DOI:
10.1039/D3SC00568B
(Edge Article)
Chem. Sci., 2023,
14, 4633-4640
Time-dependent photo-activated aminoborane room-temperature phosphorescence materials with unprecedented properties: simple, versatile, multicolor-tuneable, water resistance, optical information writing/erasing, and multilevel data encryption†
Received
2nd February 2023
, Accepted 25th March 2023
First published on 28th March 2023
Abstract
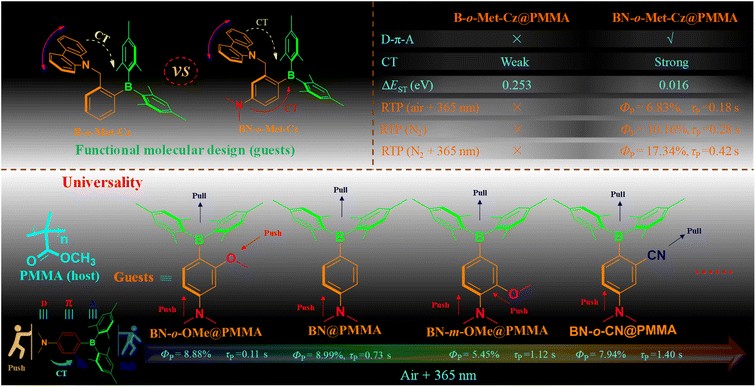
Triarylboranes-based pure organic room-temperature phosphorescence (RTP) materials are rarely investigated because of their large steric hindrance and the electron defect of the boron atom. As a result, creating functional triarylborane RTP materials is difficult. Herein, we report the first photo-activated RTP materials with lifetimes/quantum yields ≤0.18 s/6.83% based on donor (D)–π–acceptor (A) from methylene carbazole-functionalized aminoborane (BN)-doped polymethyl methacrylate (BN-o-Met-Cz@PMMA) under 365 nm UV irradiation (30 s). Incredibly, BN-o-Met-Cz@PMMA films exhibited unprecedented photo-activated RTP dual–response properties (e.g., air + 365 nm: τP = 0.18 s, ΦP = 6.83%; N2 + 365 nm: τP = 0.42 s, ΦP = 17.34%). Intriguingly, the BN (D–π–A) system demonstrated good versatility for photo-activated RTP whether the electron-donating group or electron-withdrawing group was placed in the ortho (meta)-position of the B atom. As a result, a series of photo-activated single-molecule organic RTP materials with multi-color emission, high quantum yields, and ultra-long lifetimes can be prepared rapidly. BN-X@PMMA films showed broad application prospects for information encryption, data erasure, anti-counterfeiting, and water resistance. Our method provides new strategies for the design, synthesis, and application of RTP materials, thereby enriching the types of organic RTP materials and facilitating further developments in this area.
1 Introduction
Polymer-based organic materials based on room-temperature phosphorescence (RTP) have the advantages of an amorphous state, processability, a long lifetime, low cost, and good thermal stability. They can effectively avoid the problems of stability and poor processability of organic small-molecular crystals, and have attracted extensive attention from researchers.1,2 However, due to the softness of the polymer structure, the triplet excitons of the molecule are quenched readily by oxygen and water in air, which makes polymers that have long afterglow luminescence scarce.2,3 Therefore, designing and realizing high-efficiency RTP materials of polymers has become a research “hotspot”.2,4 Hence, people began to explore and propose two construction methods: chemical synthesis and physical doping.2,5 Compared with chemical synthesis, physical doping is simple and easy to operate. The doping ratio, types of functional molecules and polymer matrix are easily adjustable, so that RTP materials can be modulated. Since then, various organic small molecule-doped polymer RTP materials have been reported.2,6 For example, commonly used polymer matrices include polymethyl methacrylate (PMMA), polylactic acid (PLA), polyacrylamide (PAM), and polystyrene (PS).7 Among them, organic RTP materials based on PMMA are popular because of their abundant carbonyl groups, good transparency, chemical stability, mechanical properties, and low permeability by oxygen, and PMMA is called “plexiglass”. In such a rigid environment, the nonradiative transition and quenching of triplet excitons can be suppressed effectively, resulting in the release of highly efficient phosphorescence.8
However, not all organic fluorescent molecules can become efficient RTP materials through simple physical doping because the structural design of functional molecules in the doping system is extremely demanding. In general, three conditions must be met: (1) The ΔEST of functional molecules should be sufficiently small (<0.6 eV);9 (2) the excited singlet (S1) and excited triplet (T1) states can give rise to effective inter-system crossing (ISC);10 (3) abundant non-covalent bonds between functional molecules and polymers are present.11 Hence, a series of novel functional organic RTP materials based on heteroatoms (N, O, S, P) or heavy atoms (Br and I) have been developed12 that use the unique spin–orbit coupling properties of atoms to overcome prohibition of singlet–triplet spin, accelerate ISC, and generate more triplet excitons.13 Unfortunately, these materials usually need to be in a crystalline state to induce RTP.14 Therefore, obtaining single-molecule phosphorescence in polymer-doped systems is difficult, which limits their further development and applications. Therefore, the design and development of simple, efficient, and universally functional organic single-molecule RTP is a great challenge. Compared with other heteroatoms, the boron atom has an empty pz orbital,15 which is a typical Lewis acid and often exhibits strong electron-deficient properties.16 Hence, it is often used as an acceptor unit in thermally activated delayed fluorescence (TADF) materials (ΔEST < 0.2 eV).17 However, few studies have been conducted on boron (B)-containing systems,18 especially triarylborane RTP materials with photo-stimulus responsiveness, which have not been reported yet. Therefore, through rational design and structural control of triarylborane, a simple, efficient, and universally applicable RTP material could be obtained. This would be of great research importance for enriching the types of organic RTP materials and expanding their applications in different fields. Inspired by this notion, we introduced a methylene carbazole functional unit to the ortho-position of aminoborane (BN), and successfully prepared a novel photo-induced RTP material based on BN-o-Met-Cz-doped amorphous polymers (namely BN-o-Met-Cz@PMMA, τp = 0.18 s; Φp = 6.83%), in which BN-o-Met-Cz was used as the RTP guest molecule, and PMMA was used as the host matrix (Fig. 1). BN-o-Met-Cz@PMMA films exhibited an unprecedented response to light stimulation under different ambient conditions. This phenomenon occurred because BN-o-Met-Cz molecules could effectively sensitize conversion of triplet oxygen to singlet oxygen under UV irradiation (365 nm), thus creating an oxygen-free environment and promoting phosphorescence emission. In addition, UV light could effectively modulate the molecular structure to promote generation of more triplet excitons and induce efficient phosphorescence emission. Interestingly, BN exhibited strong universal properties whether electron-donating or electron-withdrawing groups were positioned in the ortho (meta)-position of the B atom. For instance, it was possible to employ BN to produce several photo-activated multicolor single-molecule RTP materials with high quantum yields (BN-o-Met-Cz@PMMA: τP = 0.42 s, ΦP = 17.34%) and long lifetimes (BN-o-CN@PMMA: τP = 1.40 s, ΦP = 7.94%), which have been rarely reported. Time dependent-density functional theory (TD-DFT) theoretical calculations and experiments demonstrated that “push–pull” substituents with a D–π–A structure improved ISC and generated more triplet excitons. Incredibly, the doped films continued to produce bright photo-activated phosphorescence to the naked eye after being immersed in water for 3 months, indicating that their superior water resistance greatly expanded their practical applications. Simultaneously, these BN-X@PMMA films showed broad application prospects in information encryption, data erasure, and anti-counterfeiting.
 |
| | Fig. 1 Chemical structure of compounds B-o-Met-Cz, BN-o-Met-Cz, BN, BN-o-OMe, BN-m-OMe, and BN-o-CN. | |
2 Results and discussion
2.1. Design and synthesis of functional monomers
Heteroatomic boron shows great potential for the construction of TADF materials. The empty pz orbitals of boron atoms can be used to construct TADF materials with D–π–A structures by exploiting the unique electron-deficient properties of boron atoms, which usually have a small ΔEST.19 Therefore, the rational design and regulation of organoboron molecular structures could be employed to build new organic RTP materials. Inspired by this notion, two potential RTP emitters containing triarylborane units, BN-o-Met-Cz and B-o-Met-Cz, were designed and synthesized conveniently through two steps (Schemes S1–S3†). The starting material carbazole was prepared according to a procedure described previously.20 The molecular structure and purity of BN-o-Met-Cz and B-o-Met-Cz were demonstrated by high-performance liquid chromatography (HPLC; methanol/water (100/0, 95/5, 90/10)), nuclear magnetic resonance (NMR) spectroscopy (1H, 13C and 11B), high-resolution mass spectrometry (HRMS), and single-crystal X-ray diffraction (XRD) spectroscopy (Fig. S2 and S7, ESI†). The 11B chemical shift of BN-o-Met-Cz and B-o-Met-Cz was 71.37 ppm and 74.34 ppm, respectively, typical of triarylboranes.21 Thermogravimetric analysis (TGA) of BN-o-Met-Cz and B-o-Met-Cz showed their decomposition temperature to be 328 °C, and 300 °C, respectively. These two molecules had good thermal stability. Detailed information about synthesis and characterization is shown in Schemes S1–S6 and Fig. S1–S13.†
2.2. Photo-activated RTP behavior
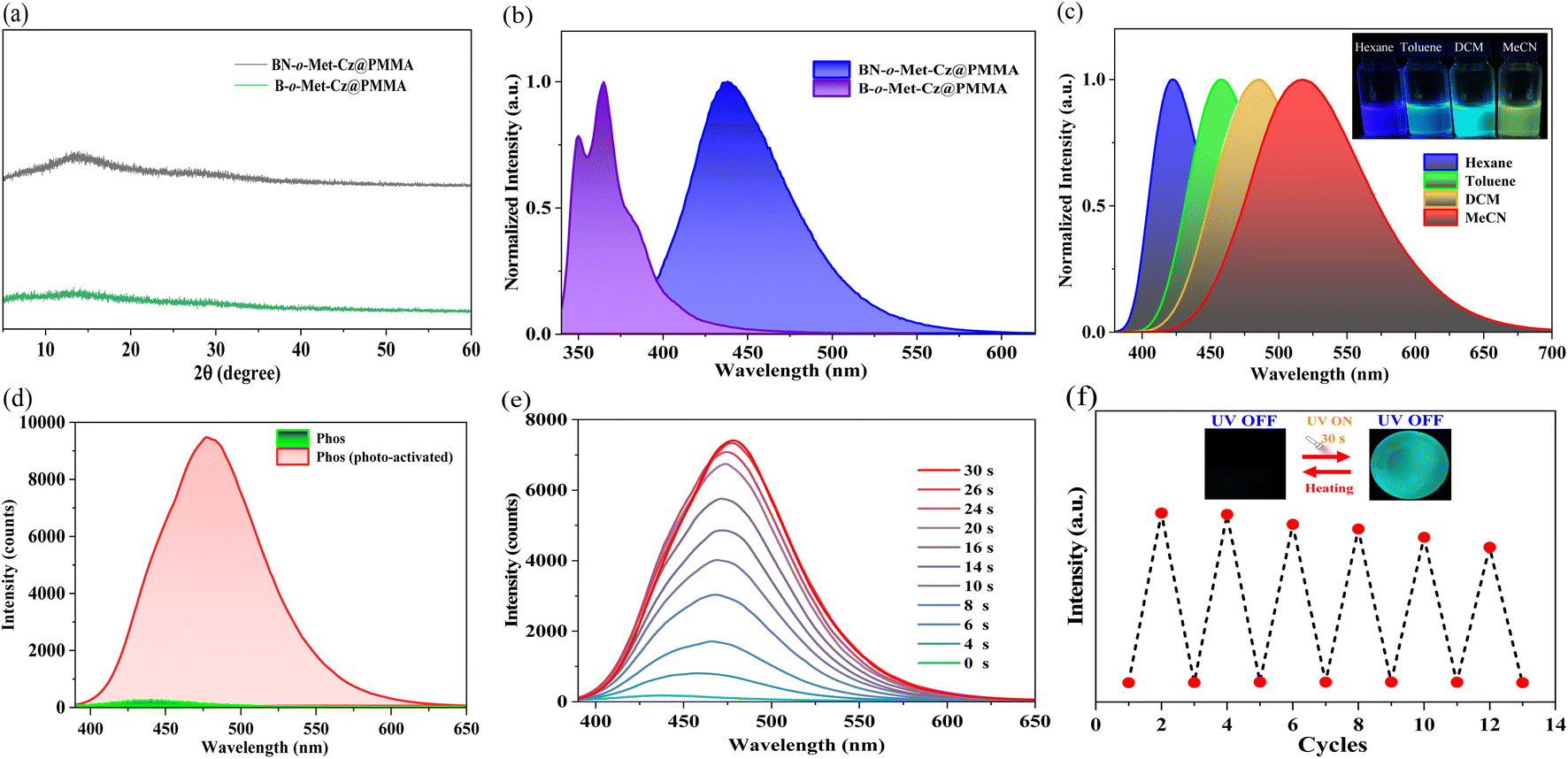
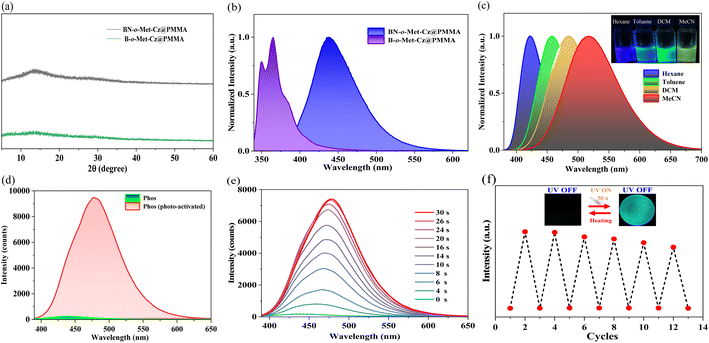
The two compounds were doped into a rigid PMMA matrix with a mass fraction of 0.5% to form BN-o-Met-Cz and B-o-Met-Cz films. Powder XRD spectroscopy suggested that they were all in an amorphous condition (Fig. 2a). Moreover, the thermal decomposition temperature of BN-o-Met-Cz@PMMA and B-o-Met-Cz@PMMA was increased by 20–40 °C compared with that of PMMA. Hence, doping triarylborane derivatives was beneficial to improve the thermal stability of PMMA (Fig. S15†). As shown in Fig. S16,† the UV absorption spectra of BN-o-Met-Cz@PMMA and B-o-Met-Cz@PMMA were distinctly different. For example, B-o-Met-Cz@PMMA had a distinct UV absorption peak at 410 nm, which was red-shifted by 20 nm from the λmax of B-o-Met-Cz@PMMA (342 nm). This peak could be attributed to an ICT from the N atom to the B unit. The UV absorption peak at 330–390 nm was attributed to an ICT from the carbazole to the boron unit. These data were verified in solvent polarity-dependent spectroscopy experiments (Fig. 2c) from hexane to acetonitrile. Simultaneously, the steady-state spectra of BN-o-Met-Cz@PMMA showed significant red-shifts with respect to those of B-o-Met-Cz@PMMA in the same state, whereas their delayed spectra were barely captured in transient spectra. Incredibly, unique photo-activated RTP emission could be observed for BN-o-Met-Cz@PMMA, from nearly non-RTP emission to strong RTP emission after UV irradiation for ∼30 s (Fig. 2d). Interestingly, this process could recover automatically to the initial state after placement in the dark environment for 1 h. Simultaneously, it took only 1 min to fully return to the initial state at 80 °C, and this process could be repeated several times (Fig. 2f). Unfortunately, the B-o-Met-Cz@PMMA films could not elicit photo-activated RTP under identical conditions. We hypothesize that this phenomenon could be connected to PMMA and the functional molecular structure. For example, the BN molecule with a D–π–A structure aids ISC under irradiation of UV light at 365 nm. This action sensitizes the conversion of triplet oxygen to singlet oxygen, and creates an oxygen-free environment that induces the triplet exciton of the molecule to undergo a radiative transition and to release phosphorescence.
 |
| | Fig. 2 (a) XRD spectra of BN-o-Met-Cz@PMMA and B-o-Met-Cz@PMMA. (b) Fluorescence spectra of BN-o-Met-Cz@PMMA (λex = 370 nm) and B-o-Met-Cz@PMMA (λex = 330 nm). (c) Fluorescence spectra of BN-o-Met-Cz in selected solvents (0.1 mM). (d) Phosphorescent spectra (delayed time: 5 ms) of BN-o-Met-Cz@PMMA before and after photoactivation by 365 nm UV light (λex = 370 nm). (e) Phosphorescent spectra of a BN-o-Met-Cz@PMMA film with the increased irradiation time (delayed time: 5 ms). (f) Reversibility of photoactivation and thermal deactivation of a BN-o-Met-Cz@PMMA film. | |
2.3. Internal mechanism of photo-activated RTP
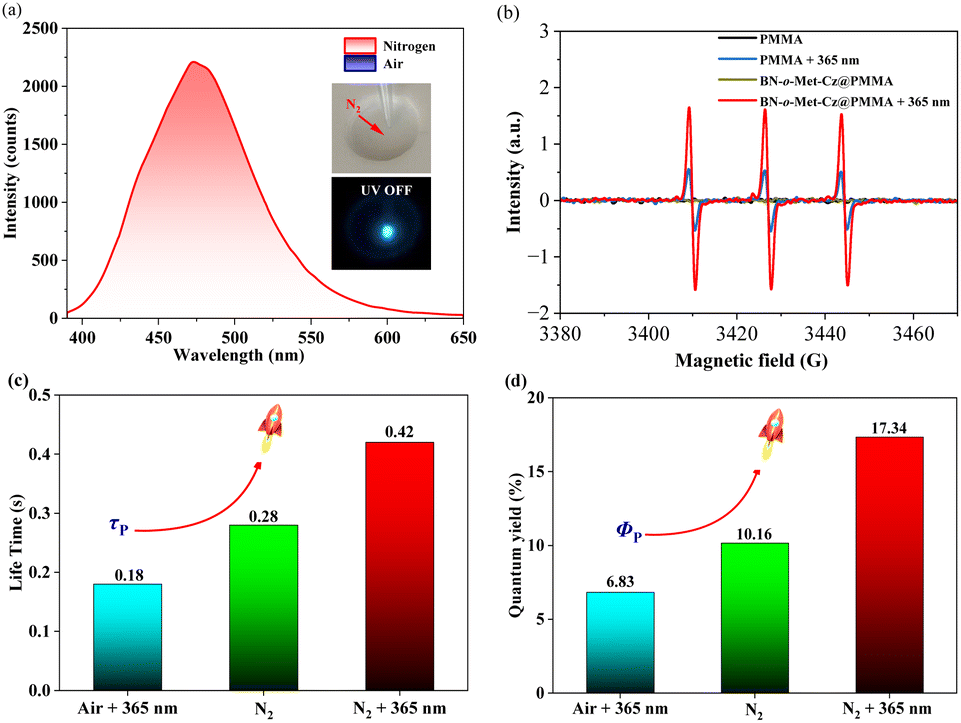
To test the reliability of our speculation, we investigated the phosphorescence properties of BN-o-Met-Cz@PMMA films under a nitrogen condition.22 As illustrated in Fig. 3a, BN-o-Met-Cz@PMMA films could elicit blue-green afterglow as seen by the naked eye after ceasing excitation (365 nm). This effect was consistent with its delay spectrum (delay time: 5 ms), indicating a trace amount of oxygen in BN-o-Met-Cz@PMMA films, which quenched the excited triplet excitons in the original state. Similarly, we captured the signal of singlet oxygen after photo-activation of BN-o-Met-Cz@PMMA films in the EPR spectrum, which was significantly stronger than the weak EPR signal generated by pure PMMA under irradiation, thereby indicating the sensitizing capability of the phosphor atoms (Fig. 3b).23 Surprisingly, BN-o-Met-Cz@PMMA exhibited an unprecedented response to light stimulation under different ambient conditions. For example, irrespective of the pretreatment of the BN-o-Met-Cz@PMMA film, its photo-activated phosphorescence quantum yield (air + 365 nm, τp = 0.18 s; Φp = 6.83%) was always inferior to that in a nitrogen atmosphere (N2: τp = 0.28 s; Φp = 10.16%). Furthermore, we found a doubled phosphorescence quantum yield (N2 + 365 nm, τp = 0.42 s; Φp = 17.34%) of BN-o-Met-Cz@PMMA films when they were photo-activated after exposure to a nitrogen atmosphere. Therefore, we hypothesized that UV light at 365 nm had a double-pronged effect for the BN-o-Met-Cz@PMMA film to obtain efficient RTP. For example, it could consume a small amount of oxygen in the BN-o-Met-Cz@PMMA film, but also regulate the molecular structure to promote ISC and induce more triplet excitons, and to release phosphorescence.
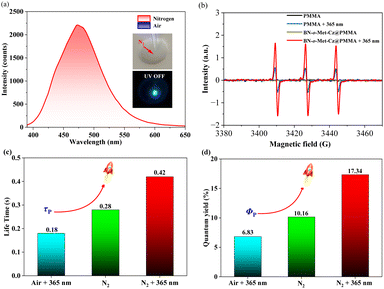 |
| | Fig. 3 (a) Phosphorescent spectra (delayed time: 5 ms) of BN-o-Met-Cz@PMMA in air and a nitrogen atmosphere. (b) Electron paramagnetic resonance (EPR) spectra of PMMA and BN-o-Met-Cz@PMMA before and after 365 nm UV-light irradiation in air. (c) RTP lifetime of BN-o-Met-Cz@PMMA under different conditions. (d) Phosphorescent quantum yield of BN-o-Met-Cz@PMMA under different conditions. | |
To further explore this double-pronged effect, we undertook single-crystal XRD spectroscopy under different environments (100 K, 298 K, 365 nm + 298 K). As shown in Fig. 4, the crystal structure changed significantly after UV irradiation at 365 nm compared with that at 298 K: the bond angle of methylene was reduced by 0.24°, and the length of the B–HMes bond was shortened by ∼0.1 Å. These phenomena were consistent with the crystal data at a low temperature of 100 K. Hence, UV light at 365 nm eliminated oxygen but also regulated the molecular structure in photo-induced RTP. Therefore, the monomer structure of BN-o-Met-Cz (365 nm + 298 K) was closer to the monomer structure at 100 K, which stabilized the excited triplet state of the monomer and released phosphorescence efficiently. This effect was verified in a dilute-solution (2-Me-THF: 0.001 mM) transient-spectroscopy experiment at 77 K. In addition, compound BN-o-Met-Cz has three dimers, all of which were formed by intermolecular C–H⋯π interactions. The C–H⋯π interaction force for D2 was reduced significantly compared with that at 298 K (from 2.747 Å to 3.240 Å), whereas D1 and D3 showed little change (Fig. S11†). These results further indicated that UV irradiation (365 nm) was beneficial to promote the transformation of aggregates into monomers, and to induce the phosphorescence emission of monomers. Furthermore, this phenomenon was confirmed in a similar experiment in the control molecule (B-o-Met-Cz). Taken together, these results verified the correctness of our previous inference. That is, UV irradiation can eliminate oxygen, but also induce the carbazole group to deviate from the center of the boron atom in a photo-activated RTP process. This action improves the push–pull electron efficiency, promotes ISC to produce more triplet excitons, and induces phosphorescence emission of the monomer. Therefore, this simple and inexpensive method could replace the low-temperature environment (77 K).
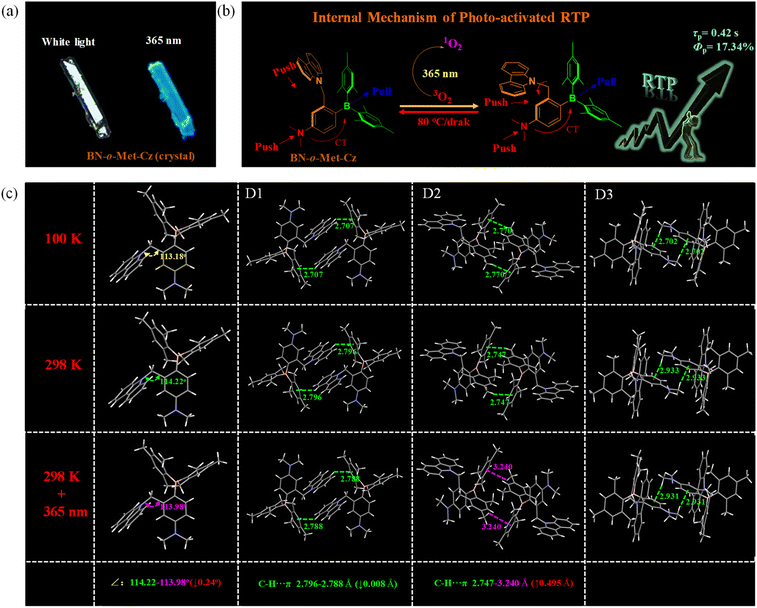 |
| | Fig. 4 (a) Photographs of BN-o-Met-Cz (crystal) under white light and UV light. (b) Internal mechanism of photo-activated RTP (schematic). (c) Crystal structure of BN-o-Met-Cz under different conditions. | |
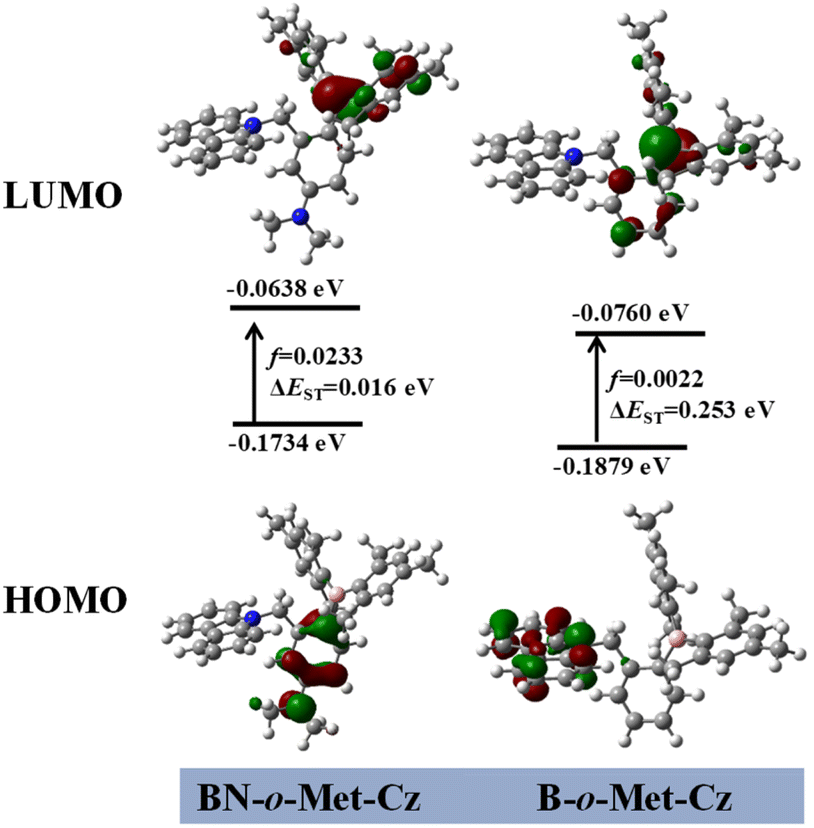
To get deeper understanding of these phenomena, TD-DFT calculations (see the ESI† for details) were carried out based on the single-crystal XRD data to investigate the electronic states of the monomers of BN-o-Met-Cz and B-o-Met-Cz.24 The highest occupied molecular orbitals (HOMOs) of the monomers of compound BN-o-Met-Cz were mainly distributed on the nitrogen atoms and benzene ring, and the lowest unoccupied molecular orbitals (LUMOs) were distributed on the boron atoms and a small amount on the benzene ring (Mes) (Fig. 5). However, the HOMO of B-o-Met-Cz was mainly concentrated on the carbazole unit, and the LUMOs were mainly distributed on the boron atoms and a small amount on the benzene ring. These results indicated a distinct ICT for both molecules, which was consistent with the solvent-dependence experiments (Fig. 2c). BN-o-Met-Cz had a smaller ΔEST and more efficient ISC channels than B-o-Met-Cz, which further indicated that molecules with a D–π–A structure could separate HOMOs and LUMOs effectively, induce a decrease in the ΔEST, and promote ISC to produce more triplet excitons. These data were consistent with the results of photo-activated RTP experiments.
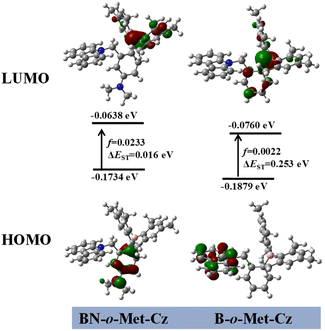 |
| | Fig. 5 Analysis of frontier molecular orbitals of the transition state for BN-o-Met-Cz and B-o-Met-Cz (B3LYP/6–31g(d) level of theory in a vacuum). | |
2.4. Photo-activated RTP behavior of BN
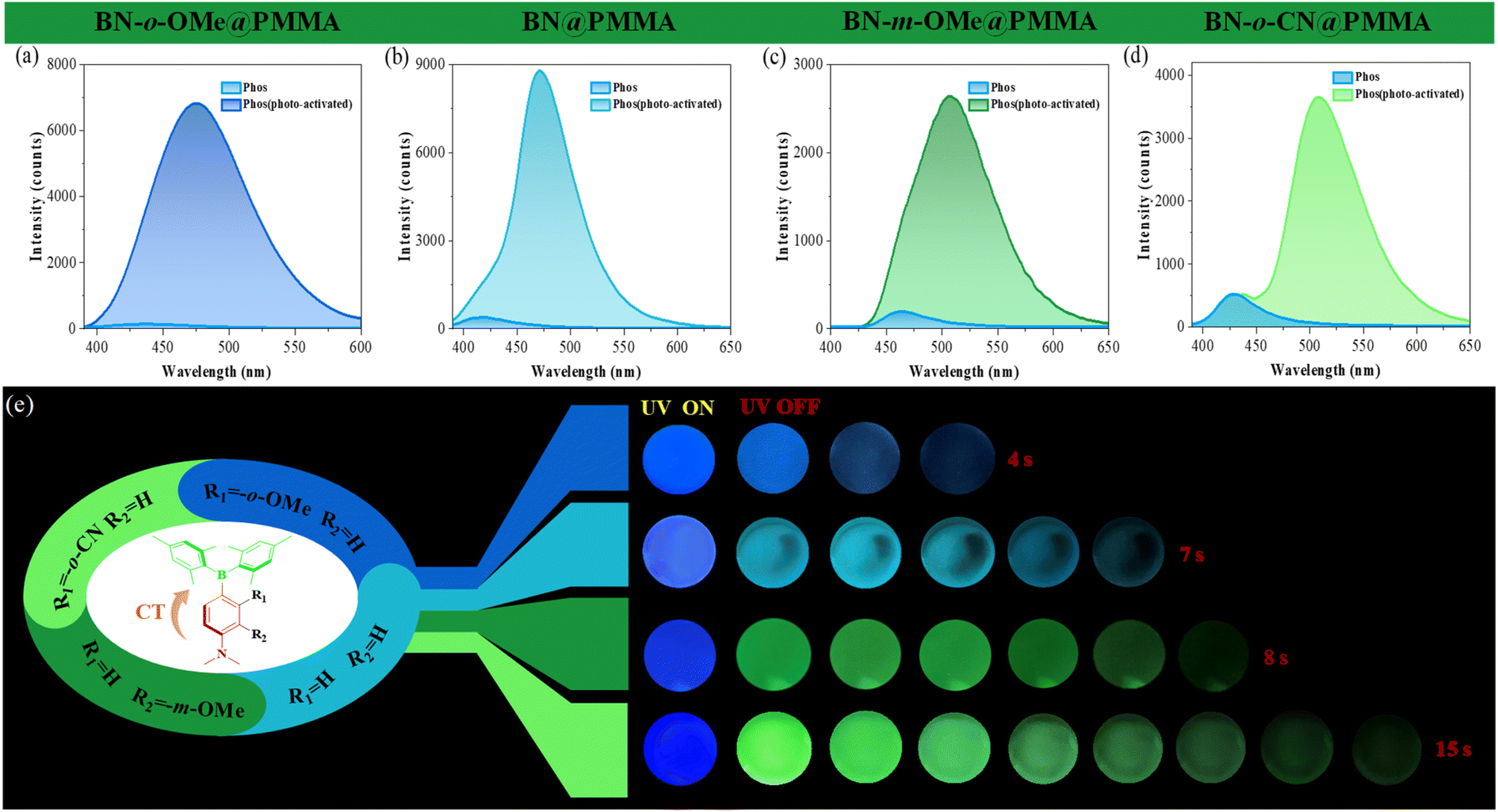
The data stated above predicted that a BN molecule with a D–π–A structure might have good pervasiveness of photo-activated RTP. To this end, a series of triarylborane derivatives with different push–pull electron substituents (BN, BN-o-OMe, BN-m-OMe, and BN-o-CN) were synthesized. Detailed information about synthesis and characterization is shown in Schemes S1–S6 and Fig. S1–S15.† As shown in Fig. 6, they all displayed clear photo-induced RTP characteristics when doped in PMMA films. For instance, their transient spectra were enhanced gradually upon extension of UV irradiation at 365 nm, exhibiting good photo-activated RTP properties, and demonstrated the universality of the BN core. Surprisingly, afterglow became more pronounced with enhancement of the properties of the push–pull electron substituents of the D–π–A molecule. For example, compound BN-o-CN had an afterglow duration ≤15 s (τP = 1.40 s, ΦP = 7.94%): such a long afterglow has rarely been reported. Interestingly, multicolor phosphorescence (from blue to cyan to yellow-green) could be modulated by changing the push–pull electron groups, which provided strong theoretical and experimental bases for preparation of single-molecule, full-color organic RTP materials.
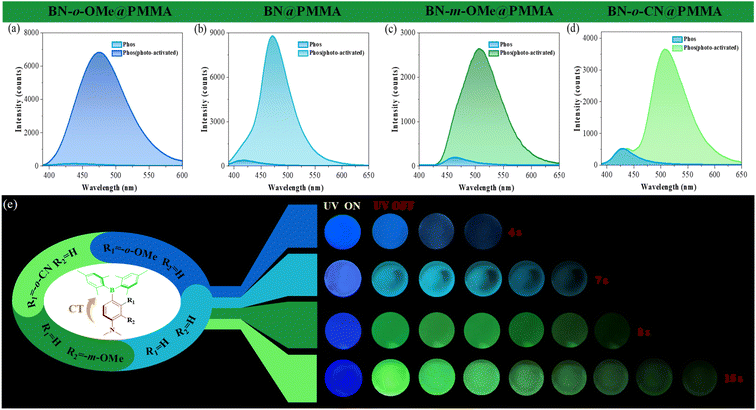 |
| | Fig. 6 Phosphorescent spectra (delayed time: 5 ms) of BN@PMMA (a), BN-o-OMe@PMMA (b), BN-m-OMe@PMMA (c), and BN-o-CN@PMMA (d) before and after photoactivation by 365 nm UV light. (e) Luminescence photographs of doped films under UV irradiation and after removal of UV irradiation with different duration times (films BN@PMMA, BN-o-OMe@PMMA, and BN-o-CN@PMMA under UV irradiation for 30 s to activate RTP, film BN-m-OMe@PMMA under UV irradiation for 60 s to activate RTP). | |
Interestingly, the photo-responsive time of BN-m-OMe@PMMA was significantly longer than that of the other molecules. We speculate that this may be related to the spatial resistance of the methoxy group (Fig. S14†). The bare methoxy group induces distortion of the adjacent dimethylamine group, thereby exposing the nitrogen atom and further enhancing the site of action of the functional molecule on oxygen. As a result, more air is adsorbed under identical conditions, so a longer light time is required to consume excess oxygen. The emission spectrum of the solid showed a clear blue-shift relative to the spectrum of the polar solvent. We speculate that this phenomenon may be related to the ICT effect of compound BN-X. Based on this, we systematically investigated the spectral properties of BN-X in undesirable solvents. As shown in Fig. S31,† the spectrum of compound BN-X shifted blue with increasing water content. These data indicated that molecules were prone to aggregation in undesirable solvents, which greatly reduced the ICT effect and exhibited a blue-shifted spectrum, which was consistent with the spectral results in non-polar solvents (hexane and toluene). In addition, we systematically investigated the photo-induced dual-response characteristics under different environments. Unfortunately, these molecules showed only the conversion of photo-induced molecular-sensitized triplet oxygen into the singlet state (Fig. 6), further indicating that methylene carbazole functional units had a crucial role in the photo-induced dual response.
2.5. Writing/erasing of optical information, water resistance, and information encryption
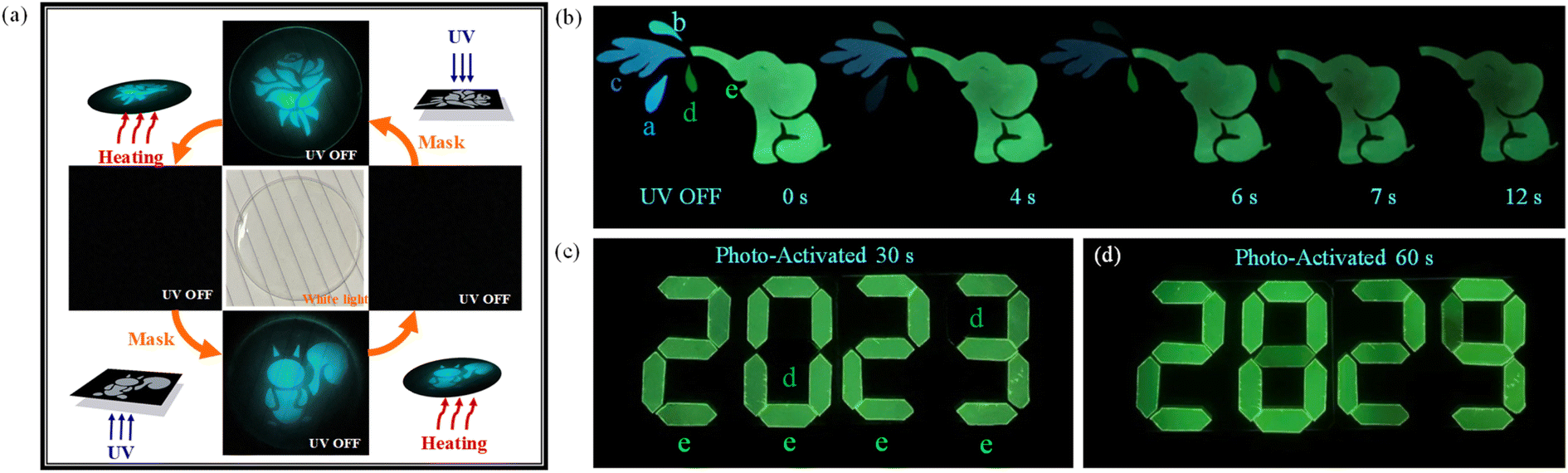
Utilizing the photo-responsive of phosphor atoms in the PMMA matrix, information could be written in these materials by light. As illustrated in Fig. 7a, the BN-o-Met-Cz@PMMA film was colorless and transparent under ambient light. When the film was irradiated with a strong UV light through a photomask for ∼30 s, a clear pattern was printed upon it. The film could regenerate by heating the film to 80 °C for 1 min. Due to excellent fatigue resistance, this writing–erasing–rewriting cycle could be repeated several times with no obvious residual information in the film. Incredibly, the BN-o-Met-Cz@PMMA film continued to have a bright, photo-activated long afterglow in water. For instance, after submerging the BN-o-Met-Cz@PMMA film in water for several days, a long afterglow that lasted for a few seconds could be seen with the naked eye after the UV lamp had been turned off. It showed good water resistance, which further improved the practicability of this material. In addition, we prepared a time-dependent phosphorescent anti-counterfeiting material based on the different luminescent colors and lifetimes of these molecules. As shown in Fig. 7b, the pattern gradually changed from a playful baby elephant squirting water to a baby elephant with a runny nose to a healthy baby elephant. It was very easy to capture the “mood” of the baby elephant in different states as the delay time increased, thus demonstrating a good anti-counterfeiting performance. In the meantime, simple application of information encryption was designed using the different photo-activated RTP times of BN-o-CN@PMMA and BN-m-OMe@PMMA. As shown in Fig. 7c, the green phosphorescence of BN-o-CN@PMMA was activated after the data encryption material had been exposed to a UV lamp at 365 nm for 30 s, which revealed that the first set of encryption codes, “2023”, came from the BN-o-CN@PMMA material. However, when we extended the 365 nm UV exposure to 60 s, the green phosphorescence of BN-m-OMe@PMMA was also activated perfectly to reveal a second set of cryptographic codes: “2829”. Such double-encrypted RTP materials have rarely been reported, and show good application prospects.
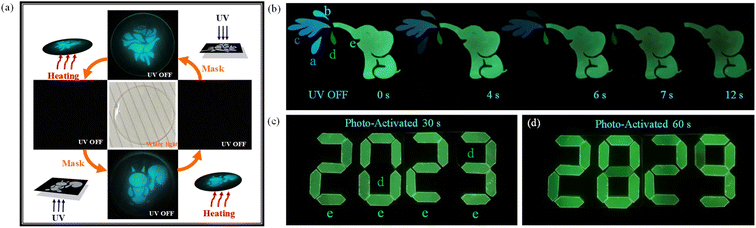 |
| | Fig. 7 (a) Application of programmable optical information writing/erasing based on BN-o-OMe@PMMA. (b) Elephant spray pattern made of films a to e. (c) Application of information encryption. After irradiation with 365 nm UV light for 30 s, the film e was activated to display information “2023”. (d) Upon extension of the irradiation time to 60 s, the film d was also activated to display the information of “2829”. Film a: BN-o-OMe@PMMA; film b: BN-o-Met-Cz@PMMA; film c: BN@PMMA; film d: BN-m-OMe@PMMA; film e: BN-o-CN@PMMA. | |
3 Conclusions
A series of triarylborane derivatives were prepared by introducing different push–pull electron groups. After incorporation into the PMMA matrix, BN with a D–π–A system had good generality as a photoinduced long-lived RTP material, such as introduction of a non-conjugated electron-donating group, methylenecarbazole, at the ortho position of the boron atom. A BN-o-Met-Cz@PMMA film exhibited different photoresponse behaviors to photoactivation in different surrounding environments (air + 365 nm: τP = 0.18 s, ΦP = 6.83%; N2 + 365 nm: τP = 0.42 s, ΦP = 17.34%). In addition, the photo-activated RTP performance was improved significantly by introducing a pseudo-halogen CN with electron-withdrawing properties in the B-atom ortho-position of the BN molecular backbone (BN@PMMA (τp = 0.73 s; Φp = 8.99%) → BN-o-CN@PMMA (τp = 1.40 s; Φp = 7.94%)). To shed light on these phenomena, a series of spectral, dilute-solution, solid-powder, crystal, and theoretical studies were undertaken to show that introduction of an electron-donating group or electron-withdrawing group to the BN skeleton could promote ICT and intramolecular interactions. Such actions could generate abundant triplet excitons, simultaneously stabilize triplet excitons thanks to the PMMA matrix, and triplet oxygen was sensitized to a singlet state under UV irradiation to release efficient RTP. Moreover, BN molecules showed broad application prospects for information encryption, data erasure, anti-counterfeiting, and water resistance. Our findings proffer new ideas for the design, synthesis, and application of organic RTP materials, enrich the types of organic RTP materials, and promote their further development.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†
Author contributions
H. Ding: conceptualization, validation, and writing (preparation of the original draft and reviewing). Y. Sun: investigation and validation. M. Tang: software use and investigation. J. Wen: validation. S. Yue: software use and investigation. Y. Peng: investigation. F. Li: resources. L. Zheng: resources. S. Wang: conceptualization. Y. Shi: supervision, conceptualization, visualization, project administration, and funding acquisition. Q. Cao: resources.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported financially by the National Natural Science Foundation of China (NSFC, 21901225, 21964020, 22164020), Yunnan Fundamental Research Projects (202001BB050016). We thank the Advanced Analysis and Measurement Center of Yunnan University for providing a sample-testing service.
Notes and references
-
(a) Kenry, C. Chen and B. Liu, Nat. Commun., 2019, 10, 2111 CrossRef CAS PubMed;
(b) W. Zhao, Z. He and B. Z. Tang, Nat. Rev. Mater., 2020, 5, 869–885 CrossRef CAS;
(c) Y. Zhang, Y. Su, H. Wu, Z. Wang, C. Wang, Y. Zheng, X. Zheng, L. Gao, Q. Zhou and Y. Yang,
et al.
, J. Am. Chem. Soc., 2021, 143, 13675–13685 CrossRef CAS PubMed;
(d) X. Yan, H. Peng, Y. Xiang, J. Wang, L. Yu, Y. Tao, H. Li, W. Huang and R. Chen, Small, 2021, 18, 2104073 CrossRef.
- T. Zhang, X. Ma, H. Wu, L. Zhu, Y. Zhao and H. Tian, Angew. Chem., Int. Ed., 2020, 59, 11206–11216 CrossRef CAS PubMed.
- L. Huang, C. Qian and Z. Ma, Chem. Eur. J., 2020, 26, 11914–11930 CrossRef CAS PubMed.
-
(a) R. Gao, M. S. Kodaimati and D. Yan, Chem. Soc. Rev., 2021, 50, 5564–5589 RSC;
(b) H. Su, K. Hu, W. Huang, T. Wang, X. Zhang, B. Chen, H. Miao, X. Zhang and G. Zhang, Angew. Chem., Int. Ed., 2023, e202218712 CAS.
-
(a) W. Zhu, H. Xing, E. Li, H. Zhu and F. Huang, Macromolecules, 2022, 55, 9802–9809 CrossRef CAS;
(b) Y. F. Zhang, J. W. Chen, Q. K. Sun, H. C. Zhang, S. F. Xue and W. J. Yang, Chem. Eng. J., 2023, 452, 139385 CrossRef CAS.
- Z. Xu, Y. He, H. Shi and Z. An, SmartMat., 2022, 4, 1–18 Search PubMed.
-
(a) G. Zhang, J. Chen, S. J. Payne, S. E. Kooi, J. N. Demas and C. L. Fraser, J. Am. Chem. Soc., 2007, 129, 8942–8943 CrossRef CAS PubMed;
(b) X. Ma, C. Xu, J. Wang and H. Tian, Angew. Chem., Int. Ed., 2018, 57, 10774 CrossRef CAS;
(c) T. Ogoshi, H. Tsuchida, T. Kakuta, T. A. Yamagishi, A. Taema, T. Ono, M. Sugimoto and M. Mizuno, Adv. Funct. Mater., 2018, 28, 1707369 CrossRef;
(d) H. Z. Wu, D. L. Wang, Z. Zhao, D. Wang, Y. Xiong and B. Tang, Adv. Funct. Mater., 2021, 31, 2101656 CrossRef CAS.
-
(a) L. Ma, S. Sun, B. Ding, X. Ma and H. Tian, Adv. Funct. Mater., 2021, 31, 2010659 CrossRef CAS;
(b) B. Chen, W. Huang, X. Nie, F. Liao, H. Miao, X. Zhang and G. Zhang, Angew. Chem., Int. Ed., 2021, 60, 16970–16973 CrossRef CAS PubMed;
(c) Z. Huang, Z. He, B. Ding, H. Tian and X. Ma, Nat. Commun., 2022, 13, 7841 CrossRef CAS PubMed;
(d) Y. Ren, W. Dai, S. Guo, L. Dong, S. Huang, J. Shi, B. Tong, N. Hao, L. Li and Z. Cai,
et al.
, J. Am. Chem. Soc., 2022, 144, 1361–1369 CrossRef CAS PubMed.
-
(a) J. Guo, C. Yang and Y. Zhao, Acc. Chem. Res., 2022, 55, 1160–1170 CrossRef CAS PubMed;
(b) Y. Zhang, X. Chen, J. Xu, Q. Zhang, L. Gao, Z. Wang, L. Qu, K. Wang, Y. Li and Z. Cai,
et al.
, J. Am. Chem. Soc., 2022, 144, 6107–6117 CrossRef CAS PubMed.
- S. Yue, H. Ding, Y. Sun, M. Tang, J. Wen, Y. Peng, L. Zheng, F. Wang, Y. Shi and Q. Cao, J. Phys. Chem. Lett., 2022, 13, 10190–10197 CrossRef CAS PubMed.
- C. Y. Shi, D. D. He, B. S. Wang, Q. Zhang, H. Tian and D. H. Qu, Angew. Chem., Int. Ed., 2023, 62, e202214422 CAS.
-
(a) X. Bi, Y. Shi, T. Peng, S. Yue, F. Wang, L. Zheng and Q. E. Cao, Adv. Funct. Mater., 2021, 31, 2101312 CrossRef CAS;
(b) H. Zhu, I. Badia-Dominguez, B. Shi, Q. Li, P. Wei, H. Xing, M. C. Ruiz Delgado and F. Huang, J. Am. Chem. Soc., 2021, 143, 2164–2169 CrossRef CAS PubMed.
-
(a) M. Kunitski, N. Eicke, P. Huber, J. Kohler, S. Zeller, J. Voigtsberger, N. Schlott, K. Henrichs, H. Sann and F. Trinter,
et al.
, Nat. Commun., 2019, 10, 1 CrossRef CAS PubMed;
(b) Z. Yin, M. Gu, H. Ma, X. Jiang, J. Zhi, Y. Wang, H. Yang, W. Zhu and Z. An, Angew. Chem., Int. Ed., 2021, 60, 2058–2063 CrossRef CAS PubMed;
(c) W. Dai, X. Niu, X. Wu, Y. Ren, Y. Zhang, G. Li, H. Su, Y. Lei, J. Xiao and J. Shi,
et al.
, Angew. Chem., Int. Ed., 2022, 61, e202200236 CAS;
(d) X. Zhang, J. Liu, B. Chen, X. He, X. Li, P. Wei, P. F. Gao, G. Zhang, J. W. Y. Lam and B. Z. Tang, Matter, 2022, 5, 3499–3512 CrossRef CAS.
- B. Zhou, Q. Zhao, L. Tang and D. Yan, Chem. Commun., 2020, 56, 7698–7701 RSC.
-
(a) Q. Hou, L. Liu, S. K. Mellerup, N. Wang, T. Peng, P. Chen and S. Wang, Org. Lett., 2018, 20, 6467–6470 CrossRef CAS PubMed;
(b) P. Li, Y. Jia, S. Zhang, J. Di, N. Zhang and P. Chen, Inorg. Chem., 2022, 61, 3951–3958 CrossRef CAS PubMed.
-
(a) S. K. Mellerup and S. Wang, Chem. Soc. Rev., 2019, 48, 3537–3549 RSC;
(b) Y.-F. Zhang, Y.-C. Wang, X.-S. Yu, Y. Zhao, X.-K. Ren, J.-F. Zhao, J. Wang, X.-Q. Jiang, W.-Y. Chang and J.-F. Zheng,
et al.
, Macromolecules, 2019, 52, 2495–2503 CrossRef CAS;
(c) Y. Wang, K. Liu, P. Chen, X. Yin, T. Peng, J. Iqbal and N. Wang, J. Mater. Chem. C., 2022, 10, 10981–10987 RSC;
(d) P. Li, D. Shimoyama, N. Zhang, Y. Jia, G. Hu, C. Li, X. Yin, N. Wang, F. Jakle and P. Chen, Angew. Chem., Int. Ed., 2022, 61, e202200612 CAS;
(e) Y. Shi, C. Li, H. Ma, Z. Cao, K. Liu, X. Yin, N. Wang and P. Chen, Org. Lett., 2022, 24, 5497–5502 CrossRef CAS PubMed;
(f) Y. Jia, P. Li, K. Liu, C. Li, M. Liu, J. Di, N. Wang, X. Yin, N. Zhang and P. Chen, Chem. Sci., 2022, 13, 11672–11679 RSC.
- X. L. Chen, J. H. Jia, R. Yu, J. Z. Liao, M. X. Yang and C. Z. Lu, Angew. Chem., Int. Ed., 2017, 56, 15006–15009 CrossRef CAS PubMed.
-
(a) Z. Wu, J. Nitsch, J. Schuster, A. Friedrich, K. Edkins, M. Loebnitz, F. Dinkelbach, V. Stepanenko, F. Wurthner and C. M. Marian,
et al.
, Angew. Chem., Int. Ed., 2020, 59, 17137–17144 CrossRef CAS PubMed;
(b) Y. Shi, Y. Zeng, P. Kucheryavy, X. Yin, K. Zhang, G. Meng, J. Chen, Q. Zhu, N. Wang and X. Zheng,
et al.
, Angew. Chem., Int. Ed., 2022, 61, e202213615 CAS;
(c) J. Jovaisaite, S. Kirschner, S. Raisys, G. Kreiza, P. Baronas, S. Jursenas and M. Wagner, Angew. Chem., Int. Ed., 2023, 62, e202215071 CrossRef CAS.
- S. Ito, M. Gon, K. Tanaka and Y. Chujo, Polym. Chem., 2021, 12, 6372–6380 RSC.
- A. W. Freeman, M. Urvoy and M. E. Criswell, J. Org. Chem., 2005, 70, 5014–9019 CrossRef CAS.
- H. J. Li, S. K. Mellerup, X. Wang and S. Wang, Org. Lett., 2019, 21, 2838–2842 CrossRef CAS PubMed.
- Y. Wang, J. Yang, M. Fang, Y. Gong, J. Ren, L. Tu, B. Z. Tang and Z. Li, Adv. Funct. Mater., 2021, 31, 2101719 CrossRef CAS.
-
(a) F. Szócs and M. Klimová, Eur. Polym. J., 1996, 32, 1087–1089 CrossRef;
(b) C. Wang, L. Qu, X. Chen, Q. Zhou, Y. Yang, Y. Zheng, X. Zheng, L. Gao, J. Hao and L. Zhu,
et al.
, Adv. Mater., 2022, 34, e2204415 CrossRef PubMed.
-
(a) J.-L. Ma, H. Liu, S.-Y. Li, Z.-Y. Li, H.-Y. Zhang, Y. Wang and C.-H. Zhao, Organometallics, 2020, 39, 4153–4158 CrossRef CAS;
(b) S. Lin, Q. Ou and Z. Shuai, ACS. Mater. Lett., 2022, 4, 487–496 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Suning
Wang
b,
Yonggang
Shi
a,
Suning
Wang
b,
Yonggang
Shi