
Mechanism of photocatalytic CO2 methanation on ultrafine Rh nanoparticles†
Xinyan
Dai
and
Yugang
Sun
 *
*
Department of Chemistry, Temple University, 1901 North 13th Street, Philadelphia, Pennsylvania 19122, USA. E-mail: ygsun@temple.edu
First published on 1st February 2024
Abstract
Selective hydrogenation of CO2 to yield CH4 relies on the appropriate catalysts that can facilitate the cleavage of CO bonds and dissociative adsorption of H2. Ultrafine Rh nanoparticles loaded on silica nanospheres were used as a class of photocatalysts to significantly improve the selectivity and reaction rate of producing CH4 from the mixture of CO2 and H2 under the illumination of a broadband visible light source. The intense light scattering resonances in the silica nanospheres generate strong electric fields near the silica surface to enhance the light absorption power in the supported ultrafine Rh nanoparticles, promoting the efficiency of hot electron generation in the Rh nanoparticles. The interaction of the hot electrons with the adsorbate species on the Rh nanoparticle surface weakens the C–O bond to facilitate the deoxygenation of CO2, favoring the production of CH4 with a unity selectivity at a faster rate in the presence of surface adsorbed hydrogen (H*). The systematic studies on reaction kinetics and diffuse reflectance infrared Fourier transform (DRIFT) spectroscopy under different conditions, including various temperatures, illumination powers, and feeding gas compositions, reveal the reaction mechanism responsible for CO2 methanation and the role of photoillumination.
New conceptsLight absorption in metal nanoparticle photocatalysts leads to the generation of heat (photothermal effect) and the generation of hot electrons (non-thermal effect), both of which can influence the catalytic reactions regarding kinetics and selectivity. The convolution of the photothermal and non-thermal effects makes it challenging to distinguish the contribution of hot-electron-driven chemistry (i.e., the non-thermal effect) and identify the corresponding mechanism. In this work, a composite catalyst of ultrafine Rh nanoparticles (with a diameter < 5 nm) dispersed on sub-micron-sized silica spheres (with a diameter of ∼400 nm) was used to catalyze CO2 hydrogenation in the presence of H2. The efficiency of generating hot electrons in metal nanoparticles under photoillumination usually increases as the nanoparticle size decreases and the strength of the excitation electric field increases. The silica spheres in the composite catalyst can exhibit intense light scattering resonances to generate enhanced electric fields near their surfaces to benefit energy absorption in the supported Rh nanoparticles, leading to a high efficiency of hot electron generation in the ultra-small Rh nanoparticles. The sub-micron size of the silica spheres allows packing a thin and loose catalyst bed on a platinum mesh to eliminate the temperature gradient and mass transport gradient in the catalyst, enabling the direct measurement of actual catalyst temperature. The ultrasmall size of Rh nanoparticles and continuous-wave xenon light source eliminates the possible local heating effect. Therefore, the non-thermal effect will make a dominant contribution to the photocatalytic performance. In addition, systematic studies on the reaction kinetics and steady-state DRIFT spectroscopy of adsorbate species allow for determination of the CO2 methanation reaction mechanism on the catalyst and the role of photoillumination on the rate-determining steps. The results highlight the potential of photocatalyst design to achieve efficient performance in both reaction kinetics and product selectivity. |
Introduction
Methanation of CO2 (Sabatier reaction) is kinetically challenging because of the difficulty in dissociating the strong C–O bond with a bond energy of 8.33 eV and the existence of a competitive reverse water gas shift (rWGS) reaction that produces CO.1 Supported Rh nanoparticles (NPs) have been widely used to catalyze the hydrogenation of CO2 due to the approximate binding of carbon-containing intermediates and hydrogen on the nanoparticles’ surface.2 Oxides represent a class of popular supports due to the low cost and ease of loading Rh NPs onto the supports using the impregnation method.3 The oxide supports are crucial to maintaining the high dispersity of Rh NPs, particularly at elevated temperatures, and promoting catalytic activity/selectivity relying on the synergistic interactions between the supports and Rh NPs. For instance, oxides of transition metals with variable oxidation numbers (e.g., semiconductor TiO2, CeO2, etc.) can be reduced by hydrogen to produce oxygen vacancies that migrate to the Rh/oxide interface to enable the dissociation of the C–O bond of intermediate species adsorbed on the Rh surface, promoting the production of CH4 against CO.4 The high activation energy of oxygen vacancy migration (e.g., 231.6 kJ mol−1 or 2.4 eV in reduced rutile TiO2)5 usually slows the overall reaction kinetics of CO2 hydrogenation.6 The surface Rh atoms interfaced with an oxide support represent only a small fraction of the overall surface Rh atoms, preventing the full capacity of the catalyst Rh NPs from achieving high reaction kinetics. The susceptibility of being reduced by hydrogen at elevated temperatures could sacrifice the stability of oxide supports and the corresponding Rh/oxide composite catalysts. On the other hand, it is promising yet challenging to enable rapid and selective production of CH4 on the Rh NPs supported on dielectric oxides (i.e., SiO2 and Al2O3) that do not react with hydrogen. However, slow CO2 hydrogenation catalyzed by the dielectric–oxide-supported Rh NPs favors the production of CO. Increasing temperature represents one effective strategy to accelerate CO2 hydrogenation, but the highly dispersed Rh NPs will suffer aggregation and fusion into larger NPs at high temperatures, lowering the availability of active surface Rh atoms. Although the selectivity of forming CH4 can be somehow improved at higher reaction temperatures, robust full methanation is still far from being achieved through thermal catalysis on dielectric-oxide-supported Rh NPs.Photocatalysis of CO2 hydrogenation provides an alternative way to lower the reaction temperature while achieving desirable reaction activity and selectivity and maintaining the stability of the catalysts. Recent examples include the use of Z-scheme semiconductor heterostructures7–9 and metal/support composites involving the photothermal effect,10–16 non-thermal effect,17–19 and metal–support interfacial interactions.20 Photocatalysis of CO2 hydrogenation on supported Rh NPs has also been recently investigated to improve the reaction kinetics and selectivity of CH4.21–24 For example, Liu and co-workers observed that both the photothermal effect and non-thermal effect were capable of accelerating full CO2 methanation on TiO2-supported Rh NPs under photoillumination.22 The origin of the non-thermal effect is unclear because the electronic coupling between the metallic Rh NPs and the semiconductor TiO2 support changes the optical responses of individual components (i.e., surface plasmon resonance absorption in Rh NPs and semiconductor interband absorption in the TiO2 support). Photoexcitation of the surface plasmon resonances in Al2O3-supported Rh nanocubes (37 nm in edge length) could simultaneously accelerate CO2 hydrogenation and increase CH4 selectivity from 50–60% to almost 100% when light intensity is high enough.21 The increased methanation selectivity is ascribed to the preferred injection of hot electrons into the antibonding orbitals of the C–O bond in the adsorbed HCO intermediates, facilitating cleavage of the C–O bond to favor the formation of CH4 with a lowered reaction activation energy under light. The interpretation is in contrast to the experimental results showing an independence of activation energy on light intensity. Kim and co-workers observed slight acceleration of the methanation reaction of low pressure CO2 and H2 (<0.04 atm) on SiO2-supported Rh NPs (<5 nm in size) under photoillumination, which was attributed to the direct photoexcitation of adsorbed CO2 molecules exhibiting a narrowed LUMO–HOMO gap.24 This explanation implies that light absorption in the Rh NPs does not influence the methanation reaction and is in apparent conflict with the superlinear dependence of reaction rate on light intensity observed under strong illumination. The reported controversial conclusions indicate the inaccuracy of the current understanding of the photocatalytic mechanism of CO2 methanation.25 For instance, cleavage of the C–O bond in intermediates may not represent the only rate-determining step (RDS) that is widely adopted in the electrochemical CO2 reduction. The inaccurate understanding of the catalytic mechanism limits the potential to maximize the activity and selectivity of photocatalytic CO2 methanation on highly dispersed Rh NPs, which exhibit a high surface area but weak optical absorption power.
Herein, we report the use of silica nanospheres (SiOx NSs) as both dielectric light antenna and support for highly dispersed Rh NPs with sizes smaller than 5 nm, in which range metal NPs usually exhibit a high efficiency of generating high-energy hot electrons for photocatalytic reactions upon photoillumination due to the size-dependent quantum behaviors.26–28 The intense light scattering resonances enhance electric fields locally near the surface of SiOx NSs to significantly boost absorption of visible light in the small Rh NPs attached to the SiOx NSs, further favoring hot-electron-driven photocatalysis.29 The synergy of optical responses in SiOx NSs and small Rh NPs makes the Rh-NP/SiOx-NS composite particles a unique class of system to maximize the contribution of hot electrons to CO2 hydrogenation even using the CW visible light source with a power density as low as 1.5 W cm−2. This merit enables us to decipher the role of photoexcited hot carriers generated in the small Rh NPs in determining the selectivity and kinetics of photocatalytic CO2 hydrogenation.
Results and discussion
Typical Rh-NP/SiOx-NS samples with Rh NPs (loading content of 2.14 wt%) of different sizes were synthesized using an in situ reduction method (ESI,† Part I including Fig. S1). X-ray photoelectron spectroscopy of the as-synthesized composite particles exhibits strong signals of Rh, revealing the high-density loading of Rh NPs on the surface of SiOx NSs (Fig. S2, ESI†). The diffuse reflectance spectra (DRS) of the composite particles in the ultraviolet-visible region exhibit distinct absorption peaks at ∼450 nm and ∼700 nm regardless of the size of Rh NPs, while the bare SiOx NSs exhibit zero absorption in the visible spectral region (Fig. S3c, ESI†), indicating that the Rh NPs are responsible for the optical absorption. On the other hand, an aqueous dispersion of Rh NPs with similar sizes shows a peak-less feature in the absorption spectrum. The spectral difference between the supported and freestanding Rh NPs highlights that the SiOx NSs enhance optical absorption in Rh NPs due to the strong light scattering resonances in the SiOx NSs.30,31 In contrast, the Rh NPs supported on alumina (Al2O3) NPs with irregular shapes (Fig. S3a, ESI†) do not show absorption peaks because of the lack of light scattering resonances in the Al2O3 NPs. The DRS spectrum of the Rh NPs loaded on mesoporous silica nanoparticles with a size of <50 nm (Fig. S3b, ESI†) also exhibits less pronounced peaks but overall higher intensity than the Rh NPs on SiOx NSs, indicating the lack of light scattering resonances in the small mesoporous silica nanoparticles and deeper penetration of light in the mesoporous nanoparticle powder. The diffuse light penetrating the powder sample exhibits random propagation directions and reduced intensity to favor the photothermal process upon absorption in the buried nanoparticles. Therefore, using SiOx NSs as a support for Rh NPs minimizes the penetration of diffuse light to weaken the photothermal process, while the strong light scattering resonance on the surface of the SiOx NSs enhances light absorption in small Rh NPs to benefit the non-photothermal process for photocatalytic CO2 hydrogenation. The catalytic performance was evaluated in a fix-bed reactor using a thin layer of Rh-NP/SiOx-NS composite particles (∼0.7 mm in thickness, ∼7 mg in mass) on a platinum mesh, which can minimize the photothermal effect.32 The loose packing of the particles favored the isotropic and uniform mass transport of gas flow and heat transfer, ensuring fast dissipation of heat generated from light absorption (i.e., photothermal effect) to eliminate significant temperature deviation from the set temperature and temperature gradient in the thin catalyst layer (ESI,† Part II and Table S1). With respect to a set temperature of the reactor, the photothermally induced temperature change in the catalyst bed increases with the light intensity but decreases with the set temperature (Fig. S4, ESI†). The temperature changes of the catalyst bed are not significant (<15 °C) using the CW visible light with a power intensity up to 1.5 W cm−2 in this work. These minor changes in temperature originated from the photothermal effect and induce only small perturbations of reaction kinetics, making it feasible to determine the presence of a non-thermal effect in the photocatalytic reactions with significantly accelerated kinetics. For example, catalytic CO2 hydrogenation on the 2.3-nm Rh-NP/SiOx-NS catalyst at 330 °C in a H2-rich atmosphere (i.e., 440 mL h−1 H2, 35 mL h−1 CO2, 85 mL h−1 Ar at 1 atm) produces CO and CH4 with a CH4 selectivity of only 71% in the dark. Illuminating the catalyst with visible light increases the steady-state production rate of both CO and CH4 with a preference for CH4, improving the CH4 selectivity (Fig. 1a). The production rate of CH4 increases by 60 times under illumination of 1.5 W cm−2 power density while the increase of CO production rate is less than one time, corresponding to a CH4 selectivity of 99%. The production rate of CH4 monotonically increases with light power density and reaches a plateau under light stronger than 1.4 W cm−2 due to the limitation of mass diffusion. In contrast, the production rate of CO increases with light power density and then decreases when the light is stronger than 1.1 W cm−2 (Fig. 1b). The volcano-type dependence of the CO production rate indicates that surface-adsorbed CO (CO*) represents the intermediate to be consumed for forming CH4. The increase in production rate under photoillumination corresponds to a decrease in the apparent activation energy of the methanation reaction (Fig. S5a, ESI†), indicating that both thermal energy and optical energy (no-thermal effect) can simultaneously determine the methanation kinetics on the Rh-NP/SiOx-NS composite catalysts. Fig. 1c and d present the production rate and selectivity of CH4 under photoillumination of different power densities and at different temperatures. Increasing both temperature and light power density favors the methanation kinetics and selectivity. The reaction kinetics is more sensitive to photoillumination at a lower reaction temperature. For example, the production rate of CH4 increases by 350 times at 290 °C under the light of 1.5 W cm−2, and the corresponding CH4 selectivity increases to 97% from 35% (i.e., the value in the dark). The temperature-dependent photosensitivity can be described by the change of reaction activation energy as a function of light power density. The activation energy of both the CO2 consumption reaction and CH4 production reaction monotonically decreases with the light power density, i.e., from 82.1 kJ mol−1 (0.85 eV) in the dark to 35.5 kJ mol−1 (0.368 eV) under light of 1.5 W cm−2 and from 115.8 kJ mol−1 (1.20 eV) in the dark to 38.6 kJ mol−1 (0.400 eV) under light, respectively (Fig. S5b, ESI†). The leveling of activation energy at a lower value leads to a larger and equivalent reaction rate for both consuming CO2 and producing CH4 under light with enough power density, resulting in a CH4 selectivity close to unity (Fig. 1d). The results in Fig. 1c and 1d highlight the potential of the non-thermal effect in the photoexcited Rh NPs to promote both the reaction kinetics and selectivity of CO2 methanation simultaneously.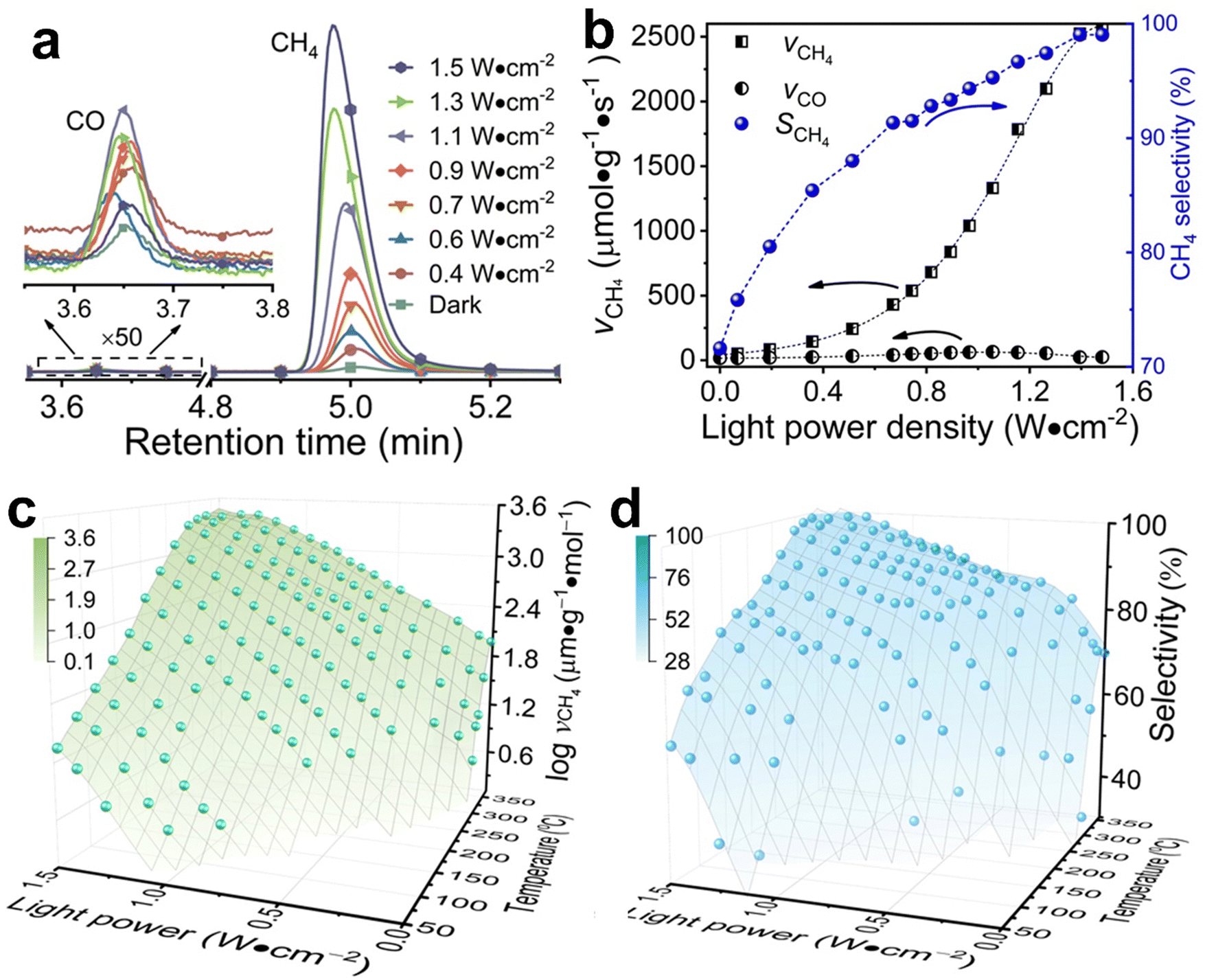 | ||
| Fig. 1 Dependence of CO2 hydrogenation reaction rate and selectivity on the light power intensity. (a) Gas chromatography (GC) graphs of the products of CO2 hydrogenation reactions at 330 °C, showing the variation of signals of CH4 and CO under different photoillumination. (b) Production rates of CH4 and CO and the selectivity for CH4 calculated from (a). (c) Production rate of CH4 and (d) selectivity for CH4 as a function of reactor temperature and light power intensity. The light power intensity refers to the corrected light absorbed in the Rh NPs with a size of 2.26 nm. The gas flow into the reactor contained 35 mL h−1 CO2, 440 mL h−1 H2, and 85 mL h−1 Ar. | ||
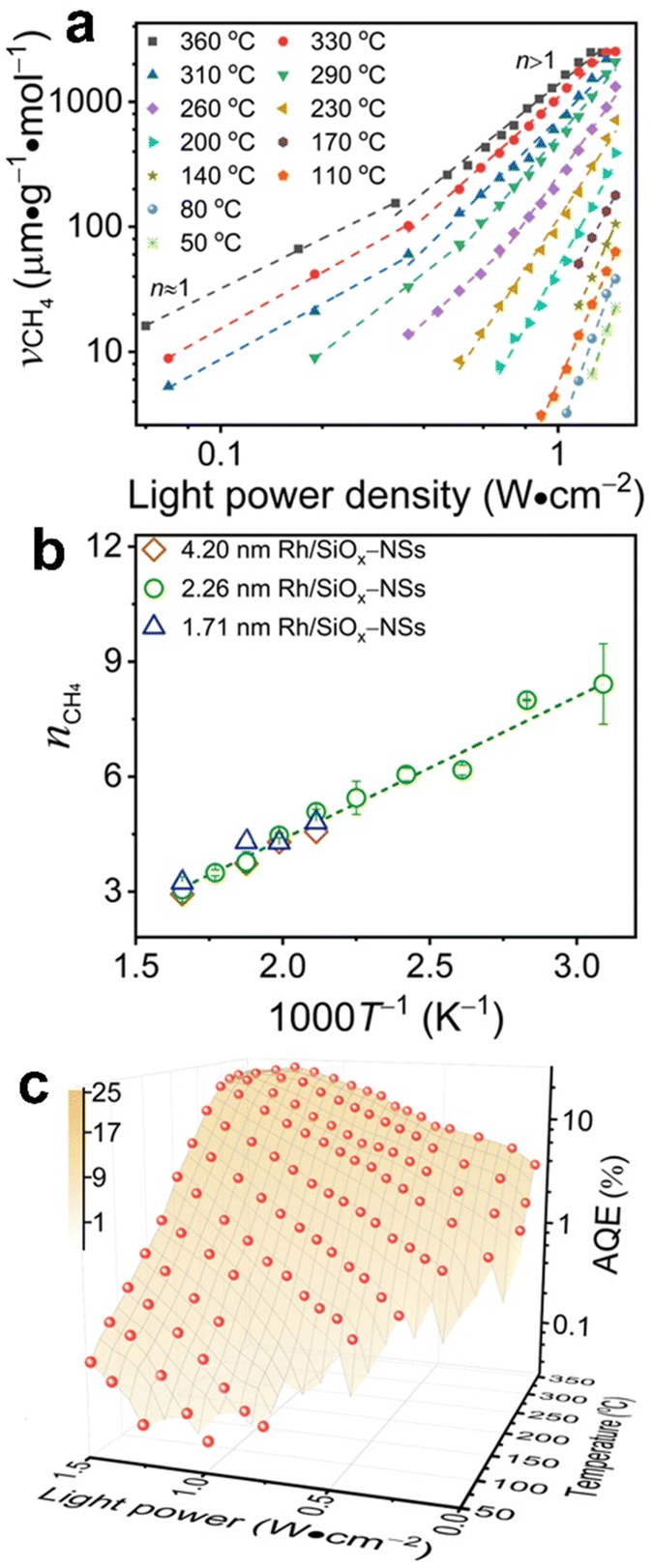
The photocatalytic methanation reaction rate (νCH4) exhibits the power law dependence on the power density (I) of incident light: νCH4 ∝ In, where n is the power law exponent (Fig. 2a). When the temperature is higher than 310 °C, the methanation reaction rate is linearly dependent on the power density of weak light (I < 0.4 W cm−2) and the value of n becomes larger than one under intense light with I > 0.4 W cm−2. The transition of power law from linear dependence to superlinear dependence, i.e., from n = 1 to n > 1, is consistent with the mechanism of photocatalytic hot-carrier-driven reactions that are ascribed to the non-thermal effect.33 This transition of power law corresponds to the change from single-electron to multiple-electron transitions that are required to excite adsorbates to accumulate sufficient vibrational energy higher than the activation barrier of the methanation reaction.34,35 As the temperature lowers, the value of n in the superlinear region increases, and the linear region even disappears at the temperature below 310 °C. The n values exhibit a strong positive correlation with the reciprocal of thermodynamic temperature (T) (Fig. 2b) regardless of the size of Rh NPs, indicating that the methanation kinetics is less sensitive to light power density at a higher temperature. The linear dependence of n on 1/T agrees with the theoretical model proposed for desorption induced by multiple electronic transitions (DIMET), in which 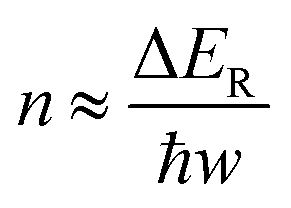 with ħw and ΔER representing the energy quantum of vibrational states and the reorganization energy of adsorbates, respectively.34 For a given adsorbate species, the energy quantum of vibrational states is defined, but the reorganization energy is smaller at a higher temperature, resulting in a smaller n for the power law.36–39 The results shown in Fig. 1c can be fitted with νCH4 = a × νCH4,dark2(T) × In(T) with n(T) = 5253/T − 5.77 (Fig. S6, ESI†), confirming the synergy of thermal energy and optical energy in promoting the methanation reaction kinetics. The minor contribution of the photothermal effect to methanation kinetics can be calculated from the Arrhenius equation with the activation energy of the dark reaction and the photothermally induced temperature increase as shown in Fig. S4b (ESI†). Subtracting the photothermal contribution from the difference in methanation reaction rates between dark and light conditions gives the quantitative contribution of the non-photothermal effect. The apparent quantum efficiency (AQE) of the non-photothermal effect is calculated by comparing the number of additional CH4 molecules formed from the non-thermal effect and the number of photons absorbed by the catalysts (ESI,† Part III and Fig. S7). The positive values of AQE confirm the existence and contribution of the non-photothermal effect to promote the methanation kinetics. The value of AQE increases with both the temperature of the catalyst bed and the power density of incident light (Fig. 2c and Fig. S8, ESI†). The AQE can reach 26% at 360 °C under light with a power density above 1 W cm−2, highlighting the high efficiency of the non-photothermal effect in driving the methanation reaction on ultrafine Rh NPs supported on SiOx NSs.
with ħw and ΔER representing the energy quantum of vibrational states and the reorganization energy of adsorbates, respectively.34 For a given adsorbate species, the energy quantum of vibrational states is defined, but the reorganization energy is smaller at a higher temperature, resulting in a smaller n for the power law.36–39 The results shown in Fig. 1c can be fitted with νCH4 = a × νCH4,dark2(T) × In(T) with n(T) = 5253/T − 5.77 (Fig. S6, ESI†), confirming the synergy of thermal energy and optical energy in promoting the methanation reaction kinetics. The minor contribution of the photothermal effect to methanation kinetics can be calculated from the Arrhenius equation with the activation energy of the dark reaction and the photothermally induced temperature increase as shown in Fig. S4b (ESI†). Subtracting the photothermal contribution from the difference in methanation reaction rates between dark and light conditions gives the quantitative contribution of the non-photothermal effect. The apparent quantum efficiency (AQE) of the non-photothermal effect is calculated by comparing the number of additional CH4 molecules formed from the non-thermal effect and the number of photons absorbed by the catalysts (ESI,† Part III and Fig. S7). The positive values of AQE confirm the existence and contribution of the non-photothermal effect to promote the methanation kinetics. The value of AQE increases with both the temperature of the catalyst bed and the power density of incident light (Fig. 2c and Fig. S8, ESI†). The AQE can reach 26% at 360 °C under light with a power density above 1 W cm−2, highlighting the high efficiency of the non-photothermal effect in driving the methanation reaction on ultrafine Rh NPs supported on SiOx NSs.
 | ||
| Fig. 2 (a) Photocatalytic CH4 production rate (i.e., the difference of the rate measured under photoillumination and in the dark) as a function of light power intensity. The log–log plots allow easy determination of the exponent n in the power law through linear fitting the data points. “n = 1” means a linear dependence of CH4 production rate on light power intensity, and “n > 1” means a superlinear dependence of CH4 production rate on light power intensity. The change from linear to superlinear dependence with an increase in the light power intensity indicates the switch of single to multiple electronic transitions in surface adsorbate induced by hot electron injection. The catalyst contained Rh NPs with a size of 2.26 nm. (b) The power n in the superlinear region as a function of the reciprocal of the reactor temperature when using catalyst Rh NPs with different sizes (i.e., 1.71 nm, 2.26 nm, and 4.20 nm). (c) Apparent quantum efficiencies (AQE) as a function of reactor temperature and light power intensity. The gas flow into the reactor contained 35 mL h−1 CO2, 440 mL h−1 H2, and 85 mL h−1 Ar. | ||
Under an atmosphere with high H2 partial pressure (pH2 = 0.786 atm), the reaction order of consuming CO2 with respect to the partial pressure of CO2 (pCO2) is 0.42 in the dark at 330 °C while the reaction order of producing CH4 is close to zero (i.e., 0.006) (Fig. 3aversusFig. 3b, black diamond symbols). The independence of CH4 production rate on pCO2 indicates that the higher chemical potential associated with a higher pCO2 is mainly responsible for producing CO. The coverage of CO* intermediate on Rh NPs remains constant to react with hydrogen of a given pressure to produce CH4. Photoillumination significantly increases the reaction rate of both CO2 consumption and CH4 production, which exhibit less difference than the dark reaction. The reaction order of producing CH4 remains close to zero, but the reaction order of consuming CO2 drops to 0.25 under visible light of 1.5 W cm−2. The comparisons indicate that photoillumination accelerates both the dissociation of CO2 and the methanation of intermediate CO*. Under an atmosphere with a low H2 partial pressure (pH2 = 0.41 atm), the reaction order of consuming CO2 with respect to pCO2 is always positive while the reaction order of producing CH4 is negative under both dark and light conditions (Fig. 3aversusFig. 3b, square symbols). The contrast value sign of the reaction orders indicates that the reactivity of hydrogen plays the most crucial role in the methanation of CO* and the dissociation of CO2 to CO* is less sensitive to hydrogen. The dependence of reaction rate on pH2 shows the corresponding orders of consuming CO2 and producing CH4 are 0.53 and 0.73, respectively, at pCO2 = 0.0625 atm and in the dark (Fig. 3c and d, black squares). The difference in reaction orders is significantly amplified under visible light of 1.5 W cm−2; for example, that of consuming CO2 remains essentially unchanged but that of producing CH4 increases to 2.55. The distinct change in reaction orders indicates that photoillumination significantly improves the reactivity of hydrogen and hydrogen-assisted deoxygenation of CO* most likely due to the increased coverage of adsorbed hydrogen atoms (H*). The higher reactivity of H* than H2 favors the surface hydrogenation reaction, and the faster consumption of H* leads to a stronger dependence on pH2 with a higher reaction order. Moreover, the consumption rate of CO2 is much higher than the production rate of CH4 at low pH2 = pCO2 = 0.0625 atm under light, i.e., 600 μmol g−1 s−1versus 11 μmol g−1 s−1, indicating the capability of photoillumination in promoting the cleavage of the C–O bond of CO2* (and possible derivatives) to form CO* on the surface of Rh NPs.
 | ||
Fig. 3 Dependence of CO2 methanation kinetics on the partial pressure of CO2 (pCO2) and H2 (pH2). (a) and (b) Influence of pCO2 on (a) consumption rate of CO2 and (b) production rate of CH4 in the dark or under photoillumination of a light power intensity of 1.5 W cm−2. pH2 was constant at either 0.410 atm or 0786 atm. (c) and (d) Influence of pH2 on (c) consumption rate of CO2 and (d) production rate of CH4 in the dark or under photoillumination. (e) Dependence of the production rate of CH4 on pH2 under photoillumination of different light power intensities. pCO2 was constant at 0.0625 atm. The total gas flow rate was 560 mL h−1 and Ar was used as the balance gas. The catalyst Rh NPs had a size of 2.26 nm. (f) Fitted plots of the data shown in (d) and (e) using the kinetic equation of 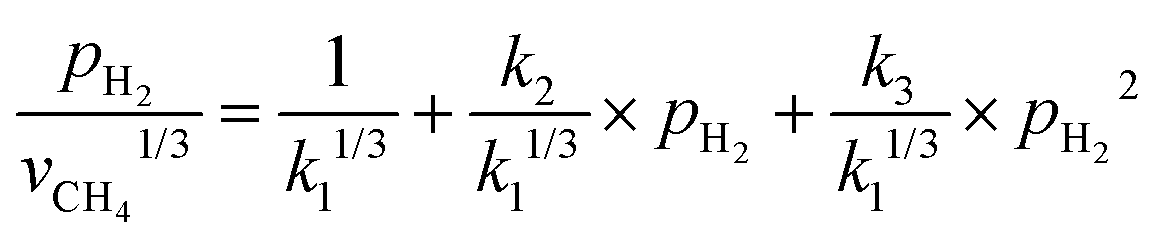 . . | ||
The results in Fig. 3a–d indicate that the non-photothermal effect (representing the major contribution of photoillumination in the current work) in Rh-NP/SiOx-NS composite catalysts can increase the adsorption coverage of H* and facilitate C–O bond cleavage to favor CO2 methanation kinetics. Light absorption in Rh NPs excites plasmons that decay to generate high-energy hot electrons with the population and energy depending on the intensity of incident light and the size of NPs.40 The non-photothermal effect originates from the photoexcited electronic structures in the Rh NPs, influencing the interactions with the surface adsorbates in CO2 hydrogenation. At a low pH2 of 0.0625 atm, the production rate of CH4 increases with the light power density up to 0.7 W cm−2 and then decreases with even stronger light. Only intense light can significantly facilitate the cleavage of the first C–O bond of CO2 to dissociate CO2 into CO, which consumes a large portion of H* atoms to lower the availability of H* for CO* methanation. When pH2 is high enough to exclude the limitation of H2 diffusion, the production rate of CH4 increases with the power density of incident light, indicating light absorption in Rh NPs favors activation of H2 and C–O cleavage in the CO-derived intermediates. Regardless of the light intensity, the production rate of CH4 increases with pH2 to reach a maximum followed by a decrease (Fig. 3e). In the bell-like dependence of CH4 production on light power density the negative correlation is ascribed to the fact that the cleavage of C–O bond in CO* methanation is not fast enough to consume H*. Shifting the maximum-point pH2 from ∼0.62 atm in the dark (black squares, Fig. 3d) to ∼0.22 atm under light of 0.5 W cm−2 (brown cycles, Fig. 3e) indicates that weak light can favor H2 activation but not for the C–O cleavage at the same level. The maximum-point pH2 is pushed back to high values under intense light (e.g., 0.42 and ∼0.7 atm for light of 1.0 and 1.5 W cm−2, respectively), implying that intense light favors C–O cleavage to accelerate methanation with fast consumption of H*.
The analysis of results in Fig. 3 reveals that the non-thermal effect induced by light absorption in Rh NPs can promote H2 activation (i.e., increase of H* coverage under a given pH2) and C–O bond cleavage of intermediate adsorbates to accelerate CO2 methanation kinetics. The C–O bond cleavage becomes predominant only under intense light although significant H2 activation can always be observed under light. Moreover, surface H* atoms react with stable surface adsorbates (i.e., CO2 and CO) to form appropriate hydrogenated intermediates to facilitate the C–O bond cleavage, promoting the formation of CH4. In situ diffuse reflectance infrared Fourier transform (DRIFT) spectroscopy has been used to identify the most possible intermediates (peaks assignments summarized in Table S2, ESI†) and the influence of light on reaction kinetics. The adsorption of CO2 on Rh NPs decreases with temperature, and the first C–O bond cleavage (aka direct dissociation of CO2) occurs to produce CO* with low yield at a high enough temperature (>300 °C) (Fig. S9a, ESI†). In the presence of H2 (pH2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :pCO2 = 1:1), the signal of CO2* significantly reduces but the signal of CO* becomes dominant even at temperatures as low as 30 °C (Fig. S9b, ESI†), confirming the critical role of hydrogen in accelerating cleavage of the first C–O bond of CO2*. The appearance of an infrared (IR) absorption peak around ∼2800 cm−1 at low temperatures (e.g., 50 °C) is ascribed to the formation of bidentate formate (bi-HCOO*).41 This peak diminishes with an increase in temperature and the peak intensity of CO* increases, indicating that bi-HCOO* is most likely the hydrogenated intermediate species to facilitate cleavage of the first C–O bond of CO2. When a gas mixture with pH2:pCO2 = 1:1 flows through the catalyst bed at 50 °C, both CO* and bi-HCOO* accumulate on the surface of Rh NPs to reach plateaus of surface coverage after ∼30 min in the dark (Fig. 4a and c). The appearance of plateaus corresponds to the steady-state reaction conditions. It becomes much faster for CO* to reach a plateau of a higher coverage under light, and the accumulation time is only ∼12 min (Fig. 4b and c). The absence of IR absorption signal of bi-HCOO* under light indicates that the dissociation of bi-HCOO* is too fast to accumulate on the surface of Rh NPs. Therefore, photoillumination promotes cleavage of one C–O bond in bi-HCOO* to produce CO* at a higher rate, resulting in a higher coverage of CO* on the surface of Rh NPs (Fig. 4c, top panel). Similar results are observed in the H2-rich atmosphere (e.g., pH2:pCO2 = 10:1) except for the non-zero signal of bi-HCOO* under light (Fig. S10, ESI†). The accumulation of bi-HCOO* under light indicates that high H2 partial pressure (corresponding to high coverage of H*) can accelerate the formation rate of bi-HCOO* intermediate.
:pCO2 = 1:1), the signal of CO2* significantly reduces but the signal of CO* becomes dominant even at temperatures as low as 30 °C (Fig. S9b, ESI†), confirming the critical role of hydrogen in accelerating cleavage of the first C–O bond of CO2*. The appearance of an infrared (IR) absorption peak around ∼2800 cm−1 at low temperatures (e.g., 50 °C) is ascribed to the formation of bidentate formate (bi-HCOO*).41 This peak diminishes with an increase in temperature and the peak intensity of CO* increases, indicating that bi-HCOO* is most likely the hydrogenated intermediate species to facilitate cleavage of the first C–O bond of CO2. When a gas mixture with pH2:pCO2 = 1:1 flows through the catalyst bed at 50 °C, both CO* and bi-HCOO* accumulate on the surface of Rh NPs to reach plateaus of surface coverage after ∼30 min in the dark (Fig. 4a and c). The appearance of plateaus corresponds to the steady-state reaction conditions. It becomes much faster for CO* to reach a plateau of a higher coverage under light, and the accumulation time is only ∼12 min (Fig. 4b and c). The absence of IR absorption signal of bi-HCOO* under light indicates that the dissociation of bi-HCOO* is too fast to accumulate on the surface of Rh NPs. Therefore, photoillumination promotes cleavage of one C–O bond in bi-HCOO* to produce CO* at a higher rate, resulting in a higher coverage of CO* on the surface of Rh NPs (Fig. 4c, top panel). Similar results are observed in the H2-rich atmosphere (e.g., pH2:pCO2 = 10:1) except for the non-zero signal of bi-HCOO* under light (Fig. S10, ESI†). The accumulation of bi-HCOO* under light indicates that high H2 partial pressure (corresponding to high coverage of H*) can accelerate the formation rate of bi-HCOO* intermediate.
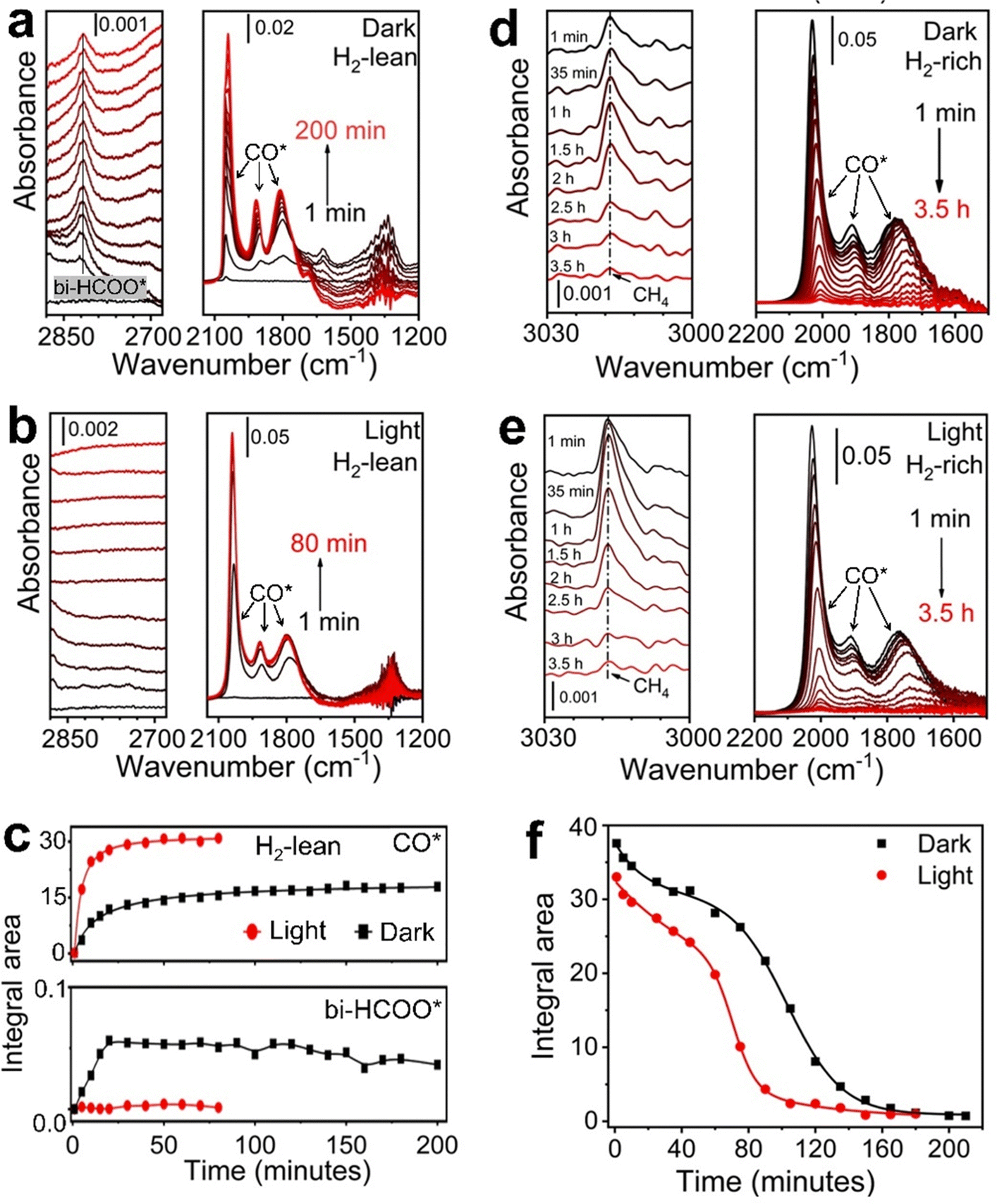 | ||
| Fig. 4 Time-dependent DRIFT spectra of adsorbate species on the Rh-NP/SiOx-NS composite catalyst. (a) and (b) DRIFT spectra recorded from CO2 hydrogenation at 50 °C (a) in the dark and (b) under photoillumination of a light power density of 1.5 W cm−2. The gas flow contained 35 mL h−1 H2, 35 mL h−1 CO2, and 960 mL h−1 Ar, providing a H2-lean condition. The three sharp peaks between 1650 cm−1 and 2100 cm−1 originate from different adsorption states of CO*. The total area of these three peaks is proportional to the overall CO* on the catalyst surface. The peak around 2800 cm−1 is ascribed to bi-HCOO* surface species. (c) Comparison of the integrated area of CO* peaks (top panel) and bi-HCOO* peak (bottom panel) in the dark and under photoillumination. The integration was derived from the spectra shown in (a) and (b). (d) and (e) DRIFT spectra recorded after CO2 hydrogenation reaction at 330 °C while exposing the catalyst to a H2-rich condition with a gas flow containing 350 mL h−1 H2 and 650 mL h−1 Ar. The pre-hydrogenation reaction was performed using conditions the same as in Fig. 1, leaving the steady-state coverage of CO* to allow studying the deoxygenation of CO*. (f) Time-dependent area of CO* derived from the spectra shown in (d) and (e), showing the faster deoxygenation kinetics under photoillumination. | ||
When temperature increases to 150 °C and above, the total signal of CO* under the steady-state condition is independent of gas composition, temperature, and photoillumination (Fig. S11 and S12, ESI†). The constant steady-state coverage of CO* on a given Rh NP catalyst suggests that the methanation of CO* has to be fast enough to match the rate of converting CO2 to CO* to achieve high CH4 selectivity. Otherwise, CO* detaches from the surface of Rh NPs to form gaseous CO. Methanation of CO* is limited by the cleavage of the C–O bond. Co-adsorption of H* redshifts the IR absorption peaks of CO* due to the slight weakening of the C–O bond. The presence of H* also favors the linear configuration of CO* neighboring with H* (i.e., rhodium carbonyl hydride) (Fig. S13, ESI†). The activation energy for directly cleaving the C–O bond of CO* on Rh catalysts decreases from 213 kJ mol−1 (2.2 eV) to 143 kJ mol−1 (1.48 eV) in the presence of one H* or 130 kJ mol−1 (1.35 eV) in the presence of two H*.42 Although the pre-adsorbed H* on Rh NPs can help reduce CO2 to CO*, a continuous supply of H2 is necessary to drive the hydrogenation of CO* into CH4 (Fig. S14, ESI†). The surface coverage of CO* is almost constant during the CO2 hydrogenation reaction regardless of the light condition at an elevated temperature (Fig. S11 and S12, ESI†). We can use the residual CO* after stopping the CO2 supply as the benchmark to evaluate the influence of light on the CO* methanation kinetics at 330 °C. A continuous flow of H2 (pH2 = 0.35 atm) can hydrogenate CO* to produce CH4 at the measurable rate in the dark (Fig. 4d), decreasing the coverage of CO* on the surface of Rh NPs until the complete consumption of CO*. Photoillumination can accelerate the methanation of CO* with a higher production rate of CH4 and fast consumption of CO* (Fig. 4e). For example, converting 80% CO* under light of 1.5 W cm−2 takes 81 min while 124 min is required in the dark (Fig. 4f). A similar difference is also observed when a continuous flow of low-pressure H2 (pH2 = 0.035 atm) is applied (Fig. S15, ESI†). It is worth noting that no CH4 formation can be detected with low-pressure H2 in the dark (Fig. S15a, ESI†). The absence of the CH4 signal might be ascribed to either that the formation rate of CH4 is too low to be measured by the GC or that the low chemical potential of H2 cannot overcome the energy barrier of cleaving the C–O bond in CO* but combine with CO* to form hydrogenated intermediate to desorb from the Rh surface. The most possible intermediate could be formaldehyde (H2CO*), which can equilibrate with rhodium carbonyl hydride (L2-CO*) (Fig. S13 and S16, ESI†). Light illumination of Rh NPs promotes dissociation of H2 to increase the adsorption coverage of H* to accelerate the formation of L2-CO* intermediate and facilitates cleavage of the C–O bond in CO* (Fig. S17, ESI†), resulting in a faster production of CH4. When Rh NPs are replaced with Ag NPs that have very weak adsorption of hydrogen (H*),43 the reduction of the CO2 reaction primarily produces CO at much lower rates (Fig. S18, ESI†), confirming that the presence of sufficient H* on the metal surface is crucial for CO2 methanation in the cleavage of both C–O bonds.
The involvement of adsorbed hydrogen in CO2 methanation is consistent with the significant kinetic dependence on the partial pressure of H2. The high reaction orders of CH4 production with respect to pH2 is not related to the mass transport limitations because of the thin catalyst bed and the stoichiometrically excess amount of H2 (ESI,† Part IV including Fig. S19). Five possible reaction paths have been proposed for CO2 methanation on catalyst surfaces based on the reported experiments and theoretical calculations: (i) CO2* → CO* → C*, (ii) CO2* → CO* → HxCO* → CHx*, (iii) CO2* → HCOO* → CO* → C*, (iv) CO2* → HCOO* → CO* → HCO* → C*/CHx*, and (v) CO2* →HCOO* → CO* → H2CO* → CHx*. By considering different rate-determining steps (RDS), the kinetic equations of the CH4 production rate as a function of pH2 can be determined (ESI,† Part V and Table S3). The results in Fig. 3d and e are then fitted with more than forty possible kinetic equations. The goodness of fitting corresponds to the possibility of the reaction mechanisms (Table S4, ESI†). The direct dissociation of CO2 and CO in the reaction paths (i, ii, and iii) cannot fit the measured data well, resulting in a value of R2 lower than 0.97 at 330 °C in the dark. The large fitting deviations could exclude the mechanisms involving the direct dissociation of CO2 and CO. In contrast, the hydrogen-assisted dissociation paths (iv and v) offer an appropriate mechanism that is possible to have fitted R2 larger than 0.99 (see the top row, Table S4, ESI†). The best-fitting equation is 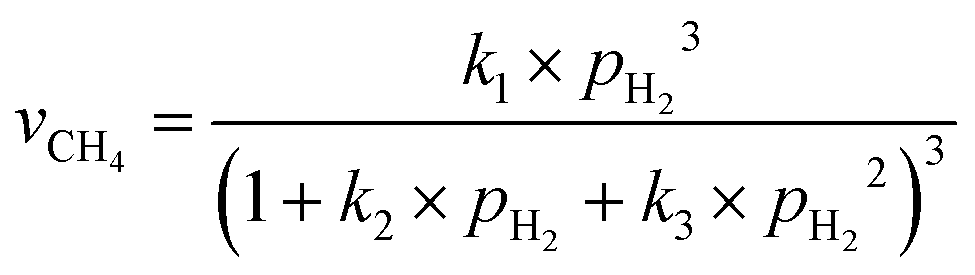 , which agrees with the experimental data regardless of the illumination condition (Fig. 3f). The corresponding methanation mechanism has the RDS of H2CO* (or L2-CO*) + 2H* → CH2* + H2O* + *, highlighting the importance of H* in the full deoxygenation of CO2 for forming CH4. Photoillumination generates hot electrons in the Rh NPs to facilitate the deoxygenation of L2-CO* in the presence of high-density H*, increasing the CH4 production rate.
, which agrees with the experimental data regardless of the illumination condition (Fig. 3f). The corresponding methanation mechanism has the RDS of H2CO* (or L2-CO*) + 2H* → CH2* + H2O* + *, highlighting the importance of H* in the full deoxygenation of CO2 for forming CH4. Photoillumination generates hot electrons in the Rh NPs to facilitate the deoxygenation of L2-CO* in the presence of high-density H*, increasing the CH4 production rate.
Conclusion
According to the results of reaction kinetics and steady-state DRIFT spectroscopy, the CO2 methanation mechanism catalyzed by the Rh NPs supported on SiOx NSs is depicted in Fig. 5. The Rh surface has an appropriate H adsorption coverage that can bind with or interact with the neighboring CO2* and CO* to facilitate the cleavage of C–O, enabling the production of CH4 even though CO* can detach from the Rh surface as a side product. Fast reaction kinetics of the hydrogen-assisted deoxygenation of CO* is crucial to minimize the release of CO side product. Photoillumination of the catalyst using a visible light source excites surface plasmon resonances in the Rh NPs to generate hot electrons, which interact with molecular orbitals of CO* to weaken the C–O bond. The weakened C–O accelerates the deoxygenation of CO* to produce methane at a higher rate in the presence of high-density H*. The deoxygenation of the first O in CO2* can also benefit from the hot-electron-induced weakening effect in the C–O bond under photoillumination to accelerate the formation rate of the intermediate CO*, which can provide enough CO* to support the accelerated CH4 production. Since the deoxygenation of the two O atoms from CO2 requires the presence of H*, the accelerated CH4 production under photoillumination requires faster adsorption kinetics for the reaction H2 → 2H*, which can generate H* at a higher rate. This correlation implies that photoillumination also favors the dissociative adsorption of H2 on the Rh NPs, which is consistent with previous studies using Au nanoparticles with strong surface plasmon resonances.44 The strategy combining the reaction kinetics and steady-state DRIFT spectroscopy allows for deconvolution of the CO2 methanation reaction mechanism on catalysts. Using SiOx NSs to support the ultrafine Rh NPs as catalysts minimizes the photothermally induced temperature change in the catalyst bed, making it feasible to evaluate the non-thermal photocatalysis. The results and conclusions shed light on photocatalyst design to achieve efficient performance in both reaction kinetics and product selectivity. For example, using Rh-NP/SiOx-NS composite catalysts under visible light of 1.5 W cm−2 at 330 °C achieved a near unity selectivity of CH4 production from CO2 hydrogenation and two orders of magnitude enhancement of production rate compared to the reaction in the dark.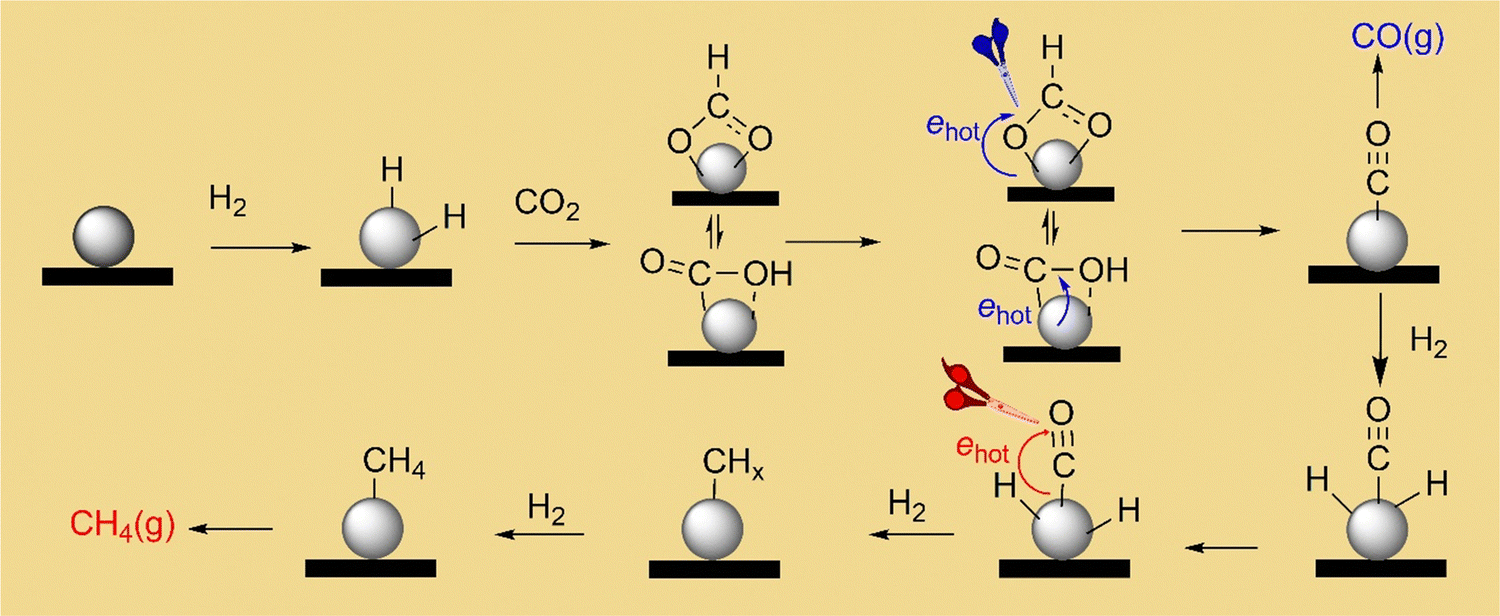 | ||
| Fig. 5 Proposed CO2 deoxygenation and hydrogenation mechanism on the Rh-NP/SiOx-NS catalyst. The primary role of the photo-generated hot electrons in the Rh NPs is to facilitate cleavage of the C–O bond. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the startup and OVPR seed grant from Temple University. Partial characterizations were performed with the use of TMI (Temple Materials Institute) facilities. We are thankful to Mr. sichuan Huang for the help with data fitting and Dr. Lauren Profitt for ICP characterizations.Notes and references
- S. Wu, S. Wu and Y. Sun, Sol. RRL, 2021, 5, 2000507 CrossRef CAS.
- A. Erdőhelyi, Catalysts, 2020, 10, 155 CrossRef.
- N. M. Martin, P. Velin, M. Skoglundh, M. Bauer and P.-A. Carlsson, Catal. Sci. Technol., 2017, 7, 1086–1094 RSC.
- É. Novák, K. Fodor, T. Szailer, A. Oszkó and A. Erdöhelyi, Top. Catal., 2002, 20, 107–117 CrossRef.
- E. Iguchi and K. Yajima, J. Phys. Soc. Jpn., 1972, 32, 1415–1421 CrossRef CAS.
- Z. Wu, W.-J. Yin, B. Wen, D. Ma and L.-M. Liu, J. Phys. Chem. Lett., 2023, 14, 2208–2214 CrossRef CAS PubMed.
- J. Hua, S. Feng, C. Ma, H. Huang, K. Wei, X. Dai, K. Wu, H. Wang and Z. Bian, J. Environ. Chem. Eng., 2023, 11, 111290 CrossRef CAS.
- M. Ni, Y. Zhu, C. Guo, D.-L. Chen, J. Ning, Y. Zhong and Y. Hu, ACS Catal., 2023, 13, 2502–2512 CrossRef CAS.
- F. Raziq, M. Z. Rahman, S. Ali, R. Ali, S. Ali, A. Zada, X. Wu, J. Gascon, Q. Wang and L. Qiao, Chem. Eng. J., 2024, 479, 147712 CrossRef CAS.
- C. J. Bueno-Alejo, A. Arca-Ramos, J. L. Hueso and J. Santamaria, Catal. Today, 2020, 355, 678–684 CrossRef CAS.
- Z. Geng, Y. Yu and J. Liu, J. Phys. Chem. C, 2023, 127, 17723–17731 CrossRef CAS.
- Z. Li, R. Shi, J. Zhao and T. Zhang, Nano Res., 2021, 14, 4828–4832 CrossRef CAS.
- K. Lorber and P. Djinović, iScience, 2022, 25, 104107 CrossRef CAS PubMed.
- D. Mateo, P. Maity, G. Shterk, O. F. Mohammed and J. Gascon, ChemSusChem, 2021, 14, 5525–5533 CrossRef CAS PubMed.
- Y. Zhou, P. Zheng, F. Wang, F. Gu, W. Xu, Q. Lu, T. Zhu, Z. Zhong, G. Xu and F. Su, Nano Res., 2023 DOI:10.1007/s12274-023-6226-5.
- R. Jiang, X. Yue, K. Wang, Z. Yang, W. Dai and X. Fu, Energy Fuels, 2022, 36, 11636–11646 CrossRef CAS.
- A. Anouar, R. García-Aboal, P. Atienzar, A. Franconetti, N. Katir, A. El Kadib, A. Primo and H. Garcia, Adv. Sustainable Syst., 2022, 6, 2100487 CrossRef CAS.
- Y. Chen, Y. Zhang, G. Fan, L. Song, G. Jia, H. Huang, S. Ouyang, J. Ye, Z. Li and Z. Zou, Joule, 2021, 5, 3235–3251 CrossRef CAS.
- X. Yue, K. Wang, Z. Yang, W. Ni, Z. Zhang, W. Dai and X. Fu, Chem. Eng. J., 2023, 458, 141491 CrossRef CAS.
- K. Wang, S. He, Y. Lin, X. Chen, W. Dai and X. Fu, Chin. J. Catal., 2022, 43, 391–402 CrossRef CAS.
- X. Zhang, X. Li, D. Zhang, N. Q. Su, W. Yang, H. O. Everitt and J. Liu, Nat. Commun., 2017, 8, 14542 CrossRef CAS PubMed.
- X. Zhang, X. Li, M. E. Reish, D. Zhang, N. Q. Su, Y. Gutiérrez, F. Moreno, W. Yang, H. O. Everitt and J. Liu, Nano Lett., 2018, 18, 1714–1723 CrossRef CAS PubMed.
- X. Li, H. O. Everitt and J. Liu, Nano Res., 2019, 12, 1906–1911 CrossRef CAS.
- C. Kim, S. Hyeon, J. Lee, W. D. Kim, D. C. Lee, J. Kim and H. Lee, Nat. Commun., 2018, 9, 3027 CrossRef PubMed.
- U. Ulmer, T. Dingle, P. N. Duchesne, R. H. Morris, A. Tavasoli, T. Wood and G. A. Ozin, Nat. Commun., 2019, 10, 3169 CrossRef PubMed.
- G. V. Hartland, L. V. Besteiro, P. Johns and A. O. Govorov, ACS Energy Lett., 2017, 2, 1641–1653 CrossRef CAS.
- K. D. Rasamani and Y. Sun, J. Chem. Phys., 2020, 152, 084706 CrossRef CAS PubMed.
- S. Stewart, Q. Wei and Y. Sun, Chem. Sci., 2021, 12, 1227–1239 RSC.
- N. Zhang, C. Han, Y.-J. Xu, J. J. Foley Iv, D. Zhang, J. Codrington, S. K. Gray and Y. Sun, Nat. Photonics, 2016, 10, 473–482 CrossRef CAS.
- L. K. Ausman and G. C. Schatz, J. Chem. Phys., 2008, 129, 054704 CrossRef PubMed.
- X. Dai, K. D. Rasamani, G. Hall, R. Makrypodi and Y. Sun, Front. Chem., 2018, 6, 494 CrossRef CAS PubMed.
- X. Bian, Y. Zhao, C. Zhou and T. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202219340 CrossRef CAS PubMed.
- L. Zhou, M. Lou, J. L. Bao, C. Zhang, J. G. Liu, J. M. P. Martirez, S. Tian, L. Yuan, D. F. Swearer, H. Robatjazi, E. A. Carter, P. Nordlander and N. J. Halas, Angew. Chem., Int. Ed., 2021, 118, e2022109118 CAS.
- T. Olsen and J. Schiøtz, Phys. Rev. Lett., 2009, 103, 238301 CrossRef PubMed.
- C. Frischkorn, Surf. Sci., 2005, 593, 67–78 CrossRef CAS.
- P. Szymanski, A. L. Harris and N. Camillone, J. Phys. Chem. A, 2007, 111, 12524–12533 CrossRef CAS PubMed.
- H. Ueba and B. N. J. Persson, J. Phys.: Condens. Matter, 2008, 20, 224016 CrossRef.
- J. Güdde and U. Höfer, J. Phys.: Condens. Matter, 2006, 18, S1409–S1424 CrossRef.
- T. Avanesian and P. Christopher, J. Phys. Chem. C, 2014, 118, 28017–28031 CrossRef CAS.
- L. V. Besteiro and A. O. Govorov, J. Phys. Chem. C, 2016, 120, 19329–19339 CrossRef CAS.
- X. Wang, H. Shi and J. Szanyi, Nat. Commun., 2017, 8, 513 CrossRef PubMed.
- R. Zhang, F. Liu, X. Zhao, B. Wang and L. Ling, J. Phys. Chem. C, 2015, 119, 10355–10364 CrossRef CAS.
- S. S. Kumar and H. Lim, Sustainable Energy Fuels, 2023, 7, 3560–3583 RSC.
- S. Mukherjee, F. Libisch, N. Large, O. Neumann, L. V. Brown, J. Cheng, J. B. Lassiter, E. A. Carter, P. Nordlander and N. J. Halas, Nano Lett., 2013, 13, 240–247 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nh00506b |
| This journal is © The Royal Society of Chemistry 2024 |