
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Putting cyaphide in its place: determining the donor/acceptor properties of the κC-cyaphido ligand†
Eric S.
Yang
 a,
Emma
Combey
a and
Jose M.
Goicoechea
*b
a,
Emma
Combey
a and
Jose M.
Goicoechea
*b
aDepartment of Chemistry, University of Oxford, Chemistry Research Laboratory, 12 Mansfield Rd., Oxford, OX1 3TA, UK
bDepartment of Chemistry, Indiana University, 800 East Kirkwood Ave., Bloomington, Indiana 47405, USA. E-mail: jgoicoec@iu.edu
First published on 6th April 2023
Abstract
The synthesis of group 9 pyridine–diimine complexes M(DippPDI)X and [M(DippPDI)L]+ (M = Co, Rh; DippPDI = 1,1′-(pyridine-2,6-diyl)bis(N-(2,6-diisopropylphenyl)ethan-1-imine); X = CP−, CCH−; L = CO, tBuNC) bearing a series of strong-field ligands, including the cyaphide ion (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) P−), is reported. A combined experimental and computational comparative study of the group 9 PDI cyaphide complexes Co(DippPDI)(CP) and Rh(DippPDI)(CP), as well as the N-heterocyclic carbene (NHC) gold(I) cyaphide complex Au(IDipp)(CP) (IDipp = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene), reveals the σ donor and π acceptor properties of the κC-cyaphido ligand, and allow us to suggest a position for this ion in the spectrochemical series.
P−), is reported. A combined experimental and computational comparative study of the group 9 PDI cyaphide complexes Co(DippPDI)(CP) and Rh(DippPDI)(CP), as well as the N-heterocyclic carbene (NHC) gold(I) cyaphide complex Au(IDipp)(CP) (IDipp = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene), reveals the σ donor and π acceptor properties of the κC-cyaphido ligand, and allow us to suggest a position for this ion in the spectrochemical series.
Introduction
The cyanide ion (CN−) is found in coordination compounds across many areas of chemistry, ranging from biological enzyme cofactors to bespoke magnetic materials and catalysts.1–4 In the end-on κC coordination mode, the cyanide ion is an archetypal strong field ligand, a good σ donor and moderate π acceptor. These properties are critical to the utility of the cyanido ligand; its uncommon ability to withdraw π electron density despite its negative charge makes it a particularly useful tool in coordination chemistry.5–7
The cyaphide ion, CP−, is a phosphorus-containing analogue of the cyanide ion. Unlike the cyanide ion, it is rare, with relatively few known examples of cyaphido metal complexes having been reported to date.8–12 As a consequence, its ligand properties are still poorly understood. However, the potential for even higher π accepting character compared to cyanide has made the cyaphide ion an attractive candidate for use as a bridging ligand in magnetic materials, where its low-lying CP π* orbitals should facilitate more effective super-exchange between open-shell metal centers.6 Conceptually, the cyaphide ion is most closely related to two isolobal congeners, the cyanide (CN−) and acetylide (CCH−) ions. While both the cyanido and acetylido ligands are known to be good σ donors, the acetylido ligand has much reduced π withdrawing character, acting as a borderline π acceptor or donor.13,14
There is scant experimental evidence available with which to probe the electronic properties of the cyaphido ligand. The only systematic study of κC-cyaphido complexes conducted to date exclusively probed the effect of trans-ligated acetylides on ruthenium(II) cyaphido complexes,15 leaving the donor/acceptor properties of the cyaphide ligand itself still somewhat of a mystery.
Recently, we reported a magnesium(II) cyaphide complex Mg(DippNacNac)(dioxane)(CP) (MgCP; DippNacNac = CH{C(CH3)N(Dipp)}2 and Dipp = 2,6-diisopropylphenyl) which, by analogy to Grignard reagents, can be used to transfer the cyaphide ion to the coordination sphere of other metal centers using simple salt metathesis reactions.16 This has enabled synthetic access to many metal cyaphide coordination complexes which can be studied to better understand the properties of this anion. To this end, we recently used the gold(I) cyaphide complex Au(IDipp)(CP) (AuCP, IDipp = 1,3-bis(diisopropylphenyl)-imidazol-2-ylidene) to prepare heterometallic complexes featuring the cyaphide ion as a bridging ligand, revealing the electrophilic, π withdrawing nature of the cyaphide ion in side-on η2-coordination to metal centers (Fig. 1).17
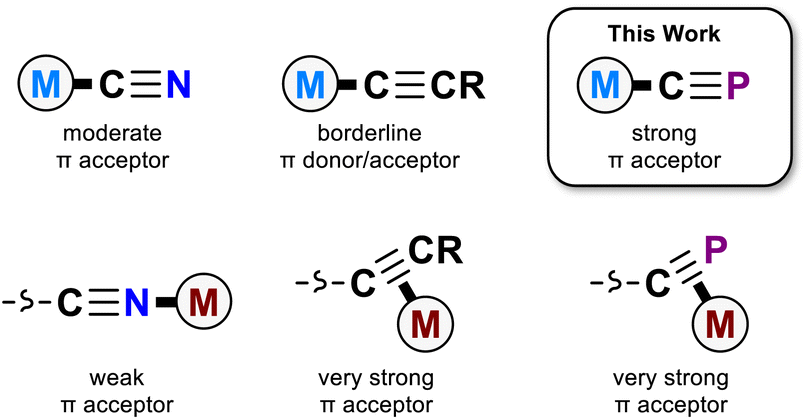 | ||
| Fig. 1 Coordination chemistry of the cyanido, acetylido, and cyaphido ligands. | ||
As traditional methods for probing ligand donor strength are currently inaccessible for the cyaphido ligand – due to a lack of [M(CP)6]x− or [M(CP)4]x− type complexes – in this study we focus on the use of both NMR spectroscopic and crystallographically determined bond metric data to do so. This allows us to deconvolute the donor/acceptor properties of the terminal κC-cyaphido ligand through comparative studies of three cyaphido metal complexes, the gold(I) complex (IDipp)Au(CP) (AuCP), the cobalt(I) complex Co(DippPDI)(CP) (CoCP; DippPDI = 1,1′-(pyridine-2,6-diyl)bis(N-(2,6-diisopropylphenyl)ethan-1-imine)), and the novel rhodium(I) complex Rh(DippPDI)(CP) (RhCP). AuCP is compared to other known gold(I) complexes Au(IDipp)X and [Au(Dipp)L]+ (AuX and AuL+; where X and L are used to describe anionic and neutral ligands, respectively, in accordance with the Covalent Bond Classification Method).18 Analysis of the gold(I) complexes allows us to determine the σ donor properties of the cyaphide ion. A systematic comparison of CoCP and RhCP with both known and novel M(DippPDI)X and [M(DippPDI)L]+ complexes (MX and ML+, M = Co, Rh) allows us to probe the π accepting character of the terminally bonded cyaphido ligand.
Results and discussion
From a theoretical standpoint, the ligand properties of the cyaphide ion are not immediately obvious. Its 2p–3p CP π bonds result both in low energy antibonding π* orbitals (6.69 eV, cf. CN−: 9.34 eV, CCH−: 8.98 eV) as well as high energy bonding π orbitals (−3.36 eV, cf. CN−: −4.47 eV, CCH−: −3.08 eV), increasing both π withdrawing and π donating ability relative to the cyanide ion. Moreover, the lower electronegativity of phosphorus relative to carbon results in the polarization of the π* orbital away from the coordinating carbon atom (41.0% C, cf. CN−: 79.3% C, CCH−: 48.5% C), in principle worsening Md–π* orbital overlap (Fig. 2, see ESI† for full computational details).
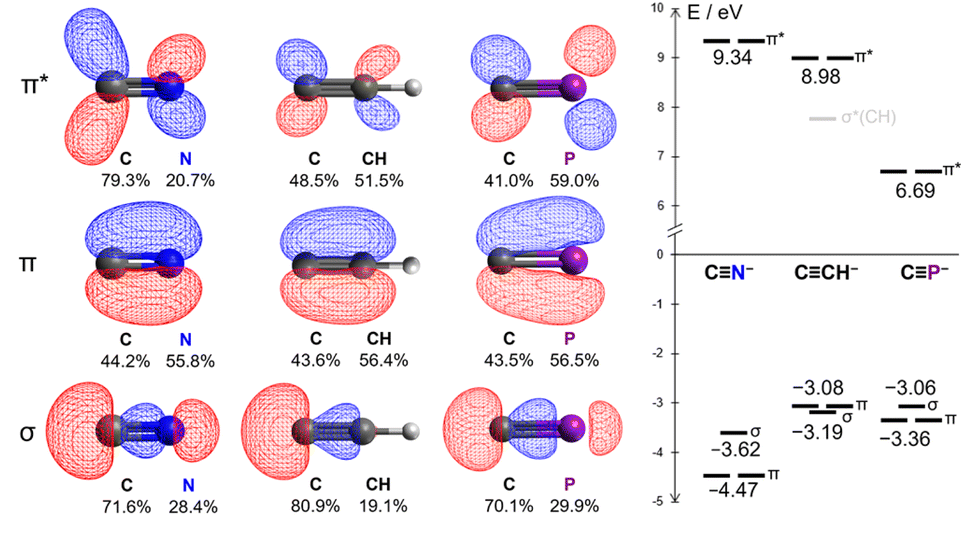 | ||
| Fig. 2 Kohn–Sham DFT frontier orbitals of the cyanide, acetylide, and cyaphide ions. | ||
It has been previously demonstrated that the N-heterocyclic carbene (NHC) carbenoid 13C{1H} NMR chemical shift (13CNHC) is sensitive to the σ basicity of trans-coordinated co-ligands.19 A similar trend can be observed in the case of gold(I) Au(IDipp)X and [Au(Dipp)L]+ complexes (AuX and AuL+) which exhibit 13CNHC chemical shifts over a wide frequency range (∼50 ppm) depending on the nature of the ligand trans to the carbene (Table S9†).16,17,20–28 Weak σ donors (such as acetonitrile) give rise to low 13CNHC chemical shifts (e.g.AuNCMe+: 13CNHC = 166.0 ppm), whereas strong donors (e.g. boryls) give rise to high shifts (e.g.AuBPin: 13CNHC = 216.7 ppm). The solid-state CNHC–Au bond lengths in such complexes are less sensitive to the nature of the trans-ligand, although they follow a similar trend, apart from strong π donors (such as chloride and hydroxide ligands) which deviate slightly. This is consistent with previous theoretical studies on gold(I) NHC complexes: while electrostatic and σ donation effects dominate for NHC–AuI bonds,29 π donating co-ligands have been shown to enable more significant AuI–NHC π backbonding.30
EDA-NOCV analysis of the L–Au bond in the aforementioned complexes allows for the theoretical examination of σ donor and π acceptor orbital interactions. Experimental 13CNHC chemical shifts correlate very well with calculated ETS-NOCV σ donation energies (Fig. 3), and conversely do not correlate well with ETS-NOCV π backdonation energies (Fig. S50†). Thus the 13CNHC chemical shift in Au(IDipp)X and [Au(Dipp)L]+ compounds can be taken as a measure of the σ donating ability of the trans ligand.
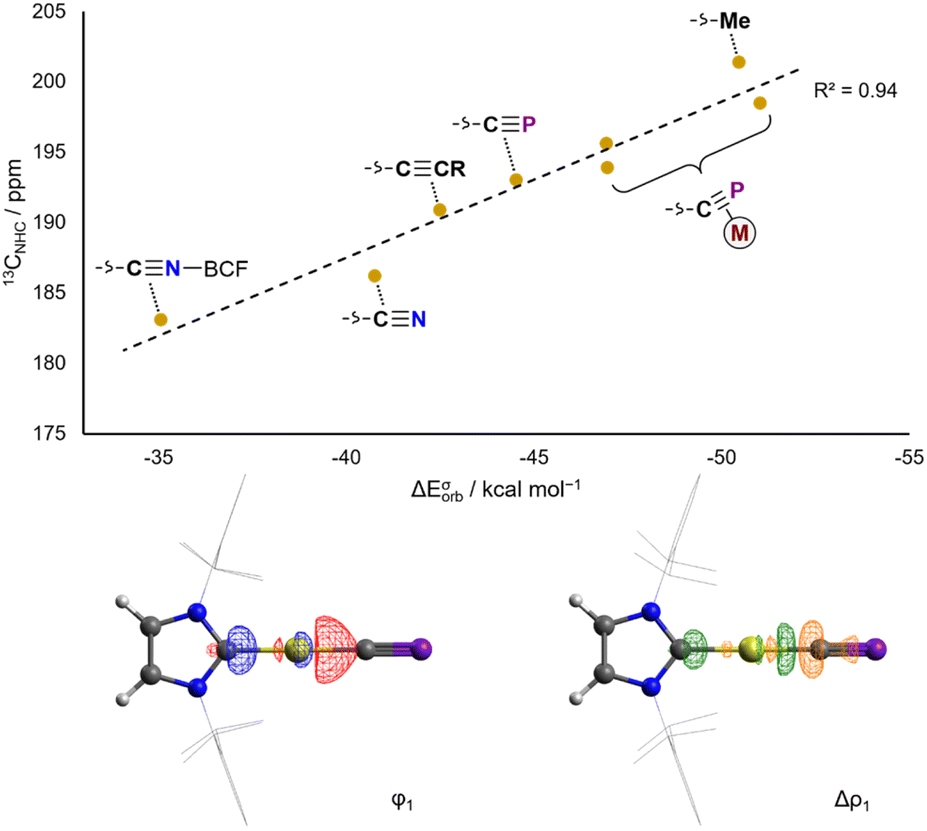 | ||
| Fig. 3 Correlation of calculated ETS-NOCV σ donation energies with experimental 13CNHC NMR spectroscopic data for AuX complexes (top). Isosurfaces for the σ bonding NOCV and difference density in AuCP (bottom). | ||
In AuCP, the 13CNHC chemical shift is 193.0 ppm, in a very similar range to other sp-hybridized carbanions such as cyanide and phenylacetylide (AuCN: 13CNHC = 186.2 ppm; AuCCPh: 13CNHC = 190.0 ppm). This is also reflected computationally in the ETS-NOCV σ donor energies: the cyaphide complex has a ΔEσorb value of −44.5 kcal mol−1 compared to −40.8 kcal mol−1 for cyanide and −42.5 kcal mol−1 for acetylide. These data show that the cyaphido ligand has a very similar σ basicity to closely related sp-carbanionic ligands, though slightly higher due to its higher energy HOMO and/or the lower electronegativity of phosphorus resulting in higher charge density at carbon.
The same analysis can be performed for the previously reported hetero- bi- and tri-metallic bridging cyaphide complexes {Au(IDipp)}{Ni(MeIiPr)2}(μ-CP), {Au(IDipp)}{Rh(Cp*)(PMe3)}(μ-CP), and {Au(IDipp)}{Rh(Cp*)(PMe3)}{W(CO)5}(μ-CP).17 The bimetallic complexes give rise to higher 13CNHC chemical shifts (13CNHC = 198.5 and 195.6 ppm, for the nickel and rhodium complexes, respectively), showing that the net electron-withdrawing nature of the η2 cyaphido-metal interaction results in heightened σ donor ability at carbon, which is also reflected in larger calculated ΔEσorb energies (ΔEσorb = −51.0 and −46.9 kcal mol−1, respectively). Coordination of a third metal to the cyaphide ion via the phosphorus lone pair results in a moderate reduction in σ donor ability, with the trimetallic complex exhibiting a 13CNHC chemical shift of 193.9 ppm.
Whereas AuCP allows for the study of the σ donor properties of the κC-cyaphido ligand, the group 9 cyaphido complexes Co(DippPDI)(CP) (CoCP) and Rh(DippPDI)(CP) (RhCP) are sensitive to its π donor/acceptor character. The tridentate pyridine diimine (PDI) ligand framework has a low lying π* orbital that gives rise to strong π accepting character and redox non-innocence. Population of this π* orbital by π backdonation or reduction results in measurable changes in the solid-state bond metrics of the PDI ligand, summarized in the parameter δ(PDI),31 which allows for convenient assessment of the electronic structure of metal PDI complexes (see Tables S10 and S11†). An NMR study of the purple, square-planar cobalt complexes Co(DippPDI)H (CoH), Co(DippPDI)Me (CoMe),32 and Co(DippPDI)Cl (CoCl),33 has previously shown that they consist of cobalt(II) centers ligated by reduced (DippPDI)˙− radical anions.34 Notably absent from this study were ligands with appreciable π accepting ability. Indeed it has been noted that the cationic dinitrogen complex [Co(DippPDI)(N2)][B(Me)(C6F5)3] has a markedly different δ(PDI) value than for the aforementioned complexes (0.132(9); cf. 0.090(9)–0.098(7) for CoH, CoMe, and CoCl) and is a deep blue color instead of purple. This suggests it is better described as having a cobalt(I) center and neutral DippPDI ligand.31
In order to provide additional useful data points against which the cobalt(I) cyaphide complex CoCP can be compared, several complexes with ligands of varying π acceptor character (CoX and CoL+) were prepared and characterized.‡ The purple cobalt acetylide complex Co(DippPDI)(CCH) (CoCCH) was prepared by salt metathesis of CoCl with ethynylmagnesium chloride (Scheme 1). Its solid-state structure was determined by single crystal X-ray diffraction (Fig. 4), showing a square-planar cobalt center with a Npy–Co bond length of 1.812(2) Å and a δ(PDI) value of 0.098(7) (Table S10†). If the reaction is performed with an excess of sodium acetylide instead of the Grignard reagent, a mixture of products forms, from which Na(THF)Co(PIEA)(CCH) (PIEA = pyridine–imine–enamine) can be isolated, containing a deprotonated PDI ligand and a sodium cation held in a close ion pair by π interactions with arene and acetylide moieties.
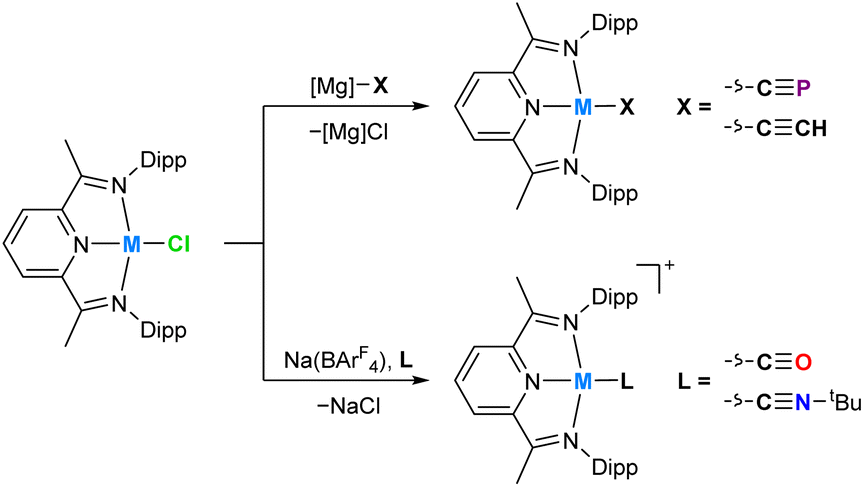 | ||
| Scheme 1 Synthesis of MX (M = Co, Rh; X = CP− CCH−) and ML+ complexes (M = Co, Rh; L = CO, CNtBu). | ||
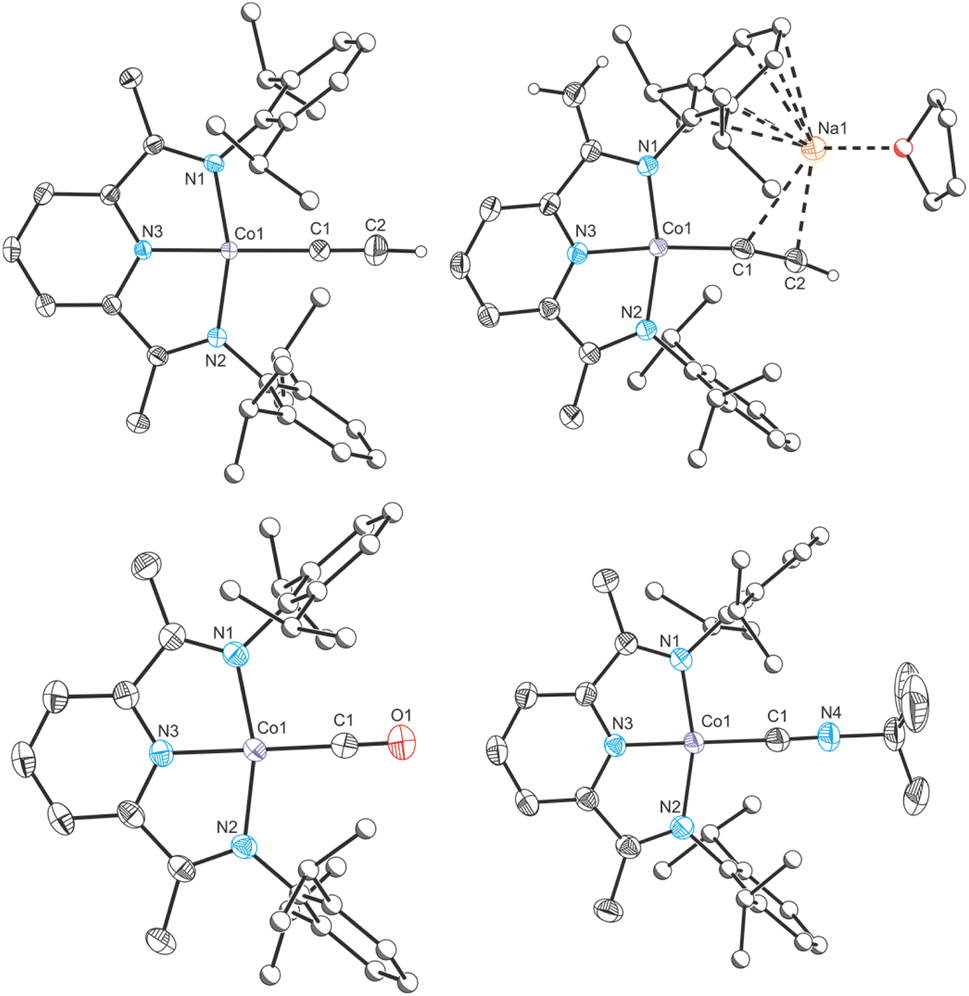 | ||
| Fig. 4 Clockwise from top left: single crystal X-ray structures of complexes CoCCH, Na(THF)Co(PIEA)(CCH), CoCNBu+ and CoCO+. Thermal ellipsoids set at 50% probability level. Hydrogen atoms (except for acetylide and terminal alkene protons) removed for clarity. Atoms of Dipp groups pictured as spheres of arbitrary radius. | ||
Cobalt carbonyl [Co(DippPDI)(CO)][BArF4] (CoCO+, BArF4− = tetrakis(3,5-bis(trifluoromethyl)phenyl)borate) and tert-butyl isocyanide [Co(DippPDI)(CNtBu)][BArF4] (CoCNBu+) complexes were synthesized by halide abstraction with Na(BArF4) in 1,2-difluorobenzene (1,2-DFB), in the presence of CO gas or tBuNC, respectively. Both complexes are blue and have solid-state structures with Npy–Co bond lengths of 1.847(2) Å (CoCO+) and 1.819(2) Å (CoCNBu+), and δ(PDI) values of 0.144(7) (CoCO+) and 0.121(4) (CoCNBu+). If two equivalents of tBuNC are used the reaction results in a different product, [Co(DippPDI)(CNtBu)2][BArF4] (Co2CNBu+), with a five-coordinate square-pyramidal cobalt center (see ESI†).
The cobalt cyanide complex Co(DippPDI)(CN) (CoCN) was also targeted, although attempts to prepare it via several methods were unsuccessful, including salt metathesis with KCN or Me3SiCN, and halide abstraction in the presence of [Bu4N]CN. This is likely due to the potential for redox disproportionation. The reaction of the cobalt(II) complex Co(DippPDI)Cl2 with sodium cyanide was previously shown to lead to disproportionation, forming Co(DippPDI)(CN)3 and other unidentified byproducts.35 An alternative decomposition pathway could involve deprotonation of the PDI ligand by the Brønsted basic cyanide ion (as observed for the reaction of CoCl with sodium acetylide; vide supra).
The cobalt cyaphide complex CoCP is blue, with a Npy–Co bond length of 1.825(3) Å and a δ(PDI) value of 0.131(9). From these data, it is clear that CoX and CoL+ (X and L are CP−, CCH−, Me−, N2, CO, CNtBu, and Cl−) can be separated into two groups with different electronic structures. The purple complexes with low δ(PDI) values are best described as Co2+(DippPDI)˙− systems, whereas the blue complexes with high δ(PDI) values are better described as Co1+(DippPDI). In order to quantify this, UV-vis spectra for all complexes were measured. As expected, the absorbance maxima (λmax) relate discontinuously with δ(PDI), with low λmax corresponding to low δ(PDI) and vice versa (Fig. 5).
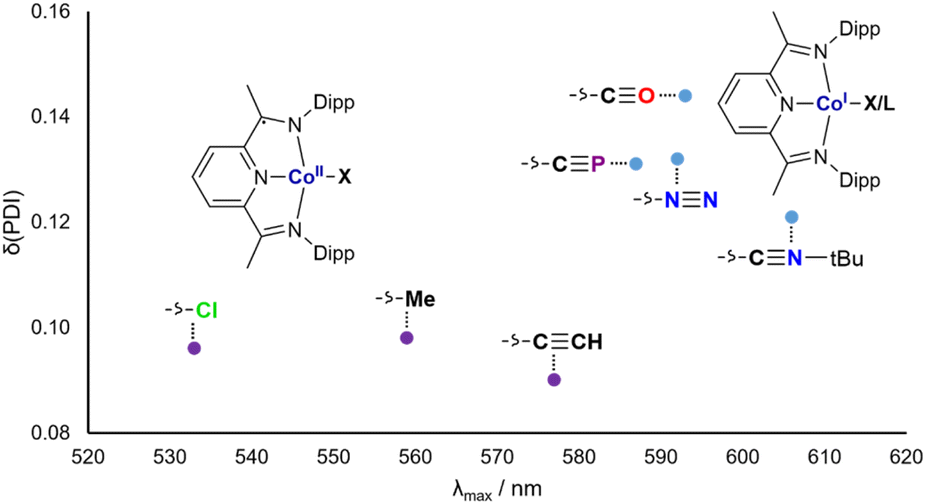 | ||
| Fig. 5 UV-vis absorbance maxima (λmax) plotted against δ(PDI) for CoX and CoL+. | ||
The group of blue Co1+(DippPDI) complexes with higher λmax and δ(PDI) values all feature π accepting ligands, with the notable inclusion of the cyaphide ion. This can be rationalized by considering the effect of the ligand on the Co(DippPDI) fragment: when it is primarily electron donating, the electron rich Co(DippPDI) fragment favors the reduced DippPDI state, whereas when the ligand is π withdrawing, the less electron rich Co(DippPDI) fragment favors the neutral DippPDI state.
In order to better quantify the π accepting ability of the cyaphido ligand, the heavier group 9 DippPDI complexes Rh(DippPDI)X and [Rh(DippPDI)L]+ (RhX and RhL+) were prepared, in which the much lower relative favorability of the rhodium(II) oxidation state should preclude the redox non-innocence of the PDI ligand. Addition of Rh(DippPDI)Cl36 (RhCl) to a toluene solution of the cyaphide transfer reagent MgCP results in the formation of the novel rhodium(I) cyaphide complex Rh(DippPDI)(CP) (RhCP) after 3 days. RhCP exhibits a doublet in its 31P{1H} NMR spectrum at 251.4 ppm (2JP–Rh = 6 Hz) corresponding to the cyaphide phosphorus atom and a doublet of doublets in its 13C{1H} NMR spectrum at 264.15 ppm (1JC–Rh = 54.8 Hz, 1JC–P = 22.9 Hz) corresponding to the cyaphide carbon atom. The CP vibrational stretching frequency was measured to be 1334 cm−1 by Raman spectroscopy (cf. 1306 cm−1 for CoCP). Its solid-state structure was determined by X-ray crystallography (Fig. 6), revealing a square-planar rhodium center, a C–P bond length of 1.542(4) Å (cf. 1.506(4) Å for CoCP), a Npy–Rh bond length of 1.943(2) Å, and a δ(PDI) value of 0.134(3).
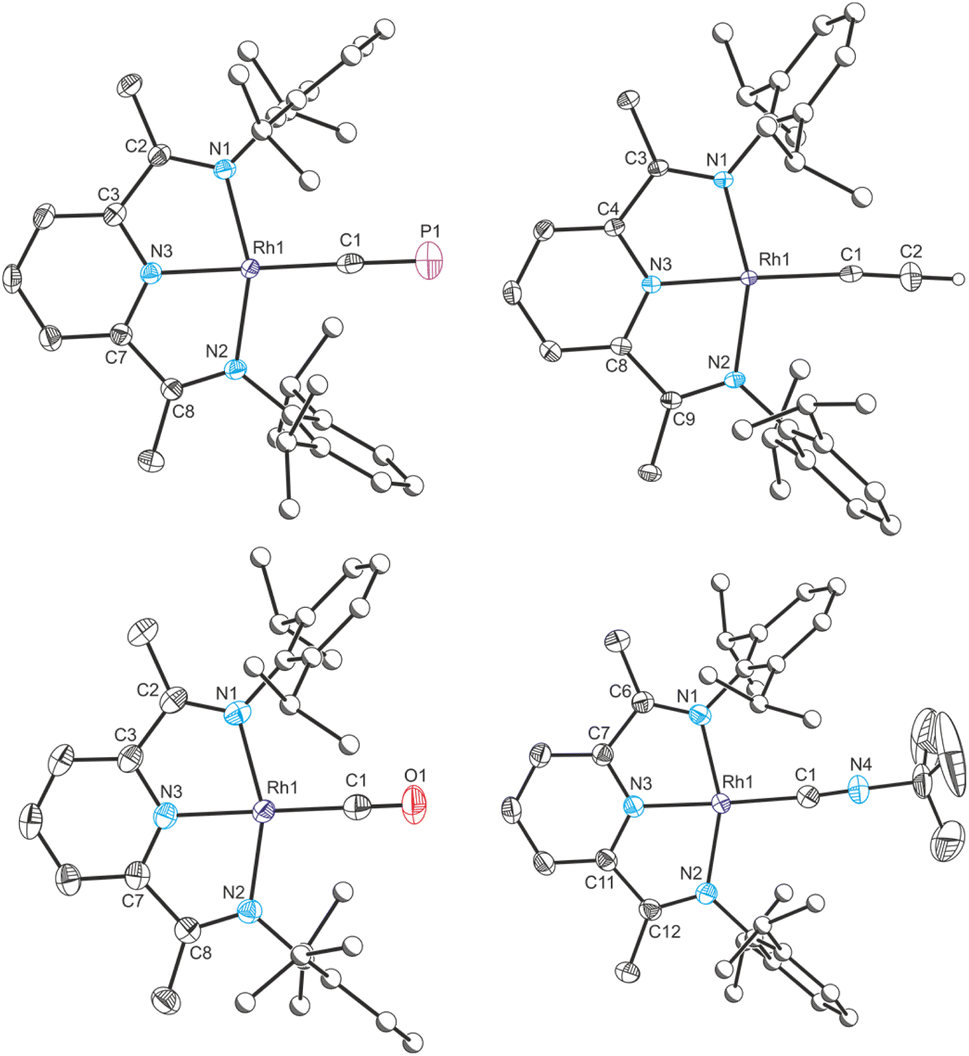 | ||
| Fig. 6 Clockwise from top left: single crystal X-ray structures of complexes RhCP, RhCCH, RhCNBu+ and RhCO+. Thermal ellipsoids set at 50% probability level. Hydrogen atoms (except for acetylide protons) removed for clarity. Atoms of Dipp groups pictured as spheres of arbitrary radius. Selected bond distances (Å) and angles (°) for RhCP: Rh1–C1 1.969(3), C1–P1 1.542(4), Rh1–N1 2.019(2), Rh1–N2 2.023(2), Rh1–N3 1.942(2), N1–C2 1.309(4), C2–C3 1.470(4), C3–N3 1.360(4), N3–C7 1.357(4), C7–C8 1.468(4), C8–N2 1.315(4); C1–Rh1–N1 101.06(11), C1–Rh1–N2 101.41(11), C1–Rh1–N3 179.24(14), N1–Rh1–N2 157.48(10), N1–Rh1–N3 78.92(10), N2–Rh1–N3, 78.59(10). | ||
The rhodium(I) acetylide complex Rh(DippPDI)(CCH) (RhCCH) could likewise be prepared by salt metathesis with ethynylmagnesium chloride. Single crystal X-ray crystallography confirms its structure, with a Npy–Rh bond length of 1.926(2) Å and a δ(PDI) value of 0.124(2).
As with CoCO+ and CoCNBu+, cationic rhodium(I) carbonyl and isocyanide complexes could be prepared by halide abstraction. Reaction of 1,2-DFB solutions of RhCl and Na(BArF4) with CO or tBuNC resulted in the formation of [Rh(DippPDI)(CO)][BArF4] (RhCO+) and [Rh(DippPDI)(CNtBu)][BArF4] (RhCNBu+), respectively. Single crystal X-ray diffraction reveals a Npy–Rh bond length of 1.969(2) Å and a δ(PDI) of 0.161(3) for RhCO+, and a Npy–Rh bond length of 1.942(3) Å and a δ(PDI) of 0.152(3) for RhCNBu+.
To provide further points of comparison, the solid-state structures of the known rhodium(I) complexes Rh(DippPDI)Cl (RhCl), Rh(DippPDI)Me (RhMe),34 and [Rh(DippPDI)(C2H4)][BArF4] (Rhethene+),37 were measured by single crystal X-ray diffraction. This allowed their Npy–Rh bond lengths and δ(PDI) values to be determined: Npy–Rh = 1.898(4) Å, and δ(PDI) = 0.108(6) for RhCl, Npy–Rh = 1.936(3) Å, and δ(PDI) = 0.110(6) for RhMe, Npy–Rh = 1.960(2) Å, and δ(PDI) = 0.152(3) for Rhethene+.
In contrast to the situation for the analogous cobalt complexes, the δ(PDI) values for all the aforementioned RhX and RhL+ complexes vary continuously with different ligands (Fig. 7). Changes to δ(PDI) represent different degrees of rhodium(I) to DippPDI π backdonation, and consequently are highly sensitive to the π donor/acceptor properties of L. The π donor ligand Cl− in RhCl results in a low δ(PDI) value (0.108(6)), whereas the strong π acceptor CO results in a high δ(PDI) value (0.161(3)) in RhCO+. Calculated ETS-NOCV π backdonation energies (ΔEπorb) correlate very well with δ(PDI), as do calculated NBO π backdonation energies from 2nd order perturbation theory (E(2)π). The Npy–Rh bond length also varies with the trans-influence of L, though is also sensitive to σ donating ability, as evidenced by the anomalously high δ(PDI) for RhMe (Fig. S58†).
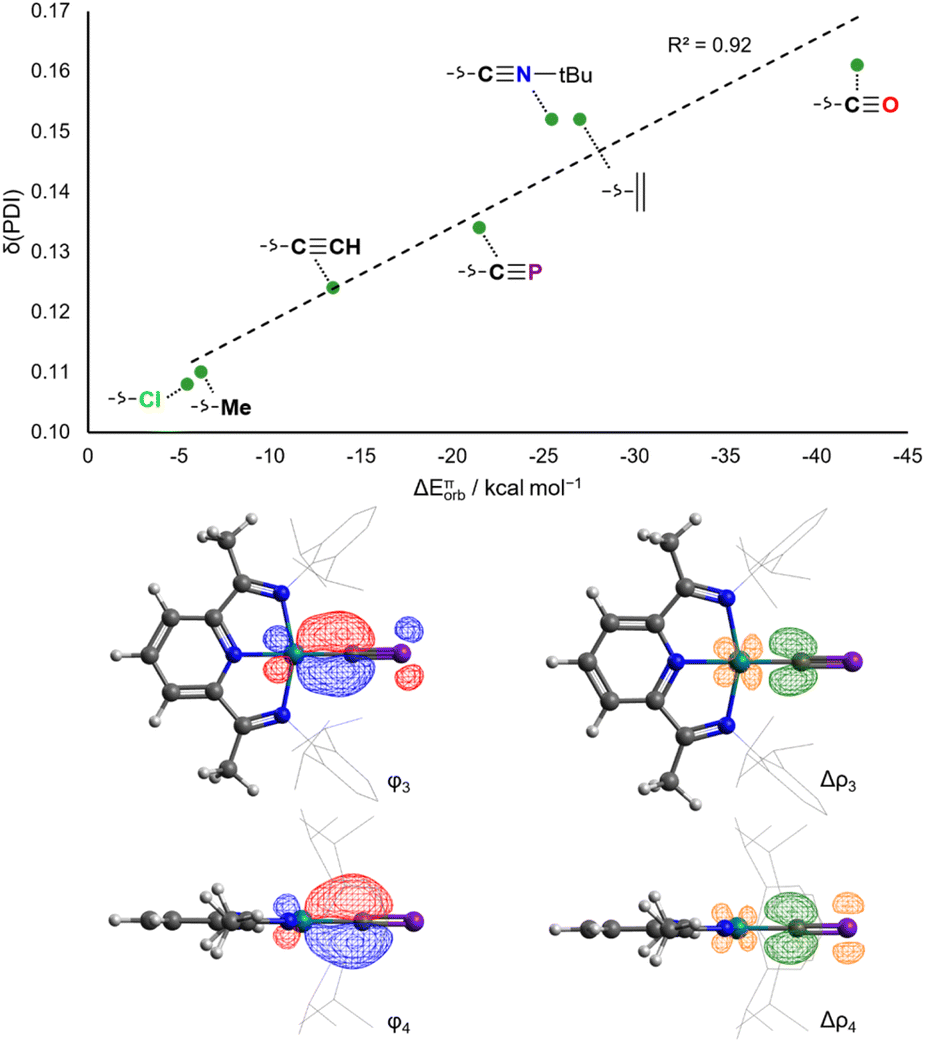 | ||
| Fig. 7 Correlation of calculated ETS-NOCV π backdonation energies with experimental δ(PDI) values for RhX and RhL+ (top). Isosurfaces for the π bonding NOCVs and difference densities in RhCP (bottom). | ||
The rhodium(I) cyaphide complex RhCP has a δ(PDI) of 0.134(3) and Npy–Rh bond length of 1.943(2) Å. This is likewise reflected in its calculated ΔEπorb (−21.5 kcal mol−1) and E(2)π (44.1 kcal mol−1) values. This places the π accepting ability of the cyaphide ion as being higher than the acetylide ion and almost as high as isocyanides, though still lower than neutral, strong π accepting ligands (e.g. CO, C2H4).
Conclusions
The cyaphide ion is both a strong σ donor and π acceptor in the terminal, κC coordination mode. The relatively low electronegativity of phosphorus makes the cyaphide ion a slightly stronger σ donor than the cyanide and acetylide ions, and its low energy 2p–3p CP π* orbitals make it a potent π acceptor despite its negative charge, with a π acidity comparable to neutral isocyanides. This will undoubtedly have consequences for using the cyaphide ion as a tool to tune the properties of coordination complexes. In particular, its strong π accepting ability in both the η1, κC coordination mode as well as the side-on η2 coordination mode will make the cyaphide ion especially suitable for use as a bridging ligand in magnetic coordination polymers, for example heavy analogues of Prussian blue.
Data availability
Crystallographic data has been deposited with the Cambridge Structural Database.Author contributions
Conceptualization: J. M. G.; experimental work: E. S. Y. and E. C.; X-ray crystallography: E. S. Y and J. M. G.; computational work: E. S. Y.; writing – original draft: E. S. Y and J. M. G.; writing & editing: all authors; supervision: E. S. Y. and J. M. G.; funding acquisition: J. M. G.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Oxford, the EPSRC, and OxICFM CDT for financial support of this research (ESY: EP/S023828/1). The University of Oxford is also acknowledged for access to Chemical Crystallography and Advanced Research Computing (ARC) facilities.Notes and references
-
E. Gail, S. Gos, R. Kulzer, J. Lorösch, A. Rubo, M. Sauer, R. Kellens, J. Reddy, N. Steier and W. Hasenpusch, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2011, pp. 673–704 Search PubMed
.
- O. Sato, T. Iyoda, A. Fujishima and K. Hashimoto, Science, 1996, 272, 704–705 CrossRef CAS PubMed
- A. Simonov, T. De Baerdemaeker, H. L. B. Boström, M. L. Ríos Gómez, H. J. Gray, D. Chernyshov, A. Bosak, H. B. Bürgi and A. L. Goodwin, Nature, 2020, 578, 256–260 CrossRef CAS PubMed
- L. M. Cao, D. Lu, D. C. Zhong and T. B. Lu, Coord. Chem. Rev., 2020, 407, 1–18 CrossRef
- E. S. Koumousi, I. R. Jeon, Q. Gao, P. Dechambenoit, D. N. Woodruff, P. Merzeau, L. Buisson, X. Jia, D. Li, F. Volatron, C. Mathonière and R. Clérac, J. Am. Chem. Soc., 2014, 136, 15461–15464 CrossRef CAS
- J. A. Valdez-Moreira, A. E. Thorarinsdottir, J. A. Degayner, S. A. Lutz, C. H. Chen, Y. Losovyj, M. Pink, T. D. Harris and J. M. Smith, J. Am. Chem. Soc., 2019, 141, 17092–17097 CrossRef CAS PubMed
- M. Lorenzi, J. Gellett, A. Zamader, M. Senger, Z. Duan, P. Rodríguez-Maciá and G. Berggren, Chem. Sci., 2022, 13, 11058–11064 RSC
- H. Jun, V. G. Young and R. J. Angelici, J. Am. Chem. Soc., 1992, 114, 10064–10065 CrossRef CAS
- M. Finze, E. Bernhardt, H. Willner and C. W. Lehmann, Angew. Chem., Int. Ed., 2004, 43, 4160–4163 CrossRef CAS
- J. G. Cordaro, D. Stein, H. Rüegger and H. Grützmacher, Angew. Chem., Int. Ed., 2006, 45, 6159–6162 CrossRef CAS
- M. C. Levis, K. G. Pearce and I. R. Crossley, Inorg. Chem., 2019, 58, 14800–14807 CrossRef CAS
- T. Görlich, D. S. Frost, N. Boback, N. T. Coles, B. Dittrich, P. Müller, W. D. Jones and C. Müller, J. Am. Chem. Soc., 2021, 143, 19365–19373 CrossRef PubMed
- D. L. Lichtenberger, S. K. Renshaw and R. M. Bullock, J. Am. Chem. Soc., 1993, 115, 3276–3285 CrossRef CAS
- J. E. McGrady, T. Lovell, R. Stranger and M. G. Humphrey, Organometallics, 1997, 16, 4004–4011 CrossRef CAS
- S. K. Furfari, M. C. Leech, N. Trathen, M. C. Levis and I. R. Crossley, Dalton Trans., 2019, 48, 8131–8143 RSC
- D. W. N. Wilson, S. J. Urwin, E. S. Yang and J. M. Goicoechea, J. Am. Chem. Soc., 2021, 143, 10367–10373 CrossRef CAS PubMed
- E. S. Yang and J. M. Goicoechea, Angew. Chem., Int. Ed., 2022, 61, e202206783 CAS
- M. L. H. Green and G. Parkin, J. Chem. Educ., 2014, 91, 807–816 CrossRef CAS
- H. V. Huynh, Y. Han, R. Jothibasu and J. A. Yang, Organometallics, 2009, 28, 5395–5404 CrossRef CAS
- P. de Frémont, N. Marion and S. P. Nolan, J. Organomet. Chem., 2009, 694, 551–560 CrossRef
- S. Gaillard, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2010, 46, 2742–2744 RSC
- C. M. Zinser, F. Nahra, L. Falivene, M. Brill, D. B. Cordes, A. M. Z. Slawin, L. Cavallo, C. S. J. Cazin and S. P. Nolan, Chem. Commun., 2019, 55, 6799–6802 RSC
- E. Y. Tsui, P. Müller and J. P. Sadighi, Angew. Chem., Int. Ed., 2008, 47, 8937–8940 CrossRef CAS PubMed
- V. J. Scott, J. A. Labinger and J. E. Bercaw, Organometallics, 2010, 29, 4090–4096 CrossRef CAS
- M. R. Fructos, T. R. Belderrain, P. de Frémont, N. M. Scott, S. P. Nolan, M. Mar Díaz-Requejo and P. J. Pérez, Angew. Chem., Int. Ed., 2005, 44, 5284–5288 CrossRef CAS PubMed
- C. Dash, P. Kroll, M. Yousufuddin and H. V. R. Dias, Chem. Commun., 2011, 47, 4478–4480 RSC
- L. Canovese, F. Visentin, C. Levi and V. Bertolasi, Organometallics, 2011, 30, 875–883 CrossRef CAS
- S. Gaillard, P. Nun, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2010, 29, 5402–5408 CrossRef CAS
- C. Boehme and G. Frenking, Organometallics, 1998, 17, 5801–5809 CrossRef CAS
- D. Marchione, L. Belpassi, G. Bistoni, A. Macchioni, F. Tarantelli and D. Zuccaccia, Organometallics, 2014, 33, 4200–4208 CrossRef CAS
- C. Römelt, T. Weyhermüller and K. Wieghardt, Coord. Chem. Rev., 2019, 380, 287–317 CrossRef
- M. J. Humphries, K. P. Tellmann, V. C. Gibson, A. J. P. White and D. J. Williams, Organometallics, 2005, 24, 2039–2050 CrossRef CAS
- V. C. Gibson, M. J. Humphries, K. P. Tellmann, D. F. Wass, A. J. P. White and D. J. Williams, Chem. Commun., 2001, 1, 2252–2253 RSC
- Q. Knijnenburg, D. Hetterscheid, T. Martijn Kooistra and P. H. M. Budzelaar, Eur. J. Inorg. Chem., 2004, 2004, 1204–1211 CrossRef
- Q. Knijnenburg, A. D. Horton, H. Van Der Heijden, T. M. Kooistra, D. G. H. Hetterscheid, J. M. M. Smits, B. De Bruin, P. H. M. Budzelaar and A. W. Gal, J. Mol. Catal. A: Chem., 2005, 232, 151–159 CrossRef CAS
- S. Nückel and P. Burger, Organometallics, 2001, 20, 4345–4359 CrossRef
- E. L. Dias, M. Brookhart and P. S. White, Organometallics, 2000, 19, 4995–5004 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2242900–2242911. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01126g |
| ‡ See ESI for full experimental details, analytical and computational data. |
| This journal is © The Royal Society of Chemistry 2023 |