10H, CH2), 0.91 (t, 3H, 3JH,H = 7.0 Hz, CH3). 13C NMR (125.7 MHz, CD3OD) δ 158.6 (CO), 75.4 (C-3), 75.1 (C-4), 72.9 (C-2), 72.3 (C-1), 68.6 (C-6), 56.9 (C-5), 50.8 (SOCH2), 32.9–23.7 (CH2), 14.4 (CH3). ESIMS: m/z 372.0 [M + Na]+. HRFABMS calcd for C15H27NO6SNa [M + Na]+ 372.1457, found 372.1452.:2 EtOAc–cyclohexane). Yield: 152 mg (30%). Rf 0.38 (1:1 EtOAc–cyclohexane). [α]D +42.3 (c 1.2 in DCM). 1H NMR (500 MHz, CDCl3) δ 5.92 (t, 1H, J2,3 = J3,4 = 10.0 Hz, H-3), 5.39 (dd, 1H, J1,2 = 7.1 Hz, H-2), 5.10 (t, 1H, J4,5 = 9.7 Hz, H-4), 4.99 (d, 1H, H-1), 4.71 (ddd, 1H, J5,6a = 9.0 Hz, J5,6b = 5.0 Hz, H-5), 4.50 (t, 1H, J6a,6b = 9.0 Hz, H-6a), 4.29 (dd, 1H, H-6b), 2.85 (ddd, 1H, 2JH,H = 13.0 Hz, 3JH,H = 9.0 Hz, 3JH,H = 6.0 Hz, SOCH2), 2.69 (ddd, 1H, SOCH2), 2.14–2.04 (3s, 9H, MeCO), 1.86–1.67 (m, 2H, SOCH2CH2), 1.50–1.20 (m, 18H, CH2), 0.88 (t, 3H, 3JH,H = 7.0 Hz, CH3). 13C NMR (125.7 MHz, CDCl3) δ 170.1–169.2 (CO ester), 156.7 (CO carbamate), 71.6 (C-4), 69.6 (C-3), 69.2 (C-2), 66.5 (C-6, C-1), 54.5 (C-5), 49.5 (SOCH2), 31.8–22.6 (CH2), 20.5 (MeCO), 14.1 (CH3). ESIMS: m/z 554.4 [M + Na]+. Anal. calcd for C25H41NO9S: C 56.48, H 7.77, N 2.63, S 6.03. Found: C 56.14, H 7.55, N 2.58, S 6.38.:2 EtOAc–cyclohexane). Yield: 225 mg (44%). Rf 0.33 (1:1 EtOAc–cyclohexane). [α]D +25.1 (c 1.1 in DCM). 1H NMR (500 MHz, CDCl3) δ 5.54 (dd, 1H, J2,3 = 7.6 Hz, J3,4 = 5.7 Hz, H-3), 5.46 (dd, 1H, J1,2 = 5.7 Hz, H-2), 5.00 (d, 1H, H-1), 4.93 (dd, 1H, J4,5 = 8.6 Hz, H-4), 4.44–4.37 (m, 2H, H-6a, H-6b), 4.05–3.99 (m, 1H, H-5), 2.90 (ddd, 1H, 2JH,H = 12.0 Hz, 3JH,H = 9.0 Hz, 3JH,H = 5.1 Hz, SOCH2), 2.76 (ddd, 1H, SOCH2), 2.10–1.98 (3s, 9H, MeCO), 1.85–1.65 (m, 2H, SOCH2CH2), 1.46–1.14 (m, 18H, CH2), 0.81 (t, 3H, 3JH,H = 7.0 Hz, CH3). 13C NMR (125.7 MHz, CDCl3) δ 168.9–168.0 (CO ester), 155.3 (CO carbamate), 71.1 (C-4), 67.9 (C-3), 66.4 (C-1, C-2), 66.2 (C-6), 53.2 (C-5), 49.4 (SOCH2), 30.9–21.6 (CH2), 19.5 (MeCO), 13.1 (CH3). ESIMS: m/z 554.4 [M + Na]+. Anal. calcd for C25H41NO9S: C 56.48, H 7.77, N 2.63, S 6.03. Found: C 56.55, H 7.81, N 2.44, S 5.79.:1 EtOAc–MeOH). [α]D +58.1 (c 1.3 in MeOH). 1H NMR (500 MHz, CD3OD) δ 4.62 (t, 1H, J5,6a = J6a,6b = 8.7 Hz, H-6a), 4.28 (dd, 1H, J5,6b = 6.4 Hz, H-6b), 4.05 (ddd, 1H, J4,5 = 9.5 Hz, H-5), 3.94 (dd, 1H, J2,3 = 9.5 Hz, J1,2 = 6.6 Hz, H-2), 3.81 (t, 1H, J3,4 = 9.5 Hz, H-3), 3.35 (t, 1H, H-4), 2.97 (ddd, 1H, 2JH,H = 13.0 Hz, 3JH,H = 9.0 Hz, 3JH,H = 7.3 Hz, SOCH2), 2.90 (ddd, 1H, SOCH2), 1.86–1.74 (m, 2H, SO2CH2CH2), 1.56–1.24 (m, 18H, CH2), 0.90 (t, 3H, 3JH,H = 7.0 Hz, CH3). 13C NMR (75.5 MHz, CD3OD) δ 159.4 (CO), 75.6 (C-3), 74.8 (C-4), 72.5 (C-1), 71.6 (C-2), 68.7 (C-6), 57.4 (C-5), 52.3 (SOCH2), 33.1–23.7 (CH2), 14.4 (CH3). ESIMS: m/z 428.4 [M + Na]+. Anal. calcd for C19H35NO6S: C 56.27, H 8.70, N 3.45, S 7.91. Found: C 55.92, H 8.63, N 3.17, S 7.54.:1 EtOAc–MeOH). [α]D +60.5 (c 0.8 in DMSO). 1H NMR (500 MHz, DMSO-d6) δ 4.57 (d, 1H, J1,2 = 5.7 Hz, H-1), 4.47 (t, 1H, J5,6a = J6a,6b = 8.3 Hz, H-6a), 4.15 (dd, 1H, J5,6b = 4.7 Hz, H-6b), 3.91 (dd, 1H, J2,3 = 8.4 Hz, H-2), 3.76 (ddd, 1H, J4,5 = 9.5 Hz, H-5), 3.73 (bt, 1H, J3,4 = 7.5 Hz, H-3), 3.24 (dd, 1H, H-4), 2.96–2.84 (m, 2H, SOCH2), 1.73–1.57 (m, 2H, SOCH2CH2), 1.45–1.19 (m, 10H, CH2), 0.85 (t, 3H, 3JH,H = 7.0 Hz, CH3). 13C NMR (125.7 MHz, DMSO-d6) δ 156.3 (CO), 73.5 (C-3), 73.3 (C-4), 71.0 (C-1), 70.9 (C-2), 66.7 (C-6), 55.2 (C-5), 48.7 (SOCH2), 31.3–22.1 (CH2), 13.9 (CH3). ESIMS: m/z 428.4 [M + Na]+. Anal. calcd for C19H35NO6S: C 56.27, H 8.70, N 3.45, S 7.91. Found: C 56.14, H 8.67, N 3.21, S 7.60.General procedure for the preparation of sulfone derivatives from sulfide precursors
To a solution of (1R)-2,3,4-tri-O-acetyl-1-octyl(dodecyl)thio-5N,6O-oxomethylidenenojirimycin (12 or 13) (0.11 mmol) in DCM (3 mL), 70% MCPBA (38 mg, 0.22 mmol) was added at 0 °C. The reaction mixture was stirred for 10 min, diluted with DCM (50 mL), washed with aqueous NaHCO3 (10 mL), brine (10 mL), dried (MgSO4) and concentrated under reduced pressure. The resulting crude was purified by column chromatography to yield the corresponding sulfones 18 and 19.:2 EtOAc–cyclohexane). Yield: 48 mg (90%). Rf 0.56 (1:1 EtOAc–cyclohexane). [α]D +26.6 (c 0.8 in DCM). 1H NMR (500 MHz, CDCl3) δ 5.83 (t, 1H, J2,3 = J3,4 = 9.0 Hz, H-3), 5.36 (d, 1H, J1,2 = 6.7 Hz, H-1), 5.23 (dd, 1H, H-2), 4.94 (t, 1H, J4,5 = 9.0 Hz, H-4), 4.50–4.43 (m, 1H, H-6a), 4.36–4.29 (m, 2H, H-6b, H-5), 3.02–2.89 (m, 2H, SO2CH2), 2.06–1.98 (3s, 9H, MeCO), 1.86–1.67 (m, 2H, SO2CH2CH2), 1.38–1.15 (m, 10H, CH2), 0.81 (t, 3H, 3JH,H = 7.0 Hz, CH3). 13C NMR (125.7 MHz, CDCl3) δ 170.1–169.1 (CO ester), 155.5 (CO carbamate), 71.8 (C-4), 68.9 (C-3), 68.1 (C-2), 66.8 (C-6), 65.6 (C-1), 52.9 (SO2CH2), 52.7 (C-5), 31.6–21.1 (CH2), 20.6–20.5 (MeCO), 14.0 (CH3). ESIMS: m/z 514.1 [M + Na]+. Anal. calcd for C21H33NO10S: C 51.31, H 6.77, N 2.85, S 6.52. Found: C 51.23, H 6.64, N 2.71, S 6.49.:1 EtOAc–cyclohexane). Yield: 23 mg (84%). Rf 0.31 (EtOAc). [α]D +30.1 (c 0.6 in MeOH). 1H NMR (500 MHz, CD3OD) δ 5.12 (d, 1H, J1,2 = 6.5 Hz, H-1), 4.63 (t, 1H, J5,6a = J6a,6b = 8.8 Hz, H-6a), 4.28 (dd, 1H, J5,6b = 6.2 Hz, H-6b), 4.14 (t, 1H, J2,3 = J3,4 = 9.5 Hz, H-3), 4.03 (td, 1H, J4,5 = 9.5 Hz, H-5), 3.82 (dd, 1H, H-2), 3.40–3.32 (m, 3H, H-4, SO2CH2), 1.85 (quint., 2H, JH,H = 7.5 Hz, SO2CH2CH2), 1.50–1.25 (m, 10H, CH2), 0.91 (t, 3H, 3JH,H = 7.0 Hz, CH3). 13C NMR (125.7 MHz, CD3OD) δ 158.4 (CO), 75.4 (C-4), 74.1 (C-3), 72.0 (C-1), 71.9 (C-2), 68.7 (C-6), 56.5 (C-5), 56.3 (SO2CH2), 32.9–22.5 (CH2), 14.4 (CH3). ESIMS: m/z 388.1 [M + Na]+. Anal. calcd for C15H27NO7S: C 49.30, H 7.45, N 3.83, S 8.77. Found: C 49.04, H 7.20, N 3.56, S 8.44.:4 EtOAc–cyclohexane). Yield: 60 mg (78%). Rf 0.50 (1:1 EtOAc–cyclohexane). [α]D +32.6 (c 1.0 in DCM). 1H NMR (500 MHz, CDCl3) δ 5.83 (t, 1H, J2,3 = J3,4 = 9.0 Hz, H-3), 5.37 (d, 1H, J1,2 = 6.7 Hz, H-1), 5.23 (dd, 1H, H-2), 4.94 (t, 1H, J4,5 = 9.0 Hz, H-4), 4.44–4.50 (m, 1H, H-6a), 4.37–4.29 (m, 2H, H-6b, H-5), 2.98 (ddd, 1H, 2JH,H = 13.8 Hz, 3JH,H = 10.2 Hz, 3JH,H = 5.7 Hz, SO2CH2), 2.93 (ddd, 1H, SO2CH2), 2.07–1.98 (3s, 9H, MeCO), 1.87–1.67 (m, 2H, SO2CH2CH2), 1.38–1.14 (m, 18H, CH2), 0.81 (t, 3H, 3JH,H = 7.0 Hz, CH3). 13C NMR (125.7 MHz, CDCl3) δ 170.2–169.1 (CO ester), 155.5 (CO carbamate), 71.8 (C-4), 68.9 (C-3), 68.1 (C-2), 66.8 (C-6), 65.6 (C-1), 52.9 (SO2CH2), 52.7 (C-5), 31.9–21.1 (CH2), 20.6–20.5 (MeCO), 14.1 (CH3). ESIMS: m/z 570.4 [M + Na]+. HRFABMS calcd for C25H41NO10SNa [M + Na]+ 570.2349, found 570.2369.:1 EtOAc–MeOH). Yield: 21 mg (83%). Rf 0.56 (9:1 EtOAc–MeOH). [α]D +13.5 (c 1.0 in DMSO). 1H NMR (300 MHz, DMSO-d6) δ 4.94 (d, 1H, J1,2 = 6.4 Hz, H-1), 4.57 (t, 1H, J5,6a = J6a,6b = 8.7 Hz, H-6a), 4.15 (dd, 1H, J5,6b = 6.1 Hz, H-6b), 3.92 (t, 1H, J2,3 = J3,4 = 9.0 Hz, H-3), 3.84–3.78 (m, 1H, H-5), 3.77 (dd, 1H, H-2), 3.40–3.20 (m, 3H, H-4, SO2CH2), 1.75–1.65 (m, 2H, SO2CH2CH2), 1.40–1.20 (m, 18H, CH2), 0.86 (t, 3H, 3JH,H = 7.0 Hz, CH3). 13C NMR (125.7 MHz, DMSO-d6) δ 155.6 (CO), 73.6 (C-4), 71.9 (C-3), 70.6 (C-1), 70.1 (C-2), 66.8 (C-6), 54.7 (C-5), 54.4 (SO2CH2), 31.3–23.0 (CH2), 13.9 (CH3). ESIMS: m/z 444.3 [M + Na]+. Anal. calcd for C19H35NO7S: C 54.13, H 8.37, N 3.32, S 7.61. Found: C 53.81, H 8.15, N 3.01, S 7.36.Acknowledgements
This work was supported by the Spanish Ministerio de Economía y Competitividad Grants SAF2012-34267 (to F.G.), SAF2011-28102 (to S.C.), SAF2013-44021R (to C.O.M.) and CTQ2010-15848 (to J.M.G.F.), the Plan Andaluz de Investigación (Proyectos de Excelencia CTS-7282 and FQM-1467), by ISCIII-Subdirección General de Redes y Centros de Investigación Cooperativa (RICET FIS Network: RD12/0018/0017), the European Union Seventh Framework Programme (FP7-People-2012-CIG), grant agreement number 333594 (to E.M.S.F., Marie Curie Reintegration Grant), the European Regional Development Fund (FEDER) and the European Social Fund (ESF). We also thank Dr Louis Maes (LMPH, University of Antwerp, Belgium) for kindly supply the axenic L. donovani amastigote line and the MRC-5 cell line. Technical assistance from the research support services of the University of Seville (CITIUS) is also acknowledged. J.M.P. thank the Instituto de Salud Carlos III (PI11/00840), and the EU Research Potential (FP7-REGPOT-2012-CT2012-31637-IMBRAIN), for financial support. G.B.P. also thanks the Obra Social La Caixa-Fundación Caja Canarias for a predoctoral grant.Notes and references
- J. Alvar, I. D. Vélez, C. Bern, M. Herrero, P. Desjeux, J. Cano, J. Jannin and M. den Boer, PLoS One, 2012, 7, e35671 CAS.
- K. Seifert, Open Med. Chem. J., 2011, 5, 31 CrossRef CAS PubMed.
- Control of the Leishmaniases, Geneva, World Health Organization, 2010, WHO Technical Report Series, no. 949.
- S. Sundar, T. K. Jha, C. P. Thakur, S. K. Bhattacharya and M. Rai, Trans. R. Soc. Trop. Med. Hyg., 2006, 100(suppl. 1), S26 CrossRef CAS PubMed.
- F. J. Pérez-Victoria, M. P. Sánchez-Cañete, K. Seifert, S. L. Croft, S. Sundar, S. Castanys and F. Gamarro, Drug Resist. Updates, 2006, 9, 26 CrossRef PubMed.
- (a) S. Sundar, P. K. Sinha, M. Rai, D. K. Verma, K. Nawin, S. Alam, J. Chakravarty, M. Vaillant, N. Verma, K. Pandey, P. Kumari, C. S. Lal, R. Arora, B. Sharma, S. Ellis, N. Strub-Wourgaft, M. Balasegaram, P. Olliaro, P. Das and F. Modabber, Lancet, 2011, 377, 477 CrossRef CAS PubMed; (b) S. Sundar, M. Rai, J. Chakravarty, D. Agarwal, N. Agrawal, M. Vaillant, P. Olliaro and H. W. Murria, Clin. Infect. Dis., 2008, 47, 1000 CrossRef CAS PubMed.
- R. Omollo, N. Alexander, T. Edwards, E. A. Khalil, B. M. Younis, A. A. Abuzaid, M. Wasunna, N. Njoroge, D. Kinoti, T. P. Dorlo, S. Eliis, M. Balasegaram and A. M. Musa, Trials, 2011, 12, 166 CrossRef CAS PubMed.
- (a) A. E. Stütz and T. M. Wrodnigg, Adv. Carbohydr. Chem. Biochem., 2011, 66, 187 CrossRef PubMed; (b) Iminosugars, from synthesis to therapeutic applications, ed. P. Compain and O. R. Martin, Wiley, Chichester, UK, 2007 Search PubMed.
- M. Liu, S. Wang, Y.-D. Zhou, T. Xiang, H. Dong, K. Yang and X.-L. Zhang, Bioorg. Med. Chem., 2012, 22, 564 CrossRef CAS PubMed.
- (a) R. J. Nash, A. Kato, C.-Y. Yu and G. W. J. Fleet, Future Med. Chem., 2011, 3, 1531 Search PubMed; (b) G. Home, X. Francis, J. Tinsley, D. H. Williams and R. Storer, Drug Discovery Today, 2011, 16, 107 CrossRef PubMed.
- (a) G. N. Wang, Y. L. Xiong, J. Ye, L. H. Zhang and X. S. Ye, ACS Med. Chem. Lett., 2011, 2, 682 CrossRef CAS PubMed; (b) G. L. Zhang, C. S. Chen, Y. L. Xiong, L. H. Zhang, J. Ye and X. S. Ye, Carbohydr. Res., 2010, 345, 780 CrossRef CAS PubMed.
- T. M. Wrodnigg, A. J. Steiner and B. J. Ueberbacher, Anti-Cancer Agents Med. Chem., 2008, 8, 77 CrossRef CAS PubMed.
- (a) D. Durantel, C. Alotte and F. Zoulim, Curr. Opin. Invest. Drugs, 2007, 8, 125 CAS; (b) P. Greimel, J. Spreitz, A. E. Stütz and T. M. Wrodnigg, Curr. Top. Med. Chem., 2003, 3, 513 CrossRef CAS PubMed; (c) S. Hussain, J. L. Miller, D. J. Harvey, Y. Gu, P. B. Rosenthal, N. Zitzmann and J. W. McCauley, J. Antimicrob. Chemother., 2015, 70, 136 CrossRef CAS PubMed.
- D. Ruhela, P. Chatterjee and R. A. Vishwakarma, Org. Biomol. Chem., 2005, 3, 1043 CAS.
- For selected examples see: (a) J. L. Jiménez Blanco, V. M. Díaz Pérez, C. Ortiz Mellet, J. Fuentes and J. M. García Fernández, Chem. Commun., 1997, 1969 RSC; (b) M. I. García-Moreno, P. Díaz-Pérez, C. Ortiz Mellet and J. M. García Fernández, J. Org. Chem., 2003, 68, 8890 CrossRef PubMed; (c) M. I. García-Moreno, D. Rodríguez-Lucena, C. Ortiz Mellet and J. M. García Fernández, J. Org. Chem., 2004, 69, 3578 CrossRef PubMed; (d) M. I. García-Moreno, C. Ortiz Mellet and J. M. García Fernández, Eur. J. Org. Chem., 2004, 1803 CrossRef; (e) M. Aguilar-Moncayo, T. M. Gloster, J. P. Turkenburg, M. I. García-Moreno, C. Ortiz Mellet, G. J. Davies and J. M. García Fernández, Org. Biomol. Chem., 2009, 7, 2738 RSC; (f) M. Aguilar-Moncayo, M. I. García-Moreno, A. Trapero, M. Egido-Gabás, A. Llebaria, J. M. García Fernández and C. Ortiz Mellet, Org. Biomol. Chem., 2011, 9, 3698 RSC; (g) B. Brumshtein, M. Aguilar-Moncayo, J. M. Benito, J. M. García Fernández, I. Silman, Y. Shaaltiel, D. Aviezer, J. L. Sussman, A. H. Futerman and C. Ortiz Mellet, Org. Biomol. Chem., 2011, 9, 4160 RSC; (h) M. Aguilar-Moncayo, P. Díaz-Pérez, J. M. García Fernández, C. Ortiz Mellet and M. I. García-Moreno, Tetrahedron, 2012, 68, 681 CrossRef CAS.
- (a) J. M. Benito, J. M. García Fernández and C. Ortiz Mellet, Expert Opin. Ther. Pat., 2011, 21, 885 CrossRef CAS PubMed; (b) Z. Luan, K. Higaki, M. Aguilar-Moncayo, H. Ninomiya, K. Ohno, M. I. García-Moreno, C. Ortiz Mellet, J. M. García Fernández and Y. Suzuki, ChemBioChem, 2009, 10, 2780 CrossRef CAS PubMed; (c) Z. Luan, K. Higaki, M. Aguilar-Moncayo, L. Li, H. Ninomiya, E. Nanba, K. Ohno, M. I. García-Moreno, C. Ortiz Mellet, J. M. García Fernández and Y. Suzuki, ChemBioChem, 2010, 11, 2453 CrossRef CAS PubMed; (d) G. Tiscornia, E. Lorenzo Vivas, L. Matalonga, I. Berniakovich, M. Barragán Monasterio, C. Eguizábal, L. Gort, F. González, C. Ortiz Mellet, J. M. García Fernández, A. Ribes, A. Veiga and J. C. Izpisua Belmonte, Hum. Mol. Genet., 2013, 22, 633 CrossRef CAS PubMed; (e) P. Alfonso, V. Andreu, A. Pino-Angeles, A. A. Moya-García, M. I. García-Moreno, J. C. Rodríguez-Rey, F. Sánchez-Jiménez, M. Pocoví, C. Ortiz Mellet, J. M. García Fernández and P. Giraldo, ChemBioChem, 2013, 14, 943 CrossRef CAS PubMed; (f) J. Rodríguez-Lavado, M. de la Mata, J. L. Jiménez-Blanco, M. I. García-Moreno, J. M. Benito, A. Díaz-Quintana, J. A. Sánchez-Alcázar, K. Higaki, E. Nanba, K. Ohno, Y. Suzuki, C. Ortiz Mellet and J. M. García Fernández, Org. Biomol. Chem., 2014, 12, 2289 RSC.
- Y. Yu, T. Mena-Barragán, K. Higaki, J. L. Johnson, J. E. Drury, R. L. Lieberman, N. Nakasone, H. Ninomiya, T. Tsukimura, H. Sakuraba, Y. Suzuki, E. Nanba, C. Ortiz Mellet, J. M. García Fernández and K. Ohno, ACS Chem. Biol., 2014, 9, 1460 CrossRef CAS PubMed.
- (a) T. Takai, K. Higaki, M. Aguilar-Moncayo, T. Mena-Barragán, Y. Hirano, K. Yura, L. Yu, H. Ninomiya, M. I. García-Moreno, Y. Sakakibara, K. Ohno, E. Nanba, C. Ortiz Mellet, J. M. García Fernández and Y. Suzuki, Mol. Ther., 2013, 21, 526 CrossRef CAS PubMed; (b) H. Suzuki, U. Ohto, K. Higaki, T. Mena-Barragán, M. Aguilar-Moncayo, C. Ortiz Mellet, E. Namba, J. M. García Fernández, Y. Suzuki and T. Shimizu, J. Biol. Chem., 2014, 289, 14560 CrossRef CAS PubMed.
- (a) E. M. Sánchez-Fernández, R. Rísquez-Cuadro, M. Chasseraud, A. Ahidouch, C. Ortiz Mellet, H. Ouadid-Ahidouch and J. M. García Fernandez, Chem. Commun., 2010, 46, 5328 RSC; (b) E. M. Sánchez-Fernández, R. Rísquez-Cuadro, C. Ortiz Mellet, J. M. García Fernández, P. M. Nieto and J. Angulo, Chem.–Eur. J., 2012, 18, 8527 CrossRef PubMed; (c) E. M. Sánchez-Fernández, R. Rísquez-Cuadro, M. Aguilar-Moncayo, M. I. García-Moreno, C. Ortiz Mellet and J. M. García Fernández, Org. Lett., 2009, 11, 3306 CrossRef PubMed.
- G. Allan, H. Ouadid-Ahidouch, E. M. Sánchez-Fernández, R. Rísquez-Cuadro, J. M. García Fernández, C. Ortiz Mellet and A. Ahidouch, PLoS One, 2013, e76411 CAS.
- W. Moreira, P. Leprohon and M. Ouellette, Cell Death Dis., 2011, 2, e201 CrossRef CAS PubMed.
- V. M. Díaz Pérez, M. I. García Moreno, C. Ortiz Mellet, J. Fuentes, J. C. Díaz Arribas, J. Cañada and J. M. García Fernández, J. Org. Chem., 2000, 65, 136 CrossRef.
- M. Nilsson, C. M. Svahn and J. Westman, Carbohydr. Res., 1993, 246, 161 CrossRef CAS PubMed.
- E. M. Sánchez, J. F. Arteaga, V. Domingo, J. F. Quílez del Moral, M. Mar Herrador and A. F. Barrero, Tetrahedron, 2008, 64, 5111 CrossRef.
- (a) M. Gorman and C. W. Ryan, in Cephalosporins and Penicillins: Chemistry and Biology, ed. E. H. Flynn, Academic Press, New York, 1972, p. 540 Search PubMed; (b) W. J. Gottstein, C. U. Kim, K. M. Shih and D. N. McGregor, J. Med. Chem., 1978, 21, 240 CrossRef CAS PubMed.
- (a) C. A. Sanhueza, A. C. Arias, R. L. Dorta and J. T. Vázquez, Tetrahedron: Asymmetry, 2010, 21, 1830 CrossRef CAS; (b) N. Khiar, Tetrahedron Lett., 2000, 41, 9059 CrossRef CAS.
- D. Liu and J. E. Uzonna, Front. Cell. Infect. Microbiol., 2012, 2, 83 Search PubMed.
- P. Wadhone, M. Maiti, R. Agarwal, V. Kamat, S. Martin and B. Saha, J. Immunol., 2009, 182, 7146 CrossRef CAS PubMed.
- On going studies confirm that the anticancer activity of sp2-iminosugars is indeed not directly associated to inhibition of human ER α-glucosidases I or II; data will be published in due curse.
- K. Seifert, S. Matu, F. J. Pérez-Victoria, S. Castanys, F. Gamarro and S. L. Croft, Int. J. Antimicrob. Agents, 2003, 22, 380 CrossRef CAS PubMed.
- (a) T. C. Chou and P. Talalay, Adv. Enzyme Regul., 1984, 22, 27 CrossRef CAS PubMed; (b) J. Topaly, W. J. Zeller and S. Fruehauf, Leukemia, 2001, 15, 342 CrossRef CAS PubMed.
- T. C. Chou, Pharmacol. Rev., 2006, 58, 621 CrossRef CAS PubMed.
- T. M. Gloster and D. Vocadlo, Nat. Chem. Biol., 2012, 8, 683 CrossRef CAS PubMed.
- R. Rísquez-Cuadro, J. M. García Fernández, J. F. Nierengarten and C. Ortiz Mellet, Chem.–Eur. J., 2013, 19, 16791 CrossRef PubMed.
- F. Dal Piaz, A. Vassallo, M. G. Chini, F. M. Cordero, F. Cardona, C. Pisano, G. Bifulco, N. D. Tommasi and A. Brandi, PLoS One, 2012, 7, e43316 CAS.
- (a) J. Castilla, R. Rísquez, D. Cruz, K. Higaki, E. Nanba, K. Ohno, Y. Suzuki, Y. Díaz, C. Ortiz Mellet, J. M. García Fernández and S. Castillón, J. Med. Chem., 2012, 55, 6857 CrossRef CAS PubMed; (b) J. Castilla, R. Rísquez, K. Higaki, E. Nanba, K. Ohno, Y. Suzuki, Y. Díaz, C. Ortiz Mellet, J. M. García Fernández and S. Castillón, Eur. J. Med. Chem., 2015, 90, 258 CrossRef CAS PubMed; (c) E. M. Sánchez-Fernández, E. Álvarez, C. Ortiz Mellet and J. M. García Fernández, J. Org. Chem., 2014, 79, 11722 CrossRef PubMed.
- P. R. Jackson, J. M. Lawrie, J. M. Stiteler, D. W. Hawkins, J. A. Wohlhieter and E. D. Rowton, Vet. Parasitol., 1986, 20, 195 CrossRef CAS PubMed.
- V. Gómez-Pérez, J. I. Manzano, R. García-Hernández, S. Castanys, J. M. Campos Rosa and F. Gamarro, Antimicrob. Agents Chemother., 2014, 58, 4103 CrossRef PubMed.
- M. De Rycker, I. Hallyburton, J. Thomas, L. Campbell, S. Wyllie, D. Joshi, S. Cameron, I. H. Gilbert, P. G. Wyatt, J. A. Frearson, A. H. Fairlamb and D. W. Gray, Antimicrob. Agents Chemother., 2013, 57, 2913 CrossRef CAS PubMed.
- K. El Fadili, M. Imbeault, N. Messier, G. Roy, B. Gourbal, M. Bergeron, M. J. Tremblay, D. Legare and M. Ouellette, Antimicrob. Agents Chemother., 2008, 52, 526 CrossRef CAS PubMed.
- M. De Rycker, I. Hallyburton, J. Thomas, L. Campbell, S. Wyllie, D. Joshi, S. Cameron, I. H. Gilbert, P. G. Wyatt, J. A. Frearson, A. H. Fairlamb and D. W. Gray, Antimicrob. Agents Chemother., 2013, 57, 2913 CrossRef CAS PubMed.
- S. Wagenpfeil, U. Treiber and A. Lehmer, Artif. Intell. Med., 2006, 37, 65 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: General procedure for the glycosidase inhibition assay, Lineweaver–Burk and double reciprocal analysis plots of 2α, 5, 7, 8, full experimental data for compounds 13β, 2β and copies of the 1H and 13C NMR spectra of all new compounds. See DOI: 10.1039/c5ra02627j |
| ‡ Both authors contributed equally to this manuscript. |
| § Equal senior investigators in this study. |
| This journal is © The Royal Society of Chemistry 2015 |