
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBinder-free germanium nanoparticle decorated multi-wall carbon nanotube anodes prepared via two-step electrophoretic deposition for high capacity Li-ion batteries†
Xuan-Manh
Pham
 ,
Syed
Abdul Ahad
,
Niraj Nitish
Patil
,
Hugh
Geaney
,
Shalini
Singh
and
Kevin M.
Ryan
*
,
Syed
Abdul Ahad
,
Niraj Nitish
Patil
,
Hugh
Geaney
,
Shalini
Singh
and
Kevin M.
Ryan
*
Department of Chemical Sciences and Bernal Institute, University of Limerick, Ireland. E-mail: Kevin.M.Ryan@ul.ie
First published on 13th February 2024
Abstract
Germanium (Ge) has a high theoretical specific capacity (1384 mA h g−1) and fast lithium-ion diffusivity, which makes it an attractive anode material for lithium-ion batteries (LIBs). However, large volume changes during lithiation can lead to poor capacity retention and rate capability. Here, electrophoretic deposition (EPD) is used as a facile strategy to prepare Ge nanoparticle carbon-nanotube (Ge/CNT) electrodes. The Ge and CNT mass ratio in the Ge/CNT nanocomposites can be controlled by varying the deposition time, voltage, and concentration of the Ge NP dispersion in the EPD process. The optimized Ge/CNT nanocomposite exhibited long-term cyclic stability, with a capacity of 819 mA h g−1 after 1000 cycles at C/5 and a reversible capacity of 686 mA h g−1 after 350 cycles (with a minuscule capacity loss of 0.07% per cycle) at 1C. The Ge/CNT nanocomposite electrodes delivered dramatically improved cycling stability compared to control Ge nanoparticles. This can be attributed to the synergistic effects of implanting Ge into a 3D interconnected CNT network which acts as a buffer layer to accommodate the volume expansion of Ge NPs during lithiation/delithiation, limiting cracking and/or crumbling, to retain the integrity of the Ge/CNT nanocomposite electrodes.
New conceptsBinder-free electrode fabrication is pitched as an emerging technology to achieve the much-needed high energy density in rechargeable batteries. Advantages such as reduction in non-active material and increase in direct electronic contact between current collector/active material help reduce cell resistance and improve cycling life. An underexplored fabrication procedure for the formation of binder-free electrodes for Li-ion applications is electrophoretic deposition (EPD), offering the potential for cost savings, simple thickness control, and enhanced uniformity compared to chemical vapor deposition (CVD), used to fabricate binder-free electrodes. Ge anode possesses high theoretical capacity and fast Li-ion diffusivity, but the limited charge transport and huge volume expansion (>300%) of Ge during the lithiation processes hinder its application in LIBs. The incorporation of Ge into a carbon nanotube (CNT) buffer layer which offers high electrical conductivity and large surface area can dramatically lower the interparticle charge transfer resistance and considerably increase the performance of Ge at high charge–discharge rates. Therefore, we report the detailed fabrication of binder-free Ge/CNT nanocomposite anodes using 2-step EPD for the first time, followed by the electrochemical test of these anodes. The promising findings represent the next stage in utilizing EPD for manufacturing binder-free electrodes with high performance in LIBs. |
Introduction
Developing lithium-ion batteries (LIBs) with high energy and power densities for uses ranging from portable electronic devices to electric vehicles is a critical scientific challenge.1–4 Materials that can give highly reversible capabilities at low voltages (vs. Li/Li+) are suitable to maximize the energy density of LIB anodes. Lithium (Li) alloying materials such as silicon (Si), tin (Sn) and germanium (Ge) have been recently receiving a lot of attention as their theoretical capacities are many times higher than those of commercially available anode materials such as graphite and lithium titanate.4–7 Among those potential anode materials, Ge has attractive properties for LIBs because it possesses a higher theoretical capacity (1384 mA h g−1) than Sn (847 mA h g−1) and has better Li+ diffusivity (400 times higher than Si) and higher electrical conductivity (104×) than Si, which has led to promising fast-charging ability and cycling stability of Ge anodes.7–11 However, the limited charge transport and huge volume expansion (>300%) of Ge during the Li+ insertion/extraction processes remain issues.2,9,10,12The most effective method for enhancing performance and reducing concerns with inadequate charge transport and degradation is to reduce Ge materials to the nanoscale.2,10,13–15 Ge nanoparticles (NPs) can relieve significant internal strain during cycling, but have a strong tendency to agglomerate, which limits the electronic conduction channels to the current collector.3,6,7 The introduction of a carbon buffer layer can dramatically lower the interparticle charge transfer resistance as well as considerably increase the performance of Ge at high charge–discharge rates, due to the enhancements in mechanical characteristics, effective contact between the active material and the current collector, electrical conductivity, and enhanced diffusivity of Li+.16,17 Carbon nanotubes (CNTs) and graphene are often used as carbon buffer layers to improve the stability and performance of alloying materials. In particular, anode materials such as Sb,18 Si,19 or Ge20,21 incorporated with multiwalled carbon nanotubes (MWCNTs) which offer high electrical conductivity and large surface area have delivered good electrochemical performances. Recent work has shown that the use of CNTs to fabricate binder-free electrodes is viable due to their lightweight, coupled with impressive mechanical and electrical characteristics, which allows high-capacity materials to be effectively supported.22 Various publications have reported the incorporation of Ge NPs on CNTs, but the enhancement of long-term cycling stability remains elusive. Additionally, this process typically requires the use of chemical vapor deposition (CVD), which is performed at high temperatures.2,6,23 An underexplored option for the formation of Ge NPs on CNTs for Li-ion applications is electrophoretic deposition (EPD). Compared to CVD, EPD offers potential for cost savings, simple thickness control, and enhanced uniformity.6,24 To date, CNT-based Ge nanocomposites have not been prepared by EPD in the literature, despite its demonstrated ability to form other high performance anode coatings.25–28
In this work, we present a straightforward method for the fabrication of binder-free Ge/CNT nanocomposite anodes by: (1) fabrication of CNT electrodes by EPD of MWCNTs directly onto the current collector and (2) decoration of Ge NPs on the as-prepared MWCNT networks by EPD at room temperature. The CNT network plays a crucial role in mitigating the volume changes of Ge NPs during lithiation/delithiation, preventing active material degradation and retaining the integrity of the Ge/CNT nanocomposite electrodes. The Ge/CNT electrodes exhibited outstanding battery performance compared to control Ge electrodes, with the optimized Ge/CNT nanocomposite electrode displaying a retained capacity of 819 mA h g−1 after 1000 cycles. In comparison, the pure Ge electrode decayed rapidly after 100 cycles. Furthermore, the Ge/CNT structure unlocked high rate cycling ability at 5C and 10C (specific capacities of 519 and 210 mA h g−1 respectively) while the control Ge electrode delivered no capacity at these rates. Post-mortem SEM and TEM investigations showed the integrity of the Ge/CNT electrodes and the effectiveness of the CNT network in alleviating the volume changes of Ge NPs during lithiation/delithiation.
Experimental section
Chemicals
All synthesis was performed in an oxygen-free, dry argon atmosphere, using standard Schlenk line and glove box techniques. Hexane (≥98.5%), isopropanol (IPA, ≥99.95%) and ethanol (≥99.5%) were purchased from Lennox, Ireland. Oleylamine (OLA, 70%), oleic acid (OA, 90%), hexamethyldisilazane (HMDS, > 99%), H2SO4 (98%), MWCNTs (>95%) and Ni(NO3)2·6H2O were bought from Sigma-Aldrich. Germanium(IV) iodide (GeI4, 99.99%) and HNO3 (65%) were purchased from Thermo Scientific Chemicals. All reagents were used as received without further purification. Cu foil (9 μm thick) was provided by Pi-Kem. Battery-grade 1.0 M LiPF6 in ethylene carbonate (EC) and diethyl carbonate (DEC), EC-DEC (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) and vinylene carbonate (VC, 97%, additives) were purchased from Sigma Aldrich.
:1 v/v) and vinylene carbonate (VC, 97%, additives) were purchased from Sigma Aldrich.
Synthesis
Ge NPs were produced using a slightly modified approach from the literature.29 GeI4 (60 mg), OLA (10 mL) and OA (0.75 mL) were added to a 20 mL scintillation vial and sealed inside a glovebox. The solution was vigorously stirred and heated at 80 °C until it was clear, and it was then transferred to a reaction vessel and evacuated at room temperature for 30 minutes and at 100 °C for another 30 minutes to remove oxygen and moisture. Next, HMDS (1 mL) was added to the vessel before being progressively heated (2 °C min−1) to 260 °C. During the heating process, the solution transformed from colorless to yellow, and it changed rapidly from yellow to dark brownish-red at 260 °C. The solution was aged for 30 minutes and then quenched using a water bath. The Ge NPs were collected and washed twice with a mixture of hexane/ethanol using a centrifuge machine. Finally, the collected Ge NPs were suspended in 10 mL hexane for further experiments.CNT treatments
MWCNTs were added to a 1:2 mixture of H2SO4 and HNO3, with a 15 mg mL−1 ratio. The mixture was then vigorously heated and refluxed at 90 °C for 40 minutes. Following the acid treatment, the MWCNTs were washed four times with deionized water and ethanol before being vacuum-dried at 80 °C.
Electrophoretic deposition
The fabrication procedure for additive-free Ge/CNT nanocomposite films for LIB electrodes is depicted schematically in Fig. 1. First, two Cu foil plates spaced approximately 2 cm apart were immersed in a MWCNT bath, and a direct current (DC) voltage of 300 V was applied using a high voltage power supply (TECHNIX SR-5-F-300) for 40 seconds. The MWCNT bath was prepared by dispersing MWCNTs in IPA (concentration: 0.2 mg mL−1) with the addition of Ni(NO3)2·6H2O (concentration: 0.1 mg mL−1) under sonication. The deposition of MWCNT was observed on the negative electrode. The electrophoretically formed MWCNT films were dried in a vacuum oven at 80 °C for 12 hours. The mass loading of CNTs on the Cu foil was between 0.1 and 0.13 mg cm−2. Afterwards, the MWCNT electrodes were immersed in a Ge NP bath, and a DC voltage of 300 V was applied. The Ge NP bath was prepared by diluting 0.4 mL of the as-synthesized Ge NPs in 10 mL of hexane. During the EPD process, the brown color of the Ge NP bath reduced in intensity after a few minutes, indicating that most of the NPs had migrated to and assembled on the electrode under the high voltage. Deposition of Ge NPs on the positive electrode was observed, and the film thickness could be controlled by varying the concentration of the Ge NP solution, the EPD time, and the EPD voltage. Ge/CNT nanocomposite electrodes had masses ranging from 0.15 to 0.35 mg of Ge NPs and contained 25–50 wt% CNTs. To make a comparison, a Ge NP film on copper foil was also prepared under the same conditions as the Ge NP film on MWCNTs. All battery electrodes had mass loadings of 0.13–0.3 mg cm−2 of the active material (Ge). The Ge/CNT nanocomposite electrodes with GeNPs/CNTs mass ratios of 1:1, 2:1, and 3:1, were denoted as Ge/CNT-1, Ge/CNT-2, and Ge/CNT-3, respectively.
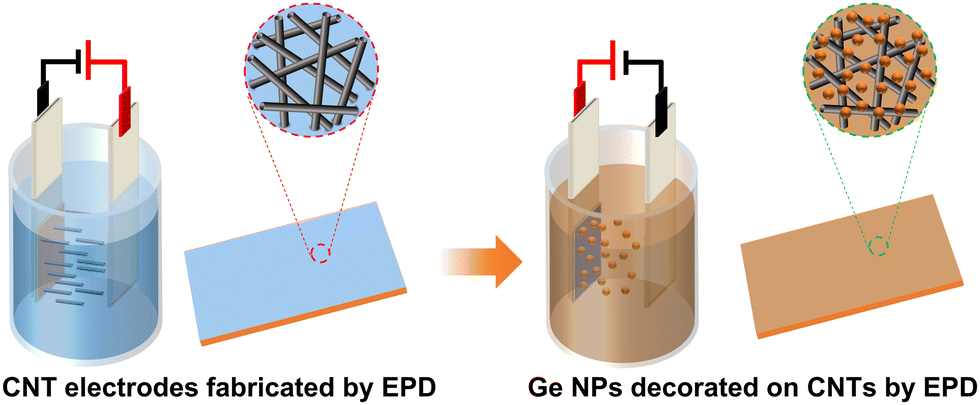 | ||
| Fig. 1 Schematic illustration of the two-step process for the fabrication of Ge/CNT nanocomposite electrodes (fabrication of CNT electrodes and decoration of Ge NPs on CNT electrodes) by using EPD. | ||
Ligand removal
The resulting films contained low conductivity long-chain organic ligands (OLA and OA). The removal of these ligands was necessary to improve film conductivity while also reducing inactive material.25,26 The electrodes were treated in a 20 mM solution of ammonium sulfide in methanol for 30 seconds, followed by a methanol rinse to remove any unreacted ammonium sulfide and free organics. After that, the electrodes were dried overnight at 70 °C before being assembled into the cell. In general, an electrode (12 mm diameter) gained around 20–30 μg due to formation of copper sulfide from the copper foil.Characterization
The crystalline phases of the resulting materials were investigated using powder X-ray diffraction (PANalytical Empyrean instrument) with Cu K radiation (λ = 1.54056 Å). Field-emission scanning electron microscopy (SEM, Helios G4 CX) was used to analyze the morphologies and structures of the materials, and transmission electron microscopy (TEM) was performed using a JEOL JEM-2100F analytical electron microscope (JEOL, Japan) operating at 200 keV. Raman spectroscopy (Horiba Labraman 300 spectrometer system) was used to characterize Ge and CNTs using 532 nm laser excitation. X-ray photoelectron spectroscopy (XPS) was performed using a Kratos AXIS ULTRA spectrometer with a mono Al Kα X-ray gun.Electrochemical characterization
Half-cells containing an active electrode, Li metal foil as a counter/reference electrode, and a polypropylene separator (Celgard 2400) soaked in electrolyte (CR 2032) were assembled in an argon filled glove box (Vigor). 1 M LiPF6 (EC:DEC, 50:50 v/v) + 3% VC (Sigma Aldrich) served as the electrolyte. Galvanostatic cycling studies were performed in a potential range of 0.01–1.5 V vs.Li/Li+ utilizing a Neware battery cycler instrument. The mass of the active material was used to calculate all current densities. Electrochemical impedance spectroscopy (EIS) was conducted at frequencies ranging from 0.1 Hz to 10 kHz, and cyclic voltammetry (CV) was conducted at a scan rate of 0.1 mV s−1 by using a biologic MPG-2 instrument.
Results and discussion
A colloidal wet-chemical synthesis method was employed to synthesize uniform Ge NPs as these methods are well-known for producing controlled shapes and sizes through the use of surfactant ligands.25,30 X-ray diffraction (XRD) of the synthesized Ge NPs (Fig. 2a) shows reflections which could be indexed to cubic Ge (JCPDS no. 04-006-9329) with no signs of any impurity phases. The characteristics of Ge NPs were further examined using Raman spectroscopy (Fig. 2b). A sharp peak at 296 cm−1 corresponds to the optical mode of crystalline Ge,15,31,32 while a weak signal at 269 cm−1 is attributed to GeO2 vibrations, which are consistent with previous studies,31,33 suggesting that the crystalline Ge surface was partially oxidized due to the reaction of Ge with oxygen. Furthermore, XPS analysis was conducted to investigate the chemical bonding of Ge and O, as shown in Fig. S1 (ESI†). Fig. S1a (ESI†) confirms the presence of Ge and O on the surface of Ge NPs. The high resolution XPS spectrum of the Ge 3d region shows 6 peaks at 29.2, 29.8, 31.0, 31.6, 32.8 and 33.4 eV (Fig. S1b, ESI†). Two peaks appearing at 31.0 and 31.6 eV correspond to Ge+2 3d5/2 and Ge+2 3d3/2, respectively, while the peaks located at 32.8 and 33.4 eV are ascribed to Ge+4 3d5/2 and Ge+4 3d3/2, respectively, indicating the occurrence of partial oxidization of Ge (GeO and GeO2) or the interaction between Ge NPs and ligands used for Ge NP synthesis. Besides, the two other binding peaks detected at 29.2 and 29.8 eV in the XPS spectrum of the Ge 3d core level are attributed to Ge0 3d5/2 and Ge0 3d3/2 (Ge metal), respectively.16,32,34 These XPS results suggest that crystalline Ge was synthesized successfully, with subsequent air exposure leading to the formation of GeO and GeO2.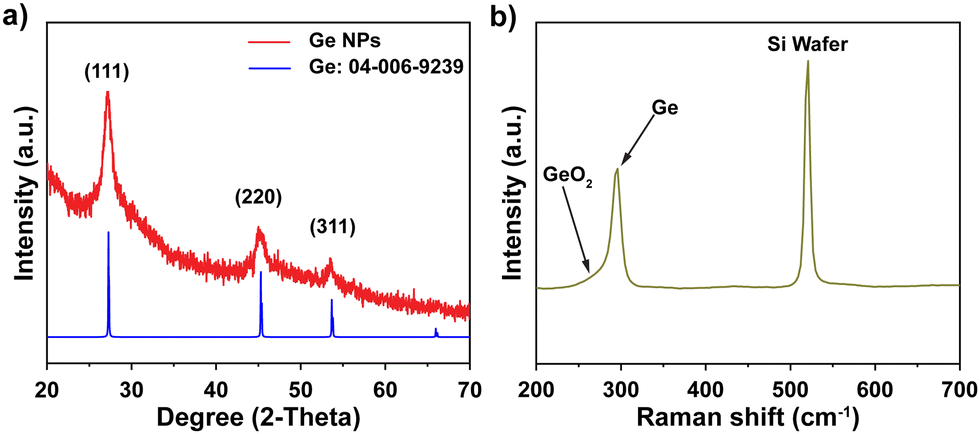 | ||
| Fig. 2 (a) XRD pattern and (b) Raman spectra of Ge NPs. | ||
The FE-SEM image shown in Fig. 3a reveals the nanoparticle morphology of the as-prepared Ge. Ge NPs had an average diameter of 10.5 nm, with a standard deviation of 5.1 nm (Fig. 3b), analyzed from 100 nanoparticles in STEM images (Fig. S2, ESI†). TEM image (Fig. 3c) and HR-TEM image (Fig. 3d) gave more specific details of the morphology of Ge NPs, confirming the successful synthesis of Ge NPs according to the adopted protocol. A distinct (111) lattice fringe with a d-spacing of 0.326 nm is consistent with the NPs being cubic Ge. The SAED pattern (inset, Fig. 3d) further verifies the crystal phase of Ge NPs, matching with the XRD results. The scanning TEM (STEM) and TEM-EDS elemental mapping were performed to showcase the distribution of Ge nanocrystals (Fig. S3, ESI†), in which Ge was identified as white dots (Fig. S3a, ESI†). Ge and O were evenly distributed throughout the sample (Fig. S3b and c, ESI†).
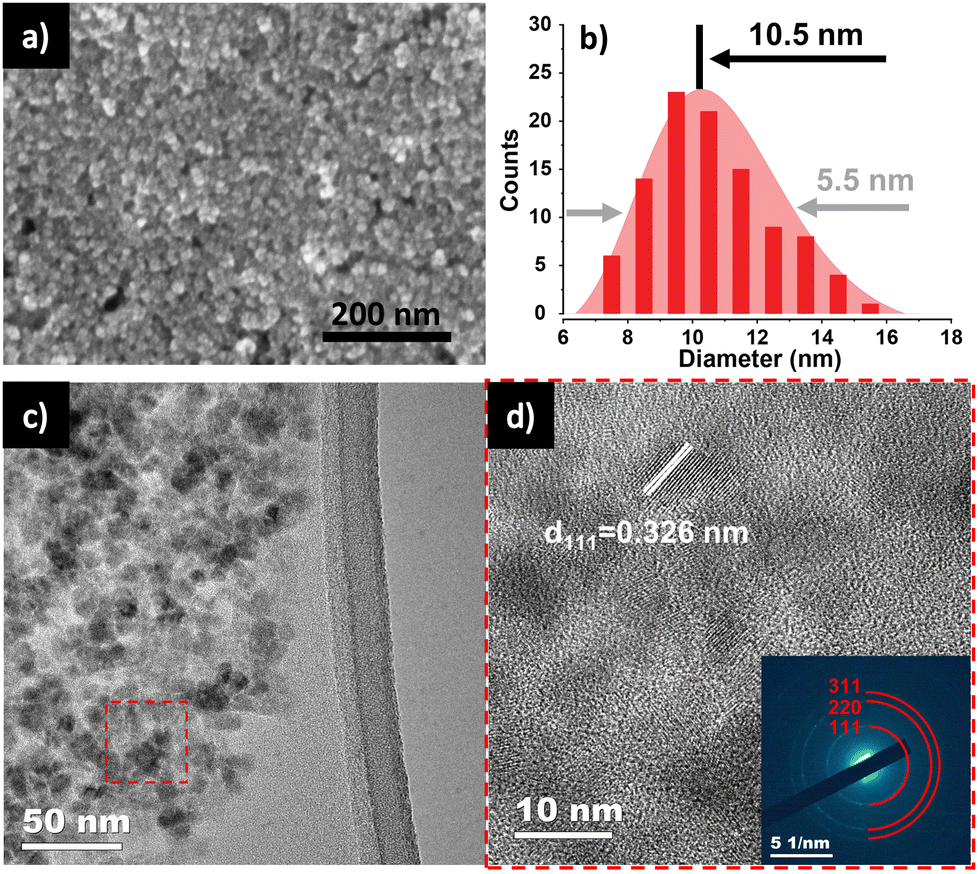 | ||
| Fig. 3 (a) HR-SEM of Ge NPs. (b) Diameter distribution of Ge NPs. (c) TEM and (d) HR-TEM with SAED pattern inset of Ge NPs. | ||
Electrophoretic deposition (EPD) was utilized to assemble colloidal Ge NPs on the electrode as this method has previously been used to efficiently prepare nanocrystal films with robust mechanical properties.25,26,35 The electrode fabrication is depicted in Fig. 1. The morphology of binder-free electrodes fabricated by EPD is shown in Fig. S4 (ESI†). A good homogeneity of Ge NPs and CNTs deposited on the copper foil can be observed in Fig. S4a (ESI†) and Fig. S4b (ESI†), respectively. A dense and uniform film is critical for application in batteries because it maintains a continuous electrically conductive network throughout the film. The Ge NPs were arranged uniformly on the CNT surfaces, within the interwoven CNT network (Fig. S4c and d, ESI†). The diffraction peaks of the Ge/CNT nanocomposite electrodes demonstrate a sharp peak at 27.29°, corresponding to the (111) of cubic Ge (JCPDS no. 04-006-9329) (Fig. S5a, ESI†), with no XRD peaks identified for the crystalline carbon of CNTs. The Raman spectra of Ge/CNT, shows two strong peaks at 1338 and 1572 cm−1, which correspond to the D and G bands of CNTs (carbon), respectively, as well as a peak of the optical mode of metallic Ge at 298 cm−1 (Fig. S5b, ESI†).
Cyclic voltammetry (CV) was used to explore the mechanism of Li storage in Ge and Ge/CNT-1 samples. Fig. 4a and b show cyclic voltammograms of Ge and Ge/CNT-1 electrodes, obtained at 0.1 mV s−1. For the Ge-containing electrode, a shoulder peak seen around 1.5 V appears to be related to the formation of the solid electrolyte interphase (SEI) layer.36–38 The magnitude of cathodic current begins to increase at potentials less than 1.0 V, related to Li alloying reactions. Two peaks were identified in the range of 0.8 to 0.1 V, which can be associated with distinct LixGe alloy formation.17,31 In the anodic scan, peaks observed between 0.25 and 1.0 V during the first cycle can be attributed to dealloying reactions, in which Li is extracted from the Li–Ge alloys to finally become Ge.3,13,31,36 The following cycles (the second and third cycles) showed overlapped curves and unchanged positions of the redox peaks, suggesting that the Ge-anodes achieved their reversible electrochemical behavior after the first cycle. The Ge/CNT-1 electrode shows similar Li storage behaviours to Ge samples with a slight difference in the first cathodic scan. The irreversible Li reactions happened earlier, about 1.7 V, and corresponded to the reaction of Li and functional groups on the CNT surface39 and the formation of the SEI layer. In the initial cycle of the Ge/CNT-1 electrode, these types of irreversible Li storage may lead to a high specific capacity and a low coulombic efficiency. The electrochemical performance of Ge and Ge/CNT-1 were assessed via charge/discharge galvanostatic cycles ranging from 0.01 to 1.5 V at C/5 (∼277 mA g−1). During the discharge process, the voltage of Ge anode (Fig. 4c) drops rapidly from the open circuit voltage to 1.5 V, and no electrochemical reaction occurs. A slope appears from 1.5 V to 0.5 V, then a plateau area is observed, and finally the voltage slowly declines to ∼0.01 V, attributed to SEI formation and Li alloying reactions, respectively. A plateau at 0.4 V can be clearly seen in the charge profile, which corresponds to the delithiation process for the formation of Ge.4,31 The discharge–charge profile of the Ge/CNT-1 sample as shown in Fig. 4d displays similar behaviours to the Ge sample and matches well with the CV data of Ge/CNT-1. The initial irreversible capacity of the Ge/CNT-1 sample, however, is more than that of the Ge sample. The irreversible capacity found in the first cycle can be attributed to the development of the SEI layer and the reaction between Li and the high surface area of CNTs, as seen in many carbon-based materials.3,39–41 Furthermore, the discharge–charge profile of CNT electrode (control sample to compare cyclic performance) confirms the high-capacity loss of the CNTs in the initial cycle (Fig. S6a, ESI†). The following discharge–charge profile of both samples exhibited similar plateau potentials for the alloying/dealloying process of Ge. However, the capacity of Ge sample decreased rapidly after 100 cycles, whereas the Ge/CNT-1 still retained a good capacity retention. The galvanostatic cycling findings of both electrodes at 0.2C provided more information on the superior performance of the Ge/CNT-1 sample over the Ge sample (Fig. 4e). Ge demonstrated a coulombic efficiency of 49% with a specific discharge capacity of 2440 mA h g−1 and a charge capacity of 1218 mA h g−1, whereas Ge/CNT-1 demonstrated an initial discharge/charge capacity of 4488 and 1306 mA h g−1, respectively, with a coulombic efficiency of 29%. The low coulombic efficiency of Ge/CNT-1 can be explained by the high irreversible capacity attributed to large SEI formation of CNTs in the initial cycle. In the first cycle, the CNT sample demonstrated a poor coulombic efficiency of 23% (Fig. S6b, ESI†). The considerable amount of Li consumed in the SEI formation of Ge/CNT and CNT electrodes can be seen obviously in Fig. S7 (ESI†). And the relative amount of Li consumed in the SEI formation is shown in Table S1 (ESI†). Nonetheless, CNTs play an important role in improving the stable cycling performance of the Ge/CNT electrode. CNTs act as a buffer layer, allowing the materials to keep their integrity by tolerating volume changes during the Li insertion/extraction process.16,31 After 1000 cycles, the Ge/CNT-1 electrode still provides a reversible charge capacity of 819 mA h g−1, which is 62.6% of the initial capacity. In comparison, the Ge electrode rapidly loses capacity after 100 cycles, reaching a capacity of 266 mA h g−1, equating to a capacity retention of 21.8% (Fig. 4e). The capacity contribution of the CNTs to Ge/CNT nanocomposite electrodes is minor as the obtained capacity of CNTs was 100 mA h g−1 after 100 cycles (Fig. S6b, ESI†), which is significantly lower than the capacity of Ge. The effect of the mass ratio between Ge and CNT in the electrodes was also investigated. The Ge/CNT-2 and Ge/CNT-3 were prepared with controlled mass ratios for Ge and CNTs of 2:1 and 3:1, respectively. The Ge/CNT-2 and Ge/CNT-3 electrodes delivered initial charge capacities of 1304, and 1357 mA h g−1, and capacity retentions of 50.1% and 30.5% after 100 cycles, respectively (Fig. S8, ESI†). These performances are lower than that of Ge/CNT-1. However, the initial coulombic efficiencies were improved when increasing the amount of Ge NPs compared to the initial coulombic of Ge/CNT-1 (Fig. 4f). The effect of CNT content to the low coulombic efficiency of Ge/CNT-1 continues in a few first cycles, then the coulombic efficiency of Ge/CNT-1 reached 97.2% after 10 cycles and maintained around 98% after that (Fig. 4f). The reduced CNT % in the Ge/CNT (an increased Ge:CNT mass ratio) can affect the cyclic stability of the Ge/CNT nanocomposite electrodes because the low CNT content may be insufficient to avoid nanosized Ge particle aggregation during cycling. The capacity of the higher ratio Ge/CNT nanocomposite electrodes may thus deteriorate because of substantial volume expansion during the alloying process.
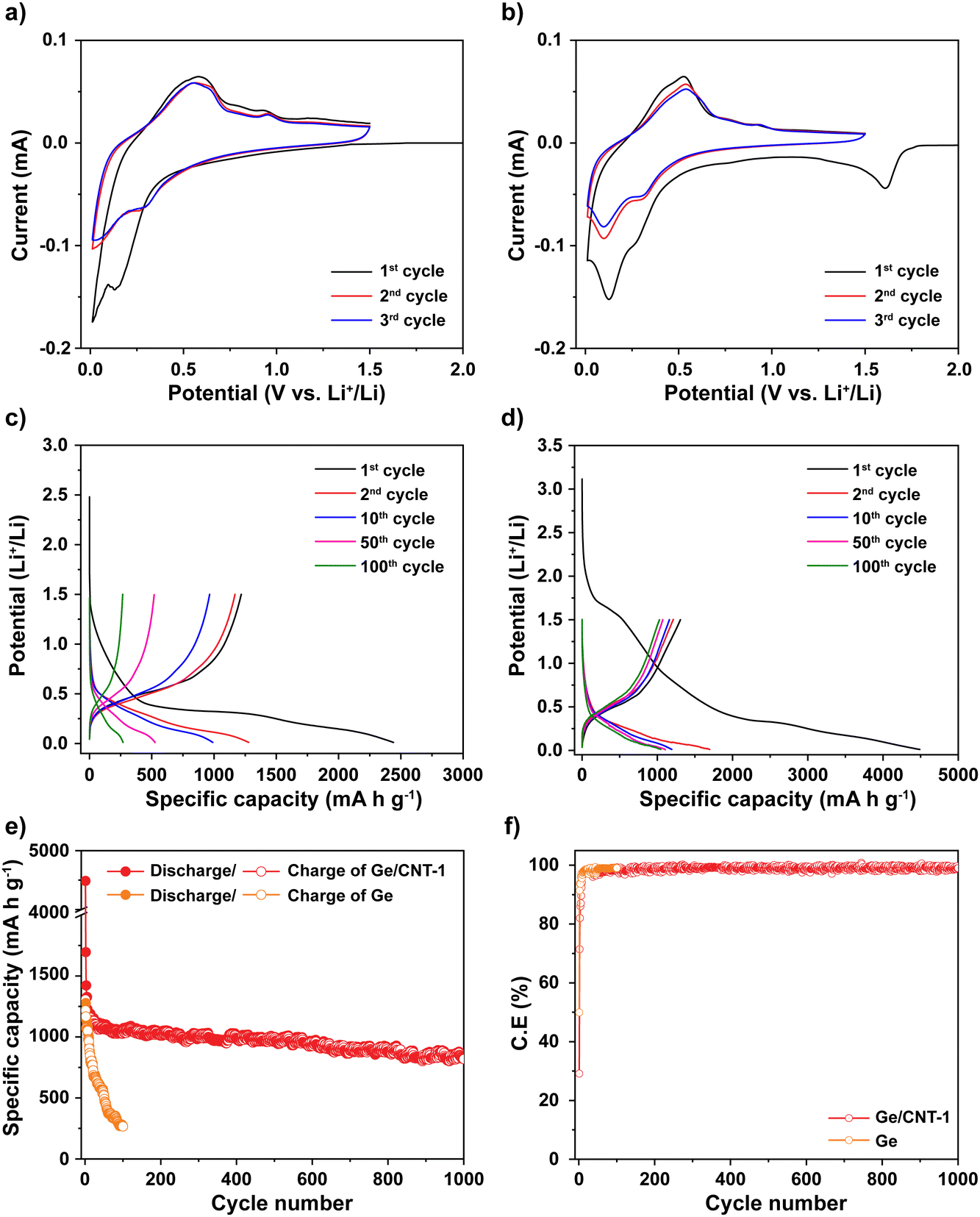 | ||
| Fig. 4 Cyclic voltammetry diagrams of (a) Ge electrode and (b) Ge/CNT-1 electrode. Discharge–charge profiles of (c) Ge electrode and (d) Ge/CNT-1 electrode. (e) Cyclability with (f) coulombic efficiency of Ge electrode and Ge/CNT-1 electrode at C/5. | ||
Rate capability testing at various current densities from C/10 to 10C was used to evaluate the reversible discharge–charge storage capacities, as illustrated in Fig. 5a. At rates of C/10, C/5, C/2, 1C, and 2C, the Ge/CNT-1 electrode exhibited specific capacities of 1269, 1190, 1122, 994, and 799 mA h g−1, respectively. The Ge/CNT-1 electrode was superior to the Ge electrode, which delivered low specific capacities at high current rates of 1C (522 mA h g−1) and 2C (253 mA h g−1). The Ge electrode did not cycle at rates of 5C and 10C while the Ge/CNT-1 electrode still exhibited specific capacities of 519 and 210 mA h g−1, respectively. When the current density reverted to the initial current rate of C/10, the specific capacity of the Ge electrode returned to 610 mA h g−1 (53.2% of the original capacities), whereas Ge/CNT-1 attained a good capacity of 1254 mA h g−1 (98.8% of the original capacities). It is believed that the contribution of CNT in Ge/CNT nanocomposite electrodes can prevent the agglomeration of Ge active material, and suppress the damage caused by the volume change of Ge NPs, thus further improving the electrode performance. Moreover, a cycling test with hundreds of cycles was performed to investigate the long-term resilience of the electrodes at high charge/discharge rates (1C∼1384 mA g−1), as shown in Fig. 5b. Before cycling at a high current rate of 1C, the electrodes were first activated at a modest current rate of C/10 for five cycles. Remarkably, Ge/CNT-1 delivered a reversible capacity of 910 mA h g−1 at the first cycle for the high current rate of 1C (overall the sixth cycle) and continued to deliver a high capacity of 686 mA h g−1 after 350 cycles (equal to 75.3% of the capacity at the 6th cycle, comparable to a capacity loss of 0.07% per cycle). The Ge electrode, in contrast, experiences a slow capacity degradation that resulted in a capacity of 122 mA h g−1 at the 100th cycle, which is equivalent to 12.5% of the capacity during the 6th cycle.
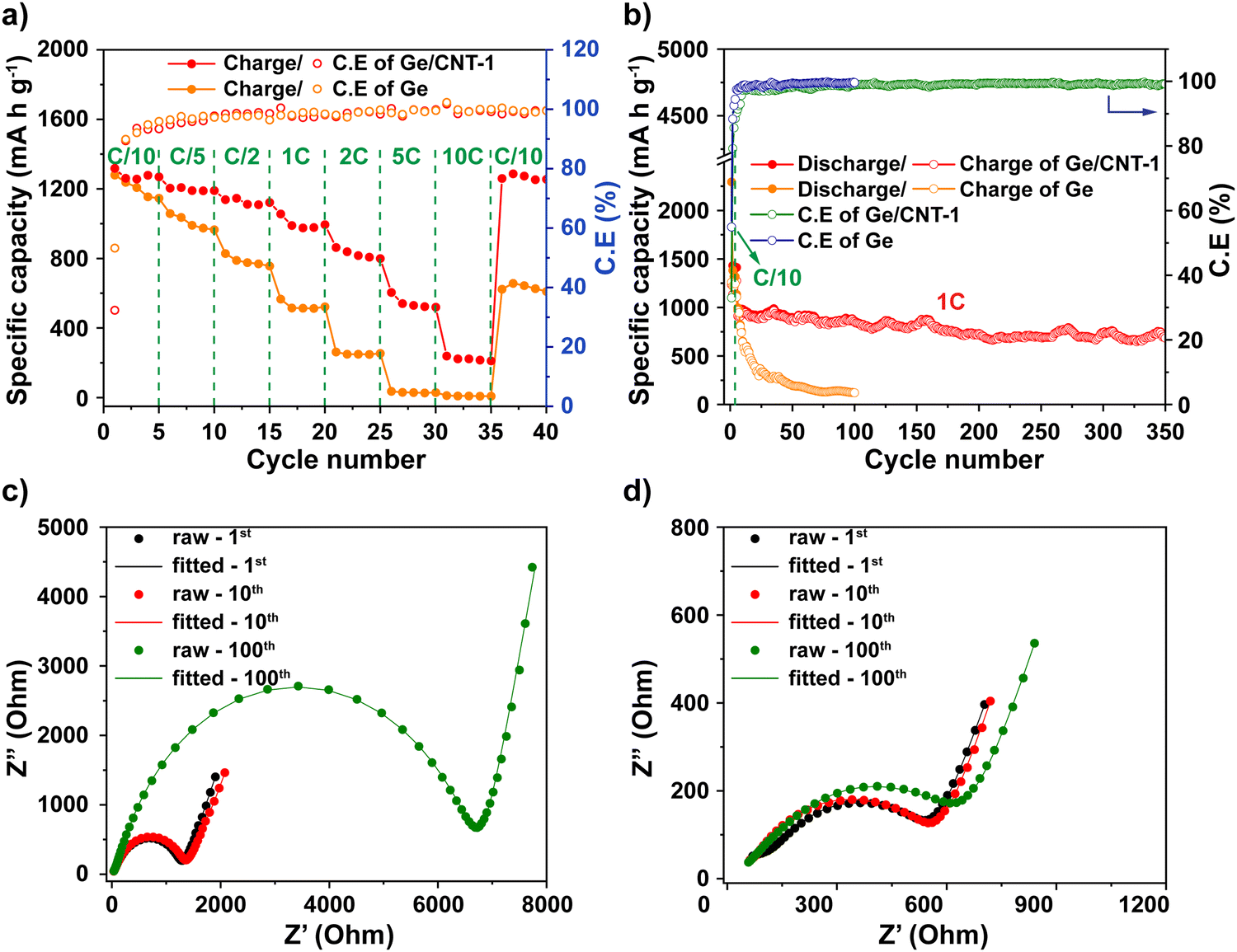 | ||
| Fig. 5 (a) Rate capability of Ge and Ge/CNT-1 electrodes at different current densities varying from C/10 to 10C. (b) Cyclability and coulombic efficiency of Ge and Ge/CNT-1 electrodes at a rate of 1C, the first 5 cycles were performed at a low current rate of C/10. Nyquist plot of (c) Ge and (d) Ge/CNT-1. | ||
Electrochemical impedance spectroscopy (EIS) was measured to clarify the remarkable electrochemical characteristics of Ge/CNT-1. An equivalent circuit shown in Fig. S9 (ESI†) was used for fitting the Nyquist plots of the Ge and Ge/CNT-1 electrodes (Fig. 5c and d, respectively). The fitted EIS data are listed in Table S2 (ESI†). According to the data shown in Table S2 (ESI†), both materials tested had comparable Rs values of 9.88 Ohm (Ge electrode) and 9.66 Ohm (Ge/CNT-1) after the first cycle, then Rs reduced gradually to 5.21 Ohm (Ge electrode) and 6.34 Ohm (Ge/CNT-1) at 100th cycle. The RSEI of Ge and Ge/CNT-1 increased over the number of cycles, signifying the development of the SEI layer during cycling. RSEI value of Ge/CNT-1 in the first cycle was higher than that of Ge, indicating that the SEI formation occurred strongly on the surface of Ge/CNT-1, mainly due to the high surface area of CNTs.42 Ge/CNT-1 electrode had a charge transfer resistance (Rct) of 457 Ohm, which is lower than 1194 Ohm of the Ge electrode after first cycling. This is explained by the greater specific area of CNTs contributing to the Ge/CNT-1 electrode, which promotes a favorable charge transfer process by providing more active Li-ion transfer sites on the electrode surface.43 The Nyquist plots of both electrodes displayed larger semicircles after long cycling (Fig. 5c and d), resulting in a larger charge transfer resistance. However, the Rct of Ge electrode increased rapidly to 6600 Ohm after 100 cycles, compared to 553.4 Ohm for the Ge/CNT-1. These results explain the reason why pure Ge exhibited a poor electrochemical performance related to the increase in the electrochemical impedance of the electrode during cycling. Furthermore, they confirm that the CNT content prevents the deterioration of the Ge/CNT electrodes and enhances the conductivity of Ge/CNT electrodes.
Post-mortem analysis of the Ge/CNT-1 used SEM, TEM and EDS mapping to understand the underlying factors that contributed to its superior cycling stability over extended periods of time. The optical photographs of the Ge/CNT-1 electrodes before and after 100 cycles did not show any obvious alterations (Fig. S10, ESI†). Even after 100 full discharge and charge cycles, the Ge/CNT-1 surface did not exfoliate, confirming the stable adhesion of the active material to the current collector and referencing the superior mechanical stability of the binder-free electrode. The surface of Ge/CNT-1 electrode has undergone restructuring after cycling with few noticeable cracks, as shown in the SEM image (Fig. 6a). However, the HR-SEM of Ge/CNT-1 observed after 100 cycles clearly shows that the Ge NPs were embedded in CNT buffer layers, which can retain the integrity of the materials by tolerating the volume changes (Fig. 6b).6,31 In contrast, for Ge electrode, noticeable agglomeration of Ge NPs was observed after cycling (Fig. S11, ESI†). Furthermore, the impact of volume expansion of both electrodes is shown in Fig. S12, ESI† (side view of electrode after cycling). The nanoscale network of 1D CNTs can offer effective electron-transport routes directly to the copper current collector during cycling, explained why Ge/CNT-1 exhibited a lower charge-transfer resistance compared to the Ge electrode over cycling.6,16,17 The TEM images further suggest the restructuring of pristine Ge NPs into Ge interconnected structures after long-term cycling (Fig. 6c and d). The Ge NPs became amorphous as evident by the SAED pattern (Fig. 6c inset) due to the repeated Li alloying/dealloying reactions. The mapping profile of Ge/CNT-1 (Fig. 6f and g), along with the scanning-TEM (STEM) image (Fig. 6e) respectively, both provide evidence that C and Ge were uniformly distributed in Ge/CNT-1 after 100 cycles. The Ge/CNT nanocomposite structure enables extended cyclability, with the electrode not being greatly impacted by the usual problems related to pulverization, agglomeration, and volume change.16,44
 | ||
| Fig. 6 (a and b) SEM images of Ge/CNT-1. (c) TEM image with the diffraction pattern (inset) of Ge/CNT-1. (d) HR-TEM image of Ge/CNT-1. (e–g) STEM image and elemental mapping of (C and Ge) of Ge/CNT-1 after 100 cycles. | ||
Conclusions
In summary, binder-free Ge/CNT nanocomposite electrodes were formed with Ge nanoparticles decorated on MWCNT networks via an EPD method. The CNTs formed an interconnected electronic network and played an important role in mitigating the effect of volume changes during lithiation/delithiation, protecting the Ge nanoparticles from cracking and/or crumbling, and retaining the integrity of the Ge/CNT nanocomposite electrodes. The Ge/CNT nanocomposite electrodes exhibited superior long-term cycle stabilities (up to 1000 cycles) and superior rate capabilities (compared to a control Ge nanoparticle anode) with the specific capacities close to the theoretical value of Ge, demonstrating the stabilising role played by CNTs in the composite anodes. Post-cycling analysis of the nanocomposite electrode using optical imaging, SEM, and TEM provided additional insights into the good electrical connection, mechanical stability, and battery performance of the nanocomposite electrode prepared via EPD. In combination, this report represents the next stage in utilizing EPD for manufacturing binder-free electrodes with high performance in LIBs.Author contributions
All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
X.-M. P. acknowledges funding from the Irish Research Council (IRC) under Grant Number IRCLA/2017/285. K. M. R. acknowledges support from Science Foundation Ireland (SFI) under the Principal Investigator Program under contract no. 16/IA/4629 and under grant no. SFI 16/MERA/3419. S. S. and N. N. P. acknowledge the funding and support from the Department of Chemical Sciences, University of Limerick. S. A. A. and H. G. acknowledge support from Science Foundation Ireland under grant no. 18/SIRG/5484. The authors would also like to thank Dr Fathima Laffir (XPS instrument scientist – Bernal Institute) for help with XPS analysis.References
- D. Howell, US Department of Energy, Washington, DC, 2010 Search PubMed.
- R. A. DiLeo, S. Frisco, M. J. Ganter, R. E. Rogers, R. P. Raffaelle and B. J. Landi, J. Phys. Chem. C, 2011, 115, 22609–22614 CrossRef CAS.
- W. Lang, C. Yue, M. Dang, G. Wang, Y. Chen, F. Hu, Z. Liu and J. Shu, J. Power Sources, 2023, 560, 232706 CrossRef CAS.
- C. Jo, B. Wen, H. Jeong, S. K. Park, Y. Son and M. De Volder, ACS Nano, 2023, 17, 8403–8410 CrossRef CAS PubMed.
- C.-M. Park, J.-H. Kim, H. Kim and H.-J. Sohn, Chem. Soc. Rev., 2010, 39, 3115–3141 RSC.
- I.-S. Hwang, J.-C. Kim, S.-D. Seo, S. Lee, J.-H. Lee and D.-W. Kim, Chem. Commun., 2012, 48, 7061–7063 RSC.
- Y. Chen, Y. Zou, X. Shen, J. Qiu, J. Lian, J. Pu, S. Li, F.-H. Du, S.-Q. Li, Z. Ji and A. Yuan, J. Energy Chem., 2022, 69, 161–173 CrossRef CAS.
- G. Uchida, K. Nagai, Y. Habu, J. Hayashi, Y. Ikebe, M. Hiramatsu, R. Narishige, N. Itagaki, M. Shiratani and Y. Setsuhara, Sci. Rep., 2022, 12, 1742 CrossRef CAS PubMed.
- X. Li, Z. Yang, Y. Fu, L. Qiao, D. Li, H. Yue and D. He, ACS Nano, 2015, 9, 1858–1867 CrossRef CAS PubMed.
- T. Kennedy, E. Mullane, H. Geaney, M. Osiak, C. O’Dwyer and K. M. Ryan, Nano Lett., 2014, 14, 716–723 CrossRef CAS PubMed.
- F.-W. Yuan, H.-J. Yang and H.-Y. Tuan, ACS Nano, 2012, 6, 9932–9942 CrossRef CAS PubMed.
- K. H. Seng, M. H. Park, Z. P. Guo, H. K. Liu and J. Cho, Angew. Chem., 2012, 124, 5755–5759 CrossRef.
- X. Liu, J. Hao, X. Liu, C. Chi, N. Li, F. Endres, Y. Zhang, Y. Li and J. Zhao, Chem. Commun., 2015, 51, 2064–2067 RSC.
- X. H. Liu, S. Huang, S. T. Picraux, J. Li, T. Zhu and J. Y. Huang, Nano Lett., 2011, 11, 3991–3997 CrossRef CAS PubMed.
- S. Abdul Ahad, S. Kilian, M. Zubair, V. A. Lebedev, K. McNamara, K. M. Ryan, T. Kennedy and H. Geaney, J. Mater. Chem. A, 2021, 9, 20626–20634 RSC.
- D. T. Ngo, R. S. Kalubarme, H. T. T. Le, J. G. Fisher, C.-N. Park, I.-D. Kim and C.-J. Park, Adv. Funct. Mater., 2014, 24, 5291–5298 CrossRef CAS.
- D. T. Ngo, H. T. T. Le, C. Kim, J.-Y. Lee, J. G. Fisher, I.-D. Kim and C.-J. Park, Energy Environ. Sci., 2015, 8, 3577–3588 RSC.
- M. C. Schulze, R. M. Belson, L. A. Kraynak and A. L. Prieto, Energy Storage Mater., 2020, 25, 572–584 CrossRef.
- W. Xiao, Y. Qiu, Q. Xu, J. Wang, C. Xie, J. Peng, J. Hu, J. Zhang and X. Li, Electrochim. Acta, 2020, 364, 137278 CrossRef CAS.
- S. Fang, L. Shen, H. Zheng and X. Zhang, J. Mater. Chem. A, 2015, 3, 1498–1503 RSC.
- J. Hao, L. Pan, H. Zhang, C. Chi, Q. Guo, J. Zhao, Y. Yang, X. Liu, X. Ma and Y. Li, Chem. Eng. J., 2018, 346, 427–437 CrossRef CAS.
- B. J. Landi, M. J. Ganter, C. D. Cress, R. A. DiLeo and R. P. Raffaelle, Energy Environ. Sci., 2009, 2, 638–654 RSC.
- G. Cui, L. Gu, N. Kaskhedikar, P. A. van Aken and J. Maier, Electrochim. Acta, 2010, 55, 985–988 CrossRef CAS.
- A. R. Boccaccini, J. Cho, J. A. Roether, B. J. C. Thomas, E. Jane Minay and M. S. P. Shaffer, Carbon, 2006, 44, 3149–3160 CrossRef CAS.
- D.-H. Ha, M. A. Islam and R. D. Robinson, Nano Lett., 2012, 12, 5122–5130 CrossRef CAS PubMed.
- G. Bree, H. Geaney, K. Stokes and K. M. Ryan, J. Phys. Chem. C, 2018, 122, 20090–20098 CrossRef CAS.
- G. Bree and C. Tong John Low, Batteries Supercaps, 2023, 6, e202200441 CrossRef CAS.
- A. Hajizadeh, T. Shahalizade, R. Riahifar, M. S. Yaghmaee, B. Raissi, S. Gholam, A. Aghaei, S. Rahimisheikh and A. S. Ghazvini, J. Power Sources, 2022, 535, 231448 CrossRef CAS.
- D. D. Vaughn, II, J. F. Bondi and R. E. Schaak, Chem. Mater., 2010, 22, 6103–6108 CrossRef.
- Y. Yin and A. P. Alivisatos, Nature, 2005, 437, 664–670 CrossRef CAS PubMed.
- D. Li, H. Wang, H. K. Liu and Z. Guo, Adv. Energy Mater., 2016, 6, 1501666 CrossRef.
- P. R. Abel, A. M. Chockla, Y.-M. Lin, V. C. Holmberg, J. T. Harris, B. A. Korgel, A. Heller and C. B. Mullins, ACS Nano, 2013, 7, 2249–2257 CrossRef CAS PubMed.
- G. Cui, L. Gu, L. Zhi, N. Kaskhedikar, P. A. van Aken, K. Müllen and J. Maier, Adv. Mater., 2008, 20, 3079–3083 CrossRef CAS.
- C.-I. Wang, T.-J. Chang, C.-Y. Wang, Y.-T. Yin, J.-J. Shyue, H.-C. Lin and M.-J. Chen, RSC Adv., 2019, 9, 592–598 RSC.
- D.-H. Ha, T. Ly, J. M. Caron, H. Zhang, K. E. Fritz and R. D. Robinson, ACS Appl. Mater. Interfaces, 2015, 7, 25053–25060 CrossRef CAS PubMed.
- K. H. Seng, M.-h Park, Z. P. Guo, H. K. Liu and J. Cho, Nano Lett., 2013, 13, 1230–1236 CrossRef CAS PubMed.
- J. Cheng and J. Du, CrystEngComm, 2012, 14, 397–400 RSC.
- D. T. Ngo, R. S. Kalubarme, M. G. Chourashiya, C.-N. Park and C.-J. Park, Electrochim. Acta, 2014, 116, 203–209 CrossRef CAS.
- K. Tang, R. J. White, X. Mu, M.-M. Titirici, P. A. van Aken and J. Maier, ChemSusChem, 2012, 5, 400–403 CrossRef CAS PubMed.
- D.-J. Xue, S. Xin, Y. Yan, K.-C. Jiang, Y.-X. Yin, Y.-G. Guo and L.-J. Wan, J. Am. Chem. Soc., 2012, 134, 2512–2515 CrossRef CAS PubMed.
- F.-W. Yuan and H.-Y. Tuan, Chem. Mater., 2014, 26, 2172–2179 CrossRef CAS.
- K. Kaneko, M. Li and S. Noda, Carbon Trends, 2023, 10, 100245 CrossRef CAS.
- H. T. T. Le, X.-M. Pham and C.-J. Park, New J. Chem., 2019, 43, 10716–10725 RSC.
- X.-M. Pham, D. T. Ngo, H. T. T. Le, P. N. Didwal, R. Verma, C.-W. Min, C.-N. Park and C.-J. Park, Nanoscale, 2018, 10, 19399–19408 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nh00501a |
| This journal is © The Royal Society of Chemistry 2024 |